

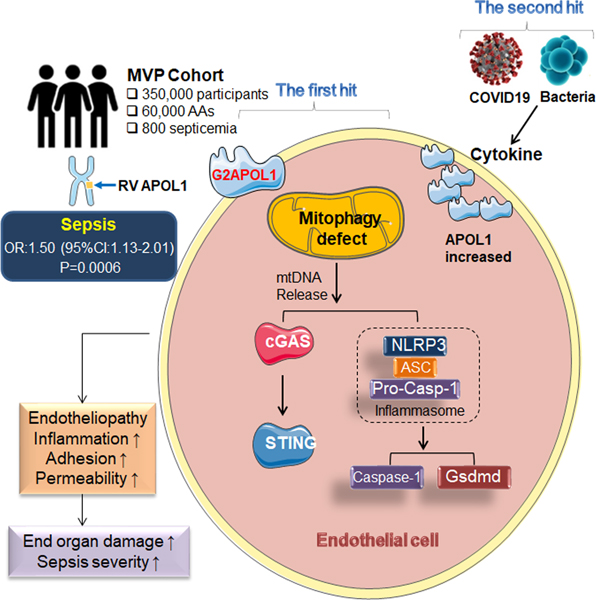
SUMMARY
The incidence and severity of sepsis is higher amongst individuals of African versus European ancestry. We found that genetic risk variants (RV) in the trypanolytic factor Apolipoprotein L1 (APOL1) present only in individuals of African ancestry were associated with increased sepsis incidence and severity. Serum APOL1 level correlated with sepsis and COVID-19 severity. Single-cell sequencing in human kidneys revealed high expression of APOL1 in endothelial cells. Analysis of mice with endothelial-specific expression of RV APOL1 and in vitro studies demonstrated that RV APOL1 interfered with mitophagy, leading to cytosolic release of mitochondrial DNA and activation of the inflammasome (NLRP3) and the cytosolic nucleotide sensing pathways (STING). Genetic deletion or pharmacological inhibition of NLRP3 and STING protected mice from RV APOL1-induced permeability defects and proinflammatory endothelial changes in sepsis. Our studies identify the inflammasome and STING pathways as potential targets to reduce APOL1-associated health disparities in sepsis and COVID-19.
Keywords: APOL1, Endothelial cell, Sepsis, Mitophagy, COVID-19
Graphical Abstract
eTOC
The incidence and severity of sepsis is increased in Black individuals versus White individuals. Wu et al. found that APOL1 risk variants specifically present in individuals of African ancestry were associated with increased sepsis incidence. Mice with endothelial cell-specific expression of risk variant APOL1 had more severe sepsis due to mitophagy defects and over-activation of the NLRP3 and STING pathways.
INTRODUCTION
Each year, at least 1.7 million adults in the United States develop sepsis, resulting in nearly 270,000 deaths. One in 3 patients who dies in a hospital has sepsis (Cates et al., 2020). Black individuals have a 67% higher severe sepsis hospitalization rate and 20% increased likelihood of dying from sepsis compared to White individuals even after adjusting for co-variates in United States (Cates et al., 2020; Suarez De La Rica et al., 2016). Similarly, COVID-19 also disproportionately affects Black individuals, with both higher infection rates and more severe disease (Alsan et al., 2020).
APOL1 is a minor apoprotein component of HDL (High-density lipoprotein), but it is also expressed by multiple cell types in the body. In the circulation, APOL1 forms a complex, known as a trypanosome lytic factor, which provides innate protection against Trypanosoma brucei infection, the causative agent of African sleeping sickness. APOL1 variants have arisen as a result of positive genetic selection, as they confer resistance against a subspecies of Trypanosome brucei rhodesiense. Close to 40% of Black individuals carry at least one of the APOL1 risk variants (RV APOL1) (Genovese et al., 2010; Tzur et al., 2010; Wasser et al., 2012). One risk allele imparts this crucial resistance against African sleeping sickness, having two risk alleles significantly increases the risk of developing chronic kidney disease (CKD) (Genovese et al., 2010; Ilboudo et al., 2012; Thomson et al., 2014; Tzur et al., 2010). These coding variants in APOL1, called G1 and G2, increase CKD and End-Stage Renal Disease (ESRD) risk by 2–20-fold (Friedman et al., 2011). Our and others’ work indicate that kidney podocyte APOL1 genotype is important for CKD development (Beckerman et al., 2017; Pays, 2020).
Since the original observation of the association of RV APOL1 and kidney disease described 10 years ago, several questions remain unanswered. First, the RV APOL1-associated phenotypic heterogeneity is poorly understood (Bajaj et al., 2017; Kopp et al., 2011). RV APOL1 have shown significant association with focal segmental glomerulosclerosis (FSGS), hypertensive kidney disease, cardiovascular disease, hypertension and preeclampsia (Reidy et al., 2018). Second, the environmental trigger for the RV APOL1 associated disease development remains poorly understood. It has been reported that disease development is strongly linked to an inflammatory trigger. Kidney disease is triggered by exogenous interferon (IFN) administration, HIV and recently COVID-19 infections (Couturier et al., 2020; Estrella et al., 2013; Fine et al., 2012; Kopp et al., 2011; Kudose et al., 2020; Lazareth et al., 2020; Oniszczuk et al., 2020; Papeta et al., 2011; Sharma et al., 2020). Finally, despite the large affected population, the mechanism of RV APOL1-induced disease pathogenesis is poorly understood limiting therapeutics development.
Here we report that RV APOL1 was associated with higher sepsis incidence and severity in Black patients even after adjusting for comorbidities. We showed that in mice endothelial cell-specific expression of APOL1 risk alleles led to an endotheliopathy, associated with increased endothelial inflammation, vascular leakage, albuminuria and increased sepsis severity. Furthermore, we demonstrated that RV APOL1 in endothelial cell induced a mitochondrial defect, including mitophagy, leakage of mtDNA into cytoplasm, and the activation of the innate immune pathways; NLRP3 and STING, which led to endothelial dysfunction. In conclusion, our study indicates the role of risk variant APOL1 in sepsis severity in both humans and mice.
RESULTS
APOL1 risk genotypes and plasma APOL1 levels correlate with sepsis and COVID-19 severity
To understand the relationship between APOL1 genotype and sepsis in Black individuals we analyzed data from the Million Veteran Program (MVP) (release 2) (Hunter-Zinck et al., 2020). We assessed the association of RV APOL1 with sepsis incidence in 57,000 Black participants. Our phenotype screen identified 800 Black participants with the diagnosis code of septicemia. Analysis adjusted for age, gender, ancestry and basic covariates indicated a statistically significant association between RV APOL1 genotype and sepsis (p=0.006, OR:1.50, 95% CI 1.13, 2.01) and prosthesis-associated sepsis (p<10-9, Figure 1A) using a recessive model. These associations remained statistically significant even after adjusting for kidney function (estimated glomerular filtration rate, eGFR) (Figure S1A). Our results suggest that high-risk genotype APOL1 was associated with increased sepsis incidence.
Figure 1. Association of APOL1 risk genotypes, plasma APOL1 levels with sepsis and COVID19 severity.
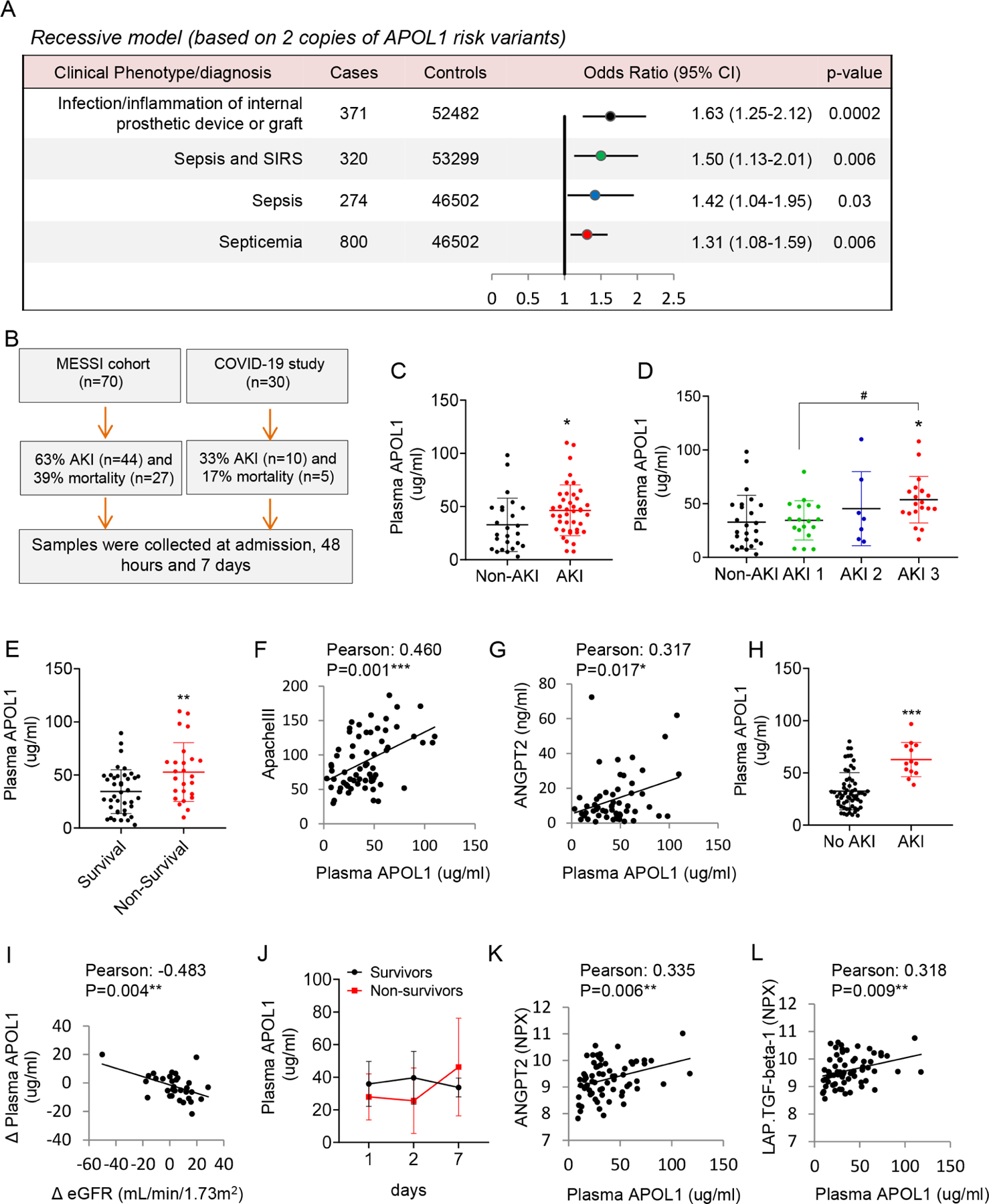
A. Association between APOL1 risk alleles and Systemic infections/Sepsis phenotypes in the MVP cohort. A recessive model was used to estimated odds ratios (ORs) and 95% CIs. All analyses were adjusted for age and sex and 10 principal components of ancestry.
B. Study flow-chart, showing AKI incidence and mortality. We enrolled n=70 participants with sepsis in the MESSI cohort (C-G) and n=30 in the COVID19 (H-L) cohorts.
C. Plasma APOL1 levels (at presentation) (y-axis) in non-AKI and AKI patients in the MESSI cohort. *p<0.05.
D. Plasma APOL1 levels (at presentation) in non-AKI, AKI stage 1 (AKI 1), AKI stage 2 (AKI 2), and AKI stage 3 (AKI 3) patients. *p<0.05 vs Non-AKI; #p < 0.05 vs. indicated group.
E. Plasma APOL1 levels in surviving and non-surviving patients at presentation. **p<0.01.
F. The correlation of plasma APOL1 levels at presentation and ApacheIII score.
G. The correlation of plasma APOL1 levels at presentation and ANGPT2.
H. Plasma APOL1 levels in participants with and without AKI in the COVID19 study. ***p<0.001.
I. The correlation of change in plasma APOL1 level (Δ APOL1) (the increase in APOL1 from admission to day 2, day 7 and day 28) with change in eGFR (Δ eGFR) (between previous measurements).
J. Plasma APOL1 levels on day 1, 2, and 7 in survivors (N=12) and non-survivors (N=6).
K. The correlation of plasma APOL1 and ANGPT2 levels in the COVID19 cohort. NPX: Normalized Protein expression.
L. The correlation of plasma APOL1 and LAP.TGF.beta-1 levels in the COVID19 cohort
The Molecular Epidemiology of Sepsis in the Intensive care unit (MESSI) cohort is a prospective cohort study of critically ill patients with sepsis at the University of Pennsylvania (Reilly et al., 2015; Shashaty et al., 2019). We analyzed plasma samples from 70 Black patients (Figure 1B) without APOL1 genotype information. Cohort characteristics are shown in Table S1. The incidence of Acute Kidney Injury (AKI) (defined by KDIGO criteria) was 63% (n=44) and the 30-day mortality was 39% (n=27). Plasma APOL1 level was higher in patients who developed AKI during the first 6 days, and APOL1 levels correlated with AKI stage (Figure 1C and 1D). APOL1 level also correlated with 30-day mortality (Figure 1E) and acute physiology and chronic health evaluation (APACHE) III score (Figure 1F). Angiopoetin-related protein 2 (ANGPTL2) is a proinflammatory protein considered a biomarker of endothelial dysfunction and sepsis severity (Fiedler et al., 2006; Lukasz et al., 2008; Reilly et al., 2018). We noticed plasma ANGPT2 level was increased in patients with AKI and its level correlated with plasma APOL1 concentration (Figure S1B and Figure 1G). The results indicated that circulating APOL1 levels are increased in patients with sepsis and APOL1 levels correlated with disease severity.
COVID-19, caused by SARS-CoV-2 infection, is associated with severe inflammation and phenotypic changes similar to sepsis, including AKI and severe vascular changes (Kellum et al., 2020; Osuchowski et al., 2021). We enrolled 40 individuals who were admitted to the Hospital of the University of Pennsylvania with COVID-19 (without APOL1 genotype information) (Figure 1B and Table S2). Blood and urine samples, along with clinical information, were collected on admission and 2, 7, and 28 days after admission. We excluded 10 participants due to underlying CKD and a total of 74 plasma samples were analyzed. We detected 13 AKI events in the cohort as defined by an increase in serum creatinine to 1.5 times baseline over ≤ 7 days (Figure S1C). Plasma APOL1 level was higher at the time of AKI compared to those without AKI (Figure 1H). As previously described, plasma APOL1 level did not correlate with baseline eGFR (Figure S1D). On the other hand, we found that the change in plasma APOL1 level correlated with the change in eGFR at different time-points (Figure 1I). Urine APOL1 level was not different when AKI and non-AKI participants were compared (Figure S1E). Higher levels of plasma APOL1 at day 7 were observed in non-survivors during the follow-up, (compared day 1 and 2) although the difference was not statistically significant (Figure 1J).
Next, we evaluated the correlation between cytokines and chemokines in plasma samples from COVID-19 patients using the O-link multiplex inflammation panel that measures 92 different proteins. Of the 92 analytes measured, 86 proteins were detected within the dynamic range of the assay. Of these 86 proteins, MCP.2, CD40.L, CD244, EGF, ANGPT2, IL6, TRAIL, CXCL9, CD8A, CAIX, MUC.16, ADA, TNFRSF12A, LAP.TGFB1, CXCL1, PDGF.B, CXCL5, HGF, Gal.9, CCL17, CXCL12, CSF.1, and VEGFR.2 were increased in AKI patients and their levels correlated with plasma APOL1 level (Figure 1K and 1L and Figure S1F). Plasma APOL1 correlated with markers of vascular damage such as ANGPT2 and LAP.TGFB1.
