

Abstract
Recent advances in analytical techniques provide the opportunity to quantify even low-abundance glycopeptides derived from complex biological mixtures, allowing for the identification of glycosylation differences between healthy samples and those derived from disease states. Herein, we discuss the sample preparation procedures and the mass spectrometry (MS) strategies that have facilitated glycopeptide quantification, as well as the standards used for glycopeptide quantification. For sample preparation, various glycopeptide enrichment methods are summarized including the columns used for glycopeptide separation in liquid chromatography separation. For mass spectrometry analysis strategies, MS1 level-based quantification and MS2 level-based quantification are described, either with or without labeling, where we have covered isotope labeling, TMT/iTRAQ labeling, data dependent acquisition, data independent acquisition, multiple reaction monitoring, and parallel reaction monitoring. The strengths and weaknesses of these methods are compared, particularly those associated with the figures of merit that are important for clinical biomarker studies and the pathological and functional studies of glycoproteins in various diseases. Possible future developments for glycopeptide quantification are discussed.
I. INTRODUCTION
Glycosylation, a non-template driven process, is one of the most abundant post-translational modification of proteins, which is common to all eukaryotes (Schjoldager et al., 2020). About 50%-70% of human proteins are glycosylated with two main types: N-linked glycosylation and O-linked glycosylation (An et al., 2009; Varki, 2017) (Figure 1). The aberrant glycosylation of proteins has been shown to be particularly important in various diseases (Cummings and Pierce, 2014; Magalhaes et al., 2021), such as inflammation, host-pathogen interaction, kidney disease, Alzheimers’ and especially in carcinogenesis (Argade et al., 2015; Magalhaes et al., 2017; Parsons et al., 2020; Zhang et al., 2020a). The detailed structure of these glycans, their various isomeric forms and their site specificities, all of which may be related to disease, have been difficult to access in the past. The development of modern mass spectrometry methods which can provide detailed structural analysis of glycan moieties on glycopeptides have recently opened up this field to great advances related to disease (Delafield and Li, 2021; Fang et al., 2021; Patabandige et al., 2021; Peng et al., 2021; Shu et al., 2021; Veillon et al., 2018; Zhu et al., 2019b).
Figure 1.
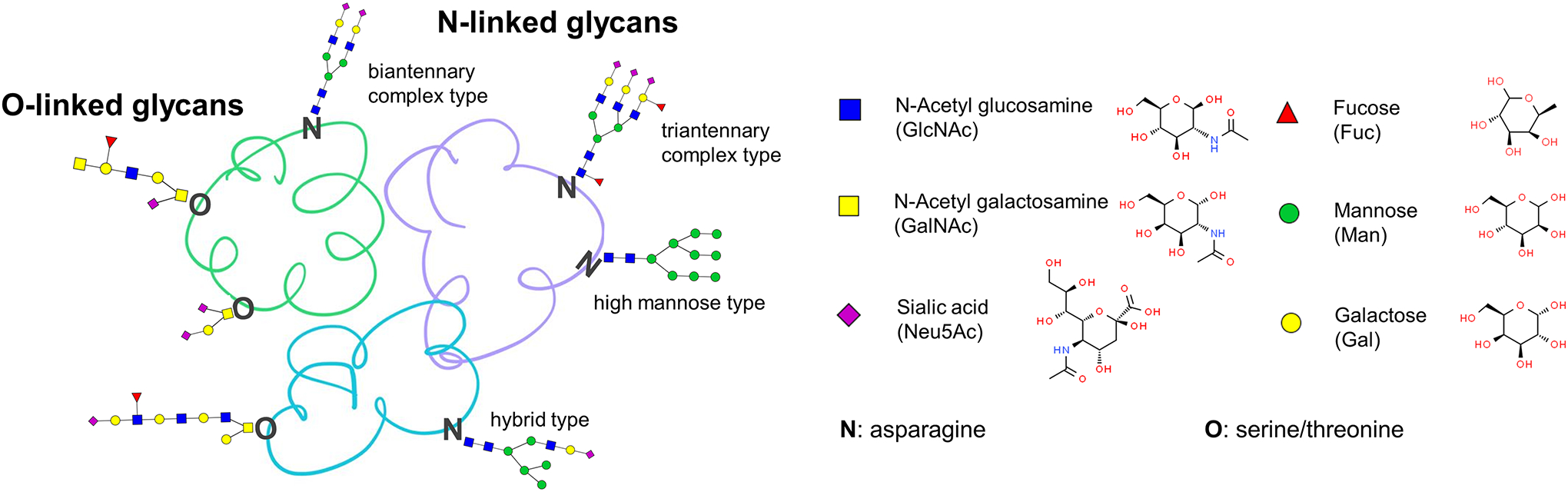
The structure of glycoproteins, glycans and glycosides. N-linked glycosylation: the reducing end of the glycan structure is linked to the primary amine group of asparagine on the peptide backbone; O-linked glycosylation: the reducing end of the glycan structure is linked to the hydroxyl group of serine/threonine on the peptide backbone.
The glycosylation changes in diseases often include the incomplete synthesis of truncated glycan structure, increased expression of complex branched N-glycans, terminal glycan sialylation, and altered fucosylation (Pinho and Reis, 2015). These changes of glycosylation may be the results of pathogenesis, based on which they can serve as biomarkers for diagnosis. The majority of clinical cancer biomarkers are glycoproteins, such as CA19–9 where SLea antigen is a serological marker for pancreatic cancer. Also, CA125 is used as a marker for ovarian cancer, and AFP as a marker for hepatocellular carcinoma (HCC) where a core fucosylated form of AFP has been proposed as an alternative marker for HCC. These changes of glycosylation may also play an important role in pathogenesis and carcinogenesis directly. For example, the increased core fucosylation of N-linked glycans of L1CAM protein on the surface of melanoma cancer cells was found to facilitate the metastasis by inhibiting its cleavage by plasmin (Agrawal et al., 2017).
The quantitative analysis of glycosylation provides a means for its functional evaluation in physiological and pathological processes (Cipollo and Parsons, 2020), for the assessment of diagnostic and prognostic values (Patwa et al., 2010; Zhu et al., 2019b), and for the exploration of possible drug targets (Alcedo et al., 2019). Mass spectrometry has been shown to be a unique tool for the study of glycosylation and its structure in both fundamental and clinical studies. The use of mass spectrometry is particularly powerful in providing the capabilities for quantitation of unique structural features of glycans and glycopeptides including structural isomers which may be essential to quantify in biological processes (Gautam et al., 2021; Zhu et al., 2020b). One aspect for glycosylation quantitation is based on the glycopeptide level with site-specific glycosylation, where this level of structural analysis has been shown to be potentially very important in biomarker and fundamental mechanistic studies.
The key considerations for glycopeptide quantitation are the sensitivity, specificity, reproducibility, precision, and throughput, which are the same as for peptide quantitation (Domon and Aebersold, 2010). Sensitivity refers to the limit of quantitation (LOQ), which, for peptides, is defined as the measurable value when S/N>6 (2013 AC, Renee). Specificity means that the ions chosen for measurement should be specific to the target, where matrix effects should be considered. Reproducibility means that the quantitation results should be reproducible with a small coefficient of variation (CV). Precision means the reliability of quantitated values, involving the generation of the calibration curve and the dynamic range of measurement, where carryover should be also evaluated. Throughput refers to the number of targets that can be quantitated within a limited time. These variables are all essential in the study of glycosylation related to biology and disease. However, glycopeptide quantitation is more complicated than peptide quantitation due to the presence of the glycan structure, especially in the aspects of sensitivity, specificity, and throughput.
Sensitivity is most important for glycopeptide quantitation. This is partly because one peptide backbone may have more than 60 glycoforms without considering the linkage differences, resulting in each glycopeptide with a much lower abundance (Liu et al., 2017a). This is especially true for the modification of sialic acid at the end of glycan structure which significantly reduces the glycopeptide signal in the mass spectrometer detection (Stavenhagen et al., 2013) compared with other glycopeptides at equal molar amount. Also, the MS intensity of sialylated glycopeptides with longer peptide chains is stronger than those with shorter peptide chains, possibly due to the enhanced ease of protonation or charge (Baerenfaenger et al., 2019). Specificity is especially important for glycopeptides because different glycopeptides have the same peptide backbone and different glycopeptides share very similar MS/MS spectra (Figure 2).
Figure 2.
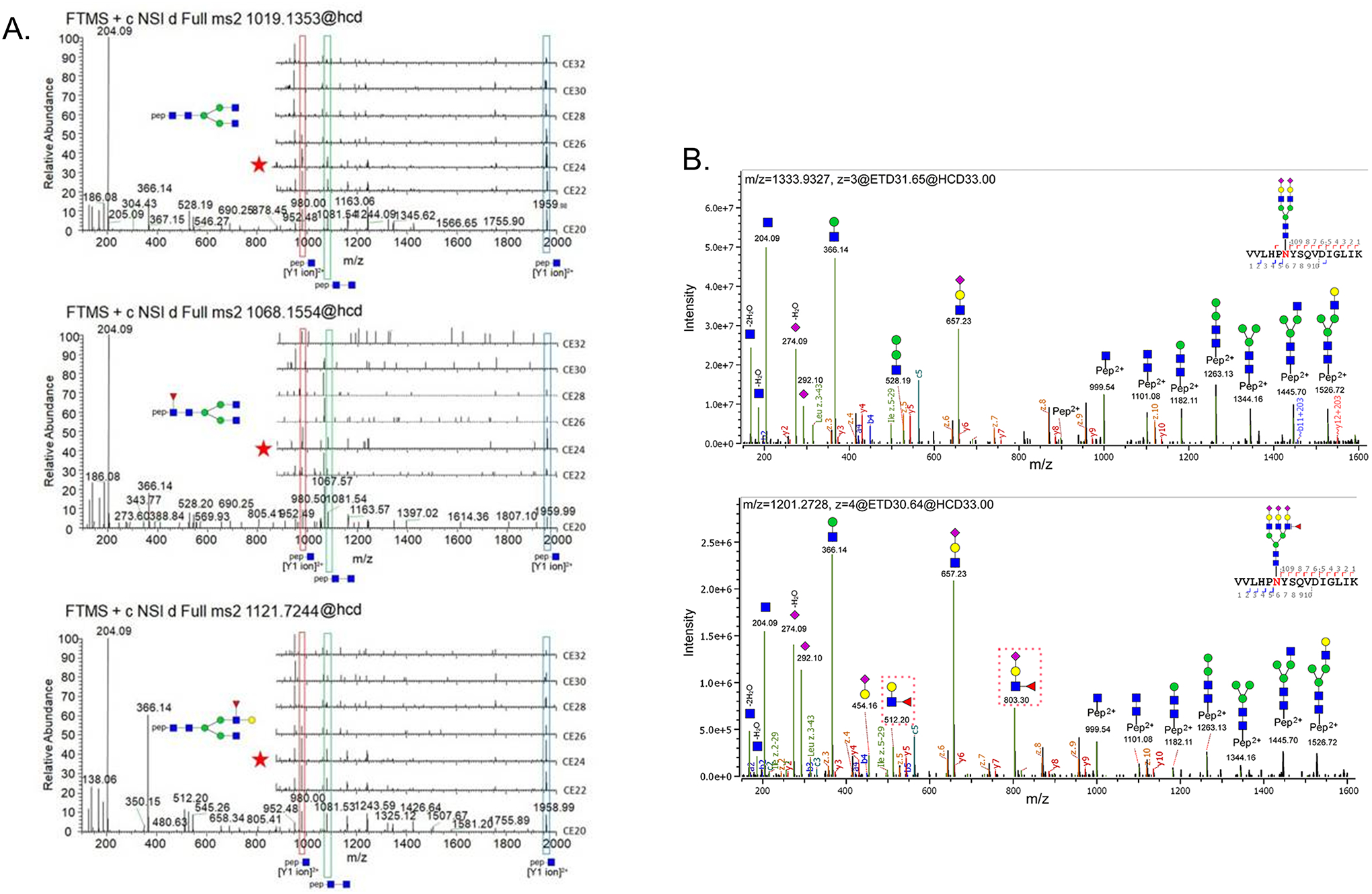
Fragmentation pattern of glycopeptides. A. HCD spectrum of three glycopeptides with the same peptide backbone and different glycan structures under various CE; B. EThcD spectrum of two glycopeptides with the same peptide backbone and different glycan structures. A Reprinted with permission from Yin. et al. (2018). Copyright © 2018 John Wiley & Sons LTD. B Reprinted with permission from Zhu. et al. (2020). Copyright © 2020 American Chemical Society.
