

SUMMARY
Sensitization of trigeminal ganglion neurons contributes to primary headache disorders such as migraine, but the specific neuronal and non-neuronal trigeminal subtypes that are involved remain unclear. We thus developed a cell atlas in which human and mouse trigeminal ganglia are transcriptionally and epigenomically profiled at single-cell resolution. These data describe evolutionarily conserved and human-specific gene expression patterns within each trigeminal ganglion cell type, as well as the transcription factors and gene regulatory elements that contribute to cell-type-specific gene expression. We then leveraged these data to identify trigeminal ganglion cell types that are implicated both by human genetic variation associated with migraine and two mouse models of headache. This trigeminal ganglion cell atlas improves our understanding of the cell types, genes, and epigenomic features involved in headache pathophysiology and establishes a rich resource of cell-type-specific molecular features to guide the development of more selective treatments for headache and facial pain.
eTOC
Trigeminal ganglion neurons are activated in migraine and related headache disorders. Yang et al. developed a cell atlas in which human and mouse trigeminal ganglia are transcriptionally and epigenomically profiled at single-cell resolution. These data provide a rich resource for understanding the cell types and genetic mechanisms underlying migraine susceptibility and for developing novel analgesics.
INTRODUCTION
Headache disorders such as migraine are among the leading causes of morbidity worldwide and are estimated to cost the US economy at least $20 billion annually in lost productivity (Steiner et al., 2020; Stewart et al., 2003). Despite recent advances (Charles and Pozo-Rosich, 2019), current headache treatments provide only partial relief to most patients (Goadsby et al., 2017; Loder and Renthal, 2019; Tfelt-Hansen and Loder, 2019). Migraine and related headache disorders involve the activation of peripheral sensory neurons whose cell bodies reside in the trigeminal ganglion (TG) (Akerman et al., 2011; Pietrobon and Moskowitz, 2013), a complex structure comprised of multiple neuronal and non-neuronal cell types (Goto et al., 2016). Improved understanding of the cell types and molecules expressed in distinct TG cell types, especially within human trigeminal nociceptors, could both help understand the complex genetic mechanisms underlying migraine susceptibility (Sutherland et al., 2019) and guide the development of novel headache and facial pain treatments.
Recent advances in single-cell transcriptomics have enabled the molecular characterization of mouse TG neurons, which in most cases correspond to previously described functional classifications (Nguyen et al., 2017; Sharma et al., 2020; von Buchholtz et al., 2021; von Buchholtz et al., 2020). For example, unique transcriptional profiles have been observed for distinct mouse dorsal root ganglion (DRG) and TG neuronal subtypes such as peptidergic and non-peptidergic nociceptors, Aβ low-threshold mechanoreceptors (LTMR) subtypes, Aδ LTMRs, C-fiber LTMRs (cLTMRs), and Sst+/Nppb+ pruriceptors (SST) (Nguyen et al., 2019; Sharma et al., 2020; Usoskin et al., 2015; von Buchholtz et al., 2021). Transcriptional profiles of non-neuronal TG cell types have yet to be characterized at single-cell resolution. It also remains unclear which TG cell types and molecules are evolutionarily conserved in human TG or are human-specific, how these cell-type-specific gene expression patterns are established epigenomically, and which of these TG cell types contribute to migraine susceptibility. These questions can be addressed by integrating single-cell transcriptomic and epigenomic analyses of mouse and human tissues to characterize cell-type-specific gene expression patterns, the putative gene regulatory elements (e.g. gene enhancers) and transcription factors (TFs) that establish these patterns, and the cell types in which disease-associated human genetic variation is likely to act (Cao et al., 2018; Cusanovich et al., 2018a; Cusanovich et al., 2018b; Lake et al., 2018; Ma et al., 2020).
Here, we present a transcriptional and epigenomic cell atlas of the mouse and human TG. We examine the range of evolutionarily conserved cell types and molecules, as well as species- and sex-specific features. Using single-nucleus epigenomics, we map putative gene regulatory elements that are likely to drive cell-type-specific gene expression within each TG cell type and leverage these epigenomic data to interpret the TG neuronal and non-neuronal cell types that may be affected by human genetic variation associated with migraine susceptibility. Finally, we identify the TG cell types that are engaged in two mouse headache models. These data provide a rich resource (available at tg.painseq.com) of cell-type-specific gene expression and epigenomic regulation in human and murine TG and improve our understanding of the cell types involved in headache disorders.
RESULTS
Neuronal enrichment of human and mouse TG nuclei for single-nucleus RNA sequencing
A technical challenge in characterizing TG neuronal subtypes is that there are many more non-neuronal cells in the TG than neurons (LaGuardia et al., 2000). This problem is further exaggerated in human tissue, where there can be significant variability in tissue procurement. To develop a method that enriches for neuronal nuclei while still sampling non-neuronal nuclei in both human and mouse TG, we compared two nuclear dissociation protocols: a commonly-used nuclear dissociation protocol (non-gradient) (Drokhlyansky et al., 2020) and a density centrifugation protocol (gradient) (Mo et al., 2015). To quantify the fraction of neuronal nuclei isolated in each protocol, we used Vglut2-Cre;Sun1-GFP reporter mice in which the nuclear membranes of >95% of sensory ganglia neurons are labeled with GFP (Renthal et al., 2020). We extracted TG nuclei using both protocols and then compared the fraction of GFP+ nuclei with fluorescence-activated cell/nucleus sorting (FACS). We observed that >30% of the nuclei recovered by the gradient method are GFP+ whereas < 6% of the nuclei recovered by the non-gradient method are GFP+ (Figure S1A). The fraction of neurons collected by FACS was 5.09 ± 1.08 (standard deviation, n = 3) times greater with the gradient protocol than the non-gradient protocol. We next performed single-nucleus RNA-sequencing (snRNA-seq) of C57BL/6 mouse TG and human TG nuclei isolated using both protocols (Figure S1B, see methods). Consistent with our FACS observations of GFP-labeled neuronal nuclei, we found that the fraction of nuclei transcriptionally classified as neurons is significantly greater with the gradient method than the non-gradient method in both mouse and human TG (Figure S1C–D). These data led us to use the gradient method to collect the remainder of our snRNA-seq data from human and mouse TG.
Human and mouse TG cell atlases
Using the gradient method, we performed snRNA-seq of 14 TG biological replicates from C57BL/6 mice (8–12 weeks old C57BL/6 mice, 11 male, 3 female, 2–3 mice per replicate) and three human donors who died of non-neurological causes (1 male, 2 female) (Figures 1A–B, S1E, Table S1). Bilateral TGs were combined and sequenced together for the mouse snRNA-seq atlas, which is comprised of 59,921 nuclei with an average of 1,851 genes detected per nucleus. Left and right TGs from human donors 1 and 2 and the left TG from donor 3 were sequenced individually for the human snRNA-seq atlas, which is comprised of 38,028 nuclei with an average of 1,974 genes detected per nucleus. We used Seurat (Butler et al., 2018) to cluster nuclei and transcriptionally classify cell types of each species separately (see methods).
Figure 1. snRNA-seq of human and mouse trigeminal ganglion.
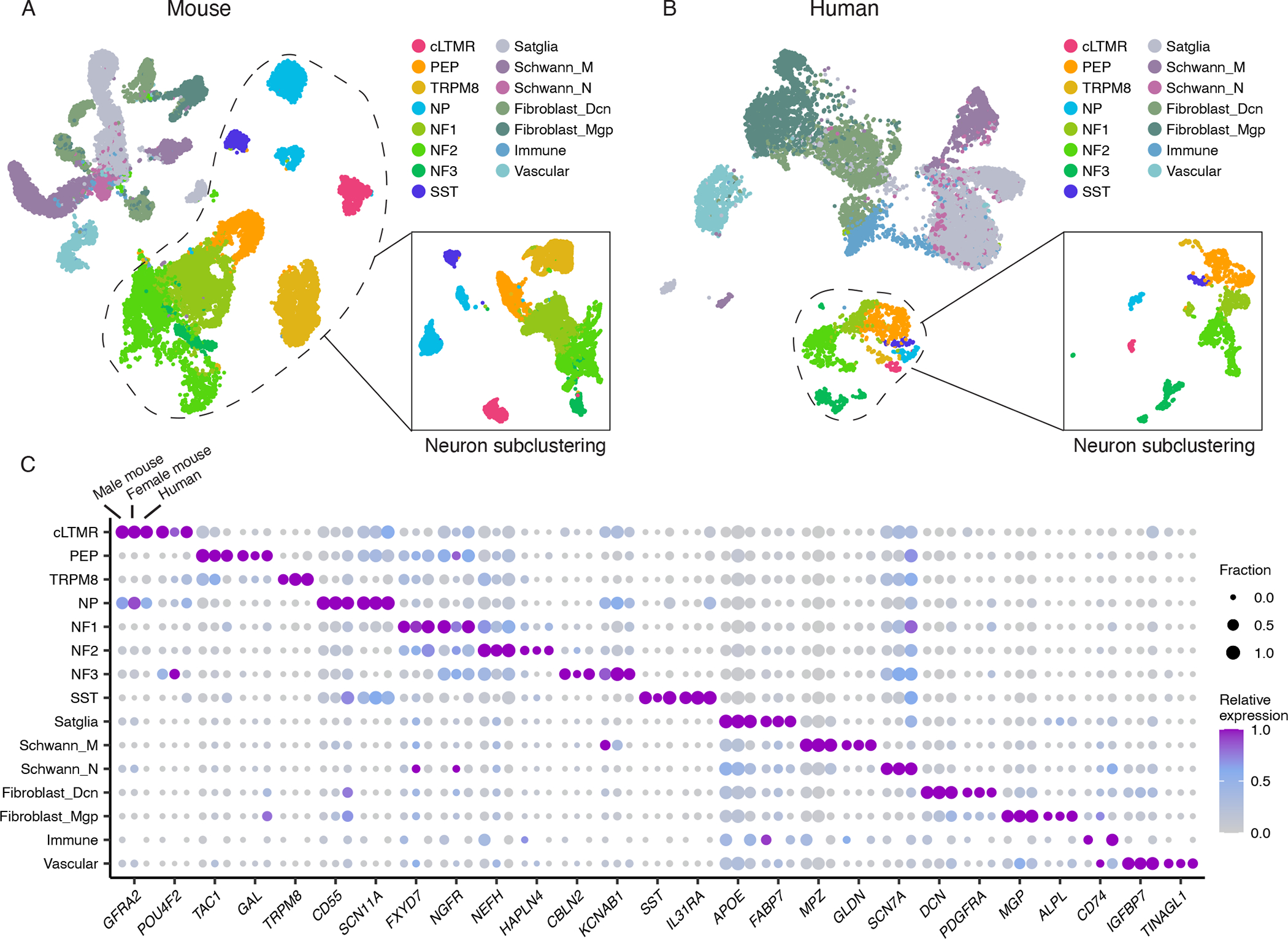
A. UMAP plot of snRNA-seq data from 59,921 mouse TG nuclei from 14 biological replicates, downsampled to display 28,000 nuclei, 2,000 per replicate (14,984 neuronal, 13,016 non-neuronal). UMAP of neuronal nuclei clustered independently is shown in the figure inset. Colors represent cell types.
B. UMAP plot of snRNA-seq data from 38,028 human TG nuclei from three donors, downsampled to display 15,000 nuclei, 5,000 per donor (1,487 neuronal, 13,513 non-neuronal). UMAP of neuronal nuclei clustered independently is shown in the figure inset. Colors represent cell types.
C. Dot plot displaying the expression of select cell-type-specific marker genes (columns) in male mice, female mice, or human TG cell types (rows). Dot size denotes the fraction of nuclei expressing a marker gene (>0 counts), and color denotes relative expression of a gene in each cell type (calculated as the mean expression of a gene relative to the highest mean expression of that gene across all cell types in the respective species and sex). Cell types with < 30 cells are not displayed.
cLTMR = c-fiber low threshold mechanoreceptor (LTMR); PEP = peptidergic nociceptor; TRPM8 = TRPM8+ cold sensitive neuron; NP = non-peptidergic nociceptor; NF1 = neurofilament+ A-LTMR enriched for A-beta-Field; NF2 = neurofilament+ A-LTMR enriched for A-beta-RA and A-beta-Field; NF3 = neurofilament+ LTMR enriched for A-delta; SST = somatostatin-positive pruriceptors; Satglia = satellite glia; Schwann_M = myelinating Schwann cells; Schwann_N = non-myelinating Schwann cells; Fibroblast_Dcn = Dcn+ meningeal fibroblasts; Fibroblast_Mgp = Mgp+ meningeal fibroblast; Immune = leukocytes; Vascular = endothelial cells.
In both mouse and human TG cell atlases, we observed 15 transcriptionally defined cell types (Figures 1A–C). These include 8 neuronal subtypes (see below) and 7 non-neuronal subtypes (Satellite glia, myelinating and non-myelinating Schwann cells [Schwann_M, Schwann_N, respectively], fibroblasts [Fibroblast_Mgp, Fibroblast_Dcn], immune cells, and vascular endothelial cells). While our study is the first report of snRNA-seq of human TG and non-neuronal cells from mouse TG, the TG neuronal subtypes we identified by snRNA-seq are highly consistent with those previously described in mice (Figures S1F–J) (Nguyen et al., 2017; Sharma et al., 2020), despite different methods being used to obtain each of these datasets. The PEP cluster is peptidergic nociceptors, TRPM8 is cold-sensitive neurons, NP is non-peptidergic nociceptors, cLTMR is c-fiber low threshold mechanoreceptors, and SST is Sst+/Nppb+ pruriceptors. NF1-3 clusters are A-fiber LTMR subtypes, and while genome-wide transcriptional profiling has not yet been performed on functionally-classified TG cell types, NF1 and NF2 neurons express S100b and likely correspond to the polymodal C4 class of Aβ neurons recently described (von Buchholtz et al., 2021). NF3 neurons express Fxyd2 and likely correspond to the brush responsive C5 Aδ neurons (von Buchholtz et al., 2021). We also compared the NF1-3 clusters to single-cell transcriptomic data from adult mouse DRG (Sharma et al., 2020), and found that NF1 is most similar to Aβ-Field DRG neurons, NF2 is similar to Aβ-RA or Aβ-Field DRG neurons, and NF3 is similar to Aδ DRG neurons (Figure S1J).
The cell-type-specific “marker” genes (defined as significantly greater expression in each TG cell type compared to all other TG cell types, see legends/methods) that are expressed in distinct TG cell types are largely conserved in both male and female mice and human (Figure 1C) and indeed label distinct populations of human TG cells as measured by in situ hybridization (Figure S2A). The TG cell types from individual libraries cluster together and are consistent across biological replicates (Figure 1A, S2B), a finding that is also observed with TG neurons from individual human donors (Figure 1B, S2B). Human non-neuronal cell types, however, appear to have a greater degree of transcriptional variability between donors than neurons (Figure S2B). We do not believe this observation is a technical batch effect of sample preparation because neuronal cell types cluster together across donors and non-neuronal cells from left and right TG of the same donor cluster together despite being prepared as individual libraries (Figure S2C). This variability in human non-neuronal TG gene expression may contribute to individual differences in TG function or simply reflect a greater sensitivity than neurons to underlying disease, medications, or post-mortem transcriptional changes.
While each of the annotated TG cell types was observed in both mouse and human atlases, the fraction of certain cell types varied between species (Figures 1A–B, S2D). Most notably, PEP neurons comprise a significantly greater fraction of the human TG neurons (23.8 – 28.2%) than mouse TG neurons (8.2 – 9.6%) sequenced (p < 0.01, two-tailed Student’s t-test) (Figure S2D). We also noted differences in the fraction of non-neuronal subtypes between species, but it is likely that technical differences in the amount of nerve and surrounding tissue included in dissections contribute to this observation. These species differences in TG prompted us to perform a more comprehensive comparison between mouse and human TG cell atlases.