In summary, we observed increased incidence of sepsis phenotypes in participants with RV APOL1. In addition, APOL1 level was higher in patients with more severe sepsis and COVID-19 and plasma APOL1 levels correlated with markers of endothelial dysfunction.
Endothelial-specific expression of RV APOL1 in mice induces vascular leakage and inflammation
Circulating APOL1, plays role in trypanosomiasis protection (Shukha et al., 2017). Podocytes express APOL1 expression is critical for kidney glomerular disease development (Beckerman et al., 2017). Here, we reexamined the cellular expression of APOL1 using single cell transcriptome and epigenome data from human kidneys. An assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-Seq) analysis of human kidney single nuclei indicated that APOL1 has the highest chromatin accessibility in endothelial cells (Figure 2A). Analysis of single cell gene expression data from human kidneys and lungs also indicated relatively high endothelial expression of APOL1 (Figure S2A and S2B). Double in situ hybridization and immunofluorescence analysis of APOL1 and the endothelial marker CD31 on human lung samples further confirmed the expression of APOL1 in endothelial cell (Figure 2B and C). The GTEx compendium of human organ specific gene expression dataset indicated that APOL1 expression is the highest in the lung (Figure S2C).
Figure 2. Endothelial-specific expression of RV APOL1 in mice induced vascular leakage and inflammation.
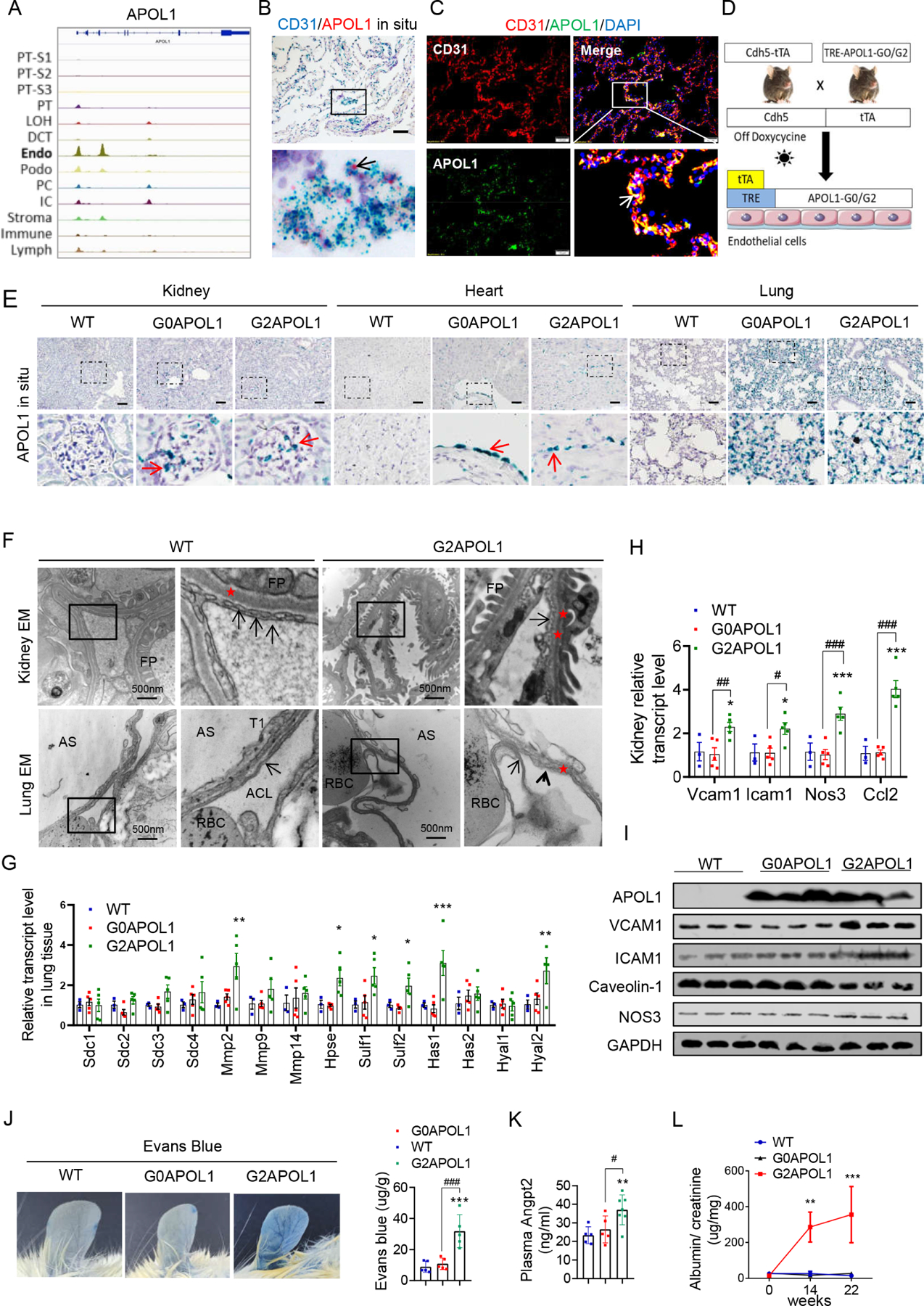
A. Genome browser view of read density in snATAC-seq clusters in human kidney cells at APOL1 locus, endo (endothelial cells).
B. Representative images of CD31 (blue) and APOL1 (red) in situ hybridization in a healthy human lung sample. Black arrows indicate co-localizations of APOL1 with CD31. Scale bar=20μm.
C. Representative images of double immunofluorescence staining of a healthy human lung sample with CD31 (red) and APOL1 (green). White arrows indicate the co-localization of APOL1 with CD31. Scale bar=10μm.
D. Experimental design for the generation of Cdh5-tTA/TREG0APOL1-GFP (EC/G0APOL1) and Cdh5-rtTA/TREG2APOL1-GFP (EC/G2APOL1) mice.
E. Representative images of APOL1 in situ hybridization in kidneys, hearts, and lungs of wild-type (WT), EC/G0APOL1 and EC/G2APOL1 mice. The red arrows indicate the expression of APOL1 mRNA. Scale bar=40μm
F. (Upper panel) Representative kidney transmission electron micrographs (TEM) of WT and EC/G2APOL1 mice reveal endothelial cells with loss of glomerular capillary cell fenestrations (arrows), and asterisks show the basement membrane, FP, podocyte foot process. (Lower panel) Representative Lung TEM from EC/G2APOL1 mice show capillary endothelial cell membrane loss (arrowheads) and delamination compared to WT mice. Arrows show the capillary endothelial cell membrane, and asterisks show the basement membrane. RBCs within the alveolar capillary lumen (ACL) are shown. AS, alveolar space; T1, alveolar type 1 epithelial cell. Scale bars, 500 nm.
G. Relative mRNA levels of glycoproteins; Syndecan 1 (Sdc1), Syndecan 2 (Sdc2), Syndecan 3 (Sdc3), Syndecan 4 (Sdc4), Metalloproteinase-2 (Mmp2), Metalloproteinase-9 (Mmp9), and Metalloproteinase-14 (Mmp14); Heparin sulfate genes Heparanase (Hpse), Sulfatase 1 (Sulf1), Sulfatase 2 (Sulf2); Hyaluronan genes Hyaluronan Synthase 1 (Has1), Hyaluronan Synthase 2 (Has2), Hyaluronidase 1 (Hyal1), and Hyaluronidase 2 (Hyal2) were evaluated in the lung of WT (N = 3), EC/G0APOL1 (N = 5) and EC/G2APOL1 (N = 5) mice. ; *p < 0.05, **p<0.01, ***p<0.001 vs WT.
H. Relative mRNA levels of vascular cell adhesion molecule 1 (Vcam1), intercellular adhesion molecule 1 (Icam1), nitric oxide synthase 3 (Nos3), and monocyte chemoattractant protein-1 (Ccl2) were evaluated in kidneys of WT (N = 3), EC/G0APOL1 (N = 5) and EC/G2APOL1 (N = 5) mice. Gapdh was used for normalization. *p < 0.05, ***p<0.001 vs WT; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. indicated group.
I. Representative western blots from lung lysates of WT, EC/G0APOL1 and EC/G2APOL1 mice showing levels of APOL1, ICAM1, VCAM1, Caveolin-1, eNOS, and GAPDH.
J. Representative ear photographs of Evan’s blue dye leakage after Evan’s blue injection into WT, EC/G0APOL1, and EC/G2APOL1 mice. Right panel, spectrophotometric analysis of the amount of extravasated Evans blue dye (N=5). ***p < 0.001, vs WT; ###p < 0.001 vs. indicated group.
K. Plasma Angiopoietin 2 (Angpt2) level in WT (N=5), EC/G0APOL1 (N=5), and EC/G2APOL1 (N=6) mice. **p<0.01 vs WT; #p < 0.05 vs. indicated group.
L. Urinary albumin/creatinine ratio (ACR) of EC/G0APOL1 and EC/G2APOL1 at baseline, 1, 14 and 22 weeks off doxycycline diet. Single transgenic littermates served as controls (WT) (N = 5, 6, 6 for WT, G0 and G2, respectively). **p<0.01, ***p<0.001, compares WT mice at the same time points.
To understand the role of APOL1 in endothelial cells, we generated mice with endothelial-specific expression of reference allele: G0APOL1 (EC/G0APOL1) and risk allele:G2APOL1 mice (EC/G2APOL1) by crossing the previously established TRE-G0APOL1 or TRE-G2APOL1 mice (APOL1 is placed under tetracycline response elements) with the Cdh5tTA mice (Figure 2D (Beckerman et al., 2017). Cdh5 (encoding Cadherin-5 or VE-cadherin) is an endothelial-specific gene ensuring endothelial specific expression. Removal of the doxycycline diet led to observable expression of APOL1 in different vascular beds such as lung, heart and kidney, which was confirmed by in situ hybridization (Figure 2E), immunohistochemical staining (Figure S2D), quantitative reverse transcriptase PCR (qRT-PCR) (Figure S2E), and immunoblotting (Figure 2I). Both mRNA and protein expression APOL1 was similar between EC/G0APOL1 or EC/G2APOL1 transgenic mice (Figure 2I and S2E).
Phenotypic characterization of EC/G2APOL1 animals indicated normal kidney function as assessed by serum BUN and creatinine levels (Figure S2F). Light microscopy did not reveal any abnormalities in the kidney, heart or lung (Figure S2G). Electron microscopical analysis indicated multifocal loss of glomerular capillary endothelial cell (GEC) fenestration in kidney samples and alveolar capillary endothelial cell delamination in lung samples of EC/G2APOL1 mice but not in wild-type (WT) mice (Figure 2F). Podocyte foot processed effacement was not detected in EC/G2APOL1 kidney, which is a key feature of nephrotic syndrome.
Further molecular profiling indicated that the transcripts for enzymes involved in endothelial glycocalyx remodeling (Figure 2G), markers of vascular inflammation, vascular tone, and adhesion molecules (Vcam1, Icam1, Nos3 and Ccl2) were increased in lung, kidney and heart samples of EC/G2APOL1 mice when compared to littermate WT mice and EC/G0APOL1 (Figure 2H, 2I, and S2H-J). Consistent with the observed molecular changes, a deeper physiological phenotyping analysis indicated increased vascular permeability in EC/G2APOL1 which was evident by increased Evans blue leakage (Figure 2J). Circulating levels of Angpt2, an important vascular permeability factor, were also elevated in EC/G2APOL1 mice (Figure 2K). Furthermore, increased endothelial permeability was evident by a mild elevation in albuminuria in EC/G2APOL1 mice (Figure 2L, Figure S2K). In summary, endothelial expression of G2APOL1 causes endotheliopathy, as indicated by a defect in the glycocalyx, increased endothelial expression of adhesion molecules, inflammation, and vascular permeability.
RV APOL1 increases sepsis severity
Endothelial permeability defect and endothelial inflammation are hallmarks of sepsis, and our data indicated an association between RV APOL1 genotype and plasma APOL1 level. Given that the liver is the main source of circulating APOL1 in humans (Shukha et al., 2017), we generated liver-specific G2APOL1-expressing animal (Liver/G2APOL1) by crossing the TRE-G2APOL1 mice with GT26 rtTA mice and albumin-Cre mice (Figure S3A). The expression of APOL1 in hepatocytes was confirmed by in situ hybridization and qRT-PCR (Figures S3B and S3C). Circulating APOL1 was higher in Liver/G2APOL1 mice compared to WT mice (Figure S3D). However, mortality, body weight and temperature changes were similar between Liver/G2APOL1 and WT mice at 24hrs after the injection of lipopolysaccharide (LPS). (Figure S3E-G).
Given the key role of endothelial dysfunction in sepsis, we analyzed sepsis severity in WT, EC/G0APOL1 and EC/G2APOL1 mice using the LPS-injection induced endotoxemia and cecal ligation and puncture (CLP) sepsis models. Although all WT and EC/G0APOL1 mice survived the LPS injection, EC/G2APOL1 mice showed at least 70% lethality by 24 hours following LPS injection (Figure 3A). Body temperature and body weight decline were more severe in EC/G2APOL1 mice (Figures 3B and 3C). Similar effects were seen in the CLP model (Figure S3H and S3I). Serum APOL1 was higher in EC/G2APOL1 mice following LPS injection or CLP as compared to EC/G0APOL1 mice (Figure S3K and S3L).
Figure 3. Endothelial RV APOL1 increases sepsis and endotoxemia severity.
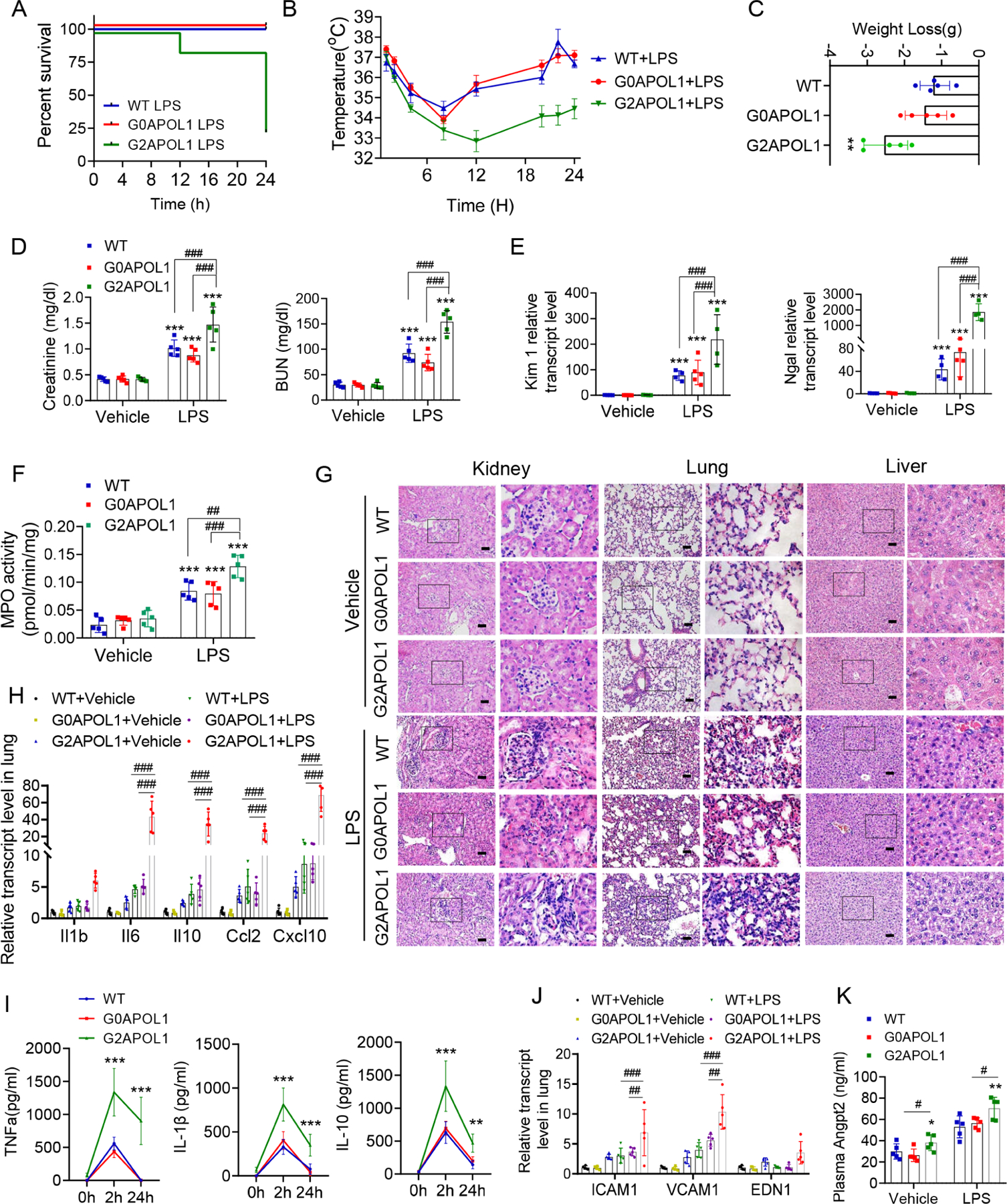
A. Survival after intraperitoneal injection of LPS (6mg/kg) in WT (N=5), EC/G0APOL1 (N=5) and EC/G2APOL1 (N=20) mice.
B. Body temperature response to LPS of WT (N=5), EC/G0APOL1 (N=5) and EC/G2APOL1 (N=5) mice.
C. Weight loss after intraperitoneal injection of LPS (6mg/kg) at 24h in WT (N=5), EC/G0APOL1 (N=5) and EC/G2APOL1 (N=5) mice. **p<0.01 vs WT.
