

Abstract
It is documented that stress activates the locus coeruleus-norepinephrine system. However, there are far few reports regarding effects of stress on the expression of dopamine β-hydroxylase, a hallmark enzyme of the noradrenergic neuron. In the present study, adult Fischer 344 rats were subjected to chronic social defeat for 4 weeks. Dopamine β-hydroxylase expressional levels in the locus coeruleus and its terminal regions were measured by in situ hybridization and western blotting. The results showed that immediately following chronic social defeat there are significantly increased mRNA and protein levels of dopamine β-hydroxylase in the locus coeruleus, and dopamine β-hydroxylase protein levels in the hippocampus, frontal cortex and amygdala, compared with those in the control. This chronic social defeat-induced upregulation of dopamine β-hydroxylase was completely abolished by adrenalectomy, and/or by treatment with corticosteroid receptor antagonists, mifepristone and spironolactone, either alone or in combination. Furthermore, treatment with desipramine, an antidepressant with specific inhibitory effects on norepinephrine transport, prevented an increased dopamine β-hydroxylase expression by chronic social defeat in the locus coeruleus and its main terminal regions such as the hippocampus, frontal cortex and amygdala. However, treatment with fluoxetine, an antidepressant with specific inhibition for serotonin transport, only selectively blocked increased dopamine β-hydroxylase protein levels in the hippocampus caused by CSD. The present findings indicate that chronic social defeat activates the locus coeruleus-norepinephrine system by upregulating the expression of dopamine β-hydroxylase, which may increase norepinephrine synthesis. This chronic social defeat induced upregulation of DBH expression was mediated through corticosterone and corticosteroid receptors, with possible interference from antidepressants.
Keywords: locus coeruleus, corticosterone, antidepressants, stress related disorders, norepinephrine
INTRODUCTION
Dopamine β-hydroxylase (DBH; EC 1.14.17.1) is an important enzyme that catalyzes the oxidation of dopamine to norepinephrine (NE) (Friedman et al., 1965). This enzyme is exclusively expressed in the noradrenergic and adrenergic neurons in the brain, as well as in the sympathetic ganglia and adrenomedullary chromaffin cells in the peripheral tissues (Goldstein et al., 1972). Because of its unique distribution in the noradrenergic system as a marker enzyme, DBH plays important roles both physiologically and pathologically. For example, DBH has been reported to be related to the control of blood pressure (Ohlstein et al., 1987), and its activity was altered in some psychiatric diseases including depression (Hornykiewicz, 1982, Nagatsu, 1986). It has been documented that a broad range of psychiatric disorders are causally related to chronic stress. An important fact is that stress can rapidly activate the noradrenergic system and maladaptation of the noradrenergic system to chronic stress increases the precipitation of psychiatric disorders (Charney, 1998). Therefore, exploring the correlation between chronic stress and alteration of the noradrenergic system would improve our understanding of the intrinsic mechanism underlying the development of these psychiatric diseases (Carrasco et al., 2003).
NE is crucial in stimulating action in response to a perceived threat, serving as an essential neurotransmitter to regulate arousal and adapt to environmental and internal stressors (Goddard et al., 2010). Increased levels of NE due to the stress stimuli may be compensated by an increase in the expression of genes encoding NE synthesizing enzymes (Sabban et al., 2001). Among these enzymes, tyrosine hydroxylase (TH) is the rate-limiting enzyme for NE synthesis (Udenfriend, 1966). Nevertheless, it has been suggested that the amount of DBH is also a key factor to determine the rate of NE synthesis (Kobayashi et al., 1994, Kim et al., 2002). It has been reported that immobilization stress elevated mRNA and protein levels of DBH in the locus coeruleus (LC) (Serova et al., 1999). Also, DBH mRNA levels were markedly elevated in adrenal medulla of stressed mice (Kubovcakova et al., 2004, Kvetnansky et al., 2004). However, compared to the intensive studies about TH activity and expression caused by stress (Melia et al., 1991, Smith et al., 1991, Watanabe et al., 1995, McDougall et al., 2005), the investigation to examine expressional alterations of DBH after chronic stress is limited, either in the intensity or diversity of stressors. Also, the investigation for effects of stress on DBH expression in the LC projection regions is lacking. Obviously, more thorough studies to explore expressional changes of DBH in the brain after chronic stress are essential.
In the previous study, we found that chronic social defeat (CSD) regimen significantly increased both mRNA and protein levels of NET in the rat LC, as well as NET protein levels in such LC terminal projection regions as the hippocampus, frontal cortex, and amygdala. This upregulated NET expression results in part from stress-induced release of corticosterone acting through corticosteroid receptors, as either adrenalectomy or treatment with the corticosteroid receptor antagonists abolished or reduced the elevation of NET expression induced by CSD. These observations indicate that CSD upregulated NET in the central noradrenergic neurons, which may be related to the development of depression.
As NET has also been considered the marker protein of the noradrenergic neurons and both NET and DBH are similarly, although not always, distributed in the noradrenergic neurons, in the present study the same rat CSD model was used to investigate effects of this chronic stressor on the expression of DBH in the LC and its major terminal regions such as the hippocampus, frontal cortex and amygdala. In this study, we also examined the potential effect of antidepressants on stress-induced alteration of DBH expression, besides corticosteroid receptor antagonists. Our results showed that CSD upregulates the expression of DBH in the LC and its projection areas, which is also mainly mediated by stress-released corticosterone. As DBH is a hallmark of the noradrenergic system, these observations reveal that chronic stress activates this system, implying that the stress-induced alteration in the noradrenergic system may play an important role during stress-related psychiatric disorders.
EXPERIMENTAL PROCEDURES
Animals
Male Fischer 344 rats, weighing 200–250 g at the beginning of the experiment, Long-Evans retired male breeder and ovariectomized female rats were purchased from Harlan Sprague Dawley, Inc. (Indianapolis, IN). All animal procedures were approved by the Animal Care and Use Committee of East Tennessee State University, and complied with the NIH Guide for the Care and Use of Laboratory Animals. Rats were maintained on a 12-h light/dark cycle (lights on at 07:00 h) with ad-libitum access to food and tap water except as specifically described below. After an acclimation period of 5 days, rats were randomly assigned to experimental groups.
Chronic social defeat paradigm and drug treatments
This paradigm is the same as reported previously (Chen et al., 2012). Briefly, the experiment began (at 9:00 am of each experimental day) by exposing an “intruder” (adult male Fischer rat) to the home cage of the “resident” (larger male Long-Evans rat) after female rats had been removed. After the “intruder” was attacked and showed the defeated behavior, the “intruder” and “resident” were immediately separated, but still kept in the same home cage for one and half hour to remain unrestricted visual, auditory, and olfactory contact with each other without physical attack. This exposure (called a session) was repeated four times in the first and fourth weeks, and two times in the second and third weeks. Some rats (as the control) were given access to the entire resident home cage when the residents have been removed (sham-exposure). Therefore, the controls are never physically attacked and defeated by the residents. One group of rats were adrenalectomized (ADX) by Harlan Laboratories Inc. before shipping to the animal facility of East Tennessee State University. The care of ADX rats was the same as reported previously (Chen et al., 2012). Briefly, ADX rats were provided with the drinking water containing 25 μg/mL corticosterone during whole experimental period. Such small replacement dose of corticosterone has been shown to be adequate for prevention of post-adrenalectomy alterations in the central nervous system (Pace et al., 2009). Trunk blood was taken when rats were sacrificed by rapid decapitation immediately after the last session of CSD on 28th day (around 11:00 am). After rats were sacrificed, brains were removed and rapidly frozen in 2-methyl-butane on dry-ice, then stored at −80°C until dissection. For the immunofluorescence staining experiments, rats were transcardially perfused under anesthesia with pentobarbital (40 mg/kg, i.p.) using 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4.
To examine whether these receptors are involved in the CSD-induced regulation of DBH expression, relatively specific glucocorticoid receptor (GR) antagonist mifepristone and mineralocorticoid receptors (MR) antagonist spironolactone were utilized in these animals. Therefore, some groups of rats were treated with mifepristone (10 mg/kg, daily, s.c.) or spironolactone (15 mg/kg, daily, s.c.), either alone or in combination (with the same dose, respectively), 30 minutes prior to each CSD session and lasted for whole CSD regimen. Also, some groups of animal were treated with antidepressants desipramine (10 mg/kg, i.p.) or fluoxetine (10 mg/kg, i.p.) 30 minutes prior to each CSD session and lasted for whole CSD regimen. All The doses of these compounds are chosen on the basis of previous reports (Ratka et al., 1989, Ni et al., 1995, Haller et al., 1998, Macunluoglu et al., 2008) and our preliminary experiments. All these compounds were purchased from Sigma-Aldrich (St. Louis, MO). Rats in the untreated control and CSD alone groups were injected with similar volumes of vehicle in the same manner.
Sucrose consumption test
This test is the same as reported previously (Chen et al., 2012). Briefly, after animals were initially exposed to two bottles (either 1% sucrose solution or tap water) for 72 h to familiarize with the test condition, water and sucrose consumption was measured by 1-h exposure (between 06:30 pm and 07:30 pm) after 10 h water deprivation as the basic rate. Then, this consumption test (deprivation in the morning and two bottles of drinking solution provided in the one hour window) was carried out weekly on the same day of the week (Thursday) throughout the four week CSD regimen.
Rat blood sampling and plasma corticosterone determination
This method is also the same as reported previously (Chen et al., 2012). Briefly, after trunk blood was quickly collected, plasma was immediately prepared and temporarily stored at −80°C. Plasma corticosterone was measured by a radioimmunoassay using a commercial kit [ImmuChem radioimmunoassay kit, MP Biomedicals, LLC in Orangeburg, NY (formerly ICN Pharmaceuticals, Costa Mesa, CA)] according to the manufacturer’s instruction with some modifications. All samples were run in a single assay with an intra-assay variance of less than 8%.
