

Abstract
During angiosperm male gametogenesis, microspores divide to produce a vegetative cell (VC) and a male germline (MG), each with distinct cell fates. The mechanism underlying determination of the MG cell/VC fate remains an important area of research, with many unanswered questions. Here, we report that H3K27me3 is essential for VC fate commitment in male Arabidopsis thaliana gametophytes; H3K27me3 erasure contributes to MG cell fate initiation. VC-targeted H3K27me3 erasure disturbed VC development and shifted the VC fate toward a gamete destination, which suggests that MG cells require H3K27me3 erasure to trigger gamete cell fate. Multi-omics and cytological analyses confirmed the occurrence of extensive cell identity transition due to H3K27me3 erasure. Therefore, we experimentally confirmed that MG cell/VC fate is epigenetically regulated. H3K27 methylation plays a critical role in guiding MG cell/VC fate determination for pollen fertility in Arabidopsis. Our work also provides evidence for two previous hypotheses: the germline cell fate is specified by the differential distribution of unknown determinants and VC maintains the default microspore program (i.e. the H3K27me3 setting) while MG requires reprogramming.
In Arabidopsis, H3K27me3 is essential for vegetative cell fate commitment in male gametophytes and H3K27me3 erasure contributes to male germline cell fate initiation.
In a Nutshell.
Background: The microspore divides asymmetrically to form a larger vegetative cell (VC) and a smaller generative cell (GC). The GC further divides to produce two sperm cells (SCs). Thus, the male gametophyte (pollen) consists of a VC and male germline (MG) cells (GC and SC twins) and is an excellent model for studying cell fate determination. MG cell fate is thought to be specified by the differential distribution of unknown determinants caused by the asymmetric division of the microspore. H3K27me3 is erased specifically in SC chromatin but persists in the VC, and DNA methylation is lost along with H3K9me2 in the VC. Therefore, certain epigenetic factors may determine the cell fate of the two cell lineages in pollen.
Question: How is germline fate suppressed in VCs but established in MG cells and do histone posttranslational modifications contribute to this process?
Findings: In mature pollen, H3K27me3 appears in VCs, but not in SCs. We generated a transgenic Arabidopsis line that expresses H3K27 demethylase REF6 (hereafter TL) to erase H3K27me3 in VCs. The VC-targeted H3K27me3 removal disturbed VC development. Pollen from strong TLs failed to germinate. The ectopic distribution of MG cell-specific markers indicated that VC-targeted H3K27me3 erasure altered TL VCs and triggered a cell fate transition toward MG cells. Multi-omics and cytological analyses confirmed the occurrence of extensive cell identity transition due to H3K27me3 erasure. Thus, we experimentally reveal that H3K27 methylation plays a critical role in guiding MG cell/VC fate specification.
Next steps: VC-targeted H3K27me3 erasure did not shift all VCs toward the MG cell fate, suggesting that other pathways or epigenetic mechanisms function in cell fate determination during male gametophyte development. How H3K27me3 erasure in the MG is controlled and how the different epigenetic mechanisms are integrated in this process remain to be elucidated.
Introduction
During sexual development in angiosperms, the factors that determine the cell fates of male germline (MG) cells and vegetative cells (VCs) remain largely unknown. Previous reports have shown that the asymmetric division of angiosperm microspores is crucial for MG cell formation and specification (Twell et al., 1998). MG cell fate is specified by unknown factors confined to the smaller daughter cell after asymmetric division of the microspore (Twell et al., 2002; Hackenberg and Twell, 2019). Mutation of RETINOBLASTOMA RELATED (RBR) in microspores causes hyperproliferation and prevents the establishment of cell identity in VCs. The VCs of rbr mutants exhibit nonspecific asymmetric division, producing an additional cell with a VC fate, MG cell fate, or mixed cell fate (Chen et al., 2009). When the MG cell type is determined, it produces male gametes that follow a developmental program that is unaffected by the cell cycle or the VC-supplied microenvironment (Chen et al., 2008; Huang et al., 2018). Notably, the MG cell-specific conserved pathway mediated by R2R3 Myb-domain protein DUO POLLEN1 (DUO1) is essential for triggering MG cell specification (Rotman et al., 2005; Berger and Twell, 2011; Borg et al., 2011; Higo et al., 2018). These studies suggest that the cell fate dichotomy of VCs and MG cells is triggered after asymmetric cell division of the microspore. However, the mechanism by which germline fate is suppressed in VCs but established in MG cells remains unclear; similarly, the factors that promote DUO1 expression in MG cells have not yet been identified. To answer these questions, epigenetic regulation has recently drawn considerable attention.
DNA methylation is erased globally in the paternal chromatin of mammals; in contrast, DNA is not demethylated at least in sequences that involve CG or CHG (where H is A, C, or T) in Arabidopsis thaliana MG cells (Calarco et al., 2012; Borg and Berger, 2015). The sperm chromatin of mammals undergoes widespread removal of histone modifications as the histones are replaced with protamines (Reik et al., 2001; Sassone-Corsi, 2002), whereas H3.1 histones are replaced by the MG cell-specific histone variant H3.10 (HTR10) in the sperm chromatin of flowering plants (Ingouff et al., 2010). H3.10 is unable to undergo H3K27me3 modifications; its incorporation into sperm chromatin coupled to the activity of the H3K27me3 demethylases RELATIVE OF ELF 6 (REF6), EARLY FLOWERING 6 (ELF6), and JUMONJI 13 (JMJ13) can lead to the global loss of H3K27me3 (Borg et al., 2020), suggesting that H3K27me3 erasure plays a critical role in MG cell fate determination. However, sperm cells (SCs) were generated in the elf6 ref6 jmj13 htr10 quadruple mutant and ectopic SWINGER (SWN, methyltransferase subunit of Polycomb repressive complex 2 [PRC2]) expression in SCs did not influence SC identity (Borg et al., 2020). Therefore, the role of H3K27 methylation in cell fate determination of two cell lineages in male gametophytes requires experimental validation. In contrast to MG cells, the chromatin of VCs undergoes global loss of the centromeric H3 variant CENH3 and the heterochromatic mark H3K9me2, both of which persist in MG cells (Schoft et al., 2009). In addition, DNA methylation is lost along with H3K9me2 in VCs, which shapes the accessible chromatin of VCs and unlocks several genes needed for pollen tube function (Borg et al., 2021; Khouider et al., 2021). In Arabidopsis, SET DOMAIN GROUP2 (SDG2) mutation disrupts H3K4me3 deposition, which leads to ectopic H3K9me2 speckle formation and chromatin condensation in VCs; however, this does not alter VC or SC fate (Pinon et al., 2017). In summary, it remains unclear whether histone posttranslational modifications guide cell fate determination in male gametophytes. Here, we experimentally confirmed that H3K27me3 is essential for maintaining VC fate and that H3K27me3 erasure resulted in VC fate shifting toward MG cell fate. Our results suggest that H3K27me3 has a critical role in guiding MG cell/VC fate determination for pollen fertility in Arabidopsis.
Results
H3K27me3 is essential for VC functional specification
In wild-type (WT) mature pollen, strong H3K27me3 immunofluorescence was detected in VC nuclei (VN), but not in SC nuclei (SN); this indicated distinct H3K27 methylation modification between VCs and SCs (Figure 1A). To investigate the role of H3K27 methylation in cell fate determination during male gametophyte development, we first observed male gametophyte development in H3K27 methyltransferase mutants. According to a previous report, methyltransferase subunits of PRC2 could not survive in the fertile double homozygous mutant of SWN and CURLY LEAF (CLF) (Lu et al., 2011). The expression pattern showed that SWN and CLF were expressed in the microspore, while only SWN was sequentially expressed in VCs after asymmetric division of the microspore (Borg et al., 2020). Thus, SWN may play a major role in H3K27me3 maintenance in VCs. Therefore, we used swn-21/– clf-29/+ in our experiments. Although the germination rate decreased as the H3K27me3 level decreased in VCs, there was no obvious morphological abnormality in the swn-21/– clf-29/+ male gametophytes (Supplemental Figure S1). Because H3K27me3 was not completely erased in the swn-21/−clf-29/+VCs (Supplemental Figure S1, C and D), there is a need to create more suitable materials to explain the role of H3K27me3 in determining the fates of distinct cell lineages in male gametophytes.
Figure 1.

VC-targeted H3K27me3 erasure in the male gametophyte. A, REF6 was expressed under the control of the Lat52 promoter to purposefully erase H3K27me3 in VCs. The transgenic lines were named TL. Whole-mount H3K27me3 antibody labeling and DAPI staining of mature pollen in WT (top) and strong TL-10 line (bottom). Dashed lines indicate VN. B, Fluorescence signal intensity of the WT (left) and TL-10 (right) VN is displayed along the direction of the long white arrow in (A). C, Statistical analysis of the average signal intensity of REF6-GFP and H3K27me3 in the VN of WT and TL-10 pollen. Statistical significance is determined by Student’s t test (***P < 0.001) and the error bars show sd. Scale bars, 2 μm (A).
