

Abstract
Bacteriolytic anti-cancer therapies employ attenuated bacterial strains that selectively proliferate within tumors. Clostridium novyi-NT spores represent one of the most promising of these agents, as they generate potent anti-tumor effects in experimental animals. We have determined the 2.55-Mb genomic sequence of C. novyi-NT, identifying a new type of transposition and 139 genes that do not have homologs in other bacteria. The genomic sequence was used to facilitate the detection of transcripts expressed at various stages of the life cycle of this bacterium in vitro as well as in infections of tumors in vivo. Through this analysis, we found that C. novyi-NT spores contained mRNA and that the spore transcripts were distinct from those in vegetative forms of the bacterium.
It has been known for over a century that severe bacterial infections in cancer patients occasionally result in the eradication of malignancy1. These clinical observations spawned attempts to use bacteria to treat tumors in laboratory animals and in cancer patients2–4. Anaerobic bacteria are particularly intriguing agents for this purpose, as the only tissues allowing the growth of such bacteria in otherwise healthy mammals are within tumors5. Indeed, a majority of human tumors contain large hypoxic regions that make them relatively insensitive to radiation or chemotherapy but provide an ideal environment for the growth of anaerobic bacteria6,7. Most previous attempts to use anaerobic bacteria for tumor therapeutics employed Clostridium sporogenes, a nonpathogenic species often used as a control for sterilization in the food industry. We developed an attenuated strain of the pathogenic clostridial species C. novyi, called C. novyi-NT, in which the phage episome containing the major systemic toxin gene was deleted8. When injected intravenously, C. novyi-NT spores produced substantial anti-tumor effects in experimental animals without excessive toxicity8–11. The spores of C. novyi-NT are very stable but the vegetative form is exquisitely sensitive to oxygen11.
C. novyi-NT lies on a distinct branch of the genus Clostridium and no previous sequencing studies of it have been published. Although a considerable number of clostridial species are of medical and biotechnological importance, only four (C. acetobutylicum12, C. difficile13, C. perfringens14 and C. tetani15) have been analyzed by complete genomic sequencing. Additionally, the genome of C. botulinum is currently under annotation (http://www.sanger.ac.uk/Projects/C_botulinum/). As judged by 16S RNA sequences, C. novyi is not highly related to any of these sequenced organisms. To better understand how clostridia function as anti-neoplastic agents, as well as to gain insight into the pathology of human clostridial diseases, we have determined the sequence of the C. novyi-NT genome and analyzed the transcriptomes of its vegetative and spore forms.
RESULTS
C. novyi-NT genome
The C. novyi-NT genome consists of a single circular chromosome, 2,547,720 base pairs (bp) in length with a G+C content of 28.9% (Fig. 1 and Table 1). Deviant G+C content and trinucleotide composition were almost completely confined to the regions that harbored the rRNA operons (Fig. 1). No extrachromosomal sequences, such as those that would be found in plasmids or phages, were identified. The general features of clostridial genomes are summarized in Table 1. The C. novyi-NT genome is smaller and contains fewer coding sequences (CDS) than the other genomes. Although atypical G+C content is tightly associated with the rRNA operons, several putative mobile elements were identified in the C. novyi-NT genome, including transposons, clustered regularly interspaced short palindromic repeats (CRISPR) and prophage elements (Table 2 and Supplementary Tables 1 and 2 online). A putative replication origin of the C. novyi-NT genome was indicated by a distinct inflection point in the coding strand, which was localized around the DnaA gene (Fig. 1, dat the top of the genome circle). Closer inspection of this region revealed multiple putative DnaA boxes (TTATCCACA) on both strands in intergenic regions between the dnaA (NT01CX0867) and rpmH (NT01CX0868) genes as well as between the dnaA and dnaN (NT01CX0866) genes. This distribution pattern of the DnaA boxes was consistent with that described for other Gram-positive bacteria16. Another inflection point in the coding strand marked a putative replication terminus located on the opposite side of the circular genome, as expected if the C. novyi-NT chromosome replicated in a bidirectional manner.
Figure 1.
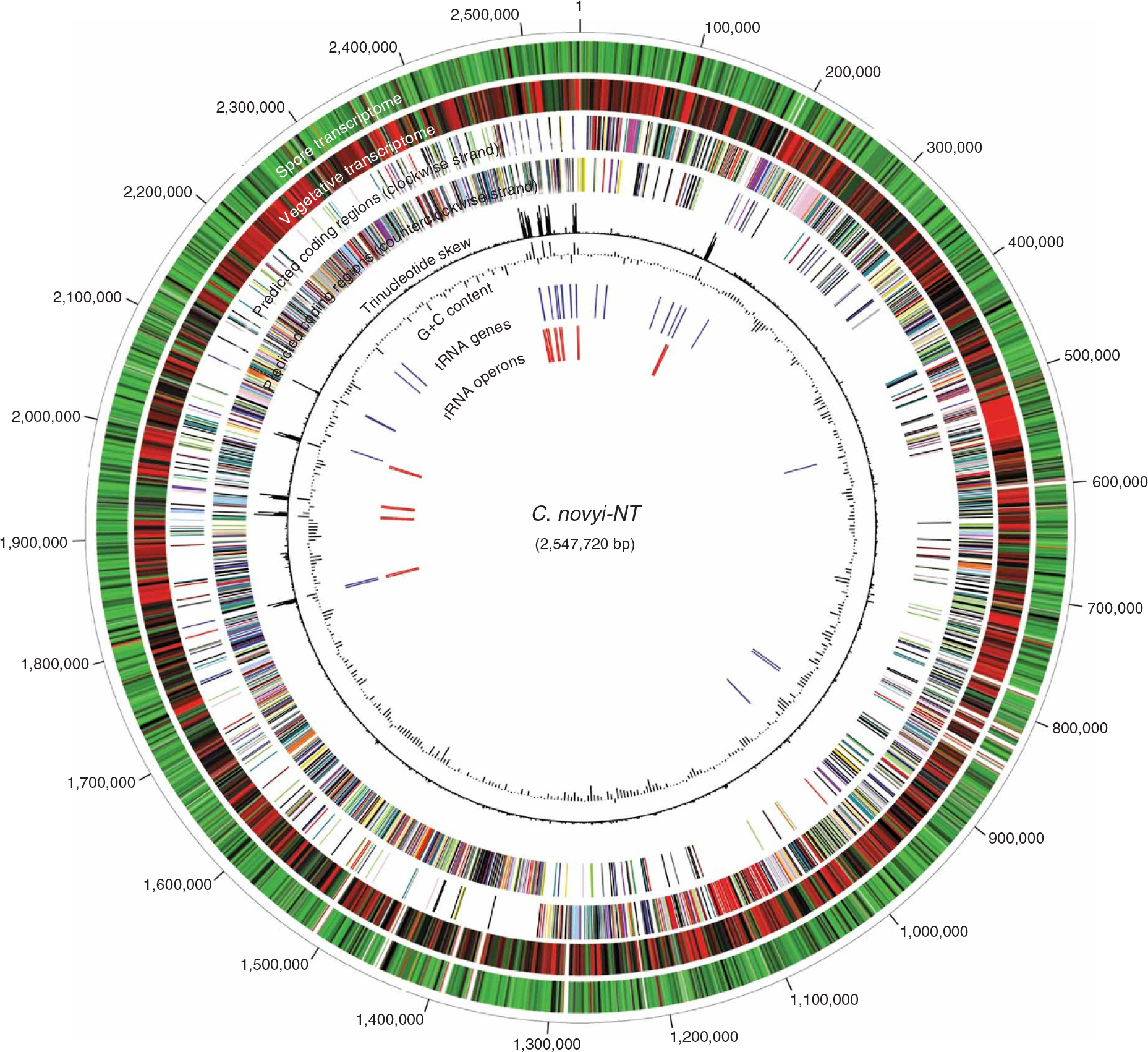
Circular representation of C. novyi-NT genome and transcriptomes. Predicted CDS are color coded based on functional classification. CDS in the transcriptome circles are also color coded, with red representing the highest mRNA abundance, green the lowest and white the intergenic regions. The G+C content and trinucleotide skew were determined using 2,000-nt windows with 1,000-nt incremental shifts.