D. Renal function of WT (N=5), EC/G0APOL1 (N=5) and EC/G2APOL1 (N=5) mice 24 hours after saline or LPS injection, assessed by serum BUN and creatinine. ***p<0.001 vs Vehicle; ##p < 0.01, ###p < 0.001 vs. indicated group.
E. Relative transcript level of AKI injury markers Kidney injury molecule-1 (Kim1), and urine neutrophil gelatinase-associated lipocalin (Ngal) in the kidneys of WT (N = 4), EC/G0APOL1 (N=5) and EC/G2APOL1 (N = 4) mice 24 hours after saline or LPS injection. ***p<0.001 vs vehicle; ###p < 0.001 vs. indicated group.
F. MPO activity was measured in lungs obtained 24 hours after administration of saline or LPS in WT (N = 5), EC/G0APOL1 (N=5) and EC/G2APOL1 (N = 5) mice. ***p<0.001 vs vehicle; ##p < 0.01, and ###p < 0.001 vs. indicated group.
G. Representative images of H&E-stained sections of kidneys, hearts, and lungs 24 hours after saline or LPS injection in WT, EC/G0APOL1 and EC/G2APOL1 mice. Scale bar=40 μm.
H. Relative mRNA levels of Il1b, Il6, Il10, Ccl2 and Cxcl10 in the lung of WT (N = 5), EC/G0APOL1 (N = 5) and EC/G2APOL1 (N = 5) mice. Gapdh was used for normalization. ###p < 0.001 vs. indicated group.
I. Serum IL10, IL-1β and TNFα of WT (N = 5), EC/G0APOL1 (N = 5) and EC/G2APOL1 (N = 5) mice at 2 and 24hr after LPS injection; **p<0.01, ***p<0.001 vs WT; ###p < 0.001 vs. indicated group.
J. Relative mRNA levels of Vcam1, Icam1, and Edn1 were evaluated in lungs of WT (N = 5), EC/G0APOL1 (N = 5) and EC/G2APOL1 (N = 5) mice; Gapdh was used for normalization. ##p < 0.01, ###p < 0.001 vs. indicated group.
K. Plasma Angpt2 of WT (N = 5), EC/G0APOL1 (N = 5) and EC/G2APOL1 (N = 5) mice 24 hours after saline or LPS injection; *p<0.05, **p<0.001 vs WT; #p < 0.05 vs. indicated group.
We observed more severe endotoxemia-induced end-organ damage in EC/G2APOL1 mice. BUN, creatinine, kidney expression of acute renal injury markers (Kim1, and Ngal) were higher in EC/G2APOL1 mice (Figure 3D and 3E). We also detected enhanced kidney disease severity (Figure S3M and S3N) in the CLP model. Increase in lung injury severity was evidenced by the higher level of lung myeloperoxidase (MPO) activity in both LPS (Figure 3F) and CLP models (Figure S3O). Histological analysis indicated more severe inflammatory changes in lungs, kidneys and livers of LPS-treated EC/G2APOL1 mice compared to controls and EC/G0APOL1 mice (Figure 3G).
Gene expression of markers of inflammation, such as Il1, Il6, Il10 and Cxcl10 and Ccl2, were higher in the lungs of EC/G2APOL1 mice when compared the control or EC/G0APOL1 after LPS injection (Figure 3H). Serum levels of IL-10, IL-1β, and TNFα were also higher in EC/G2APOL1 mice (Figure 3I). Markers of endothelial inflammation and damage such as Icam1, Vcam1, and Edn1 were elevated in lung tissue of septic EC/G2APOL1 mice compared to control and EC/G0APOL1 animals (Figure 3J). Inflammatory markers were also increased in the CLP sepsis model (Figure S4P and S4Q). Serum Angpt2 was higher in EC/G2APOL1 mice, consistent with the increased permeability (Figure 3K). In summary, our data indicated that endothelial expression of the risk variant G2APOL1 leads to increased endotoxemia or sepsis severity.
Single cell profiling of EC/G2APOL1 highlighted endothelial inflammation
To understand molecular changes in endothelial cells, we performed droplet-based single-cell RNA sequencing (scRNA-seq) on kidney samples from WT and EC/G2APOL1 mice (Figure 4A). After sequencing and alignment, we identified 53,495 high quality cells. We next performed dimensionality reduction by uniform manifold approximation and projection (UMAP) and graph-based clustering (Figure 4B and 4C). The analysis recognized 13 distinct clusters. These clusters were annotated based on previously published marker genes (Park et al., 2018).
Figure 4. Single cell profiling of EC/G2APOL1 highlighted endothelial inflammation.
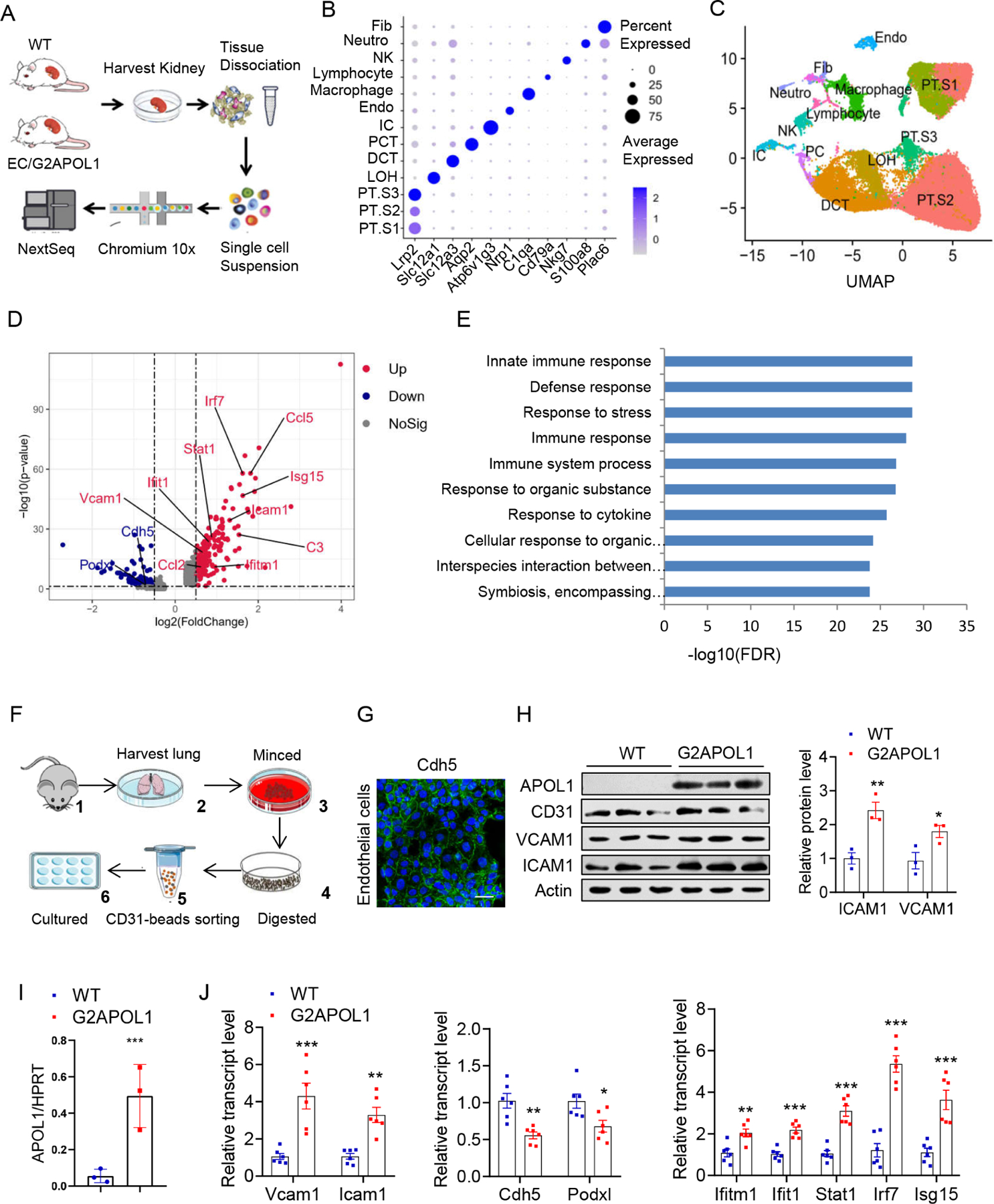
A. The diagram summarizes the process of cell isolation and single cell RNA-seq analysis of the WT and EC/G2APOL1 mouse kidney using the 10X Genomics platform.
B. Bubble plots showing the expression levels of representative marker genes across the 13 main clusters. Fib, fibroblast; Neutro, neutrophil; NK, natural killer cell; lymph, lymphocyte; Macro, macrophage; Endo, containing endothelial; IC, intercalated cell; PCT, proximal convoluted tubule; DCT, distal convoluted tubule; LOH, ascending loop of Henle; PT.S3, proximal tubule S3; PT.S2, proximal tubule S2; PT.S1, proximal tubule S1. PC, principal cells
C. UMAP dimension reduction of single cell RNAseq of mouse kidney samples from WT and EC/G2APOL1 mice.
D. Volcano plot showing the difference of the mean expression between WT and G2APOL1 ECs in the single cell data.
E. GO (gene ontology) functional analysis of DEGs when WT and G2APOL1 ECs were compared. The 10 most significantly (P<0.05) enriched GO terms in biological process are presented (p was negative 10-base log transformed). DEGs, differentially expressed genes; GO, gene ontology.
F. Schematic representation of primary lung endothelial cell isolation and enrichment.
G. Representative images of Cdh5 immunofluorescence stain in isolated primary pulmonary microvascular endothelial cells. Scale bar=20μm.
H. Protein levels of APOL1, CD31, VCAM1, ICAM1, and Actin were analyzed by immunoblots in WT and G2APOL1 ECs. (Right panel) densitometric quantification of levels ICAM1 and VCAM1 normalized to GAPDH. *p<0.05 and **p<0.01 vs WT.
I. Relative APOL1 transcript level in ECs from WT (N = 3) and EC/G2APOL1 (N = 3) mice.
APOL1 mRNA level is normalized to HPRT. ***p<0.05 vs WT.
J. Relative mRNA levels of Vcam1, Icam1, Ifitm1, Ifit1, Stat1, lrf7, Isg15, Cdh5 and Podxl in WT (N=6) and G2APOL1 (N=6) ECs. *p<0.05, **p<0.01, and ***p<0.001 vs WT.
We next compared gene expression changes in WT and EC/G2APOL1 mice, and identified 534 differentially expressed genes (DEGs) in endothelial cells (with adjusted p<0.05). The top DEGs included chemokines such as Ccl2, Ccl5, Cxcl10, complement genes, IFN signature genes (Stat1, Ifitm1, Isg15, Irf7 and Ccl5), and endothelial cell adhesion molecules (Icam and Vcam1). Expression of cell junction genes such as Cdh5 and Podxl, that are required for vascular barrier integrity, was lower in EC/G2APOL1 mice (Figure 4D). Next, we performed pathway analysis of the identified DEGs. Gene ontology analysis indicated strong enrichment for innate immune and cytokine pathways in endothelial cells as analyzed by single cell RNA-Seq (Figure 4E).
We next isolated primary lung endothelial cells (ECs) from WT and EC/G2APOL1 mice (Figure 4F). We confirmed the expression of Cadherin-5 by immunostaining (Figure 4G) and CD31 by immunoblotting (Figure 4H). Removal of doxycycline from the medium resulted in an increase in APOL1 expression in endothelial cells (Figure 4H and 4I). Consistent with the scRNA-seq data, expression of adhesion molecules such as Vcam1 and Icam1 were higher in cultured G2APOL1 ECs in absence of doxycycline (Figure 4H and 4J). Genes associated with inflammation and IFN, such as Ifitm1, Ifit1 and Stat1 were higher in cultured G2APOL1 ECs. Expression of cell-junction genes (Cdh5 and Podxl) (Figure 4J) were lower in G2APOL1 ECs, which was consistent with the scRNA-seq results.
In summary, scRNA-seq and in vitro analysis of cultured endothelial cells highlighted the role of inflammation and IFN-driven immune response in G2APOL1 endothelial cell.
RV APOL1 induces autophagy and mitophagy defects in endothelial cells
APOL1 cytotoxicity is linked to its ion conducting properties both in trypanosomes and in human cells (Molina-Portela Mdel et al., 2005; Thomson and Finkelstein, 2015). Differences in APOL1 channel activity are believed to be the result of differential trafficking and membrane insertion rather the genotype per se (Kruzel-Davila et al., 2017) (Giovinazzo et al., 2020). Because mitochondria are implicated to play a key role in cytotoxicity (Datta et al., 2020; Granado et al., 2017; Ma et al., 2017; Shah et al., 2019; Vanwalleghem et al., 2015), we examined cytosolic and mitochondrial changes in G2APOL1 ECs.
Electron microscopy of ECs from control or EC/G2APOL1 mice showed that mitochondria were enclosed by autophagosome/autolysosomes and displayed an increased number of cellular degradative compartments, such as lysosomes, autolysosomes and amphisomes (Figure 5A), indicating a potential defect in mitochondria and mitophagy. We therefore examined mitochondrial alterations and whether such changes are responsible for the observed proinflammatory gene expression differences observed in our transcriptomic analyses. We specifically analyzed whether G2APOL1 interfered with mitophagy using the mitophagy reporter Cox8-mCherry/GFP. In this assay, healthy mitochondria have both green and red fluorescence, and thus appear yellow. Mitochondria within acidic compartments, such as lysosomes, show red-only fluorescence, due to the selective sensitivity of EGFP fluorescence to low pH (Ward et al., 2016). We found minimal red only puncta in WT ECs indicating low basal mitophagy. We were able to stimulate mitophagy by serum and nutrient withdrawal (starvation in HBSS) or CCCP (2 hour), (([(3-chlorophenyl) hydrazono] malononitrile), which uncoupled oxidative phosphorylation, induced reactive oxygen generation and enhanced mitophagy in control and G0APOL1 ECs. In contrast, we observed fewer red-only puncta in G2APOL1 ECs after nutrient withdrawal or CCCP treatment, suggesting a decrease of mitophagy (Figure 5B and 5C). Total mitochondrial mass, as analyzed by mitochondrial DNA (mtDNA) (Figure 5D), mitochondrial membrane protein COX4 and molecular chaperone HSP60 were increased in G2APOL1 ECs, indicating accumulation of mitochondria, consistent with a mitophagy defect (Figure S4A and S4B). The PTEN-induced putative kinases 1 (Pink1) and Parkin have been linked to early steps of mitophagy. PINK1 recruits Parkin in response to mitochondrial membrane potential loss (Vives-Bauza et al., 2010). We observed G2APOL1 ECs had higher baseline Parkin levels in mitochondrial fraction, and the expression of Parkin was further induced by CCCP but not starvation. In WT ECs, both starvation and CCCP treatment markedly increased the expression of Parkin (Figure S4C and S4D).