Because the dynamic equilibrium of histone lysine methylation is maintained by histone lysine methylases and demethylases (Liu et al., 2010), we generated a transgenic Arabidopsis line that expresses REF6, the first H3K27 demethylase identified in plants (Lu et al., 2011; Cui et al., 2016). REF6 was placed under control of the Lat52 promoter to erase H3K27me3 in VCs. The transgenic line was named REF6-Transferred Line (REF6-TL, hereafter TL). The H3K27me3-specific fluorescent signal was scarcely detectable in VNs of the strong TL-10 line, indicating the virtual disappearance of H3K27me3 (Figure 1). Homozygous strong TL lines could not be obtained (TL-09/+, -10/+, -18/+, and -30/+) (Supplemental Figures S2, A–D and S3). One function of VCs is to germinate and form a pollen tube to deliver SCs to the embryo sac for double fertilization; therefore, we examined this function in various TL lines. No obvious differences in viability or morphology were observed in the strong TL line pollen compared with the control; however, the mature pollen from strong TL lines showed abnormal vesicle organization and failed to germinate (Figure 2, A–J; Supplemental Figures S2, D–I and S3, A and B). In addition, similar to swn-21/-clf-29/+, normal pollen morphologies with pleiotropic pollen germination phenotypes were observed in various weak homozygous TL lines (TL-13/–, -21/–, and -25/–). In the TL-13 line, ∼60% of pollen lost the ability to germinate, while ∼37% of pollen tubes burst immediately after germination; only ∼3% of tubes remained intact, but these elongated slowly compared with the control. Thus, the TL-13 line produced few seeds. Similar defects were observed in other weak TL lines (Supplemental Figures S2, B and J and S3, C–E and K–M). Notably, H3K27me3 removal disrupted the formation of the male germ unit (MGU), which is a typical feature of a functional VC (Figure 2, K–M). These results suggest that H3K27me3 is essential for VC developmental stability and functional specification.
Figure 2.

H3K27me3 is essential for VC identity and functional specification. A and B, Normal morphology in control VCs from the ProHTR10:HTR10-RFP line (A) and ProACTIN11:H2B-GFP ProHTR10:HTR10-RFP double-labeled line (B). Dashed lines and black arrows indicate VN. C and D, The morphology of the strong TL VCs at different development stages. SC formation and VC viability were unaffected, but the abnormal vesicle organization and ectopic HTR10-RFP signal were detected in the mature pollen from TL-10 (C) and TL-09 (D) lines. Dotted lines indicate the ectopic HTR10-RFP signal in VN. E and F, In vitro pollen germination assay of the ProHTR10:HTR10-RFP line (E) and TL-10/+ carrying the HTR10-RFP marker (F). Arrows indicate the pollen unable to germinate. G and H, Comparison of the in vitro pollen germination rate between control and TL-10/+. Results are from three independent experiments. I and J, In vivo–in vitro pollen germination assay following pollination of WT pistils with control pollen (ProACTIN11:H2B-GFP ProHTR10:HTR10-RFP double-labeled line [I]) and TL-10/+ pollen (J), respectively. Dotted box in (I) displays the MGU in pollen tubes. Dotted box in (J) indicates that TL-10 pollen failed to form tubes and all the observed tubes are from normal pollen. K, Statistical analysis of the abnormal MGU construction in control and TL lines. Results are from three independent experiments. Statistical significance is determined by Student’s t test (***P < 0.001) and the error bars show sd. TEM of WT (L) with normal MGU and TL-10 (M) pollen with abnormal MGU construction. Scale bars, 5 μm (A–D), 50 μm (E, F), 200 μm (I, J), and 2 μm (L, M). LBC, late bicellular; ETC, early tricellular; MP, mature pollen.
VC-targeted H3K27me3 erasure shifts VC cell fate toward MG
We found that the aberrant phenotypes of the strong TL pollen grains were similar to a reported VC-ectopic DUO1 expression line (ProLat52:DUO1) (Brownfield et al., 2009). Thus, the VC-targeted H3K27me3 erasure presumably changed the attributes of the strong TL VCs and triggered a cell fate transition to MG. To test the TL VC cell identity, we examined MG cell-specific markers in strong TL pollen. As expected, the HTR10-RFP marker was activated in ∼11% VN of TL-09, -10, -18, and -30 pollen; this was not observed in WT VN (Figures 2, A–D and 3, A and D; Supplemental Figure S2, H and I). DUO1 localization was examined using DUO1-RFP. Ectopic DUO1 signals were detected in ∼16% VN of TL-09, -10, -18, and -30 pollen (Figure 3, B and D). These findings suggest that the targeted removal of H3K27me3 in VCs induced a shift in cell fate from VC to MG cells. In plants, the H3K9me2 modification is enriched in hypermethylated heterochromatin. H3K9me2 and methylated CHG interact in a self-enforcing positive feedback loop that acts on constitutive heterochromatin and may contribute to extreme chromatin condensation in the SN (Jackson et al., 2002; Johnson et al., 2002; Schoft et al., 2009; Borg and Berger, 2015). Here, we found that the H3K9me2 marker was present only in the SN of WT pollen, whereas H3K9me2 was detected in both SN and VN in 9.90% (±0.80) of TL-10 pollen (Figure 3, C and D); the chromatin H3K9me2 and H3K27me3 status of TL-10 VN was similar to the status of SN, suggesting cell identity transition. Therefore, our results suggest that MG cell fate is regulated epigenetically and that H3K27me3 erasure is important for inducing MG cell fate.
Figure 3.

VC-targeted H3K27me3 erasure triggers the VC–SC fate transition. A–C, The MG cell-specific markers in control and TL-10 pollen. Fluorescence images of the MGU stained with DAPI, also indicated by ProLat52:REF6-GFP and ProHTR10:HTR10-RFP (A), ProDUO1:DUO1-RFP (B), and H3K9me2 signals (C), in control and TL-10. Type ① and type ② indicate TL-10 VN without or with MG cell-specific marker signals, respectively. Dotted lines indicate the ectopic HTR10-RFP (A), DUO1-RFP (B) signals, and arrow indicates the ectopic H3K9me2 signals in VN (dashed line) (C). D, Statistical analysis of the ectopic MG cell-specific marker signal in VN. Statistical significance was determined by Student’s t test (***P < 0.001) and the error bars show sd. Scale bars, 2 μm (A–C).
H3K27me3 erasure leads to chromatin remodeling in TL VCs
To further investigate the impact of the H3K27me3 erasure on TL VC cell fate, we isolated pure VN and SN from the broken pollen mixture using fluorescence-activated nuclei sorting (FANS) for H3K27me3 cleavage under targets and tagmentation (CUT&Tag), as well as assays for transposase accessible chromatin (ATAC)-Seq analyses. Six CUT&Tag and eight ATAC-Seq libraries were constructed and sequenced (Supplemental Figures S4 and S5 and Supplemental File). Principal component analysis (PCA) of the CUT&Tag and ATAC-Seq data sets showed that profiles of the same cell lineage were consistent and clearly separated from each other (Figure 4, A and B). H3K27me3 is generally enriched in chromatin areas containing silenced genes. The CUT&Tag results revealed 6,950 H3K27me3 peaks in WT VCs, while WT SCs generated 694 peaks, indicating the absence of H3K27me3 in SCs, which is consistent with the reported H3K27me3 ChIP-Seq data regarding WT SCs (Borg et al., 2020); TL VCs generated 141 peaks, indicating that H3K27me3 modifications were effectively erased in TL VCs, and that the H3K27me3 state of TL VCs closely resembled the state of WT SCs (Figure 4, C–F and Supplemental Figure S4, E–G). For the ATAC-Seq analysis, ∼16,000 peaks were identified in WT and TL SCs, and ∼21,000 peaks were detected in WT and TL VCs; the data quality was therefore sufficient for the following analyses.
Figure 4.

H3K27me3 erasure leads to chromatin remodeling in TL VC. A and B, PCA of H3K27me3 CUT&Tag (A) and ATAC-Seq data (B), illustrating high reproducibility of replicates and the relationship among cell lineages. TL-10/+ was used in experiments. C, Sample fragment length distribution of H3K27me3 CUT&Tag library reads. D, The size distribution (in kb) of WT VC, TL VC, and WT SC H3K27me3 domains. The peak numbers were 6,950 in WT VCs, 694 in WT SCs, and 141 in TL VCs (q < 0.01). E and F, CUT&Tag profiles over H3K27me3 domains of WT VC in three cell lineages, respectively, (E) and H3K27me3 signals over peak-annotated genes (F) suggest that H3K27me3 is maintained in WT VCs but lost in WT SCs. Similar to WT SCs, H3K27me3 levels are low in TL VCs. CUT&Tag data were generated from two biological replicates. G, Overlap analysis of downregulated (left) and upregulated (right) ATAC-Seq peaks between WT SC versus WT VC (WT SC compared with WT VC) and TL VC versus WT VC. The overlapping regions indicate VC-required peaks (VC-peaks) and MG-required peaks (MG-peaks). H and I, ATAC-Seq profiles over VC-peaks (H) and chromatin accessibility signals over peak-annotated genes (VC-genes) (I) in four cell lineages. J, H3K27me3 signals over the VC-genes annotated by VC-peaks. K and L, ATAC-Seq profiles over MG-peaks (K) and chromatin accessibility signals over peak-annotated genes (MG-genes) (L) in four cell lineages. M, H3K27me3 signals over the MG-genes annotated by MG-peaks in promoter transcriptional start sites (promoter-TSS), gene bodies (exons and introns), and transcription termination sites (TTS), indicating that these genes are repressed by H3K27me3 in WT VCs and activated by H3K27me3 erasure in WT SCs and TL VCs. N and O, Genome browser views of merged ATAC-Seq data sets from WT VC, TL VC, and WT SC; chromatin accessibility was low in VC-related genes (N) and high in MG cell-related genes (O) of TL VCs. The chromatin accessibility of MG cell-related genes changed near H3K27me3 domains (light blue shading) along gene bodies.