Comparison of human and mouse TG
To more directly compare snRNA-seq data from human and mouse TG, we used Seurat to anchor the human dataset to the mouse dataset and assigned an ‘anchored’ cell type classification to each of the human nuclei based on their transcriptional similarity to the mouse cell types (see methods). We found that the mouse and human data clustered together (Figures 2A–B) and resulted in highly similar cell type assignments to those made when analyzing the species separately (86.8 ± 14.5 % overlap) (Figure 2C). Consistently, we observed similar cell-type-specific gene expression patterns between species (Figure 2D, Table S2). We also observed similar gene expression patterns between mouse and human TG cell types of key transcription factors, neuropeptides, ion channels, G-protein coupled receptors (GPCRs), and functional pathways (Figures 2E, S2E, Tables S2–3) involved in sensory ganglia function (Chiu et al., 2012; Geppetti et al., 2015; Moehring et al., 2018; Zheng et al., 2019). For example, in both mouse and human, TAC1 is highly enriched in PEP nociceptors and SCN11A is highly enriched in NP nociceptors compared to other cell types (Figures 1C, 2D–E). Moreover, human and mouse also express overlapping sets of ligand-receptor pairs between distinct TG cell types and between TG neurons and the meningeal cells to which many of them project (Figure 2F, S2F, Table S3). In both mouse and human TG, satellite glia and fibroblasts express the greatest number of ligands with receptor pairs in other TG cell types (e.g. NGF, APOE, SLIT2) (Figure 2F), and PEP nociceptors and SST neurons express the greatest number of ligands with receptor pairs in meningeal cell types (e.g. Adcyap1 [PACAP] from PEP neurons is paired with the Adcyap1r [PAC1] receptor in meningeal mast cells) (Figure S2F, Table S3). Single-cell atlases do not yet exist of the trigeminal nucleus caudalis; characterizing these cells and their ligand-receptor interactions with the TG neurons from which they receive input are important future directions. The evolutionary conservation of key cell-type-specific gene expression profiles, functional pathways, and ligand-receptor pairs in the TG suggests that mouse models may be useful in many cases for characterizing novel human pain therapeutics that target these conserved pathways.
Figure 2. Evolutionary conservation of mouse and human TG cell types.
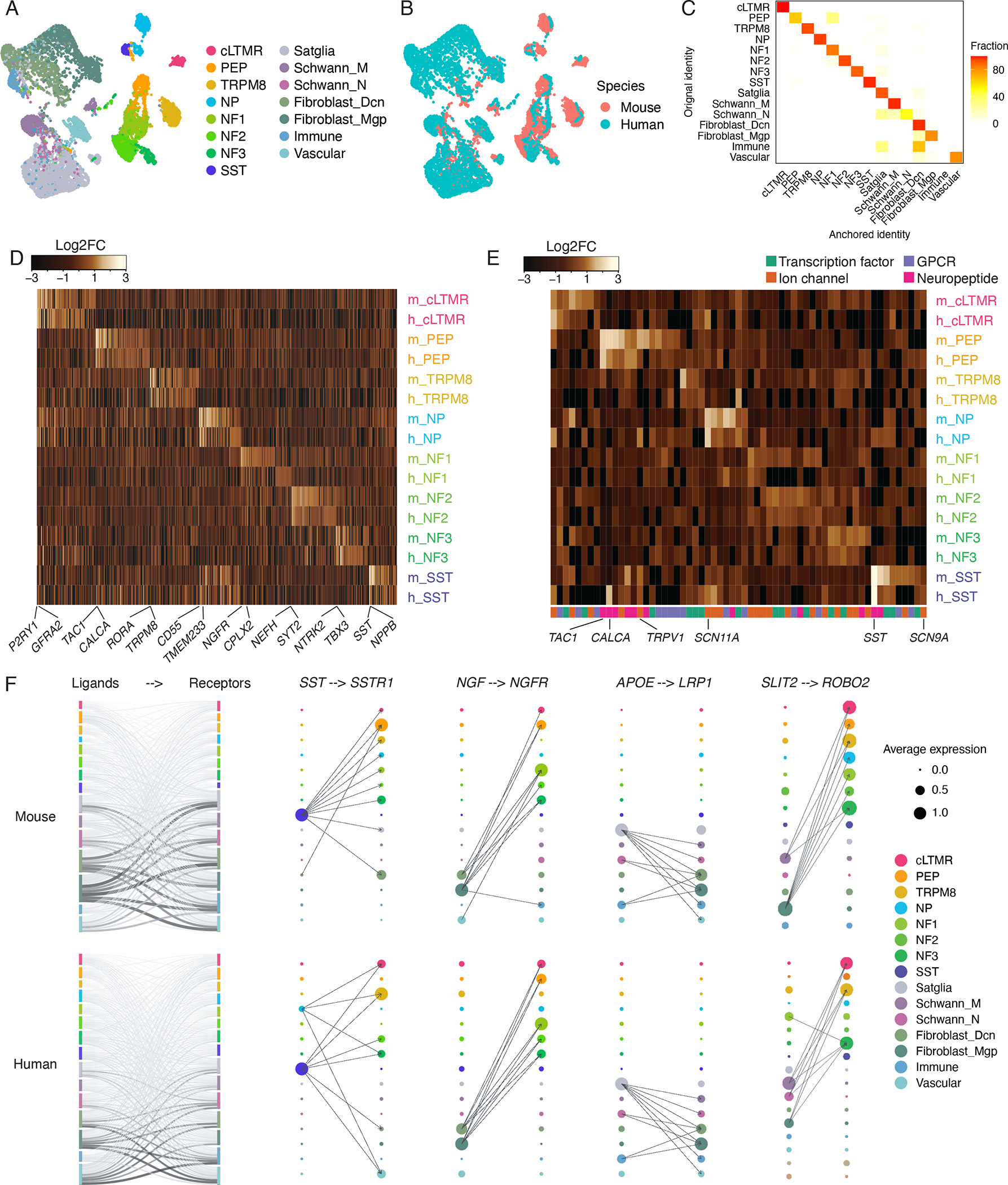
A-B. UMAP plots of human TG snRNA-seq data anchored to the mouse TG snRNA-seq data (see methods). Each species is downsampled to display 5,000 nuclei. (A) Colors represent cell type classifications determined from clustering each species separately prior to anchoring (as in Figures 1A–B). (B) Colors represent species.
C. Overlap of human TG cell types between the initial classifications (as in Figure 1B) and the classifications assigned by anchoring human TG to the male mouse TG reference (see methods). Plot displays fraction of nuclei within the initial cell type assignment that is assigned to each TG cell type after anchoring to the mouse TG reference.
D. Heatmap of evolutionarily conserved cell-type-specific gene expression (columns) in mouse and human TG cell types (rows, m = mouse, h = human). Cell-type-specific genes in each species are included in the heatmap if they are significantly enriched in a cell type compared to all other cell types (FDR < 0.01, top 50 genes by log2FC per cell type, Table S2).
E. Heatmap of select gene expression patterns (columns) in mouse and human TG cell types (rows, m = mouse, h = human). Genes are included in the heatmap if they are significantly enriched in a cell type compared to all other cell types (FDR < 0.05, log2FC > 0.5, Table S2).
F. Ligand-receptor interactions in mouse and human TG. Left) Putative interactions between ligands and receptors within mouse (top row) and human (bottom row) TG cell types. Vertical bars are colored by cell type and the height of bars depict the number of ligands (left column) and receptors (right column) in the given cell type. The thickness of connecting lines is proportional to the number of total ligand-receptor interactions between the two connecting cell types. Right) Dot size denotes relative expression of a gene in each cell type, and colors indicate cell type. Arrows between cell types denote the 10 highest ligand-receptor scores (The full set of ligand-receptor pairs can be found in Table S3).
While cell-type-specific gene expression patterns between human and mouse TG exhibit a high degree of similarity, we also identified genes that are differentially expressed between species (Figures 3A, S3A, Table S4). For example, SST neurons in humans but not mice express high levels of CALCA (Figure 3B–E, S3A), which encode calcitonin gene-related peptide (CGRP). While SST neurons in DRG are critical mediators of itch in mice (Huang et al., 2018; Stantcheva et al., 2016), the high expression of CALCA in human TG suggests that this cell type may also contribute to the known role of CGRP in head pain and migraine pathophysiology (Karsan and Goadsby, 2015). Indeed, SST neurons (Sst+/Nppb+) are well positioned to do so, as they have been observed to extensively innervate the meninges (von Buchholtz et al., 2020). The gene that encodes serotonin receptor 1F (HTR1F) is another migraine-associated gene that displays differential expression in mouse and human, and suggests that PEP nociceptors may be a key cell type through which the FDA-approved HTR1F agonist Lasmiditan aborts migraine headaches (Figure S3B–C). These species-specific gene expression patterns and ligand-receptor pairs point to opportunities for novel analgesic design that have been previously overlooked from molecular study of rodents.
Figure 3. Species-specific features of TG cell types.
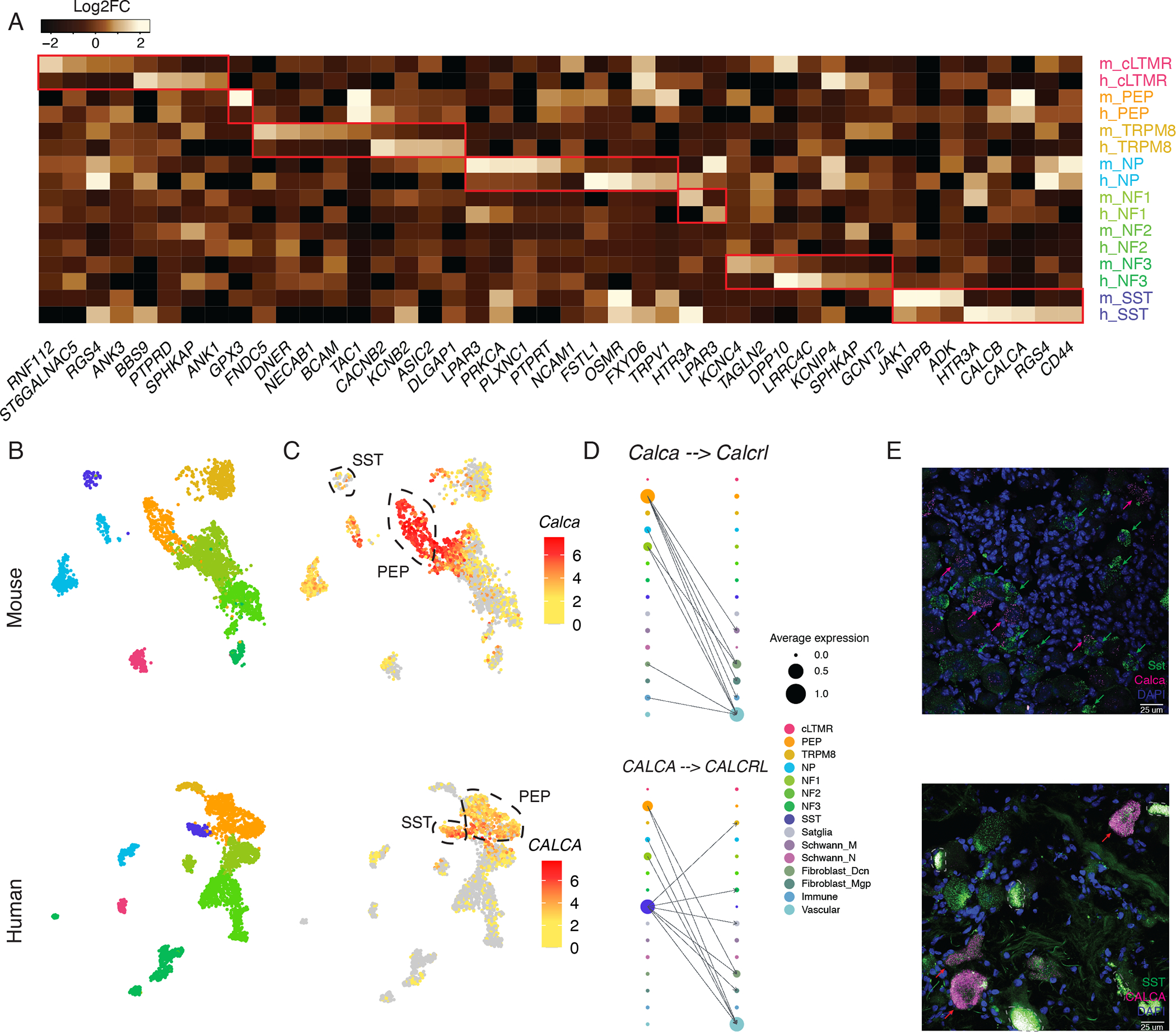
A. Heatmap of human and mouse TG cell-type-specific genes. Genes (columns) are included in the heatmap if they are both significantly enriched in a cell type (rows, m = mouse, h = human) compared to all other cell types (FDR < 0.01, log2FC > 1, see Table S3) and expressed significantly more in either human or in mouse (FDR < 0.01, top 5 genes by log2FC between human and mouse).
B-C. UMAP plots of snRNA-seq data of 3,000 nuclei downsampled from 15,303 TG neurons (top) or 3,000 nuclei downsampled from 3,873 human (bottom) TG neurons. B). Nuclei are colored by cell type (same colors as in Figure 3A). C). Nuclei are colored by log2 expression of Calca/CALCA. Peptidergic nociceptors (PEP) and Sst-expressing pruriceptors (SST) are circled.
D. Ligand-receptor interactions in mouse (top) and human (bottom) TG cell types. The dot size denotes relative expression of a gene in each cell type, and the color indicates cell type. Arrows between cell types denote the 10 highest ligand-receptor scores (The full set of ligand-receptor pairs can be found in Table S3).
E. Florescent in situ hybridization images of mouse TG (top) and human TG (donor 3, bottom) stained with probes against Sst/SST (magenta) and Calca/CALCA (green). Magenta arrows point to examples of human SST neurons that co-express CALCA (bottom) or mouse SST neurons that do not co-express Calca (top). Lipofuscin is circled by grey dotted lines in human slides. Scale bars = 25μm.
Sex differences in gene expression in TG cell types
The prevalence of migraine and chronic pain are several fold higher in females than males (Greenspan et al., 2007), which has prompted intense investigation into potential sex differences that may contribute to this observation (Avona et al., 2021; Mogil, 2020; Renthal et al., 2020; Sorge et al., 2015; Tavares-Ferreira et al., 2022a; Yu et al., 2020). We thus compared gene expression profiles of distinct TG cell types between male and female mice. As mentioned above, male and female TGs contain the same cell types that express highly similar cell-type-specific genes (Pearson’s r = 0.95 – 0.98 per cell type between males and females) (Figures 1C, S3D). While the transcriptional identity of each TG cell type appears to exhibit few sex differences, differential gene expression analysis comparing nuclei of the same cell type between male and female mice did reveal 123 genes that are significantly (Log2FC > 1, FDR < 0.05) more highly expressed in males than females and 180 genes that are more highly expressed in females than males (Figure S3E, Table S5). The most dramatic sex differences in gene expression are known sex-specific genes involved in X-inactivation (e.g., Xist, Tsix) or are Y chromosome genes (e.g., Uty, Ddx3y). However, we also observed differential gene expression between male and female mice such as Ptgds (prostaglandin D2 synthase) and Prl (prolactin). Ptgds and Prl are expressed more highly in females than males in both TG and DRG (Avona et al., 2021; Renthal et al., 2020; Tavares-Ferreira et al., 2022a), and have been shown to contribute to sex differences in pain-related behaviors (Avona et al., 2021; Tavares-Ferreira et al., 2022a). While there are few differences in cell-type-defining gene expression patterns between the male and female TG cell types sequenced here, the gene expression differences we do observe may have important functional consequences.