In this review, we will first briefly summarize the enrichment methods for glycopeptides that were involved in glycopeptide quantification. We will then summarize the HPLC columns used for separations of glycopeptides which can be integrated online with MS, including C18, HILIC, and PGC. We will focus mainly on the comparison of different strategies for glycopeptide quantitation for clinical biomarker studies and the pathological and functional studies of glycoproteins in various diseases. We will discuss MS1 level-based quantification including labeling based and label free quantification, and MS2 level-based quantification including non-targeted labeling methods (TMT/iTRAQ), non-targeted non-labeling methods (DDA, DIA), and targeted methods (MRM, PRM) (Figure 3). Other recent reviews have summarized the methods to improve the quantitative glycoprotein coverage (Chang and Zaia, 2021).
Figure 3.
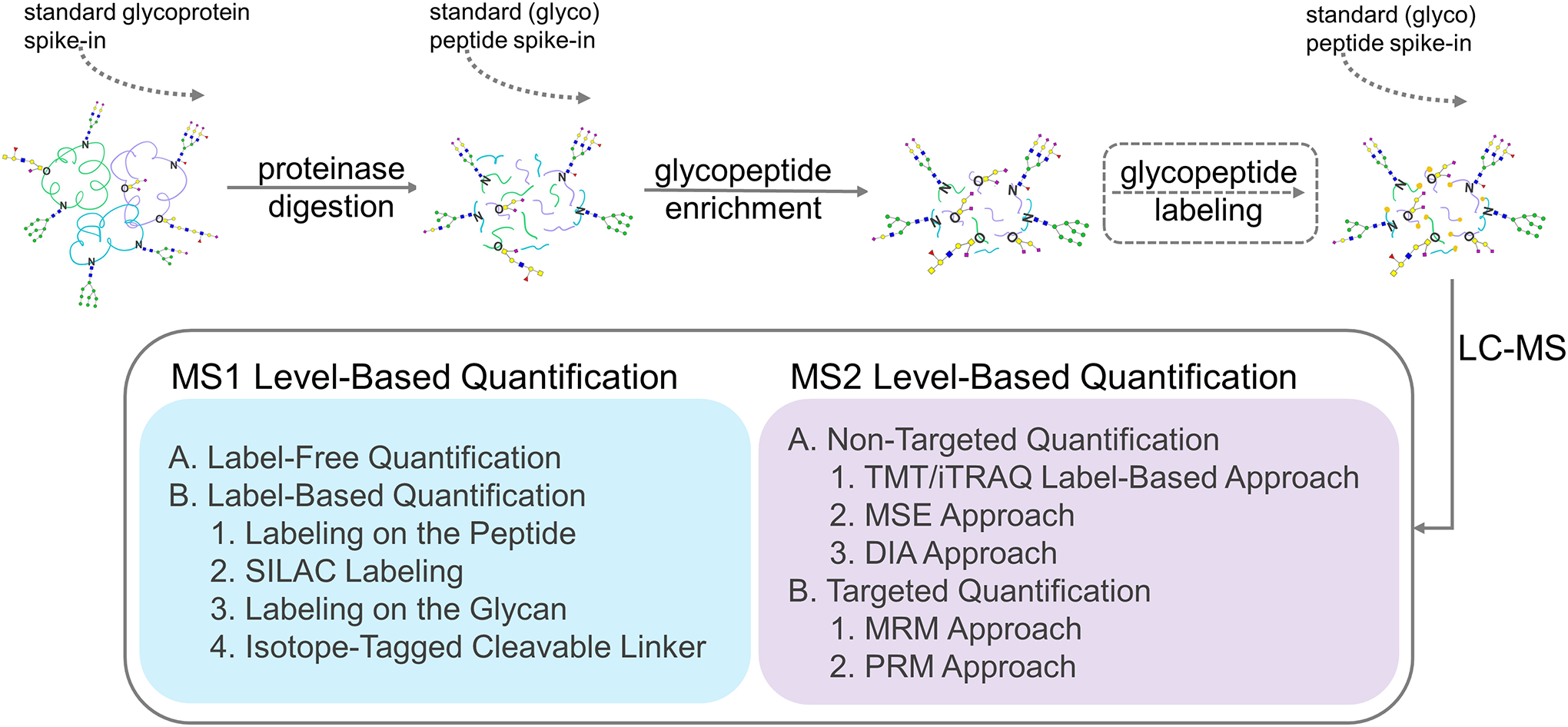
Strategies for glycopeptide quantification by mass spectrometry. The standard glycoprotein or (glyco)peptide can be optionally used as spike-in for glycopeptide quantitation.
Herein, we will focus on the comparison of the benefits and the trade-offs of different strategies for glycopeptide analysis based on liquid separations and mass spectrometry and the advantages of using various mass analyzers for this analysis. We will also summarize the glycopeptide standards which have been used for quantitation. Various MS/MS collision methods (i.e. EThcD, AI-ETD, CID, HCD and stepped HCD) for glycopeptide identification and softwares for glycopeptide analysis have been reviewed elsewhere (Chang and Zaia, 2021; Klein and Zaia, 2020; Ruhaak et al., 2018), including glycan library dependent software (such as, GPQuest (Toghi Eshghi et al., 2015); SweetNET (Nasir et al., 2016); GPfinder (Strum et al., 2013), pGlyco (Liu et al., 2017a)) and glycan library independent software (such as MSFragger-Glyco (Polasky et al., 2020)). Ultimately, this review will serve to summarize the advantages of mass spectrometry technology for quantitative analysis of glycopeptides and its applications to specific problems.
II. ENRICHMENT AND SEPARATION OF GLYCOPEPTIDES
Due to the low abundance of glycopeptides compared to peptides in protein digests, the complexity/microheterogeneity of glycosylation, and ion suppression effects from the co-eluting peptides in LC-MS analysis, specific sample preparation such as glycopeptide/glycoprotein enrichment is necessary prior to MS analysis of glycopeptides. The stationary phases for enrichment include ligand immobilization or chemical bonding strategies on solid supports to selectively enrich N- or O-glycopeptides in either physical or chemical modes of binding (Huang et al., 2014). The major methods and materials for the enrichment of glycoproteins/glycopeptides have been reviewed recently (Pujic and Perreault, 2021; Suttapitugsakul et al., 2020; Xiao et al., 2019; Yu et al., 2018; Zhu et al., 2019b), including the methodologies that involve the use of microarray platforms (Patwa et al., 2010). Herein, we mainly summarize the enrichment strategies incorporated into quantitative glycoproteomic studies as well as column selection for liquid separation of glycopeptides.
2.1. Enrichment Methods for Glycopeptides
Enrichment methods, including lectin affinity chromatography, HILIC solid phase enrichment, boronate affinity, and hydrazide chemistry, have been widely employed to advance the identification and quantitation of glycopeptides in complex biological samples. Based on the type of enrichment, they can be grouped into two categories: glyco-motif targeted enrichment and universal enrichment.
2.1.1. Glyco-Motif Targeted Enrichment
Lectins are carbohydrate-binding proteins that bind to sugars with specific moieties. Among them, sambucus nigra lectin (SNA) preferentially binds to sialic acid residues, aleuria aurentia lectin (AAL) specifically recognizes terminal α-linked fucose, and lens culinaris agglutinin (LCA) has strong preference for core α1,6-fucose, which are often used for enrichment of glycopeptides with sialic acid or fucose moieties to determine changes in sialylated or fucosylated glycopeptides/glycoproteins correlated with cancer/diseases. Concanavalin A (Con A) and wheat germ agglutinin (WGA) are widely used for glycopeptide enrichment (Miyamoto et al., 2016; Pap et al., 2018; Riley et al., 2019) since they recognize α-linked mannose and terminal GlcNAcβ residues, respectively, the two glyco-structures common to many glycoproteins. Typically, lectins are immobilized onto a solid support, such as agarose beads, which can be packed into a spin/centrifuge column for solid-phase extraction of glycopeptides/glycoproteins (Zhao et al., 2007; Zhao et al., 2006).
The use of lectin-affinity columns represents an important advance in targeted enrichment of glycopeptides with specific glyco-motifs for quantitative analysis of glycopeptides as potential cancer biomarkers. To study changes in fucosylated glycopeptides/glycoproteins, AAL- and LCA-affinity columns are frequently used (Kaji et al., 2013; Liu et al., 2010; Tanabe et al., 2016; Yin et al., 2015a; Zhu et al., 2012). For example, Lubman and coworkers integrated LCA enrichment into a method for quantitative analysis of site-specific core-fucosylated (CF) peptides in patient sera between HCC and cirrhosis (Yin et al., 2015a). The strategy involved depletion of high-abundance serum proteins, trypsin digestion, iTRAQ labeling of the peptides, LCA enrichment of CF peptides, and endoglycosidase F3 digestion prior to mass spectrometry analysis. In total, 1300 CF peptides from 613 glycoproteins were identified and quantified, where 15 CF peptides were found over-expressed in ALC-related HCCs and 12 CF peptides in HCV-related HCCs compared to their corresponding cirrhosis patients. In another work by the Lubman group, LCA enrichment was applied to improve the quantitation of CF sites in serum glycoproteins among 13 sets of serum samples from pancreatic cancer, chronic pancreatitis, and healthy controls, respectively (Tan et al., 2015). In total, 630 CF sites were quantified from 322 glycoproteins in pancreatic cancer, revealing 8 differentially expressed CF peptides in pancreatic cancer.
