

Abstract
Our kidneys receive about one-fifth of the cardiac output at rest and have a low oxygen extraction ratio, but may sustain, under some conditions, hypoxic injuries that might lead to chronic kidney disease. This is due to large regional variations in renal blood flow and oxygenation, which are the prerequisite for some and the consequence of other kidney functions. The concurrent operation of these functions is reliant on a multitude of neuro-hormonal signaling cascades and feedback loops that also include the regulation of renal blood flow and tissue oxygenation. Starting with open questions on regulatory processes and disease mechanisms, we review herein the literature on renal blood flow and oxygenation. We assess the current understanding of renal blood flow regulation, reasons for disparities in oxygen delivery and consumption, and the consequences of disbalance between O2 delivery, consumption, and removal. We further consider methods for measuring and computing blood velocity, flow rate, oxygen partial pressure, and related parameters and point out how limitations of these methods constitute important hurdles in this area of research. We conclude that to obtain an integrated understanding of the relation between renal function and renal blood flow and oxygenation, combined experimental and computational modeling studies will be needed.
Keywords: Kidney, Perfusion, Autoregulation, Oxygenation
Introduction
The kidneys fulfill a multitude of functions that can be described, in very general terms, as blood conditioning. To this end, mammalian kidneys receive approximately 20% of the cardiac output under resting conditions, which is more than they need to meet their own metabolic demand, as evinced by the low oxygen extraction ratio of 10–15%. At the same time, there is marked heterogeneity of both blood perfusion and oxygen consumption in renal tissues, which renders parts of the kidney susceptible to hypoxia despite overall excess of oxygen. While the partial pressure of oxygen (pO2) in renal cortical tissue is in the range of 20–60 mmHg, the outer and inner medulla show characteristic pO2 values of 15–30 mmHg and < 15 mmHg, respectively [1]. Such regional variations in pO2 are owed, in part, to the renal vascular architecture.
As interlobular arteries ascend from the cortico-medullary junction to the renal capsule, they supply the afferent arterioles of juxtamedullary glomeruli at lower levels and of cortical glomeruli at higher levels. Thus, the cortical and medullary vasculatures are connected in series. The efferent arterioles of superficial glomeruli give rise to the peritubular capillaries that surround proximal and distal tubules in the cortex, while the efferent arterioles of juxtamedullary glomeruli give rise to descending vasa recta that supply the medulla, and that turn back at varying depths to form ascending vasa recta. These observations already imply that blood flow regulation in the kidney is more complex than in organs where metabolic demand is the primary determinant. Exceeding regulatory limits or dysfunction of renal blood flow (RBF) regulation can lead to renal ischemia, which is an important factor in both the genesis of acute kidney injury and the development of chronic kidney disease. To investigate the mechanisms of renal pathogenesis, and for clinical decision-making, understanding the determinants of RBF and renal tissue oxygenation is required. However, despite recent advances, obtaining an integrated picture remains challenging.
The ability of the kidney to perform multiple functions simultaneously is sustained by a large number of neuro-hormonal agents that interact in numerous ways, with signaling cascades and feedback loops that are difficult to disentangle. Many questions, ranging from fundamental regulatory processes to specific disease mechanisms, have yet to be resolved: To what extent are cortical and medullary oxygenation independently regulated? What is the role of spatial oxygen gradients in renal tissue in the regulation of erythropoiesis? What are the relative contributions of vasa recta and juxtamedullary arterioles to the control of medullary blood flow (MBF) in vivo? What is the role of MBF in pressure natriuresis? What role do changes in regional hemodynamics and tissue oxygenation play in renal disease and kidney injury? How do age and sex influence the regulation of RBF and tissue oxygenation?
In the following, we review the current literature on RBF and oxygenation beginning with the regulation of blood flow. We consider reasons for disparities between oxygen delivery and consumption, as well as the consequences of the disbalance between O2 delivery, consumption, and removal. Finally, we describe present hurdles in studying renal blood flow and oxygenation and close the circle by showing how the open questions listed above are linked to limitations of available methods for measuring and computing blood flow rate, velocity, renal tissue oxygenation, and related parameters.
Renal blood flow
Renal blood flow or renal blood flow rate may refer both to flow through the entire organ, or through parts thereof. At the highest level, cortical and medullary blood flow needs to be differentiated. Total RBF is maintained approximately constant over a wide pressure range, e.g., from about 100 to 160 mmHg in rats [2, 3]. Current understanding is that the medulla receives approximately 10% of the total RBF [4, 5] through juxtamedullary efferent arterioles that give rise to vasa recta, which are surrounded by pericytes that allow for independent control of medullary blood flow by vasoactive substances. Since MBF is small compared to total renal blood flow, moderate changes in MBF may not have a measurable effect on total RBF. The autoregulation of RBF is achieved by intrinsic mechanisms that modulate the resistance of (mostly) preglomerular vessels, and is thereby coupled to the autoregulation of the glomerular filtration rate (GFR). RBF is also regulated by extrinsic mechanisms, some of which differentially alter the resistance of afferent versus efferent arterioles, allowing for the partial decoupling of GFR and RBF, and/or differentially impact the cortical versus medullary circulation. While RBF autoregulation affects renal oxygenation, control of O2 delivery is not its primary role.
Intrinsic mechanisms of renal blood flow regulation
The autoregulation of RBF is mediated primarily by two mechanisms, the fast myogenic response and the slower macula densa (MD) tubulo-glomerular feedback (TGF). Two other autoregulatory mechanisms have been proposed, one that operates at a low frequency [6] and one that involves crosstalk between the connecting tubule and the afferent arteriole [7], but their contributions appear smaller and they remain poorly understood [8]. Modeling studies have significantly expanded our understanding of these intrinsic regulatory mechanisms, including their relative contributions and interactions and how they are affected by specific pathological conditions [9, 10].
Myogenic mechanism
Preglomerular vessels constrict when subjected to an increase in transmural pressure difference, a response known as the myogenic mechanism; the resulting increase in vascular resistance lowers perfusion. Thus, the myogenic response acts to stabilize RBF and GFR and to protect the glomerulus from increases in systolic blood pressure.
The myogenic response is initiated by mechano-receptors that transduce the mechanical signal into transmembrane currents, which then activate cytosolic signaling cascades that amplify the signal and ultimately cause smooth muscle cell contraction. The first step, i.e., mechanical-electrical transduction, may involve cell surface integrin receptors [11], mechanosensitive ion channels such as ENaC/degenerins [12], transient receptor potential channels, and stretch-activated G-protein coupled receptors [13]. These pathways, the relative contributions of which remain to be fully elucidated, depolarize the membrane, thereby activating voltage-gated Ca2+ channels and triggering Ca2+ influx. The resulting increase in cytosolic Ca2+ triggers the release of Ca2+ from sarcoplasmic reticulum stores, all of which stimulate myosin light chain kinase and inhibit myosin light chain phosphatase, thus eliciting cross-bridge formation and muscle contraction.
Tubulo-glomerular feedback
TGF modulates the resistance of the afferent arteriole in response to changes in NaCl delivery to the macula densa, a small cluster of tubular epithelial cells between the cortical thick ascending limb and the distal convoluted tubule. Specifically, an increase in tubular NaCl concentration at the MD, reflecting a higher rate of NaCl filtration and/or reduced reabsorption in pre-MD segments, triggers contraction of the afferent arteriole, thus reducing blood flow and the filtration rate. Conversely, a decrease in NaCl concentration at the MD leads to dilation of the arteriole.
The mechanisms underlying TGF are not completely understood. Elevations in NaCl delivery to the MD stimulate the apical uptake of Na+, K+, and Cl− via the Na-K-2Cl cotransporter NKCC2, thereby raising intracellular Cl− levels and altering the basolateral membrane potential of MD cells. One or both of these signals activate the release of ATP and/or its breakdown product adenosine into extra-glomerular mesangial cells and adjacent vascular smooth muscle cells (VSMCs) on the afferent arteriole. The binding of ATP and adenosine to, respectively, purinergic P2X and adenosine A1 receptors on VSMCs triggers signaling cascades that raise intracellular Ca2+ levels, thereby activating contraction and reducing the diameter of the arteriole. The subsequent reduction in the filtration rate in turn reduces NaCl delivery to the MD, thus closing the negative feedback loop. Note that MD signaling may also involve vasoactive factors, including prostaglandins and nitric oxide [13].