Latent alphaherpes virus expression in human TG
Alphaherpes viruses (e.g. herpes simplex virus 1, varicella zoster) lie dormant in human sensory neurons for decades in ~50% of the population (Gilden et al., 2007). When reactivated, these viruses can cause significant morbidity, such as painful ulcers, keratitis/vision loss, and more rarely, encephalitis. The TG cell types and gene regulatory mechanisms affected by alphaherpes viruses remain incompletely understood. We thus searched our human TG snRNA-seq data for reads that map to HSV1 latency-associated transcript (HSV1-LAT), HSV2-LAT, and VZV latency-associated transcript (VZV-LAT), transcripts expressed by these viruses in their latent state (Kennedy et al., 2015, LaPaglia et al., 2018). While we did not observe any reads that map to HSV2-LAT or VZV-LAT, we did identify 115 nuclei with reads that mapped to HSV1-LAT across the three human TG donors (0.185 – 0.325% per donor). Consistent with the literature, neurons are the primary cell types in which HSV1-LAT is detected (Figure 4A) (Gilden et al., 2007). The rare detection of HSV1-LAT in non-neuronal cells may be from secondary infection of these cells that occurs post-mortem or background RNA during single-cell encapsulation. The most common HSV1-LATpositive neuronal subtypes are NF1 and PEP nociceptors, followed by SST neurons (Figure 4B). The preference of HSV1 to lay dormant in neuronal subtypes that mediate nociception and neurogenic inflammation (NF1, PEP, and SST are all CALCA+ subtypes) is consistent with the clinical observation of herpes reactivation (Gilden et al., 2007).
Figure 4. Latent alphaherpes virus expression in human TG neurons.
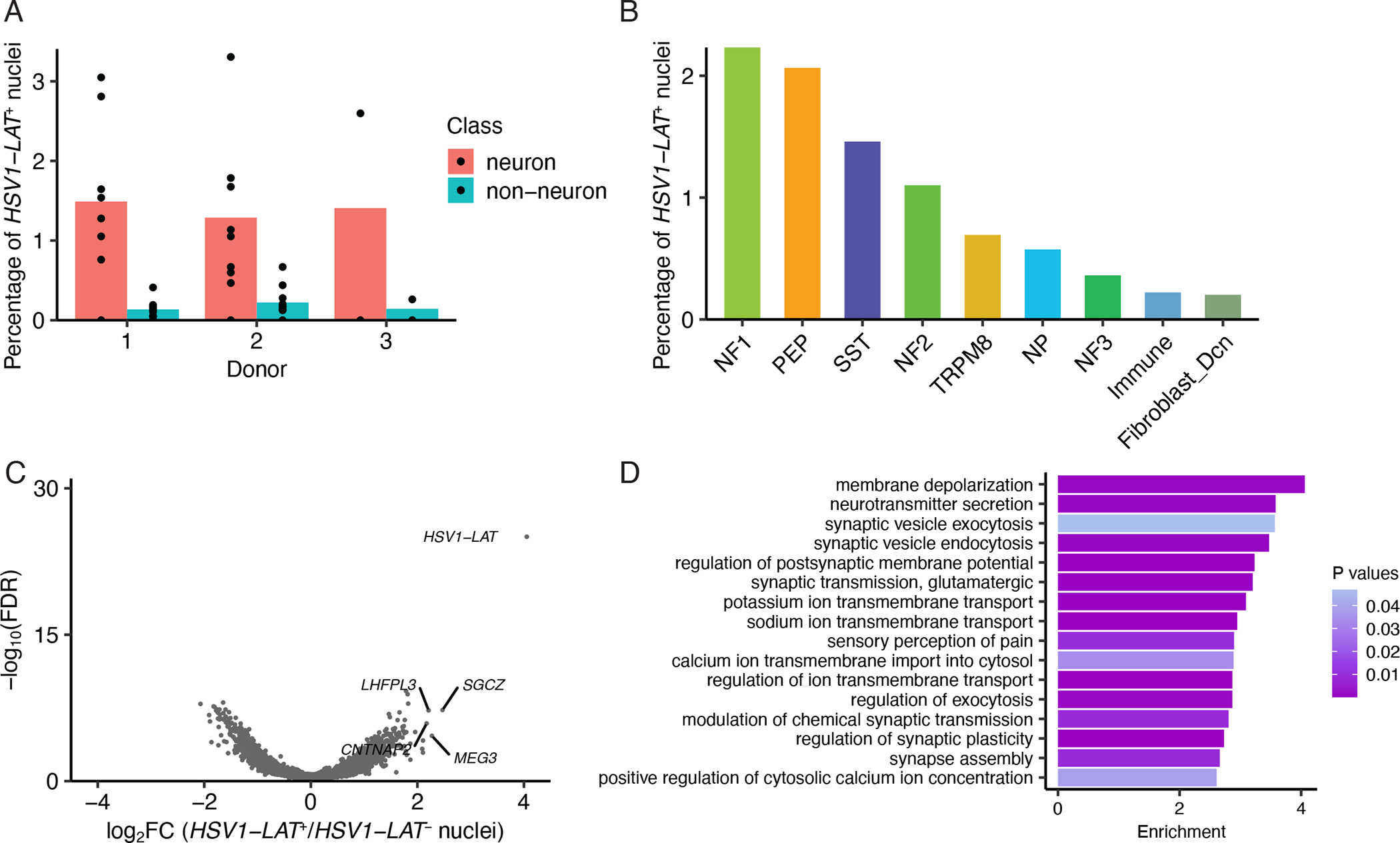
A. Percentage of HSV1_LAT+ neurons or non-neuronal cells in each donor. Dots show percentage of HSV1_LAT+ nuclei in each library and bars indicate the average across libraries.
B. Percentage of each neuronal subtype that are HSV1_LAT+ across all donors.
C. Volcano plot displaying differential gene expression between HSV1_LAT+ and HSV1_LAT- nuclei. HSV1-LAT (n = 115) and the same number of randomly selected cells of the same cell type distribution without detectable expression of HSV1_LAT. Significance is displayed on the Y-axis as -log10 False Discovery Rate (FDR); magnitude of differences is displayed on X-axis as log2 Fold Change (FC).
D. Gene ontology analysis of differentially expressed genes (Log2FC > 1, FDR < 0.05) between HSV1_LAT+ and HSV1_LAT- nuclei. Enrichment is the number of times an ontology term is observed in the differentially expressed gene set over a random gene set of expressed genes.
Bar shows enrichment and color shows P-value.
We next asked how HSV latency alters gene expression patterns within host TG cell types by performing differential gene expression analysis between HSV1-LAT positive and negative nuclei. We observed that there were 343 upregulated genes and 298 down-regulated genes (Log2FC > 1 or < −1, FDR < 0.05) in HSV1-LAT positive nuclei compared to HSV1-LAT-negative nuclei (Figure 4C, Table S5). Notably, these dysregulated genes are associated with pathways implicated in sensory perception of pain, synaptic vesicle exocytosis, and neurotransmitter secretion (Figure 4D). These findings suggest that neurons are primed for hyperactivity when stress, immunosuppression, or other triggers for reactivation occur.
Epigenomic mechanisms that drive cell-type-specific gene expression
We were particularly struck by both conserved and species-specific cell-type-specific gene expression patterns in the TG and reasoned that improved understanding of these gene regulatory mechanisms might provide new opportunities for understanding the function of disease-associated genetic variation within these regulatory regions and for designing cell-type-specific genetic tools (e.g., transgenic mice or gene therapy vectors).
As cell-type-specific gene regulation is thought to be largely mediated by the action of distal gene regulatory elements (e.g. gene enhancers) (Heinz et al., 2015), we turned to the assay for transposase-accessible chromatin (ATAC-seq) to characterize these putative gene regulatory elements in mouse and human TG at single-cell resolution (Buenrostro et al., 2015). In mice, we first enriched our cell population for neurons by sorting fluorescently labeled TG neuronal nuclei from male and female Vglut2-cre;Sun1-GFP mice prior to performing snATAC-seq. From three biological replicates (Table S1), we obtained 316 million unique transposase-sensitive fragments in the expected nucleosomal size distribution (Figure S4A–B), generating a dataset of 8,064 TG nuclei with an average sequencing depth of 31,559 transposase-sensitive fragments per nucleus. These snATAC-seq fragments formed 306,222 peaks when aggregated across all TG nuclei. The peaks most frequently mapped to gene distal genomic regions (68.4%, within 200kb of TSS, not overlapping with gene body or promoter). The remainder of fragments mapped to intragenic (17.6%), promoter (5.4%, ≤1,000bp upstream or ≤100bp downstream of TSS) and intergenic (8.6%, >200kb from any genes) regions (Figure 5A).
Figure 5. Single-nucleus epigenomic analysis of mouse trigeminal ganglion.
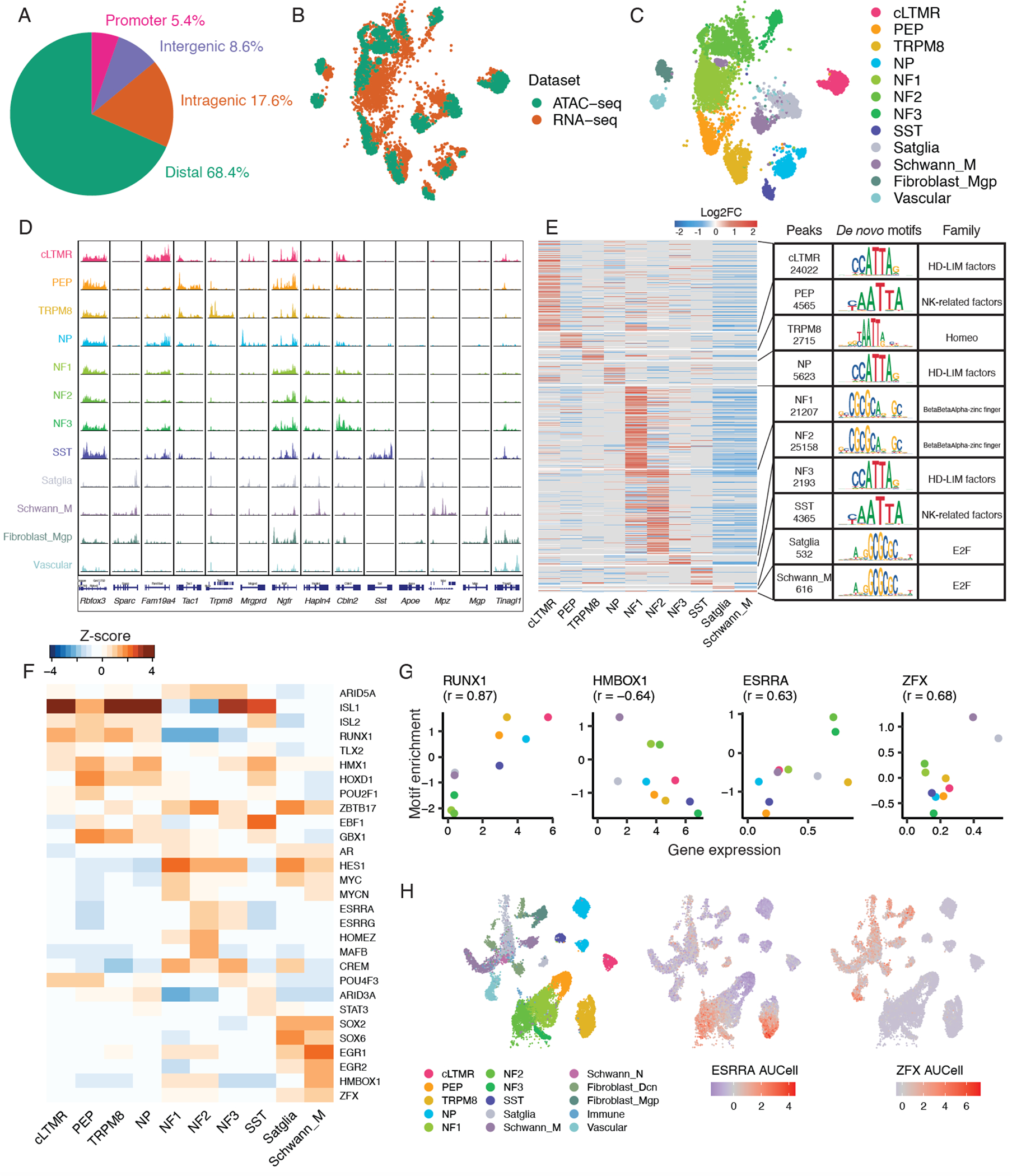
A. Fraction of peaks in snATAC-seq data that map to promoter regions (−1,000bp to +100bp of transcription start site [TSS]), intragenic regions (within gene body excluding promoter region), distal regions (<200 kb upstream or downstream of TSS excluding promoter and intragenic regions), and intergenic regions (>200kb upstream or downstream of TSS) across 3 biological replicates of Vglut2-cre;Sun1-GFP+ TG nuclei.
B-C. UMAP plots of 3,519 mouse TG nuclei profiled by snATAC-seq anchored to 5,584 male naive mouse TG nuclei profiled by snRNA-seq. Nuclei are colored by B) single-nucleus profiling technique or C) cell type classification. Cell types that are present in snRNA-seq data but not in snATAC-seq data are not shown.
D. For each TG cell type (rows), chromatin accessibility is displayed at cell-type-specific genes (columns). snATAC-seq data is displayed as the average frequency of sequenced DNA fragments per cell for each cell type, grouped by 50 bins per displayed genomic region; Y-axis is scaled for each gene (column).
E. Differential chromatin accessibility analysis of 90,996 cell-type-specific peaks in each TG cell type compared to all other TG cell types (Log2FC > 0.5, FDR < 0.05, Table S8). Heatmap displays log2FC for each peak (rows) in the respective TG cell type (columns). Transcription factor (TF) DNA binding motifs that are most significantly enriched within each cell type’s differentially accessible peaks (Log2FC > 0, FDR < 0.05) compared to randomly selected peaks (Table S9) are shown. The most enriched motif and its TF family are displayed for each cell type. Cell types with ≤ 500 differentially accessible peaks are not shown.
F. Heatmap of TFs whose DNA binding motifs are significantly enriched (Log2FC > 0, FDR < 0.05) within each cell type’s differentially accessible peaks compared to randomly selected peaks (see methods). Top 5 TFs by motif enrichment per cell type are included in the heatmap. Heatmap shows the Z-score (column-scaled) of motif fold enrichment.
G. Scatter plots of average normalized expression of cell-type-specific TF mRNA and its TF motif fold enrichment (Z-score as in Figure 5F) in each mouse TG cell type. Pearson’s r between gene expression and motif enrichment in each cell type is displayed.
H. UMAP plots of 59,921 naive mouse TG nuclei profiled by snRNA-seq downsampled to display 28,000 nuclei (as in Figure 1A). Nuclei are colored by cell type (left) or by AUCell regulon scores of ESRRA (middle) and ZFX (right) using SCENIC (see methods).
We next assigned cell types to each epigenomically profiled nucleus by multi-omic anchoring them to our annotated mouse TG snRNA-seq data (Figure 5B, see methods) (Butler et al., 2018). Anchoring enabled us to assign known cell types to 3,519 out of 8,064 epigenomically profiled TG nuclei (Figure 5C, Figure S4C), yielding 73.6% neurons and 26.4% non-neurons. Chromatin accessibility near cell-type-specific marker genes was consistent with the expected cell types (Figure 5D); for example, chromatin accessibility near Rbfox3, a neuronal marker gene, was preferentially accessible in neurons. Likewise, chromatin accessibility near Sparc, a non-neuronal marker gene, was preferentially accessible in non-neuronal cells. Cell-type-specific marker genes for neuronal subtypes such as Tac1, Trmp8, and Sst also displayed preferential accessibility at these genomic loci in their respective cell types (Figure 5D). Consistent with the broad transcriptional similarity in male and female TG cell types, genome-wide chromatin accessibility is also highly correlated between male and female mice (Pearson’s r = 0.98). That said, we did observe some significant sex differences in chromatin accessibility at known sex-specific genes (e.g., Xist, Uty) (Figure S4D), as well as several other regions that may contribute to or reflect sex differences in gene expression (Table S6).
To characterize the extent to which chromatin accessibility is conserved between mice and human, we next performed snATAC-seq data from three human TGs (Table S1). We obtained 11,346 nuclei with an average sequencing depth of 5,931 transposase-sensitive fragments per nucleus that form 124,619 peaks (Figures S4E–F). While multi-omic anchoring could only confidently classify 46 neurons and 4,989 non-neuronal nuclei (see methods) (Figures S4G–H), 58.4% of the human snATAC-seq peaks occur in genomic loci that correspond to regions in mice where we also observed peaks of chromatin accessibility, indicating highly significant overlap between human and mouse epigenomic profiles (p < 1 × 10−155, hypergeometric test, Table S7).