2.1.2. Universal Enrichment
The universal enrichment methods for glycopeptides include HILIC, boronate affinity, hydrazide chemistry, etc. HILIC materials are usually packed into columns or pipette tips to enrich glycopeptides based on the increased hydrophilicity by glycans (Qing et al., 2020). Boronate affinity is based on the strong covalent interaction between boronic acid and sugars, which enables universal capture of glycopeptides, while the reversible property allows the release of glyco-species without side effects (Chen et al., 2014; Wang et al., 2013; Xiao et al., 2018). Hydrazide chemistry for glycopeptide enrichment is based on the covalent bond formation between the hydrazide groups and the aldehyde groups on oxidized glycan motifs (Zhang et al., 2003). All these enrichment methods take advantage in unbiased binding to glycopeptides for comprehensive characterization of the glycosylation landscape and quantitation of glycopeptides in complex biological samples.
1). HILIC
HILIC displays broad applications and outstanding performance in comprehensive glycoproteomic analyses of biological samples (Liu et al., 2021; Shu et al., 2020; Wang et al., 2020; Zacharias et al., 2016; Zhu et al., 2020a). The Lubman group developed an integrated workflow with HILIC enrichment for differentially quantitative analysis of the microheterogeneity of site-specific intact N-glycopeptides of serum haptoglobin (Hp) between early HCC and liver cirrhosis (Zhu et al., 2019a; Zhu et al., 2020a). Hp was immunopurified from 20 μL of serum, followed by trypsin/GluC digestion, glycopeptide enrichment with HILIC tips, and LC-EThcD-MS/MS analysis. In total, 70 NASH patients (37 HCC and 33 cirrhosis cases) were analyzed, where the differential quantitation analysis revealed that five N-glycopeptides at sites N184 and N241 in serum Hp were significantly elevated in HCC compared to cirrhosis (p < 0.05). The 5 independent replicates of an Hp standard showed the Pearson correlation coefficient R2 value for the binary comparison of the 5 replicates were from 0.955 to 0.995. The relative standard deviation (RSD) of the most abundant glycopeptide VVLHPN241YSQVD_A2G2S2 was 7.73% across the 5 replicates, and the low abundant glycopeptides of VVLHPN241YSQVD_A4G4S4 and VVLHPN241YSQVD_A4G4F1S4 had an RSD of 10.20% and 15.23%, respectively, showing good reproducibility of the method (Zhu et al., 2020a).
In a large-scale glycopeptide quantitative study by the Lubman group, HILIC enrichment was also incorporated into an LC-Stepped HCD-MS/MS workflow for broad-scale marker discovery for HCC (Lin et al., 2021). In this work, 10 μL of depleted serum was used for trypsin digestion, followed by glycopeptide enrichment using HILIC tips, offline fractionation, and LC-Stepped HCD-MS/MS analysis for differential determination of changes in site-specific glycopeptides in whole serum between HCC and cirrhosis. As a result, a panel of N-glycopeptides were identified as potential biomarker candidates for early HCC, where the 65 glycopeptide biomarker candidates were further targeted quantitated among 78 patients (40 cirrhosis and 38 HCCs) by LC-Stepped HCD-PRM-MS/MS. The method reproducibility was evaluated by three independent experiments from a random serum sample, where the Pearson correlation coefficient R2 value was found from 0.9630 to 0.9998, indicating good reproducibility of the method.
2). Boronate Affinity
The Wu group has employed a boronic acid-based chemical method to universally enrich glycopeptides (Chen et al., 2014; Xiao et al., 2018). By combining boronic acid enrichment with PNGase F treatment in heavy-oxygen water and LC-MS/MS, they were able to identify 816 N-glycosylation sites in 332 yeast proteins. The Li group (Chen et al., 2021) has developed an N-glycoproteomic approach with sequential enrichment of N-glycopeptides by HILIC and boronic acid enrichment, followed by EThcD for large-scale intact N-glycopeptide analysis. With this approach, they were able to identify a total of 2893 intact N-glycopeptides from 511 N-glycosites and 285 N-glycoproteins in human cerebrospinal fluid (CSF) samples, which is the largest site-specific N-glycoproteome dataset reported for CSF to date (Chen et al., 2021).
3). Hydrazide Chemistry and Others
Sun et al. developed a solid phase based labeling approach by integration of glycopeptide enrichment and stable isotope labeling on hydrazide beads for differential quantitation of glycopeptides between HCC versus normal serum samples (Sun et al., 2012). This approach showed good linearity range with 2 orders of magnitude for quantification of glycopeptides. Compared to dimethyl labeling performed in solution, this approach has better enrichment recovery (10–330% improvement) and high detection sensitivity in which 42% of annotated glycosites (vs 26%) still can be quantified using only 10 μg of glycoprotein mixtures. The Ye group developed a chemoenzymatic method to analyze mucin-type core-1 O-glycosylation in human serum where the oxidized O-GalNAcylated peptides were captured by hydrazide beads and eluted with methoxylamine for LC-MS/MS analysis (You et al., 2018).
There are also some other glycopeptide enrichment methods, such as a unique EXoO method developed by the Zhang group to enrich glycopeptides with Tn antigen (Yang et al., 2020b; Yang et al., 2020c) and a chemoenzymatic method named IsoTaG to enrich isotope-labeled N- and O-glycopeptides from whole cell proteomes (Woo et al., 2017; Woo et al., 2015). In addition, monolithic columns were also used for glycopeptide enrichment including Borate-monolithic column (Chen et al., 2009), LCA-monolithic column (Bedair and Oleschuk, 2006; Feng et al., 2009) and HILIC monolithic columns (Jiang et al., 2016).
2.1.3. Comparison of Enrichment Methods
Among the enrichment methods, lectin affinity can be used to capture a specific glycosylation structure due to the carbohydrate specificity of the lectin, in particular sialylation or fucosylation, which are the two most important glycosylations correlated to cancer and diseases. HILIC and covalent binding are often used for universal enrichment of glycopeptides when full characterization of N- or O-glycosylation in biological samples is required.
Chen et al. evaluated three commonly used enrichment methods for N-glycopeptides, including HILIC, lectin affinity, and boronic acid in their work of site-specific analysis of N-glycoproteome in human CSF in Alzheimer’s disease (Chen et al., 2021). They found that the boronic acid approach outperformed the other two methods regarding the number of N-glycopeptide identifications. However, HILIC enrichment showed the highest identification number of sialylated glycopeptides, a significant modification in many biological systems. The preferential enrichment of sialylated N-glycopeptides is probably because sialic acid increases the hydrophilic interactions between the glycopeptides and HILIC (Chen et al., 2021).
Wohlgemuth et al. compared HILIC, hydrazide chemistry, and titanium dioxide (for capturing sialylated glycopeptides) in quantitative analysis of site-specific glycopeptides (Wohlgemuth et al., 2009). Quantitative analysis demonstrated that glycopeptides could be enriched by ZIC-HILIC without bias for particular glycan structures and without significant losses. Sialylated glycopeptides could be efficiently enriched by titanium dioxide and in addition to HILIC both methods enable a comprehensive analysis of protein glycosylation by MS. However, enrichment of N-glycopeptides by hydrazine chemistry resulted in lower peptide recovery using a more complex enrichment scheme (Wohlgemuth et al., 2009).
The Zhang group compared ZIC-HILIC and strong anion exchange (SAX) cartridges in the yield of enrichment of intact N- and O-glycopeptides from 1 mg of serum or tissue protein digests as well as the effectiveness to enrich isobarically labeled glycopeptides for glycoproteomic analysis. The result showed that the enrichment effect of SAX and ZIC-HILIC was similar but the recovery of SAX was much higher than ZIC-HILIC resin, and SAX was better for O-glycopeptides enrichment. Furthermore, isobaric tag labeled glycopeptides after C18 desalting could be readily enriched by SAX cartridges but not by HILIC to enable quantitative glycoproteomics (Yang et al., 2017b). The same group recently developed one-step glycopeptide enrichment method where C18 beads and Oasis MAX beads (Waters) were filled in one cartridge for glycopeptide enrichment and desalting, which greatly simplifies the sample preparation for large scale clinical sample analysis (Chen et al., 2020; Yang et al., 2020a).
It should be noted that the cartridge-based platform is only applicable for large sample amount. When handling limited starting materials, a tip-based platform could efficiently minimize sample losses. In addition, these enrichment methods can be applied as a single enrichment strategy or coupled together to maximize the glycoproteome of biological samples.
2.2. Column Selection for Glycopeptide LC Separation
The chromatography separation before MS analysis could greatly enhance the MS sensitivity which is decreased due to the micro-heterogeneity of glycopeptides with various glycan structures. The separation is important for the reproducibility, precision, and throughput of the analysis. When conjugating with MS using ESI (electrospray), the flow rate, directly correlated to the inner diameter of the columns, strongly affects the ionization process. For peptides, the sensitivity gain increases exponentially from an analytical flow rate (0.1–0.4 mL/min for 2.1 mm ID column) to capillary flow rate (1–15 μL/min for 0.1–0.3 mm ID column) to nanoflow rate (300 nL/min for 75 μm ID column) (Wilm and Mann, 1996); lower flow rates also enhance the MS signal of neutral glycans without sialic acid (Bahr et al., 1997; Schmidt et al., 2003). Other column parameters include column length and particle size. The most commonly used particle sizes are 5 μm, 3 μm and 1.7 μm. In general, a smaller particle size results in a better separation but also a higher column pressure. However, the improvement from using a longer column, 30 cm or 50 cm compared with 15 cm column, is negligible with a 3 μm particle but has slightly better separation with a 1.7 μm particle. Columns with a smaller flow rate require a longer time to complete a run, reducing the overall throughput (Liu et al., 2007). These parameters of columns are expected to be similar for glycopeptide analysis, but there has not been such a detailed study on glycopeptides yet.
As discussed earlier, glycopeptides with sialic acid modification may have one tenth of the signal of its peptide backbone in equimolar amounts. The better separation of glycopeptides with microheterogeneity would alleviate the burden to quantitate many glycopeptides in a short time window. Based on the stationary phase, the columns for glycopeptide separation can be divided into 3 categories as shown in Table 1, including C18 column, HILIC column, and PGC column. Only commercially available columns are included in Table 1. Homemade columns are summarized in the text only.
Table 1.