Changes in NaCl delivery to the MD may result from variations in the filtration pressure and/or in proximal reabsorption. Thus, TGF acts not only to regulate glomerular pressure and flow, but also stabilizes fluid and electrolyte delivery to the distal nephron, which facilitates the hormone-mediated fine-tuning of reabsorption and secretion therein [14]. TGF may be modulated by the connecting tubule-glomerular feedback, a positive feedback mechanism that dilates the afferent arteriole when high concentrations of sodium are detected in the connecting tubule, thereby increasing filtration [15]. The connecting tubule-glomerular feedback is abolished when the epithelial channel ENaC is inhibited, and there is evidence that it contributes to TGF resetting during high salt intake [16].
Extrinsic mechanisms of renal blood flow regulation
RBF is also regulated by systemic and paracrine factors. Cortical versus medullary differences in vascular structure, receptor expression, and hormone synthesis rates allow for the differential regulation of cortical (CBF) versus medullary blood flow and may help to prevent medullary hypoxia even when RBF is reduced [1].
Hormonal signals
Renal vascular tone is controlled by many vasoconstrictors (including angiotensin II, endothelins [17], norepinephrine, vasopressin [18], and reactive oxygen species [19]) and vasodilators (including nitric oxide, adenosine, prostaglandins [20], and bradykinin [21]). Many of these factors also affect tubular function, i.e., they modulate both O2 supply and O2 demand (see below). Of particular importance are angiotensin II (Ang II), nitric oxide (NO), and adenosine.
Ang II is the most potent effector of the renin-angiotensin-aldosterone system. Its effects are mediated by two receptors, AT1 (predominantly expressed) and AT2. The binding of Ang II to AT1 elicits vasoconstriction and stimulates tubular sodium reabsorption, whereas binding to AT2 causes vasodilation and favors natriuresis. In the renal vasculature, Ang II (via AT1) augments the resistance of efferent arterioles more than that of afferent arterioles, thereby decreasing RBF while maintaining GFR [22].
While Ang II reduces CBF [23], it can induce a paradoxical increase in MBF owing to its indirect effects. As reviewed by Evans et al. [24], Ang II directly constricts medullary vessels but it also activates the release of vasodilator paracrine factors (such as prostaglandins and NO) from vascular, tubular, and interstitial cells. The net impact of Ang II on medullary blood flow depends on the relative levels of these effects, as well as the source of Ang II (systemic versus intra-renal).
NO is a vasodilator that is produced by both tubular and vascular cells at significantly higher rates in the medulla than in the cortex [25]. NO synthase inhibition studies have shown that NO plays an important role in maintaining MBF and medullary O2 supply under both acute and chronic conditions [26]. Subpressor doses of Ang II stimulate tubular NO synthesis, and the subsequent diffusion of NO towards adjacent pericytes, referred to as tubulo-vascular crosstalk, promotes vasodilation and thereby buffers the vasoconstrictor actions of Ang II [27].
Adenosine is a breakdown product of ATP, and, like NO, it is more concentrated in the medulla than in the cortex [28]. Adenosine acts a paracrine signal via two receptors, A1AR and A2AR [29]. Activation of A1AR constricts afferent arterioles and lowers single nephron GFR and is an essential component of TGF (see above). In contrast, activation of A2AR dilates postglomerular vessels. While A1AR-mediated constriction is the dominant effect of adenosine in superficial nephrons, A2AR-mediated vasodilation prevails in juxtamedullary nephrons, where it acts to enhance MBF [28]. Of note, the release of adenosine by cells of the medullary thick ascending limb (mTAL) is transport-dependent and stimulated by hypoxia [30].
While Ang II stimulates the (counteracting) release of NO, Ang II and adenosine cooperate in inducing afferent arteriole contraction [29]. Moreover, other vasoactive agents, such as endothelin-1 and prostaglandin E2, contribute to tubulo-vascular crosstalk in the renal medulla [31]. Even though a full description is beyond the scope of this review, it is important to recognize that the hormonal and neural factors that regulate renal perfusion (and tubular transport, see below) do not act independently but instead interact in synergistic and/or antagonistic ways.
Neural signals
Renal blood flow is also modulated by renal sympathetic nerve activity. Renal afferent nerves, situated mainly in the pelvis, are activated by mechanical and chemical signals (including Ang II) and strongly regulate central sympathetic outflow [32]. Renal efferent nerves are found predominantly in the nephron segments and vasculature of the renal cortex and outer medulla [33]. They release norepinephrine, which provokes contraction of the afferent and efferent arterioles via α1 and α2 adrenoreceptors, thereby reducing RBF and GFR. A role for sympathetic nerve-derived ATP in modulating vasa recta diameter and regulating MBF specifically has also been proposed [34].
Studies compiled by Evans et al. [35] suggest that, with some exceptions, vasoconstrictors appear to decrease CBF more than MBF, whereas most vasodilators appear to increase MBF more than CBF. Aside from regional differences in hormone production rates and receptor expression, another possible factor may be that juxtamedullary arterioles are significantly larger than cortical arterioles, so that their resistance is less impacted, in relative terms, by vasoconstrictor-induced reductions in diameter [35].
Renal oxygenation
The interplay between renal O2 delivery and consumption is complex. Oxygen delivery is proportional to total RBF, and thus directly dependent on blood flow regulation. Oxygen consumption is primarily driven by tubular demand, which, in turn, is dependent on GFR. Increase in total RBF leads to a proportional increase in GFR under normal physiologic conditions [36]. Regional variations in O2 delivery and consumption produce spatial pO2 gradients that are of relevance for the regulation of erythropoietin (EPO) production [37]. At the same time, areas with lower pO2 under normal conditions are at increased risk of hypoxic damage when the balance between O2 delivery and consumption shifts. We note that the term “O2 delivery” can be ambiguous: it can refer to the O2 carried by the blood supplying a given renal region, or it can refer to the O2 that has exited the blood stream and diffused into tissue. We will use “O2 delivery to tissue” to refer to the latter.
Determinants of regional pO2
Local pO2 is determined by the rate of O2 delivery, consumption, and removal, and by local tissue and blood properties (Fig. 1). Increase in O2 demand and removal, or decrease in the rate of delivery to tissue, will reduce local pO2. The same will occur when the affinity of hemoglobin for O2 is increased due to, e.g., alkalosis, or due to pathologic changes in tissue properties such as fibrosis. In the medulla, O2 delivery is limited by the requirement for low blood flow through the vasa recta to maintain the osmolality gradients produced by the countercurrent arrangement of descending and ascending vessels and tubules [38]. The process in the kidney with the highest O2 demand is synthesis of ATP. Only about 5% of ATP is produced anaerobically [39]. Most of the ATP is utilized to drive tubular Na+ reabsorption primarily in the proximal tubule and the thick ascending limb (TAL) of the loop of Henle, where O2 consumption is, consequently, high. The efficiency of O2 utilization for Na+ reabsorption varies between tubular segments. It is higher in the proximal tubule than in the TAL [40]. Also, the distribution of Na+ reabsorption is dependent on the level of dietary Na+ intake. Increase in GFR leads to higher O2 consumption due to increased delivery and consequent higher reabsorption of Na+. Elevation of blood glucose levels leads to increased reabsorption of glucose and Na+ in the proximal tubule by sodium–glucose cotransporters, and thereby to increased O2 consumption [41].
Fig. 1.