We next asked which TFs and putative gene enhancers contribute to cell-type-specific gene expression patterns in distinct TG cell types. We addressed most of these questions using mouse snATAC-seq data because of the greater number of neuronal nuclei profiled. To identify which TFs are likely to mediate cell-type-specific gene expression in TG, we identified 91,269 genomic regions that are preferentially accessible within 12 distinct TG cell types (Log2FC > 0.5, FDR < 0.05, comparing accessibility within one TG cell type to that of all others) (Figure 5E, Table S8) and the transcription factor binding motifs that are enriched within these sites (Log2FC > 0, FDR < 0.05) (Figures 5E–F, Table S9). We identified TFs that have been previously implicated in the function of sensory ganglion cells such as ISL1 (Sun et al., 2008), whose motif is enriched across most sensory neuron subtypes, RUNX1 (Chen et al., 2006), whose motif is enriched in unmyelinated C-fibers, and SOX2 and SOX6 (Cantone et al., 2019; Parrinello et al., 2010), whose motifs are enriched in satellite glia and Schwann cells (Figure 5F). We also implicate several new cell-type-specific TFs in TG function such as the ISL LIM Homeobox 2 (ISL2) and AT-Rich Interaction Domain 3A (ARID3A) in C-fibers and ARID5A and estrogen related receptor alpha (ESRRA) in A-fibers.
To explore whether cell-type-specific TFs likely function to activate or repress gene expression within their respective cell types, we next correlated the gene expression measured by snRNA-seq to the motif enrichment measured by snATAC-seq for each TF across all cell types. We found that the neuronal-specific TFs such as RUNX1 and ESRRA, and non-neuronal TFs such as ZFX likely function to activate gene expression because the expression of these TFs is positively correlated with their motif enrichment across cell types (Figure 5G). Consistent with these findings, we observed that expression of the predicted downstream target genes (regulons) of both ESRRA and Zinc Finger Protein X-Linked (ZFX) are preferentially enriched in similar mouse TG snRNA-seq cell types (PEP and TRPM8 for ESRRA and Schwann cells and satellite glia for ZFX) to those predicted by motif analysis of cell-type-specific regions of chromatin accessibility (snATAC-seq) (Figure 5H). We also identified TFs such as Homeobox Containing 1 (HMBOX1) that are likely to function as cell-type-specific repressors of gene expression, as the expression of these TFs and their motif enrichment is negatively correlated across cell types (Figure 5G).
As cell-type-specific TFs regulate gene expression through gene regulatory elements located in promoter and distal enhancer regions, we next characterized these putative regulatory sites genome-wide. To prioritize peaks of chromatin accessibility that are likely to function as gene enhancers, we correlated the chromatin accessibility of cell-type-specific peaks with expression of cell-type-specific genes in each cell type (see methods). We found 43,885 positively correlated (r > 0) cell-type-specific peak-gene pairs that are distributed with a median distance of 33,717 bp upstream of their putative target genes (Figure S5A). These cell-type-specific peaks are significantly more correlated with the expression of cell-type-specific genes than all genes on the same chromosome (Figure S5B); indeed, among those positively correlated pairs of cell-type-specific peaks and genes, 29,881 (68.1%) have a Pearson’s r > 0.5.
To assign these putative enhancers to their most likely target gene, we calculated the Activity-By-Contact (ABC) score (Fulco et al., 2019), an experimentally validated enhancer prediction tool, for each peak of accessible chromatin genome-wide (Table S10). For example, ABC predicted that non-coding genomic regions ~37 – 56 kb downstream of Calca, ~19 – 24 kb upstream of Scn11a, and ~24 – 46 kb upstream of Ngfr are likely to regulate the expression of their respective genes (Figure 6A, Table S10). We also identified 1,080 ABC-predicted gene enhancers that exhibit cell-type-specific chromatin accessibility that is highly correlated (r > 0.5) with their predicted target gene expression (Figure 6B). These findings provide new insight into the gene regulatory mechanisms of distinct TG cell types and may be of particular use for developing tools such as viral vectors or reporter mice that can enable genetic access to distinct sensory ganglia cell types.
Figure 6: Gene regulatory mechanisms in distinct trigeminal ganglion cell types.
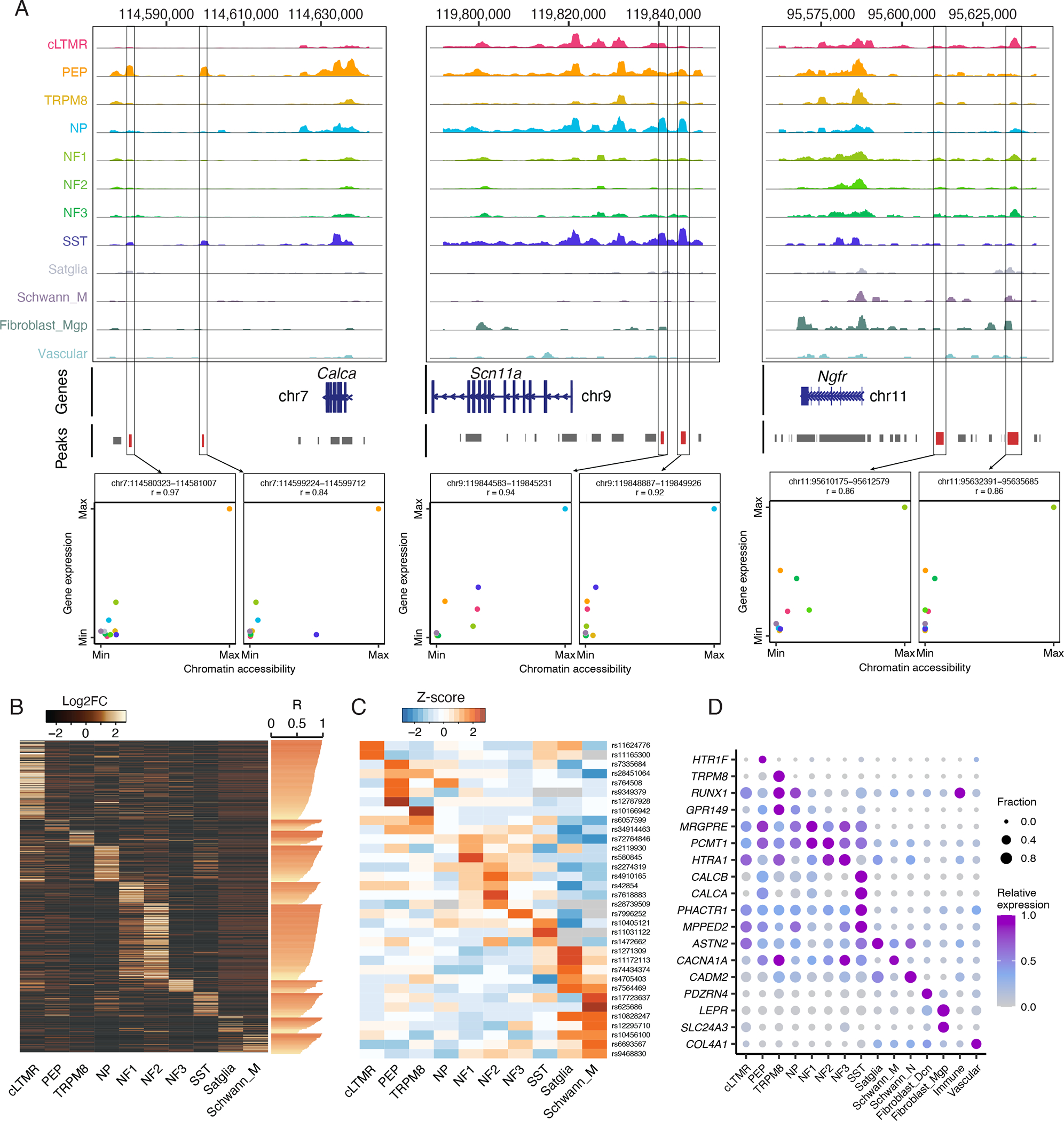
A. Chromatin accessibility is displayed at the genomic loci of Calca, Scn11a, or Ngfr. At each genomic locus, chromatin accessibility is displayed as the average fraction of transposase-sensitive fragments per nucleus at that region (grouped by 50 bins per displayed genomic region). Accessibility at each locus (Y-axis) is scaled to the max value across all cell types (column). Peaks are annotated as grey bars and differentially accessible peaks are colored red. Scatter plot shows the correlation of their chromatin accessibility of a given snATAC-seq peak and expression of a nearby gene in each cell type (chromatin accessibility and gene expression are normalized to their max values). Colors indicate cell types.
B. Heatmap displays Log2FC of 1,080 cell-type-specific snATAC-seq peaks (rows, Log2FC > 0.5, FDR < 0.05) whose chromatin accessibility is highly correlated (Pearson’s r > 0.5, values are displayed as horizontal bars) with the expression of cell-type-specific genes (Log2FC > 1, FDR < 0.05, comparing gene expression in one TG cell type to all other cell types) in the respective cell type. These peaks are positioned within 200 kb upstream of the respective cell-type-specific gene’s TSS and are associated with its regulation by ABC score.
C. Migraine-associated genomic variants exhibit preferential chromatin accessibility within distinct TG cell types. Heatmap displays the Z-score (row scaled) of the fraction per nucleus of transposase-sensitive fragments that overlap with a 1 kb window around the genomic locus that corresponds to migraine-associated genomic variants (rows).
D. Dot plot displays the expression of migraine-associated genes (rows) in human TG cell types (columns) as measured by snRNA-seq. Dot size denotes the fraction of nuclei expressing a marker gene (>0 counts), and color denotes relative expression of a gene in each cell type.
Genetic susceptibility to migraine can occur in cell-type-specific putative enhancers
Epigenomic cell atlases have recently been used to help localize the cell types in which disease-associated genomic variation drives pathophysiology (Cusanovich et al., 2018a). This approach is particularly useful for interpreting the potential function of non-coding genomic variants because chromatin accessibility at these loci can be matched with gene expression in a cell-type-specific fashion. We reasoned that our catalogue of chromatin accessibility in TG cell types could provide a new opportunity for predicting the genes and cell types affected by genomic variants associated with migraine, a heritable condition that involves disabling head pain and sensitization of TG neurons (Ashina et al., 2019). Recent GWAS have identified 123 single nucleotide polymorphisms (SNPs) that are significantly associated with migraine susceptibility, but it remains unclear which genes and TG cell types are affected by these variants and how they contribute to migraine susceptibility (Gormley et al., 2016; Hautakangas et al., 2022). Of the 123 migraine-associated SNPs, 70 have corresponding genomic coordinates in the mouse and of these, over half occur in peaks of accessible chromatin, and 14 exhibit significant enrichment of chromatin accessibility in specific TG cell types compared to all TG cell types (Figure 6C, Table S11). For example, rs10166942 is selectively accessible in TRPM8 neurons and is located ~1kb upstream of the TRPM8 gene, rs7564469 is selectively accessible in satellite glia and located within a ZEB2 intron, and rs10828247 is selectively accessible in Schwann cells and located within an MLLT10 intron. We also observed peaks of chromatin accessibility at several migraine-associated loci in our human TG snATAC-seq data (Figure S5C), including rs580845 (located within an intron of neuronal TF, NFIB) with ~10-fold greater accessibility in TG neurons compared to non-neuronal cell types in both human and mice (Figures 6C, S5C).
Current migraine therapeutic target genes (e.g., CALCA, CALCB, HTR1F) as well as several genes implicated by GWAS exhibit cell-type-specific gene expression patterns in human TG (Figure 6D). These findings suggest that multiple TG cell types such as PEP and NP nociceptors, SST neurons, and satellite glia may contribute to migraine pathophysiology and highlight the potential of integrating GWAS with single-cell transcriptional and epigenomic data. To comprehensively assess which of these TG cell types are engaged during head pain, we turned to two animal models of headache: inflammatory soup (IS) and cortical spreading depression (CSD). The IS model involves the application of IS to dura, which acutely causes migraine-like headache behaviors by activating and sensitizing trigeminal meningeal nociceptors (Burgos-Vega et al., 2019; De Felice et al., 2013; Strassman et al., 1996). CSD, the neurophysiological correlate of migraine aura, is also known to acutely activate and sensitize trigeminal meningeal nociceptors (Ashina et al., 2019; Bolay et al., 2002; Zhang et al., 2010; Zhao and Levy, 2016) and promote migraine-like pain (Charles, 2017; Harriott et al., 2021). For IS, bilateral TGs were isolated from male mice 1h, 6h, or 24h after dural application of IS, female mice 1h after IS, and mice injected with PBS. For CSD, mice were cannulated a week prior to induction of CSD by pinprick; TG ipsilateral to the pinprick were collected 1.5h or 6h after induction of CSD. In total, we sequenced 15 replicates containing 37,012 nuclei across both headache models (Table S1). Co-clustering of those nuclei from headache models and nuclei from naive mice led to the identification of the same 15 TG neuronal and non-neuronal cell types described above (Figure 7A, Figure S6A).
Figure 7. Activation of multiple TG cell types in mouse models of headache.
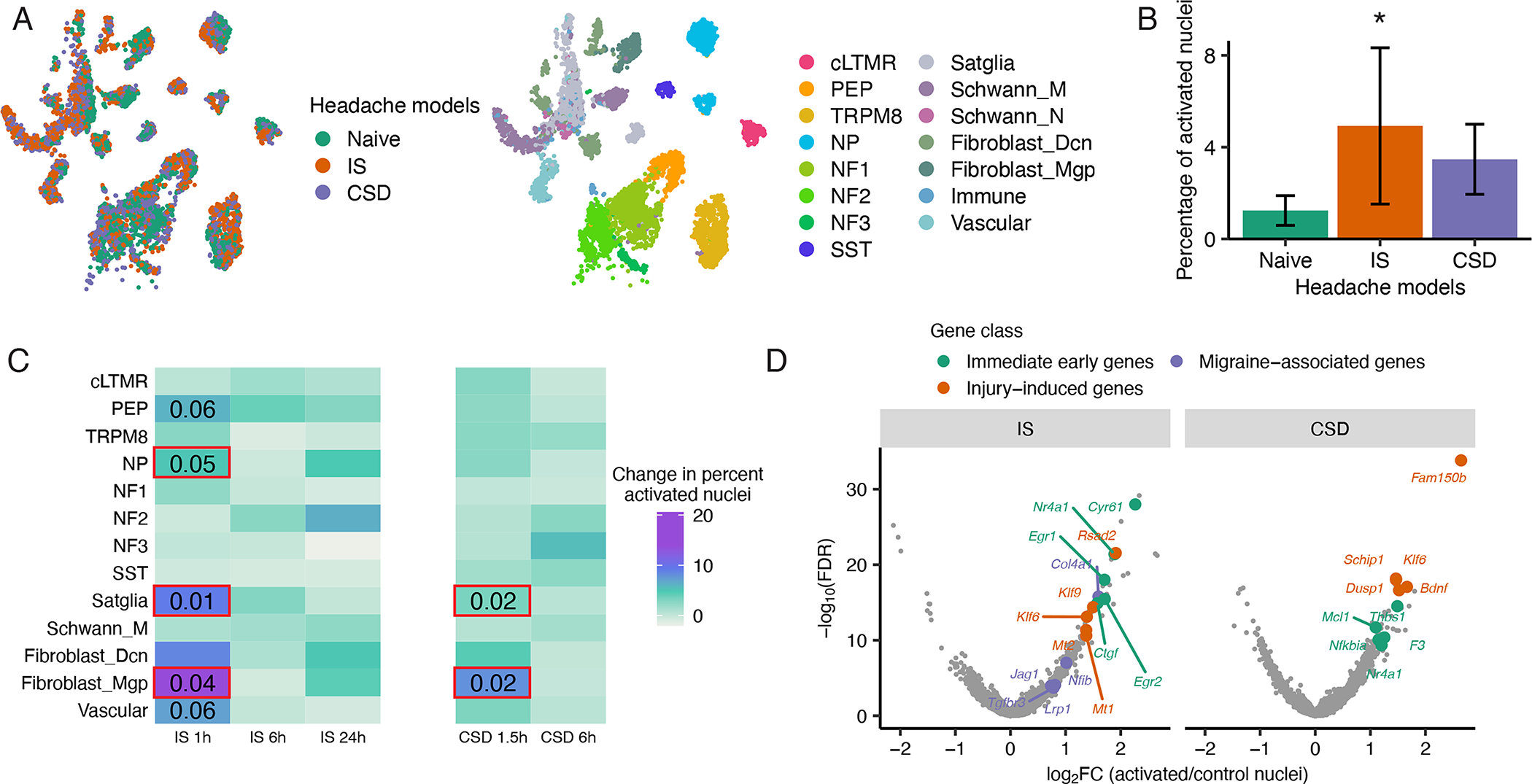
A. UMAP plots of snRNA-seq data from 96,933 mouse TG nuclei from both naive and headache models (downsampled to display 3,000 naive, 3,000 IS, and 3,000 CSD). Nuclei are colored by condition (left) or by cell type (right).