Commercially available columns used for glycopeptide analysis.
| Stationary phase | UPLC/nanoLCa | Brand name | Stationary phase structure | Property of packing material | Column IDb | Reference example |
|---|---|---|---|---|---|---|
| C18 | C18 nano LC column | Acclaim PepMap C18 (Thermo Fisher) |
|
particle size: 3/5 μm pore size: 100 Å |
75/100 μm | (Li et al., 2020) |
| Nano cHiPLC C18 (Sciex) | particle size: 3/5 μm pore size: 120/250/300 Å |
75 μm | (Kozlik et al., 2017) | |||
| PGC | PGC UPLC column | Hypercarb (Thermo Fisher) |
|
particle size: 3/5/7 μm pore size: 250 Å |
1/2.1/3/4.6 mm | (Zhu et al., 2020b) |
| PGC nano LC column | 75/100 μm (discontinued) | NA | ||||
| PGC nano LC column | PGC-chip (Agilent) | particle size: 3 μm pore size: 250 Å |
43 mm chip with 40 nL trap column | (Alley et al., 2009) | ||
| HILIC | HALO nano LC column | cHiPLC HALO HILIC (Eksigent) |
|
particle size: 3 μm pore size: 120/300 Å |
75 μm | (Kozlik et al., 2017) |
| HALO HILIC (Advanced Materials Technology) | particle size: 2.7 μm pore size: 90 Å |
75 μm | (Kozlik et al., 2018) | |||
| Zwitterionic UPLC column | SeQuant ZIC-HILIC column (EMD Millipore) |
|
particle size: 3.5/5 μm pore size: 100/200 Å |
2.1/4.6 mm | (Furuki and Toyo’oka, 2017) | |
| SeQuant ZIC-cHILIC column (EMD Millipore) |
|
particle size: 3 μm pore size: 100 Å |
2.1 mm | (Furuki and Toyo’oka, 2017) | ||
| Amide UPLC column | BEH Ethylene Bridged Hybrid amide (Waters) |
|
particle size: 1.7 μm pore size: 130 Å |
2.1/3/4.6 mm | (Furuki and Toyo’oka, 2017) | |
| Accucore Amide (Thermo Fisher) |
|
particle size: 2.6 μm pore size: 150 Å |
2.1/3/4.6 mm | (Camperi et al., 2020) | ||
| Amide nano LC column | Accucore Amide (Thermo Fisher) | 75 μm | NA |
NanoLC columns are marked with underline
Most columns provide column with length: 5/10/15/25 cm; C18 columns provide extralong column with length 50 cm
2.2.1. C18 column
The C18 column is the most widely used column for glycopeptide separation based on the hydrophobicity of glycopeptides in high aqueous phase. Glycopeptides with longer peptide chains bind stronger with C18 column, where glycopeptides with microheterogeneity on the same glycosylation site elute in clusters based on the number of sialic acids, and within the same cluster the glycopeptides with more glycan units elute earlier (Liu et al., 2017a) (Figure 4A). Nanoflow C18 columns have been used in glycopeptide profile and quantitation analysis (Darebna et al., 2017; Kim et al., 2019; Yin et al., 2020; Yu et al., 2017b; Yuan et al., 2018; Zhu et al., 2019a; Zhu et al., 2020a), whereas analytical flow C18 columns have been used in quantitation analysis (Hong et al., 2013; Miyamoto et al., 2018). C18 column has been found to be able to separate some glycopeptide isomers, where higher temperature and none-positive charged peptide ending seem to enhance the separation efficiency. The Yoo group found that high column temperatures enhanced the separation of sialylated O- and N-linked glycopeptide isomers after trypsin digestion with positive charged ending (Ji et al., 2019b). Yin et al. found glycopeptides from chymotrypsin digested alpha-1 antitrypsin have various isomers on C18 column at room temperature, possibly due to the none-positive charged ending from chymotrypsin digestion that enhanced the binding variation of glycopeptides to the C18 column (Yin et al., 2020). The Mechref group further employed a long C18 column that was 50 cm in length at high temperature 60°C for the separation of glycopeptide isomers after trypsin and Glu-C digestion, where 72 isomeric structures corresponding to 42 glycopeptide isoforms of haptoglobin were separated and accurately quantified, compared to 35 glycopeptides without isomer separation using C18 column with regular length at low temperature (Reyes CDG, 2021). The stability of C18 columns is much better than PGC and HILIC columns, especially in nano-flow scale. Also, the peak width on C18 is usually narrower than on PGC and HILIC columns. However, HILIC and PGC could separate those glycopeptides which elute in clusters on C18 columns.
Figure 4.
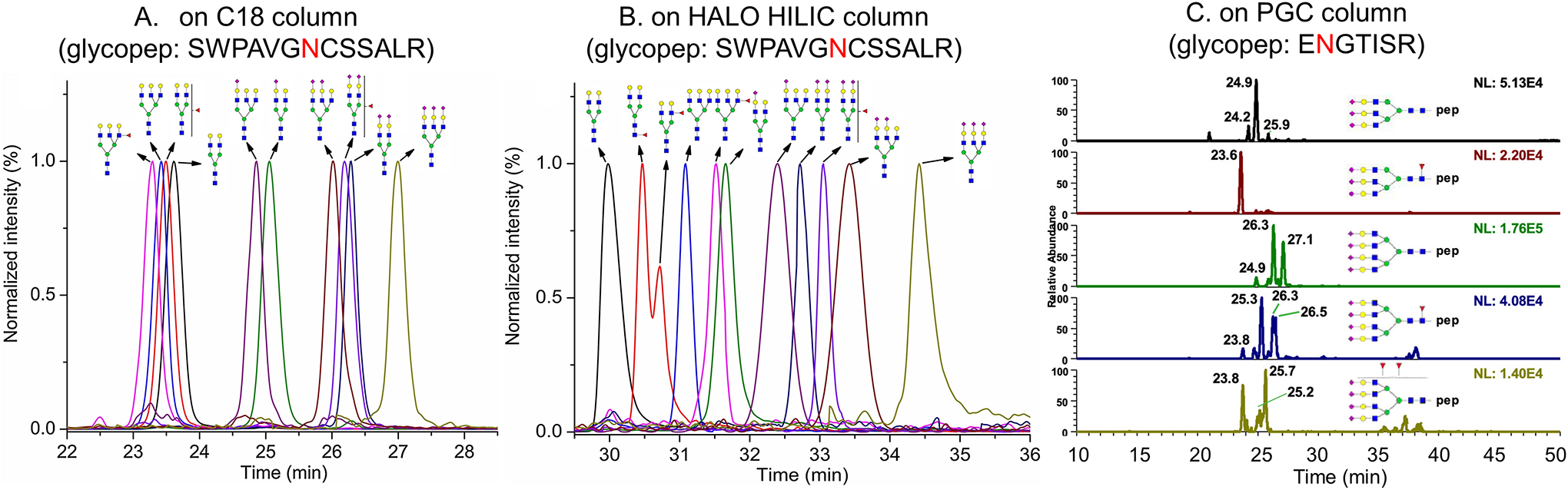
Separation of glycopeptides on various columns. A. on C18 column (glycopep: SWPAVGNCSSALR); B. on HALO HILIC column (glycopep: SWPAVGNCSSALR); C. on PGC column (glycopep: ENGTISR). A&B Reprinted with permission from Kozlik P. et al. (2017). Copyright © 2017 Elsevier. C Reprinted with permission from Zhu R. et al. (2020). Copyright © 2020 American Chemical Society.
2.2.2. HILIC column
HILIC columns have become more popular for glycopeptide analysis, which separates glycopeptides based on the hydrophilicity of glycopeptides in high organic phase. Glycopeptides with shorter peptide chains bind stronger with HILIC column, where glycopeptides with microheterogeneity on one glycosylation site separates much better than C18 column and may expand to the elution region of other glycopeptides. There are various types of HILIC columns with various HILIC stationary phases, including ZIC-HILIC column with zwitterionic functional groups (tetramethyl ammonium group +sulphonic acid group /phosphoryl group), amide column with primary amine groups, and HALO HILIC column with multiple hydroxyl group (as shown in Table 1). The elution order of glycopeptides and separation vary among these different HILIC columns, where the HALO penta-HILIC column showed the best separation compared with amide columns or ZIC-HILIC columns (Molnarova and Kozlik, 2020). The average separation of glycopeptides on a nano HILIC column (cHiPLC, HALO HILIC column from Eksigent) is better than in a nano C18 column (cHiPLC, C18 column from Eksigent) (Kozlik et al., 2017) (Figure 4B). Also, the separation of HILIC columns changes with different pairing reagents. FA is the most MS compatible pairing reagent in HILIC column separation, but the separation of glycopeptides are worse than that with TFA (Furuki and Toyo’oka, 2017). Therefore, for LC analysis only, TFA is a better choice, whereas for LC-MS analysis, only FA can be used. ZIC-HILIC columns are designed with analytical flow rate only. Amide columns have both analytical flow rate column and nanoflow columns. HALO HILIC columns only have nanoflow columns, including cHiPLC HALO HILIC columns from Eksigent and HALO HILIC columns from Advanced Materials Technology. HALO HILIC provides overall the best separation compared with other HILIC columns. Yet, an important disadvantage of HILIC separation is that some glycopeptides may do not dissolve well in high organic phase.
2.2.3. PGC column
PGC columns are filled with porous graphitized carbon packing material. It is a complementary stationary phase to octadecylsilane (C18), specifically for the separation of closely related substances and polar analytes (Toernkvist, 2003). PGC columns were initially made by Wolfson Unit in Edinburgh University, UK, then transferred to Thermo Scientific Hypersil-Keystone HyperCarb (Hypersil Hypercarb Information Brochure). Agilent developed PGC-Chip which is composed of a 9 × 0.075 mm i.d. enrichment column and a 43 × 0.075 mm i.d. analytical column with particle size 5 μm.
PGC columns have been widely used for glycan separation (Ashwood et al., 2019; Ruhaak et al., 2013; Seo et al., 2019; Zhang et al., 2020b; Zhou et al., 2017). It has recently been applied for the separation of glycopeptides. The Mechref group used PGC column at high temperature 75°C to separate glycopeptide isomers with different sialylation linkages (Zhu et al., 2020b) (Figure 4C), where no isomer separation was found at 50°C and a significant signal intensity decrease at 100°C. The Liang group combined a home-made click TE-Cys HILIC column and PGC column into a 2D system for glycopeptide separation of ribonuclease B (Lu et al., 2017), where glycopeptides with 5–9 mannoses were fully separated on TE-Cys HILIC column and some isomers were resolved on PGC column. Yet, PGC cannot be applied for glycopeptides with long peptide backbones (>4–6 amino acids) or high hydrophobicity (Zhu et al., 2020b). The Wuhrer group developed integrated C18 and in house packed PGC column for glycopeptide identification after pronase digestion, where the more hydrophobic glycopeptides were retained on C18 and more hydrophilic glycopeptides (C18-unbound glycopeptides) were separated by a downstream PGC column, where glycopeptides with various peptide backbone lengths were analyzed within a single analysis (Stavenhagen et al., 2017). Pronase digestion cut proteins into smaller pieces but its non-specificity for digestion restricts the application for glycopeptide quantitation. Furthermore, the nanoflow PGC column from Thermo Scientific has been discontinued due to column fouling, instability, and lack of reproducibility, leaving only capillary flow PGC columns as being still available.