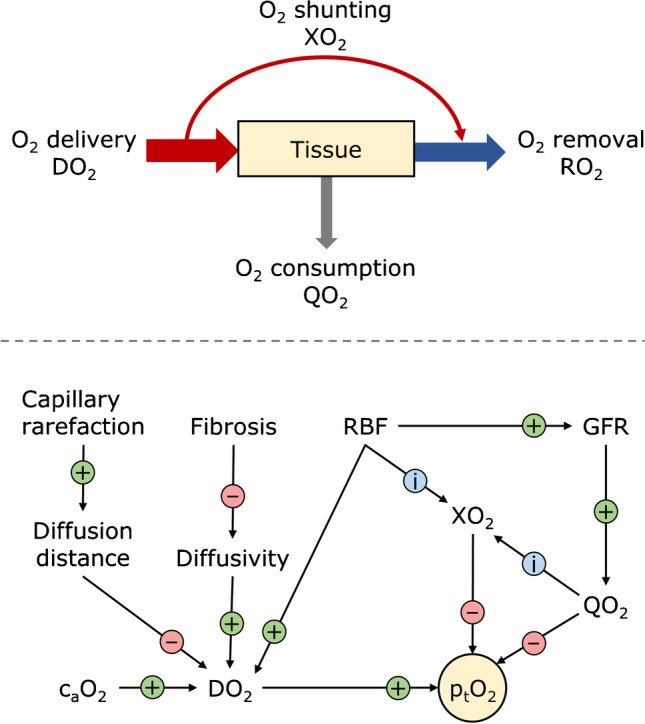
Key factors determining oxygen partial pressure in tissue. Top panel: Tissue oxygenation is a function of O2 delivery (DO2), consumption (QO2), and removal (RO2). About 10–15% of O2 delivered to the kidney is consumed under normal physiologic conditions. Not all renal tissues are supplied equally, which is, in part, due to arterial-to-venous oxygen shunting (XO2). O2 not consumed by the kidney is removed by venous efflux. Bottom panel: Tissue partial pressure of oxygen (ptO2) is dependent on factors that influence O2 delivery, consumption, and removal. Circled + and − signs indicate the effect of an increase in the factor upstream of the corresponding arrow on the parameter pointed by the arrowhead under the assumption that everything else remains the same. Circled i indicates that an increase in the respective factor will influence the indicated parameter. Only selected factors are shown. An increase in renal blood flow (RBF) increases DO2 and glomerular filtration rate (GFR), and may influence XO2. Whether XO2 increases is dependent on the location of the tissue under observation, among other factors. XO2 is also influenced by QO2. Since QO2 produces the arterial-to-venous pO2 gradients necessary for XO2, an increase in consumption will likely, but not necessarily, increase shunting; the local arrangement of O2 sinks and sources plays a role as well. More shunting leads to reduced ptO2. Increased GFR leads to higher QO2, as O2 demand for Na+ reabsorption increases, and thereby to reduced ptO2. DO2 increases with increased arterial blood oxygen concentration (caO2) and partial pressure. Capillary rarefaction and fibrosis reduce oxygen delivery to tissue due to increased diffusion distance and reduced diffusivity, respectively
Oxygen is removed by venous outflow. Not all O2 travels along the full length of the renal vasculature: O2 can be shunted radially from one vessel or vessel segment to another through adjacent tissue, provided that they are within diffusion distance of each other and that there is a driving pO2 gradient. This is the case in the vasa recta and in parts of the preglomerular vasculature. As blood flows through the descending vasa recta towards the inner medulla, O2 is delivered to the surrounding tissue, where it is, in part, consumed by the nephrons. The remaining O2 diffuses into the ascending vasa recta and is transported back in direction outer medulla. This shunting from descending to ascending vasa contributes to medullary hypoxia [42], in particular when reduced perfusion and/or elevated O2 consumption increase O2 gradients favorable to shunting, thereby further reducing O2 delivery to tissue [1]. Complicating matters, blood does not have to follow the full length of the vasa recta, but may take a shortcut from descending to ascending vasa recta through the peritubular capillary plexus. In general, the intricate three-dimensional vascular organization of the outer medulla adds a layer of complexity to the understanding of shunting mechanisms [43–45].
Regulation mechanisms
In other organs, the end-products of metabolism regulate O2 supply without affecting O2 consumption. In the kidney, since GFR determines the rate of sodium reabsorption (TNa), increases in RBF and GFR raise O2 consumption concomitantly with O2 supply, a positive feedback loop that is ill-suited to match O2 supply and demand (Fig. 2). However, two general mechanisms dissociate O2 consumption from O2 supply, namely, (i) the decoupling of GFR from RBF via differential modulation of pre- and postglomerular resistance, which alters the filtration fraction and (ii) changes in metabolic efficiency, as reflected by the Na+ transport-to-O2 consumption ratio (TNa/QO2) [46]. This ratio is altered by shifts in TNa to segments with higher/lower passive reabsorption capacity, changes in paracellular permeability, and variations in the efficiency of ATP production in mitochondria. We note that there is little evidence that the kidney purposely regulates TNa/QO2 in order to maintain kidney oxygenation when pO2 increases or decreases, such as during ischemia [47].
Fig. 2.
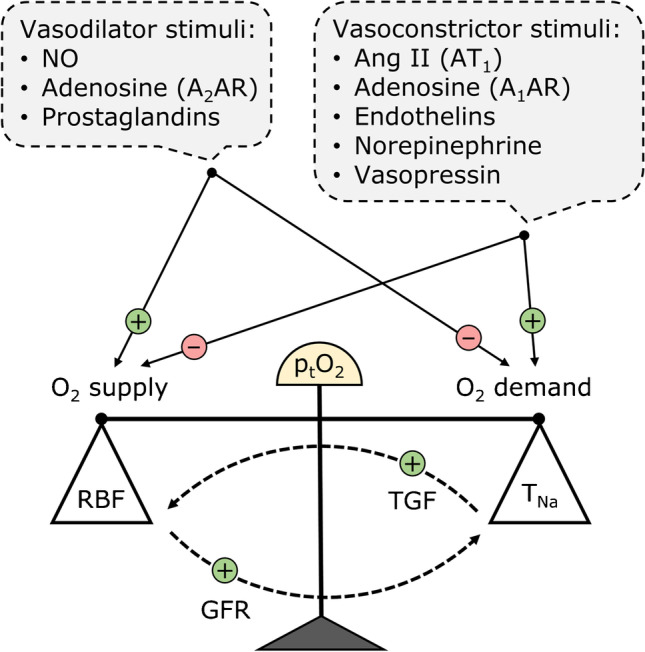
Regulation of tissue pO2 (ptO2) by neuro-hormonal agents. The vasoconstrictor factors that reduce RBF and O2 delivery (DO2) generally also stimulate sodium reabsorption (TNa) and increase O2 consumption (QO2). Conversely, the vasodilator factors that increase RBF and DO2 may also act to reduce TNa and QO2. However, these effects are blunted by two mechanisms: increasing RBF also raises GFR and therefore TNa, whereas increases in TNa in the proximal tubule and the ascending limb may reduce NaCl delivery to the macula densa and raise RBF via tubulo-glomerular feedback (TGF). Hence, the effectiveness of ptO2 regulation also depends on how neuro-hormonal agents modulate the coupling between RBF and GFR (i.e., by changing the filtration fraction), or between TNa and QO2 (i.e., by changing the metabolic efficiency of Na+ transport). These effects are not explicitly shown in the figure (see text). Not shown either are the synergistic and antagonistic effects between various neuro-hormonal agents
Many of the neuro-humoral factors that regulate blood flow and O2 supply also modulate the filtration fraction and TNa/QO2, in particular Ang II, NO, and adenosine. As described above, Ang II preferentially constricts efferent arterioles, which raises the filtration fraction and reduces O2 supply relative to O2 consumption, thereby lowering cortical pO2 [23]. Moreover, Ang II stimulates Na+ transporter abundance and activity, particularly in the distal nephron [48] where TNa/QO2 is lower, which also raises O2 consumption. Palm and colleagues recently demonstrated that activation of the renin-angiotensin-aldosterone system by a low Na+ diet increased QO2 and lowered cortical pO2 but raised medullary pO2 [49]; a similar low-salt diet-induced reversal of pO2 gradients was previously observed [50]. Of note, AT1 receptor blockade restored pO2 without reducing QO2, confirming the major impact of Ang II on renal O2 delivery [49].
Conversely, NO acts both to increase MBF and to lower QO2 by inhibiting distal Na+ transport and enhancing the efficiency of Na+ reabsorption. NO reduces NKCC2 activity in the mTAL [51], and it reduces TNa and vasopressin-stimulated water permeability in the collecting ducts; its effects in the proximal tubule remain controversial [52, 53]. Computational studies suggest that under basal conditions, the NO–induced decrease in QO2 and increase in O2 supply contribute to a similar extent to the maintenance of medullary pO2 [54]. In addition, NO inhibits respiration in renal tubules [55] by blocking complex IV in mitochondria. Accordingly, NO synthase inhibition reduces renal perfusion and preferentially decreases medullary pO2, both acutely and chronically [56].
Adenosine formation is activated by low ATP levels. A1AR activation stimulates Na+ and fluid reabsorption in the proximal tubule but inhibits TNa in the mTAL, thereby shifting O2 consumption towards the cortex where O2 is more plentiful [29]. In addition, A2AR-mediated enhancement of MBF increases O2 supply to the medulla. Thus, similar to NO, adenosine plays an important role in preserving medullary oxygenation [57].
Norepinephrine released by renal efferent nerves also modulates Na+ and water reabsorption, as well as intra-renal renin release [33]. Thus, renal sympathetic nerve activity likely also affects O2 consumption and TNa/QO2 in the kidney.
Impact of dysregulation
A shift in the balance of O2 delivery, consumption, and removal can be caused by acute and chronic pathologies, but may also lead to such. While in the healthy kidney all regions receive enough O2 to maintain their respective normal physiologic pO2, loss of cortical peritubular capillaries may critically reduce O2 delivery [58]. Together with the inherent increase in diffusion distances, such capillary rarefaction may cause hypoxia.