Figure 5. RV APOL1 in endothelial cells induces autophagy and mitophagy defect.
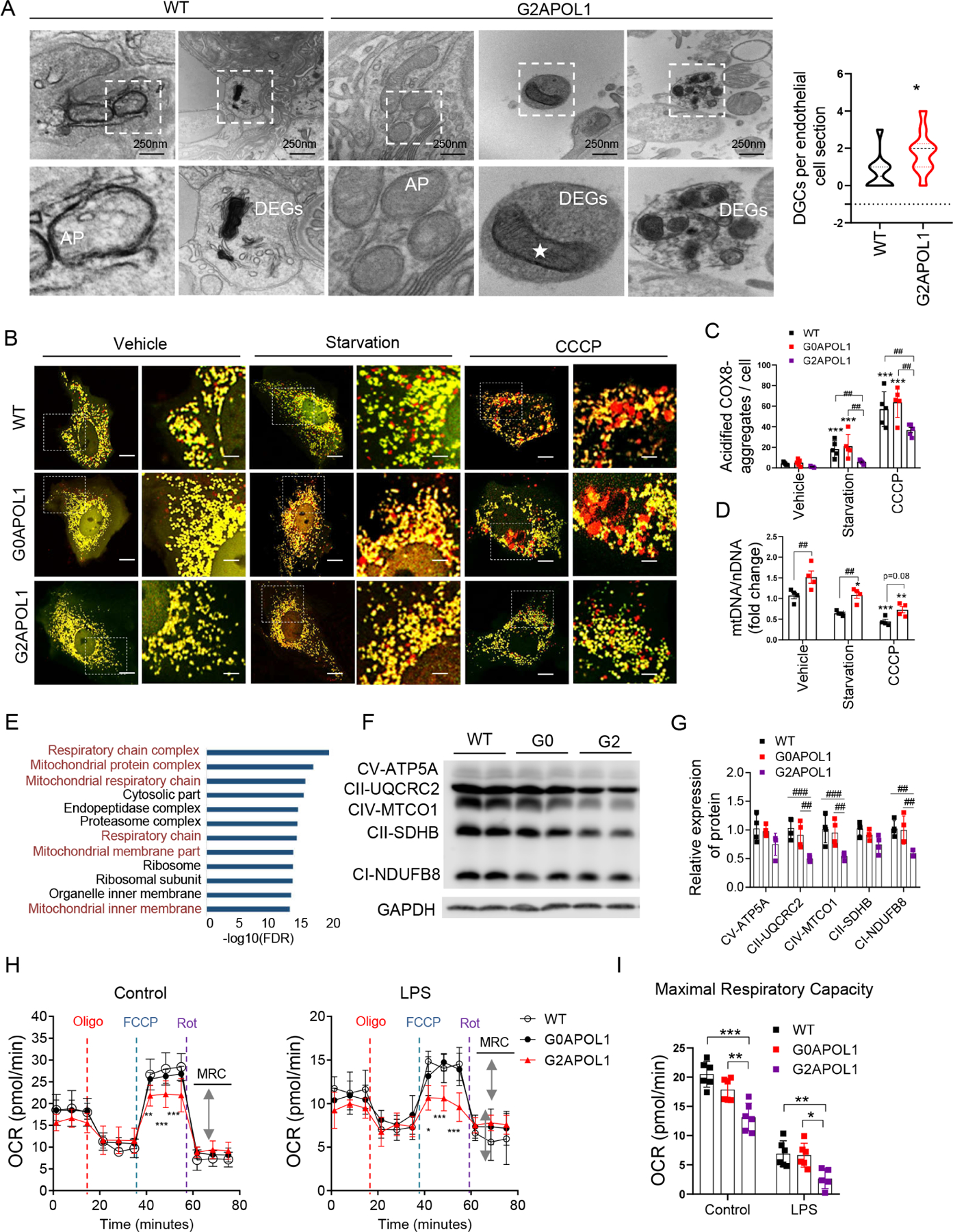
A. Representative EM images of the autophagosomes (AP) and DGCs in the kidney of WT and EC/G2APOL1 mice. White star, Autophagosome / autolysosome-engulfed mitochondria. Scale bars: 250 nm. (Right panel) Quantification of the number of DGCs per cell section; DGCs, cellular degradative compartments; *p < 0.05 vs. WT
B. Mitophagy was examined in cells transiently expressing Cox8-EGFP-mCherry. WT, G0APOL1 or G2APOL1 ECs treated with vehicle, starvation (HBSS, 4h), or CCCP (40uM, 2h). The red punctate represent mitochondrial contents within acidic compartments. Scale Bar = 10 µm.
C. Quantification of red-only punctate in panel (B). ***p<0.001 vs. Vehicle; ##p < 0.01 vs. indicated group. 30 cells in 6 fields were counted in each group.
D. Mitochondrial mass determined by qRT-PCR analysis of the mtDNA/nuclear DNA (nDNA) ratio in WT (N=6) or G2APOL1 (N=6) ECs treated as in B; **p<0.01 and ***p<0.001 vs. Vehicle. ##p < 0.01 vs. indicated group.
E. Gene ontology analysis (molecular function) of genes differentially expressed by endothelial cells (G2 vs WT) using DAVID. The 12 most significantly (P<0.05) enriched GO terms in cellular component branches are presented (p is negative 10-base log transformed).
F. Western blots of WT, G0APOL1 and G2APOL1 ECs, showing levels of OXPHOS proteins (CV-ATP5A, CII-UQCRC2, CIV-MTCO1, CII-SDHB and CI-NDUFB8), and GAPDH.
G. Densitometric quantification of OXPHOS proteins normalized to GAPDH. N=3 independent experiments; ##p < 0.01, ###p < 0.001 vs. indicated group.
H. Real-time changes in the OCR of WT, G0APOL1 and G2APOL1 ECs after treatment with oligomycin (Oligo), FCCP, and rotenone (Rot) in the presence or absence of LPS (100ng/ml, 24h). MRC, maximal respiratory capacity (double-headed arrow). *p<0.05, **p<0.01, ***p<0.001, compares WT at the same time points.
I. Maximal respiratory capacity of ECs measured by real-time changes in OCR. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. indicated group.
Compromised mitochondria and mitophagy lead to accumulation of defective mitochondria. Gene Ontology analysis of the scRNA-Seq indicated strong enrichment for impaired mitochondrial genes in G2APOL1 ECs (Figure 5E and S4L), which was confirmed by OXPHOS immunoblotting and qRT-PCR (Figure 5F, 5G and S4M). We next analyzed oxygen consumption (OCR), as a measure of mitochondrial function in control, G0APOL1 and G2APOL1 ECs at baseline and following LPS treatment. The G2APOL1 cells had lower basal OCR and lower maximal respiratory capacity (MRC) when compared to WT and G0APOL1 ECs. LPS treatment resulted in marked reduction in basal OCR and MRC in G2APOL1 ECs (Figure 5H and 5I). To investigate whether the altered mitochondrial metabolic profile was caused by abnormal mitochondrial function, we next analyzed mitochondrial membrane potential and ROS generation as a functional read-out for mitochondrial defect. We observed higher mitochondria ROS production by mitoSOX in G2APOL1 ECs (Figure S4G). Mitochondrial membrane potential was evaluated by JC-1 stain (Figure S4H). G2APOL1 ECs had higher green fluorescence intensity indicating loss of negative membrane potential.
To investigate potential defects in autophagy, in addition to the observed mitophagy defects, we measured the levels of autophagosomal marker LC3-II. G2APOL1 ECs had higher baseline LC3-II level. In control ECs, LC3-II expression was markedly increased in response to chloroquine (an inhibitor of the lysosomal compartment) indicating a healthy autophagy flux. On the other hand, G2APOL1 ECs showed attenuated changes in LC3-II when induced by starvation or chloroquine (Figure S4E and S4F and S4K). Analysis of p62 expression confirmed changes seen by LC3-II (Figure S4E and S4F and S4K). In summary, these data indicate a defect in mitochondria, mitophagy and autophagy in RV APOL1 ECs.
RV APOL1-mediated mitophagy defect in endothelial cells causes cytosolic mtDNA leakage and inflammasome and STING activation leading to an inflammatory, pro-adhesive endothelial phenotype
We hypothesized that the defect in mitochondria and mitophagy could result in cytosolic leak of mtDNA. To test this, we isolated cytosolic, mitochondrial, and nuclear extracts from cultured endothelial cells and measured the mtDNA content in the cytosolic fraction. The purity of the extracts was confirmed by immunoblotting (Figure S4I). qRT-PCR analysis of the ratio of mitochondrial gene (mt-Atp6) and nuclear (Rpl13a) genes indicated 3- to 5-fold higher mtDNA content in the cytosolic fraction in G2APOL2 ECs when compared to WT endothelial cells (Figure S4J).
The cytosol is normally free of nucleic acids, and the presence of DNA in the cytosol is generally considered a sign a pathogenic infection, but can also result from injured mitochondria (Shimada et al., 2012). Cytosolic DNA is sensed by the inflammasome and by the cytosolic DNA sensor, cGAS, which activates STING. Downstream of STING is TBK1 which then activates transcription factors such as IRF3/7 and NF-κB, leading to cytokine release. We observed increased ICAM1, VCAM1, cGAS and increased STING phosphorylation in G2APOL1 ECs when compared with WT and G0APOL1 ECs (Figure S5A-5C). we observed NLRP3 activation and cleavage of caspase 1 (Casp1) and gasdermin D (Gsdmd) (Figure S5A-5C). The mitochondrial defect was responsible for cGAS/STING and GSDMD activation as depletion of cells of mtDNA by ethidium bromide treatment or mitophagy activation by CCCP (Figure 6B) ameliorated the NLRP3 and cGAS/STING and downstream TBK, IRF3 and NF-kB activation. Ethidium bromide or CCCP treatment suppressed the expression of ICAM1 and VCAM1 (Figure 6A, S5D and S5E). Activation of cGAS and STING was upstream of inflammation, as cGAS and STING siRNA prevented TBK1, IRF3, NFkB activation and lowered ICAM1 and VCAM1 expression (Figure 6C, 6D, S5F and S5G). Our data suggest the expression of RV APOL1 in endothelial cells results in increased cytosolic mtDNA, and subsequent activation of STING and NLRP3.
Figure 6. Mitophagy defect in RV APOL1 ECs causes cytosolic mtDNA leakage and inflammasome and STING activation leading to an inflammatory, pro-adhesive endothelial phenotype.
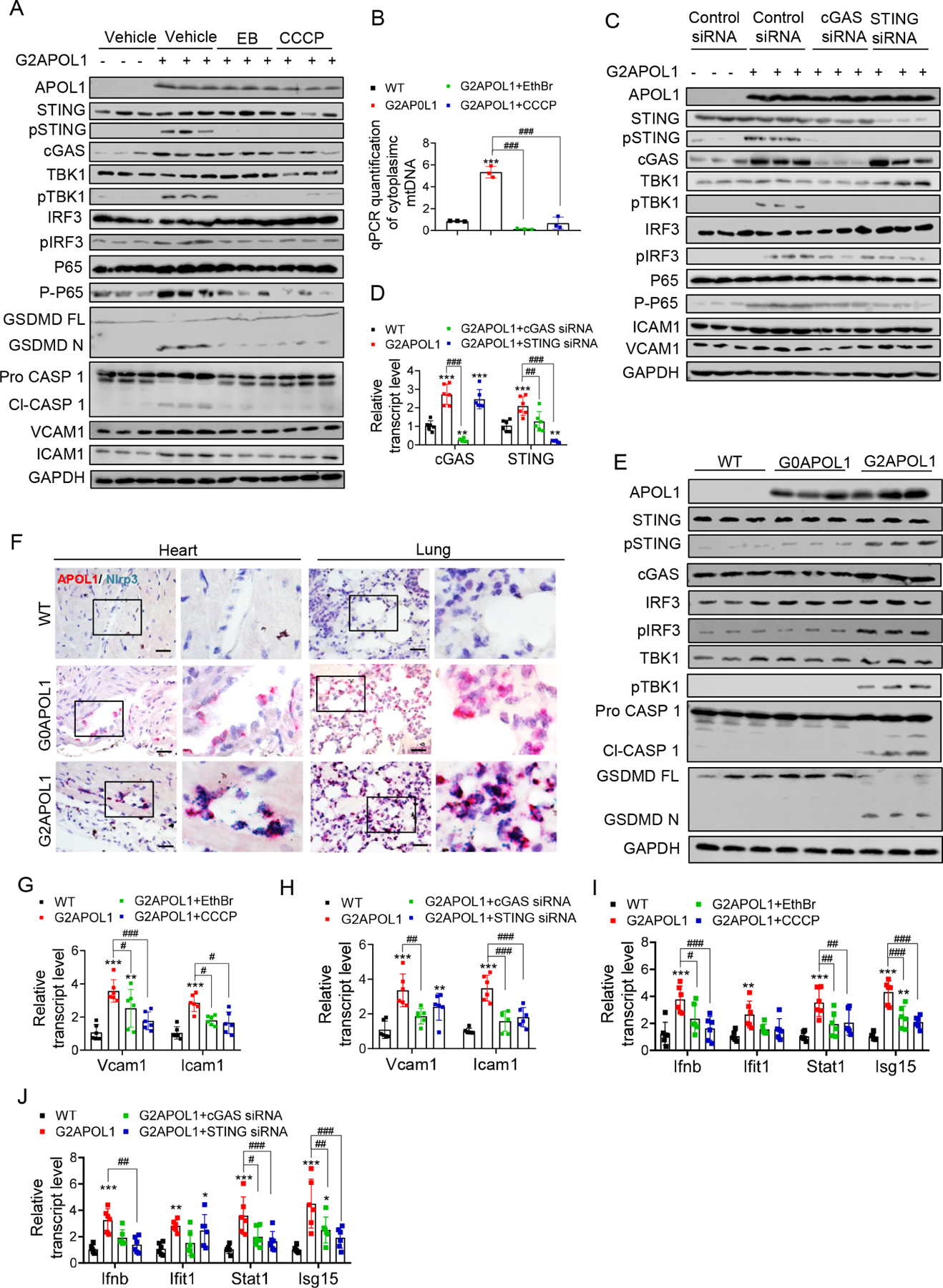
A. Protein expression of APOL1, VCAM1, ICAM1, inflammasome and cytosolic nucleotide sensors (STING, pSTING, cGAS, TBK1, pTBK1, IRF3, pIRF3, P65, P-P65, GSDMD and Caspase 1) were analysed on immunoblots in WT, and G2APOL1 ECs treated with Vehicle, ethidium bromide (EthBr) (150ng/ml 48hours), or CCCP (40uM, 2hours).
B. Total DNA was harvested from cytosolic and nuclear fractions of WT or G2APOL1 ECs treated as (A) and analysed by qRT-PCR. Cytosolic mtDNA genes were normalized to respective nuclear Rpl13a; ***p<0.001 vs WT; ###p < 0.001 vs. indicated group.