2.2.4. MGC column
As an alternative, the Mechref group recently developed a mesoporous graphitized carbon (MGC) column, which has a similar pore diameter of between 2 and 50 nm as PGC Hypercarb packing material (25 nm), but with particle size less than 500 nm, much smaller than that of PGC Hypercarb packing material (3/5/7 μm). The MGC column showed much higher separation capacity, where the MGC column with 1 cm length separated permethylated glycan isomers better than the PGC Hypercarb nanoLC column with 10 cm length (Gautam et al., 2021). The application of MGC on glycopeptides has not been published.
III. MS1 LEVEL-BASED QUANTIFICATION
Recent advances in mass spectrometry provide the opportunity to identify and quantify even low-abundant glycopeptides in complex biological mixtures, allowing for quantification of site-specific glycosylation changes between disease states. Various fragmentation strategies have been developed and applied for glycopeptide characterization (Riley et al., 2020b), including CID/HCD (Lee et al., 2016), stepped HCD (Yin et al., 2018), CID/ETD (Mechref, 2012), HCD/ETD (Singh et al., 2012), and EThcD (Chen et al., 2018; Glover et al., 2018; Yu et al., 2017a), which have facilitated accurate glycopeptide quantification. Software tools are available for relative quantitation of glycopeptides (Cao et al., 2021a), such as pGlyco 2.0 (Liu et al., 2017a), Integrated GlycoProteome Analyzer (I-GPA) (Park et al., 2016), and LaCyTools (Jansen et al., 2016), which are applicable for CID/HCD data, as well as a combination of Byonic/Byologic (Protein Metrics Inc.) that can analyze all types of MS tandem data with CID, HCD, ETD, or EThcD fragmentation (Bern et al., 2012). Recent advances in analytical approaches for glycan and glycopeptide quantitation have been summarized (Delafield and Li, 2021). Currently, a widely used approach of quantitative glycoproteomics is based on quantifying the MS1 signal of glycopeptides, i.e., MS1-based quantification. This section covers the methods of glycopeptide quantification at MS1-level, including label-free quantitation and label-based quantification with labeling on the glycan motif or the peptide backbone.
3.1. Label-Free Quantification
3.1.1. Applications of Label-Free Approach
The label-free quantification of glycopeptides has been applied for analysis of complex biological samples as well as purified glycoproteins (Rebecchi et al., 2009). Based on extracted ion chromatogram (XIC) peaks of precursor ions from MS1 scan, the peak area of a given glycopeptide can be integrated and quantitated, followed by normalization against the sum of peak areas of all glycopeptides identified in an MS run. The label-free quantitation of glycopeptides can be used to evaluate changes in glycosylation in two ways: the overall glycopeptide level in a complex sample and site-specific glycosylation level on a given glycoprotein.
The Manfred group profiled IgG using TFA for LC separation and the propionic acid containing sheath-liquid for TFA gas-phase ion-pairing suppression, enabling the relative quantitation of 8 glycoforms of IgG subclasses (Selman et al., 2012). The Qian group recently reported an integrated MS data processing strategy for fast identification, in-depth, and reproducible label-free quantification of protein O-glycosylation in a large cohort of human urine samples (Zhao et al., 2020). The strategy integrates glycoform-specific database searching, reference library-based MS1 feature matching, and MS2 identification propagation, leading to a 30%-40% enhanced intact O-glycopeptide quantification in individual samples with an improved reproducibility.
3.1.2. Biomarker Studies with Label-Free Approach
In other work by the Lubman group, Zhu et al. achieved label-free quantitation of N-glycopeptides in a site-specific manner for differential analysis of glycosylation changes in purified haptoglobin from patient serum by LC-EThcD-MS/MS (Zhu et al., 2019a; Zhu et al., 2020a). EThcD glycopeptide data were interpreted by Byonic software and then quantified by Bylogic (Protein Metrics Inc.), where the peak area of XIC of each glycopeptide was automatically quantitated, followed by normalization at total glycopeptide level and individual glycosite level, respectively. Based on this method, 101 site-specific N-glycopeptides of serum haptoglobin were identified and relatively quantified (Zhu et al., 2019a). With the EThcD MS/MS-based label-free quantitation platform, Zhu et al. further performed a comprehensive screening of site-specific N-glycopeptide biomarkers in serum haptoglobin among 70 nonalcoholic steatohepatitis (NASH) patients (37 HCC and 33 cirrhosis cases) (Zhu et al., 2020a). In total, 140 MS datasets were collected using LC-EThcD-MS/MS where the relative abundance of N-glycopeptides was quantified at glycosite level using Byologic. Differential quantitation analysis revealed that 5 N-glycopeptides at sites N184 and N241 of serum haptoglobin were significantly elevated during the progression from NASH cirrhosis to HCC (p<0.05). Receiver operating characteristic (ROC) curve analysis demonstrated that the N-glycopeptides MVSHHN184LTTGATLINE and VVLHPN241YSQVDIGLIK, bearing a monofucosylated tri-antennary glycan A3G3F1S3, had the best diagnostic performance in detection of early NASH HCC (Zhu et al., 2020a).
Zhang et al. reported a label-free quantitative analysis of intact N- glycopeptides of plasma IgGs in both subclass-specific and site-specific N-glycosylation manners using pGlyco 2.0 and MaxQuant software (Zhang et al., 2020c). Plasma IgGs were purified from plasma among 51 prostate carcinoma (PCa) and 45 benign prostatic hyperplasia (BPH) patients followed by HILIC enrichment and high- resolution LC-MS/MS analysis. They identified and quantified the relative abundance of 24 glycoforms of IgG1, 32 glycoforms of IgG2, 4 glycoforms of IgG3 and 12 glycoforms of IgG4. As a result, they found the N-glycopeptide IgG2-GP09 (EEQFNSTFR (H5N5S1)) was dramatically elevated in plasma from PCa patients, compared with that in BPH patients (PCa/BPH ratio = 5.74, p = 0.001) (Zhang et al., 2020c). In another work, the Yoo group reported a interlaboratory study of site-specific N-glycopeptide isoforms of α-1-acid glycoprotein (AGP) using CID- and HCD-MS/MS (Lee et al., 2016), where site-specific AGP N-glycopeptides were automatically identified by Integrated GlycoProteome Analyzer (I-GPA) (Park et al., 2016) and label-free quantitative analysis was performed for the 10 major abundant N-glycopeptides based on the peak area of XICs. Quantitative analysis showed that the coefficient of variation in four laboratories was <25% for all test samples (Lee et al., 2016).
3.2. Label-Based Quantification
Label-based MS1 quantification methods are also available for relative quantitation of glycopeptides in biological samples. Labeling can be performed on the glycan motif or the peptide backbone, via enzyme labeling (i.e., trypsin catalyzed 18O labeling), chemical labeling (i.e., dimethyl labeling), and metabolic labeling (i.e., IsoTaG). Isotope labeling has been achieved using 2H, 13C, 15N or 18O, as heavy isotopes (Boersema et al., 2009). Glycopeptides from different samples can be isotope labeled, mixed, and then combined for a single MS run, where the quantitative results can be obtained simultaneously by comparing the abundance of the isotopes (Zhang et al., 2019). Among them, enzymatic 18O labeling only requires to be in the presence of 18O-water, without extra reagents, additional steps, side reactions, and chromatographic isotope effects (Capelo et al., 2010).
3.2.1. Labeling on the Peptide
Isotope labeling can be performed on the peptide backbone via enzyme labeling (i.e., trypsin catalyzed 18O labeling) and chemical labeling (i.e., dimethyl labeling). The Lu group has reported an N-glycopeptide quantitative method based on 18O/16O C-terminal labeling to obtain 82 comparisons of serum from patients with HBV-related HCC and liver cirrhosis (Zhang et al., 2019). The 16O/18O labeled N-glycopeptides were identified using pGlyco 2.0 and quantified by pQuant which calculates 18O/16O glycopeptides ratio based on a pair of least interfered isotopic chromatograms (Zhang et al., 2019). With the 16O/18O C-terminal labeling quantification method, Cao et al. assessed the alteration of site-specific N-glycopeptides of serum paraoxonase 1 (PON1) for distinguishing AFP-negative HCC from LC patients (n = 64) (Cao et al., 2021b). In this work, PON1 was immunopurified from patient serum, followed by tryptic digestion with H216O/H218O, and the 16O- and 18O-labeled digests were pooled before LC-MS/MS analysis. Two glycopeptides HAN253WTLTPLK bearing the glycan H5N4S2 and H5N4S1, respectively, were found significantly increased in AFP-negative HCC patients as compared with cirrhosis patients.
Kurogochi et al. developed a sensitive method for quantitative glycopeptide profiling using stable isotope labeling and MALDI-TOF MS. They synthesized benzoic acid-d0 N-succinimidyl ester (BzOSu) and benzoic acid-d5 N-succinimidyl ester (d-BzOSu) as light and heavy isotope reagents for stable isotope quantification for the comparative analysis of glycopeptides (Kurogochi and Amano, 2014).
Alternately, stable isotope dimethyl labeling has also been applied for glycopeptide MS1-based quantitation, which is based on the reaction of peptide primary amines with formaldehyde to generate a Schiff base that is reduced by the addition of cyanoborohydride to the mixture (Boersema et al., 2009). Xiao et al. characterized the differentially expressed N-glycosylation in HCC HepG2 cells relative to LO2 cells using isotopic dimethyl labeling and 2D LC–MS/MS analysis (Xiao and Tian, 2019). They also developed an N-glycopeptide search engine GPSeeker and the GPSeeker-centered quantitative structural N-glycoproteomics pipeline. In total, 5,405 and 1,081 intact N-glycopeptides with putative linkage structures were identified and quantified, where microheterogeneity with different differentially expression was observed on 183 out of 231 quantified N-glycosites (Xiao and Tian, 2019).
3.2.2. SILAC Labeling
The Mann group used the SILAC strategy to label glycoproteins with heavy labeled amino acids in cell culture to quantify the N-glycosylated secretome during breast cancer progression, where PNGase F was used to remove the N-glycan and the quantitation was done based on MS1 peak area integration (Boersema et al., 2013). The Hattori group used the SILAC strategy to monitor the over glycosylation of collagen in isolated skin fibroblast cells from a newborn Osterogenensis Imperfecta patient, where the identification of short glycan structure was done via variable modification searching and quantitation was done at the MS1 level (Taga et al., 2013).
The Parker group used SILAC to investigate the membrane glycoprotein upon TNF-alpha-induced insulin resistance in adipocytes (Parker et al., 2016). They used the DDA approach on an Orbitrap Fusion MS with Byonic software for identification and for MS1 level quantitation where a de-glycosylated peptide database was constructed beforehand. A total of 1,580 unique N-glycopeptides were identified which covered 332 unique peptide sequences on 154 proteins, of which 883 were quantified in two out of three biological replicate cell lines. A method for the stoichiometry analysis of glycosylation has been developed using SILAC labeling with PNGase F (Yang et al., 2017a).
Further work on SILAC for glycopeptides has not been pursued after these studies. A possible reason is that MS1 precursors of glycopeptides are already complex and SILAC would double the complexity, which would make the quantitation of glycopeptides even more difficult.