Hypoxia has been proposed as a key element in the development and progression of chronic kidney disease (CKD) [59]: Starting from an initial glomerular injury by any mechanism, postglomerular peritubular vascular insufficiency develops, leading to reduced blood flow and reduced O2 delivery to tissue. The ensuing hypoxia causes tubular injury and inflammation. This also impacts adjacent capillaries, damaging their respective glomeruli which had previously been unharmed. From there, the cycle repeats. However, conclusive evidence for such a causal link between hypoxia and CKD is lacking [60, 61], even though there are clinical indications for an association between hypoxia and CKD progression [62]. Hypoxia has also been proposed as one of the mechanisms that can lead to acute kidney injury (AKI), which is increasingly recognized as a risk factor for CKD [63]. At least for some forms of AKI, the link to hypoxia appears to be clearer than in the case of CKD: When a decrease in RBF occurs, e.g., due to surgical intervention, O2 delivery is temporarily reduced, putting particularly the mTAL with its high O2 demand and low medullary blood flow in danger of hypoxic damage [64]. In this case, the S3 segment of the proximal tubule is also at risk, although to a lesser extent. When both O2 delivery and transport function temporarily cease without hypothermia, as in warm ischemia reflow animal experiments, the S3 segment is primarily affected [65]. In contrast to the TAL, which can maintain itself by anaerobic glycolysis under such conditions, the proximal tubule is dependent on O2 [38].
In hypertension, a shift of Na+ reabsorption towards the energetically less efficient TAL appears to contribute, together with other factors, to reduced tissue pO2. In diabetes mellitus, O2 consumption is increased due to mitochondrial dysfunction, reduced Na+ reabsorption efficiency, and glomerular hyperfiltration [66, 67]. In general, conditions that lead to glomerular hyperfiltration have the potential to contribute to hypoxia if dysregulation ensues. An area where the contribution of dysregulation of blood flow and oxygenation is particularly difficult to quantify is the development of anemia in CKD. Renal EPO-producing (REP) cells are specialized tubulo-interstitial cells located, under normal conditions, at the cortico-medullary border where there is a steep spatial gradient in pO2. It is hypothesized that REP cells integrate, among other cues, information on O2 availability and consumption to regulate EPO [37]. As CKD develops, these cells stop producing EPO and dedifferentiate. However, to what extent changes in the spatial pO2 gradient at the cortico-medullary border may contribute remains unclear.
Hurdles in studying renal blood flow and oxygenation
As in other areas of physiology, kidney research has progressed in tandem with technological innovations. To advance our understanding of renal function, we are dependent on accurate measurements, ideally ones that can be carried out in vivo in a noninvasive fashion to minimize confounding factors. Limitations of available methods for measuring RBF and oxygenation constitute hurdles that must be overcome or bypassed before many of the open questions in this area can be answered. For instance, to understand the role of renal tissue oxygen gradients in the regulation of erythropoiesis, we need to accurately quantify local differences in pO2, which is currently not possible [68]. Likewise, to evaluate the role that changes in regional hemodynamics and tissue oxygenation play in renal disease and kidney injury, reliable, quantitative information on RBF and pO2 with high spatial resolution is needed. However, shortcomings of measurement methods, including the comparably low resolution of noninvasive approaches, and introduction of local perturbations by invasive techniques, leave us with sparse data.
Most questions are open not only because of limitations in measurement methods, but also due to a lack of tools that would enable the integration of complex data into a bigger picture. For example, studies in rats have shown that medullary pO2 may decrease during moderate to severe cortical ischemia even as MBF is maintained [69], possibly as the result of the complex architecture of the renal vasculature [1]. Yet to reliably quantify to what extent the regulation of cortical and medullary oxygenation is independent, a quantitative model is needed; mental representations are not sufficient. Such models could contribute to quantifying the relative contributions of vasa recta and juxtamedullary arterioles to the control of MBF. The use of computational methods in renal physiology is not new but has only spread slowly. For instance, such methods have been employed to investigate the role of MBF in pressure natriuresis, indicating that the modulation of interstitial composition by MBF strongly affects hydration status and the excretion of water, but not that of NaCl [70]. In contrast, observations in rats suggested that changes in medullary perfusion may modulate both sodium and water excretion and thereby alter effective circulating volume [19, 71]. However, recent findings do not support a key role of MBF in regulating blood pressure in humans [72, 73].
In the following sections, we discuss the capabilities and limitations of current methods for measuring and calculating RBF and renal oxygenation, differentiating between invasive, noninvasive, and computational approaches. While blood flow on the whole organ level lends itself well to noninvasive acquisitions, localized high-resolution measurements necessitate invasive methods. Renal blood flow or renal blood flow rate is measured in units of volume per time, e.g., milliliters or liters of blood per minute. In contrast, perfusion is a volume (or mass) averaged metric, representing the volume of blood supplied to a unit volume (or unit mass) of tissue per unit time. Blood velocity gives the direction and speed of blood at a specific location in units of distance traveled per time. When both blood velocity and vessel morphology are known, the blood flow rate can be calculated by spatial integration of the velocity field. The average blood velocity in a vessel is then defined as the blood flow rate through that vessel divided by the vessel’s cross-sectional area.
Methods for noninvasive measurements
Ultrasound and magnetic resonance imaging (MRI) are used clinically and in animal studies to acquire RBF data. Phase-contrast MRI (PC-MRI) can produce images with controlled sensitivity to flow. In combination with magnitude images to capture anatomy, it allows for the acquisition of spatially resolved blood velocity fields in larger vessels such as the renal artery [74]. While not inherently complicated to use, PC-MRI requires strict adherence to validated protocols for producing reliable results. In contrast to PC-MRI, arterial spin labeling (ASL) MRI can yield data on renal perfusion, i.e., distribution of arterial blood flow throughout the kidney, but with limited spatial resolution. Although ASL is not widely employed in the clinical setting, it is expected that this method will gain in importance [75]. For assessment of renal oxygenation, blood oxygenation level–dependent (BOLD) MRI can be used. It is currently the only clinically relevant noninvasive method for assessing renal oxygenation status. The BOLD signal is not only oxygen-dependent, but also influenced by factors such as hydration level, hematocrit, and pH value. Therefore, validated protocols for acquisition and data analysis must be strictly followed [76]. Oxygenation can also be measured with fluorine-19 MRI [77]. This method is employed in pre-clinical studies, requiring the application of perfluorinated hydrocarbons as contrast agents.
Doppler ultrasound is a widely available, inexpensive method for measuring blood velocity with high temporal resolution. However, it has two important shortcomings: Firstly, the measured velocity is dependent on the angle of insonation, which can neither be precisely controlled nor measured, as ultrasound transducers are generally hand-held and not registered to a reference coordinate frame. Secondly, the calculation of blood flow rate from the measured velocities is error prone, as it is generally based on the assumption of exclusively parallel flow along the longitudinal axis of the vessel. Consequently, traditional Doppler ultrasound measurements offer relatively low accuracy outside idealized settings [78]. Newer methods [79], e.g., ones based on a combination of high frame rate and speckle decorrelation [80], promise more reliable estimation of blood flow, but they have yet to be established in the clinic.
Methods for invasive measurements
A reliable and easy-to-use method for measuring total RBF is transit time ultrasound via intraoperative application or chronic implantation of an ultrasound probe [81]. For intraoperative measurements, electromagnetic probes can be employed as well. They require close contact with the blood vessel, which excludes chronic implantation due to possible vessel wall damage [82]. Microcirculatory perfusion can be assessed by laser Doppler flowmetry using fiber optic probes. However, the obtained signal only provides a relative measure of perfusion, as calibration is tissue-dependent [83]. Dilution indicator and microsphere methods for blood flow measurement have been largely displaced [84].
Renal oxygenation can be assessed with electrochemical sensors [85], luminescence-based optical sensors [86], intravital fluorescence and phosphorescence lifetime microscopy [87], photoacoustic microscopy [88], and electron paramagnetic resonance spectroscopy [89]. Of these, electrochemical and luminescence sensors are the most widely used in the clinical setting. The best-known electrochemical oxygen sensor is the Clark electrode. Formerly considered the reference standard, it is prone to drift and has limited accuracy at low pO2 due to oxygen consumption as the basis of its operating principle. Luminescence sensors do not show these issues. However, they have a larger sampling volume and are bigger than Clark microelectrodes, which may lead to more tissue damage [90]. Electron paramagnetic resonance spectroscopy requires the placement of a paramagnetic material in the kidney at the location of intended pO2 measurement. The actual measurements are noninvasive, allowing for longitudinal observations [89]. Intravital fluorescence and phosphorescence microscopy requires the use of oxygen-sensitive molecular probes. It yields spatially resolved data, but has lower accuracy and is less robust than electrochemical and luminescence sensor-based methods. Using fiber optic systems, fluorescence imaging is also possible in deep tissue [91].