C. Western blot analysis of APOL1, VCAM1, ICAM1 and cGAS-STING proteins (STING, pSTING, cGAS, TBK1, pTBK1, IRF3, pIRF3) in WT and G2APOL1 ECs treated with vehicle, STING or cGAS siRNA.
D. Relative transcript level of cGAS and STING in WT and G2APOL1 ECs treated as (C). **p<0.01, ***p<0.001 vs WT; ###p < 0.001 vs. indicated group.
E. Immunoblot analysis using antibodies against STING, pSTING, cGAS, TBK1, pTBK1, IRF3, pIRF3, Caspase 1, GSDMD and Actin in lung tissue from WT, EC/G0APOL1 and EC/G2APOL1 mice.
F. Representative images of Nlrp3 (blue) and APOL1 (red) in situ hybridization in heart and lung of WT, EC/G0APOL1 and EC/G2APOL1 mice. Scale bar=20μm.
G. Relative transcript level of Icam1 and Vcam1 in WT and G2APOL1 ECs treated as (A); Gapdh was used for normalization. **p<0.01, ***p<0.001 vs WT; #p < 0.05, ###p < 0.001 vs. indicated group.
H. Relative transcript level of Icam1 and Vcam1 in WT and G2APOL1 ECs treated as (C); Gapdh was used for normalization. **p<0.01, ***p<0.001 vs WT; ##p < 0.01, ###p < 0.001 vs. indicated group.
I. Relative transcript level of Ifnb, Ifitm1, Stat1 and Isg15 in WT and G2APOL1 ECs treated as (A); **p<0.01, ***p<0.001 vs WT; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. indicated group.
J. Relative transcript level of Ifnb, Ifitm1, Stat1 and Isg15 in WT and G2APOL1 ECs treated as (C); **p<0.01, ***p<0.001 vs WT; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. indicated group.
Next, we adopted a well-established in vitro model of NLRP3 inflammasome activation, by adenosine triphosphate (ATP) treatment, which drives cleavage of caspase 1 in LPS-primed cells (Shimada et al., 2012). Upon ATP stimulation, we observed, NLRP3 activation and an increase in cleaved caspase 1 and secreted IL-1β levels by Western blot and ELISA, with G2 APOL1 ECs having the largest increase (Figure S5J, S5K and S5L). The level of pSTING was unaffected after LPS/ATP stimulation.
To show that self-damaged cytosolic DNA is responsible for STING activation, we isolated DNA from hydrogen peroxide treated endothelial cells for transfection into WT, G0APOL1 and G2APOL1 ECs (Luo et al., 2020; Shimada et al., 2012). Genomic DNA transfection resulted in robust expression of pSTING, NLRP3, cleaved caspase-1 and increase in secreted IL-1β (Figure S5J, S5K and S5L). G2APOL1 endothelial cells had substantially higher levels of pSTING, cleaved caspase 1 and secreted IL-1β compared to G0APOL1 ECs. Increased activation of NLRP3 and cGAS/STING was also confirmed in vivo by immunoblotting in lung tissue samples of WT, EC/G0APOL1 and EC/G2APOL1 mice (Figure 6E, S5H and S5I). Furthermore, the endothelial expression of NLRP3 in vivo was validated by double in situ hybridization in lung and heart sections obtained from EC/G2APOL1 mice (Figure 6F).
Treatment with CCCP, ethidium bromide, or siRNA silencing of cGAS and STING ameliorated the increased transcript levels of Vcam1, Icam1, Ifnb, Ifit1, Stat1, and Isg15 in cultured G2APOL1 ECs (Figure 6G-J), indicating that cGAS/STING and NLRP3 activation was responsible for the observed proinflammatory phenotype. The G2APOL1-induced cGAS-STING activation was cell-type dependent, as we did not observe it in hepatocytes (Figure S5M) but was evident in podocytes (data not shown).
In summary, our results indicate RV APOL1-induced mitophagy defects in endothelial cells led to cytosolic mtDNA leakage, which resulted in the activation of the inflammasome and nucleotide sensing pathways and downstream cytokine release.
Genetic or pharmacological disruption of NLRP3 and STING ameliorated endothelial RV APOL1 induced endotheliopathy and sepsis
Our in vitro experiments indicated that activation of the inflammasome and nucleotide sensing pathways is critical for EC/RVAPOL1 induced endotheliopathy, including inflammation and permeability. To test the role of NLRP3 and STING during sepsis in EC/RVAPOL1 mice, we crossed global Nlrp3–/–, Gsdmd–/–, Casp 1/11–/– or STING–/– mice with EC/G2APOL1 mice, to generate Cdh5tTA/TREG2APOL Nlrp3–/– (G2APOL1/Nlrp3–/–), Cdh5tTA/TREG2APOL1 Gsdmd–/– (G2APOL1/Gsdmd–/–), Cdh5tTA/TREG2APOL1 Casp 1/11–/– (G2APOL1/Casp1/11–/–) and Cdh5tTA/TREG2APOL1 STING–/– (G2APOL1/STING–/–) mice. We confirmed the genetic deletion of Nlrp3, Gsdmd, Casp1/11 and STING by lung tissue western blotting and qRT-PCR (Figure S6A and S6B).
To understand the role of the inflammasome and the cytosolic nucleotide sensing pathway in G2APOL1 endothelial permeability defect we investigated endothelial resistance in contact-inhibited ECs. Cultured ECs cell-cell contacts mature into adherent and tight junctions, a process required for ECs to form and maintain a barrier between the intravascular and extravascular space. ECs obtained from EC/G2APOL1 mice showed impaired cell-cell contact solidification, as reflected in their inability to achieve continuous cell-cell contact (Figure S6C). This defect was consistent with in vivo gene expression (loss of Cdh5) and permeability results. Consistent with these effects, the trans-endothelial electrical resistance (TEER) across a monolayer of ECs (Szulcek et al., 2014) (Figure 7A) was reduced in G2APOL1 ECs, demonstrating impaired barrier function. This defect was rescued by the genetic deletion of Nlrp3, Gsdmd, Casp1/11 or STING (Figure 7B, S6E, and S6F).
Figure 7. Genetic targeting of the NLRP3 and STING ameliorated endothelial RV APOL1 induced endotheliopathy and sepsis with pharmacological therapeutic potential.
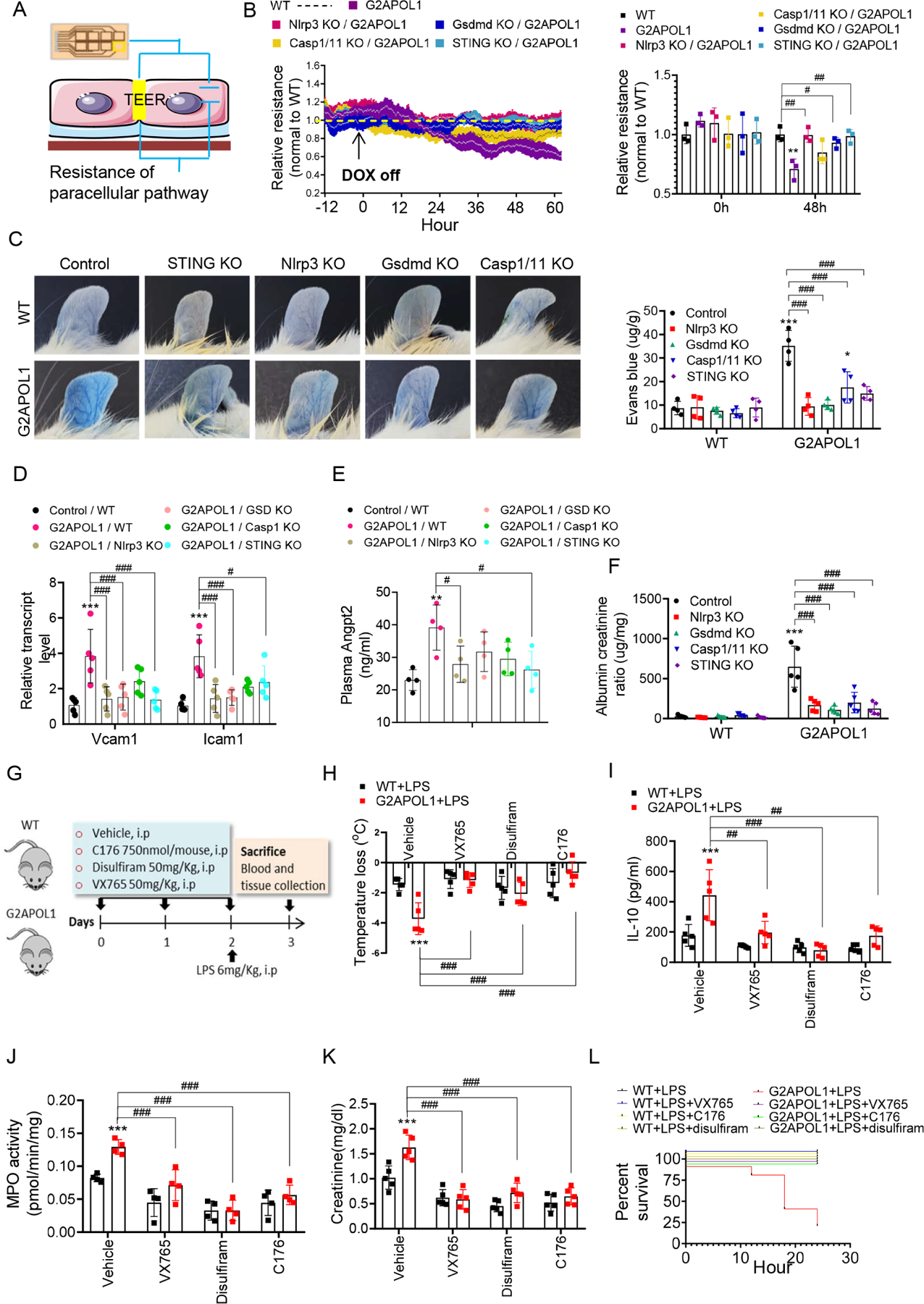
A. TEER: Trans-endothelial electrical resistance measurement of endothelial function
B. (Left panel) TEER measured in real time in ECs from WT, EC/G2APOL1, Nlrp3−/−/G2APOL1, Gsdmd−/− /G2APOL1, Casp 1/11−/−/G2APOL1 and STING−/−/G2APOL1 mice. At each individual time point, TEER values were normalized to WT ECs. (Right panel) Relative TEER at 0 hr and 48 hr after removal of DOX. **p<0.01 vs. WT; #p < 0.05, ##p < 0.01 vs. indicated group.
C. Representative photographs of Evan’s blue dye leakage in ears. (Right panel) Spectrophotometric analysis of the amount of extravasated Evans blue dye from mouse ears. *p<0.05, **p<0.01 vs. WT; ###p < 0.001 vs. indicated group.
D. Relative mRNA levels of Vcam1 and Icam1 in indicated groups. ***p<0.001 vs. WT; #p < 0.05, ###p < 0.001 vs. indicated group.
E. Plasma Angpt2 level in indicated groups. **p<0.01 vs. WT; #p < 0.05 vs. indicated group.
F. Urinary albumin/creatinine ratio (ACR) of indicated groups at 10 weeks off doxcycyline diet. ***p<0.001 vs. WT; ###p < 0.001 vs. indicated group.
G. Experimental design: Pharmacological targeting of the inflammasome and nucleotide sensing pathways in WT and EC/G2APOL1 mice.
H. Body temperature response to saline or LPS at 12h in group as shown in Panel (G).
I. Serum IL10 of mice as shown in Panel (G) 24h after saline or LPS injection; ***p<0.001 vs WT+LPS; ##p < 0.01, ###p < 0.001 vs. indicated group.
J. MPO activity was measured in the lung obtained 24 hr after the administration of saline or LPS in in group as shown in Panel G; **p<0.01, ***p<0.001 vs WT+LPS; ##p < 0.01 vs. indicated groups
K. Renal function of mice as shown in Panel G at 24h after saline or LPS injection, assessed by creatinine. ***p<0.001 vs WT+LPS; ###p < 0.001 vs. indicated group.
L. Survival after intraperitoneal injection of LPS (6mg/kg) in group as shown in Panel G.
To understand the role of Nlrp3, Gsdmd, Casp1/11 and STING in G2APOL1-induced vascular permeability defect we examined in vivo barrier function in G2APOL1 knock-out animals. Genetic deletion of the inflammasome or the intracellular nucleotide sensing pathway (STING) markedly improved barrier function as analyzed by Evans blue leakage (Figure 7C). Vcam1 and Icam1 expression in the lung (Figure 7D, S6G and S6H), and plasma Angpt2 (Figure 7E) were also improved in mice with deletion of inflammasome components. Albuminuria, another marker of vascular leakage, improved following inflammasome deletion in EC/G2APOL1 mice (Figure 7F).
To understand the role and therapeutic potential of NLRP3/STING targeting in RV APOL1-associated sepsis, we injected EC/G2APOL1 mice with small molecular inhibitors of CASP1 (VX654), GSDMD (disulfuram) and STING (C176). We induced endotoxemia by LPS injection (Figure 7G). Weight loss and hypothermia were less severe in inhibitor-treated mice, compared to LPS injected EC/G2APOL1 mice (Figure 7H, and Figure S7A). Serum concentrations of IL-10, IL-1β and TNF-α, as well as lung MPO level were reduced in inhibitor treated mice, compared to EC/G2APOL1 mice (Figure 7I ,7J, S7B and S7C). mRNA expression of cytokines such as Il1, Il6, Il10, Ccl2, Cxcl10 was markedly higher in the LPS injected EC/G2APOL1 mice (Figure S7D-H). Overall, Il6 and Il1 expression was lower in VX654 and disulfiram treated animals, while the effect of C176 on Il6 level was modest. As an estimate of end organ damage, serum creatinine was higher in LPS-induced sepsis in EC/G2APOL1 mice and all 3 inhibitors effectively ameliorated renal damage (Figure 7K). Finally, all 3 inhibitors showed protection from mortality (Figure 7L), indicating the potential role of inflammasome and downstream CASP1, GSDMD and STING in LPS-induced endotoxemia development in EC/G2APOL1 mice.
In summary, genetic deletion and pharmacological inhibitors indicate a key role for the inflammasome and cytosolic nucleotide sensing pathways in endothelial RV APOL1-induced endotheliopathy and sepsis severity.
DISCUSSION
In this study, we show that Black race-specific RV APOL1 genotype is associated with increased sepsis susceptibility. The inflammatory milieu in sepsis and in COVID-19 via increasing APOL1 expression likely serves as a second hit (in addition to the genetic variant) for exacerbating disease severity. G2APOL1 induces mitochondrial dysfunction, mitophagy defect and activates the inflammasome and intracellular cytosolic nucleotide sensing pathways. Our results could help to explain the increased sepsis and COVID-19 severity amongst Black individuals and can open new avenues for genetic and precision medicine targeting that is very much needed in minority populations.
We propose a two-hit model, such as the underlying genetic susceptibility conferred by RV APOL1 and an inflammatory trigger (in sepsis and COVID-19) increasing RV APOL1 expression. This two-hit model is similar to that observed in patients with APOL1-associated glomerulopathy, where in addition to the genetic susceptibility by RV APOL1, inflammation and the increase in RV APOL1 plays a key role in disease development (May et al., 2021; Nichols et al., 2015). In vitro, the RV APOL1-induced cytotoxicity showed strong genotype and dose dependency (Beckerman et al., 2017; Datta et al., 2020). For example, APOL1 transcript level was higher in glomeruli of subjects with kidney disease compared to those without. We show that APOL1 is also higher in plasma samples of patients with sepsis-associated AKI. As the APOL1 gene is not present in mice, we used an inducible overexpression system. Our data indicates that circulating APOL1 level in these animals is much lower than observed in patients, indicating that the observed phenotypes are not caused by supraphysiological transgene expression. The most prominent regulators of APOL1 transcription are STAT and IRF transcription factors (Nichols et al., 2015). In cultured cells, IFN-γ, TNF-a and in some studies even LPS increases APOL1 expression (Nichols et al., 2015). Given that such cytokine levels are higher in patients with sepsis, they likely contribute to the increase in APOL1 expression in sepsis and COVID-19.