B. Fraction of nuclei in each headache model that display a transcriptionally activated state after IS or CSD as defined by the expression of a panel of immediate early genes (IEG) (see Act-seq methods). There are significantly more transcriptionally activated nuclei after IS than in naive (*P < 0.05, two-tailed Student’s t-test, error bars are SEM).
C. Heatmap showing change in percentage of transcriptionally activated nuclei in each TG cell type after IS or CSD compared naive. Significant effects of IS were observed in NP, satellite glia and fibroblasts (*P < 0.05 1-way ANOVA, see Figure S6C for ANOVA statistics); strong trends were observed in PEP and vascular cells. Significant effects of CSD were observed in satellite glia and fibroblasts (*P < 0.05, 1-way ANOVA, see Figure S6D for ANOVA statistics).
D. Volcano plot of differentially expressed genes between transcriptionally activated nuclei in Left) IS or Right) CSD and the same number of randomly selected control cells of the same cell type distribution. Significance is displayed on the Y-axis as -log10 False Discovery Rate (FDR); magnitude of gene expression differences is displayed on X-axis as log2 Fold Change (FC). Top five genes by log2FC in each gene class are labeled. Immediate early genes (IEGs) are orange, genes that are activated by axonal injury (see methods) are green, and migraine-associated genes are purple.
To characterize the TG cell types engaged in these headache models, we used an snRNA-seq approach (Act-seq) that identifies “activated” cells by the expression of immediate early genes (see methods) (Wu et al., 2017). We observed a significantly greater fraction of activated nuclei overall in the IS model (4.9%) than CSD (3.5%) and naive (1.2%) (Figure 7B), with the fraction of activated NP nociceptors, satellite glia, and fibroblasts 1h after IS significantly increased compared to naive mice (p < 0.05, one-way ANOVA) (Figures 7C, S6B). A trend of increased activation is also observed in PEP nociceptors and vascular cells 1h after IS (p = 0.06, one-way ANOVA). We also observed significant increases in the fraction of activated TG fibroblasts and vascular cells after IS compared to naive mice in females (p < 0.001, permutation test) (Figure S6C), but future studies are needed to sufficiently power sex comparisons of less abundant TG neuronal cell types.
We next asked whether the TG cell types that are activated by CSD overlap with those which are activated by IS. We observed significant increases in the fraction of activated satellite glia and fibroblasts 1.5h after CSD compared to naive mice (p < 0.05, one-way ANOVA) (Figures 7C, S6D), which suggests that there is some convergence at the level of TG cell type activation between distinct mouse models of headache. This cell type convergence between headache models is particularly relevant for our interpretation of IS because TG cells can be activated by the acute surgical process in this model (Figure S6E).
To characterize the transcriptional program induced in TG cells that are activated by headache, we performed differential expression analysis between the activated and non-activated nuclei defined previously by Act-seq. We found that IS significantly upregulates 96 genes and CSD significantly upregulates 72 genes in nuclei that are classified as activated compared to the same number of nuclei that are not transcriptionally activated (Log2FC > 1, FDR < 0.05) (Figure 7D, Table S12). These headache-induced transcriptional programs are associated with pathways involved in gene regulation, axon guidance, inflammation (Figures S6F–G, Table S12) and may contribute to the peripheral sensitization that is thought to underlie headache disorders such as migraine (Strassman et al., 1996). Indeed, several of these headache-activated genes (e.g., Nfib, Lrp1, Col4a1, Tgfbr3, and Jag1) are near to and/or are predicted to be affected by migraine-associated SNPs (Figures 6C–D, S5C). Future studies are aimed at characterizing precisely how this genetic variation drives susceptibility to migraine.
DISCUSSION
The transcriptional and epigenomic TG cell atlas presented here identified evolutionarily conserved and species-specific gene expression patterns within distinct TG cell types, epigenomic features that may establish these cell-type-specific gene expression patterns, and the TG cell types in which migraine-associated genetic variation may contribute to disease. These data also provide a resource for studying other TG functions, including the sensory innervation of cornea, nasal mucosa, and teeth. To facilitate access to these data, we have built a searchable web resource for the research community at tg.painseq.com.
Our studies of human and mouse sensory ganglia are particularly relevant as the field of pain medicine is trying to overcome decades of difficulty with the translation of novel analgesic targets from mice to humans. These difficulties raise important questions about the similarities and differences between mouse and human pain processing and led us to build a TG cell atlas in both species. We observed eight neuronal subtypes and seven non-neuronal subtypes in both species. For a given TG cell type, we observed similar cell-type-specific patterns of gene expression between mice (male and female) and human including most cell-type-specific marker genes (Figures 1C, 2D, Table S2). These similarities extend to conserved expression of ion channels that are critical for pain processing (e.g., SCN9A, SCN11A, TRPV1) and support the utility of rodent models for studying the function of these channels in the same cell types in which they are expressed in humans (Figure 2E, Table S2).
We found that the transcriptional identity of TG cell types is largely conserved not only between species, a finding that is consistent with recent cross-species comparisons of DRG (Kupari et al., 2021; Nguyen et al., 2021; Tavares-Ferreira et al., 2022b), but also between TG and DRG. Indeed, with a notable exception of proprioceptors (Figure S1J), the same general classes of cell types are present in both TG and DRG, and they express highly similar patterns of cell-type-specific genes in both mouse and human (Figures S7A–G). Thus, while we did detect gene expression differences between TG and DRG cell types in both mouse (Figure S7C, Table S13) and human (Figure S7G, Table S13), deeper sequencing is likely necessary to identify differences in additional genes (e.g., lowly-expressed GPCRs) that could guide the development of ganglion-specific therapeutic strategies.
While the transcriptional identities of broad cell types are largely conserved between species, we also observed notable gene expression differences that may have implications for pain processing and underscore the importance of including studies of human cells in the design of next-generation pain therapeutics (Renthal et al., 2021). For example, Calca, the gene that encodes the neuropeptide CGRP, is predominantly expressed in PEP nociceptors and NF1 neurons in mouse TG, but in human, it is expressed in SST neurons in addition to PEP and NF1. As CGRP is inhibited by new classes of FDA-approved migraine therapeutics, SST neurons, which have recently been shown to innervate the meninges (von Buchholtz et al., 2020), may play an important role in migraine pathophysiology through CGRP’s downstream actions. Our ligand-receptor analyses suggest that while we do observe subtle CGRP receptor expression in Aδ-fibers (Edvinsson et al., 2019; Melo-Carrillo et al., 2017), CGRP may exert stronger effects on non-neuronal cells such as vascular cells or fibroblasts in the TG or in the meninges (Levy et al., 2019). Future studies are needed to better characterize the roles of CGRP+ SST and PEP neurons in migraine pathophysiology.
As gene expression profiles are largely determined by distinct epigenomic signatures at gene promoters and enhancers, we mapped these elements in the TG across species using single-cell epigenomics, linked these putative regulatory elements with their most likely target genes (Table S10), and identified TFs whose consensus binding motifs are enriched within these elements (Table S9).
In addition to providing new insight into the gene regulatory mechanisms underlying TG cell-type-specific gene expression patterns, the TG transcriptional and epigenomic cell atlas presented here also enables important future applications. For example, ongoing work is focused on identifying the putative gene regulatory elements identified in our epigenomic data that are sufficient to drive TG cell-type-specific gene expression in the setting of a viral vector (e.g., enhancer viruses). Such an application has recently been reported for CNS interneuron subtypes using ATAC-seq data (Hrvatin et al., 2019; Mich et al., 2021; Vormstein-Schneider et al., 2020), so we are optimistic that this approach will lead to the development of nociceptor-specific AAVs for the treatment of pain.
We also leveraged the TG epigenomic data presented here to guide the interpretation of non-coding genomic variants associated with migraine, a heritable condition in which pathological sensitization of TG meningeal nociceptors occurs through mechanisms that remain unclear. Indeed, we found that over half of the index variants that are significantly associated with migraine occur within peaks of chromatin accessibility, several of which are predicted to regulate genes that are expressed preferentially in TG nociceptors and satellite glia (Figure 6D). These findings prompted us to perform a comprehensive assessment of trigeminal cell types that are engaged in two distinct animal models of headache. We found that satellite glia and fibroblasts are transcriptionally activated in both IS and CSD headache models, which suggests that these distinct models mechanistically converge on the activation of these cell types. We also observed PEP and NP nociceptors and vascular cells are activated after IS, which is consistent with IS being a stronger acute inflammatory stimulus than CSD. While neither IS nor CSD model all aspects of migraine pathophysiology, some of the TG cell types that are activated in these models are consistent with the cell types implicated in our analyses of human genetic variation associated with migraine susceptibility (e.g. NP cells, satellite glia, fibroblasts). Future studies are still needed to clarify the mechanisms by which specific genetic variants increase migraine susceptibility, but our data can guide these studies toward the cell types in which each variant is most likely to cause dysfunction.
Together, the human and mouse TG transcriptional and epigenomic cell atlases presented here provide a wealth of new data that can be used to identify more specific headache and pain therapeutic targets and begin to interrogate the complex biology of migraine and other headache and facial pain disorders. We imagine that the cellular and molecular resolution of the resource presented here will continue to improve as additional human donors and TG cells are sequenced.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, William Renthal (wrenthal@bwh.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Processed snRNA-seq data are available at tg.painseq.com. Raw and processed snRNA-seq data were also deposited within the Gene Expression Omnibus (GEO) repository (www.ncbi.nlm.nih.gov/geo) with an accession number (GSE197289).
Custom R scripts are available on https://github.com/Renthal-Lab/.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
8–12-week-old C57BL6/J male and female mice were obtained from the Jackson Laboratory (JAX) (strain #000664). Sun1-GFP (strain #021039) and Vglut2-Cre (strain #016963) male and female mice were also obtained from JAX. All animal experiments were conducted according to institutional animal care and safety guidelines at Brigham and Women’s Hospital.
Headache models
The inflammatory soup (IS) model was performed as described in (Strassman et al., 1996) with minor modifications. Briefly, C57BL6/J mice were anesthetized by isoflurane and head fixed in a stereotaxic device. Frontal and parietal dura were exposed bilaterally by four burr holes (from bregma, 1.5mm anterior +/− 2mm lateral, 3.75mm posterior +/− 2mm lateral) and IS (capsaicin 0.5mM, bradykinin 1mM, histamine 1mM, PGE2 0.1mM) or vehicle control (PBS) were slowly applied topically (10uL per burr hole). Mice remained anesthetized for 15 min prior suturing the scalp incision. Mice were observed closely after surgery and sacrificed at 1h, 6h, or 24h after application of IS or control. Bilateral TGs were collected for each mouse; two mice were pooled per biological replicate. The cortical spreading depression model (CSD) was performed as described in previously (Zhao and Levy, 2018) with minor modifications. Briefly, C57BL6/J mice were anesthetized by isoflurane and a 0.5mm bur hole was made above the left frontal cortex (1.5 mm lateral, 1.5 mm anterior of bregma) and sealed with a silicon elastomer (Kwik-Cast Silicone Sealant). After 1 week of recovery, mice were briefly anesthetized with isoflurane, the sealant removed, and CSD was induced by briefly inserting a glass micropipette (50μm diameter) ~1mm deep into the cortex for 2 seconds. This approach has been demonstrated to reliably induce CSD and prolonged meningeal nociceptor activation and sensitization (Zhao and Levy, 2016). Mice were sacrificed 1.5h or 6h after CSD induction and the right TG (CSD) and left TG (control) were collected separately. TG from two mice were pooled per biological replicate. For both IS and CSD models, TG nuclei were processed using the gradient method followed by inDrops snRNA-seq (see below).
METHOD DETAILS
Human and mouse TG dissection
Human TGs were obtained from consented donors using a rapid autopsy protocol at Mass General Brigham (IRB#2017P000757). After removal of the brain for neuropathological analysis, Meckle’s cave was identified in the skull base and manually dissected by one of the authors (JKL). After visualizing the ganglia, the V1-3 nerve branches and the cranial nerve 5 root were cut as they emerged from the ganglion. Right and left ganglia were dissected, flash-frozen in liquid nitrogen, and subsequently stored individually at −80C. The date and time of death, the time of dissection, and freezing time were recorded by pathology staff and donor information was anonymized for downstream processing. None of the donors carried a diagnosis of migraine. TGs from experimental mice were dissected from the skull base after removal of the brain and direct visualization. V1-3 and the proximal projection were severed as close to the ganglia as possible, and TGs were snap frozen on dry ice.
Single-nuclei isolation from human and mouse TG
Single-nuclei suspensions of human and mouse TG were collected using a non-gradient protocol modified from one described previously (Slyper et al., 2020) or a Gradient protocol modified from ones described previously (Yang et al., 2021). Human and mouse TGs were processed similarly except for the initial homogenization step. Human TGs were initially pulverized on dry ice and approximately 0.5–1cm3 of powder was placed into homogenization buffer (non-gradient method = 73 mM NaCl, 0.5 mM CaCl2, 10 mM MgCl2,5 mM Tris-HCl pH 7.5, 1% BSA, and 0.1 U/ul RNase inhibitor (Promega); gradient method = 0.25 M sucrose, 25 mM KCl, 5 mM MgCl2, 20 mM tricine-KOH, pH 7.8, 1 mM DTT, 5 μg/mL actinomycin, 0.04% BSA, and 0.1 U/ul RNase inhibitor) for ~15 seconds on ice. Frozen mouse TGs were placed directly into cold homogenization buffer, incubated for 15 seconds on ice, and briefly homogenized (~5 seconds) using a Tissue-Tearor. After the brief incubation on ice (human TG) or the Tissue-Tearor homogenization (mouse TG), samples were transferred to a Dounce homogenizer for an additional 10 strokes with a tight pestle in a total volume of 5 mL homogenization buffer. After 10 strokes with a tight pestle, the non-gradient method involves adding IGPAL (Sigma) to a final concentration of 0.2% followed by five additional strokes with the tight pestle. The gradient method involves adding IGEPAL to a final concentration of 0.32% and five additional strokes with the tight pestle. The tissue homogenate was then passed through a 40 μm filter, and diluted 1:1 with either homogenization buffer (non-gradient method) or working solution (Gradient method, 50% iodixanol, 25 mM KCl, 5 mM MgCl2, and 20 mM tricine-KOH, 0.04% BSA, and 0.1 U/ul RNase inhibitor). Nuclei extracted with the non-gradient method were then centrifugated at 500g for 10 minutes at 4C and resuspended in 1X PBS, 0.04% BSA, and 0.1 U/ul RNase inhibitor. Nuclei extracted using the gradient method were layered onto an iodixanol gradient after homogenization and ultracentrifuged as described previously (Yang et al., 2021). After ultracentrifugation, nuclei were collected between the 30 and 40% iodixanol layers and diluted for microfluidic encapsulation of individual nuclei in barcoded droplets (see below). TG nuclei were prepared using the gradient method followed by inDrops unless otherwise specified.