Computational approaches
Where measurements do not suffice, mathematical or computational approaches based on first principles, i.e., on fundamental laws of physics, may fill some of the gaps. For instance, on the simplest level, the law of mass conservation dictates that the overall mass of blood supplied to the kidney has to be equal to the mass exiting the kidney as venous blood, urine, and lymph. Therefore, if arterial inflow and venous and urinary outflows can be measured, lymphatic outflow can be calculated. On a more complex level, global or coarse local measurements of RBF and renal tissue oxygenation can be used in conjunction with quantitative data on the renal anatomy to infer local perfusion and oxygen delivery values. Models of biochemical reactions and biophysical actions of, e.g., epithelial membrane transporters can be employed to estimate the local oxygen consumption rate. Combining both types of models allows, in principle, for the calculation of pO2 values throughout the kidney.
For an overview of modeling of renal oxygenation, including a general introduction to the application of computational models in renal pathophysiology, we refer the reader to the recent review of Evans and co-workers [92]. Here, we give two examples of how computational models have helped fill in gaps not accessible by measurements. The first one pertains to the question of whether preglomerular arterial-to-venous oxygen shunting plays a role in the oxygenation of renal tissue. This question cannot be answered by direct measurement, as it would require acquisition of oxygen fluxes along the full length of pairs of arterial and venous vessels. In a series of computational models with increasing complexity [93–96], the conclusion was reached that preglomerular oxygen shunting is negligible under normal physiologic conditions, but may have to be accounted for in specific situations, e.g., during ischemia [97]. The second example addresses the question of how enough oxygen can reach the papilla given the countercurrent architecture. A model representing the detailed three-dimensional structural organization of the outer medulla suggested that the segregation of vasa recta within vascular bundles limits O2 escape from descending vasa recta in the inner stripe and helps to preserve O2 delivery to the inner medulla [98, 99].
The main limitations of current computational models of renal blood flow and oxygenation are related to the multiple length and time scales of kinetic, transport and regulation processes, the sparsity of available quantitative data to characterize these processes, the complex spatial architecture of the kidney, and the high computational power needed to run three-dimensional, time-resolved, multiscale models. To capture time-dependent processes, the equations describing the modeled system are commonly solved in a stepwise fashion, where each step moves the model forward in time, and requires a quantum of computer power. Processes at the cellular level tend to be fast, requiring small time steps to be captured adequately. At organ scale, process cycles tend to be longer and characteristic time constants larger, allowing for the use of bigger time steps. When both scales are modeled concurrently, the computational power needed to compute long cycles with small time steps can exceed the available resources by orders of magnitude. For a discussion of further challenges of and progress in multi-scale modeling, we refer to reader to the review by Auffray and co-workers [100].
Adequately representing the complex organization of vessels and tubules also remains challenging. In addition to the intricate vascular geometry described above, recent studies based on optical clearing methods have revealed great variability in the position, size, shape, and tortuosity of nephrons [101]. Representing each individual structure is prohibitively expensive from a computational perspective, but selecting representative populations that preserve the key features of this anatomical arrangement is difficult, particularly given that the functional significance of this variability remains to be fully understood.
Added to this structural complexity is the complexity of biological processes that determine the local pO2 at any given moment in time. A complete model of renal oxygenation may need to accurately represent O2 dissociation from hemoglobin, which depends on the acid-base environment, as well as O2 diffusion across multiple barriers, the permeabilities of which remain poorly characterized. It should also take into account the efficiency of O2 utilization by mitochondria together with ATP-consuming processes other than active transport, all of which are strongly modulated by hormones, neural signals, and local factors. Finally, it should also account for physiologic differences due to sex as well as age-induced changes.
The high number of structures and processes involved in kidney function raise the question whether machine learning approaches such as deep neural networks might not be better suited for modeling renal blood flow and oxygenation than conventional methods. The two main problems associated with machine learning approaches are that they require large amounts of data for training purposes and that they reproduce, rather than provide, functional insight into the modeled system. A new class of neural networks—physics [102], biology [103], or otherwise “informed” neural networks—seeks to merge conventional methods for physiological modeling with machine learning. It is too early to say what impact it will have on kidney research.
Conclusion
Renal function is both dependent on and the cause of regional variations in renal blood flow and oxygenation. A multitude of interactions between neural, endocrine, paracrine, and autocrine factors work in concert to regulate RBF and pO2. To advance our understanding of the interplay between RBF, tissue oxygen, and renal function, spatially resolved measurements are necessary. Currently available measurement methods have limitations in that they introduce confounding factors, have low resolution, or cannot reach deep tissue. Major breakthroughs in this area will likely require hybrid approaches with both experimental methods and computational modeling tools.
Funding
Open access funding provided by University of Zurich. Financial support for this work was provided by the Swiss National Science Foundation through NCCR Kidney.CH (to VK) and project 205321_153523 (to VK). The authors have no relevant financial or non-financial interests to disclose.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
This article is part of the special issue on Kidney Control of Homeostasis in Pflügers Archiv—European Journal of Physiology
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.O'Connor PM. Renal oxygen delivery: matching delivery to metabolic demand. Clin Exp Pharmacol Physiol. 2006;33:961–967. doi: 10.1111/j.1440-1681.2006.04475.x. [DOI] [PubMed] [Google Scholar]
- 2.Arendshorst WJ. Autoregulation of renal blood flow in spontaneously hypertensive rats. Circ Res. 1979;44:344–349. doi: 10.1161/01.res.44.3.344. [DOI] [PubMed] [Google Scholar]
- 3.Arendshorst WJ, Finn WF, Gottschalk CW. Autoregulation of blood flow in the rat kidney. Am J Physiol. 1975;228:127–133. doi: 10.1152/ajplegacy.1975.228.1.127. [DOI] [PubMed] [Google Scholar]
- 4.Damkjaer M, Vafaee M, Moller ML, Braad PE, Petersen H, Hoilund-Carlsen PF, Bie P. Renal cortical and medullary blood flow responses to altered NO availability in humans. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1449–R1455. doi: 10.1152/ajpregu.00440.2010. [DOI] [PubMed] [Google Scholar]
- 5.Pallone TL, Robertson CR, Jamison RL. Renal medullary microcirculation. Physiol Rev. 1990;70:885–920. doi: 10.1152/physrev.1990.70.3.885. [DOI] [PubMed] [Google Scholar]
- 6.Siu KL, Sung B, Cupples WA, Moore LC, Chon KH. Detection of low-frequency oscillations in renal blood flow. Am J Physiol Renal Physiol. 2009;297:F155–F162. doi: 10.1152/ajprenal.00114.2009. [DOI] [PubMed] [Google Scholar]