Table 1.
General features of Clostridia senomesa
| C. novb | C. aceb,c | C. difb | C. perb,c | C. tetb | |
|---|---|---|---|---|---|
| Size (bp) | 2,547,720 | 4,132,880 | 4,290,252 | 3,085,740 | 2,799,251 |
| G+C content (%) | 28.9 | 30.9 | 29.1 | 28.5 | 28.7 |
| Average CDS length (bp) | 952 | 841 | 943 | 935 | 886 |
| Total number of CDS | 2,325 | 4,273 | 3,776 | 2,749 | 2,784 |
| Number of tRNA genes | 81 | 73 | 87 | 96 | 54 |
| Number of rRNA operons | 10 | 11 | 11 | 10 | 6 |
Based on the TIGR annotations (http://cmr.tigr.org/tigr-scripts/CMR/shared/Genomes.cgi) of the indicated genomes with the exception of C. difficile, which was annotated using Artemis software.
C. nov, C. novyi-NT; C. ace, C. acetobutylicum ATCC824; C. dif, C. difficile str. 630; C. per, C. perfringens str. 13; C. tet, C. tetani E88.
Data were analyzed for both genome and plasmid sequences.
Table 2.
Target sites of C. novyi-NT insertion sequence elements
| Gene ID | 5′ Surrounding sequencea | ISE terminal inverted repeat | 3′ Surrounding sequenceb |
|---|---|---|---|
| Clostridium novyi-NT | |||
| NT01CX0036 | agagtaaagttaaactttactct | cag … … … … … … … … … … … … … ctg | agagtaaagttaaactttactct |
| NT01CX0169 | aatataaaataaaaatgattataattattaattgtaatcatttttattttatatt | cag … … … … … … … … … … … … … ctg | aatataaaataaaaatgattataattattaattgtaatcatttttattttatatt |
| NT01CX0268 | aaaccaccctttaggtggttt | cag … … … … … … … … … … … … … ctg | aaaccaccctttaggtggttt |
| NT01CX0396 | atgaaatcacctctacattattgtgtagaggtgatttca t | cag … … … … … … … … … … … … … agg | atgaaatcacctctacattattgtgtagaggtgatttcat |
| NT01CX0448 | taacagcctagtttttaggctgttt | cag … … … … … … … … … … … … … ctg | taacagcctagtttttaggctgttt |
| NT01CX0499 | agtcagtaatcataatgattactgact | cag … … … … … … … … … … … … … ctg | agtcagtaatcataatgattactgact |
| NT01CX0691 | agctaccttaaataaggtggct | cag … … … … … … … … … … … … … ctg | agctaccttaaataaggtggct |
| NT01CX0704/0705 | agaaactattagaaaaatctaatagtttct | cag … … … … … … … … … … … … … ttt | agaaaccattagaaaaatctaatagtttct |
| NT01CX0769 | aaacttcctagcatataggaagttt | cag … … … … … … … … … … … … … ctg | aaacttcctagcatataggaagttt |
| NT01CX0938c | atttcatttgaataagattaaagccttattttataaggct | cag … … … … … … … … … … … … … ctg | gggagatactttttgtatctccttttaatttttctaaata |
| NT01CX0971/0972 | aataaaataaacctcattcacttagtgaatgaggtttattttata | cag … … … … … … … … … … … … … ctg | aataaaataaacctcattcacttagtgaatgaggtttattttata |
| NT01CX1212 | agggtaagctttaataaaaagcttaccct | cag … … … … … … … … … … … … … ctg | agggtaagctttaataaaaagcttaccct |
| NT01CX1343 | agcttctttaatctaaagaagct | cag … … … … … … … … … … … … … ctg | agcttctttaatctaaagaagct |
| NT01CX1370 | aataaaacctacaaccataaaacatggttgtaggttttact | cag … … … … … … … … … … … … … ctg | aataaaacctacaaccataaaacatggttgtaggttttact |
| NT01CX1406 | actagaatttatcaataattctagt | cag … … … … … … … … … … … … … ctg | actagaatttatcaataattctagt |
| NT01CX1460 | agaaaatagctttaaagctattttct | cag … … … … … … … … … … … … … ctg | agaaaatagctttaaagctattttct |
| NT01CX1493 | aaagcagagcctacgctctgcttt | cag … … … … … … … … … … … … … ctg | aaagcagagcctacgctctgcttt |
| NT01CX1539 | agaacattcatccaagaatgttct | cag … … … … … … … … … … … … … ctg | agaacattcatccaagaatgttct |
| NT01CX1577 | aggtaatccatt tcaaat aatggattacct | cag … … … … … … … … … … … … … ctg | aggtaatccatttcaaataatggattacct |
| NT01CX1666/1667 | actagaattatttttttataattctagt | cag … … … … … … … … … … … … … ctg | actagaattatttttttataattctagt |
| NT01CX1930 | aacgttgacaaaatgtcaacgtt | cag … … … … … … … … … … … … … ctg | aacgttgacaaaatgtcaacgtt |
| NT01CX1968 | agaaatatctatttaagatatttct | cag … … … … … … … … … … … … … ctg | agaaatatctatttaagatatttct |
| NT01CX2015 | agaaaaagtatagcaataaaatgctatactttttct | cag … … … … … … … … … … … … … ctg | agaaaaagtatagcaataaaatgctatactttttct |
| NT01CX2038 | aaaatataagtgttataaaaataacactt | cag … … … … … … … … … … … … … ctg | aaaatataagtgttataaaaataacactt |
| NT01CX2055 | agctttcactatgtgaaagct | cag … … … … … … … … … … … … … ctg | agctttcactatgtgaaagct |
| NT01CX2090 | acatggaattaatccatgt | cag … … … … … … … … … … … … … ctg | acatggaattaatccatgt |
| NT01CX2331 | agagtatttcaattaaatactct | cag … … … … … … … … … … … … … ctg | agagtatttcaattaaatactct |
| Burkholderia pseudomallei K96243 (NC_006350) d | |||
| BPSL1910 | agccgcccgagggcggct | cagattgctgacaaaccc … … gggttcgtcagcagtctg | agccgcccgagggcggct |
| BPSL2296 | aagccccgcgaatgcggggctt | cagactgctgacgaaccc … … gggtttgtcagcaatctg | aagccccgcgaatgcggggctt |
| Thermoanaerobacter tengcongensis MB4 (AE013051) d | |||
| TTE0846 | agctagagaggtcttctctagct | cagactgttgacaaa … … … … tttgtcaacaaactg | agctagagaggtcttctctagct |
| TTE0865 | agagggattagaattttaatccctct | cagactgttgacaaa … … … … tttgtcaacaaactg | agagggattagaattttaatccctct |
| Streptococcus pyogenes (CP000003; NC_003485) d | |||
| M6_Spy0973 | aaatgagtagtcaactgactactcattt | cagactgaagacaaa … … … … …tttgtcttcaatctg | aaatgagtagtcaattgactactcattt |
| spyM18_0536 | aagccacccgttttcacgggtgcttt | cagactgaagacaaa … … … … …tttgtcttcaatctg | aagccacccgttttcacgggtggttt |
DNA sequences adjacent to the 5′ end of the ISE, with complementary nucleotides in the inverted repeats bolded and italicized.