We then examined whether the chromatin regions were altered during MG cell specification and how the alterations were related to targeted H3K27me3 erasure. Pairwise comparisons of the ATAC-Seq data sets revealed 1,794 differentially accessible regions (DARs) with increased openness (up) and 2,547 DARs with decreased openness (down) in WT SCs compared with WT VCs (WT SC versus WT VC). In TL VCs compared with WT VCs, 357 DARs had less accessibility and 4,195 DARs had significantly greater accessibility (Supplemental Figure S5, F and G). Gene ontology (GO) analysis revealed that the genes with peaks “WT SC versus WT VC down” or “TL VC versus WT VC down” were related to pollen tube development, including pollen tube growth, pH regulation, and receptor kinase signaling. Furthermore, the genes with peaks “WT SC versus WT VC up” or “TL VC versus WT VC up” were related to SC differentiation, cell–cell signaling, and cell fate commitment (Supplemental Figure S5H). This suggests that the chromatin accessibility regions of TL VCs are modified due to cell fate transition.
Next, we combined the ATAC-Seq data from the WT SC versus WT VC and TL VC versus WT VC comparisons, which yielded 102 VC-required peaks (VC-peaks) and 467 MG-required peaks (MG-peaks) related to cell lineage specificity in male gametophytes (Figure 4, G, H, and K). The annotated genes associated with VC-peaks were less accessible in WT SCs and TL VCs than in WT VCs. In comparison, the annotated genes associated with MG-peaks tended to be more open in WT SCs and TL VCs than in WT VCs (Figure 4, I and L and Supplemental Figure S6F). In WT VCs, the annotated genes of the VC-peaks harbored fewer H3K27me3 modifications; the annotated genes of the MG-peaks were broadly suppressed by H3K27me3, especially in the gene body and promoter-transcription start sites (Figure 4, J and M and Supplemental Figure S6E). In addition, the large number of new peaks in TL VCs (Figure 4G) may indicate TF redistribution to a wide range of genomic regions and also the activation of specific genes for the cell fate transition, which can only be observed in the TL VCs.
Previous reports identified essential VC and MG cell-related genes, which we used as markers in the following work. The well-established VC-enriched genes ANX1/2, BUPS1/2, RALF4, PRK1/2A/3/6, and VGD1, which are involved in pollen tube growth and guidance (Jiang et al., 2005; Boisson-Dernier et al., 2009; Takeuchi and Higashiyama, 2016; Ge et al., 2017), were more accessible in WT VCs than in WT SCs; they were less accessible in TL VCs (Figure 4N). Conversely, the MG cell-specific genes, which include BNB2, DUO1, HTR10, GCS1, SSP, and IMPa8 (Bayer et al., 2009; Borg et al., 2011; Yamaoka et al., 2018), had less accessible chromatin in WT VCs than in WT SCs, whereas their accessibility was greater in TL VCs (Figure 4O). In WT VCs, the VC-enriched genes were not marked with H3K27me3, but the MG cell-specific genes (with the exception of DUO1) were marked with H3K27me3 in the closed chromatin region (Figure 4, N and O and Supplemental Figure S4H); these findings suggested that H3K27me3 participates in VC identity maintenance and that its erasure may trigger MG cell differentiation. Although DUO1 itself was not modified by H3K27me3, its potential upstream regulator BNB1/2 (Yamaoka et al., 2018) was marked by H3K27me3 (Supplemental Figure S4H). Thus, H3K27me3 erasure may activate, directly or indirectly, the transcriptional pathways of MG cell specification through a pleiotropic mechanism. The VC-targeted H3K27me3 erasure therefore led to VC-related chromatin closure and MG cell-related chromatin opening in TL VCs. In addition, the presence of H3K27me3 targeting VC-enriched genes in WT SCs (Supplemental Figure S4H) suggests that H3K27me3 is selectively retained in SCs and required to repress the expression of VC-enriched genes; thus, SC identity can be established without interruption from those VC-enriched genes. These findings might indicate both sides of the same process necessary for MG fate establishment.
H3K27me3 erasure induces ectopic activation of sperm-specific transcription in TL VCs
To determine which genes were regulated in VC and MG cell fate determination, VN and SN were isolated for RNA-Seq; 12 transcriptome libraries were constructed and sequenced (Supplemental Figure S6A and Supplemental File). In total, ∼20,000 genes were detected in the transcriptomes of WT/TL SCs and WT VCs, while ∼27,000 genes were detected in the transcriptome of TL VCs. PCA of the transcriptomic data sets showed that the profiles of the same cell lineage were consistent; PC1 and PC2 were sufficient to separate the samples into four groups. Although TL VCs and WT VCs were clearly separated, TL SCs and WT SCs were clustered more closely (Figure 5, A–C). Moreover, correlation matrices showed that TL VCs were more similar to WT SCs than to WT VCs at the transcriptional level (Figure 5B), implying a transcriptome transition from VCs to SCs because of the targeted H3K27me3 erasure. These results suggest that H3K27me3 erasure altered the nature of TL VCs but did not significantly affect the identity of TL SCs; this conclusion was supported by unsupervised hierarchical clustering (UHC) analysis of the transcriptomic data sets (Supplemental Figure S6D). Because the Lat52 promoter weakly drives REF6 expression at the late microspore stage, six transcriptome libraries from the polarized microspore (PMS) of both WT and TL-10 were sequenced to assess the possible effects of weak REF6 expression on subsequent cell lineage specialization (Supplemental Figures S2, D–G and S6B and Supplemental File). The correlation matrices and UHC analysis showed that the WT PMS and TL PMS transcriptomic data sets clustered together (Supplemental Figure S6, C and D), indicating that the weak REF6 expression did not significantly affect TL-10 PMS development, which was consistent with the phenotypic analysis.
Figure 5.

H3K27me3 erasure induces transcriptome reconstruction in TL VCs. A, PCA of WT VC, WT SC, TL VC, and TL SC transcriptomes, illustrating the high reproducibility of replicates and the relationship among the four cell lineages. TL-10/+ was used in experiments. B, Pearson correlation matrix of the WT VC, WT SC, and TL VC transcriptomes shows that TL VC is more similar to WT SC, compared with WT VC. C, Pearson correlation matrix of the WT VC, WT SC, and TL SC transcriptomes shows that there is no significant difference between TL SC and WT SC. D and E, Scatterplot showing the numbers of up- and downregulated genes in WT SC versus WT VC (D) and TL VC versus WT VC (E). F and G, Overlap analysis of the down- (F) and upregulated (G) genes between WT SC versus WT VC and TL VC versus WT VC. The overlapping regions indicate VC-required genes (F) and MG-required genes (G), respectively. H and I, Expression profiles of VC-peak annotated genes (H) and MG-peak annotated genes (I) in four cell lineages, showing that TL VCs have similar characteristics to those of WT SCs at the transcriptional level. J and K, Expression profiles of VC-required genes (J) and MG-required genes (K) in four cell lineages, showing that TL VCs have similar characteristics to those of WT SCs at the transcriptional level. Blue (H, J) or red (I, K) lines indicate the mean expression levels of all genes in the different groups. L and M, GO enrichment analysis of VC-required genes (J) and MG-required genes (K) in the transcriptome. ∞ indicates P = 0. N and O, Relative expression levels of VC-enriched genes (N) and MG cell-specific genes (O) in WT and TL-10/+ pollen. Total RNA was extracted from whole mature pollen. Results are an average of three independent experiments. Statistical significance is determined by Student’s t test (**P < 0.01, ***P < 0.001) and the error bars show sd.