Our study indicates that the phenotypic heterogeneity observed with RV APOL1 is related to the cell type of expression. Most prior studies have focused on podocyte RV APOL1 expression, which plays a key role in a rare form of glomarular disease (Freedman et al., 2021). Circulating APOL1 (which mostly originates from the liver) is important for trypanosome defense. Here we propose that endothelial APOL1 is a key determinant of sepsis. In addition, endothelial RV APOL1 could also explain the observed association between RV APOL1 and typical endothelial diseases such as hypertension, hypertensive kidney disease and preeclampsia. Data from GTEx and from single cell analysis indicates that endothelial cells express high level of APOL1, and they may even contribute to circulating APOL1 expression. Future studies are warranted to analyze the role of RV in these diseases.
Endothelial APOL1 could also have a potential critical evolutionary role in trypanosomiasis protection. At early disease stage, the parasite is only present in the blood (hemolymph). Endothelial cell invasion and the ensuing vascular permeability defect is critical for the blood-brain barrier invasion and for the development of the devastating neurological defect. Cell adhesion molecules such as Icam1, Vcam1 and proinflammatory cytokines such as interferon, Cxcl10 and Il10 are critical for the development of the neuroinflammatory stage of the trypanosomiasis (Tiberti et al., 2013) . Future studies shall focus on characterizing the role of endothelial APOL1 in the vascular invasion of Trypanosome.
On the molecular level, our study further highlights the critical role of APOL1 trafficking and mitochondria in endothelial dysfunction. In vivo single cell sequencing, in vitro cultured endothelial cells, genetic and pharmacological models collectively indicated the key role of mitochondrial defect, activation of the inflammasome and cGAS/STING in RV APOL1 induced disease development. Our findings indicate the RV APOL1 could explain an important racial disparity observed in sepsis incidence and severity amongst Black individuals. Furthermore, our work implies that identification of subjects with HR APOL1 genotype might be important for disease risk prediction and the use of specific therapeutics for subjects with HR genotype.
In summary, our work identifies the role of endothelial RV APOL1 in sepsis amongst Black individuals, highlights the key role of mitochondrial dysfunction and activation of the inflammasome and cGAS and STING and emphasizing critical therapeutic targets for intervention.
Limitations of the Study
An important limitation of our study remains that the APOL1 gene is missing from the mouse genome, making it difficult to generate animal models that recapitulate the human condition. Here we compared humanized mice expressing reference allele to those expressing risk variant. Defining the exact molecular mechanism of RV APOL1 induced cytotoxicity has been challenging. Future studies shall address RV-induced changes in intracellular trafficking and changes in autophagy. In addition, future larger genetic studies shall examine the correlation between risk variant APOL1 and sepsis and COVID-19 severity in larger cohorts and correlate inflammatory changes with APOL1 level.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to, and will be fulfilled by, the lead contact Katalin Susztak (ksusztak@pennmedicine.upenn.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Caspase-1 | Santa Cruz | Cat#SC56036 |
| Anti-GAPDH | CST | Cat#2118 |
| Anti-GSMD | Abcam | Cat#ab209845 |
| Anti-APOL1 | Proteintech | Cat#66124–1-Ig |
| Anti-APOL1 | Genentech | Rabmab 5.17D12 |
| Anti-GFP | Abcam | Cat#AB6673 |
| Anti-LC3II | CST | Cat#2775 |
| Anti-CD31 | Abcam | Cat#ab24590 |
| Anti-Icam1 | Abcam | Cat#ab179707 |
| Anti-Vcam1 | Abcam | Cat#ab134047 |
| Anti-Caveolin-1 | CST | Cat#3267 |
| Anti-Enos | Abcam | Cat#ab5589 |
| Anti-Hsp60 | CST | Cat#12165 |
| Anti-Cox4 | R&D Systems | Cat#MAB6980 |
| Anti-STING | CST | Cat#13647s |
| Anti-phosphorylated STING | CST | Cat#19781s |
| Anti-cGAS | CST | Cat#31659S |
| Anti-TBK1 | CST | Cat#3504S |
| Anti-phosphorylated TBK1 | CST | Cat#5483 |
| Anti-IRF3 | CST | Cat#11904T |
| Anti-phosphorylated IFR3 | CST | Cat#37829 |
| Anti-P65 | CST | Cat#5483 |
| Anti-Phospho-NF-κB p65 | CST | Cat#8242 |
| Monoclonal Anti-β-Actin−Peroxidase antibody | Sigma Aldrich | Cat#A3854 |
| Anti-VE-cadherin | Santa Cruz | Cat#sc-6458 |
| OXPHOS antibody cocktail | Abcam | Cat#ab110413 |
| Anti-p62 | CST | Cat#5114 |
| Donkey anti-Rabbit IgG (H+L) Alexa Fluor 555 | Invitrogen | Cat#A31572 |
| Chicken anti-Mouse IgG (H+L), Alexa Fluor 488 | Invitrogen | Cat#21200 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LPS (Escherichia. coli O26:B6) | Sigma-Aldrich | Cat#L8274 |
| VX765 | Cayman | Cat#CAYM-28825–50 |
| Disulfiram | Cayman | Cat#97–77-8 |
| C176 | Bio Vision | Cat#B2341 |
| Evans blue dye | Sigma | Cat#E2129 |
| CD31 MicroBeads, mouse | miltenyi biotec | Cat#130–097-418 |
| TET system approved FBS | clontech | Cat#631106 |
| Microvascular EBM-2 | Lonza | Cat#cc-3162 |
| Hydrogen peroxide solution 30% | Sigma | H1009–500ML |
| Trizol | Invitrogen | Cat#15596018 |
| cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368813 |
| Power SYBR® Green PCR Master Mix | Applied Biosystems | Cat#4367659 |
| RIPA buffer | Cell signaling | Cat#9806 |
| Protease cocktail | Roche | Cat#11836153001 |
| BCA protein assay | Thermo Scientific | Cat#23225 |
| Western Blotting Substrate | Pierce | Cat#32106 |
| Lipofectamine 3000 reagents | Thermo Fisher Scientific | Cat#11668019 |
| LysoTracker™ Blue DND-22 | Thermo Fisher Scientific | Cat#L7525 |
| ECIS Cultureware Disposable Electrode Arrays | Applied BioPhysics | 8W10E+ (8 Well PET) |
| l-cysteine | Sigma | Cat#C7352–25G |
| Gelatin solution | Sigma | Cat#G1393–100ML |
| JC-1 | Invitrogen | Cat#T3168 |
| Digitonin | EMD Millipore | Cat#11024–24-1 |
| DNeasy Blood and Tissue kit | QIAGEN | Cat#69506 |
| COX8-EGFP-mCherry plasmid | Addgene | Cat#78520 |
| Hs-APOL1 -C probe | bio-techne | Cat#459791 |
| APOL1-No-XMm-C2 | bio-techne | Cat#459798-C2 |
| Mm-Nlrp3 | bio-techne | Cat#439571 |
| Hs-PECAM1-O1 | bio-techne | Cat#487381 |
| Critical Commercial Assays | ||
| Mouse albumin specific ELISA | Bethyl Laboratories | Cat#E99–134 |
| Creatinine reagent | Diazyme | Cat#DZ072B |
| RNAscope 2.5 HD Duplex Detection Kit | bio-techne | Cat#322436 |
| Multi Tissue dissociation kit 1 | Miltenyi | Cat#130–110-2011 |
| APOL1 ELISA Kit | Proteintech | Cat#KE00047 |
| Mouse IL-1 beta ELISA Kit | R&D Systems | MLB00C |
| Mouse TNF-alpha | R&D Systems | MTA00B |
| Il-10 ELISA Kit | R&D Systems | Cat#M1000B |
| Angpt2 ELISA Kit | R&D Systems | Cat#MANG20 |
| Myeloperoxidase assay kit | Sigma | Cat#MAK068–1KT |
| Expermental Models:Organisms/Strains | ||
| Cdh5 tTA | Jackson Laboratory | Stock#013585 |
| GT26.rtTA | Jackson Laboratory | Stock#5670 |
| Nlrp3KO | Jackson Laboratory | Stock#21302 |
| STING KO | Jackson Laboratory | Stock#025805 |
| Caspase1/11 KO | Jackson Laboratory | Stock#16621 |
| Gsdmd KO | Jackson Laboratory | Stock#032410 |
| Alb-Cre | Jackson Laboratory | Stock#003574 |
| TREG0APOL1 | N/A | |
| TREG2APOL1 | N/A | |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO:GSE181671 |
| Seqence-based Reagents | ||
| Primers see Table S2 | This paper | N/A |
| siRNA targeting sequence: Tmem173 (Duplex 1): GAAUCGGGUUUAUUCCAACAGCGTC |
IDT | N/A |
| siRNA targeting sequence: Tmem173 (Duplex 2): UUCUUAGCCCAAAUAAGGUUGUCGCAG |
IDT | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij |
| Prism 8 | Graphpad Software | https://www.graphpad.com/scientific-software/prism |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animal Studies
TRE-APOL1 mice were previously generated in Susztak lab by cloning APOL1 (G0/G2) cDNA into the pBI-EGFP vector containing tetracycline response element (TRE). The transgenic construct was injected into FVB/N (Beckerman et al., 2017).
The Cdh5 tTA (Stock#013585), Alb-Cre (Stock#003574), GT26.rtTA (Stock#5670), Nlrp3-/- (Stock#21302), STING-/- (Stock#025805), Caspase1/11-/- (Stock#16621), Gsdmd-/- (Stock#032410) mice were obtained from Jackson lab, bred, and genotyped according to JAX genotyping protocols.
Cdh5 tTA mice were crossed to TRE-G0APOL1 and TRE-G2APOL1 mice to generate Cdh5tTA/G0APOL1 (EC/G0APOL1) and Cdh5tTA/G2APOL1 (EC/G2APOL1) mice. APOL1 transgene expression was induced by taking off doxycycline containing food (200mg/kg, Bio-Serv).
Alb-Cre mice were crossed to GT26.rtTA and TREG2APOL1 mice to generate Alb-Cre/GT26/TREG2APOL1 mice. APOL1 transgene expression was induced by feeding doxycycline containing food (200mg/kg, Bio-Serv)
Nlrp3-/-, STING-/-, Caspase1/11-/-, and Gsdmd-/- mice were crossed to Cdh5tTA/G2APOL1 mice to generate Nlrp3-/- Cdh5tTA/G2APOL1, STING-/- Cdh5tTA/G2APOL1, Caspase1/11-/- Cdh5tTA/G2APOL1 and Gsdmd-/- Cdh5tTA/G2APOL1 mice, respectively.
For LPS endotoxemia, mice were injected intraperitoneally with LPS (6mg/kg) derived from Escherichia. coli O26:B6 (L8274, Sigma-Aldrich) dissolved in PBS and sacrificed after 24 h.
Cecal ligation and puncture was performed following a modification of a previously published method (Rittirsch et al., 2009). Briefly, mice were anesthetized with isoflurane. A 1- to 2-cm midline laparotomy was made to isolate and expose cecum. The cecum was ligated at 20% of its total length, and then punctured with a 20-gauge needle. A small amount of feces was extruded to ensure patency. The peritoneal wall and skin were closed. The sham group underwent the same surgery without cecum ligation and puncture. Pain control for CLP and sham mice was achieved after surgery.
We used the following inhibitors and doses for in vivo studies: VX765 (50mg/kg, Cayman), disulfiram (50mg/kg, Cayman), C176 (750nm per mouse, Bio Vision) or DMSO dissolved in 85ul corn oil were administered intraperitoneally at 2h, 24h, and 48h before the injection of LPS (6mg/kg) or PBS, all mice were sacrificed 24h after the injection of LPS.
All mice were provided food and water ad libitum and monitored daily.
Human participants
MESSI cohort: We included patients presenting to the emergency department and admitted to the medical ICU with plasma samples available at presentation and approximately 48 h later. MESSI patients met American College of Chest Physicians/Society of Critical Care Medicine consensus criteria for severe sepsis or septic shock (Levy et al., 2003). We defined AKI by KDIGO criteria (Kellum JA, 2012). Mortality was determined at 30 days after admission. Cohort characteristics are shown in Table S1.
COVID cohort: We included 30 symptomatic patients with positive SARS-CoV-2 PCR test admitted to Hospital of the University of Pennsylvania, US. Patients were recruited after hospitalization. We excluded chronic kidney disease (CKD) patients, pregnant patients and patients younger than 18 years old. Electronic medical records (EMR) were used to collect the data on all enrolled patients. Blood and urine were collected at admission time (n=30), 48 hours (n=27), 7 days (n=17), and 28 days (n=6). The survival was determined at 28 days. AKI was defined by KDIGO criteria (Kellum JA, 2012). eGFR was calculated using the Chronic Kidney Disease Epidemiology Collaboration equation. (Levey et al., 2009). Cohort characteristics are shown in Table S2.
MVP cohort: The Million Veteran Program is a longitudinal cohort study with clinical EHR data containing inpatient and outpatient data linked with genomic data. The Million Veteran Program recruits from approximately 63 Veterans Affairs facilities across the United States. Inclusion criteria in the MVP include age of 18 years or older, having a valid mailing address, and having the ability to provide informed consent. At recruitment, individuals completed baseline and lifestyle questionnaires, including self-reported race/ethnicity, and provided blood samples for genotyping and biomarker studies. Although MVP has genetic information in 660,000 individuals, the current analysis was done using pre-existing data available for another study in MVP release 2. In release 2 there were 57,000 Black participants.
Genotyping in MVP: The 2 APOL1 risk allele variants G1 (rs73885319 p.S342G; rs60910145 p.I384M) and G2 (rs71785313, a 6–base pair deletion that removes amino acids N388 and Y389) were directly genotyped on the Affymetrix Axiom Biobank Array platform on DNA that was extracted from whole blood. Individuals who carried G1S342G alleles (the functional missense variant in the G1 allele) in the absence of the G1I384M variant were considered to be G1 allele carriers. Participants were defined as 2 risk allele carriers if they were homozygotes for G1/G1, homozygotes for G2/G2, or compound heterozygotes for G1/G2 (Bick et al., 2019).
Study approval
Human kidney and lung samples were procured with approval from the Institutional Review Boards (IRBs) of the University of Pennsylvania.
For mouse studies, the protocol for use of all mice used in this study was approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
METHOD DETAILS
Renal phenotype analysis of mice
Urine albumin was determined using mouse albumin specific ELISA (Bethyl Laboratories) and creatinine by reagent set (Diazyme, DZ072b-KY1), per manufacturer’s protocol. Urine was subjected to SDS-PAGE and Coomassie brilliant blue staining (Biotium, 21003).
Histological Analysis
For histological analysis, kidneys, lungs, hearts and livers were fixed in 4% paraformaldehyde overnight, dehydrated, embedded into paraffin blocks and sectioned onto glass slides. Sections were stained with H&E. For in situ hybridization, paraffin-embedded tissue samples were processed using RNAscope 2.5 HD Duplex Detection Kit (bio-techne, 322436) protocol. The Hs-APOL1-C probe cat# 459791, Hs-APOL1-No-XMm-C2 cat# 459791-C2, Mm-Nlrp3 cat# 439571, Hs-PECAM1-O1 cat# 487381 were used. Immunofluorescence analyses were performed to visualize the expression of proteins in the lung. Briefly, paraffin-embedded sections were deparaffinized, rehydrated, and incubated with indicated primary antibodies. APOL1 antibody (Rabmab 5.17D12) applied in Figure 2C was a gift from Dr. Scales (Genentech) (Scales et al., 2020) . Slides were incubated with fluorescent conjugated secondary antibodies, counterstained and mounted with DAPI (nuclear stain).