DNA sequences adjacent to the 3′ end of the ISE, with complementary nucleotides in the inverted repeats bolded and italicized.
No inverted repeat was found surrounding this ISE and the insertion site for this ISE was not duplicated.
Examples of other genomes that carry the ISE.
Coding sequences
A total of 2,325 CDS were predicted from the genomic sequence, with an average length of 952 base pairs. We assigned putative functions to 1,620 (70%) of the CDS. Of the remaining 705 CDS, 566 (24%) showed similarity to CDS or hypothetical proteins of unknown function annotated in other genomes, whereas 139 (6%) had no substantial similarity to other CDS (Supplementary Table 3 online). Several multicopy CDS were found in the C. novyi-NT genome, most notably a 27-copy insertion sequence element (ISE; Table 2). Other multicopy genes included nine copies encoding a conserved hypothetical protein (NT01CX0251, NT01CX0272, NT01CX0341, NT01CX0774, NT01CX0782, NT01CX0799, NT01CX0816, NT01CX0858, NT01CX1052) and two copies encoding each of the following proteins: 3-hydroxybutyryl-coA dehydrogenase (NT01CX0470 and NT01CX0604), glycerol uptake facilitator protein (NT01CX0606 and NT01CX0609), electron transfer flavoprotein betasubunit (NT01CX0472 and NT01CX2323), translation elongation factor Tu (NT01CX1100 and NT01CX1113) and a hypothetical protein (NT01CX0106 and NT01CX0114).
Although 704 C. novyi-NT CDS were shared with all the other sequenced clostridial genomes, 551 were present only in C. novyi-NT (Supplementary Table 4 online). These 551 included the above-noted 139 genes that had no homologs in any prokaryote. When the CDS were classified by putative function, it was apparent that most CDS involved in the biosynthesis of proteins and nucleic acids were shared by all analyzed clostridia. Seventy percent (110/157) of the CDS involved in protein synthesis were shared by all five genomes, whereas only 3.2% (5/157) were C. novyi-NT specific. Similarly, 59% (36/61) of the CDS involved in base, nucleoside and nucleotide metabolisms were found in all five genomes whereas only 1.6% (1/61) were unique to C. novyi-NT. Conversely, hypothetical CDS and those in mobile elements dominated the CDS specific to C. novyi-NT. Those classified in the category of cell envelope and the category of transport and binding proteins also seemed to be enriched in the C. novyi-NT–specific CDS (Supplementary Table 4 online).
Palindrome-specific transposition
Twenty-seven copies of an ISE17 were identified in the C. novyi-NT genome. These ISEs encoded an enzyme homologous to a family of transposases found in a variety of bacterial species (Supplementary Table 5 online). The most interesting aspect of the C. novyi-NT ISEs was that their 27 insertion sites did not share any sequence similarities. Instead, all but one were composed of inverted repeats varying from 7 to 27 bp in size (Table 2). Though not previously recognized, we found that similar insertion sites were present in other bacterial genomes harboring the ISBma2 family of ISE (examples shown in Table 2). The direct repeats of varied palindromic sequences at the borders of these ISEs suggested that target site duplication was part of the transposition process and that the transposase recognized target sites based on secondary structure rather than recognizing a particular sequence motif. Alternatively, an enzyme that targets cruciform DNA structures might be recruited as part of the transposition machinery. It is intriguing in this regard that one of the insertion sequences, which harbors NT01CX0938, was located next to a resolvase gene (NT01CX0939). This ISE was the only copy whose target site was not duplicated and did not contain a cruciform structure. It is thus tempting to speculate that NT01CX0938 was the ancestor and that the 26 other ISEs distributed throughout the genome were descendants.
Extracellular proteins that may interact with the host
One hundred fifty-three of the proteins were predicted to be either cell-surface associated or secreted (Supplementary Table 1 online). In addition, several proteins were identified that were potentially cytolytic because of their predicted ability to degrade lipids or proteins (Table 3). These proteins were of special interest because of the tumor-lytic properties of C. novyi-NT. One of the lipid-degrading proteins was phospholipase C (NT01CX0979), with 61% identity over 397 amino acids to its homolog in C. perfringens (ABA64009). This relatedness is consistent with previous biochemical studies showing that the C. novyi phospholipase C has similar activities to the C. perfringens phospholipase C18. These activities include hemolysis and the activation of the arachidonic acid cascade, which in turn triggers a series of host inflammatory responses18,19. The C. novyi-NT phospholipase C gene may thereby contribute to the tumor destruction and the induction of host-mediated anti-tumor immunity that result from treatment with C. novyi-NT spores9. Two putative lipases (NT01CX2047 and NT01CX0630) were other potentially interesting lipid-degrading proteins discovered through this analysis. The C. novyi phospholipase C and the two lipases were highly expressed in tumors as well as in vitro, as discussed below.
Table 3.
Extracelluar degradative proteins
| Gene ID | Gene name | mRNA abundancea |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Early log | Mid log | Late log | Spore | HCT116 (infected) |
CT26 (infected) |
HCT116 (uninfected) |
CT26 (uninfected) |
||
| NT01CX0021 | Serine protease, subtilase family | 10,497 | 16,727 | 16,861 | 67 | 161 | 154 | 74 | 47 |
| NT01CX0173 | Protease/transglutaminase | 1,013 | 1,058 | 1,037 | 64 | 45 | 52 | 38 | 47 |
| NT01CX0283 | Zn-dependent peptidase, insulinase family | 303 | 310 | 446 | 102 | 99 | 196 | 137 | 107 |
| NT01CX0478 | Metalloendopeptidase | 2,005 | 1,839 | 1,986 | 365 | 427 | 927 | 440 | 525 |
| NT01CX0560 | Thermolysin metallopeptidase | 318 | 373 | 276 | 223 | 228 | 271 | 128 | 63 |
| NT01CX0630 | Lipase | 4,212 | 7,759 | 14,830 | 61 | 3,187 | 848 | 130 | 101 |
| NT01CX0944 | Serine protease, subtilase family | 786 | 527 | 659 | 134 | 272 | 303 | 121 | 125 |
| NT01CX0979 | Phospholipase C | 7,419 | 28,960 | 29,177 | 140 | 5,793 | 2,191 | 114 | 155 |
| NT01CX1195 | Clostridiopeptidase B | 2,659 | 13,541 | 12,452 | 167 | 451 | 236 | 58 | 92 |
| NT01CX1295 | Collagenase | 942 | 1,556 | 2,589 | 53 | 68 | 77 | 52 | 58 |
| NT01CX1541 | Protease/transglutaminase | 491 | 2,325 | 1,158 | 57 | 43 | 49 | 53 | 57 |
| NT01CX1544 | Protease/transglutaminase | 2,784 | 12,774 | 14,905 | 132 | 844 | 271 | 93 | 140 |
| NT01CX2047 | Lipase | 4,939 | 18,684 | 20,366 | 108 | 8,047 | 3,040 | 59 | 81 |
Determined by microarray analysis; values normalized across arrays as described in Methods.