Fluorescence activated cell/nucleus sorting (FACS)
For samples prepared using the non-gradient method or gradient method followed by 10X Gene Expression Assay and ATAC-seq Assay (Table S1), FACS was carried out to remove cellular debris. Nuclei were counterstained with Hoechst 33258 and sorted using a 70 μm nozzle and a flow rate of 3 on a BD FACSARIA II into a 1.5 ml microcentrifuge tube containing 15 ul of 1X PBS,0.04% BSA, and 0.1 U/ul RNase inhibitor. Size gates need to be customized for the non-gradient and Gradient buffers and can significantly affect the fraction of neurons obtained after FACS. The mouse snATAC-seq nuclei samples were prepared from Vglut2-Cre Sun1-GFP mouse TG using the non-gradient method, FACS was performed to enrich the GFP+ population. Nuclei were sorted based on size and native GFP fluorescence. GFP+ nuclei (FACS gate determined using TG nuclei from wild-type mice as a negative control) were collected using a 70 μm nozzle and a flow rate of 3 on a BD FACSARIA II into a 1.5 ml microcentrifuge tube containing 50 ul of 1X PBS,0.04% BSA, and 0.1 U/ul RNase inhibitor.
Single-nucleus RNA and ATAC sequencing
Nuclei suspensions that were sequenced using 10X Genomics assays (Table S1) were resuspended and loaded onto the 10X Genomics Chromium device for either snRNA-seq (10X Genomics, V3.1 Gene Expression Assay) or snATAC-seq (10X Genomics, V1.1 ATAC-seq Assay) according to the manufacturer’s protocol aiming to encapsulate 10,000 nuclei per library. Library preparations for snRNA-seq and snATAC-seq were performed according to the manufacturer’s protocol. Nuclei prepared for snATAC-seq were derived from sorted Vglut2-cre;Sun1-GFP mice and were thus enriched for neurons and depleted for non-neuronal subtypes. Libraries were sequenced on an Illumina Nextseq 500 (human and mouse TG) or Novaseq 6000 (human TG) [see Table S1 for sequencing depth per sample]. Sequencing data was processed and mapped to the mouse (GRCm38) or human (GRCh38) genome using 10X Genomics cellranger V6.0.0 and cellranger-ATAC V1.1.0. To identify reads that map to alphaherpes virus genomes, we re-mapped the human snRNA-seq data to a modified human reference genome that included HSV-1 (GenBank: X14112.1), HSV-2 (NCBI Reference Sequence: NC_001798.2), and VZV (NCBI Reference Sequence: NC_001348.1). HSV-1 was the only alphaherpesvirus to which reads from human snRNA-seq mapped. Nuclei suspensions that were sequenced using inDrops (see Table S1), were encapsulated into droplets along with unique oligonucleotide barcodes as described previously (Klein et al., 2015). Libraries were sequenced on an Illumina Nextseq 500 to a depth of 500 million reads per ~30,000 droplets collected. Sequencing data was processed and mapped to the mouse genome GRCm38 using the pipeline described in https://github.com/indrops/indrops (Klein et al., 2015). The web resource used to present our data at tg.painseq.com was built using R shiny apps and ShinyCell (Ouyang et al., 2021).
Pre-processing, clustering, visualization, and annotation of snRNA-seq data
As we used the gradient method to perform snRNA-seq (inDrops) for most of the mouse TG samples in our study (Table S1), we used these nuclei to generate a reference against which we could compare other datasets (e.g., non-gradient method, human TG). Nuclei from all mice that were extracted using the gradient method were analyzed together. Nuclei with > 400 unique genes, < 15,000 total UMIs, and < 5% of the counts deriving from mitochondrial genes were included for analysis. Seurat package (version 3.2.0) in R (version 4.0.1, R Core Team, 2018) was used to perform clustering of these nuclei as previously described (Butler et al., 2018; Stuart et al., 2019). Briefly, raw counts were scaled to 10,000 transcripts per nucleus to control the sequencing depth between nuclei. Counts were centered and scaled for each gene. The effects of total UMI and percent of mitochondrial genes in each nucleus were regressed out using the ScaleData() function. Highly variable genes were identified using the FindVariableFeatures(). The top 20 principal components were retrieved with the RunPCA() function. Nuclei clustering was performed using FindClusters() based on the top 20 principal components, with resolution at 1.5. For dimension reduction and visualization, Uniform Manifold Approximation and Projection (UMAP) coordinates were calculated based on the top 20 principal components by using the implemented function runUMAP() in Seurat.
After clustering all nuclei that passed initial quality control, clusters enriched for the expression of the neuronal marker gene Rbfox3 were classified as neuronal clusters, and clusters enriched for the expression of the non-neuronal marker gene Sparc were classified as non-neuronal clusters. Neuronal and non-neuronal nuclei were clustered separately as described above. Differential expression analysis was run using FindAllMarkers() in Seurat comparing nuclei in one cluster to all other nuclei. Doublet or low-quality clusters were identified as clusters which were significantly enriched for at least two mitochondrial genes (Log2FC > 0.5, FDR < 0.05), have no significantly enriched cluster marker genes (FDR < 0.05, log2FC > 1) other than Rgs11. Nuclei in those clusters were excluded from the dataset. The remaining neuronal and non-neuronal nuclei were clustered again separately, and differential expression analysis was run as described above. Significant enrichment (FDR < 0.05, log2FC > 0.5) of known neuronal subtype marker genes (peptidergic nociceptors (PEP) = Tac1, non-peptidergic nociceptors (NP) = Cd55, pruriceptors (SST) = Sst, cLTMR = Fam19a4, A-LTMR (NF) = Nefh) within a cluster of nuclei compared to all other neuronal nuclei was used to assign the neuronal subtype to each neuronal cluster. Significant enrichment (FDR < 0.05, log2FC > 0.5) of known non-neuronal subtype marker genes (Satglia = Apoe, Schwann cells = Mpz, fibroblasts = Dcn or Mgp, immune cells = Cd74, and vascular = Igfbp7) was used to assign the non-neuronal subtype to each cluster in the non-neuronal clusters. Nuclei with subtype annotations were then combined into a single counts table to visualize neuronal and non-neuronal nuclei on the same UMAP. Variable genes were identified from the merged dataset, and PCA and UMAP were run to generate new UMAP coordinates. All functions were used with default parameters except where otherwise specified. In the text, expression is defined as the raw counts that were scaled to 10,000 transcripts per nucleus, log2 expression is log2 transformed expression.
To annotate the mouse TG nuclei sequenced using 10X Genomics Gene Expression Assay, we used Seurat to anchor these nuclei (query data) to the mouse snRNA-seq (inDrops) samples (reference data). FindTransferAnchors(reduction = “cca”) in Seurat was used to identify anchors between the two datasets. TransferData() was used to transfer subtype labels to each nucleus in the query data. Query nuclei with anchoring prediction score < 0.5 were excluded from the dataset. Two datasets were then combined, and variable genes were identified from the merged dataset. PCA and UMAP were run to generate new UMAP coordinates for all mouse TG nuclei. A cluster of mouse TG nuclei representing the injured states were identified and were excluded from the UMAP.
Human nuclei extracted using the gradient method that have > 1000 unique genes, < 15,000 total UMIs, and < 5% of the counts deriving from mitochondrial genes were included for analysis. Clustering, visualization, doublet removal, and annotation of human data were carried out in Seurat as described above. The same subtype marker genes that were used to assign mouse subtypes were also used to assign subtypes to each cluster in the human snRNA-seq data.
Anchoring of human snRNA-seq data to mouse snRNA-seq data
To directly compare the mouse and human TG, we used Seurat to anchor the human snRNA-seq data to the mouse snRNA-seq data (mouse naive male replicates 1 – 4). First, mouse genes were converted to human orthologs and genes that existed in both human and mouse data were retained for the analysis. FindTransferAnchors(reduction = “cca”) in Seurat was then used to identify anchors between human and mouse data. TransferData() was used to transfer mouse subtype labels to each nucleus in the human data. Human TG nuclei with anchoring prediction score < 0.5 were excluded from the dataset. Variable genes were identified from the merged dataset, and PCA and UMAP were run to generate new UMAP coordinates.
Analysis and annotation of TG snRNA-seq samples prepared using the non-gradient method To compare the non-gradient and Gradient nuclei extraction methods, nuclei prepared using the non-gradient methods that have > 400 unique genes for mouse (or > 1,000 unique genes for human), < 15,000 total UMIs, and < 5% of the counts deriving from mitochondrial genes were included for analysis. To annotate classes of either neurons or non-neurons, we anchored the nuclei from the non-gradient sample to the nuclei from the gradient samples of the same species in Seurat as described above. Specifically, mouse non-gradient sample was anchored to naive male mouse replicates 1 – 4, and human non-gradient sample was anchored to all human TG nuclei (Figure 1B). It is important to note that several factors can affect the fraction of neurons obtained after snRNA-seq analysis from sample preparation to analysis. As discussed in the text, the nuclear isolation protocol clearly improves the fraction of neurons collected during FACS (see Figure S1A). The fraction of neurons sequenced can also be affected by the FACS size gates. These gates are important to customize to the buffer being used. Failure to do this can be observed in our IS samples that were sequenced by 10X (Table S1), which had comparatively poorer neuronal enrichment than other samples we sequenced. We have also consistently observed that the gradient method yields a higher fraction of neurons if sequenced directly without FACS than if they were first cleaned with FACS, which may be related to the encapsulation and sequencing of axonal/neuronal fragments with sufficiently high RNA content to be included in the analysis. We generally recommend removing debris by FACS, however, to reduce the fraction of doublets observed. Finally, nUMI/nGene analysis threshold selected for snRNA-seq analysis can also dramatically affect the fraction of neuronal nuclei obtained as we have observed that neuronal nuclei tend to have more genes per cell than non-neuronal nuclei. Thus, raising the nUMI/nGene threshold will increase the apparent neuron to non-neuron ratio.
Integration of TG data with other published TG and DRG data
Nyguen et al. mouse TG snRNA-seq data is downloaded from GEO (GSE131272), and Sharma et al. mouse TG scRNA-seq data is acquired by personal communication. These datasets were analyzed and annotated as described above. To compare these datasets and our mouse TG snRNA-seq data in the same UMAP space, we integrated the two datasets with male naive mouse replicates 1 – 4 using FindIntegrationAnchors() and IntegrateData() in Seurat with default settings.
Nyguen et al. human DRG snRNA-seq data was downloaded from GEO (GSE168243), and this dataset was analyzed and annotated as described above. Renthal et al. mouse DRG snRNA-seq data was annotated as described previously by us. To compare the gene expression profile of TG and DRG, we anchored our TG snRNA-seq data to the DRG data of the same species. Specifically, TG nuclei from naive mice in our study were anchored to the DRG nuclei from C57 naive mice from Renthal et al., and human TG nuclei in our study were anchored to the human DRG nuclei from Nyguen et al. After anchoring, DRG cell type labels were transferred to the TG nuclei and PCA and UMAP were run as described above for each dataset.
Differential gene expression analysis
To identify genes that are enriched in distinct mouse TG cell types, differential expression analysis was performed using findAllMarkers() in Seurat, comparing nuclei from one cell type to all other nuclei (Log2FC > 1, FDR < 0.05). The same process was conducted on the human TG snRNA-seq data to identify the genes that are enriched in distinct in human TG cell types compared to all other TG cell types.
To identify genes that are differentially expressed in HSV1_LAT+ human TG nuclei compared to HSV1_LAT- human TG nuclei, HSV1_LAT+ population was first identified as nuclei with HSV1_LAT counts > 0. The HSV_LAT- population used for differential expression analysis was selected by randomly sampling the same number of HSV_LAT- nuclei of the same cell type distribution as HSV1_LAT+ nuclei. Gene counts of the group with higher sequencing depth were downsampled to match the average number of UMIs in the group of lower sequencing depth. Differential expression analysis was performed using edgeR (version 3.24.3) as described previously (Renthal et al., 2020). Selection of HSV_LAT- population and differential expression analysis was repeated ten times and median Log2FC and FDR for each gene is reported.
To identify genes that are differentially expressed in female and male mice, naive samples sequenced using 10X Genomics Gene Expression Assay were used. Differential expression analysis was performed using edgeR (version 3.24.3) as described above to compare nuclei from female mice to male mice for each cell type.
Ligand receptor pair analysis
Ligand receptor pair analysis was performed using the R package SingleCellSignalR (version: 0.0.1.8) (Cabello-Aguilar et al., 2020). We filtered the ligand receptor database provided by SingleCellSignalR for ligand receptor pairs that are annotated in the database to have literature support in PubMed. We also filtered the mouse to human orthology table from the package to exclude mouse genes without human orthologues. We used the scaled counts table as input to predict cell-cell interaction. The function cell_signaling() in the package was used to predict potential interactions by setting parameter ‘tol’ to be 1, ‘s.score’ to be 0 and ‘int.type’ to be paracrine. The sankeyplots were generated using R package networkD3(version: 0.4), which represented the putative cell-cell interactions. Ligand receptor pairs are shown in Figure 2F if ligand receptor scores are > 0.5, average ligand expression in each cell type > 0.5, and average receptor expression in each cell type > 0.2. Ligand receptor dot plots were generated using R package ggplot2. For each ligand and receptor, the cell-cell interactions from the top 10 highest ligand receptor scores are plotted. Single-cell RNA-seq data of mouse Meningeal dura mater (Brioschi et al., 2021) was merged with human and mouse snRNA seq data separately using merge() from Seurat. The genes in human snRNA-seq were converted into mouse homolog using biomaRt (version: 2.44.1). Ligand receptor pairs are shown in Fig. S2F if the ligands belong to 250 marker genes ranked by Log2FC in each cell type.
Gene ontology (GO) analysis
GO analysis was performed using topGO (version: 2.40.0) in R (https://bioconductor.org/packages/topGO/). Marker genes (adjusted p value < 0.01 with log2FC > 0.5 from Seurat differentially expression analysis) were used as the input gene list. For comparison, the background gene list included all genes with average expression > 0.5 in the respective nuclei being analyzed. R package org.Mm.eg.db (version 3.11.4) was used as the genome wide annotation database for Mus musculus. R package org.Hs.eg.db (version3.11.4) was used as the genome wide annotation database for Homo sapiens. Genes were annotated for their biological process and associated gene ontology terms. Enrichment is defined as the number of annotated genes observed in the input list divided by the number of annotated genes expected from the background list. GO terms with > 5 annotated genes and enrichment P-value < 0.05 were returned except otherwise specified.
snATAC-seq analysis
Three mouse TG snATAC-seq libraries were aggregated by running cellranger-ATAC aggr function using default settings. Accessible peaks were identified using cellranger-ATAC by analyzing the combined fragment signal across all cells in the dataset. The peak-cell matrix was generated by counting the fragments overlapping each peak in each nucleus. Mouse nuclei with > 600 fragments overlapping with peaks were included for downstream analysis. To annotate the mouse snATAC-seq data, a pseudo gene expression matrix was generated from snATAC-seq data by counting fragments overlapping within the 2kb region upstream of TSS of each gene using CreateGeneActivityMatrix() in Seurat. Using the pseudo gene expression matrix, mouse nuclei profiled by snATAC-seq data were anchored to snRNA-seq data (mouse naive male replicates 1 – 4, including doublets and low-quality nuclei in those samples that were excluded from final dataset) using FindTransferAnchors(reduction = “cca”) in Seurat. TransferData() was used to transfer mouse snRNA-seq subtype labels to each nucleus in the mouse snATAC-seq data. Nuclei profiled by snATAC-seq with anchoring prediction score < 0.5 were excluded from downstream analyses. snATAC-seq nuclei that were anchored to the doublet and low-quality nuclei in snRNA-seq were also excluded. Visualization of snATAC-seq data by UMAP was generated using the pseudo gene expression matrix. Nuclei from human TG snATAC-seq data were analyzed in a similar fashion; briefly, three human TG snATAC-seq libraries were aggregated and accessible peaks were called using cellranger-ATAC aggr. Nuclei with > 600 fragments overlapping with peaks were included. Anchoring was performed in Seurat to human TG snRNA-seq data and cell type labels were transferred. snATAC-seq nuclei with anchoring prediction score < 0.5 or snATAC-seq nuclei anchored to the doublet and low-quality nuclei in snRNA-seq nuclei were excluded from downstream analyses. Due to the low cell number, human snATAC-seq nuclei that were labeled as individual neuronal cell types were combined and labeled as ‘neuron’.