In situ hybridization
The in situ hybridization method is the same as described before (Zhu et al., 2002). Briefly, after rats were sacrificed, brains were removed and rapidly frozen in 2-methyl-butane on dry-ice, then stored at −80°C until slicing. Sections (16 μm) around the pons-brain stem LC region were cut on a cryostat, mounted on SuperFrost Plus slides (Fisher Scientific; Pittsburg, PA), and stored at −80°C. When hybridization was performed, the slides were fixed with 4% (w/v) paraformaldehyde followed by acetylation with acetic anhydride. Lipids were extracted by washing with increasing concentrations of alcohol (50, 70, 95, and 100% [vols]). The [35S]-labeled cRNA probes (Perkinelmer, MA) were transcribed in vitro from cDNAs for rat DBH (1.4 kb, which was hydrolyzed about to 0.5 kb after riboprobe preparation for proper binding) in pGEM-3Zf vectors with T7 RNA polymerase. Prehybridized sections were incubated with hybridization solution containing the radiolabeled probes at 55°C for 3–5 h. The brain tissue sections were then washed extensively and apposed to Biomax autoradiographic films (Kodak; Rochester, NY). For higher-resolution studies, sections were also dipped in Kodak NTB2 emulsion (Fisher, Pittsburgh, PA) and quantitatively analyzed with the Bioquant Nova program (R.M. Biometrics, Nashville, TN). The specificity of cRNA probes was tested using three criteria. First, sense probes synthesized from each cDNA were used to perform in situ hybridization in parallel with antisense probes. There were no specific signals on these slides. Second, antisense probes were used on control slides from the cerebellum and cortex and no hybridization signals were detected. Third, antisense probes were hybridized to slides that were treated with RNase A (20 μg/mL) and no hybridization signal was detected. For analysis of in situ hybridization results, three sections from each rat and bilateral LC regions from each section were quantitated. A mean value was obtained from these six measurements and represented for each rat.
Western blotting
The LC, hippocampus, frontal cortex and amygdala of rats were punched from frozen coronal sections, guided by the rat brain atlas of Paxinos and Watson (Paxinos et al., 2005). Protein levels of DBH in these multiple regions were determined by western blotting as described before (Chen et al., 2012). Briefly, these samples were prepared and equal amounts of samples (10 μg of protein per lane) were loaded on 10% SDS-polyacrylamide gels for electrophoresis. After electro-blotting, the membranes were incubated with primary antibody against DBH (1:330 dilution; Alpha Diagnostic Intl. Inc, San Antonio, TX) overnight at 4°C, and then further incubated with secondary anti-bodies (horseradish peroxidase-conjugated anti-rabbit IgG, 1:3000; Amersham Biosciences, Little Chalfont, UK). Immunoreactive bands were visualized by enhanced chemiluminescence (ECL, Amersham; Piscataway, NJ), and then quantified by imaging software (Molecular Dynamics IQ solutions, Molecular Dynamics, Inc, Sunnyvale, CA). A linear standard curve was created from optical densities (ODs) of bands with a dilution series of total proteins prepared from brain tissues. OD values of DBH signals were compared and normalized with β-actin immunoreactivities, which were determined on the same blot, to assess equal protein loading. Normalized values were then averaged for all replicated gels and used to calculate the relative changes of the same gel.
Immunofluorescence staining for DBH
For the immunofluorescence staining experiments, the immunofluorescence staining method is similar to that described previously (Fan et al., 2011). Briefly, brain sections were pre-incubated in 5% bovine serum albumin in phosphate-buffered saline supplemented with 0.2% Triton-X 100 for 1 h at room temperature, followed by incubation with the primary polyclonal antibody against DBH raised from rabbit (1:500; Protos Biotech Corp, New York, NY) overnight at 4°C. Then sections were incubated with the secondary antibody (Alexa Fluor 488-conjugated goat anti-rabbit IgG, Invitrogen, Carlsbad, CA) for 2 h at room temperature. Immunofluorescence labeling was observed and acquired under a Leica TCS SP2 confocal microscope system (Leica Microsystems, Bannock-burn, IL). Images were semiquantitatively analyzed by using ImageJ software (Rasband, US National Institutes of Health, Bethesda, http://rsbweb.nih.gov/ij, 2010). Reference background levels were obtained from non-immunoreactive portions of brain sections adjacent to the LC region by determining the optical density on a 0–255 grayscale (0 being white and 255 black). The area fraction of immunofluorescence was quantified at three levels of each section and two sections from each rat.
Statistics
All experimental data are presented in the text and graphs as the mean ± SEM. Data in Figure 1B were analyzed by the repeated measure analysis of variance (ANOVA) and data in other figures were analyzed by one way ANOVA (SigmaStat, Systat Software, Richmond, VA) when multiple treatment groups were compared in all experiments. Then post-hoc Student-Newman-Keuls tests were performed for planned comparisons.
Fig. 1.
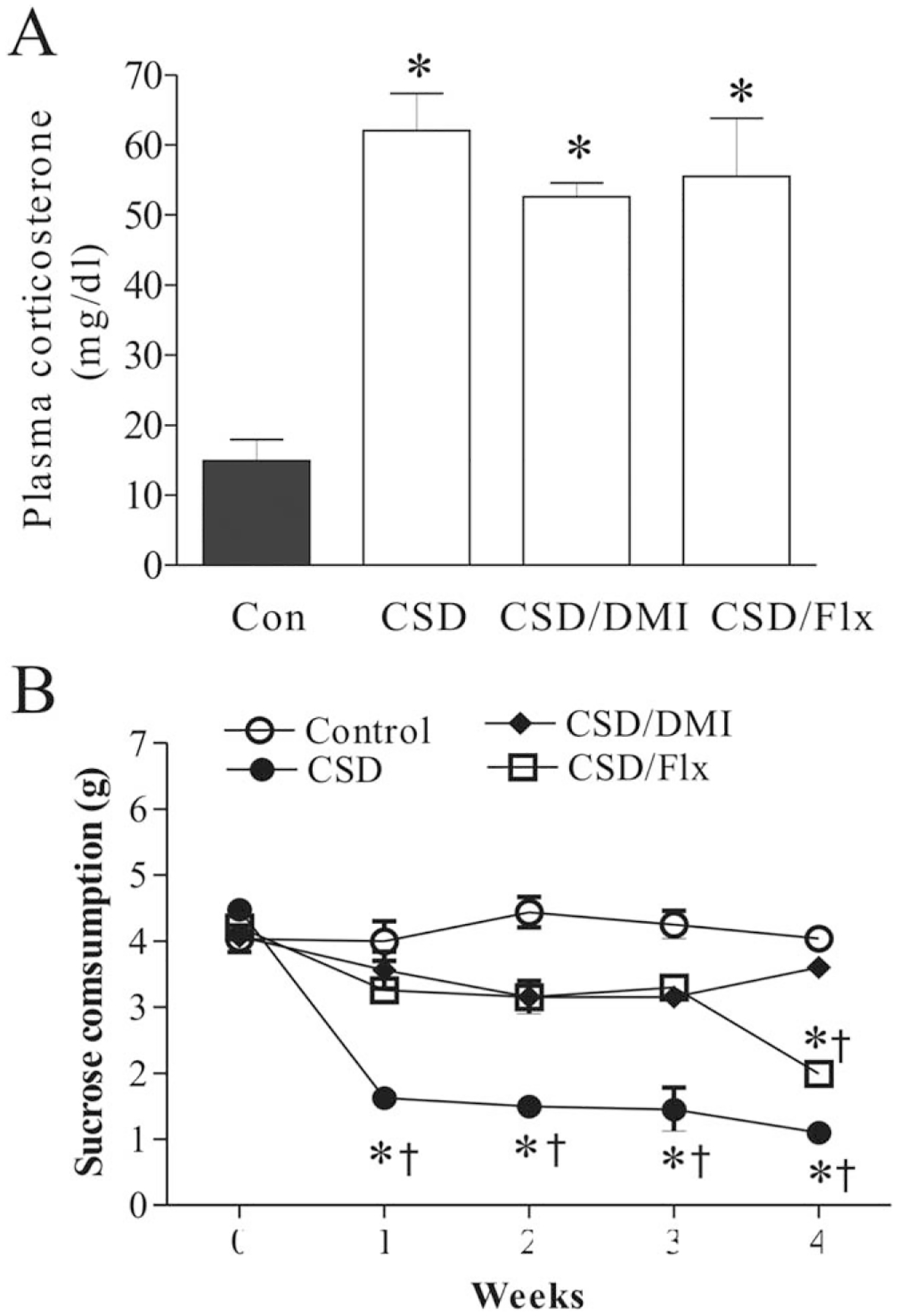
Effects of chronic social defeat (CSD) stress, and CSD plus treatment with desipramine or fluoxetine on plasma corticosterone concentrations (A, n = 8/group) and sucrose consumption (B, n = 8/group). The trunk blood was collected on the 28th day immediately after the end of last session of CSD. *P < 0.01, compared with the control; † P < 0.01, compared with the corresponding time point in the control group. Con, control; CSD/DMI, CSD plus treatment with desipramine; CSD/Flx, CSD plus treatment with fluoxetine.
The photomicrographs were produced by following procedures and softwares: images were taken using MCID CORE 7.0 program (Imaging Research; Linton, England) and pasted to CorelDRAWx4 software Corel, Mountain View, CA) without modification of contrast and brightness or any parameter except for sizing of images and addition of labels.
RESULTS
Plasma corticosterone measurement and sucrose consumption test
Plasma corticosterone levels were measured to define the efficacy of chronic stress in inducing hormonal modification in samples collected at the end of the 4-week stress. CSD regime significantly affected plasma corticosterone levels (F3,31=13.60, P < 0.001, Fig. 1A). Post hoc test revealed that plasma corticosterone levels were significantly elevated in the rats subjected to CSD (P < 0.01), as compared with control rats. However, treatment with desipramine or fluoxetine failed to change CSD-released higher corticosterone levels significantly, although the corticosterone levels in these two groups were lower than that of the CSD group.
The sucrose consumption test was performed to measure anhedonia, a core symptom of depression. As shown in Figure 1B, the intake of the sucrose solution was not significantly changed in nonstressed animals over the whole duration of the experiment (F4,34 = 1.13, P > 0.05). However, it was significantly changed by CSD regime (F4,34 =52.02, P < 0.001). Further tests showed that the sucrose consumption was significantly reduced from the first to fourth week of stress exposure in CSD rats, compared to the basic level (0 time) (all P < 0.01). Also, the sucrose intake in the first week to fourth week was significantly lower than those in the corresponding time in the nonstressed animals (all P < 0.01). Furthermore, there is no significant difference for the sucrose intakes between CSD rats treated with desipramine and the control group. In CSD rats treated with fluoxetine the sucrose intakes were similar to those of desipramine-treated rats in the first three week of CSD exposure. However, in the fourth week of stress exposure the sucrose consumption was markedly reduced (P < 0.01), as compared with the basic level (0 time), which was also significant lower compared to that in the nonstressed animals (P < 0.05).