The transcriptomes of growing bacteria
With the C. novyi-NT genome in hand, we were able to evaluate transcription patterns of cells at various stages in their life cycle. Custom oligonucleotide arrays representing all 2,325 predicted CDS were constructed and hybridized with labeled cDNA prepared from bacteria at early-, mid- and late-log growth phases. It was apparent that many transcripts were regulated in growth phase–specific patterns (Supplementary Table 1 online), among which 259 (11% of all CDS) were enriched in a single growth phase by at least twofold (examples in Fig. 2). We categorized these growth phase–specific genes by their functions (Supplementary Tables 6 and 7 online). Several genes involved in energy metabolism and biosynthesis of cofactors, such as vitamins, were preferentially expressed in the early-log growth phase. Notably, the expression of several genes responsible for the biosynthesis or transport of amino acids and other precursor molecules was upregulated in mid-log phase. These differences presumably reflect the evolutionary development of the most efficient strategy to maximize growth. In this regard, it was intriguing that 20 conserved genes of unknown function were preferentially expressed in mid-log phase, suggesting a previously unrecognized component of growth regulation that may be widespread among bacteria. There were only seven genes that were preferentially expressed in late-log phase. These included one signaling molecule, one transcription factor, one hypothetical protein and four enzymes involved in energy metabolism. The transcription factor, an extracytoplasmic function sigma-70 factor (NT01CX0693), was transcribed early, disappeared in mid-log phase and was expressed again in late-log phase at higher levels. The late-log phase hypothetical gene (NT01CX0692) showed precisely the same expression pattern and was apparently organized in the same operon with NT01CX0693, as these two adjacent genes were oriented in the same direction but opposite to that of the flanking genes.
Figure 2.
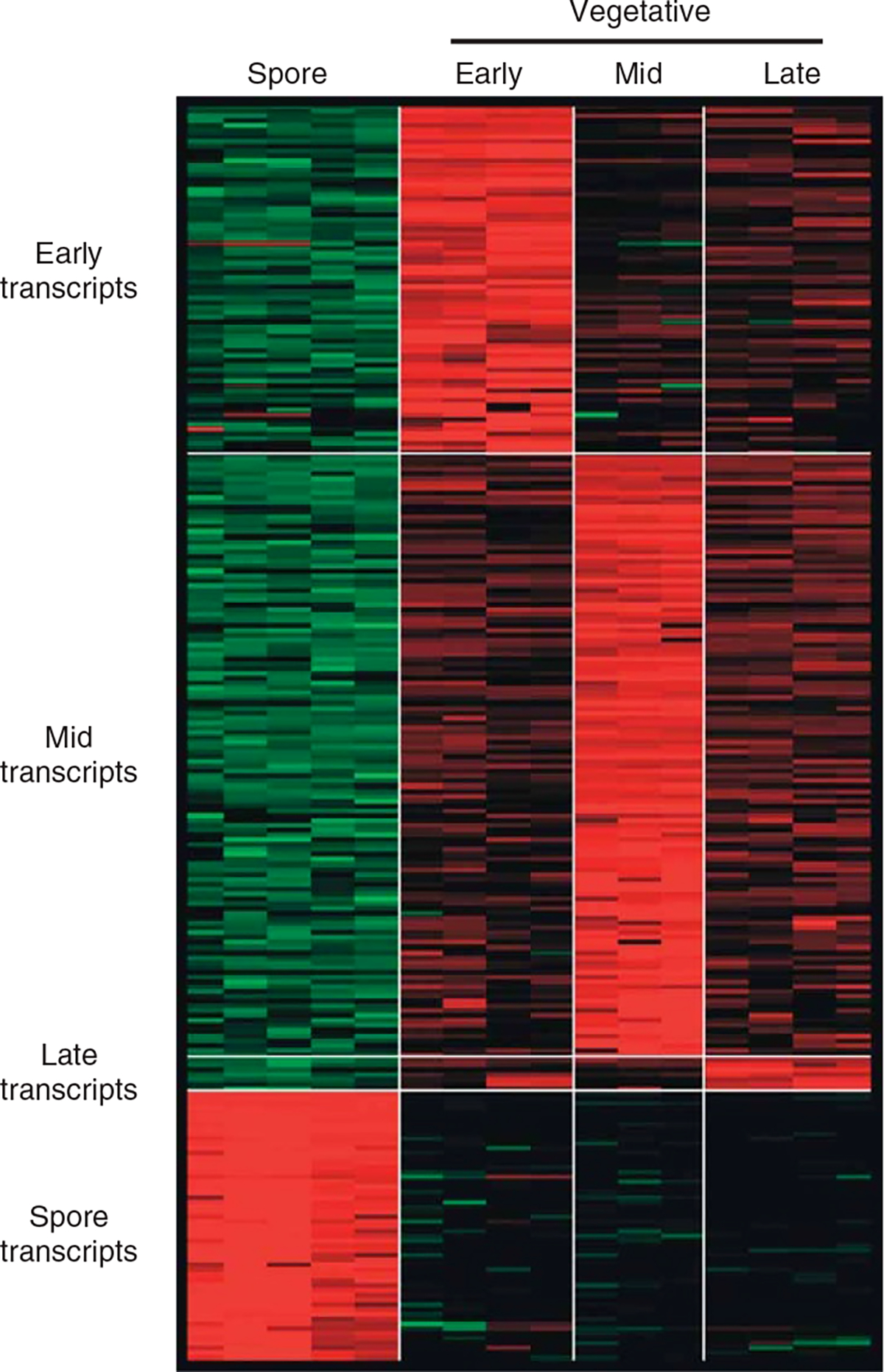
Transcriptomes of C. novyi-NT in various growth states. Each column represents a different microarray experiment performed with RNA from spores or vegetative bacteria growing at early-, mid-, or late-log phase. Each row represents a gene that was differentially expressed in the indicated phase. Red and green indicate high and low relative mRNA abundance, respectively. This figure shows only genes expressed in a growth phase at levels at least twofold higher than any of the other growth phases; these genes are described in Supplementary Table 7 online.
The transcriptome of spores
Though some early studies suggested that bacterial spores contain mRNA20,21, it is generally believed that they do not22. This explains why there have been no attempts to identify the specific transcripts present in the mature spores of any bacterial species, though several studies have assessed the RNA in sporulating bacteria22–26. To definitively address whether C. novyi-NT spores contained mRNA, we prepared spores that had matured in sporulation medium for >14 d, long past the active growth phase. We then rigorously purified the spores through consecutive Percoll gradients to remove any vegetative or sporulating bacteria. Phase contrast microscopy as well as cytochemical staining demonstrated that spore purity was >99.9%. To prepare RNA from the purified spores, unusually harsh treatments had to be used.