- 7.Romero CA, Carretero OA. A novel mechanism of renal microcirculation regulation: connecting tubule-glomerular feedback. Curr Hypertens Rep. 2019;21:8. doi: 10.1007/s11906-019-0911-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Just A. Mechanisms of renal blood flow autoregulation: dynamics and contributions. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1–17. doi: 10.1152/ajpregu.00332.2006. [DOI] [PubMed] [Google Scholar]
- 9.Liu R, Layton AT. Modeling the effects of positive and negative feedback in kidney blood flow control. Math Biosci. 2016;276:8–18. doi: 10.1016/j.mbs.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sgouralis I, Layton AT. Mathematical modeling of renal hemodynamics in physiology and pathophysiology. Math Biosci. 2015;264:8–20. doi: 10.1016/j.mbs.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balasubramanian L, Ahmed A, Lo CM, Sham JS, Yip KP. Integrin-mediated mechanotransduction in renal vascular smooth muscle cells: activation of calcium sparks. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1586–R1594. doi: 10.1152/ajpregu.00025.2007. [DOI] [PubMed] [Google Scholar]
- 12.Grifoni SC, Chiposi R, McKey SE, Ryan MJ, Drummond HA. Altered whole kidney blood flow autoregulation in a mouse model of reduced beta-ENaC. Am J Physiol Renal Physiol. 2010;298:F285–F292. doi: 10.1152/ajprenal.00496.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlstrom M, Wilcox CS, Arendshorst WJ. Renal autoregulation in health and disease. Physiol Rev. 2015;95:405–511. doi: 10.1152/physrev.00042.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volker V. Tubuloglomerular feedback and the control of glomerular filtration rate. Physiology. 2003;18:169–174. doi: 10.1152/nips.01442.2003. [DOI] [PubMed] [Google Scholar]
- 15.Ren Y, Garvin JL, Liu R, Carretero OA. Crosstalk between the connecting tubule and the afferent arteriole regulates renal microcirculation. Kidney Int. 2007;71:1116–1121. doi: 10.1038/sj.ki.5002190. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, D'Ambrosio MA, Garvin JL, Ren Y, Carretero OA. Connecting tubule glomerular feedback in hypertension. Hypertension. 2013;62:738–745. doi: 10.1161/HYPERTENSIONAHA.113.01846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Miguel C, Speed JS, Kasztan M, Gohar EY, Pollock DM. Endothelin-1 and the kidney: new perspectives and recent findings. Curr Opin Nephrol Hypertens. 2016;25:35–41. doi: 10.1097/MNH.0000000000000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cowley AW Jr (2000) Control of the renal medullary circulation by vasopressin V1 and V2 receptors in the rat. Exp Physiol 85 Spec No:223S-231S. 10.1111/j.1469-445x.2000.tb00027.x [DOI] [PubMed]
- 19.Cowley AW, Jr, Abe M, Mori T, O'Connor PM, Ohsaki Y, Zheleznova NN. Reactive oxygen species as important determinants of medullary flow, sodium excretion, and hypertension. Am J Physiol Renal Physiol. 2015;308:F179–F197. doi: 10.1152/ajprenal.00455.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hao CM, Breyer MD. Physiological regulation of prostaglandins in the kidney. Annu Rev Physiol. 2008;70:357–377. doi: 10.1146/annurev.physiol.70.113006.100614. [DOI] [PubMed] [Google Scholar]
- 21.Rhaleb NE, Yang XP, Carretero OA. The kallikrein-kinin system as a regulator of cardiovascular and renal function. Compr Physiol. 2011;1:971–993. doi: 10.1002/cphy.c100053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denton KM, Anderson WP, Sinniah R. Effects of angiotensin II on regional afferent and efferent arteriole dimensions and the glomerular pole. Am J Physiol Regul Integr Comp Physiol. 2000;279:R629–R638. doi: 10.1152/ajpregu.2000.279.2.R629. [DOI] [PubMed] [Google Scholar]
- 23.Emans TW, Janssen BJ, Pinkham MI, Ow CP, Evans RG, Joles JA, Malpas SC, Krediet CT, Koeners MP. Exogenous and endogenous angiotensin-II decrease renal cortical oxygen tension in conscious rats by limiting renal blood flow. J Physiol. 2016;594:6287–6300. doi: 10.1113/JP270731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans RG, Head GA, Eppel GA, Burke SL, Rajapakse NW. Angiotensin II and neurohumoral control of the renal medullary circulation. Clin Exp Pharmacol Physiol. 2010;37:e58–e69. doi: 10.1111/j.1440-1681.2009.05233.x. [DOI] [PubMed] [Google Scholar]
- 25.Zou AP, Cowley AW., Jr Nitric oxide in renal cortex and medulla. An in vivo microdialysis study. Hypertension. 1997;29:194–198. doi: 10.1161/01.hyp.29.1.194. [DOI] [PubMed] [Google Scholar]
- 26.Cowley AW, Jr, Mori T, Mattson D, Zou AP. Role of renal NO production in the regulation of medullary blood flow. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1355–R1369. doi: 10.1152/ajpregu.00701.2002. [DOI] [PubMed] [Google Scholar]
- 27.Dickhout JG, Mori T, Cowley AW., Jr Tubulovascular nitric oxide crosstalk: buffering of angiotensin II-induced medullary vasoconstriction. Circ Res. 2002;91:487–493. doi: 10.1161/01.res.0000035243.66189.92. [DOI] [PubMed] [Google Scholar]
- 28.Zou AP, Nithipatikom K, Li PL, Cowley AW., Jr Role of renal medullary adenosine in the control of blood flow and sodium excretion. Am J Physiol. 1999;276:R790–R798. doi: 10.1152/ajpregu.1999.276.3.R790. [DOI] [PubMed] [Google Scholar]
- 29.Vallon V, Osswald H (2009) Adenosine receptors and the kidney. Handb Exp Pharmacol:443–470. 10.1007/978-3-540-89615-9-15 [DOI] [PMC free article] [PubMed]
- 30.Beach RE, Watts BA, 3rd, Good DW, Benedict CR, DuBose TD., Jr Effects of graded oxygen tension on adenosine release by renal medullary and thick ascending limb suspensions. Kidney Int. 1991;39:836–842. doi: 10.1038/ki.1991.105. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy-Lydon TM, Crawford C, Wildman SS, Peppiatt-Wildman CM. Renal pericytes: regulators of medullary blood flow. Acta Physiol (Oxf) 2013;207:212–225. doi: 10.1111/apha.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frame AA, Carmichael CY, Wainford RD. Renal afferents. Curr Hypertens Rep. 2016;18:69. doi: 10.1007/s11906-016-0676-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sata Y, Head GA, Denton K, May CN, Schlaich MP (2018) Role of the sympathetic nervous system and its modulation in renal hypertension. Front Med 510.3389/fmed.2018.00082 [DOI] [PMC free article] [PubMed]
- 34.Crawford C, Wildman SS, Kelly MC, Kennedy-Lydon TM, Peppiatt-Wildman CM. Sympathetic nerve-derived ATP regulates renal medullary vasa recta diameter via pericyte cells: a role for regulating medullary blood flow? Front Physiol. 2013;4:307. doi: 10.3389/fphys.2013.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans RG, Eppel GA, Anderson WP, Denton KM. Mechanisms underlying the differential control of blood flow in the renal medulla and cortex. J Hypertension. 2004;22:1439–1451. doi: 10.1097/01.hjh.0000133744.85490.9d. [DOI] [PubMed] [Google Scholar]
- 36.Smith HW, Chasis H, Goldring W, Ranges HA. Glomerular dynamics in the normal human kidney. J Clin Invest. 1940;19:751–764. doi: 10.1172/JCI101180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nolan KA, Wenger RH. Source and microenvironmental regulation of erythropoietin in the kidney. Curr Opin Nephrol Hypertens. 2018;27:277–282. doi: 10.1097/MNH.0000000000000420. [DOI] [PubMed] [Google Scholar]
- 38.Brezis M, Rosen S. Mechanisms of disease - hypoxia of the renal medulla - its implications for disease. New England J Med. 1995;332:647–655. doi: 10.1056/Nejm199503093321006. [DOI] [PubMed] [Google Scholar]
- 39.Soltoff SP. ATP and the regulation of renal cell function. Annu Rev Physiol. 1986;48:9–31. doi: 10.1146/annurev.ph.48.030186.000301. [DOI] [PubMed] [Google Scholar]
- 40.Udwan K, Abed A, Roth I, Dizin E, Maillard M, Bettoni C, Loffing J, Wagner CA, Edwards A, Feraille E. Dietary sodium induces a redistribution of the tubular metabolic workload. J Physiol. 2017;595:6905–6922. doi: 10.1113/JP274927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vallon V, Thomson SC. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat Rev Nephrol. 2020;16:317–336. doi: 10.1038/s41581-020-0256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Edwards A. Oxygen transport across vasa recta in the renal medulla. Am J Physiol Heart Circ Physiol. 2002;283:H1042–H1055. doi: 10.1152/ajpheart.00074.2002. [DOI] [PubMed] [Google Scholar]