Single cell sequencing
We followed our previously established protocol (Park et al., 2018). Kidneys were digested using Multi Tissue dissociation kit 1 (Miltenyi:130–110-2011). Debris removed using Miltenyi Debris Removal Solution and dead cells using the Miltenyi Dead Cell Removal kit. Cell number and viability were analyzed using Countess AutoCounter. Single cell sequencing was performed using the 10xGenomics Chromium 3’ Single Cell Controller System using a V3 chemistry.
Single cell sequencing analysis
Raw fastq files were aligned to the mm10 (Ensembl GRCm38.93) reference genome and quantified using CellRanger v3.1.0. Seurat R package v3.0 was used for data quality control, preprocessing, and dimensional reduction analysis. The WT and G2APOL1 data matrices were merged and poor-quality cells with <200 or >3000 expressed genes and mitochondrial gene percentages >50 were excluded. We identified 53,495 high quality cells. Seurat was used for further processing, dimension reduction and differential expression analysis.
Vascular permeability assay
30 mg/kg Evans blue dye (Sigma) in normal saline was intravenously injected by mice tail vein. After the injection, 5% mustard oil (Sigma) diluted in mineral oil was applied to the surfaces of the ears; 30 minutes later, mice were euthanized by CO2 inhalation, photographs were taken. For quantification of the vascular leakage, ears were collected and weighted, the Evans blue was extracted from the ears with formamide overnight at 55°C. The concentration of Evans blue dye was measured by spectrophotometer at 600nm and quantified according to a standard curve.
Isolation of primary pulmonary endothelial cells
We followed a previously described protocol to isolate mouse lung vascular endothelial cells (van Beijnum et al., 2008). Briefly, lungs were harvested, digested with 0.5 mg/ml collagenase I for 30 min at 37°C, and filtered through 40um cell strainer. Cells were centrifuged and incubation with anti-CD31-coated magnetic beads (Dynabeads, Invitrogen) for 15 min at room temperature, followed by magnetic separation per manufacturers’ instructions. Endothelial cells were then cultured in endothelial growth media (Microvascular EBM-2; Lonza, Basel, Switzerland) containing 5% Tet system approved FBS (Clontech) in a humidified atmosphere containing 5% CO2 at 37°C. APOL1 expression was inhibited by 300ng/ml doxycycline and induced by removing the doxycycline. Cells from passages 1–3 were used for all experiments.
Plasmids, short interfering RNA, and transfection
The COX8-EGFP-mCherry plasmid was obtained from Addgene (78520). STING, cGAS, and control siRNA were purchased from Integrated DNA Technologies (IDT). For transfection, cells were seeded in 6-well plates, grown for overnight until 60–70% confluency, and then transfected with 2500ng DNA, or 50 nM (final concentration) siRNA using Lipofectamine 3000 reagents (Thermo Fisher Scientific, 11668019). Cells were harvested 48 hours post infection under different conditions.
Electron microscopy
Dissected kidneys and lungs were fixed with 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, overnight at 4 °C, washed, dehydrated, and embedded in epoxy resin for sectioning according to standard procedures. Samples were examined using a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera and AMT Advantage image-capture software (at the UPenn Electron Microscopy Core). Scoring of the intracellular vesicular compartments was performed without information of sample identity (blinded). Statistical analysis of the number of structures of interest per cell profile was performed by randomly selecting and analyzing 30 cell sections from 2 independent grids.
RNA extraction and qRT-PCR
RNA was isolated from mouse tissue or primary cells using Trizol reagent (Invitrogen) following manufacturer’s protocol. RNA was transcribed into cDNA using cDNA Archival Kit (Life Technologies). cDNA was used in the qPCR reaction in a 384 well plate with SYBR green dye (Applied Biosystems) and gene-specific primer pairs. qPCR reaction was carried out in the ViiA 7 System (Life Technologies). The data were normalized and analyzed using the ΔΔCt method with indicated reference gene. APOL 1 and HPRT Taqman primers were from Life Technologies, other primers used are listed in Table S3.
Protein extraction and Western Blotting
All solutions, tubes, and centrifuges were maintained at 4°C. RIPA buffer (Cell signaling, #9806) with 1% SDS and protease inhibitor (Complete Mini, Roche) was used to extract total protein lysates from tissues or cells according to the manufacturer’s instructions. Protein concentrations were measured using BCA protein assay (Pierce, #23225). Lysates were heated (95°C) for 10 minutes in Laemmli sample buffer (Bio-Rad). Proteins were then separated by electrophoresis in acrylamide gels (8–15%) and transferred using a Bio-Rad western system to nitrocellulose (Bio-Rad) membranes. Transferred blots were blocked for 1h in 5% non-fat milk in Tris-buffered saline. Membranes were incubated with specific primary antibodies at 4°C overnight, followed by incubation with a horseradish peroxidase-conjugated anti-mouse antibody (1:5,000), or anti-rabbit antibody (1:5,000) at 25°C for 1 h. Resulting immunoblots were visualized using ECL Western Blotting Substrate (Pierce) in LI-COR chemiluminescence imager (LI-COR), according to the manufacturers’ instructions.
Immunofluorescence for cultured cells
Cells were seeded on glass coverslips, fixed with 75% cold ethanol, washed and blocked with blocking buffer (PBS, 10% bovine serum albumin). Primary antibodies were diluted in the blocking buffer and incubated for 2 hours, and then slides were incubated with fluorescent conjugated secondary antibodies, and mounted with DAPI (nuclear stain) for fluorescence microscopy.
LysoTracker labeling
Cells were incubated with 50 nM LysoTracker (Thermo Fisher Scientific) for 1min and cells were visualized using under an inverted fluorescence microscope.
Trans-endothelial electrical resistance (TEER).
Real time analysis of trans-endothelial impedance was measured by seeding 105 endothelial cells onto 8W10E+ 8 well arrays (Applied BioPhysics) pretreated with 10 mM l-cysteine and 1% gelatin. The 8W10E+ 8 well arrays were then put into a chamber at 37°C with 5% CO2. Resistance changes were measured at 4,000 Hz by an electric cell-substrate impedance sensor (ECIS) system (Applied BioPhysics). The results were then normalized to the WT and expressed as relative resistance.
Oxygen consumption measurements
For real-time analysis of the extracellular acidification rate (ECAR) and the oxygen consumption rate (OCR), endothelial cells were analyzed using an XF-96 Extracellular Flux Analyzer (Seahorse Bioscience). In brief, cells were plated in XF-96 cell culture plates (7ⅹ 104 cells/well in 70 µL) and either left unstimulated or stimulated with indicated LPS. At the indicated time points, cells were washed and analyzed in XF Running Buffer following manufacturer’s instructions to obtain real-time measurements of OCR and ECAR. Where indicated, ECAR and/or OCR were analyzed in response to 1µM oligomycin, 1.5µM fluorocarbonyl cyanide phenylhydrazone (FCCP) and 100 nM rotenone plus 1µM antimycin A.
Mitochondrial membrane potential (Δψm) measurement
Δψm was measured according to the manufacturer’s instructions (T3168, Invitrogen). Briefly, cells were washed twice with PBS, and then incubated with 5 µg/ml JC-1 for 10 min at 37 °C and 5% CO2. After washing with PBS twice, cells were analyzed for mitochondrial membrane potential by measuring the green: red ratio with an inverted fluorescence microscopy (Nikon, Japan).
ELISA quantification
APOL1 (Proteintech, KE00047), Il-10 (R&D Systems, M1000B), Il-1 beta (R&D Systems, MLB00C), TNFα (R&D Systems, MTA00B) and Angpt2 (R&D Systems, MANG20) concentration in serum samples were measured by specific sandwich ELISA kits according to the manufacturer’s instructions.
Subcellular fractionation
Quantification of mitochondrial DNA in the cytosol was adapted from Lauren D et al (Aarreberg et al., 2019). Briefly, cells were lysed by mild Digitonin buffer containing 150mM NaCl, 50mM HEPES pH7.4, 25ug/ml Digitonin (EMD Chemicals), Protease and phosphatase inhibitors and incubated on ice for 10 min then centrifuged at 2,000xg for 10 min at 4℃. Supernatants were centrifuged three times at 17,000xg for 10 min at 4℃ to yield cytosolic fraction free of nuclear and mitochondrial contamination.
The pellet from the first spin was washed and resuspended in NP-40 buffer containing 150mM NaCl, 50mM HEPES pH7.4, 1% NP-40, protease and phosphatase inhibitors and incubated on ice for 30 min then centrifuged at 7,000xg for 10 min at 4℃ to yield the crude mitochondria fraction (supernatant) and nuclear fraction (pellet). DNA was then isolated from these pure cytosolic and nuclear fractions using DNeasy Blood and Tissue kit (QIAGEN). The qPCR was performed on the cytosolic and nuclear fraction using nuclear DNA primer Rpl13 and mtDNA primer mtATP6. The CT values of Rpl13 obtained from the respective nuclear fraction served as normalization controls.
Measurements of MPO activity
MPO activity was determined using a myeloperoxidase assay kit (Sigma, MAK068–1KT) according to manufacturer’s protocol. Briefly, 50 μg of snap frozen lung from animals was homogenized in 200ul of MPO Assay Buffer and centrifuged at 4°C for 10 min at 13,000 rpm. MPO activity was determined in duplicate using development reagent. Activity was measured at 412 nm.
Plasma protein profiling
Plasma samples, collected from patients with COVID-19 were inactiveted with 1% TritonX (T9284, Sigma) before analyzed using an O-link Inflammation panels (OLINK Bioscience, Uppsala, Sweden). The microtiter plate provides measurements for 92 protein biomarkers, with data presented as Normalized Protein expression (NPX) values.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless noted otherwise, statistical significance was determined by one-way analysis of variance (ANOVA) with a Bonferroni multiple comparison post-test for multiple comparisons. Differences between two groups were analyzed using Student’s 2-tailed t-test with unequal variance. p < 0.05 was significant. All data are presented as mean ± SD and other details such as the number of replicates and the level of significance is mentioned in figure legends.
MVP Statistical analysis: We used a restricted PheWAS approach to determine the association of APOL1 risk alleles and infection events in the MVP cohort. We used logistic regression to test the associations between APOL1 risk alleles and the outcomes of interest: sepsis, septicemia, SIRS and infection inflammation of prosthetic devices. We used a recessive model and estimated odds ratios (ORs) and 95% CIs. All analyses were adjusted for (1) age and sex and 10 principal components of ancestry and eGFR. Analyses were performed using R, version 3.4.4 (R Project for Statistical Computing).
Supplementary Material
Highlights:
APOL1 risk variant (RV) is associated with increased sepsis incidence and severity
Mice with endothelial-specific RV APOL1 expression have increased sepsis severity
RV APOL1 interferes with mitophagy, leading to cytosolic release of mitochondrial DNA
Deletion or inhibition of NLRP3 and STING protects against RV APOL1-induced defects
Acknowledgement
Work in the Susztak lab is supported by the NIH NIDDK R01DK076077, R01 DK087635, and R01 DK105821. JW was supported by National Natural Science Foundation of China (81870487). AMH and HCC were supported by VA CSR&D MVP grant CX001897.
This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration. This publication does not represent the views of the Department of Veteran Affairs or the United States Government. Acknowledgement of the MVP leadership and staff contributions can be found in the Supplementary note 1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests:
The Susztak lab is supported by Boehringer Ingelheim, Regeneron, Bayer, GSK, Novartis and Novo Nordisk for work that is not related to the current manuscript. Dr Susztak is on the advisory board of Jnana.