Using these procedures, we first found that the ribosomal RNA (rRNA) species of spores and vegetative bacteria were different. A new rRNA species, slightly smaller than the normal 23S rRNA, was found in the spores (Supplementary Fig. 1a online). Mapping experiments showed that this transcript resulted from a deletion of ~300 nt in the 5′ region of the normal 23S rRNA. Specifically fragmented rRNA has been detected in other bacteria, but has never been described to be associated with specific processes such as sporulation27. Stimulated by this observation, we determined whether a similar species of rRNA could be detected in other spores and found that it was indeed present in the spores of two strains of Bacillus subtilis (Supplementary Fig. 1a online).
In addition to the spore-specific rRNA, there were striking differences in the mRNA species of spores compared to that in growing cells. (A pictorial representation of these data is presented as the two outer circles in Fig. 1 and complete data are provided in Supplementary Table 1 online.) The 50 transcripts most abundant in any stage of growing bacteria were usually involved in protein synthesis (ribosomal proteins or translation factors; Supplementary Table 8 online). In striking contrast, there was no overlap between these genes and those found to be most abundant in spores. Instead, 60% of the spore transcripts had no known function, and many of the others were predicted to encode proteins with redox activity (Supplementary Table 8 online). The differential expression observed in the microarrays was confirmed by conventional or real-time RT-PCR in 58 of 59 genes evaluated (examples in Supplementary Fig. 2 online).
Genes with redox activity were especially interesting in that C. novyi-NT spores are stable at ambient oxygen concentrations for years, whereas vegetative cells cannot survive exposure to even minute concentrations of oxygen11. This behavior is essential for the tumor-specific effects of C. novyi-NT, as it eliminates the possibility of spores germinating in normal tissues. Redox genes whose transcripts were present at relatively high levels in spores included those encoding a thioredoxin reductase (NT01CX2374), a glutaredoxin (NT01CX2375), a glutathione peroxidase (NT01CX2376) and a rubredoxin (NT01CX1169). Glutathione peroxidases have been implicated as major scavengers for hydrogen peroxide as well as for a variety of organic hydroperoxides in other organisms28,29. The C. novyi-NT glutathione peroxidase gene (NT01CX2376) was organized into an operon containing an NADPH thioredoxin reductase (NT01CX2374) and a glutaredoxin-like protein (NT01CX2375). NT01CX2374 is likely to reduce NT01CX2375 using NADPH as electron donor, whereas the reduced NT01CX2375 may serve as an electron donor for the glutathione peroxidase in the same redox chain. Notably, mRNAs transcribed from this operon were present at relatively high levels in spores but were barely detectable in vegetative cells (Supplementary Table 1 and Supplementary Fig. 2 online). The redox-related transcripts preferentially present in spores were a specific subset of the total redox-related proteins. Other redox-related transcripts were present in vegetative cells but not detectable in spores (e.g., a Fe/Mn superoxide dismutase (NT01CX0194), two ferredoxins (NT01CX0589 and NT01CX0756), a flavodoxin (NT01CX1654), a flavodoxin oxidoreductase (NT01CX1854) and a thioredoxin (NT01CX2304); Supplementary Table 1 and Supplementary Fig. 2 online).
Bacterial transcriptomes in experimental infections
To determine whether the patterns of gene expression in vitro differed from those resulting from infection of tumors in vivo, we purified RNA from two infected tumor models—human HCT116 colorectal cancer xenografts growing in nude mice and murine CT26 colorectal cancers growing in syngeneic BALB/c mice. C. novyi-NT spores were intravenously injected into mice bearing subcutaneous tumors and the RNA isolated 18 h later, just after germination became evident. When separated by electrophoresis, the 23S, 16S bacterial rRNA and the spore-specific truncated 23S rRNA were observable, whereas the 28S mammalian rRNA was partially degraded, presumably as a result of C. novyi-NT–mediated tumor destruction (Supplementary Fig. 1b online). The cDNA generated from infected-tumor RNA was labeled and hybridized to the C. novyi-NT microarrays described above. To control for cross hybridization with mammalian mRNAs, we extracted total RNA from uninfected tumors and hybridized it to parallel arrays. The hybridizations revealed that the bacterial mRNAs in the infected tumors were derived from both vegetative bacteria (e.g., those encoding ribosomal proteins) and spores (e.g., those encoding small acid-soluble spore proteins) (Supplementary Tables 1 and 8 online). This was consistent with the presence of the spore-specific rRNA in tumors (Supplementary Fig. 1b online) and with previous microbiological observations indicating that sporulation occurred in the tumor microenvironment11. These results suggested that germination and sporulation are two arms of a dynamic process in tumors in vivo, reflecting a continuous struggle between the bacteria and their host.
Though most transcripts found in tumors in vivo were present at levels roughly similar to those found in either spores or growing bacteria in vitro, there were several bacterial transcripts that were expressed at much higher levels in both the CT26 and HCT116 tumors than in vitro (Supplementary Tables 1 and 8 online). These included transcripts of three genes involved in spore coat formation or degradation (NT01CX0987, NT01CX0986 and NT01CX1338), two involved in glycerol metabolism (glycerol kinase, NT01CX0605 and glycerol uptake facilitator protein, NT01CX0606), and three of unknown function (NT01CX0988, NT01CX1634, NT01CX2395). We also noted that a number of genes involved in fatty acid and lipid metabolism (NT01CX0470, NT01CX0471, NT01CX0472, NT01CX0473, NT01CX0474, NT01CX0538, NT01CX0603, NT01CX0604, NT01CX0605, NT01CX0606, NT01CX0608, NT01CX0630 and NT01CX2047) were among the most abundant transcripts in tumors in vivo. The copious amounts of biological membranes and plasma exudates in infected tissues represent a rich carbon/energy source for C. novyi-NT. It is known that bacteria can use fatty acids and glycerol as carbon and energy sources and the enzymes in the relevant pathways can be induced when these substrates are present in culture media30,31.
DISCUSSION
The completion of the C. novyi-NT genome will facilitate understanding of the biology of C. novyi-NT and related species32–34. Bacteriolytic therapy with C. novyi-NT has emerged as a promising approach to treat solid tumors8–11,35, though the mechanisms involved in tumor destruction by such bacteria have not been explored. The combined genomic/transcriptomic analysis reported herein allowed us to identify genes that may be important for tumor lysis. Several genes encoding extracellular proteins were found to be expressed at relatively high levels in C. novyi-NT–infected tumors (Table 3 and Supplementary Table 8 online). Most notable among these were three lipid-degrading proteins—a phospholipase C (NT01CX0979) and two lipases (NT01CX2047 and NT01CX0630). Although phospholipase C is well known for its hemolytic activity, lipases are usually not considered cytolytic, as their natural substrates are long-chain triacylglycerides36. However, we have recently characterized one of the C. novyi lipases (NT01CX2047) and shown that in addition to its expected lipase activity, it was able to alter the structure of lipid bilayers, change membrane permeability and thereby be potentially cytotoxic51. Therefore, lipid-degradative proteins may be major players in the potent antitumor effect of C. novyi-NT. Additionally, the ability of phospholipases to activate inflammatory responses18,19,37 and induce anti-tumor immunity could have a major effect on the battle between host and bacteria9. A related group of genes highly expressed in the infected tumors encoded enzymes involved in fatty acid and lipid metabolism (Supplementary Table 8 online). These gene expression profiles may reflect the adaptation of the bacteria to the membrane-rich environment at the infection site. Spore-specific transcripts were also highly represented in the transcriptomes of infected tumors, suggesting extensive sporulation as a result of the struggle within a hostile host environment. These data provide insights into the processes affecting the success of and toxicity associated with intentional or accidental infections by clostridia.