CSD upregulated DBH mRNA levels in the LC of rats
Ex vivo in situ hybridization was performed to measure effects of CSD and related treatments on mRNA levels of DBH in rat LC. First, whether stress-released corticosterone is involved in the CSD-caused alteration of DBH mRNA levels was investigated. As shown in Figure 2A, immediately after CSD regime, there are markedly influenced DBH mRNA levels (F3,31=4.56, P < 0.01). Post hoc tests revealed that mRNA levels of DBH in sham group (sham for ADX) were similar to those of the control rats. However, CSD significantly increased mRNA levels of DBH in the LC region by 75.5% (P < 0.05), compared with those in the control. This CSD-induced elevation in DBH mRNA levels was abolished by ADX (P < 0.01), indicating the stress-released corticosterone plays a role in this regulation.
Fig. 2.
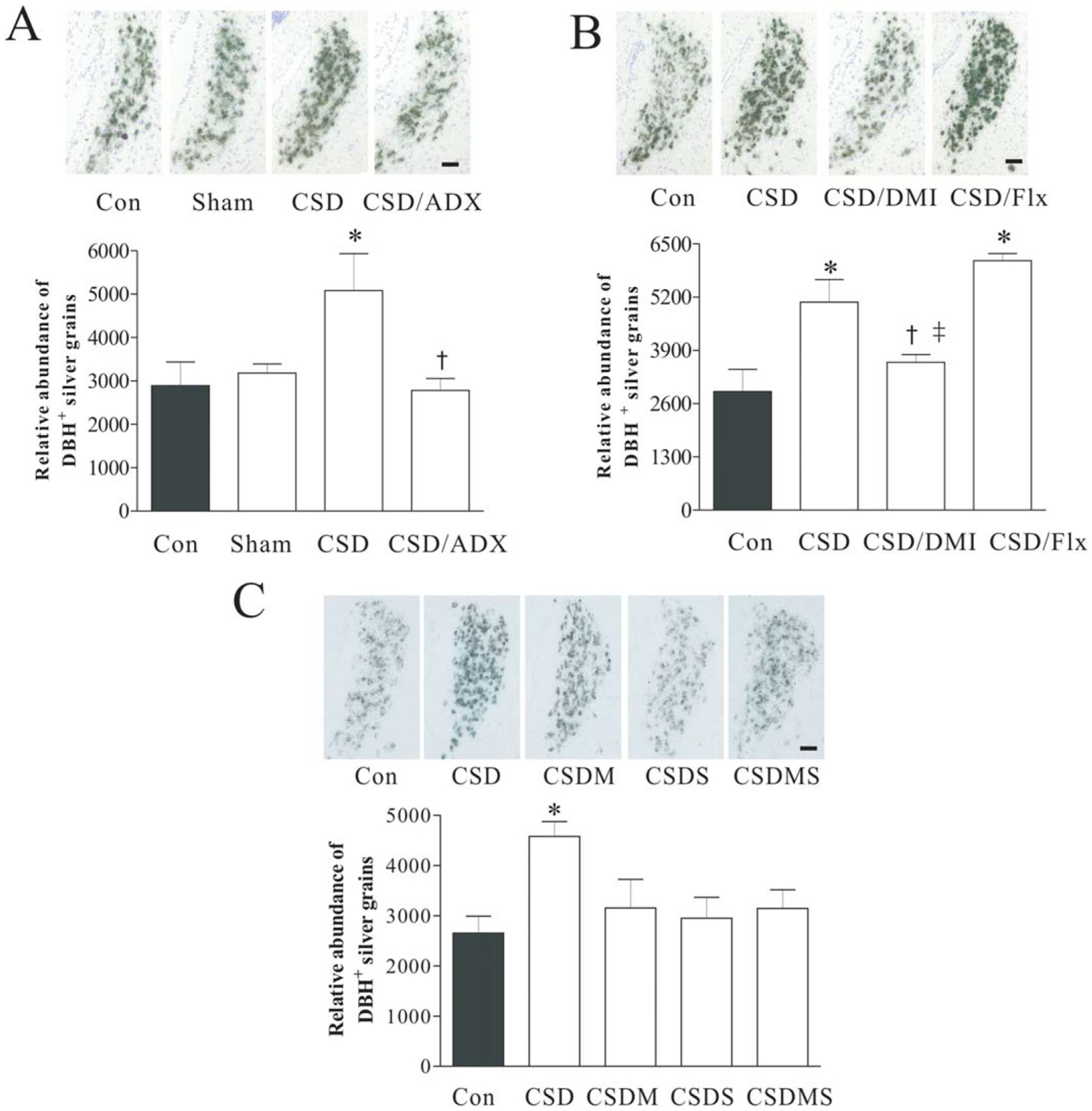
Effects of CSD, adrenalectomy (A), and treatment with desipramine or fluoxetine (B), as well as with corticosteroid receptor antagonists (C) on DBH mRNA in the LC. Upper panel: DBH mRNA in LC tissues of rats detected by in situ hybridization (n = 7/group). Coronal brain sections were taken at 9.7 mm posterior from bregma (corresponding to Plate 58 in the brain atlas) (Paxinos and Watson, 2005). Lower panel: Quantitative analyses of mRNA in slides. * P < 0.05, compared with the control; † P < 0.05, compared with the CSD group;‡ P < 0.05, compared with the CSD/Flx group. ADX, adrenalectomy; Con, control; Sham, sham operation; CSD/ADX, CSD plus adrenalectomy; CSD/DMI, CSD plus treatment with desipramine; CSD/Flx, CSD plus treatment with fluoxetine; CSDM, CSD plus treatment with mifepristone; CSDS, CSD plus treatment with spironolactone; CSDMS, CSD plus treatment with mifepristone and spironolactone. Scale Bar: 50 μm.
Next, we investigated whether antidepressants have any influence on CSD-induced upregulation of DBH mRNA levels in the LC. Rats were treated with NE reuptake inhibitor desipramine or selective serotonin reuptake inhibitor fluoxetine 30 minutes prior to CSD regimen. Analysis of mRNA measurement results showed that these treatments had significant effects on DBH mRNA levels (F3,23 = 6.93, P < 0.01). While desipramine abolished CSD-induced elevation of DBH mRNA levels (P < 0.05), treatment with fluoxetine did not show similar result. Instead, DBH mRNA levels in the group treated with fluoxetine were significantly higher than either those in the control, or in the group treated with desipramine (Fig. 2B).
Similarly, whether the altered DBH mRNA levels in the LC caused by CSD regime were mediated by corticosteroid receptors was investigated. Before exposure to each CSD session, rats were injected with corticosteroid antagonist mifepristone and spironolactone, alone or in combination. In situ hybridization results demonstrated that treatment with corticosteroid receptor antagonists markedly affected CSD-induced alteration of DBH mRNAs (F3,39=3.90, P < 0.05). Although the reduction caused by the treatment with corticosteroid antagonists did not reach the significant level compared to those in the CSD group, treatment with either mifepristone or spironolactone alone, or combination of both prevented the CSD-induced increase of DBH mRNAs in the LC (Fig. 2C).
CSD upregulated DBH protein levels in the LC of rats
In another set of experiments, the LC region was punched-out for western blotting to determine the effect of CSD on DBH protein levels. Similar to those revealed by in situ hybridization, the protein assay demonstrated that CSD immediately and significantly increased DBH protein levels in the LC (F3,27 = 7.60, P = 0.01), and ADX almost completely abrogated CSD’s effect on DBH protein levels (Fig. 3A). The treatment with antidepressants showed that desipramine prevented CSD-induced elevation of DBH protein levels in the LC, but treatment with fluoxetine did not show such action (Fig. 3B). Similar to the findings of DBH mRNA levels from the LC, treatment with corticosteroid receptor antagonists markedly affected upregulation of DBH protein levels caused by CSD regime (F3,34= 3.09, P < 0.05). The Newman-Keuls test revealed that CSD significantly increased DBH protein levels by 95.4% (P < 0.05), while treatment with mifepristone or spironolactone alone blocked CSD-induced elevation of DBH protein levels. Furthermore, treatment with combination of mifepristone and spironolactone caused a reduced DBH protein level, which was significantly different from those of the CSD group (P < 0.05, Fig. 3C). To confirm the result of western assays, immunofluorescence staining was performed in the brain samples from the parallel experiment. Results showed that CSD significantly affected DBH immunoreactivity in the LC (F4,29= 4.56, P = 0.01). Post hoc tests exhibited that while CSD significantly increased DBH immunoreactivity in the LC (P < 0.01), treatments with spironolactone or combination of mifepristone and spironolactone attenuated CSD-induced enhancement of DBH immunoreactivities (Fig. 4).
Fig. 3.
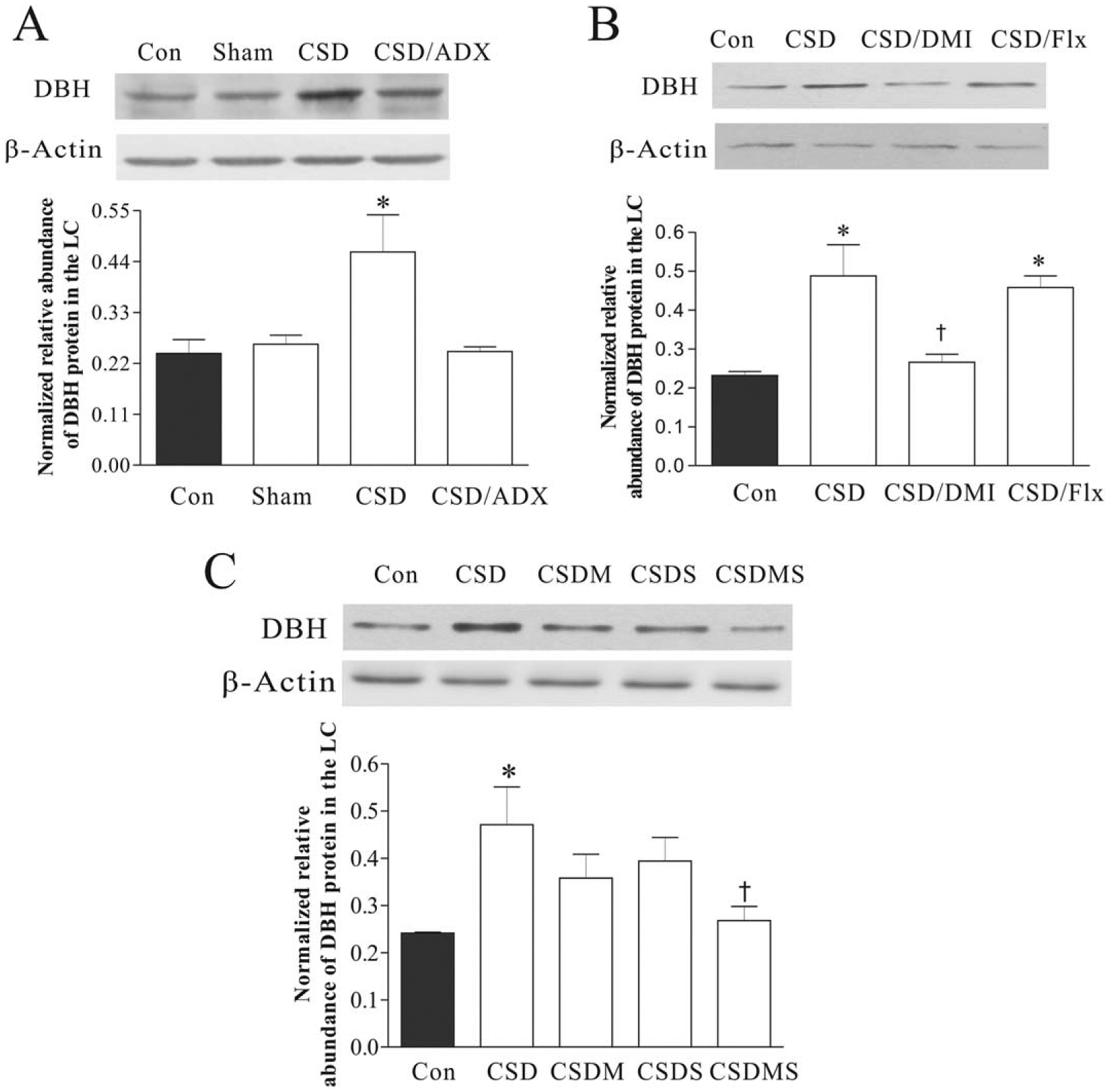
Effects of CSD, adrenalectomy (A), treatment with desipramine or fluoxetine (B), and with corticosteroid receptor antagonists (C) on DBH proteins in the LC. The upper figures in A, B, and C show autoradiographs obtained by western blotting of DBH in the LC (n = 6–8/group). The lower graphs in A, B, and C show quantitative analysis of band densities. Values of DBH bands were normalized to those of β-actin probed on the same blot. *P < 0.05, compared with the control group; † P < 0.05, compared with the CSD group. See Figure 2 for abbreviations.