3.2.3. Labeling on the Glycan
Metabolic labeling of glycans with unnatural sugar analogs coupled with click chemistry have become a powerful method to study glycans and protein glycosylation (Xiao et al., 2019). The Bertozzi group has performed pioneering work on using unnatural sugar analogs to label glycoproteins (Breidenbach et al., 2010; Woo et al., 2015). A benchmark study, isotope-targeted glycoproteomics (IsoTaG), has been introduced for characterization of intact, metabolically labeled glycopeptides at the whole-proteome scale (Woo et al., 2017; Woo et al., 2015). In IsoTaG, metabolic labeling of the glycoproteome is combined with (i) chemical tagging and enrichment using an isotopic recoding affinity probe, (ii) directed tandem MS and (iii) mass-independent assignment of intact glycopeptides. They structurally assigned 32 N-glycopeptides and over 500 O-glycopeptides in human cancer cell lines (Woo et al., 2015). A later study that incorporated alkyne-sugars (Ac4GalNAz or Ac4ManNAz) rather than azide-sugars revealed 1375 N- and 2159 O-glycopeptides cross 15 cell lines. The effort was enabled by a new high-fidelity pattern-searching and glycopeptide validation algorithm termed IsoStamp v2.0, as well as by novel stable isotope probes (Woo et al., 2017).
3.2.4. Isotope-Tagged Cleavable Linker
Qin et al. reported a detailed O-glycopeptide analysis through an isotope-tagged cleavable linker (isoTCL) and quantitation using MaxQuant (Qin et al., 2018). With isotopic labeling of O-GlcNAc through bioorthogonal conjugation of affinity tags, the authors demonstrated the application of the isoTCL in mapping and quantification of O-GlcNAcylation sites in HeLa cells. To eliminate the harsh solution conditions associated with acid labile chemical probes, Li et al. developed a photocleavable biotin tag for O-GlcNAcylated glycopeptide quantification (Li et al., 2019a). This probe enabled selective tagging and isotopic labeling of O-GlcNAcylated proteins in one step from complex cellular mixtures. Cells were treated with ThiaMet-G where the cell lysates were chemoenzymatically tagged with GalNAz and further reacted with the 1H7 coded and 2D7 coded probes, respectively. The ‘light’ and ‘heavy’ samples were mixed, proteolytically digested, captured, and analyzed by LC-MS/MS. A mass shift of 7 Da between 1H7-coded and 2D7-coded peptides was observed, where the XICs were used for quantitative analysis. The authors identified 419 and 276 O-GlcNAcylation sites from sorafenib-sensitive and sorafenib-resistant HepG2 cells, respectively. A total of 94 and 150 O-GlcNAcylated peptides were quantified from at least two replicates, via CID and ETD, respectively (Liu et al., 2017b).
IV. MS/MS LEVEL BASED QUANTIFICATION
Besides the above widely used MS1 level-based quantification, there have been several methods that have been developed for MS2 (or MS/MS) level-based quantification of glycopeptides, aiming for better specificity and higher throughput.
4.1. Non-Targeted Quantification
There are three non-targeted MS/MS-based methods for glycopeptide quantitation: TMT/iTRAQ label-based approach, MSE approach and DIA approach. For the quantitation of non-glycosylated peptides, the main advantage of MS/MS-based methods over MS1-based methods is the higher precision. However, for the quantitation of glycopeptides, these approaches have some additional advantages.
4.1.1. TMT/iTRAQ Label-Based Approach
1). MS2/MS3 Based Quantitation
For TMT labeling on non-glycopeptides, both MS2 based and MS3 based quantitation can be used, where MS2 based methods have more quantifiable peptides and the MS3 based methods have more precise quantitation. For TMT labeling on glycopeptide, however, the Yoo group found that MS2 based quantitation for glycopeptides did not work (Lee et al., 2014). Their results showed that the MS2 HCD normalized collision energy (NCE) should be set high at 70% to obtain the signal from reporter ions where not many other fragments can be observed from glycopeptides, but at conventional NCE such as 25% no reporter ions could be found, indicating that the TMT bonds are much stronger than glycosidic bonds.
Instead, the Urlaub group showed that MS3 based quantitation worked well for TMT labeled glycopeptides (Fang et al., 2020). As shown in Figure 5, at low NCE MS2 HCD (~25%), TMT labeled glycopeptides produced B-series ions (oxonium ions) and Y-series ions with peptide backbones. The 10 most abundant fragments in the range of 700–2000 m/z were then co-selected and co-fragmentated with higher NCE MS3 HCD (35–40%) to generate peptide fragments and TMT reporter ions. Finally, fragments from MS2 and MS3 were combined as the fragments from the same glycopeptide precursor for identification. This strategy enhanced glycopeptide identification by 3.6 times compared to MS2 HCD alone. In this study, they identified and quantified over 5,300 unique glycopeptides of 528 glycoproteins with 855 glycosites in a lymphoma cell line. The MS3 strategy for glycopeptide quantitation not only used MS3 to generate reporter ion for more precise quantitation, but also facilitated the identification of glycopeptides. A minor drawback is that extra MS3 results in a longer cycle time, where low abundance glycopeptides may fail to be selected and identified.
Figure 5.
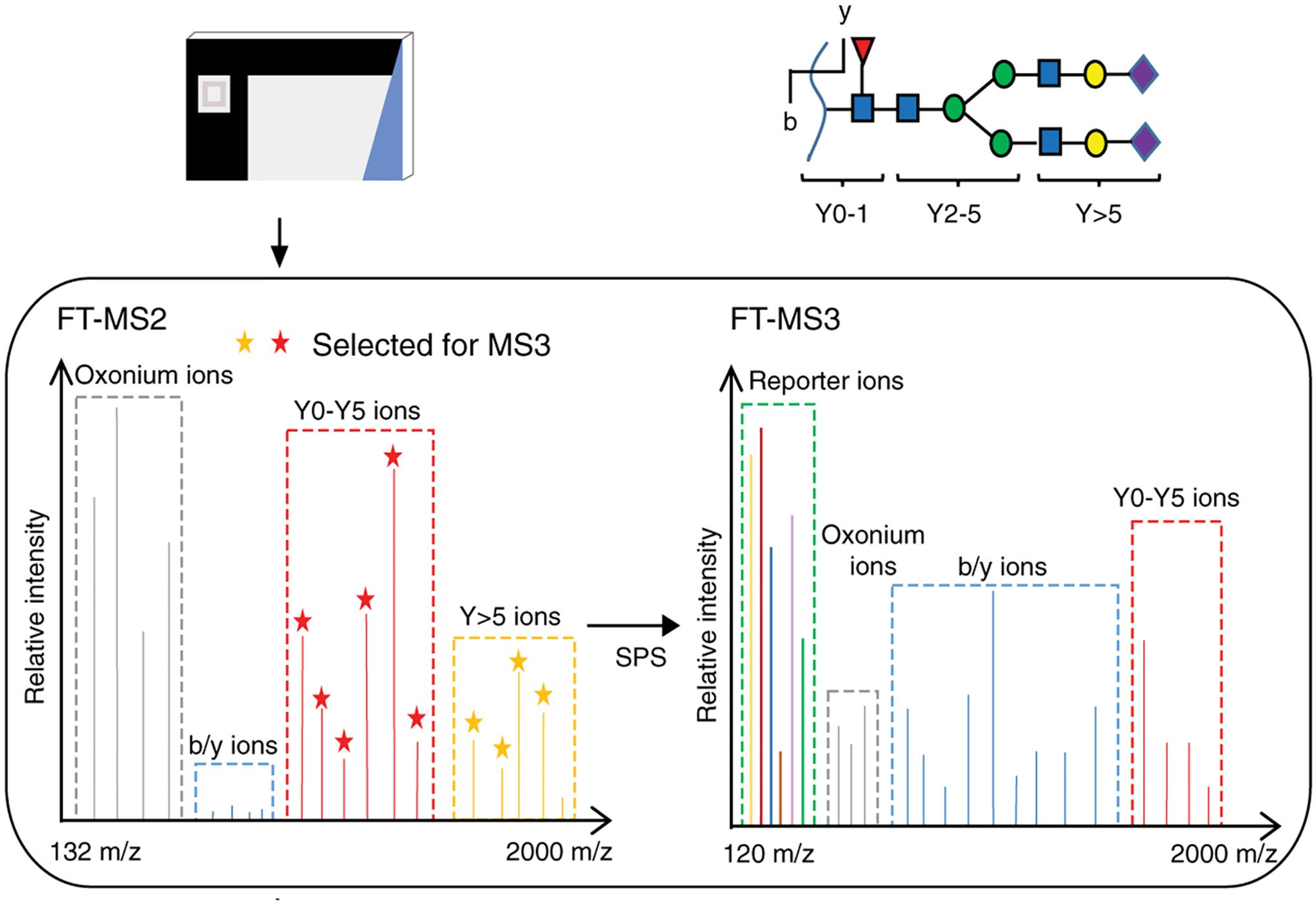
Quantitation of glycopeptides with TMT labeling by multi-notch MS3 acquisition on a Orbitrap Fusion Lumos mass spectrometer. Reprinted with permission from Fang et al. (2020). Open access.
2). Quantitation of Glycoproteins via Non-Glycosylated Peptides
The Lubman group used TMT and iTRAQ to quantitate glycoproteins via non-glycosylated peptides after lectin-based enrichment of glycoproteins. Their results showed that labeling at the protein level or at the peptide level provides comparable results where TMT labeling and iTRAQ labeling showed close efficacy (Nie et al., 2013). They further applied TMT labeling at the protein level for pancreatic cancer marker screening, where about 240 serum glycoproteins were quantified. A panel of α-1-antichymotrypsin (AACT), thrombospondin-1 (THBS1), and haptoglobin (HPT) outperformed CA 19−9 in distinguishing pancreatic cancer from normal controls (AUC = 0.95), diabetes (AUC = 0.89), cyst (AUC = 0.82), and chronic pancreatitis (AUC = 0.90) (Nie et al., 2014). The Zhang group used iTRAQ to label glycopeptides and quantified glycopeptides after enrichment and glycan truncation, where over 6000 proteins and about 500 glycoproteins from non-small cell lung carcinoma adenocarcinoma (ADC) and squamous cell carcinoma (SqCC) tissues were quantified (Yang et al., 2017a).
3). Quantitation of Truncated Glycopeptides
The Lubman group also tried to use iTRAQ to label glycopeptides with truncated glycan structure for serum marker screening of pancreatic cancer and HCC, where over 500 core-fucosylation sites from over 300 serum glycoproteins were quantified and the core-fucosylation level of some glycopeptides were found to be much higher in cancer states (Tan et al., 2015; Yin et al., 2015b).
4.1.2. MSE Approach
MSE is a data acquisition method used on the Q-TOF/TOF, which allows collision of all precursors and collects all generated fragments in each MS/MS scan. It has been successfully used for identification of glycopeptides (An et al., 2015; Montacir et al., 2018). However, glycopeptides with glycan structure microheterogeneity at one glycosylation site usually elute at similar retention times and share similar fragment profiles, where transition ions that are unique to specific glycopeptides, are difficult to observe. Therefore, although theoretically possible, MSE has so far not been successfully applied for glycopeptide quantitation.