One hundred and thirty-nine hypothetical CDS with no homologs to those in other organisms and no known function (Supplementary Tables 1 and 3 online) were identified in the C. novyi-NT genome. Though the predictive algorithms used to define hypothetical CDS can sometimes be misleading, our approach provided experimental evidence that validated a major fraction of these genes. Among the 139 hypothetical genes, 113 were shown to be expressed at substantial levels under at least one growth condition. As we weren’t able to capture all growth conditions or developmental stages, 113 represents an underestimate of the number of hypothetical genes that are transcribed. Furthermore, the combined genomic/transcriptomic approach provided some insights into these unique C. novyi genes as well as conserved genes that lack known function. One example involves the late-log phase genes NT01CX0692 and NT01CX0693, mentioned previously. Another involves the cotJ operon. NT01CX0988 is a hypothetical CDS coexpressed with NT01CX0986 (cotJC) and NT01CX0987 (cotJB) at high levels only in experimental infections (Supplementary Tables 1 and 8 online). These three CDS are colocalized and arranged in the same orientation, opposite to that of flanking CDS. Therefore, these genes appear to be organized in an operon and NT01CX0988 is likely to be involved in spore coat assembly.
In addition to the many novel genes, the studies described above document a novel form of transposition. Unlike conventional transposition38, the C. novyi ISE transposition apparently involves an endonuclease activity that recognizes the stem-loop structure formed at palindromic sequences and cleaves at its root. The C. novyi ISEs then insert specifically into these cruciform structures. On this basis, we named the ISEs ‘crucitrons’. As rho-independent transcriptional termination in bacteria also involves stem-loop structures, it is conceivable that crucitrons preferentially integrate into these sites. A potential biological advantage for such a preference would be that the insertion event would not disrupt an essential host gene. In fact, the insertion would not even disrupt the transcriptional termination site, as these sites are perfectly duplicated during transposition. Consequently, crucitrons would be flanked on both sides by transcriptional terminators and would be insulated from the expression of the surrounding host genes. Crucitrons may provide a unique mechanism to ensure that transposition neither threatens the host nor is influenced by host transcription.
One of the most interesting observations made in the current study was that spores contain RNA transcripts, many of which were highly abundant. Spores are among the most resilient cells on earth; they can lie dormant for millions of years and do not actively express either RNA or protein39,40. Accordingly, it is generally thought that spores do not contain mRNA. Our studies show that this assumption is incorrect and, moreover, that the transcripts in spores are strikingly different from those present in growing cells. The role of these mRNAs is not yet clear. One possibility is that they are simply trapped within the 7 spore during the final stages of sporulation. Any mRNAs so trapped must be tightly bound within the spore itself rather than in the outer layer (exosporium), because removal of the exosporium with high concentrations of urea plus dithiothreitol (DTT) did not alter the mRNA composition of spores (Supplementary Fig. 3 online). Furthermore, the mRNA composition found in spores is not that predicted if the transcripts were assumed to be regulated by forespore (part of the bacterial cell that will eventually become the core of an endospore) transcription factors (Supplementary Table 9 online). It is also notable that the spore transcripts were very stable, as the mRNA composition was retained after incubation of spores at 37 °C for 14 d followed by storage at 4 °C for a year. In light of these features, it is tempting to postulate that some of the spore mRNAs may have a functional role. Specifically, we speculate that the mRNA stored in spores could allow them to rapidly synthesize proteins that are required for successful germination. This hypothesis would explain why spore mRNA is enriched for enzymes that can detoxify reactive oxygen species, as these are undoubtedly encountered during the germination process22. These proposed functions are speculative, however, and only genetic disruption of such genes can provide unequivocal definition of their function. Finally, we note that the majority of the most abundant spore mRNAs have no known homologs; studying them may provide new insights into the mechanisms underlying the remarkable resilience of these peculiar cellular forms.
METHODS
Bacterial culture.
C. novyi-NT culture and spore preparation were performed essentially as described8. When bacteria in defined growth phases were required, overnight cultures were diluted into fresh deoxygenated culture medium and anaerobic incubation continued until bacteria entered early-(OD600, 0.2), mid- (OD600, 0.6) or late- (OD600, 0.8) log phases. For spore preparation, the bacteria were cultured in sporulation medium for at least 2 weeks to ensure maximum yield of mature spores. Mature spores were purified through two consecutive continuous Percoll gradients followed by four washes and resuspensions in PBS. Purity of the spore preparations was determined to be >99.9% using phase contrast microscopy as well as by light microscopy after staining with malachite green and eosin Y following the procedure described in http://pb.merck.de/servlet/PB/show/1235100/115942en.pdf. The denuded spores were prepared by suspending spores in 8 M Urea and 20 mM DTT. They were then vigorously mixed by vortexing for 15 s, incubated at ~22 °C for 20 min and washed twice with PBS. The two B. subtilis strains were cultured and sporulated in Difco Nutrient Broth (BD). Mature spores were purified as described above.
Genomic DNA isolation.
C. novyi-NT bacteria were collected from 150 ml of late-log–phase culture by centrifugation at 3,000–5,000g for 5–10 min at ~22 °C. Genomic DNA was purified using aQiagen Genomic DNA Buffer system (Qiagen) followed by phenol/chloroform extraction and ethanol precipitation.
Construction of genomic libraries, DNA sequence determination and genome assembly.
Four genomic DNA libraries containing various insert sizes were constructed and sequenced by a shotgun approach. The first library (insert size, 1–2 kb) was generated by cloning a Tsp 509 I partial digest into the pZero vector (Invitrogen). The second (3–4 kb) and third libraries (9.5–10.5 kb) were generated by mechanical shearing followed by cloning into the pSMART vector (Lucigen). The fosmid library (~40 kb) was constructed in the pCC1FOS vector (Epicentre). The first library was constructed and sequenced at Johns Hopkins, using a 384-capillary sequencing instrument (Spectrumedix). The other libraries were constructed and sequenced by Agencourt. More than 12-fold coverage was obtained and assembled using a combination of Phred/ Phrap/Consed41–43 andParacel Genome Assembler software (Paracel). After the initial sequencing and contig assembly, two different approaches were used to close gaps. The first was a modified form of PCR-assisted contig extension (PACE)44. In brief, for each PACE cycle, first-round PCR was performed using a set of eight random primers in combination with a sequence-specific primer located ~140 bp from the end of the contig. A second sequence-specific primer ~40 bp closer to the end of the contig was then used in conjunction with the same set of random primers for a subsequent semi-nested PCR, employing 1 μl of a 1:100 dilution of the first-round PCR product as template. The resulting PACE product was sequenced and the extended contig subjected to the next round of PACE. The second approach involved combinatorial PCR using sequence-specific primers located at the ends of the 23 enlarged contigs generated by PACE. The 46 primers were randomly assigned into eight groups, each containing 5–6 primers. Each group of primers was then mixed with one of the other groups and the mixture of 10–12 primers was used in an initial PCR. If a PCR product was generated from this mixture, a subsequent PCR with single primer pairs was used to define the pair that gave rise to the PCR product. Direct sequencing of the PCR product was carried out thereafter. To ensure the fidelity of the final assembly, 495 overlapping regions (5–6 kb in size) covering the entire genome were amplified and the PCR products resolved by agarose gel electrophoresis to confirm the predicted size of each fragment; one incorrectly assembled segment was discovered using this approach. The 10 rRNA operons were amplified using specific flanking primers, sequenced and assembled individually. Finally, to ensure that every base in the genome was correctly assigned, all nucleotides with Phred scores <40 were resequenced using an independent PCR fragment as template. The sequencing error rate was thereby estimated to be <1:10,000.