Pairwise comparisons of the transcriptomic data sets were then performed to identify differentially expressed genes (DEGs). We found that 5,082 genes were significantly upregulated and 2,465 genes were significantly downregulated in WT SCs relative to WT VCs (WT SC versus WT VC). Furthermore, 11,962 genes were upregulated and 3,932 genes were downregulated in TL VCs relative to WT VCs (Figure 5, D and E). We then combined the RNA-Seq data sets of WT SC versus WT VC and TL VC versus WT VC; this revealed 1,892 VC- and 3,123 MG-required genes that, respectively, contribute to VC or MG cell lineage specificity in male gametophytes (Figure 5, F and G). Analogous to VC- and MG-required genes, the mean expression levels of the VC-peak annotated genes were downregulated and the MG-peak annotated genes were upregulated in TL VCs relative to WT VCs, which is consistent with the changes in chromatin accessibility reported above (Figures 4, I and L and 5, H–K; Supplemental Figure S6, G and H). Similar to the ATAC-Seq results, GO analysis revealed that VC-required genes were enriched in biological processes related to pollen tube development and signal transduction. Examples of VC-required genes include PRK6, ANX1, and BUPS1. MG-required genes, including DUO1 and BNB2, were mainly involved in gene expression and organelle organization (Figure 5, L and M). Real-time quantitative polymerase chain reaction (RT-qPCR) confirmed that ANX1/2, BUPS1/2, RALF4/19, PRK1/2A/6, and VGD1 were downregulated, while BNB1/2 and DUO1 were upregulated, in TL-10/+ pollen (Figure 5, N and O); these findings mutually corroborated the transcriptome data (Supplemental Figure S6I). Taken together, these results are consistent with H3K27me3 erasure-induced chromatin remodeling and provide a molecular basis for the VC–MG cell fate transition, suggesting that the developmental program was extensively altered in TL VCs after H3K27me3 erasure.
TL VCs exhibit MG cell ultrastructural features
Next, we analyzed the ultrastructure of TL-10 VCs to examine how subcellular organization was affected by the cell fate transition induced by H3K27me3 erasure. Compared with the highly condensed chromatin exhibited by WT SN, chromatin was highly diffuse in WT VN with no distinct nucleolus. Abnormally condensed chromatin speckles were observed in TL-10 VN (Figure 6, C, G, and I and Supplemental Figure S7). Furthermore, the lipid body, an organelle found only in WT VCs (Park and Twell, 2001), was not present in TL-10 VCs (Figure 6, B, D, and H). In WT pollen, small spherical vacuoles exist in SCs; whole vacuoles are no longer visible in VCs at the mature pollen stage (Huang et al., 2018). However, vacuoles of various sizes were present in TL VCs (Figure 6, A, B, G, H, J, O, and R). The ultrastructural discrepancy between WT VCs and TL VCs was also observed in the simplification of organelle structure. Many regular cristae were present in the mitochondria of WT VCs; in contrast, shortened or absent cristae were observed in TL VCs, similar to SCs (Figure 6, E, K, M, N, S, T, W, and X). In WT VCs, the Golgi body consisted of long, narrow, stacked lamellar vesicles, with smaller vesicles at each end. In TL VCs, the vesicular structures of the Golgi bodies were abnormally short and expanded. The endoplasmic reticulum had degenerated in TL VCs. These structures are analogous to the structures in SCs (Figure 6, F, L, P, Q, U, V, Y, and Z). The membrane-bound structures with fibrillar substances and Golgi vesicles that are considered polysaccharide particles were absent from TL VCs, similar to the findings in SCs (Figure 6, D, H, M, and R). Overall, there were no obvious differences in morphology or structure between WT SCs and TL SCs. The ultrastructural features of TL VCs were highly similar to the features of WT and TL SCs, suggesting that the TL VCs had switched cell fate to SCs and developed sperm-like ultrastructural features.
Figure 6.

TL VCs exhibit similar ultrastructural morphology to SC. A–F, WT VC ultrastructure. A, Panoramic image of WT VC. B, Cytoplasm of WT VC displays the abundant organelles. C, Highly diffused chromatin lacking a distinct nucleolus in VN. D, Golgi vesicle, membrane-bound structures with fibrillar substance, and VC-specific lipid body. E, Mitochondria with numerous, regular-sized cristae. F, Golgi body consisting of long, narrow, stacked lamellar vesicles with small vesicles at both ends. G–L, TL-10 VC ultrastructure. G, Panoramic image of TL-10 VC. H, Cytoplasm of TL-10 VC displays the simplification of organelles. I, The giant nucleolus and compact chromatin speckles of TL-10 VN. J, Vacuoles of varying sizes. K, Mitochondria with shortened or absent cristae. L, The shortened, abnormally expanded vesicular structure in Golgi bodies. M–V, Ultrastructure of WT SCs (M–Q) and TL-10 SCs (R–V). M and R, Panoramic images display scarce organelles and highly condensed chromatin in SCs. Black boxes show mitochondria (M) and vacuoles (R) in SCs, respectively. N, S, and T, The cristae are reduced or absent in SC mitochondria. O, Small spherical vacuoles in SCs. P and U, Golgi body in SCs. Q and V, Endoplasmic reticulum in SCs. Q, Higher magnification view of the regions enclosed in white Dashed boxes in (M). W, Number analysis of the mitochondrial cristae in WT VC, TL-10 VC, and WT SC. X, Length analysis of mitochondrial cristae in WT VC, TL-10 VC, and WT SC. The average length of the cristae is the sum of all cristae divided by the number of cristae in a mitochondrion. n = 48 in WT VCs, 75 in TL VCs, and 41 in WT SCs. Y, Statistical analysis of length/width of Golgi body saccules in WT VC, TL-10 VC, and WT SC. Z, Statistical analysis of width of Golgi body saccules in WT VC, TL-10 VC, and WT SC. n = 64 in WT VCs, 119 in TL VCs, and 83 in WT SCs. Statistical significance is determined by Student’s t test (*P < 0.05, ***P < 0.001) and the error bars show sd. Scale bars, 5 μm (A, G), 1 μm (B, C, H, I, M, R), 0.5 μm (D–F, J–L), 0.2 μm (N–Q, S–V). Mt, mitochondrion; Gb, Golgi body; Er, endoplasmic reticulum; Lb, lipid body; Va, vacuole; star (⋆), highly compacted chromatin speckle and nucleolus; *, Golgi vesicle.
Discussion
This work provides supporting evidence that H3K27me3 is essential for VC identity maintenance; H3K27me3 erasure in MG cells contributes to SC fate establishment. By combining a multi-omics approach with cytology techniques, we demonstrated that VC-targeted H3K27me3 erasure triggers a shift in the typical VC fate to a SC fate, thus experimentally demonstrating that H3K27me3 is essential for VC fate commitment and proper pollen development. Recently, H3K27me3 erasure was reported to be critical for MG differentiation (Borg et al., 2020). Our work provides further experimental support for this conclusion; it shows the efficiency of H3K27me3 erasure in establishing MG cell fate. Importantly, the differential distribution of unknown cell fate determinants has been hypothesized to explain how cell fate is established in pollen (Twell et al., 1998). Here, we revealed that H3K27me3 acts as an epigenetic determinant during pollen development to mediate VC and MG cell fates; it ensures the successful formation of the two divergent cell lineages in Arabidopsis male gametophytes. An earlier study also proposed that after microspore asymmetric division, VCs maintain the “default setting” of the microspore developmental program (Twell et al., 1998), while MG cells require reprograming to induce their gamete cell fate specification (Berger and Twell, 2011). Our results also reveal that H3K27me3 is a “default setting”; specifically, the VC inherits the default H3K27me3 status to guarantee development and functional specification for fertilization, whereas the MG cell undergoes H3K27me3 reprogramming to determine SC fate (Figure 7).
Figure 7.
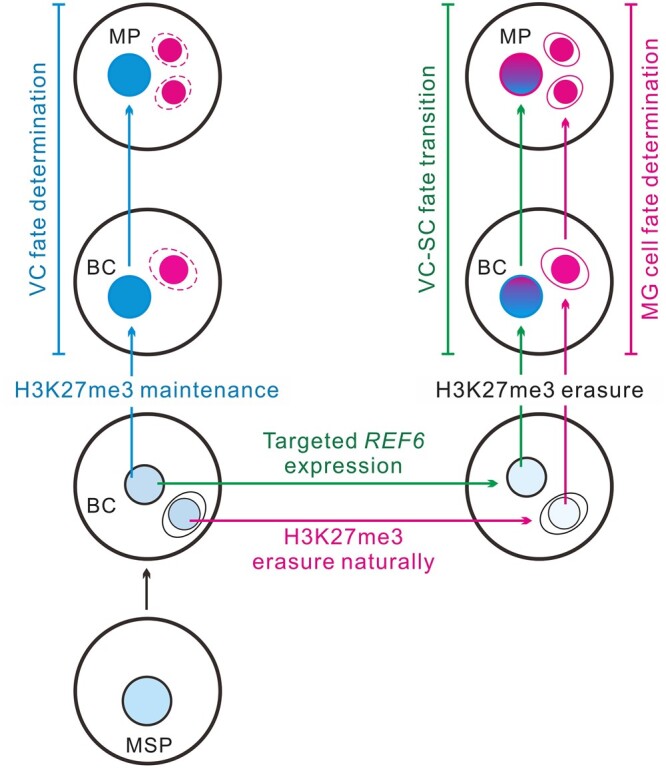
Putative model of how H3K27 methylation guides distinct cell fates of two cell lineages in the male gametophyte. In angiosperms, the microspore undergoes mitosis to generate two daughter cells. The VC maintains the H3K27me3 setting from its mother cell microspore and then differentiates into a functional male gametophyte (left, indicated in blue). In contrast, H3K27me3 erasure in the MG triggers the gamete cell fate in Arabidopsis (right, indicated in carmine). The artificial VC-targeted H3K27me3 erasure mediated by REF6 expression leads to VC developmental reprogramming toward a MG destination, that is the VC shifts to the SC fate (right, indicated in green). Thus, H3K27me3 acts as an epigenetic switch that mediates distinct germline or VC fate in male gametophytes. To highlight the VC formation pathway (left, blue arrows), MG fate specification in the same male gametophyte is showed by red dashed lines. MSP, microspore; BC, bicellular; and MP, mature pollen.