Fig. 4.
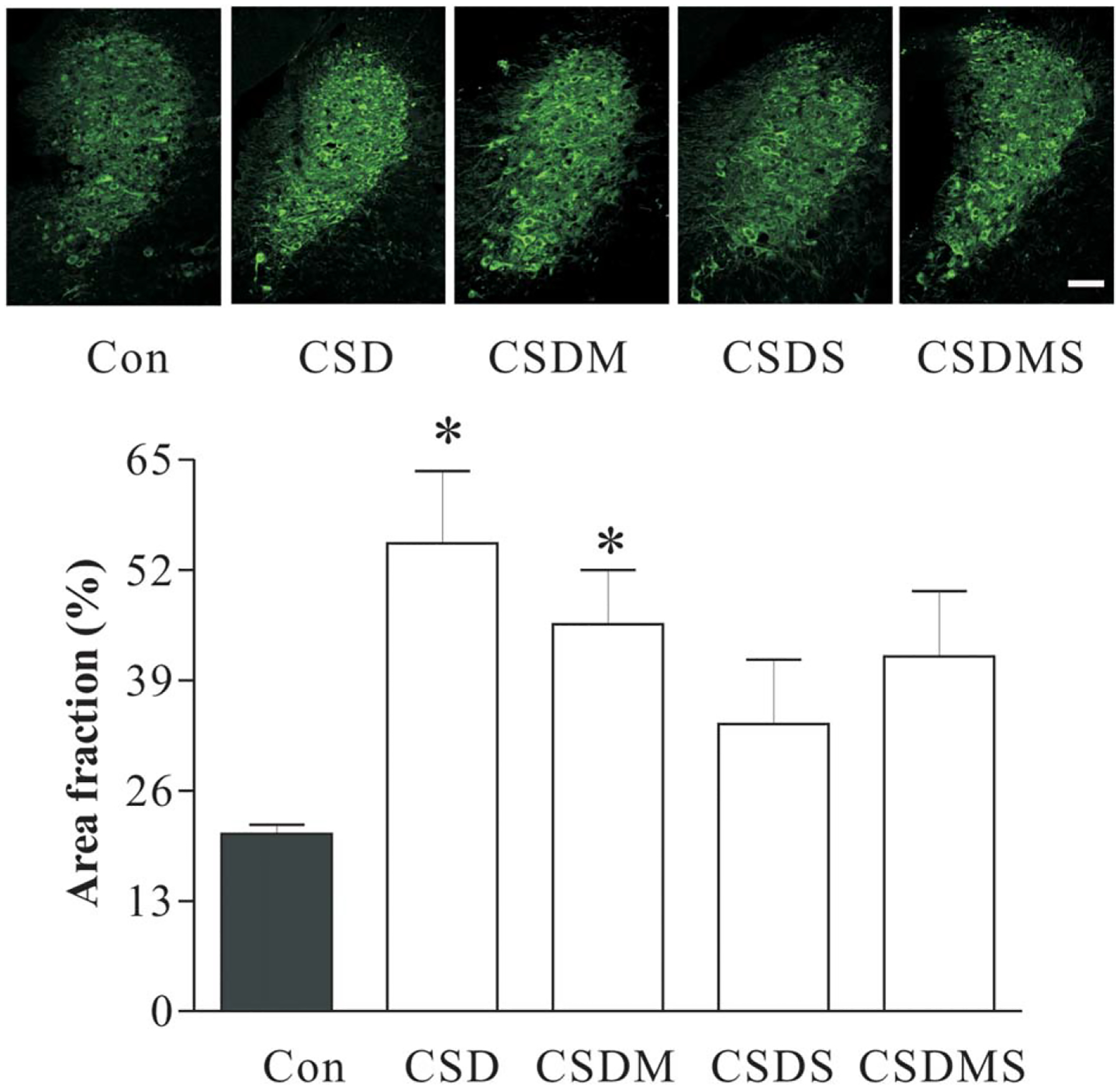
Effects of CSD and treatment with corticosteroid receptor antagonists on DBH-immunoreactivity in the LC. The top panels are representative micrographs of DBH immunofluorescence in the LC region from experimental rats. The bottom panels show measurements of the area fraction of DBH immunofluorescence in the LC region (n = 6/group). *P < 0.01, compared with the control. See Figure 2 for abbreviations. Scale Bar: 50 μm.
Effects of CSD on DBH protein levels in the terminal regions of the LC
To investigate whether CSD changes DBH protein levels in the terminal regions of the LC, the hippocampus, frontal cortex and amygdala were dissected for western blotting. As shown in Figure 5, there are no significant differences in DBH protein levels between the control and ADX sham group in the hippocampus, frontal cortex, and amygdala, indicating that the surgery procedure had no marked effect on DBH protein levels in these regions. However, CSD significantly increased DBH protein levels in these regions (F3,31= 10.32, P < 0.001 for the hippocampus; F3, 31=7.42, P < 0.01 for the frontal cortex; F3, 23=7.82, P < 0.01 for the amygdala). ADX completely abolished CSD-induced increase of DBH protein levels in all these regions. The DBH protein levels in CSD/ADX group were significantly lower than those in the CSD group (all P < 0.05).
Fig. 5.
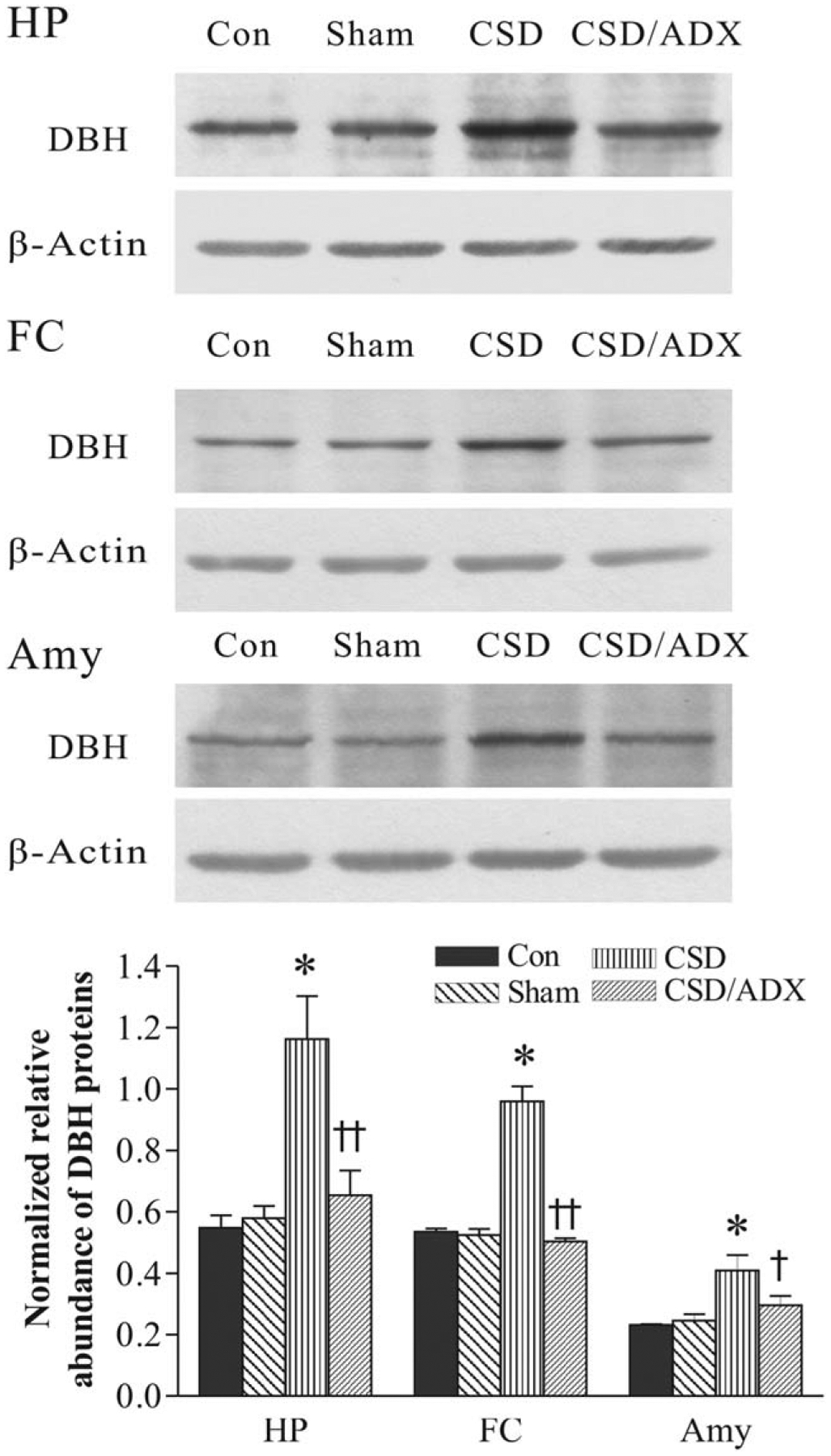
Effects of CSD and adrenalectomy (ADX) on DBH protein levels in the hippocampus (HP), frontal cortex (FC) and amygdala (Amy). The image figures show autoradiographs obtained by western blotting of DBH in different regions (n = 6–8/group). The lower graph shows quantitative analysis of band densities. Values of DBH bands were normalized to those of β-actin probed on the same blot. *P < 0.01, compared with the control group. †P < 0.05, ††P < 0.01, compared with the CSD group. See Figure 2 for abbreviations.