Genome annotation.
CDS were predicted by TIGR’s Annotation Engine Service (http://www.tigr.org/edutraining/training/annotation_engine.shtml) and errors generated by the automatic analysis were corrected manually with the assistance of TIGR’s Manatee software. G+C content and trinucleotide composition were calculated by TIGR algorithms as described45. The C. novyi-NT hypothetical genes were identified by comparing the C. novyi-NT genome with other genomes that were annotated by TIGR and made available in TIGR’s database (http://cmr.tigr.org/tigr-scripts/CMR/CmrHomePage.cgi). The C. novyi-NT genes orthologous to those in the other analyzed Clostridia genomes were identified by reciprocal best matches using BLASTP with a cutoff value of e−20. The cellular localization of all putative proteins was predicted with the aid of bioinformatic programs PSORTb, SignalP, LipoP, ScanProsite.
Establishing intratumoral C. novyi-NT infections.
All animal experiments were overseen and approved by the Animal Welfare Committee of The Johns Hopkins University and were in compliance with University standards. We inoculated 6- to 8-week-old athymic nu/nu or BALB/c mice (Harlan) subcutaneously with 5 million HCT116 human or CT26 murine colon cancer cells, respectively9,10. Three hundred million C. novyi-NT spores were administered by tail-vein injection once the tumor volumes reached ~500 mm3. Germination was generally observed to initiate within 12 h.
RNA preparation.
Vegetative bacteria or spores were collected by centrifugation and homogenized by vortexing in RNAwiz buffer (RiboPure-Bacteria kit, Ambion) containing 250 μl of Zirconia beads (Biospec) for 10 min at 4 °C, followed by a 5-min break. This cycle was repeated twelve times to ensure disruption of the spores (as assessed by phase contrast microscopy and RNA yield). RNA was purified with the RiboPure-Bacteria kit following the manufacturer’s instructions.
RNA was prepared from infected tumors by homogenizing (50 strokes) the dissected tumors in a glass homogenizer containing a tenfold volume of the RNAwiz buffer on ice. The homogenate was aliquoted into 0.5-ml RNase-free tubes. The samples were then processed exactly as described above, using a 12-cycle homogenization with Zirconia beads. RNA was quantified with a UV spectrophotometer (Spectra MAX Plus, Molecular Devices) and separated through a precast 1.25% agarose gel (Sigma-Aldrich) to examine its integrity. Purified RNA samples were stored at −80 °C.
To map the truncated 23s rRNA found in spores, northern blots containing RNA from spores or growing cells were prepared as described46. The blots were probed with 32P-labeled oligonucleotides from the rRNA gene, washed and autoradiographed. A fragment from the 5′ region (nucleotide 244–276) of the gene did not hybridize to the spore-specific 23S rRNA but did hybridize to the normal 23S rRNA, whereas a fragment from the 3′- terminal end (nucleotide 2499–2533) hybridized to both the spore-specific and normal 23S rRNA species. Based on this hybridization pattern and the size of the spore-specific 23S rRNA, we estimated that it was missing ~300 nucleotides in the 5′ region.
Microarray analysis.
C. novyi-NT transcriptomes in six developmental stages or experimental conditions were established, as described in the text. Three to five replicates for each condition were used in independent hybridizations. RNA prepared from uninfected HCT116 and CT26 tumors was also used to assess cross hybridization between the bacteria and mammalian genes (‘crossspecies hybridization’). Custom high-density oligonucleotide arrays for the 2,325 C. novyi-NT CDS were constructed by NimbleGen using the Nimble-Screen 12 System. Sample labeling and hybridization were done as previously described, with some modifications47. Briefly, total RNA was reverse transcribed with SuperScript II Reverse Transcriptase (Invitrogen). The first-strand cDNA was fragmented by DNase I (Promega) and labeled with biotinylated ddATP through a terminal transferase end-labeling reaction. Hybridization with the end-labeled samples was carried out at 45 °C for 16 h in 100 mM MES, 1 M NaCl, 20 mM EDTA, 0.01% Tween 20 and 10.5% glycerol. After washing, the arrays were stained with Cy3-Streptavidin, then scanned with an Axon 4000B scanner. The features were extracted by using the NimbleScan software.
The analysis of the microarray data followed this overall plan: (i) normalization across RNA types; (ii) quantile normalization across chips of the same RNA type; (iii) subtraction of estimated nonspecific binding using the crossspecies hybridization data; (iv) ANOVA analysis of differences across RNA types. For this analysis, we used a combination of well-established preprocessing and analysis approaches48, taking into consideration the special nature of this experiment, which involved RNA from two different organisms. The NimbleScreen 12 chips incorporated five probes per gene. Log-absolute probe intensities without background subtraction were used for the analyses49. Steps 1–3 were performed on the data from each of the five probes representing a gene to generate a median expression level for that gene. This median is reported as ‘mRNA abundance’ in all figures and tables. Step 4 was then carried out using these gene-level data and an ANOVA model as implemented in the Limma software50. For hypothetical genes, ‘expression’ was defined as a microarray value (‘mRNA abundance’) from any of the six test conditions (early log, mid log, late log, spore, infected HCT116, or infected CT26) threefold higher than the highest value observed in hybridizations with RNA from uninfected HCT116 or CT26 tumors.
Reverse transcription-PCR (RT-PCR) and real-time PCR.
First-strand cDNA was synthesized from total RNA using random oligonucleotides as primers and the SuperScript First-Strand Synthesis System (Invitrogen). RT-PCR was done with gene-specific primers and Platinum Taq DNA polymerase (Invitrogen) for 30 cycles at 94 °C for 10 s, 57 ° for 15 s, and 70 °C for 30 s. Real-time PCR was carried out with gene-specific primers, Platinum Taq DNA polymerase, and SYBR green I (Invitrogen) in an iCycler (Bio-Rad) at 94 °C for 20 s, 57 °C for 20 s and 70 °C for 20 s.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Peter Setlow for his insightful suggestions about the nature of spore mRNA, Rachel Green for her comments on rRNA fragmentation, Anca Segall for her comments on transposition, and Tanja Davidsen, Michelle Gwinn Giglio and Nikhat Zafar of TIGR for their expert assistance with the genomic bioinformatic analysis. The authors also thank Brent Ewing, Phil Green and David Gordon for kindly providing the Phred/Phrap and Consed software package. The B. subtilis strains were generously provided by Peter Mullany. This work was supported by the Virginia and D.K. Ludwig Fund for Cancer Research, the Commonwealth Foundation, the Miracle Foundation, National Science Foundation grant DMS034211 and National Institutes of Health grant CA062924.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
Accession codes. GenBank: the annotated genome, CP000382. NCBI Gene Expression Omnibus (GEO): microarray data, GSE6087.