In addition, DUO1 was the first reported regulator of GC division and MG cell fate establishment (Durbarry et al., 2005; Rotman et al., 2005; Brownfield et al., 2009; Borg et al., 2011). Here, we found that DUO1 itself was not directly repressed by H3K27me3 in VCs; however, after VC-targeted H3K27me3 removal, DUO1 and its downstream target gene HTR10 were expressed ectopically and aberrant phenotypes similar to the VC-ectopic DUO1 expression line (pLat52:DUO1) (Brownfield et al., 2009) were present in strong TL pollen grains. These findings suggest that although H3K27me3 does not directly repress DUO1 expression in VCs, it is a critical upstream regulator of DUO1. BNB1 and BNB2, which transiently accumulate in germ cell progenitors and are required for GC specification, are pollen-specific VIIIa subfamily bHLH transcription factors that potentially regulate DUO1 (Yamaoka et al., 2018). Notably, we found that BNB1 and BNB2 were repressed by H3K27me3 in WT VCs and expressed in TL VCs. Thus, H3K27me3 erasure is a primary upstream signal that activates BNB1/2 expression during early MG cell fate determination, which then activates DUO1 expression.
During evolution, intricate multilayer strategies have been established in both animals and plants to ensure that cell fate is determined in a precise manner. In mouse, the enforced expression of a core set of transcription factors swiftly converted pluripotent stem cells into oocyte-like cells (Hamazaki et al., 2021). The direct conversion of differentiated cells into another cell type is considerably more difficult than dedifferentiation to induce a stem-cell state. Here, we successfully triggered a differentiated VC shift toward an MG cell fate in a one-step manipulation through VC-targeted H3K27me3 erasure; we created a cell model as a useful tool to elucidate the molecular mechanism underlying male gametogenesis, which can serve as a reference for research concerning animal and plant reproductive biology.
As shown here, VC-targeted H3K27me3 erasure caused approximately 15% of VCs to shift toward the MG cell fate, suggesting that other pathways or epigenetic mechanisms regulate cell fate determination in male gametophytes. During male gametogenesis in Arabidopsis, H3K4me3 is present in both VCs and SCs, while H3K9me2 and CENH3 persist only in SCs (Schoft et al., 2009; Pinon et al., 2017). The SDG2-mediated H3K4me3 deposition in VCs prevents ectopic heterochromatic H3K9me2 speckle formation and facilitates VN chromatin decondensation. The loss of H3K4me3 scarcely affects the fates of VCs and MG cells, but causes substantial compaction of chromatin in VCs, along with the obstruction of pollen germination and pollen tube elongation. The H3K4me3 reduction in MG cells partly disturbs the pollen mitotic cell cycle, particularly the second mitosis that establishes the two SCs (Pinon et al., 2017). These results suggest that H3K4me3 is involved in the functional specialization of VC and MG cells through a pleiotropic mechanism. Omics analysis shows that H3K4me3 and H3K27ac selectively accumulate at the Polycomb target genes in SCs, implying that they collaborate with H3K27me3 erasure to facilitate SC chromatin remodeling and the transcription of genes essential for male gametogenesis (Borg et al., 2020). Therefore, further exploration is needed to fully elucidate the epigenetic regulation network underlying cell fate determination in the male gametophyte.
Materials and methods
Plant material, growth conditions, and transformation
All A. thaliana lines were in the Columbia background. Seeds were sown on half-strength Murashige and Skoog plates with 1.0% (w/v) sucrose and then placed in an incubator under long-day conditions (16-h light/8-h dark) at 22°C. Light is provided by GreenPower TLED (Philips, 9290008431) at 100 μmol m–2 s–1. About a week later, the seedlings were grown in a greenhouse with the same diurnal cycle of photoperiod and temperature.
The HTR10-RFP marker is a gift from Prof. Frédéric Berger and swn-21/− clf-29/+ is a gift from Prof. Xiaofeng Cao. The ProACTIN11:H2B-GFP reporter was obtained by the fusion of the ACTIN11 promoter with the HISTONE2B-GFP. The DUO1-RFP marker line was constructed as described previously (Rotman et al., 2005). The ProLat52:REF6-GFP overexpression construct was generated via two steps. Firstly, the Lat52 promoter from the plasmid P092 (Wu et al., 2012) was amplified by PCR to introduce BstXI and SacI flanking restriction sites. The BstXI-SacI-digested Lat52 promoter fragment was then cloned into the BstXI-SacI-digested Q108 vector (modified from the PART27 vector, carrying a GFP and a hygromycin-resistance gene). Next, a WT genomic sequence containing the REF6 (AT3G48430) open-reading frame was PCR-amplified from genomic DNA and was then cloned into the KpnI and NotI sites following the Lat52 promoter. The gene construct was transferred into Agrobacterium tumefaciens strain GV3101, which was used to transform Arabidopsis plants by the floral dip method (Clough and Bent, 1998). For hygromycin (Merck) selection, the medium was supplemented with 50 μg mL–1 hygromycin. Different independent transgenic lines were selected according to their hygromycin resistance and subsequent phenotypic analysis. All the transgenic lines contained a single copy of the transgene and had a single insertion site.
Pollen cytological visualization
The pollen and VC morphology were examined using a Leica SP8 confocal laser scanning microscope (CLSM, Leica Microsystems) with differential interference contrast. Mature pollen grains were dissected rapidly in Alexander’s dye (25% glycerin, 10% ethyl alcohol, 0.01% malachite green, 0.01% acid fuchsin, 0.01% Orange G, and 4% acetic acid) to evaluate the substance accumulation and viability of the VC. Images were collected using an Olympus IX71 microscope. Unless otherwise indicated in the figure legends, the images are representative of at least three independent experiments. For every experiment, pollen grains from at least five flowers of the control and TL lines were collected and stained.
Pollen germination assays
The in vitro pollen germination was performed according to a previous method (Wu et al., 2012). The pollen grains were harvested from fresh self-dehiscing anthers and placed onto pollen germination medium (PGM: 18% sucrose, 1-mM CaCl2·2H2O, 1-mM Ca(NO3)2·4H2O, 1-mM MgSO4·7H2O, 1% H3BO3, pH 7.0, containing 0.9% agarose) and cultured at 22°C for 4 h. For the in vivo–in vitro pollen growth assay, WT styles were cut horizontally at the shoulder region of the ovaries after hand-pollination by control and TL pollen grains, respectively. Then, the pollinated styles were incubated on PGM for 6 h. Unless otherwise indicated in figure legends, the images are representative of at least three independent experiments. The materials from control and different TL lines were placed in the same culture dish for every experiment to guarantee the consistency of the experimental conditions.
Cryo-fixation, freeze-substitution, and transmission electron microscopy
The dehiscing anthers encased with mature pollen grains were dissected in the high-pressure freezer specimen holder and then embedded with the external cryo-protectant (2% low melting point agarose, Sigma, A9539). During the manipulation, any air bubbles were carefully removed, to avoid collapse under high pressure and disruption to the tissues. Samples were rapidly freeze fixed using the Lene and stoica EM ICE and stored in liquid nitrogen.
Fixative was prepared as follows. Stock solutions of 4% osmium tetroxide (OsO4) in acetone were prepared and stored in liquid nitrogen. OsO4 crystals were dissolved in a special glass container. Three specimen holders carrying samples (three biological repeats) were combined in a standard 1.8 mL cryo-tube for the freeze substitution. Samples at liquid nitrogen temperature were transferred to fixative vials at an identical temperature and then placed in the Leica EM ASF2, where they were warmed to −78.5°C. Substitution was completed in 2.5 days and then the samples were warmed according to the following schedule: 2 h at –20°C (standard freezer), 2 h at 0–4°C (refrigerator), and 2 h at 23°C (room temperature [RT]). Samples were separated carefully from the holders and rinsed three times in dried acetone. After epoxy propane gradient replacement, samples were embedded in Spurr resin. Ultra-thin section preparation and subsequent transmission electron microscopic (TEM) observation were performed following a previous description (Huang et al., 2019). Ultra-thin sections (70 nm) were stained with 2% (w/v) uranyl acetate and 2.6% (w/v) lead citrate aqueous solution and then examined with a JEM-1400 plus TEM (JEOL) at 100 kV.
Pollen grain collection
Fresh flowers were cut and placed in a 50-mL centrifuge tube containing 25-mL pollen buffer (18% sucrose, 1% H3BO3, 1-mM CaCl2·2H2O, 1-mM Ca(NO3)2·4H2O, 1-mM MgSO4·7H2O, pH 7.0). Oscillate violently for 5–10 min to ensure that the pollen grains could be washed out. Filter the pollen suspension through a 75-μm nylon mesh and then pellet pollen grains at RT, 1,500 rpm, 3 min.