REFERENCES
- Aarreberg LD, Esser-Nobis K, Driscoll C, Shuvarikov A, Roby JA, and Gale M Jr. (2019). Interleukin-1beta Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol Cell 74, 801–815 e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsan M, Stantcheva S, Yang D, and Cutler D (2020). Disparities in Coronavirus 2019 Reported Incidence, Knowledge, and Behavior Among US Adults. JAMA Netw Open 3, e2012403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj A, Susztak K, and Damrauer SM (2017). APOL1 and Cardiovascular Disease: A Story in Evolution. Arterioscler Thromb Vasc Biol 37, 1587–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerman P, Bi-Karchin J, Park AS, Qiu C, Dummer PD, Soomro I, Boustany-Kari CM, Pullen SS, Miner JH, Hu CA, et al. (2017). Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 23, 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick AG, Akwo E, Robinson-Cohen C, Lee K, Lynch J, Assimes TL, DuVall S, Edwards T, Fang H, Freiberg SM, et al. (2019). Association of APOL1 Risk Alleles With Cardiovascular Disease in Blacks in the Million Veteran Program. Circulation 140, 1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cates J, Lucero-Obusan C, Dahl RM, Schirmer P, Garg S, Oda G, Hall AJ, Langley G, Havers FP, Holodniy M, et al. (2020). Risk for In-Hospital Complications Associated with COVID-19 and Influenza - Veterans Health Administration, United States, October 1, 2018-May 31, 2020. MMWR Morb Mortal Wkly Rep 69, 1528–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier A, Ferlicot S, Chevalier K, Guillet M, Essig M, Jaureguiberry S, Collarino R, Dargelos M, Michaut A, Geri G, et al. (2020). Indirect effects of severe acute respiratory syndrome coronavirus 2 on the kidney in coronavirus disease patients. Clin Kidney J 13, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Kataria R, Zhang JY, Moore S, Petitpas K, Mohamed A, Zahler N, Pollak MR, and Olabisi OA (2020). Kidney Disease-Associated APOL1 Variants Have Dose-Dependent, Dominant Toxic Gain-of-Function. J Am Soc Nephrol 31, 2083–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrella MM, Wyatt CM, Pearce CL, Li M, Shlipak MG, Aouizerat BE, Gustafson D, Cohen MH, Gange SJ, Kao WH, et al. (2013). Host APOL1 genotype is independently associated with proteinuria in HIV infection. Kidney Int 84, 834–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, Gale NW, Witzenrath M, Rosseau S, Suttorp N, et al. (2006). Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med 12, 235–239. [DOI] [PubMed] [Google Scholar]
- Fine DM, Wasser WG, Estrella MM, Atta MG, Kuperman M, Shemer R, Rajasekaran A, Tzur S, Racusen LC, and Skorecki K (2012). APOL1 risk variants predict histopathology and progression to ESRD in HIV-related kidney disease. J Am Soc Nephrol 23, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman BI, Kopp JB, Sampson MG, and Susztak K (2021). APOL1 at 10 years: progress and next steps. Kidney Int 99, 1296–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DJ, Kozlitina J, Genovese G, Jog P, and Pollak MR (2011). Population-based risk assessment of APOL1 on renal disease. J Am Soc Nephrol 22, 2098–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, et al. (2010). Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovinazzo JA, Thomson RP, Khalizova N, Zager PJ, Malani N, Rodriguez-Boulan E, Raper J, and Schreiner R (2020). Apolipoprotein L-1 renal risk variants form active channels at the plasma membrane driving cytotoxicity. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granado D, Muller D, Krausel V, Kruzel-Davila E, Schuberth C, Eschborn M, Wedlich-Soldner R, Skorecki K, Pavenstadt H, Michgehl U, et al. (2017). Intracellular APOL1 Risk Variants Cause Cytotoxicity Accompanied by Energy Depletion. J Am Soc Nephrol 28, 3227–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter-Zinck H, Shi Y, Li M, Gorman BR, Ji SG, Sun N, Webster T, Liem A, Hsieh P, Devineni P, et al. (2020). Genotyping Array Design and Data Quality Control in the Million Veteran Program. Am J Hum Genet 106, 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilboudo H, Berthier D, Camara M, Camara O, Kabore J, Leno M, Keletigui S, Chantal I, Jamonneau V, Belem AM, et al. (2012). APOL1 expression is induced by Trypanosoma brucei gambiense infection but is not associated with differential susceptibility to sleeping sickness. Infect Genet Evol 12, 1519–1523. [DOI] [PubMed] [Google Scholar]
- Kellum JA, L. N, Aspelin P, Barsoum RS, Burdmann EA, Goldstein SL, Herzog CA, Joannidis M, Kribben A, Levey AS, MacLeod AM, Mehta RL, Murray PT, Naicker S, Opal SM, Schaefer F, Schetz M, Uchino S (2012). Kidney disease: Improving global outcomes (KDIGO) acute kidney injury work group KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl 2, 1–138. [Google Scholar]
- Kellum JA, Nadim MK, and Forni LG (2020). Sepsis-associated acute kidney injury: is COVID-19 different? Kidney Int 98, 1370–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, Friedman D, Briggs W, Dart R, Korbet S, et al. (2011). APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 22, 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruzel-Davila E, Shemer R, Ofir A, Bavli-Kertselli I, Darlyuk-Saadon I, Oren-Giladi P, Wasser WG, Magen D, Zaknoun E, Schuldiner M, et al. (2017). APOL1-Mediated Cell Injury Involves Disruption of Conserved Trafficking Processes. J Am Soc Nephrol 28, 1117–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudose S, Batal I, Santoriello D, Xu K, Barasch J, Peleg Y, Canetta P, Ratner LE, Marasa M, Gharavi AG, et al. (2020). Kidney Biopsy Findings in Patients with COVID-19. J Am Soc Nephrol 31, 1959–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazareth H, Pere H, Binois Y, Chabannes M, Schurder J, Bruneau T, Karras A, Thervet E, Rabant M, Veyer D, et al. (2020). COVID-19-Related Collapsing Glomerulopathy in a Kidney Transplant Recipient. Am J Kidney Dis 76, 590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, et al. (2009). A new equation to estimate glomerular filtration rate. Ann Intern Med 150, 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, and Ramsay G (2003). 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 31, 1250–1256. [DOI] [PubMed] [Google Scholar]
- Lukasz A, Hellpap J, Horn R, Kielstein JT, David S, Haller H, and Kumpers P (2008). Circulating angiopoietin-1 and angiopoietin-2 in critically ill patients: development and clinical application of two new immunoassays. Crit Care 12, R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Wang Y, Zhang L, Ren P, Zhang C, Li Y, Azares AR, Zhang M, Guo J, Ghaghada KB, et al. (2020). Critical Role of Cytosolic DNA and Its Sensing Adaptor STING in Aortic Degeneration, Dissection, and Rupture. Circulation 141, 42–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Chou JW, Snipes JA, Bharadwaj MS, Craddock AL, Cheng D, Weckerle A, Petrovic S, Hicks PJ, Hemal AK, et al. (2017). APOL1 Renal-Risk Variants Induce Mitochondrial Dysfunction. J Am Soc Nephrol 28, 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May RM, Cassol C, Hannoudi A, Larsen CP, Lerma E, Haun RS, Braga JR, Hassen SI, Wilson J, VanBeek C, et al. (2021). A multi-center retrospective cohort study defines the spectrum of kidney pathology in Coronavirus 2019 Disease (COVID-19). Kidney Int [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Portela Mdel P, Lugli EB, Recio-Pinto E, and Raper J (2005). Trypanosome lytic factor, a subclass of high-density lipoprotein, forms cation-selective pores in membranes. Mol Biochem Parasitol 144, 218–226. [DOI] [PubMed] [Google Scholar]
- Nichols B, Jog P, Lee JH, Blackler D, Wilmot M, D’Agati V, Markowitz G, Kopp JB, Alper SL, Pollak MR, et al. (2015). Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int 87, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oniszczuk J, Moktefi A, Mausoleo A, Pallet N, Malard-Castagnet S, Fourati S, El Karoui K, Sahali D, Stehle T, Boueilh A, et al. (2020). De Novo Focal and Segmental Glomerulosclerosis After COVID-19 in a Patient With a Transplanted Kidney From a Donor With a High-risk APOL1 Variant. Transplantation [DOI] [PubMed] [Google Scholar]
- Osuchowski MF, Winkler MS, Skirecki T, Cajander S, Shankar-Hari M, Lachmann G, Monneret G, Venet F, Bauer M, Brunkhorst FM, et al. (2021). The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir Med 9, 622–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papeta N, Kiryluk K, Patel A, Sterken R, Kacak N, Snyder HJ, Imus PH, Mhatre AN, Lawani AK, Julian BA, et al. (2011). APOL1 variants increase risk for FSGS and HIVAN but not IgA nephropathy. J Am Soc Nephrol 22, 1991–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, and Susztak K (2018). Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pays E (2020). APOL1 variant-associated kidney disease: from trypanosomes to podocyte cytoskeleton. Kidney Int 98, 1373–1377. [DOI] [PubMed] [Google Scholar]
- Reidy KJ, Hjorten RC, Simpson CL, Rosenberg AZ, Rosenblum SD, Kovesdy CP, Tylavsky FA, Myrie J, Ruiz BL, Haque S, et al. (2018). Fetal-Not Maternal-APOL1 Genotype Associated with Risk for Preeclampsia in Those with African Ancestry. Am J Hum Genet 103, 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly JP, Anderson BJ, Mangalmurti NS, Nguyen TD, Holena DN, Wu Q, Nguyen ET, Reilly MP, Lanken PN, Christie JD, et al. (2015). The ABO Histo-Blood Group and AKI in Critically Ill Patients with Trauma or Sepsis. Clin J Am Soc Nephrol 10, 1911–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly JP, Wang F, Jones TK, Palakshappa JA, Anderson BJ, Shashaty MGS, Dunn TG, Johansson ED, Riley TR, Lim B, et al. (2018). Plasma angiopoietin-2 as a potential causal marker in sepsis-associated ARDS development: evidence from Mendelian randomization and mediation analysis. Intensive Care Med 44, 1849–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittirsch D, Huber-Lang MS, Flierl MA, and Ward PA (2009). Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 4, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales SJ, Gupta N, De Maziere AM, Posthuma G, Chiu CP, Pierce AA, Hotzel K, Tao J, Foreman O, Koukos G, et al. (2020). Apolipoprotein L1-Specific Antibodies Detect Endogenous APOL1 inside the Endoplasmic Reticulum and on the Plasma Membrane of Podocytes. J Am Soc Nephrol 31, 2044–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SS, Lannon H, Dias L, Zhang JY, Alper SL, Pollak MR, and Friedman DJ (2019). APOL1 Kidney Risk Variants Induce Cell Death via Mitochondrial Translocation and Opening of the Mitochondrial Permeability Transition Pore. J Am Soc Nephrol 30, 2355–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma Y, Nasr SH, Larsen CP, Kemper A, Ormsby AH, and Williamson SR (2020). COVID-19-Associated Collapsing Focal Segmental Glomerulosclerosis: A Report of 2 Cases. Kidney Med 2, 493–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashaty MGS, Reilly JP, Faust HE, Forker CM, Ittner CAG, Zhang PX, Hotz MJ, Fitzgerald D, Yang W, Anderson BJ, et al. (2019). Plasma receptor interacting protein kinase-3 levels are associated with acute respiratory distress syndrome in sepsis and trauma: a cohort study. Crit Care 23, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukha K, Mueller JL, Chung RT, Curry MP, Friedman DJ, Pollak MR, and Berg AH (2017). Most ApoL1 Is Secreted by the Liver. Journal of the American Society of Nephrology 28, 1079–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez De La Rica A, Gilsanz F, and Maseda E (2016). Epidemiologic trends of sepsis in western countries. Ann Transl Med 4, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulcek R, Bogaard HJ, and van Nieuw Amerongen GP (2014). Electric cell-substrate impedance sensing for the quantification of endothelial proliferation, barrier function, and motility. J Vis Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson R, and Finkelstein A (2015). Human trypanolytic factor APOL1 forms pH-gated cation-selective channels in planar lipid bilayers: relevance to trypanosome lysis. Proc Natl Acad Sci U S A 112, 2894–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson R, Genovese G, Canon C, Kovacsics D, Higgins MK, Carrington M, Winkler CA, Kopp J, Rotimi C, Adeyemo A, et al. (2014). Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci U S A 111, E2130–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiberti N, Matovu E, Hainard A, Enyaru JC, Lejon V, Robin X, Turck N, Ngoyi DM, Krishna S, Bisser S, et al. (2013). New biomarkers for stage determination in Trypanosoma brucei rhodesiense sleeping sickness patients. Clin Transl Med 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, Bekele E, Bradman N, Wasser WG, Behar DM, et al. (2010). Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 128, 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beijnum JR, Rousch M, Castermans K, van der Linden E, and Griffioen AW (2008). Isolation of endothelial cells from fresh tissues. Nat Protoc 3, 1085–1091. [DOI] [PubMed] [Google Scholar]
- Vanwalleghem G, Fontaine F, Lecordier L, Tebabi P, Klewe K, Nolan DP, Yamaryo-Botte Y, Botte C, Kremer A, Burkard GS, et al. (2015). Coupling of lysosomal and mitochondrial membrane permeabilization in trypanolysis by APOL1. Nat Commun 6, 8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107, 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward C, Martinez-Lopez N, Otten EG, Carroll B, Maetzel D, Singh R, Sarkar S, and Korolchuk VI (2016). Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim Biophys Acta 1861, 269–284. [DOI] [PubMed] [Google Scholar]
- Wasser WG, Tzur S, Wolday D, Adu D, Baumstein D, Rosset S, and Skorecki K (2012). Population genetics of chronic kidney disease: the evolving story of APOL1. J Nephrol 25, 603–618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Caspase-1 | Santa Cruz | Cat#SC56036 |
| Anti-GAPDH | CST | Cat#2118 |
| Anti-GSMD | Abcam | Cat#ab209845 |
| Anti-APOL1 | Proteintech | Cat#66124–1-Ig |
| Anti-APOL1 | Genentech | Rabmab 5.17D12 |
| Anti-GFP | Abcam | Cat#AB6673 |
| Anti-LC3II | CST | Cat#2775 |
| Anti-CD31 | Abcam | Cat#ab24590 |
| Anti-Icam1 | Abcam | Cat#ab179707 |
| Anti-Vcam1 | Abcam | Cat#ab134047 |
| Anti-Caveolin-1 | CST | Cat#3267 |
| Anti-Enos | Abcam | Cat#ab5589 |
| Anti-Hsp60 | CST | Cat#12165 |
| Anti-Cox4 | R&D Systems | Cat#MAB6980 |
| Anti-STING | CST | Cat#13647s |
| Anti-phosphorylated STING | CST | Cat#19781s |
| Anti-cGAS | CST | Cat#31659S |
| Anti-TBK1 | CST | Cat#3504S |
| Anti-phosphorylated TBK1 | CST | Cat#5483 |
| Anti-IRF3 | CST | Cat#11904T |
| Anti-phosphorylated IFR3 | CST | Cat#37829 |
| Anti-P65 | CST | Cat#5483 |
| Anti-Phospho-NF-κB p65 | CST | Cat#8242 |
| Monoclonal Anti-β-Actin−Peroxidase antibody | Sigma Aldrich | Cat#A3854 |
| Anti-VE-cadherin | Santa Cruz | Cat#sc-6458 |
| OXPHOS antibody cocktail | Abcam | Cat#ab110413 |
| Anti-p62 | CST | Cat#5114 |
| Donkey anti-Rabbit IgG (H+L) Alexa Fluor 555 | Invitrogen | Cat#A31572 |
| Chicken anti-Mouse IgG (H+L), Alexa Fluor 488 | Invitrogen | Cat#21200 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LPS (Escherichia. coli O26:B6) | Sigma-Aldrich | Cat#L8274 |
| VX765 | Cayman | Cat#CAYM-28825–50 |
| Disulfiram | Cayman | Cat#97–77-8 |
| C176 | Bio Vision | Cat#B2341 |
| Evans blue dye | Sigma | Cat#E2129 |
| CD31 MicroBeads, mouse | miltenyi biotec | Cat#130–097-418 |
| TET system approved FBS | clontech | Cat#631106 |
| Microvascular EBM-2 | Lonza | Cat#cc-3162 |
| Hydrogen peroxide solution 30% | Sigma | H1009–500ML |
| Trizol | Invitrogen | Cat#15596018 |
| cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368813 |
| Power SYBR® Green PCR Master Mix | Applied Biosystems | Cat#4367659 |
| RIPA buffer | Cell signaling | Cat#9806 |
| Protease cocktail | Roche | Cat#11836153001 |
| BCA protein assay | Thermo Scientific | Cat#23225 |
| Western Blotting Substrate | Pierce | Cat#32106 |
| Lipofectamine 3000 reagents | Thermo Fisher Scientific | Cat#11668019 |
| LysoTracker™ Blue DND-22 | Thermo Fisher Scientific | Cat#L7525 |
| ECIS Cultureware Disposable Electrode Arrays | Applied BioPhysics | 8W10E+ (8 Well PET) |
| l-cysteine | Sigma | Cat#C7352–25G |
| Gelatin solution | Sigma | Cat#G1393–100ML |
| JC-1 | Invitrogen | Cat#T3168 |
| Digitonin | EMD Millipore | Cat#11024–24-1 |
| DNeasy Blood and Tissue kit | QIAGEN | Cat#69506 |
| COX8-EGFP-mCherry plasmid | Addgene | Cat#78520 |
| Hs-APOL1 -C probe | bio-techne | Cat#459791 |
| APOL1-No-XMm-C2 | bio-techne | Cat#459798-C2 |
| Mm-Nlrp3 | bio-techne | Cat#439571 |
| Hs-PECAM1-O1 | bio-techne | Cat#487381 |
| Critical Commercial Assays | ||
| Mouse albumin specific ELISA | Bethyl Laboratories | Cat#E99–134 |
| Creatinine reagent | Diazyme | Cat#DZ072B |
| RNAscope 2.5 HD Duplex Detection Kit | bio-techne | Cat#322436 |
| Multi Tissue dissociation kit 1 | Miltenyi | Cat#130–110-2011 |
| APOL1 ELISA Kit | Proteintech | Cat#KE00047 |
| Mouse IL-1 beta ELISA Kit | R&D Systems | MLB00C |
| Mouse TNF-alpha | R&D Systems | MTA00B |
| Il-10 ELISA Kit | R&D Systems | Cat#M1000B |
| Angpt2 ELISA Kit | R&D Systems | Cat#MANG20 |
| Myeloperoxidase assay kit | Sigma | Cat#MAK068–1KT |
| Expermental Models:Organisms/Strains | ||
| Cdh5 tTA | Jackson Laboratory | Stock#013585 |
| GT26.rtTA | Jackson Laboratory | Stock#5670 |
| Nlrp3KO | Jackson Laboratory | Stock#21302 |
| STING KO | Jackson Laboratory | Stock#025805 |
| Caspase1/11 KO | Jackson Laboratory | Stock#16621 |
| Gsdmd KO | Jackson Laboratory | Stock#032410 |
| Alb-Cre | Jackson Laboratory | Stock#003574 |
| TREG0APOL1 | N/A | |
| TREG2APOL1 | N/A | |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO:GSE181671 |
| Seqence-based Reagents | ||
| Primers see Table S2 | This paper | N/A |
| siRNA targeting sequence: Tmem173 (Duplex 1): GAAUCGGGUUUAUUCCAACAGCGTC |
IDT | N/A |
| siRNA targeting sequence: Tmem173 (Duplex 2): UUCUUAGCCCAAAUAAGGUUGUCGCAG |
IDT | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij |
| Prism 8 | Graphpad Software | https://www.graphpad.com/scientific-software/prism |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.