- 43.Kuo W, Le NA, Spingler B, Wenger RH, Kipar A, Hetzel U, Schulz G, Muller B, Kurtcuoglu V. Simultaneous three-dimensional vascular and tubular imaging of whole mouse kidneys with X-ray muCT. Microsc Microanal. 2020;26:731–740. doi: 10.1017/S1431927620001725. [DOI] [PubMed] [Google Scholar]
- 44.Kuo W, Rossinelli D, Schulz G, Wenger RH, Hieber S, Müller B, Kurtcuoglu V (2021) Terabyte-scale supervised 3D training and benchmarking dataset of the mouse kidney. arXiv preprint 2108.02226, https://arxiv.org/abs/2108.02226 [DOI] [PMC free article] [PubMed]
- 45.Zdora MC, Thibault P, Kuo W, Fernandez V, Deyhle H, Vila-Comamala J, Olbinado MP, Rack A, Lackie PM, Katsamenis OL, Lawson MJ, Kurtcuoglu V, Rau C, Pfeiffer F, Zanette I. X-ray phase tomography with near-field speckles for three-dimensional virtual histology. Optica. 2020;7:1221–1227. doi: 10.1364/Optica.399421. [DOI] [Google Scholar]
- 46.Singh P, Thompson SC, McDonough AA. Metabolic basis of solute transport. In: Glenn Chertow GM, Luyckx VA, Marsden PA, Skorecki K, Taal MW, Yu AS, Brenner BM, editors. Brenner and Rector's The Kidney. Philadelphia: Elsevier; 2019. pp. 133–155. [Google Scholar]
- 47.Evans RG, Eppel GA, Michaels S, Burke SL, Nematbakhsh M, Head GA, Carroll JF, O'Connor PM. Multiple mechanisms act to maintain kidney oxygenation during renal ischemia in anesthetized rabbits. Am J Physiol Renal Physiol. 2010;298:F1235–F1243. doi: 10.1152/ajprenal.00647.2009. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen MT, Lee DH, Delpire E, McDonough AA. Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol. 2015;305:F510–F519. doi: 10.1152/ajprenal.00183.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patinha D, Carvalho C, Persson P, Pihl L, Fasching A, Friederich-Persson M, O'Neill J, Palm F. Determinants of renal oxygen metabolism during low Na(+) diet: effect of angiotensin II AT1 and aldosterone receptor blockade. J Physiol. 2020;598:5573–5587. doi: 10.1113/JP280481. [DOI] [PubMed] [Google Scholar]
- 50.Stillman IE, Brezis M, Heyman SN, Epstein FH, Spokes K, Rosen S. Effects of salt depletion on the kidney: changes in medullary oxygenation and thick ascending limb size. J Am Soc Nephrol. 1994;4:1538–1545. doi: 10.1681/ASN.V481538. [DOI] [PubMed] [Google Scholar]
- 51.Herrera M, Ortiz PA, Garvin JL. Regulation of thick ascending limb transport: role of nitric oxide. Am J Physiol Renal Physiol. 2006;290:F1279–F1284. doi: 10.1152/ajprenal.00465.2005. [DOI] [PubMed] [Google Scholar]
- 52.Ortiz PA, Garvin JL. Role of nitric oxide in the regulation of nephron transport. Am J Physiol Renal Physiol. 2002;282:F777–F784. doi: 10.1152/ajprenal.00334.2001. [DOI] [PubMed] [Google Scholar]
- 53.Satoh N, Nakamura M, Suzuki A, Tsukada H, Horita S, Suzuki M, Moriya K, Seki G. Effects of nitric oxide on renal proximal tubular Na(+) transport. Biomed Res Int. 2017;2017:6871081. doi: 10.1155/2017/6871081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fry BC, Edwards A, Layton AT. Impact of nitric-oxide-mediated vasodilation and oxidative stress on renal medullary oxygenation: a modeling study. Am J Physiol Renal Physiol. 2016;310:F237–F247. doi: 10.1152/ajprenal.00334.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koivisto A, Pittner J, Froelich M, Persson AE. Oxygen-dependent inhibition of respiration in isolated renal tubules by nitric oxide. Kidney Int. 1999;55:2368–2375. doi: 10.1046/j.1523-1755.1999.00474.x. [DOI] [PubMed] [Google Scholar]
- 56.Emans TW, Janssen BJ, Joles JA, Krediet CTP. Nitric oxide synthase inhibition induces renal medullary hypoxia in conscious rats. J Am Heart Assoc. 2018;7:e009501. doi: 10.1161/JAHA.118.009501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dinour D, Brezis M. Effects of adenosine on intrarenal oxygenation. Am J Physiol. 1991;261:F787–F791. doi: 10.1152/ajprenal.1991.261.5.F787. [DOI] [PubMed] [Google Scholar]
- 58.Evans RG, Smith DW, Lee CJ, Ngo JP, Gardiner BS (2020) What makes the kidney susceptible to hypoxia? Anatomical record-advances in integrative anatomy and evolutionary biology 303:2544–2552. 10.1002/ar.24260 [DOI] [PubMed]
- 59.Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics. Kidney Int. 2008;74:867–872. doi: 10.1038/ki.2008.350. [DOI] [PubMed] [Google Scholar]
- 60.Faivre A, Scholz CC, de Seigneux S. Hypoxia in chronic kidney disease: towards a paradigm shift? Nephrol Dial Transplant. 2021;36:1782–1790. doi: 10.1093/ndt/gfaa091. [DOI] [PubMed] [Google Scholar]
- 61.Ow CPC, Ngo JP, Ullah MM, Hilliard LM, Evans RG. Renal hypoxia in kidney disease: cause or consequence? Acta Physiol (Oxf) 2018;222:e12999. doi: 10.1111/apha.12999. [DOI] [PubMed] [Google Scholar]
- 62.Zhou H, Yang M, Jiang Z, Ding J, Di J, Cui L. Renal hypoxia: an important prognostic marker in patients with chronic kidney disease. Am J Nephrol. 2018;48:46–55. doi: 10.1159/000491551. [DOI] [PubMed] [Google Scholar]
- 63.Kurzhagen JT, Dellepiane S, Cantaluppi V, Rabb H. AKI: an increasingly recognized risk factor for CKD development and progression. J Nephrol. 2020;33:1171–1187. doi: 10.1007/s40620-020-00793-2. [DOI] [PubMed] [Google Scholar]
- 64.Heyman SN, Gorelik Y, Zorbavel D, Rosenberger C, Abassi Z, Rosen S, Khamaisi M. Near-drowning: new perspectives for human hypoxic acute kidney injury. Nephrol Dial Transplant. 2020;35:206–212. doi: 10.1093/ndt/gfz016. [DOI] [PubMed] [Google Scholar]
- 65.Heyman SN, Rosenberger C, Rosen S. Experimental ischemia-reperfusion: biases and myths-the proximal vs. distal hypoxic tubular injury debate revisited. Kidney Int. 2010;77:9–16. doi: 10.1038/ki.2009.347. [DOI] [PubMed] [Google Scholar]
- 66.Hansell P, Welch WJ, Blantz RC, Palm F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin Exp Pharmacol Physiol. 2013;40:123–137. doi: 10.1111/1440-1681.12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hesp AC, Schaub JA, Prasad PV, Vallon V, Laverman GD, Bjornstad P, van Raalte DH. The role of renal hypoxia in the pathogenesis of diabetic kidney disease: a promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int. 2020;98:579–589. doi: 10.1016/j.kint.2020.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wenger RH, Kurtcuoglu V, Scholz CC, Marti HH, Hoogewijs D. Frequently asked questions in hypoxia research. Hypoxia (Auckl) 2015;3:35–43. doi: 10.2147/HP.S92198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O'Connor PM, Kett MM, Anderson WP, Evans RG. Renal medullary tissue oxygenation is dependent on both cortical and medullary blood flow. Am J Physiol Renal Physiol. 2006;290:F688–F694. doi: 10.1152/ajprenal.00275.2005. [DOI] [PubMed] [Google Scholar]
- 70.Moss R, Layton AT. Dominant factors that govern pressure natriuresis in diuresis and antidiuresis: a mathematical model. Am J Physiol Renal Physiol. 2014;306:F952–F969. doi: 10.1152/ajprenal.00500.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mattson DL. Importance of the renal medullary circulation in the control of sodium excretion and blood pressure. Am J Physiol Regul Integr Comp Physiol. 2003;284:R13–R27. doi: 10.1152/ajpregu.00321.2002. [DOI] [PubMed] [Google Scholar]
- 72.Assersen KB, Hoilund-Carlsen PF, Olsen MH, Greve SV, Gam-Hadberg JC, Braad PE, Damkjaer M, Bie P. The exaggerated natriuresis of essential hypertension occurs independently of changes in renal medullary blood flow. Acta Physiol (Oxf) 2019;226:e13266. doi: 10.1111/apha.13266. [DOI] [PubMed] [Google Scholar]
- 73.Sadowski J, Badzynska B. Altered renal medullary blood flow: a key factor or a parallel event in control of sodium excretion and blood pressure? Clin Exp Pharmacol Physiol. 2020;47:1323–1332. doi: 10.1111/1440-1681.13303. [DOI] [PubMed] [Google Scholar]