Quantitative real-time PCR assay
Total RNA of mature pollen grains was extracted using the TaKaRa MiniBEST Plant RNA Extraction Kit (9769) according to the manufacturer’s protocol. The subsequent process was performed following a previous description (Huang et al., 2018, 2019). The first strand of cDNA was synthesized using PrimeScript RT Master Mix (Perfect Real Time) (TaKaRa). Quantitative real-time PCR (qPCR) was performed using FastStart Essential DNA Green Master (Roche) on a CFX Connect Real-Time System (Bio-Rad). Each experiment was repeated three times and samples were normalized according to UBQ10 expression. Data were acquired using Bio-Rad CFX Manager software; the relative expression levels were measured using the 2(–ΔΔCt) analysis method (Huang et al., 2018, 2019). The RT-qPCR primers used in this study are listed in Supplemental Data Set S1.
Whole-mount immunofluorescence
Whole-mount immunofluorescence of histone modifications in the pollen grains was adapted from a procedure described previously (She et al., 2018). Pollen grains were immediately fixed with 1% formaldehyde containing 0.1% Triton X-100 for 30 min with gentle rotation and then rinsed two times in 1× TTBS buffer (Tris Buffered Saline [TBS] buffer wtih 0.1% Triton X-100). Before antibody labeling, tissue clarification (ethanol-xylene [1:1], RT, 30 min; rinsed 2 × 10 min in 1× TTBS buffer), cell wall digestion (1.5% cellulose R-10, 0.75% macerozyme R-10, 28°C, 4 h), and blocking (TBS, 0.1% Triton X-100, 5% BSA, RT, 1 h) were performed. Following these pretreatments, samples were incubated with primary antibodies (anti-H3K27me3, Millipore 07-449, 1:100; and anti-H3K9me2, Abcam ab1220, 1:100) for 24 h at 4°C and washed twice in 1× TTBS for 1 h each time. Then samples were incubated with either anti-rabbit Alexa Fluor 647 (Invitrogen, A32733, 1:1,000, for anti-H3K27me3) or anti-rabbit Alexa Fluor 488 (Invitrogen, A32731, 1:1,000, for anti-H3K27me3), or anti-mouse Alexa Fluor 647 (Invitrogen, A32728, 1:1,000, for anti-H3K9me2) for 24 h at 4°C and washed twice in 1× TTBS for 1 h each time. Finally, pollen grains were spotted onto slides and mounted in Vectashield mounting medium with DAPI (Vector Laboratories). Images were taken using a Leica SP8 CLSM.
Nuclei isolation and FANS
FANS was performed according to a previous procedure (Borges et al., 2012) with modification. The TL-10/+ line with the HTR10-RFP marker and the control (ProACTIN11:H2B-GFP ProHTR10:HTR10-RFP double-labeled line) carry GFP and RFP fluorescent markers in the VN and SN, respectively, to facilitate sorting of viable VN and SN (Supplemental File). The collected pollen grains were re-suspended in Galbraith buffer [30-mM trisodium citrate dehydrate, 45-mM MgCl2, 20-mM MOPS, 0.1% [v/v] Triton X-100, adjust pH to 7.0] and then transferred to a 1.5-mL tube containing glass beads (Sigma, diam 2 mm, Z273627), before vortexed continuously at 2,000 rpm for 3 min. Samples were then filtered through a 10-μm nylon mesh in a 2-mL tube to acquire the nucleus mixture.
Fluorescence-activated SN and VN sorting was performed in a BD FACSAria III cell sorter. The nuclei can be obtained with high purity and integrity by running a 100-μm nozzle with 330-kPa sheath pressure. GFP (488 nm) and RFP (561 nm) were used to gate VN population (GFP-positive) and SN population (RFP-positive), respectively. Both VN and SN were also gated with a uniform side scatter (SSC) profile, which removed debris and incomplete nuclei. The sorted nuclei were collected directly into Galbraith buffer and kept on ice for the subsequent experiments. The FANS plot analysis was performed using FACSDiva software (BD Biosciences).
Microspore isolation and fluorescence-activated cell sorting
Flower buds at the late microspore stage were picked from TL-10/+ carrying the HTR10-RFP marker and control (ProHTR10:HTR10-RFP line) plants. The sample preparation and fluorescence-activated cell sorting (FACS) of the PMSs on a BD FACSAria III cell sorter were preformed according to the method described previously (Santos et al., 2017) with minor modifications. The sorter was run in standard configuration with a 100-μm nozzle, and gate for microspores based on their green autofluorescence (488 nm). Microspores were also gated with the RFP (561 nm) channel to exclude bicellular pollen. The collected microspores were maintained on ice and RNA was extracted immediately.
CUT&Tag and data analysis
A CUT&Tag assay was employed to profile H3K27me3 as described previously with minor modifications (Kaya-Okur et al., 2019). The FANS-purified nuclei must be preserved on ice to remain viable, but never frozen. About 106 nuclei per sample were collected and then centrifuged at 500g at 4°C for 10 min. Aliquots of nuclei were washed twice in 0.5-mL wash buffer (20-mM HEPES, pH 7.5; 150-mM NaCl; 0.5-mM Spermidine; and 1× Protease inhibitor cocktail) by gentle pipetting, and then centrifuged at 500g at 4°C for 10 min to remove the supernatant. Concanavalin A-coated magnetic beads (Bangs Laboratories) were washed twice with binding buffer (20-mM HEPES, pH 7.5, 10-mM KCl, 1-mM MnCl2, and 1-mM CaCl2) for activation and then 10 μL of the activated beads was added per sample and subsequently incubated at RT (25°C) for 15 min. Bead-bound cells were re-suspended in 50-μL Dig-wash buffer (20-mM HEPES, pH 7.5; 150-mM NaCl; 0.5-mM Spermidine; 1× Protease inhibitor cocktail; and 0.05% Digitonin) containing 2-mM EDTA and a 1:50 dilution of primary antibody (anti-H3K27me3, Millipore), and subsequently incubated overnight at 4°C with slow rotation. Samples were centrifuged at 500g at 4°C for 10 min to remove primary antibody thoroughly and secondary antibody (Santa Cruz) diluted in 50-μL Dig-wash buffer (1:100) was added, and then samples were incubated at RT (25°C) for 1 h. Then, nuclei were washed using magnet stand (Promega) three times for 5 min each time in 1-mL Dig-wash buffer to remove the unbound antibodies. A 1:100 dilution of pA-Tn5 adapter complex (TTE mix, 4 μM, Novoprotein) was prepared in Dig-med buffer (20-mM HEPES, pH 7.5, 300-mM NaCl, 0.5-mM Spermidine, 1× Protease inhibitor cocktail, and 0.05% Digitonin). Nuclei were incubated with 100-μL 0.04-μM TTE mix at RT for 1 h with gentle vortexing and then washed three times for 5 min each time in 1-mL Dig-med buffer to remove the unbound pA-Tn5 protein. The nuclei were then re-suspended in 300-μL tagmentation buffer (10-mM MgCl2 in Dig-med buffer) and incubated at 37°C for 1 h. To terminate tagmentation, 10-μL 0.5 M EDTA, 2.5-μL 20-mg mL–1 Proteinase K, and 3-μL 10% SDS were added and the samples were incubated at 55°C for 1 h. DNA was purified using phenol–chloroform–isoamyl (25:24:1, Sigma, P3803) extraction and ethanol precipitation, as well as RNaseA (Roche, 10109142001) treatment.
For library amplification, 21 μL of the purified DNA was mixed with 2 μL of universal i5 and a uniquely barcoded i7 primer (Buenrostro et al., 2015), and then 25-μL NEB Next HiFi 2× PCR Master mix was added and the samples were mixed. Then, 50-μL sample was placed in a PCR thermal cycler with a heated lid and subjected to the following program: 72°C for 5 min; 98°C for 30 s; 16 cycles of 98°C for 10 s; and 63°C for 30 s; final extension at 72°C for 1 min and hold at 4°C. To purify PCR products, 1.5× volumes of AMPure XP beads (Beckman Coulter) were added and incubated for 15 min at RT. Next, the libraries were washed twice gently in 80% ethanol and eluted in 30-μL 10-mM Tris pH 8.0. The size distribution of libraries was determined by Agilent 4200 TapeStation analysis and all libraries underwent Paired-end Illumina sequencing on an Illumina NovaSeq platform.
Before mapping of pair-end reads, clean reads were obtained from the raw reads by removing the adaptor sequences and then the reads were aligned to the Arabidopsis genome reference sequence (TAIR10) using the BWA program. We calculated the fragment sizes for read pairs using the BAM file generated during paired-end sequencing. Several regions were sampled to estimate the summary statistics on the fragment lengths. Properly paired reads were used for computation. The BAM file generated from the unique mapped reads was used as an input file, using MACS2 software for callpeak with a cutoff q < 0.05 and the parameters “macs2 callpeak–nomodel-f BAMPE–keep-dup 1-q 0.05 -B-SPMR” (Zhang et al., 2008). Reads distributions from bigwig across peaks and genes were presented as an average plot (average signals of reads across all peaks and genes) using the deeptools tool. The peak annotation and genomic distribution of CUT&Tag peaks were analyzed by HOMER (Heinz et al., 2010) and we counted the annotation results and plotted the distribution results using R software. The peaks of two biological replicates were merged to obtain a peak matrix. The reads of each replicate on the peak matrix were analyzed and then Pearson correlation coefficient was obtained according to the previous description (Kaya-Okur et al., 2019). The irreproducible discovery rate (IDR) was presented by IDR software (v.2.0.4.2). CUT&Tag signal tracks were presented by Integrative Genomics Viewer software (Robinson et al., 2011).