Treatment with antidepressants also had significant effects on DBH protein levels in the hippocampus, frontal cortex, and amygdala (F3,31= 4.44, P < 0.05 for the hippocampus; F3,31=4.42, P < 0.05 for the frontal cortex; F3,23=3.92, P < 0.05 for the amygdala). Post hoc tests revealed that while both desipramine and fluoxetine abrogated CSD-induced increase of DBH protein levels in the hippocampus, only desipramine markedly reduced DBH protein levels in the frontal cortex and amygdala. Although DBH protein levels in the frontal cortex and amygdala in the group treatment with fluoxetine were relatively lower than those in the CSD group, they did not reach the significant level (Fig. 6).
Fig. 6.
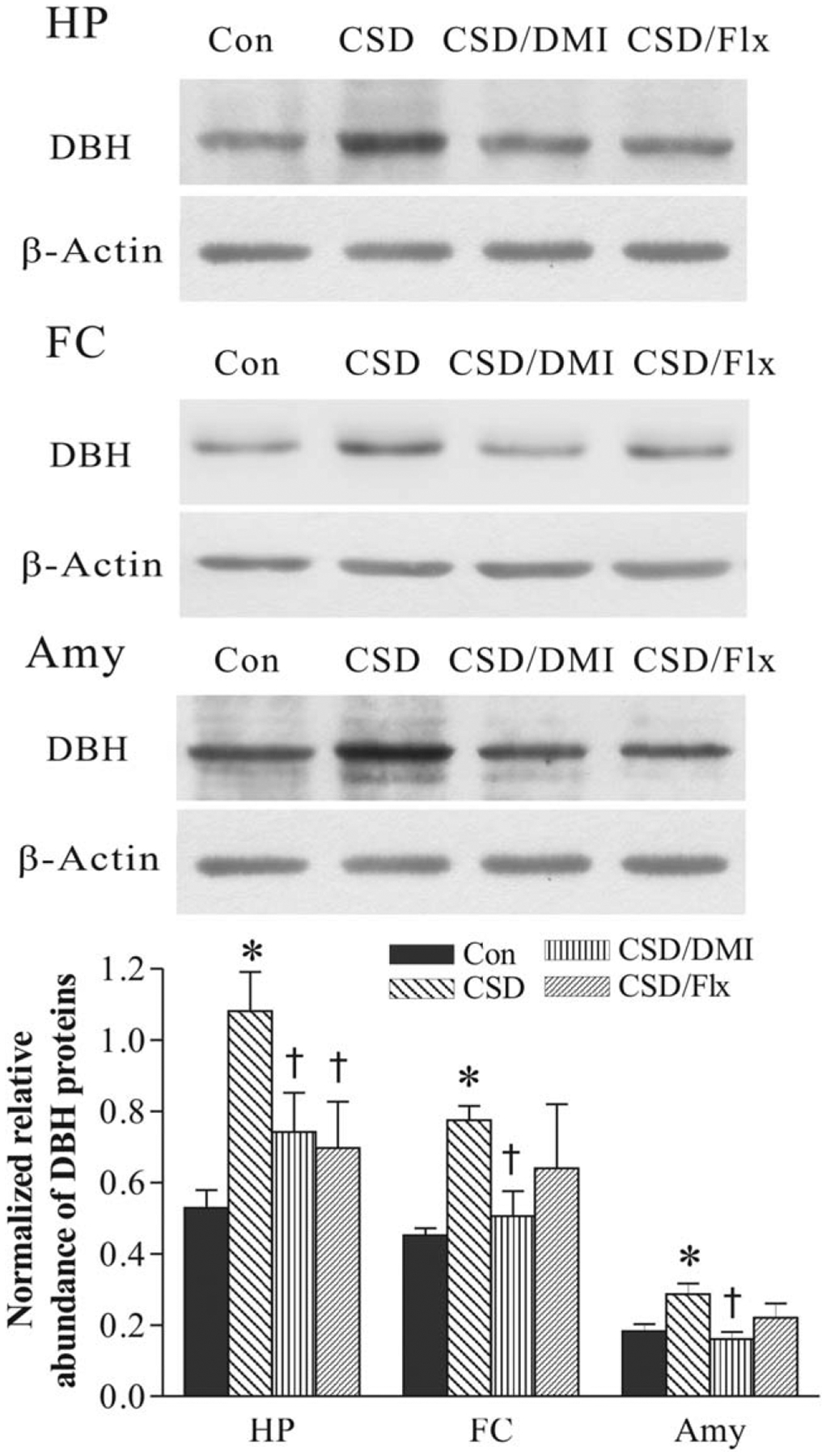
Effect of CSD and treatment with desipramine or fluoxetine on DBH protein levels the hippocampus (HP), frontal cortex (FC) and amygdala (Amy). The image figures show autoradiographs obtained by western blotting of DBH in different regions (n = 6–8/group). The lower graph shows quantitative analysis of band densities. Values of DBH bands were normalized to those of β-actin probed on the same blot. *P < 0.01, compared with the control group. †P < 0.05, compared with the CSD group. See Figure 2 for abbreviations.
Similarly, treatment with corticosteroid receptor antagonists significantly affected CSD-induced upregulation of DBH protein levels in all three terminal regions (F3,39= 22.52, P < 0.001 for the hippocampus; F3,39=3.45, P < 0.05 for the frontal cortex; F3,29=4.13, P < 0.01 for the amygdala; Fig. 7). Further tests revealed that CSD analogously increased DBH protein levels in these regions (all P < 0.01). Treatment with mifepristone alone or combination of mifepristone and spironolactone almost completely blocked CSD-induced elevation of DBH protein levels (P < 0.01 for the hippocampus and frontal cortex; P < 0.05 for the amygdala). However, treatment with spironolactone did not have any significant effects on up-regulated DBH protein levels in the hippocampus and frontal cortex, although this treatment prevented increase of DBH protein levels in the amygdala caused by CSD regime.
Fig. 7.
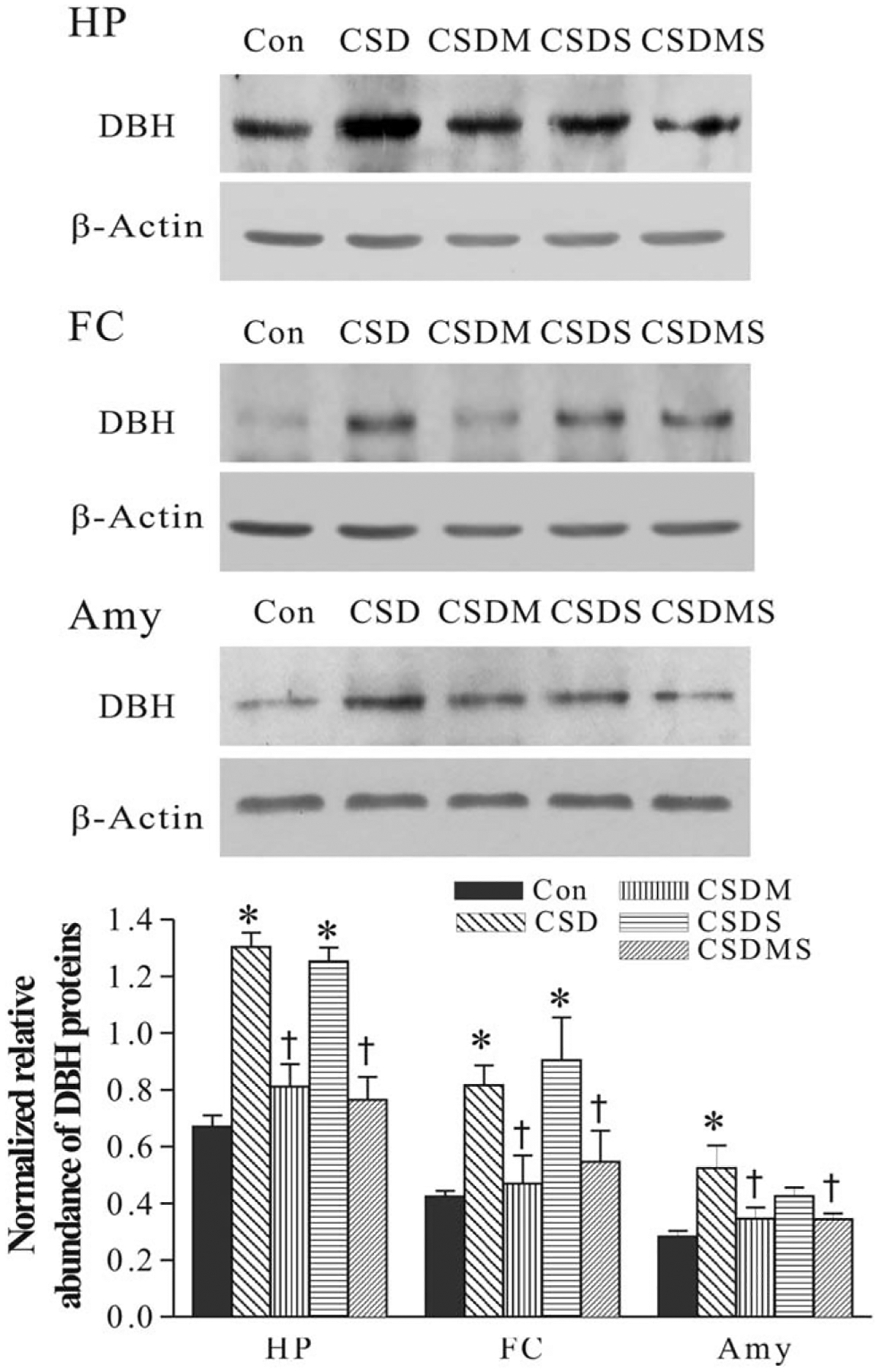
Effect of CSD and treatment with corticosteroid receptor antagonists on DBH protein levels the hippocampus (HP), frontal cortex (FC) and amygdala (Amy). The image figures show autoradiographs obtained by western blotting of DBH in different regions (n = 6–8/group). The lower graph shows quantitative analysis of band densities. Values of DBH bands were normalized to those of β-actin probed on the same blot. *P < 0.01, compared with the control group. †P < 0.01, compared with the CSD group. See Figure 2 for abbreviations.