Differential chromatin accessibility analysis
To identify cell-type-specific peaks of transposase-sensitive chromatin, the raw peak-cell matrix from the mouse snATAC-seq data was first scaled to 10,000 fragments per nucleus to control the sequencing depth between nuclei. Counts were centered and scaled for each peak. Differential chromatin accessibility analysis was performed using findAllMarkers() in Seurat, comparing nuclei from one cell type to all other TG nuclei. Cell-type-specific snATAC-seq peaks are reported for each subtype if Log2FC > 0.5 and FDR < 0.05.
Sex-specific peak identification
Sex labels were assigned to each nucleus in mouse TG snATAC-seq data based on their chromatin accessibility around sex-specific genomic regions. FractionCountsInRegion() in Signac was used to calculate the fraction of counts in each nucleus that map to either the Xist locus or to the chromosome Y. Based on the distribution of fractions of counts at the Xist locus or across chromosome Y in all nuclei, nuclei with fraction of counts mapping to the Xist locus > 3e-05 was labeled female nuclei, and nuclei with fraction of counts mapping to chromosome Y > 2e-04 was labeled male. In total, 185 nuclei were labeled female only and 702 were labeled male only, with a double positive rate of 3.2%. To identify sex-specific peaks, differential chromatin accessibility analysis was performed using findAllMarkers() comparing female nuclei to male nuclei. Sex-specific snATAC-seq peaks are reported Log2FC > 0.25 and FDR < 0.05.
Transcription factor motif enrichment analysis
To identify enriched motifs in each mouse subtype, we used FindMotifs() in Signac (Stuart et al., 2020) to test for overrepresentation of each DNA motif in the JASPAR database. This analysis compares the motifs present in cell-type-specific snATAC-seq peaks to those in a background set of the same number of GC-content-matched and width-matched snATAC-seq peaks present in the same subtype. The background set was randomly sampled, and the motif enrichment analysis was repeated ten times. The median enrichment and median FDR for each motif in each subtype were used to identify enriched motifs (enrichment > 1, FDR < 0.05). Fibroblast_Mgp and Vascular cells were excluded from these analyses because they have < 500 cell-type-specific accessible peaks. To identify transcription factors whose motifs are enriched in the mouse TG snRNA-seq data and their regulons (e.g., target genes), SCENIC (version 1.1.1) in R was used as described previously (Aibar et al., 2017; Renthal et al., 2020).
Putative gene regulatory element identification
To identify snATAC-seq peaks that are correlated with gene expression in cis and may act as gene regulatory elements, we computed the Pearson correlation coefficient r between the snATAC-seq peaks (e.g., counts of transposase-sensitive chromatin) and the expression of each gene on the same chromosome. Distance between each snATAC-seq peak and gene was calculated between the center of the peak and the transcription start site of each gene. To identify putative gene regulatory elements that may direct cell-type-specific gene expression, we identified pairs of cell-type-specific snATAC-seq peaks (Log2FC > 0.5, FDR < 0.05, comparing accessibility in one TG cell type to all others) and cell-type-specific genes (Log2FC > 0.5 FDR < 0.05, comparing gene expression in one TG cell type to all others) in the same cell type. We further required the gene-peak pairs to have a distance < 200 kb and r > 0.5. To identify snATAC-seq peaks that are likely to act as gene regulatory elements and their most likely target genes genome-wide, we calculated the ABC scores for each snATAC-seq peak. To do this, we re-called snATACseq peaks using MACS2 (version: 2.1.1.20160309) (Zhang et al., 2008). We returned all peaks with p-value < 0.1 based on MACS2 peak calling. After peak calling, we resized the peaks to 500 bp centered on the peak summit. The MACS2 called peaks and promoter regions 500 bp centered on the transcription start site were defined as the candidate elements for Activity-By-Contact (ABC) analysis (Fulco et al., 2019). ABC scores were calculated separately in each cell type using snATAC-seq reads in each candidate element and powerlaw estimated Hi-C data (https://github.com/broadinstitute/ABC-Enhancer-Gene-Prediction). ABC scores ≤ 0.015 were excluded from downstream analysis.
Analysis of chromatin accessibility at migraine-associated gene variants
123 independent risk loci that are significantly associated with migraine were obtained from the most recent migraine GWAS (Hautakangas et al., 2022). To assess chromatin accessibility at each migraine SNP in our human TG snATAC-seq data, we calculated the fraction of snATACseq counts within each nucleus that overlap with a region 500 bp up and downstream of each migraine SNP. As our mouse snATAC-seq data had greater cell type resolution, we also analyzed chromatin accessibility at migraine-associated SNPs present in the mouse gene. To estimate the coordinates of migraine-associated SNPs in the mouse genome, we used the UCSC web-based tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver) to liftover all human SNP genomic regions (40 bp up and downstream) from genome assembly GRCh37 to genome assembly mm 10m with minimum ratio of bases set to 0.05. The center of each liftover genomic region was assigned as the coordinate of the migraine-associated SNP in the mouse genome. The chromatin accessibility at migraine-associated SNPs in mouse snATAC-seq data was calculated as above.
Activity sequencing of TG after inflammatory soup (IS)
To identify nuclei that are activated after IS and CSD, a set of 139 immediate early genes (IEG) was retrieved from (Wu et al., 2017). AddModuleScore() in Seurat was run to generate a IEG score representing the aggregated expression of those 139 IEGs for each nucleus. A nucleus was considered transcriptionally “activated” if its IEG score is 2 standard deviations higher than the average IEG scores across all nuclei of the same cell type. To identify genes that are differentially expressed in activated nuclei, differential expression analysis was performed between activated nuclei for IS and CSD to a control population of nuclei. The control population of nuclei was randomly selected from the uninduced nuclei from naive animals (the same number of nuclei and cell type distribution as the activated cell population). Gene expression counts of the group with higher sequencing depth were downsampled to match the average number of UMIs in the group of lower sequencing depth. Differential expression analysis was performed using edgeR (version 3.24.3) as described previously (Renthal et al., 2020). Selection of the control population and differential expression analysis was repeated ten times and median Log2FC and FDR for each gene were reported.
RNAScope in situ hybridization
RNAscope fluorescence in situ hybridization (FISH) experiments were performed according to the manufacturer’s instructions, using the RNAscope Fluorescent Multiplex kit (Advanced Cell Diagnostics (ACD)) for fresh frozen tissue, as previously described (Zeisel et al., 2018). Briefly, human TG was frozen in OCT and sectioned into 15 μm sections using a cryostat. RNAscope probes against the following genes were ordered from ACD and multiplexed as described in the text. Human: CD55 (Cat# 426551), TAC1 (Cat# 310711-C2), SST (Cat# 310591-C3), GFRA2 (Cat# 463011), KCNS1 (Cat# 487141), and CALCA (Cat# 605551-C2); mouse: Sst (Cat# 404631-C3) and Calca (Cat# 404631-C3). Following FISH, sections were imaged using a 40x oil immersion objective on a Zeiss LSM710 confocal microscope.
FISH quantification
Three non-consecutive sections per probe set were stained and used for quantification. Regions of interest (ROI) that showed puncta in any of the probe channels and had an area > 200 um2 were manually segmented using ImageJ. Four background regions per section were selected and background intensity was calculated for each channel by averaging the fluorescence intensity across all pixels in the background regions. An ROI was identified as positive for a given probe if the average intensity per pixel in that ROI was > twice the background intensity for any probes visualized in the green channel, five times of that in the red channel, and 10 times of that in the far-red channel. Lipofuscin autofluorescence was identified if, in ROI, large globular structures were observed in the same pattern across all three channels; lipofuscin was excluded from the analysis.
Data visualization
Plots were generated using R version 4.0.1 with ggplot2 package (version 3.2.0) (https://ggplot2.tidyverse.org). Heatmaps were generated using gplots package (version 3.0.1.1) (https://github.com/talgalili/gplots). Figures were made using Adobe Illustrator (Adobe Systems; RRID: SCR_010279).
Quantification and Statistical Analysis
Statistical analyses including the number of animals or cells (n) and P values for each experiment are noted in the figure legends. Statistics were performed using R version 4.0.1. Hypergeometric tests were used to test the significance of overlap between two gene sets. It was conducted by calling phyper() function in R version 4.0.1.
ADDITIONAL RESOURCES
Supplementary Material
Table S13. Comparisons of gene expression between TG and DRG in mouse and human. Related to Figure 1.
Table S12. Gene regulation in TG 1h after IS or 1.5h after CSD. Related to Figure 7.
Table S11. Analysis of migraine-associated genomic loci in mouse and human TG cell types. Related to Figure 6.
Table S10. Enhancer predictions of mouse TG cell-type-specific peaks of chromatin accessibility. Related to Figure 6.
Table S9. Motif enrichment in mouse TG cell-type-specific peaks of chromatin accessibility. Related to Figure 5.
Table S8. Cell-type-specific peaks of chromatin accessibility in mouse TG. Related to Figure 5.
Table S7. Overlapping peaks of chromatin accessibility in mouse and human TG. Related to Figure 4.
Table S6. Sex differences in chromatin accessibility in mouse TG. Related to Figure 4.
Table S5. Sex differences in gene expression in mouse TG cell types. Related to Figure 3.
Table S4. Differential expression analyses between human and mouse TG cell types. Related to Figure 3.
Table S3. Cell-type-specific GO terms and ligand-receptor pairs in mouse and human TG. Related to Figure 2.
Table S2. Cell-type-specific gene expression in mouse and human TG. Related to Figure 2.
Table S1. Characteristics of samples sequenced in this study. Related to Figure 1.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| RNase inhibitor | Promega | Cat#N2611 |
| 60% Iodixanol solution | Sigma | Cat#D1556 |
| IGEPAL CA-630 | Sigma | Cat#I8896 |
| 35% Bovine Serum Albumin solution | Sigma | Cat#A7979 |
| Sucrose | Millipore | Cat#573113 |
| KCl | Thermo Fisher Scientific | Cat#AM9640G |
| MgCl2 | Thermo Fisher Scientific | Cat#AM9530G |
| PBS | Thermo Fisher Scientific | Cat#10010023 |
| Tris-HCl | Thermo Fisher Scientific | Cat#AM9850G, Cat#AM9855G |
| Tricine | Sigma | Cat#T0377 |
| DTT | Sigma | Cat#646563 |
| Actinomycin | Sigma Aldrich | Cat#A9415 |
| Hoechst 33258 | Invitrogen | Cat#H3569 |
| Capsaicin | Tocris | Cat#0462 |
| Prostaglandin E2 | Tocris | Cat#2296 |
| Bradykinin | Tocris | Cat#3004 |
| Histamine | Sigma | Cat#H7125 |
| KOH | Sigma | Cat#221465 |
| ProLong Gold Antifade Mountant with DAPI | Thermo Fisher | Cat#P36931 |
| RNAscope® Probe-Hs-CD55 | ACD Bio | Cat#426551 |
| RNAscope® Probe-Hs-TAC1 | ACD Bio | Cat#310711-C2 |
| RNAscope® Probe-Hs-SST | ACD Bio | Cat#310591-C3 |
| RNAscope® Probe-Hs-GFRA2 | ACD Bio | Cat#463011 |
| RNAscope® Probe-Hs-KCNS1 | ACD Bio | Cat#487141 |
| RNAscope® Probe-Hs-CALCA | ACD Bio | Cat#605551-C2 |
| RNAscope® Probe-Mm-Sst | ACD Bio | Cat#404631-C3 |
| RNAscope® Probe-Mm-Calca | ACD Bio | Cat#404631-C3 |
| Critical commercial assays | ||
| RNAscope Fluorescent Multiplex Assay | ACD Bio | Cat#320850 |
| Chromium Next GEM Chip G Single Cell Kit | 10X Genomics | Cat#1000127 |
| Chromium Next GEM Single Cell 3' GEM, Library & Gel Bead Kit v3.1 | 10X Genomics | Cat#1000128 |
| Single Index Kit T Set A | 10X Genomics | Cat#1000213 |
| Chromium Next GEM Chip H Single Cell Kit | 10X Genomics | Cat#1000162 |
| Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1 | 10X Genomics | Cat#1000176 |
| 1000212 | 10X Genomics | Cat#1000212 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE197289 |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains f | ||
| Mouse: C57BL/6 | The Jackson Laboratory | Cat#000664 |
| Mouse: Sun1-GFP | The Jackson Laboratory | Cat#021039 |
| Mouse: Vglut2-Cre | The Jackson Laboratory | Cat#016963 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| custom R scripts | This paper | https://github.com/Renthal-Lab/ |
| Fiji | ImageJ | RRID:SCR_002285 |
| Adobe Illustrator CC | Adobe Systems | RRID: SCR_010279 |
| Indrops mapping | Klein et al., 2015 | https://github.com/indrops/indrops |
| Cellranger | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Cellranger-atac | 10X Genomics | https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/what-is-cell-ranger-atac |
| R for statistical computing- 4.0.1 | R Core Team, 2018 | https://www.r-proiect.org/ |
| Seurat – 3.2.0 | Satija et al., 2015 | https://satiialab.org/seurat/ |
| Signac – 1.1.0 | Stuart et al., 2020 | https://satiialab.org/signac/ |
| SingleCellSignalR – 0.0.1.8 | Cabello-Aguilar et al., 2020 | https://www.bioconductor.org/packages/release/bioc/html/SingleCellSignalR.html |
| SCENIC – 1.2.2 | Aibar et al., 2017 | https://aertslab.org/#scenic |
| edgeR – 3.30.3 | Robinson et al., 2009 | http://bioinf.wehi.edu.au/edgeR/ |
| ggplot2 – 3.3.2 | Wickham, 2016 | https://ggplot2.tidyverse.org/ |
| gplots – 3.0.4 | Warnes et al., 2009 | https://github.com/talgalili/gplots |
| topGO – 2.40.0 | Alexa, 2018 | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| Other | ||
| Resource website for this publication | This paper | https://tg.painseq.com |
Highlights.
A human and mouse trigeminal ganglion (TG) cell atlas
Conservation of cell-type-specific gene expression between human and mouse TG
Epigenomic maps of TG offer insight into migraine-associated genetic variants
Neuronal and non-neuronal cell types are engaged in two mouse headache models
ACKNOWLEDGEMENTS
We posthumously thank R.J.C for infecting us with his passion for understanding alphaherpes virus latency. We also thank the donors (Figure S1E) who helped advance our understanding of human trigeminal neurons. Helpful discussions with Ivan Tochitsky, Clifford Woolf, Charles Jennings, Michael Greenberg, and Andrea Harriott also significantly improved this study. This work was primarily supported by the Migraine Research Foundation (W.R.) and the Burroughs Wellcome Fund (W.R.). W.R. also receives support from National Institute of Neurological Disorders and Stroke K08NS101064 and R01NS119476, National Institute of Drug Abuse DP1DA054343, Teva Pharmaceuticals, BWH Women’s Brain Initiative and Neurotechnology studio. D.L. receives support from National Institute of Neurological Disorders and Stroke R01NS115972 and R01NS078263.
Footnotes
DECLARATION OF INTERESTS
W.R. receives research funding from Teva Pharmaceuticals and is on an Abbvie scientific advisory board.