ATAC-Seq and data analysis
ATAC-Seq is a method for mapping chromatin accessibility genome-wide and used to determine the regions of increased accessibility. Transposase integration reactions and sequencing library preparations were manipulated as a procedure reported previously (Buenrostro et al., 2015). FACS-purified fresh nuclei to be used for ATAC-Seq must be placed on ice prior to the transposase integration reaction but should never be frozen. Approximately 106 nuclei per sample were collected and pelleted by centrifugation at 500g at 4°C for 10 min. The supernatant was then removed, leaving ∼50 µL in the tube. The nuclei were re-suspended and washed with 1× phosphate-buffered saline (PBS) and then centrifuged using the above settings. The supernatant was discarded. Next, the Tn5 reaction (25 µL 2× TD buffer, 2.5 µL TDE1 [Nextera Tn5 Transposase], and 22.5 µL nuclease-free H2O) was prepared using Nextera reagents (Illumina, FC-121-1030) and mixed well with nuclei. The reaction was placed in a thermal cycler at 37°C for 30 min. DNA fragments were purified immediately using the Minelute PCR purification kit (Qiagen) and eluted in 10-µL elution buffer (10-mM Tris–HCl pH 8), and each sample was amplified using NEB Next High-Fidelity 2× PCR Master Mix for 12 PCR cycles (program: 72°C for 5 min; 98°C for 30 s; 12 cycles of 98°C for 10 s; and 63°C for 30s). All amplified ATAC-Seq libraries were purified using AMPure XP beads (Beckman Coulter), quantified by a Qubit Fluorometer, and analyzed on an Agilent 2100 Bioanalyzer prior to pooling and subsequently sequenced using Illumina NovaSeq.
Mapping of pair-end reads, callpeak, peaks annotation, and genomic distribution of ATAC-Seq peaks were analyzed as described in CUT&Tag analysis. PCA was performed on the signals of merged peaks from all samples and PlotPCA was used to sort the principal components according to the variability of the data. Based on this, a plot that shows the eigenvalues of the top two principal components was obtained. The peak files of all replicate samples from each cell type were merged using Bedtools software and the differential accessible regions (DARs) were assessed using DESeq2 (Love et al., 2014) according to an absolute value of log2 fold change (FC) of 1 (|log2FC| > 1) at P < 0.05. DARs were annotated using the annotatePeak function of ChIPseeker (Yu et al., 2015). The IDR was presented by IDR software (v.2.0.4.2). The distributions of the reads across genes were presented using deeptools based on the BED file.
RNA-seq and data analysis
Messenger RNA (mRNA) isolation and RNA-Seq library construction were performed as described previously (Zhao et al., 2019). mRNA was extracted from the FANS-isolated VN and SN of control and TL-10/+ (about 106 nuclei per sample) with independent biological replicates using a Dynabeads mRNA DIRECT Micro Purification Kit (Life Technologies). Total RNA of late microspore (about 106 microspores per sample) was extracted using a RNeasy Plant Mini Kit (QIAGEN). Subsequent cDNA synthesis and amplification were performed using a SMART-Seq HT Kit (Takara) and the amplified cDNA was then purified using an Agencourt AMPure Purification Kit (Beckman Coulter). Purified cDNA was quantified using a Qubit 3.0 Fluorometer (Life Technologies) and validated using a Bioanalyzer 2100 system (Agilent Technologies). Sequencing libraries were constructed using the VAHTS Universal V6 RNA-Seq Library Kit for MGI (Vazyme) following the manufacturer’s recommendations with unique index codes. The abundance of the libraries was quantified using a Qubit 3.0 Fluorometer and Bioanalyzer 2100 system. Subsequently, libraries were sequenced on an MGI-SEQ 2000 platform by Frasergen Bioinformatics Co., Ltd. (Wuhan, China) and about 6 Gb of raw sequence data was obtained per library.
Low quality reads, such as reads with adaptor sequences, reads with >5% N, or >20% bases and with quality <Q20 (percentage of sequences with sequencing error rates <1%), were removed using perl script. The clean reads filtered from raw reads were mapped to the Arabidopsis genome (TAIR10) using HISAT2 (Li and Dewey, 2011) and gene expression levels were quantified and normalized as fragments per kilo-base per million bases measurements using RSEM (Kim et al., 2019). RNA-Seq heatmaps were generated using the pheatmap package in R (https://github.com/ raivokolde/pheatmap). DESeq2 (Love et al., 2014) was used to identify DEGs between sample groups, and only the genes with a FC value ≥ 1 (|log2FC|≥1) and false discovery rate (FDR) significance score (padj) < 0.01 were considered as significant DEGs. Hierarchical clustering of the gene expression data was carried out using the Agnes function in the Cluster package based on the value of the Pearson correlation coefficient between different samples.
GO analysis
GO analysis was performed to facilitate elucidating the biological implications of the unique genes (Ashburner et al., 2000). We downloaded the GO terms for the genes from The Arabidopsis Information Resource (TAIR) (https://www.arabidopsis.org/). Fisher’s exact test (ATAC-Seq) or hypergeometric distribution (RNA-Seq) was applied to identify the GO categories and the FDR method was used to correct the P values.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (version 7.0) and Microsoft Excel 2003. Statistical significance is determined by two-tailed Student’s t test. Results of statistical analyses are listed in Supplemental Data Set S2.
Accession numbers
Sequence data from this article can be found in the Arabidopsis TAIR database (www.arabidopsis.org) under the following accession numbers: REF6 (AT3G48430). High-throughput sequencing data that support the findings of this study are available in the NCBI Gene Expression Omnibus under accession code GSE162640.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Comparison of male gametophyte development and function between WT and clf-29/+ swn-21/– double mutant.
Supplemental Figure S2. TL line selection and H3K27me3 is essential for VC development.
Supplemental Figure S3. H3K27me3 is indispensable for VC functional specification.
Supplemental Figure S4. H3K27me3 CUT&Tag profiling of Arabidopsis WT VC, WT SC, and TL VC.
Supplemental Figure S5. ATAC-Seq profiling of Arabidopsis WT VC, WT SC, TL VC, and TL SC and global dynamics of chromatin accessibility during VC and MG lineage development.
Supplemental Figure S6. Transcriptome reproducibility, correlation among cell lineages and transcriptome dynamics during VC and MG lineage development.
Supplemental Figure S7. Abnormal condensation of chromatin in TL-10 VN.
Supplemental File. FANS strategies and quality control of ATAC-Seq, CUT&Tag, RNA-Seq.
Supplemental Data Set S1. The primers used in this study.
Supplemental Data Set S2. Summary of statistical analyses.
Supplementary Material
Acknowledgments
We thank Prof. Xiaofeng Cao (Institute of Genetics and Developmental Biology, Chinese Academy of Sciences) for the swn-21/– clf-29/+ seed; Prof. Frédéric Berger (Gregor Mendel Institute, Austrian Academy of Sciences) for the HTR10-RFP marker seed and careful reading of the manuscript; Dr Wenxuan Zou (College of Life Science, Wuhan University) for Cryo-fixation, freeze-substitution, and TEM; Dr Ying Liu (Medical Research Institute, Wuhan University) for FANS; Shanghai Jiayin Biotechnology for H3K27me3 CUT&Tag and ATAC-Seq library construction, sequencing, and data analysis; and Wuhan Frasergen Bioinformatics Co., Ltd. for RNA-Seq library construction, sequencing, and data analysis.
Funding
This work was supported by the National Natural Science Foundation of China (31991201, 31800265, and 32130031).
Conflict of interest statement. None declared.
Contributor Information
Xiaorong Huang, State Key Laboratory of Hybrid Rice, College of Life Science, Wuhan University, Wuhan 430072, China.
Meng-Xiang Sun, State Key Laboratory of Hybrid Rice, College of Life Science, Wuhan University, Wuhan 430072, China.
X.H. designed and performed the research, analyzed the data, and wrote the draft. M.-X.S. designed the research, analyzed the data, and wrote the paper.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) is: Meng-Xiang Sun (mxsun@whu.edu.cn).