- 74.Villa G, Ringgaard S, Hermann I, Noble R, Brambilla P, Khatir DS, Zollner FG, Francis ST, Selby NM, Remuzzi A, Caroli A. Phase-contrast magnetic resonance imaging to assess renal perfusion: a systematic review and statement paper. MAGMA. 2020;33:3–21. doi: 10.1007/s10334-019-00772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Odudu A, Nery F, Harteveld AA, Evans RG, Pendse D, Buchanan CE, Francis ST, Fernandez-Seara MA. Arterial spin labelling MRI to measure renal perfusion: a systematic review and statement paper. Nephrol Dial Transplant. 2018;33:ii15–ii21. doi: 10.1093/ndt/gfy180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pruijm M, Mendichovszky IA, Liss P, Van der Niepen P, Textor SC, Lerman LO, Krediet CTP, Caroli A, Burnier M, Prasad PV. Renal blood oxygenation level-dependent magnetic resonance imaging to measure renal tissue oxygenation: a statement paper and systematic review. Nephrol Dial Transplant. 2018;33:ii22–ii28. doi: 10.1093/ndt/gfy243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu S, Shah SJ, Wilmes LJ, Feiner J, Kodibagkar VD, Wendland MF, Mason RP, Hylton N, Hopf HW, Rollins MD. Quantitative tissue oxygen measurement in multiple organs using 19F MRI in a rat model. Magn Reson Med. 2011;66:1722–1730. doi: 10.1002/mrm.22968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wan L, Yang N, Hiew CY, Schelleman A, Johnson L, May C, Bellomo R. An assessment of the accuracy of renal blood flow estimation by Doppler ultrasound. Intensive Care Med. 2008;34:1503–1510. doi: 10.1007/s00134-008-1106-8. [DOI] [PubMed] [Google Scholar]
- 79.Zhou X, Zhou X, Leow CH, Tang MX. Measurement of flow volume in the presence of reverse flow with ultrasound speckle decorrelation. Ultrasound Med Biol. 2019;45:3056–3066. doi: 10.1016/j.ultrasmedbio.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tang J, Postnov DD, Kilic K, Erdener SE, Lee B, Giblin JT, Szabo TL, Boas DA. Functional ultrasound speckle decorrelation-based velocimetry of the brain. Adv Sci (Weinh) 2020;7:2001044. doi: 10.1002/advs.202001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schoenberg SO, Bock M, Kallinowski F, Just A. Correlation of hemodynamic impact and morphologic degree of renal artery stenosis in a canine model. J Am Soc Nephrol. 2000;11:2190–2198. doi: 10.1681/ASN.V11122190. [DOI] [PubMed] [Google Scholar]
- 82.Hartman JC, Olszanski DA, Hullinger TG, Brunden MN. In vivo validation of a transit-time ultrasonic volume flow meter. J Pharmacol Toxicol Methods. 1994;31:153–160. doi: 10.1016/1056-8719(94)90078-7. [DOI] [PubMed] [Google Scholar]
- 83.Rajan V, Varghese B, van Leeuwen TG, Steenbergen W. Review of methodological developments in laser Doppler flowmetry. Lasers Med Sci. 2009;24:269–283. doi: 10.1007/s10103-007-0524-0. [DOI] [PubMed] [Google Scholar]
- 84.Young LS, Regan MC, Barry MK, Geraghty JG, Fitzpatrick JM. Methods of renal blood flow measurement. Urol Res. 1996;24:149–160. doi: 10.1007/BF00304078. [DOI] [PubMed] [Google Scholar]
- 85.Li CM, Dong H, Cao X, Luong JH, Zhang X. Implantable electrochemical sensors for biomedical and clinical applications: progress, problems, and future possibilities. Curr Med Chem. 2007;14:937–951. doi: 10.2174/092986707780362970. [DOI] [PubMed] [Google Scholar]
- 86.Wolfbeis OS. Luminescent sensing and imaging of oxygen: fierce competition to the Clark electrode. Bioessays. 2015;37:921–928. doi: 10.1002/bies.201500002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mik EG, van Leeuwen TG, Raat NJ, Ince C. Quantitative determination of localized tissue oxygen concentration in vivo by two-photon excitation phosphorescence lifetime measurements. J Appl Physiol. 2004;97(1985):1962–1969. doi: 10.1152/japplphysiol.01399.2003. [DOI] [PubMed] [Google Scholar]
- 88.Sun N, Zheng S, Rosin DL, Poudel N, Yao J, Perry HM, Cao R, Okusa MD, Hu S. Development of a photoacoustic microscopy technique to assess peritubular capillary function and oxygen metabolism in the mouse kidney. Kidney Int. 2021;100:613–620. doi: 10.1016/j.kint.2021.06.018. [DOI] [PubMed] [Google Scholar]
- 89.Khan N, Williams BB, Hou H, Li H, Swartz HM. Repetitive tissue pO2 measurements by electron paramagnetic resonance oximetry: current status and future potential for experimental and clinical studies. Antioxid Redox Signal. 2007;9:1169–1182. doi: 10.1089/ars.2007.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Leong CL, O'Connor PM, Eppel GA, Anderson WP, Evans RG. Measurement of renal tissue oxygen tension: systematic differences between fluorescence optode and microelectrode recordings in anaesthetized rabbits. Nephron Physiol. 2008;108:p11–p17. doi: 10.1159/000114203. [DOI] [PubMed] [Google Scholar]
- 91.Flusberg BA, Cocker ED, Piyawattanametha W, Jung JC, Cheung EL, Schnitzer MJ. Fiber-optic fluorescence imaging. Nat Methods. 2005;2:941–950. doi: 10.1038/nmeth820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gardiner BS, Smith DW, Lee CJ, Ngo JP, Evans RG. Renal oxygenation: from data to insight. Acta Physiol (Oxf) 2020;228:e13450. doi: 10.1111/apha.13450. [DOI] [PubMed] [Google Scholar]
- 93.Gardiner BS, Smith DW, O'Connor PM, Evans RG. A mathematical model of diffusional shunting of oxygen from arteries to veins in the kidney. Am J Physiol Renal Physiol. 2011;300:F1339–F1352. doi: 10.1152/ajprenal.00544.2010. [DOI] [PubMed] [Google Scholar]
- 94.Gardiner BS, Thompson SL, Ngo JP, Smith DW, Abdelkader A, Broughton BR, Bertram JF, Evans RG. Diffusive oxygen shunting between vessels in the preglomerular renal vasculature: anatomic observations and computational modeling. Am J Physiol Renal Physiol. 2012;303:F605–F618. doi: 10.1152/ajprenal.00186.2012. [DOI] [PubMed] [Google Scholar]
- 95.Olgac U, Kurtcuoglu V. Renal oxygenation: preglomerular vasculature is an unlikely contributor to renal oxygen shunting. Am J Physiol Renal Physiol. 2015;308:F671–F688. doi: 10.1152/ajprenal.00551.2014. [DOI] [PubMed] [Google Scholar]
- 96.Olgac U, Kurtcuoglu V. The Bohr effect is not a likely promoter of renal preglomerular oxygen shunting. Front Physiol. 2016;7:482. doi: 10.3389/fphys.2016.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee CJ, Gardiner BS, Ngo JP, Kar S, Evans RG, Smith DW. Accounting for oxygen in the renal cortex: a computational study of factors that predispose the cortex to hypoxia. Am J Physiol Renal Physiol. 2017;313:F218–F236. doi: 10.1152/ajprenal.00657.2016. [DOI] [PubMed] [Google Scholar]
- 98.Chen J, Layton AT, Edwards A. A mathematical model of O-2 transport in the rat outer medulla. I. Model formulation and baseline results. Am J Physiol-Renal Physiol. 2009;297:F517–F536. doi: 10.1152/ajprenal.90496.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fry BC, Edwards A, Sgouralis I, Layton AT. Impact of renal medullary three-dimensional architecture on oxygen transport. Am J Physiol Renal Physiol. 2014;307:F263–F272. doi: 10.1152/ajprenal.00149.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Auffray C, Noble D, Nottale L, Turner P. Erratum to: progress in integrative systems biology, physiology and medicine: towards a scale-relative biology. Eur Phys J A. 2020;56:170. doi: 10.1140/epja/s10050-020-00137-5. [DOI] [Google Scholar]
- 101.Blanc T, Goudin N, Zaidan M, Traore MG, Bienaime F, Turinsky L, Garbay S, Nguyen C, Burtin M, Friedlander G, Terzi F, Pontoglio M. Three-dimensional architecture of nephrons in the normal and cystic kidney. Kidney Int. 2021;99:632–645. doi: 10.1016/j.kint.2020.09.032. [DOI] [PubMed] [Google Scholar]
- 102.Raissi M, Perdikaris P, Karniadakis GE. Physics-informed neural networks: a deep learning framework for solving forward and inverse problems involving nonlinear partial differential equations. J Computational Phys. 2019;378:686–707. doi: 10.1016/j.jcp.2018.10.045. [DOI] [Google Scholar]
- 103.Lagergren JH, Nardini JT, Baker RE, Simpson MJ, Flores KB. Biologically-informed neural networks guide mechanistic modeling from sparse experimental data. PLOS Computational biology. 2020;16:e1008462. doi: 10.1371/journal.pcbi.1008462. [DOI] [PMC free article] [PubMed] [Google Scholar]