4.1.3. DIA Approach
An improved approach of MSE strategy is DIA (Data Independent Acquisition) or SWATH (Sequential Window acquisition of all theoretical fragment ions) (the latter is a registered name by SCIEX, in the following text DIA is used), where, instead of collecting fragments from all precursors in one MS/MS as in MSE, the MS repeatedly cycles through 30–40 consecutive precursor isolation windows (for example, 25 m/z as isolation window), and collects all generated fragments of all precursors within that window, achieving almost complete peptide fragment-ion coverage for precursors in the entire mass range (Gillet et al., 2012) (Figure 6). This provides special challenges and benefits for glycopeptide quantitation using DIA.
Figure 6.
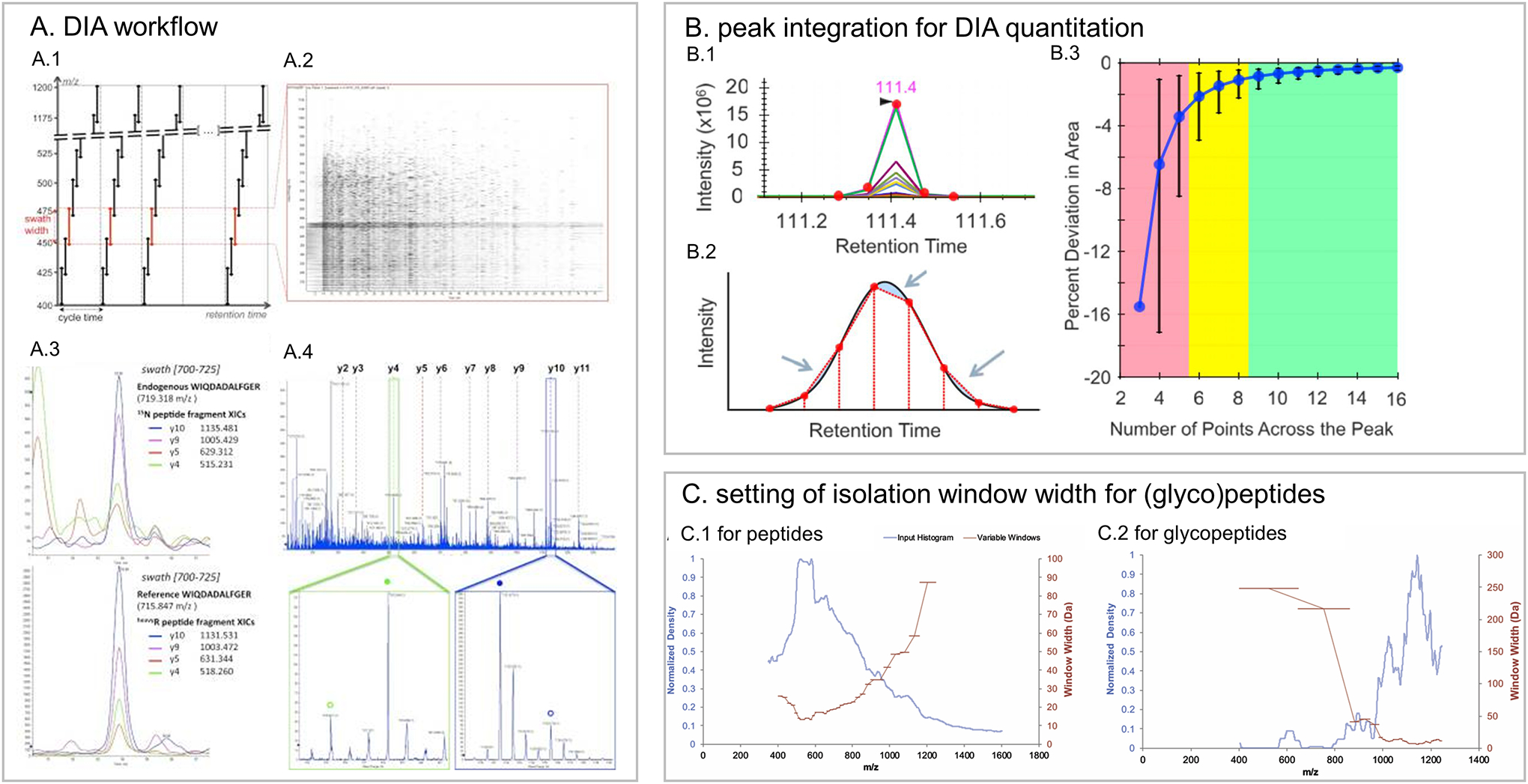
DIA approach. A. DIA workflow; B. peak integration for DIA quantitation; C. setting of isolation window width for peptides and glycopeptides. A Reprinted with permission from Gillet et al. (2012). Open access. B Reprinted with permission from Pino LK. et al. (2020). Open access. C Reprinted with permission from Zhou C. (2020) Copyright © 2020 Elsevier.
1). DIA for De-glycosylated Glycosite-containing Peptides
The Aebersold group first applied DIA for the quantitation of de-glycosylated glycosite-containing peptides using the TripleTOF 5600 (SCIEX) (Liu et al., 2013). They profiled the peptide backbone of glyocopeptides after glycan removal by PNGase F, where the glycan binding N (asparagine) is deamidated to D (aspartic acid) (mass difference +0.9840 Da, close to the mass of one neutron 1.0087 Da). The software originally designed for peptide quantitation failed to distinguish the two, where it outputs two precursor ions with both non-glycosylated precursor and the deaminated precursor.
The Schulz’s group tackled this problem by using various enzymes (Xu et al., 2015). They used glycosidase Endo H for yeast glycoproteins (yeast has high mannose glycan type which Endo H can cut, with one GlcNAc left on the peptide), leading to a much larger m/z difference between glycosylated and non-glycosylated peptides. As Endo H does not cut hybrid and complex glycosylation types, they then used AspN+PNGase F digestion on mammalian samples, where deglycosylated peptides with aspartic acid could be digested, resulting in a much larger difference between de-glycosylated peptide and non-glycosylated peptide. Alternatively, the Nesvizhskii group solved this problem by evaluating the isotope envelops of the two in DIA-Umpire software (Tsou et al., 2015).
2). DIA approach for Glycopeptide Identification and Quantitation
In proteomics studies, the ion library for interrogation of peptides in the DIA method is routinely built from peptide identification data obtained through DDA workflows, where DIA could not quantitate more peptides than DDA (Gillet et al., 2012; Rost et al., 2014). Glycopeptides which are highly branched and contain sialic acids, usually have a much weaker signal, and would not pass the intensity cut-off requirements for MS/MS by DDA. However, their precursor-transition-retention time information are indiscriminately collected by DIA, which may be potentially useful for the identification of glycopeptides. The difficult aspect is the use of this information for glycopeptide identification. Early studies used manual inspection to use this information.
The Goldman group first employed DIA for the analysis of glycopeptides with long glycan chains in 2016 (Sanda and Goldman, 2016). They used the TripleTOF 5600 with nano-LC for analysis of IgG glycopeptides from unenriched human plasma digests. The previously known precursor-Y ion (peptide+partial glycan structure) pairs generated by “soft” CID were used for quantitation, resulting in the quantitation of 26 glycoforms of IgG1, 22 glycoforms of IgG 2/3, and 19 glycoforms of IgG4 glycopeptides from 1 μg of human plasma sample (Sanda and Goldman, 2016), much more than previously discussed MS1 level based quantification where 8 glycoforms of Ig G subclasses were quantified (Selman et al., 2012). The Urlaub group deployed DIA for Ig M glycopeptide identification by manually inspecting the parent ion-Y ion pairs based on the results from DDA, where 31 glycopeptides were identified by DIA compared with 3 by DDA (Pan et al., 2017). The Packer group further developed this approach by manual inspection of the chromatogram of at least three Y ions of an unknown precursor: if those match with an identified glycopeptide in DDA, the unknown precursor is considered to have the same peptide backbone with the known glycopeptide, from which the glycan composition could be deducted (Lin et al., 2018). In this way, they identified 21 glycopeptides from human serum Ig G1 compared with 3 by DDA. Further optimization is needed for this strategy for the false positive error rate control and for full automated data-processing.
The Vakhrushev group further expanded this idea to identify low abundant glycopeptides in a complex O-glycoproteome system. They used a silico-boosted glycopeptide library for the identification of glycopeptides in DIA approach, defined as “glyco-DIA” (Figure 7) (Ye et al., 2019). They first used the DDA approach to generate the simplest spectral libraries for the core1 (GalNAc+Gal) and GalNAc O-glycoproteome of SimpleCell (which only has the two simple glycosylation types) and wild-type (WT) HepG2, then they added mono-sialic and di-sialic O-glycans to this spectral library in silico and generated 5 libraries with various retention time shifts, which contains 11,452 O-glycopeptides (2,076 O-glycoproteins). With this silico-boosted library they managed to identify and quantify almost twice the number of glycopeptides in DIA than in DDA, with over 2400 O-glycopeptides in lectin enriched HepG2 WT and over 250 O-glycopeptides in unfractionated serum. The idea of above studies is similar, all of which used the fragments of more abundant glycopeptides in DDA to identify other low abundant glycopeptides in DIA. This can be a promising strategy to make full use of the information collected by DIA for glycopeptide identification, for both types of O-glycosylation and for N-glycosylation.
Figure 7.