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer M, Nawy T, Giglione C, Galli M, Meinnel T, Lukowitz W (2009) Paternal control of embryonic patterning in Arabidopsis thaliana. Science 323: 1485–1488 [DOI] [PubMed] [Google Scholar]
- Berger F, Twell D (2011) Germline specification and function in plants. Annu Rev Plant Biol 62: 461–484 [DOI] [PubMed] [Google Scholar]
- Borg M, Brownfield L, Khatab H, Sidorova A, Lingaya M, Twell D (2011) The R2R3 MYB transcription factor DUO1 activates a male germline-specific regulon essential for sperm cell differentiation in Arabidopsis. Plant Cell 23: 534–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg M, Berger F (2015) Chromatin remodeling during male gametophyte development. Plant J 83: 177–188 [DOI] [PubMed] [Google Scholar]
- Borg M, Jacob Y, Susaki D, LeBlanc C, Buendía D, Axelsson E, Kawashima T, Voigt P, Boavida L, Becker J, et al. (2020) Targeted reprogramming of H3K27me3 resets epigenetic memory in plant paternal chromatin. Nat Cell Biol 22: 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg M, Papareddy RK, Dombey R, Axelsson E, Nodine MD, Twell D, Berger F (2021) Epigenetic reprogramming rewires transcription during the alternation of generations in Arabidopsis. eLife 10: e61894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges F, Gardner R, Lopes T, Calarco JP, Boavida LC, Slotkin RK, Martienssen RA, Becker JD (2012) FACS-based purification of Arabidopsis microspores, sperm cells and vegetative nuclei. Plant Methods 8: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson-Dernier A, Roy S, Kritsas K, Grobei MA, Jaciubek M, Schroeder JI, Grossniklaus U (2009) Disruption of the pollen-expressed FERONIA homologs ANXUR1 and ANXUR2 triggers pollen tube discharge. Development 136: 3279–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownfield L, Hafidh S, Borg M, Sidorova A, Mori T, Twell D (2009) A plant germline-specific integrator of sperm specification and cell cycle progression. PLoS Genet 5: e1000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ (2015) Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523: 486–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (2015) ATAC-Seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 5: 21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Hafidh S, Poh SH, Twell D, Berger F (2009) Proliferation and cell fate establishment during Arabidopsis male gametogenesis depends on the retinoblastoma protein. Proc Natl Acad Sci USA 106: 7257–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Jeanie LH, Mathieu I, Venkatesan S, Berger F (2008) Chromatin assembly factor 1 regulates the cell cycle but not cell fate during male gametogenesis in Arabidopsis thaliana. Development 135: 65–73. [DOI] [PubMed] [Google Scholar]
- Calarco JP, Borges F, Donoghue MT, Van Ex F, Jullien PE, Lopes T, Gardner R, Berger F, Feijó JA, Becker JD, et al. (2012) Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 151: 194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16: 735–743 [DOI] [PubMed] [Google Scholar]
- Cui X, Lu F, Qiu Q, Zhou B, Gu L, Zhang S, Kang Y, Cui X, Ma X, Yao Q, et al. (2016) REF6 recognizes a specific DNA sequence to demethylate H3K27me3 and regulate organ boundary formation in Arabidopsis. Nat Genet 48: 694–699 [DOI] [PubMed] [Google Scholar]
- Durbarry A, Vizir I, Twell D (2005) Male germ line development in Arabidopsis. Duo pollen mutants reveal gametophytic regulators of generative cell cycle progression. Plant Physiol 137: 297–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Z, Bergonci T, Zhao Y, Zou Y, Du S, Liu MC, Luo X, Ruan H, García-Valencia LE, Zhong S, et al. (2017) Arabidopsis pollen tube integrity and sperm release are regulated by RALF-mediated signaling. Science 358: 1596–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenberg D, Twell D (2019) The evolution and patterning of male gametophyte development. Curr Top Dev Biol 131: 257–298 [DOI] [PubMed] [Google Scholar]
- Hamazaki N, Kyogoku H, Araki H, Miura F, Horikawa C, Hamada N, Shimamoto S, Hikabe O, Nakashima K, Kitajima TS, et al. (2021) Reconstitution of the oocyte transcriptional network with transcription factors. Nature 589: 264–269 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo A, Kawashima T, Borg M, Zhao M, López-Vidriero I, Sakayama H, Montgomery SA, Sekimoto H, Hackenberg D, Shimamura M, et al. (2018) Transcription factor DUO1 generated by neo-functionalization is associated with evolution of sperm differentiation in plants. Nat Commun 9: 5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Peng X, Xie F, Mao W, Chen H, Sun M-X (2018) The stereotyped positioning of the generative cell associated with vacuole dynamics is not required for male gametogenesis in rice pollen. New Phytol 218: 463–469 [DOI] [PubMed] [Google Scholar]
- Huang X, Run M, Sun M-X (2019) OsGCD1, a novel player in rice intine construction. J Genet Genomics 46: 359–362 [DOI] [PubMed] [Google Scholar]
- Ingouff M, Rademacher S, Holec S, Soljić L, Xin N, Readshaw A, Foo SH, Lahouze B, Sprunck S, Berger F (2010) Zygotic resetting of the HISTONE 3 variant repertoire participates in epigenetic reprogramming in Arabidopsis. Curr Biol 20: 2137–2143 [DOI] [PubMed] [Google Scholar]
- Jackson JP, Lindroth AM, Cao X, Jacobsen SE (2002) Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 416: 556–560 [DOI] [PubMed] [Google Scholar]
- Jiang L, Yang SL, Xie LF, Puah CS, Zhang XQ, Yang WC, Sundaresan V, Ye D (2005) VANGUARD1 encodes a pectin methylesterase that enhances pollen tube growth in the Arabidopsis style and transmitting tract. Plant Cell 17: 584–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L, Cao X, Jacobsen S (2002) Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr Biol 12: 1360–1367 [DOI] [PubMed] [Google Scholar]
- Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, Henikoff S (2019) CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10: 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khouider S, Borges F, LeBlanc C, Ungru A, Schnittger A, Martienssen R, Colot V, Bouyer D (2021) Male fertility in Arabidopsis requires active DNA demethylation of genes that control pollen tube function. Nat Commun 12: 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37: 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Lu F, Cui X, Cao X (2010) Histone methylation in higher plants. Annu Rev Plant Biol 61: 395e420. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Cui X, Zhang S, Jenuwein T, Cao X (2011) Arabidopsis REF6 is a histone H3 lysine 27 demethylase. Nat Cenet 43: 715–719 [DOI] [PubMed] [Google Scholar]
- Park SK, Twell D (2001) Novel patterns of ectopic cell plate growth and lipid body distribution in the Arabidopsis gemini pollen1 mutant. Plant Physiol 126: 899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinon V, Yao X, Dong A, Shen WH (2017) SDG2-mediated H3K4me3 is crucial for chromatin condensation and mitotic division during male gametogenesis in Arabidopsis. Plant Physiol 174: 1205–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J (2001) Epigenetic reprogramming in mammalian development. Science 293: 1089–1093. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thoryaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotman N, Durbarry A, Wardle A, Yang WC, Chaboud A, Faure JE, Berger F, Twell D (2005) A novel class of MYB factors controls sperm cell formation in plants. Curr Biol 15: 244–248. [DOI] [PubMed] [Google Scholar]
- Santos MR, Bispo C, Becker JD (2017) Isolation of Arabidopsis pollen, sperm cells, and vegetative nuclei by fluorescence-activated cell sorting (FACS). Methods Mol Biol 1669: 193–210 [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P (2002) Unique chromatin remodeling and transcriptional regulation in spermatogenesis. Science 296: 2176–2178 [DOI] [PubMed] [Google Scholar]
- Schoft VK, Chumak N, Mosiolek M, Slusarz L, Komnenovic V, Brownfield L, Twell D, Kakutani T, Tamaru H (2009) Induction of RNA directed DNA methylation upon decondensation of constitutive heterochromatin. EMBO Rep 10: 1015–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- She W, Baroux C, Grossniklaus U (2018) Cell-type specific chromatin analysis in whole-mount plant tissues by immunostaining. Methods Mol Biol 1675: 443–454 [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Higashiyama T (2016) Tip-localized receptors control pollen tube growth and LURE sensing in Arabidopsis. Nature 531: 245–248 [DOI] [PubMed] [Google Scholar]
- Twell D, Park SK, Lalanne E (1998) Asymmetric division and cell-fate determination in developing pollen. Trends Plant Sci 3: 305–310 [Google Scholar]
- Twell D, Park SK, Hawkins TJ, Schubert D, Schmidt R, Smertenko A, Hussey PJ (2002) MOR1/GEM1 has an essential role in the plant-specific cytokinetic phragmoplast. Nat Cell Biol 4: 711–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JJ, Peng XB, Li WW, He R, Xin HP, Sun M-X (2012) Mitochondrial GCD1 dysfunction reveals reciprocal cell-to-cell signaling during the maturation of Arabidopsis female gametes. Dev Cell 23: 1–16 [DOI] [PubMed] [Google Scholar]
- Yamaoka S, Nishihama R, Yoshitake Y, Ishida S, Inoue K, Saito M, Okahashi K, Bao H, Nishida H, Yamaguchi K, et al. (2018) Generative cell specification requires transcription factors evolutionarily conserved in land plants. Curr Biol 8: 479–486 [DOI] [PubMed] [Google Scholar]
- Yu GC, Wang LG, He Q-Y (2015) ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31: 2382–2383 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008) Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Zhou XM, Shen K, Liu ZZ, Cheng TH, Liu DN, Cheng YB, Peng XB, Sun M-X (2019) Two-step maternal-to-zygotic transition with two-phase parental genome contributions. Dev Cell 49: 1–12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.