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERNCES
- Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman S, Holland PR, and Goadsby PJ (2011). Diencephalic and brainstem mechanisms in migraine. Nat Rev Neurosci 12, 570–584. [DOI] [PubMed] [Google Scholar]
- Ashina M, Hansen JM, Do TP, Melo-Carrillo A, Burstein R, and Moskowitz MA (2019). Migraine and the trigeminovascular system-40 years and counting. Lancet Neurol 18, 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avona A, Mason BN, Burgos-Vega C, Hovhannisyan AH, Belugin SN, Mecklenburg J, Goffin V, Wajahat N, Price TJ, Akopian AN, et al. (2021). Meningeal CGRP-Prolactin Interaction Evokes Female-Specific Migraine Behavior. Ann Neurol 89, 1129–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, and Moskowitz MA (2002). Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 8, 136–142. [DOI] [PubMed] [Google Scholar]
- Brioschi S, Wang WL, Peng V, Wang M, Shchukina I, Greenberg ZJ, Bando JK, Jaeger N, Czepielewski RS, Swain A, et al. (2021). Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Vega CC, Quigley LD, Trevisan Dos Santos G, Yan F, Asiedu M, Jacobs B, Motina M, Safdar N, Yousuf H, Avona A, et al. (2019). Non-invasive dural stimulation in mice: A novel preclinical model of migraine. Cephalalgia 39, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabello-Aguilar S, Alame M, Kon-Sun-Tack F, Fau C, Lacroix M, and Colinge J (2020). SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res 48, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantone M, Kuspert M, Reiprich S, Lai X, Eberhardt M, Gottle P, Beyer F, Azim K, Kury P, Wegner M, et al. (2019). A gene regulatory architecture that controls region-independent dynamics of oligodendrocyte differentiation. Glia 67, 825–843. [DOI] [PubMed] [Google Scholar]
- Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, Daza RM, McFaline-Figueroa JL, Packer JS, Christiansen L, et al. (2018). Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles A (2017). Migraine. N Engl J Med 377, 1698–1699. [DOI] [PubMed] [Google Scholar]
- Charles A, and Pozo-Rosich P (2019). Targeting calcitonin gene-related peptide: a new era in migraine therapy. Lancet 394, 1765–1774. [DOI] [PubMed] [Google Scholar]
- Chen CL, Broom DC, Liu Y, de Nooij JC, Li Z, Cen C, Samad OA, Jessell TM, Woolf CJ, and Ma Q (2006). Runx1 determines nociceptive sensory neuron phenotype and is required for thermal and neuropathic pain. Neuron 49, 365–377. [DOI] [PubMed] [Google Scholar]
- Chiu IM, von Hehn CA, and Woolf CJ (2012). Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci 15, 1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, Filippova GN, Huang X, Christiansen L, DeWitt WS, et al. (2018a). A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174, 1309–1324 e1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Reddington JP, Garfield DA, Daza RM, Aghamirzaie D, Marco-Ferreres R, Pliner HA, Christiansen L, Qiu X, Steemers FJ, et al. (2018b). The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 555, 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice M, Eyde N, Dodick D, Dussor GO, Ossipov MH, Fields HL, and Porreca F (2013). Capturing the aversive state of cephalic pain preclinically. Ann Neurol 74, 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drokhlyansky E, Smillie CS, Van Wittenberghe N, Ericsson M, Griffin GK, Eraslan G, Dionne D, Cuoco MS, Goder-Reiser MN, Sharova T, et al. (2020). The Human and Mouse Enteric Nervous System at Single-Cell Resolution. Cell 182, 1606–1622 e1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson JCA, Warfvinge K, Krause DN, Blixt FW, Sheykhzade M, Edvinsson L, and Haanes KA (2019). C-fibers may modulate adjacent Adelta-fibers through axon-axon CGRP signaling at nodes of Ranvier in the trigeminal system. J Headache Pain 20, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, et al. (2019). Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat Genet 51, 1664–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppetti P, Veldhuis NA, Lieu T, and Bunnett NW (2015). G Protein-Coupled Receptors: Dynamic Machines for Signaling Pain and Itch. Neuron 88, 635–649. [DOI] [PubMed] [Google Scholar]
- Gilden DH, Mahalingam R, Cohrs RJ, and Tyler KL (2007). Herpesvirus infections of the nervous system. Nat Clin Pract Neurol 3, 82–94. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Reuter U, Hallstrom Y, Broessner G, Bonner JH, Zhang F, Sapra S, Picard H, Mikol DD, and Lenz RA (2017). A Controlled Trial of Erenumab for Episodic Migraine. N Engl J Med 377, 2123–2132. [DOI] [PubMed] [Google Scholar]
- Gormley P, Anttila V, Winsvold BS, Palta P, Esko T, Pers TH, Farh KH, Cuenca-Leon E, Muona M, Furlotte NA, et al. (2016). Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet 48, 856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto T, Oh SB, Takeda M, Shinoda M, Sato T, Gunjikake KK, and Iwata K (2016). Recent advances in basic research on the trigeminal ganglion. J Physiol Sci 66, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan JD, Craft RM, LeResche L, Arendt-Nielsen L, Berkley KJ, Fillingim RB, Gold MS, Holdcroft A, Lautenbacher S, Mayer EA, et al. (2007). Studying sex and gender differences in pain and analgesia: a consensus report. Pain 132 Suppl 1, S26–S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriott AM, Chung DY, Uner A, Bozdayi RO, Morais A, Takizawa T, Qin T, and Ayata C (2021). Optogenetic Spreading Depression Elicits Trigeminal Pain and Anxiety Behavior. Ann Neurol 89, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautakangas H, Winsvold BS, Ruotsalainen SE, Bjornsdottir G, Harder AVE, Kogelman LJA, Thomas LF, Noordam R, Benner C, Gormley P, et al. (2022). Genome-wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype-specific risk alleles. Nat Genet 54, 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, and Glass CK (2015). The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 16, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrvatin S, Tzeng CP, Nagy MA, Stroud H, Koutsioumpa C, Wilcox OF, Assad EG, Green J, Harvey CD, Griffith EC, et al. (2019). A scalable platform for the development of cell-type-specific viral drivers. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Polgar E, Solinski HJ, Mishra SK, Tseng PY, Iwagaki N, Boyle KA, Dickie AC, Kriegbaum MC, Wildner H, et al. (2018). Circuit dissection of the role of somatostatin in itch and pain. Nat Neurosci 21, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsan N, and Goadsby PJ (2015). Calcitonin gene-related peptide and migraine. Curr Opin Neurol 28, 250–254. [DOI] [PubMed] [Google Scholar]
- Kennedy PG, Rovnak J, Badani H, and Cohrs RJ (2015). A comparison of herpes simplex virus type 1 and varicella-zoster virus latency and reactivation. J Gen Virol 96, 1581–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupari J, Usoskin D, Parisien M, Lou D, Hu Y, Fatt M, Lonnerberg P, Spangberg M, Eriksson B, Barkas N, et al. (2021). Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain. Nat Commun 12, 1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGuardia JJ, Cohrs RJ, and Gilden DH (2000). Numbers of neurons and non-neuronal cells in human trigeminal ganglia. Neurol Res 22, 565–566. [DOI] [PubMed] [Google Scholar]
- Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, et al. (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPaglia DM, Sapiosessualita MR, Burbero PD, Thierry-Miei J, Thierry-Miei D, Raithel SJ, Ramadan CE, Iadarola MJ, Mannes AJ (2018). RNA-Seq investigations of human post-mortem trigeminal ganglia” Cephalalgia 38, 912–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Labastida-Ramirez A, and MaassenVanDenBrink A (2019). Current understanding of meningeal and cerebral vascular function underlying migraine headache. Cephalalgia 39, 16061622. [DOI] [PubMed] [Google Scholar]
- Loder E, and Renthal W (2019). Calcitonin Gene-Related Peptide Monoclonal Antibody Treatments for Migraine. JAMA Intern Med 179, 421–422. [DOI] [PubMed] [Google Scholar]
- Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, Ding J, Brack A, Kartha VK, Tay T, et al. (2020). Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 183, 1103–1116 e1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo-Carrillo A, Strassman AM, Nir RR, Schain AJ, Noseda R, Stratton J, and Burstein R (2017). Fremanezumab-A Humanized Monoclonal Anti-CGRP Antibody-Inhibits Thinly Myelinated (Adelta) But Not Unmyelinated (C) Meningeal Nociceptors. J Neurosci 37, 1058710596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mich JK, Graybuck LT, Hess EE, Mahoney JT, Kojima Y, Ding Y, Somasundaram S, Miller JA, Kalmbach BE, Radaelli C, et al. (2021). Functional enhancer elements drive subclass-selective expression from mouse to primate neocortex. Cell Rep 34, 108754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. (2015). Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 86, 1369–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring F, Halder P, Seal RP, and Stucky CL (2018). Uncovering the Cells and Circuits of Touch in Normal and Pathological Settings. Neuron 100, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS (2020). Qualitative sex differences in pain processing: emerging evidence of a biased literature. Nat Rev Neurosci 21, 353–365. [DOI] [PubMed] [Google Scholar]
- Nguyen MQ, Le Pichon CE, and Ryba N (2019). Stereotyped transcriptomic transformation of somatosensory neurons in response to injury. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MQ, von Buchholtz LJ, Reker AN, Ryba NJ, and Davidson S (2021). Single-nucleus transcriptomic analysis of human dorsal root ganglion neurons. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MQ, Wu Y, Bonilla LS, von Buchholtz LJ, and Ryba NJP (2017). Diversity amongst trigeminal neurons revealed by high throughput single cell sequencing. PLoS One 12, e0185543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang JF, Kamaraj US, Cao EY, and Rackham OJL (2021). ShinyCell: Simple and sharable visualisation of single-cell gene expression data. Bioinformatics. [DOI] [PubMed] [Google Scholar]
- Parrinello S, Napoli I, Ribeiro S, Wingfield Digby P, Fedorova M, Parkinson DB, Doddrell RD, Nakayama M, Adams RH, and Lloyd AC (2010). EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell 143, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D, and Moskowitz MA (2013). Pathophysiology of migraine. Annu Rev Physiol 75, 365–391. [DOI] [PubMed] [Google Scholar]
- Renthal W, Chamessian A, Curatolo M, Davidson S, Burton M, Dib-Hajj S, Dougherty PM, Ebert AD, Gereau R.W.t., Ghetti A, et al. (2021). Human cells and networks of pain: Transforming pain target identification and therapeutic development. Neuron 109, 1426–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Tochitsky I, Yang L, Cheng YC, Li E, Kawaguchi R, Geschwind DH, and Woolf CJ (2020). Transcriptional Reprogramming of Distinct Peripheral Sensory Neuron Subtypes after Axonal Injury. Neuron 108, 128–144 e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Flaherty K, Lezgiyeva K, Wagner DE, Klein AM, and Ginty DD (2020). The emergence of transcriptional identity in somatosensory neurons. Nature 577, 392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, Smillie C, Smith-Rosario G, Wu J, Dionne D, et al. (2020). A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat Med 26, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, et al. (2015). Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 18, 1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stantcheva KK, Iovino L, Dhandapani R, Martinez C, Castaldi L, Nocchi L, Perlas E, Portulano C, Pesaresi M, Shirlekar KS, et al. (2016). A subpopulation of itch-sensing neurons marked by Ret and somatostatin expression. EMBO Rep 17, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner TJ, Stovner LJ, Jensen R, Uluduz D, Katsarava Z, and Lifting The Burden: the Global Campaign against H (2020). Migraine remains second among the world’s causes of disability, and first among young women: findings from GBD2019. J Headache Pain 21, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Ricci JA, Chee E, and Morganstein D (2003). Lost productive work time costs from health conditions in the United States: results from the American Productivity Audit. J Occup Environ Med 45, 1234–1246. [DOI] [PubMed] [Google Scholar]
- Strassman AM, Raymond SA, and Burstein R (1996). Sensitization of meningeal sensory neurons and the origin of headaches. Nature 384, 560–564. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Srivastava A, Lareau C, and Satija R (2020). Multimodal single-cell chromatin analysis with Signac. bioRxiv. [Google Scholar]
- Sun Y, Dykes IM, Liang X, Eng SR, Evans SM, and Turner EE (2008). A central role for Islet1 in sensory neuron development linking sensory and spinal gene regulatory programs. Nat Neurosci 11, 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland HG, Albury CL, and Griffiths LR (2019). Advances in genetics of migraine. J Headache Pain 20, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares-Ferreira D, Ray PR, Sankaranarayanan I, Mejia GL, Wangzhou A, Shiers S, Uttarkar R, Megat S, Barragan-Iglesias P, Dussor G, et al. (2022a). Sex Differences in Nociceptor Translatomes Contribute to Divergent Prostaglandin Signaling in Male and Female Mice. Biol Psychiatry 91, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares-Ferreira D, Shiers S, Ray PR, Wangzhou A, Jeevakumar V, Sankaranarayanan I, Cervantes AM, Reese JC, Chamessian A, Copits BA, et al. (2022b). Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci Transl Med 14, eabj8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tfelt-Hansen P, and Loder E (2019). The Emperor’s New Gepants: Are the Effects of the New Oral CGRP Antagonists Clinically Meaningful? Headache 59, 113–117. [DOI] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling-Leffler J, Haeggstrom J, Kharchenko O, Kharchenko PV, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci, 18 (2015), pp. 145–153 [DOI] [PubMed] [Google Scholar]
- von Buchholtz LJ, Ghitani N, Lam RM, Licholai JA, Chesler AT, and Ryba NJP (2021). Decoding Cellular Mechanisms for Mechanosensory Discrimination. Neuron 109, 285–298 e285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Buchholtz LJ, Lam RM, Emrick JJ, Chesler AT, and Ryba NJP (2020). Assigning transcriptomic class in the trigeminal ganglion using multiplex in situ hybridization and machine learning. Pain 161, 2212–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vormstein-Schneider D, Lin JD, Pelkey KA, Chittajallu R, Guo B, Arias-Garcia MA, Allaway K, Sakopoulos S, Schneider G, Stevenson O, et al. (2020). Viral manipulation of functionally distinct interneurons in mice, non-human primates and humans. Nat Neurosci 23, 1629–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YE, Pan L, Zuo Y, Li X, and Hong W (2017). Detecting Activated Cell Populations Using Single-Cell RNA-Seq. Neuron 96, 313–329 e316. [DOI] [PubMed] [Google Scholar]
- Yang L, Tochitsky I, Woolf CJ, and Renthal W (2021). Isolation of Nuclei from Mouse Dorsal Root Ganglia for Single-nucleus Genomics. Bio Protoc 11, e4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Liu H, Hamel KA, Morvan MG, Yu S, Leff J, Guan Z, Braz JM, and Basbaum AI (2020). Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat Commun 11, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G, et al. (2018). Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014 e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, and Burstein R (2010). Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci 30, 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, and Levy D (2016). Cortical Spreading Depression Promotes Persistent Mechanical Sensitization of Intracranial Meningeal Afferents: Implications for the Intracranial Mechanosensitivity of Migraine. eNeuro 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, and Levy D (2018). Dissociation between CSD-Evoked Metabolic Perturbations and Meningeal Afferent Activation and Sensitization: Implications for Mechanisms of Migraine Headache Onset. J Neurosci 38, 5053–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Liu P, Bai L, Trimmer JS, Bean BP, and Ginty DD (2019). Deep Sequencing of Somatosensory Neurons Reveals Molecular Determinants of Intrinsic Physiological Properties. Neuron 103, 598–616 e597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S13. Comparisons of gene expression between TG and DRG in mouse and human. Related to Figure 1.
Table S12. Gene regulation in TG 1h after IS or 1.5h after CSD. Related to Figure 7.
Table S11. Analysis of migraine-associated genomic loci in mouse and human TG cell types. Related to Figure 6.
Table S10. Enhancer predictions of mouse TG cell-type-specific peaks of chromatin accessibility. Related to Figure 6.
Table S9. Motif enrichment in mouse TG cell-type-specific peaks of chromatin accessibility. Related to Figure 5.
Table S8. Cell-type-specific peaks of chromatin accessibility in mouse TG. Related to Figure 5.
Table S7. Overlapping peaks of chromatin accessibility in mouse and human TG. Related to Figure 4.
Table S6. Sex differences in chromatin accessibility in mouse TG. Related to Figure 4.
Table S5. Sex differences in gene expression in mouse TG cell types. Related to Figure 3.
Table S4. Differential expression analyses between human and mouse TG cell types. Related to Figure 3.
Table S3. Cell-type-specific GO terms and ligand-receptor pairs in mouse and human TG. Related to Figure 2.
Table S2. Cell-type-specific gene expression in mouse and human TG. Related to Figure 2.
Table S1. Characteristics of samples sequenced in this study. Related to Figure 1.
Data Availability Statement
Processed snRNA-seq data are available at tg.painseq.com. Raw and processed snRNA-seq data were also deposited within the Gene Expression Omnibus (GEO) repository (www.ncbi.nlm.nih.gov/geo) with an accession number (GSE197289).
Custom R scripts are available on https://github.com/Renthal-Lab/.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.