Note: Supplementary information is available on the Nature Biotechnology website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Ryan RM, Green J & Lewis CE Use of bacteria in anti-cancer therapies. Bioessays 28, 84–94 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Minton NP Clostridia in cancer therapy. Nat. Rev. Microbiol 1, 237–242 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Pawelek JM, Low KB & Bermudes D Bacteria as tumour-targeting vectors. Lancet Oncol. 4, 548–556 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Barbe S, Van Mellaert L & Anne J The use of clostridial spores for cancer treatment. J. Appl. Microbiol 101, 571–578 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Theys J et al. Tumor-specific gene delivery using genetically engineered bacteria. Curr. Gene Ther 3, 207–221 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Jain RK & Forbes NS Can engineered bacteria help control cancer? Proc. Natl. Acad. Sci. USA 98, 14748–14750 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerar A, Zidar N & Vodopivec B Colorectal carcinoma in endoscopic biopsies; additional histologic criteria for the diagnosis. Pathol. Res. Pract 200, 657–662 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Dang LH, Bettegowda C, Huso DL, Kinzler KW & Vogelstein B Combination bacteriolytic therapy for the treatment of experimental tumors. Proc. Natl. Acad. Sci. USA 98, 15155–15160 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agrawal N et al. Bacteriolytic therapy can generate a potent immune response against experimental tumors. Proc. Natl. Acad. Sci. USA 101, 15172–15177 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bettegowda C. et al. Overcoming the hypoxic barrier to radiation therapy with anaerobic bacteria. Proc. Natl. Acad. Sci. USA 100, 15083–15088 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diaz LA Jr et al. Pharmacologic and toxicologic evaluation of C. novyi-NT spores. Toxicol. Sci 88, 562–575 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Nolling J et al. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J. Bacteriol 183, 4823–4838 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sebaihia M et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet 38, 779–786 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Shimizu T et al. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc. Natl. Acad. Sci. USA 99, 996–1001 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruggemann H et al. The genome sequence of Clostridium tetani, the causative agent of tetanus disease. Proc. Natl. Acad. Sci. USA 100, 1316–1321 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moriya S, Imai Y, Hassan AK & Ogasawara N Regulation of initiation of Bacillus subtilis chromosome replication. Plasmid 41, 17–29 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Segall AM & Craig NL New wrinkles and folds in site-specific recombination. Mol. Cell 19, 433–435 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Songer JG Bacterial phospholipases and their role in virulence. Trends Microbiol. 5, 156–161 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Titball RW, Naylor CE & Basak AK The Clostridium perfringens α-toxin. Anaerobe 5, 51–64 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Chambon P, Deutscher MP & Kornberg A Biochemical studies of bacterial sporulation and germination. X. Ribosomes and nucleic acids of vegetative cells and spores of Bacillus megaterium. J. Biol. Chem 243, 5110–5116 (1968). [PubMed] [Google Scholar]
- 21.Jeng YH & Doi RH Messenger ribonucleic acid of dormant spores of Bacillus subtilis. J. Bacteriol 119, 514–521 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu H et al. Formation and composition of the Bacillus anthracis endospore. J. Bacteriol 186, 164–178 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Setlow P Spore germination. Curr. Opin. Microbiol 6, 550–556 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Fawcett P, Eichenberger P, Losick R & Youngman P The transcriptional profile of early to middle sporulation in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 97, 8063–8068 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alsaker KV & Papoutsakis ET Transcriptional program of early sporulation and stationary-phase events in Clostridium acetobutylicum. J. Bacteriol 187, 7103–7118 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang ST et al. The forespore line of gene expression in Bacillus subtilis. J. Mol. Biol 358, 16–37 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Evguenieva-Hackenberg E Bacterial ribosomal RNA in pieces. Mol. Microbiol 57, 318–325 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Arthur JR The glutathione peroxidases. Cell. Mol. Life Sci 57, 1825–1835 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore TD & Sparling PF Interruption of the gpxA gene increases the sensitivity of Neisseria meningitidis to paraquat. J. Bacteriol 178, 4301–4305 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weeks G, Shapiro M, Burns RO & Wakil SJ Control of fatty acid metabolism. I. Induction of the enzymes of fatty acid oxidation in Escherichia coli. J. Bacteriol 97, 827–836 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kunau WH, Dommes V & Schulz H beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: a century of continued progress. Prog. Lipid Res 34, 267–342 (1995). [DOI] [PubMed] [Google Scholar]
- 32.Ward N & Fraser CM How genomics has affected the concept of microbiology. Curr. Opin. Microbiol 8, 564–571 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Coenye T, Gevers D, Van de Peer Y, Vandamme P & Swings J Towards a prokaryotic genomic taxonomy. FEMS Microbiol. Rev 29, 147–167 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Subramanian G, Mural R, Hoffman SL, Venter JC & Broder S Microbial disease in humans: a genomic perspective. Mol. Diagn 6, 243–252 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Dang LH et al. Targeting vascular and avascular compartments of tumors with C. novyi-NT and anti-microtubule agents. Cancer Biol. Ther 3, 326–337 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Jaeger KE, Dijkstra BW & Reetz MT Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu. Rev. Microbiol 53, 315–351 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Forsdahl K & Larsen TS Phospholipid degradation in hypoxic/reoxygenated cardiomyocytes in response to phospholipase C from Bacillus cereus. J. Mol. Cell. Cardiol 27, 893–900 (1995). [DOI] [PubMed] [Google Scholar]
- 38.Craig NL Target site selection in transposition. Annu. Rev. Biochem 66, 437–474 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Cano RJ & Borucki MK Revival and identification of bacterial spores in 25- to 40-million-year-old Dominican amber. Science 268, 1060–1064 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Vreeland RH, Rosenzweig WD & Powers DW Isolation of a 250 million-year-old halotolerant bacterium from a primary salt crystal. Nature 407, 897–900 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Ewing B, Hillier L, Wendl MC & Green P Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Ewing B & Green P Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8, 186–194 (1998). [PubMed] [Google Scholar]
- 43.Gordon D, Abajian C & Green P Consed: a graphical tool for sequence finishing. Genome Res. 8, 195–202 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Carraro DM et al. PCR-assisted contig extension: stepwise strategy for bacterial genome closure. Biotechniques 34, 626–628, 630–622 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Seshadri R et al. Genome sequence of the PCE-dechlorinating bacterium Dehalococcoides ethenogenes. Science 307, 105–108 (2005). [DOI] [PubMed] [Google Scholar]
- 46.El-Deiry WS et al. WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 (1993). [DOI] [PubMed] [Google Scholar]
- 47.Nuwaysir EF et al. Gene expression analysis using oligonucleotide arrays produced by maskless photolithography. Genome Res. 12, 1749–1755 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parmigiani G, Garrett ES, Irizarry R & Zeger SL The analysis of gene expression data: methods and software. (Springer, New York, 2003). [Google Scholar]
- 49.Scharpf R, Iacobuzio-Donahue CA, Sneddon JB & Parmigiani G When should one subtract background fluorescence in cDNA microarrays? Biostatistics (in the press). [DOI] [PubMed] [Google Scholar]
- 50.Smyth GK. Limma in Bioinformatics and Computational Biology Solutions Using R and Bioconductor (eds. Gentleman V, Carey S, Dudoit R & Irizarry WH) 397–420 (Springer, New York, 2005). [Google Scholar]
- 51.Cheong I et al. A bacterial protein enhances the release and efficacy of liposomal cancer drugs. Science, in press (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.