DISCUSSION
The major findings of this study were that DBH mRNA and protein levels in the LC, as well as DBH protein levels in the hippocampus, frontal cortex, and amygdala were upregulated immediately following CSD regime. Similarly, this upregulated DBH expression was completely abolished or prevented by adrenalectomy and treatment with corticosteroid receptor antagonists, indicating the involvement of stress-released corticosterone in these actions. Furthermore, treatment with desiprimine, an antidepressant with specific inhibition for NE reuptake, also blocked CSD-induced elevation of DBH expression in the LC and its major terminal regions. However, treatment with fluoxetine, an antidepressant with specific inhibition for serotonin reuptake, selectively prevented upregulated DBH protein levels in the terminal regions, but not in the LC. Taken together with the parallel study reported previously (Chen et al., 2012), in which CSD regime also caused an upregulated expression of NET in the LC and its main projection regions via corticosteroid receptors, it indicates that chronic stress activates the central LC-NE system by upregulation of the expression of the noradrenergic phenotype in the brain. This activation is not only needed for the functional adaptation of the body response to stress, but also may causatively account for pathogenic alteration of chronic stress seen in some psychiatric diseases.
NE is a major monoamine neurotransmitter that has widespread expression across multiple brain regions to regulate arousal and stress responses. The diversity of targets that receive NE input suggests that changes in NE activity can globally influence a wide range of psychobiologic functions. Therefore, dysregulation of the NE system may turn a homeo-static stress response into a pathological result, which has been implicated in the pathogenesis of psychiatric diseases including anxiety and depression (Goddard et al., 2010). Animal studies demonstrated that stress induced activation of the LC-NE system (Abercrombie et al., 1987) includes extensive release of NE and its metabolisms in the noradrenergic neuronal neurons (Glavin et al., 1983, Pacak et al., 1995, Smagin et al., 1997). As a result, stress can reduce cellular NE levels, which can subsequently be compensated for with increased NE biosynthesis by elevated gene expression and activity of NE biosynthetic enzymes. TH as a rate-limiting enzyme plays an important role for the biosynthesis of NE. However, DBH also is a key factor to determine the rate of NE synthesis (Kobayashi et al., 1994, Kim et al., 2002), as disruption of the DBH gene has been reported to block the synthesis of NE (Sabban 2007, Kvetnansky et al., 2008). While stress-induced upregulation of TH expression has been intensively studied (Zigmond et al., 1974, Smith et al., 1991, Melia et al., 1992) and TH expression is also increased by CSD regime (unpublished data), there are very few studies about effects of stress on DBH expression, which used limited stressors. It was reported that acute and repeated immobilization stress increased mRNA and protein levels DBH in the LC, which parallels to an increased TH mRNA levels (Serova et al., 1999). Also, stress-induced DBH upregulation was found in the stellate ganglia (Gavrilovic et al., 2009), adrenal medulla (Nankova et al., 1999, Spasojevic et al., 2010) and sympathetic ganglia (Kvetnansky et al., 2004). Consistent to those observations, the present study demonstrated that CSD regimen markedly increased mRNA and protein levels of DBH in the LC, and DBH protein levels in the hippocampus, frontal cortex and amygdala. These results indicate that in response to chronic stress, upregulated expression of DBH in the LC and its terminal regions, together with TH, may account for the increased synthesis of NE. Given the LC and these terminal regions play an important role in the attention, emotion, learning and memory (Robbins et al., 1995, Usher et al., 1999, Swaab et al., 2005, Howland et al., 2008), therefore, in addition to altered TH, stress-induced elevation of DBH expression may also contribute to the development of stress-related disturbances, such as depression, posttraumatic stress disorder, and attention deficit hyperactivity disorder (Biederman et al., 1999, Southwick et al., 1999, Carrasco and Van de Kar, 2003).
Generally, both acute and chronic stress can increase expression of the noradrenergic phenotypes in the LC. However, they showed different characteristics. Acute stress-induced enhancement in DBH (and TH) is rapid and transient (Nankova et al., 1994). In contrast, the increased expression of the noradrenergic phenotypes by chronic stress lasted longer (Baruchin et al., 1990, Nankova et al., 1994). Furthermore, different mechanism and signaling pathways are involved in these different durations of stress. For example, acute stress-induced rapid transcription may be related to a non-neuronally mediated mechanism (Nankova et al., 1994) and the posttranslational activation of pre-existing factors such as CREB or cJUN (Kovacs 1998). Conversely, transcriptional activation may account for chronic stress-induced upregulation of these phenotypes and the cAMP pathway is related (Nestler et al., 1999, Sabban & Kvetnansky 2001). Likely, this molecular mechanism may also underlie the stress-trigged upregulation of DBH expression. Structurally, the rat DBH promoter contains a multifunctional region (DB1 enhancer), which is essential for basal induction, as well as for cAMP- and phorbol ester-mediated induction of DBH transcription (Sabban & Kvetnansky 2001). In addition, it has an overlapping or adjacent CRE/AP1 motif which may bind AP1 or CREB. Also, the DBH promoter contains potential glucocorticoid regulatory elements (McMahon et al., 1992, Wong et al., 1994). By run-on assays of isolated nuclei from the LC, an increase in the relative rate of DBH gene transcription has been observed after exposure of rats to immobilizations daily for seven days (Nankova et al., 1999, Serova et al., 1999). This increased transcription rate and steady-state mRNA levels of DBH are sustained for at least one day after the last episode of stress (Nankova et al., 1999). Our previous observation that the protein kinases A, C and CREB were involved in the CSD-induced upregulation of NET (Chen et al., 2012) also support the involvement of these signal pathways in the CSD regime. Consequently, chronic stress-induced alteration in the noradrenergic phenotype may be crucial for adaption to a stressful situation, as well as pathogenic results.
There are some reports that DBH may be involved in the antidepressant effects. For example, in the Dbh−/− mice desipramine and reboxetine, both as the blocker of NET, failed to exhibit antidepressant-like behavioral effects. On the other hand, there was not any difference in the expression levels of the NET between Dbh−/− and Dbh+/−. It indicates that the inability of these antidepressants in these mice is not due to any alterations in the levels of NET but rather to a loss of the endogenous ligand itself (Cryan et al., 2001). Furthermore, the effects of some specific serotonin reuptake inhibitors (SSRIs) including fluoxetine were also absent or severely attenuated in these Dbh−/− mice (Cryan et al., 2004). The reduced sensitivity to these antidepressants in those mice may be related to NE function, a potential relevance to DBH. Hence, in the present study desipramine and fluoxetine were administrated to examine whether these antidepressants influence CSD-upregulated DBH expression in the central noradrenergic system. Firstly, treatment with desipramine almost completely blocked CSD-induced elevation in DBH mRNA in the LC, and DBH protein levels in the LC, hippocampus, frontal cortex and amygdala (Figs. 2, 3, and 5). These findings are partially consistent to our previous study for another enzyme for NE synthesis, TH, in which treatment of intact rats with desipramine for 3 or 14 days significantly reduced TH protein levels in the LC, although it was accompanied by an increased TH mRNA levels (Zhu et al., 2005). It indicates that desipramine has a similar effect on NE biosynthesis enzymes in the LC and its main terminal regions, especially in the stressful status. While the increase in TH mRNA levels in the previous study could be explained as a compensatory effect for reduced TH protein levels, the discrepancy in different effects of desipramine treatment on TH (previous study) and DBH expression (present study) may be accounted by the differences in animals used and the time course. That is, the previous study used intact rats, but the current study used stress-model animals. Also, the relatively short (3 or 14 days) and longer periods (4 weeks) were another difference between these two studies.
Second, treatment with fluoxetine did not show significant effect on CSD-induced upregulation of DBH in the LC. However, it did block an increased DBH protein level in the hippocampus and prevented CSD-induced increase of DBH protein levels in the frontal cortex and amygdala (Fig. 6). Currently we do not have satisfactory explanation for the different effects of fluoxetine on DBH expression in the noradrenergic cell bodies and projection regions. However, an important fact is that while the LC has been implicated in many physiological functions, these projection regions are actually the noradrenergic effector to fulfill many LC function. Therefore, the present observation did indicate that fluoxetine’s antidepressant effects may be related to the NE transformation in the brain, especially in the noradrenergic terminal regions. This notion is consistent to previous reports, in which certain SSRIs, in addition to increasing serotonin, may also alter NE neurotransmission in vivo. For example, administration of fluoxetine increases brain extracellular concentration of NE, in addition to that of serotonin (Jordan et al., 1994, Hughes et al., 1996, Gobert et al., 1997, Perry et al., 1997, Zhang et al., 2000, Bymaster et al., 2002). Similar results have also been observed after administration of other SSRIs (Thomas et al., 1998, Hajos-Korcsok et al., 2000, Beyer et al., 2002), which implicated that the action of SSRI antidepressants, to some extent, is at the expense of the NE (Frazer 2000). Taken together, the present observation that administration of fluoxetine blocked CSD-induced increase of DBH protein levels in these LC projection regions would mean that fluoxetine acts by inhibiting NE synthesis in these brain regions. It indicates that the therapeutic action of antidepressants may reverse overactive noradrenergic system by reversing DBH expression in these regions. More studies are needed to elucidate real regulatory effects of fluoxetine on NE synthesis enzymes in the brain.
Similar to our previous observation (Chen et al., 2012), CSD regime diminished sucrose consumption, a measure of anhedonia which is a core symptom of depression (Papp et al., 1991, Rygula et al., 2005). The present study also showed that while treatment with desipramine almost reversed CSD-induced reduction of sucrose consumption, treatment with fluoxetine failed to do so in the fourth week of CSD exposure. At present we do not have satisfactory explanation for the loss of fluoxetine to prevent anhedonia in the fourth week of CSD exposure. However, it is consistent with the observation that fluoxetine failed to block CSD-induced enhance of DBH in the LC. Nevertheless, it is unknown whether there is a causative relationship between its failures in preventing stress-induced elevation of DBH in the LC and anhedonia. More experiments are needed to clarify this question. It is worth to notice that this observation about fluoxetine’s effect on sucrose consumption appears contradictory to some previous reports, which showed an almost complete reversion of stress-induced reduction in sucrose consumption by treatment with fluoxetine (Rygula et al., 2006, Banasr et al., 2007, Yang et al., 2009, Recamier-Carballo et al., 2012). This discrepancy may be at least partly accounted for by the use of different stressors in our study and in three of those reports, in which chronic unpredictable mild stress or chronic mild stress were performed (Banasr et al., 2007, Yang et al., 2009, Recamier-Carballo et al., 2012). In another report (Rygula et al., 2006), although similar CSD was used, fluoxetine was orally administrated, and CSD regime was performed daily. It is well known that repeated exposure to a homotypic stressor generally results in habituation, a desensitization of the hypothalamic-pituitary-adrenal axis in response to stressor (Hashimoto et al., 1988, Hauger et al., 1990, Armario 2006). In the present study, CSD regime was performed unpredictably enough to prevent the habituation. Synergistic effect in several CSD episodes may likely explain the reason for fluoxetine to lose its ability to reverse reduced sucrose consumption.
In conclusion, the present study demonstrates that CSD regime significantly upregulated DBH expression in the LC and its main terminal regions and it was mediated by corticosteroid receptors, a similar phenomenon to that of NET as reported previously (Chen et al., 2012). Taken together with the well-documented reports that stress-induced upregulation of TH expression (Zigmond et al., 1974, Smith et al., 1991, Melia et al., 1992), it indicates that chronic stress activates the LC-NE system, which may be related to the pathophysiology of stress-precipitated psychiatric disorders. This notion is supported by further observation that such upregulated expression of DBH can be abolished by desipramine. However, treatment with fluoxetine resulted in different action on CSD-induced elevation of DBH expression in the cell bodies and terminal regions of the noradrenergic neurons, which requires more experiments to elucidate molecular mechanisms underlying the regulation of DBH expression by serotonergic reuptake inhibitors.
ACKNOWLEDGMENTS
The authors thank Mr. Hobart Zhu for his helpful comments on the manuscript. The authors declare no conflict of interest regarding the work reported here.
Contract grant sponsor:
NIH; Contract grant number: MH080323.
REFERENCES
- Abercrombie ED, Jacobs BL. 1987. Single-unit response of noradrenergic neurons in the locus coeruleus of freely moving cats. I. Acutely presented stressful and nonstressful stimuli. J Neurosci 7:2837–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armario A 2006. The hypothalamic-pituitary-adrenal axis: What can it tell us about stressors? CNS Neurol Disord Drug Targets 5:485–501. [DOI] [PubMed] [Google Scholar]
- Banasr M, Valentine GW, Li XY, Gourley SL, Taylor JR, Duman RS. 2007. Chronic unpredictable stress decreases cell proliferation in the cerebral cortex of the adult rat. Biol Psychiatry 62:496–504. [DOI] [PubMed] [Google Scholar]
- Baruchin A, Weisberg EP, Miner LL, Ennis D, Nisenbaum LK, Naylor E, Stricker EM, Zigmond MJ, Kaplan BB. 1990. Effects of cold exposure on rat adrenal tyrosine hydroxylase: An analysis of RNA, protein, enzyme activity, and cofactor levels. J Neurochem 54:1769–1775. [DOI] [PubMed] [Google Scholar]
- Beyer CE, Boikess S, Luo B, Dawson LA. 2002. Comparison of the effects of antidepressants on norepinephrine and serotonin concentrations in the rat frontal cortex: An in-vivo microdialysis study. J Psychopharmacol 16:297–304. [DOI] [PubMed] [Google Scholar]
- Biederman J, Spencer T. 1999. Attention-deficit/hyperactivity disorder (ADHD) as a noradrenergic disorder. Biol Psychiatry 46:1234–1242. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Zhang W, Carter PA, Shaw J, Chernet E, Phebus L, Wong DT, Perry KW. 2002. Fluoxetine, but not other selective serotonin uptake inhibitors, increases norepinephrine and dopamine extracellular levels in prefrontal cortex. Psychopharmacology (Berl) 160:353–361. [DOI] [PubMed] [Google Scholar]
- Carrasco GA, Van de Kar LD. 2003. Neuroendocrine pharmacology of stress. Eur J Pharmacol 463:235–272. [DOI] [PubMed] [Google Scholar]
- Charney DS. 1998. Monoamine dysfunction and the pathophysiology and treatment of depression. J Clin Psychiatry 59(Suppl 14):11–14. [PubMed] [Google Scholar]
- Chen P, Fan Y, Li Y, Sun Z, Bissette G, Zhu MY. 2012. Chronic social defeat up-regulates expression of norepinephrine transporter in rat brains. Neurochem Int 60:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, Dalvi A, Jin SH, Hirsch BR, Lucki I, Thomas SA. 2001. Use of dopamine-beta-hydroxylase-deficient mice to determine the role of norepinephrine in the mechanism of action of antidepressant drugs. J Pharmacol Exp Ther 298:651–657. [PubMed] [Google Scholar]
- Cryan JF, O’Leary OF, Jin SH, Friedland JC, Ouyang M, Hirsch BR, Page ME, Dalvi A, Thomas SA, Lucki I. 2004. Norepinephrine-deficient mice lack responses to antidepressant drugs, including selective serotonin reuptake inhibitors. Proc Natl Acad Sci USA 101:8186–8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Huang J, Duffourc M, Kao RL, Ordway GA, Huang R, Zhu MY. 2011. Transcription factor Phox2 upregulates expression of norepinephrine transporter and dopamine beta-hydroxylase in adult rat brains. Neuroscience 192:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer A 2000. Norepinephrine involvement in antidepressant action. J Clin Psychiatry 61(Suppl 10):25–30. [PubMed] [Google Scholar]
- Friedman S, Kaufman S. 1965. 3,4-Dihydroxyphenylethylamine Beta-Hydroxylase: A Copper Protein. J Biol Chem 240:PC552–PC554. [PubMed] [Google Scholar]
- Gavrilovic L, Spasojevic N, Dronjak S. 2009. Psychosocial stress-related changes in gene expression of norepinephrine biosynthetic enzymes in stellate ganglia of adult rats. Autonomic Neurosci 150:144–146. [DOI] [PubMed] [Google Scholar]
- Glavin GB, Tanaka M, Tsuda A, Kohno Y, Hoaki Y, Nagasaki N. 1983. Regional rat brain noradrenaline turnover in response to restraint stress. Pharmacol Biochem Behav 19:287–290. [DOI] [PubMed] [Google Scholar]
- Gobert A, Rivet JM, Cistarelli L, Melon C, Millan MJ. 1997. Alpha2-adrenergic receptor blockade markedly potentiates duloxetine- and fluoxetine-induced increases in noradrenaline, dopamine, and serotonin levels in the frontal cortex of freely moving rats. J Neurochem 69:2616–2619. [DOI] [PubMed] [Google Scholar]
- Goddard AW, Ball SG, Martinez J, Robinson MJ, Yang CR, Russell JM, Shekhar A. 2010. Current perspectives of the roles of the central norepinephrine system in anxiety and depression. Depress Anxiety 27:339–350. [DOI] [PubMed] [Google Scholar]
- Goldstein M, Fuxe K, Hokfelt T. 1972. Characterization and tissue localization of catecholamine synthesizing enzymes. Pharmacol Rev 24:293–309. [PubMed] [Google Scholar]
- Hajos-Korcsok E, McTavish SF, Sharp T. 2000. Effect of a selective 5-hydroxytryptamine reuptake inhibitor on brain extracellular noradrenaline: Microdialysis studies using paroxetine. Eur J Pharmacol 407:101–107. [DOI] [PubMed] [Google Scholar]
- Haller J, Millar S, Kruk MR. 1998. Mineralocorticoid receptor blockade inhibits aggressive behaviour in male rats. Stress 2:201–207. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Suemaru S, Takao T, Sugawara M, Makino S, Ota Z. 1988. Corticotropin-releasing hormone and pituitary-adrenocortical responses in chronically stressed rats. Regul Pept 23:117–126. [DOI] [PubMed] [Google Scholar]
- Hauger RL, Lorang M, Irwin M, Aguilera G. 1990. CRF receptor regulation and sensitization of ACTH responses to acute ether stress during chronic intermittent immobilization stress. Brain Res 532:34–40. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O 1982. Brain catecholamines in schizophrenia—A good case for noradrenaline. Nature 299:484–486. [DOI] [PubMed] [Google Scholar]
- Howland JG, Wang YT. 2008. Synaptic plasticity in learning and memory: Stress effects in the hippocampus. Prog Brain Res 169:145–158. [DOI] [PubMed] [Google Scholar]
- Hughes ZA, Stanford SC. 1996. Increased noradrenaline efflux induced by local infusion of fluoxetine in the rat frontal cortex. Eur J Pharmacol 317:83–90. [DOI] [PubMed] [Google Scholar]
- Jordan S, Kramer GL, Zukas PK, Moeller M, Petty F. 1994. In vivo biogenic amine efflux in medial prefrontal cortex with imipramine, fluoxetine, and fluvoxamine. Synapse 18:294–297. [DOI] [PubMed] [Google Scholar]
- Kim CH, Zabetian CP, Cubells JF, Cho S, Biaggioni I, Cohen BM, Robertson D, Kim KS. 2002. Mutations in the dopamine beta-hydroxylase gene are associated with human norepinephrine deficiency. Am J Med Genet 108:140–147. [PubMed] [Google Scholar]
- Kobayashi K, Morita S, Mizuguchi T, Sawada H, Yamada K, Nagatsu I, Fujita K, Nagatsu T. 1994. Functional and high level expression of human dopamine beta-hydroxylase in transgenic mice. J Biol Chem 269:29725–29731. [PubMed] [Google Scholar]
- Kovacs KJ. 1998. c-Fos as a transcription factor: A stressful (re)view from a functional map. Neurochem Int 33:287–297. [DOI] [PubMed] [Google Scholar]
- Kubovcakova L, Tybitanclova K, Sabban EL, Majzoub J, Zorad S, Vietor I, Wagner EF, Krizanova O, Kvetnansky R. 2004. Catecholamine synthesizing enzymes and their modulation by immobilization stress in knockout mice. Ann NY Acad Sci 1018:458–465. [DOI] [PubMed] [Google Scholar]
- Kvetnansky R, Krizanova O, Tillinger A, Sabban EL, Thomas SA, Kubovcakova L. 2008. Regulation of gene expression of catecholamine biosynthetic enzymes in dopamine-beta-hydroxylase- and CRH-knockout mice exposed to stress. Ann NY Acad Sci 1148:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvetnansky R, Micutkova L, Rychkova N, Kubovcakova L, Mravec B, Filipenko M, Sabban EL, Krizanova O. 2004. Quantitative evaluation of catecholamine enzymes gene expression in adrenal medulla and sympathetic Ganglia of stressed rats. Ann NY Acad Sci 1018:356–369. [DOI] [PubMed] [Google Scholar]
- Macunluoglu B, Arikan H, Atakan A, Tuglular S, Ulfer G, Cakalagaoglu F, Ozener C, Akoglu E. 2008. Effects of spironolactone in an experimental model of chronic cyclosporine nephrotoxicity. Transplant Proc 40:273–278. [DOI] [PubMed] [Google Scholar]
- McDougall SJ, Widdop RE, Lawrence AJ. 2005. Differential gene expression in WKY and SHR brain following acute and chronic air-puff stress. Brain Res Mol Brain Res 133:329–336. [DOI] [PubMed] [Google Scholar]
- McMahon A, Sabban EL. 1992. Regulation of expression of dopamine beta-hydroxylase in PC12 cells by glucocorticoids and cyclic AMP analogues. J Neurochem 59:2040–2047. [DOI] [PubMed] [Google Scholar]
- Melia KR, Duman RS. 1991. Involvement of corticotropin-releasing factor in chronic stress regulation of the brain noradrenergic system. Proc Natl Acad Sci USA 88:8382–8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melia KR, Nestler EJ, Duman RS. 1992. Chronic imipramine treatment normalizes levels of tyrosine hydroxylase in the locus coeruleus of chronically stressed rats. Psychopharmacology (Berl) 108:23–26. [DOI] [PubMed] [Google Scholar]
- Nagatsu T 1986. Neurotransmitter Enzymes: Neuromethods. Humana Press, Clifton, NJ. [Google Scholar]
- Nankova B, Kvetnansky R, McMahon A, Viskupic E, Hiremagalur B, Frankle G, Fukuhara K, Kopin IJ, Sabban EL. 1994. Induction of tyrosine hydroxylase gene expression by a nonneuronal nonpituitary-mediated mechanism in immobilization stress. Proc Natl Acad Sci USA 91:5937–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nankova BB, Tank AW, Sabban EL. 1999. Transient or sustained transcriptional activation of the genes encoding rat adrenomedullary catecholamine biosynthetic enzymes by different durations of immobilization stress. Neuroscience 94:803–808. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Alreja M, Aghajanian GK. 1999. Molecular control of locus coeruleus neurotransmission. Biol Psychiatry 46:1131–1139. [DOI] [PubMed] [Google Scholar]
- Ni H, Mune T, Morita H, Daidoh H, Hanafusa J, Shibata T, Yamakita N, Yasuda K. 1995. Inhibition of aldosterone turn-off phenomenon following chronic adrenocorticotropin treatment with in vivo administration of antiglucocorticoid and antioxidants in rats. Eur J Endocrinol 133:578–584. [DOI] [PubMed] [Google Scholar]
- Ohlstein EH, Kruse LI, Ezekiel M, Sherman SS, Erickson R, DeW-olf WE Jr, Berkowitz BA. 1987. Cardiovascular effects of a new potent dopamine beta-hydroxylase inhibitor in spontaneously hypertensive rats. J Pharmacol Exp Ther 241:554–559. [PubMed] [Google Scholar]
- Pacak K, Palkovits M, Kopin IJ, Goldstein DS. 1995. Stress-induced norepinephrine release in the hypothalamic paraventricular nucleus and pituitary-adrenocortical and sympathoadrenal activity: In vivo microdialysis studies. Front Neuroendocrinol 16:89–150. [DOI] [PubMed] [Google Scholar]
- Pace TW, Gaylord RI, Jarvis E, Girotti M, Spencer RL. 2009. Differential glucocorticoid effects on stress-induced gene expression in the paraventricular nucleus of the hypothalamus and ACTH secretion in the rat. Stress 12:400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp M, Willner P, Muscat R. 1991. An animal model of anhedonia: Attenuation of sucrose consumption and place preference conditioning by chronic unpredictable mild stress. Psychopharmacology (Berl) 104:255–259. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. 2005. The rat brain in stereotaxic coordinates. Oxford, UK: Elsevier. [DOI] [PubMed] [Google Scholar]
- Perry KW, Fuller RW. 1997. Fluoxetine increases norepinephrine release in rat hypothalamus as measured by tissue levels of MHPG-SO4 and microdialysis in conscious rats. J Neural Transm 104:953–966. [DOI] [PubMed] [Google Scholar]
- Ratka A, Sutanto W, Bloemers M, de Kloet ER. 1989. On the role of brain mineralocorticoid (type I) and glucocorticoid (type II) receptors in neuroendocrine regulation. Neuroendocrinology 50:117–123. [DOI] [PubMed] [Google Scholar]
- Recamier-Carballo S, Estrada-Camarena E, Reyes R, Fernandez-Guasti A. 2012. Synergistic effect of estradiol and fluoxetine in young adult and middle-aged female rats in two models of experimental depression. Behav Brain Res 233:351–358. [DOI] [PubMed] [Google Scholar]
- Robbins T, Everitt B. 1995. Central norepinephrine neurons and behavior. In: Bloom F, Kupfer D, editors. Neuropsychopharmacology: The fourth generation of progress. New York: Raven Press. [Google Scholar]
- Rygula R, Abumaria N, Domenici E, Hiemke C, Fuchs E. 2006. Effects of fluoxetine on behavioral deficits evoked by chronic social stress in rats. Behav Brain Res 174:188–192. [DOI] [PubMed] [Google Scholar]
- Rygula R, Abumaria N, Flugge G, Fuchs E, Ruther E, Havemann-Reinecke U. 2005. Anhedonia and motivational deficits in rats: Impact of chronic social stress. Behav Brain Res 162:127–134. [DOI] [PubMed] [Google Scholar]
- Sabban EL. 2007. Catecholamines in stress: Molecular mechanisms of gene expression. Endocr Regul 41:61–73. [PubMed] [Google Scholar]
- Sabban EL, Kvetnansky R. 2001. Stress-triggered activation of gene expression in catecholaminergic systems: Dynamics of transcriptional events. Trends Neurosci 24:91–98. [DOI] [PubMed] [Google Scholar]
- Serova LI, Nankova BB, Feng Z, Hong JS, Hutt M, Sabban EL. 1999. Heightened transcription for enzymes involved in norepinephrine biosynthesis in the rat locus coeruleus by immobilization stress. Biol Psychiatry 45:853–862. [DOI] [PubMed] [Google Scholar]
- Smagin GN, Zhou J, Harris RB, Ryan DH. 1997. CRF receptor antagonist attenuates immobilization stress-induced norepinephrine release in the prefrontal cortex in rats. Brain Res Bull 42:431–434. [DOI] [PubMed] [Google Scholar]
- Smith MA, Brady LS, Glowa J, Gold PW, Herkenham M. 1991. Effects of stress and adrenalectomy on tyrosine hydroxylase mRNA levels in the locus ceruleus by in situ hybridization. Brain Res 544:26–32. [DOI] [PubMed] [Google Scholar]
- Southwick SM, Bremner JD, Rasmusson A, Morgan CA III, Arnsten A, Charney DS. 1999. Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biol Psychiatry 46:1192–1204. [DOI] [PubMed] [Google Scholar]
- Spasojevic N, Gavrilovic L, Dronjak S. 2010. Effects of repeated maprotiline and fluoxetine treatment on gene expression of catecholamine synthesizing enzymes in adrenal medulla of unstressed and stressed rats. Autonomic Autacoid Pharmacol 30:213–217. [DOI] [PubMed] [Google Scholar]
- Swaab DF, Bao AM, Lucassen PJ. 2005. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev 4:141–194. [DOI] [PubMed] [Google Scholar]
- Thomas DN, Nutt DJ, Holman RB. 1998. Sertraline, a selective serotonin reuptake inhibitor modulates extracellular noradrenaline in the rat frontal cortex. J Psychopharmacol 12:366–370. [DOI] [PubMed] [Google Scholar]
- Udenfriend S 1966. Tyrosine hydroxylase. Pharmacol Rev 18:43–51. [PubMed] [Google Scholar]
- Usher M, Cohen JD, Servan-Schreiber D, Rajkowski J, Aston-Jones G. 1999. The role of locus coeruleus in the regulation of cognitive performance. Science 283:549–554. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, McKittrick CR, Blanchard DC, Blanchard RJ, McEwen BS, Sakai RR. 1995. Effects of chronic social stress on tyrosine hydroxylase mRNA and protein levels. Brain Res Mol Brain Res 32:176–180. [DOI] [PubMed] [Google Scholar]
- Wong DL, Wang W. 1994. Neural control of dopamine beta-hydroxylase in vivo: Acute and chronic effects. Brain Res Mol Brain Res 25:57–66. [DOI] [PubMed] [Google Scholar]
- Yang C, Wang G, Wang H, Liu Z, Wang X. 2009. Cytoskeletal alterations in rat hippocampus following chronic unpredictable mild stress and re-exposure to acute and chronic unpredictable mild stress. Behav Brain Res 205:518–524. [DOI] [PubMed] [Google Scholar]
- Zhang W, Perry KW, Wong DT, Potts BD, Bao J, Tollefson GD, Bymaster FP. 2000. Synergistic effects of olanzapine and other antipsychotic agents in combination with fluoxetine on norepinephrine and dopamine release in rat prefrontal cortex. Neuropsychopharmacology 23:250–262. [DOI] [PubMed] [Google Scholar]
- Zhu MY, Kim CH, Hwang DY, Baldessarini RJ, Kim KS. 2002. Effects of desipramine treatment on norepinephrine transporter gene expression in the cultured SK-N-BE(2)M17 cells and rat brain tissue. J Neurochem 82:146–153. [DOI] [PubMed] [Google Scholar]
- Zhu MY, Wang WP, Baldessarini RJ, Kim KS. 2005. Effects of desipramine treatment on tyrosine hydroxylase gene expression in cultured neuroblastoma cells and rat brain tissue. Brain Res Mol Brain Res 133:167–175. [DOI] [PubMed] [Google Scholar]
- Zigmond RE, Schon F, Iversen LL. 1974. Increased tyrosine hydroxylase activity in the locus coeruleus of rat brain stem after reserpine treatment and cold stress. Brain Res 70:547–552. [DOI] [PubMed] [Google Scholar]