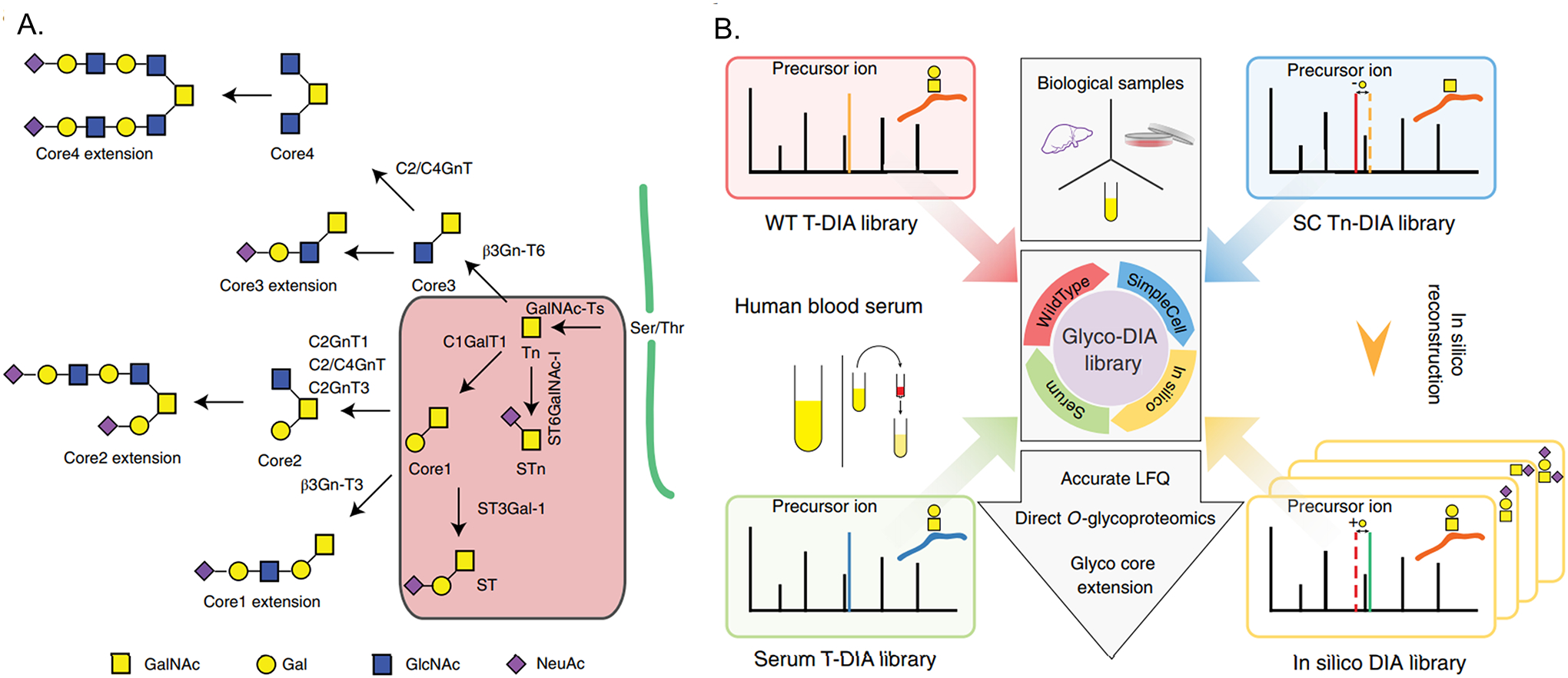
Library construction in DIA approach for O-glycopeptide quantitation using a silico-boosted glycopeptide library. (A) The biosythetic steps involved for core1-4 O-glycan structures; (B) The construction of Glyco-DIA libraries. Reprinted with permission from Ye ZL et al. (2019). Copyright © 2019 Springer Nature.
A spectrum-library-free approach for identification of unmodified peptides from DIA data has become possible (Searle et al., 2018; Tsou et al., 2015). Following those studies, the Schulz group developed DIALib, a software for spectrum-library-free glycopeptide quantitation (Phung et al., 2020). They attempted to generate a silico-theoretical library for glycopeptides without any information from DDA, with a broad precursor window, theoretical b, y, Y ion transitions, and broad retention time window. Yet, this was found not to be successful, with worse identification than the DDA library-based approach.
3). DIA Development to Improve Glycopeptides Quantitation with Glycan Micro-heterogeneity
Another challenge for glycopeptide analysis with the DIA approach is that glycopeptides with glycan micro-heterogeneity have close retention times, similar MS/MS spectra, and may have close precursor m/z. Fragments of the two glycopeptides from the same isolation window are difficult to assign to the correct precursor. A possible solution is to separate glycopeptides with glycan microheterogeneity into different windows by setting smaller isolation windows. Pan et al. (2017) discovered that two Y1 ions from two glycopeptides with a m/z difference of 5 Da could be resolved when a 4 Da isolation window was applied. The Schulz group developed variable window DIA for glycopeptide quantitation where a variable mass window was applied from 6.4 Da to 248.2 Da with 34 windows covering a 400–1250 m/z scan range, based on a roughly equal density of precursor ions in each window (Figure 6) (Zhou and Schulz, 2020). With variable window DIA, more glycopeptides from yeast cell wall proteins could be identified and more precisely quantified.
In summary, compared with DDA, the DIA strategy is appealing with better coverage of glycopeptides, less stochastic selection for precursor fragmentation, and resilient matrix suppression for glycopeptides with weaker signal. Still, DIA has several challenges. The data processing algorithm for the DIA strategy needs further optimization for automatic identification and quantitation. Co-eluted and co-isolated glycopeptides cannot be correctly identified or quantified, requiring mass spectrometers with higher scan speed for more isolation windows with variable window sizes. Although a full spectrum-library-free DIA quantitation strategy has not been successful, the silico-boosted construction of a spectrum library based on a partial DDA spectrum library for glycopeptide identification is a promising future of DIA quantitation for glycopeptides.
4.2. Targeted Quantification
Targeted quantitation strategies have not been well explored for intact glycopeptide analysis due to the complexity of glycopeptides composed of both peptide sequence and heterogeneous glycan structures. There are mainly two types of targeted quantitation strategies using mass spectrometers, Multiple Reaction Monitoring (MRM) and Parallel Reaction Monitoring (PRM) (Figure 8). SRM (Selected Reaction Monitoring), is very similar to MRM, but selects a single daughter transition ion for quantitation where MRM selects multiple daughter transitions. SRM and MRM are discussed together as MRM in the following text.
Figure 8.
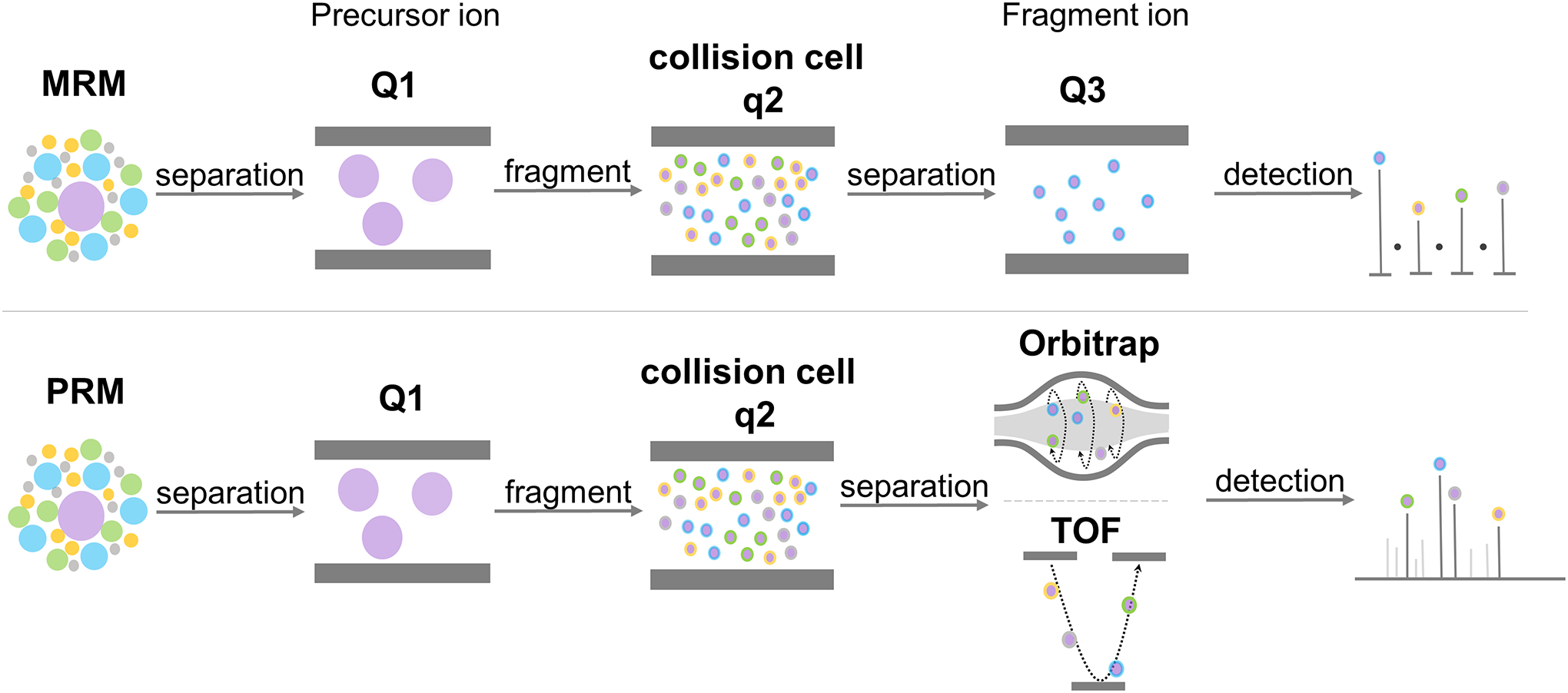
MRM and PRM strategies for targeted quantification.
4.2.1. MRM Approach
In MRM approach, the first quadruple selects the fixed parent ion, the second quadruple is used for collisions for fragmentation and then the third quadrupole or ion trap is used for daughter transition ions for quantitation (Figure 8). Prior to setting up an MRM method, the precursor ion-transition ion pair m/z, cone voltage for the parent ion and optimal CE for each transition ion should be available first, preferably with the retention time to reduce the cycle time and the optimal dwell time of each transition, among which, cone voltage for parent ion has a minor effect on the transition signal while CE has a major effect. The cycle time, which is the time spent monitoring all transitions in one duty cycle, depends on the dwell time of each transition ion and the number of transitions monitored at the same retention time window. The cycle time restricts the data points needed for the peak area integration. The information about precursor-transition ion pair m/z and the retention time are usually obtained from the standards or from a sample pre-run on a high-resolution MS; and the optimal cone voltage and CE for each transition were priorly obtained from the optimization process on a triple quadrupole mass analyzer. The quantitation of glycopeptide using MRM is similar to that of non-glycosylated peptides, but it has some specific difficulties: 1. Glycopeptides usually do not have standards (difficult or expensive to synthesize), therefore, the MRM required information needs to be obtained from a sample pre-run on a high-resolution MS; 2. The fragments of glycopeptides are biased with stronger non-specific B ions and weaker specific Y ions, the relative intensity of which is CE dependent, making the selection of transition ions trickier than non-glycosylated peptides; 3. Glycopeptides on C18 columns elute in clusters within a narrow retention time window, resulting in a longer cycle time and restricting the number of glycopeptides can be monitored at a time.
1). MRM for the Quantitation of Truncated Glycopeptide
In early investigations, glycopeptides were truncated first for MRM quantitation, which not only reduced the bias during fragmentation, but also enhanced the sensitivity significantly by increasing their ionization in MS and reducing the heterogeneity. The Qian group developed MRM for the quantitation of endoF3 truncated glyocopeptides (Zhao et al., 2011). Endo F digested glycopeptides cut the main glycan structure off the peptide backbone leaving the innermost GlcNAc with or without fucose, the fragmentation profile of which under high collision energy is similar to that of peptides. CE was optimized using the glycopeptide mixture and fragments with the highest intensity were used as transitions for quantitation. The Goldman group used a combination of endoF1–3 to expand the digestion specificity and applied this strategy for biomarker screening of liver fibrosis and cirrhosis (Ma et al., 2018). In this study, they managed to quantitate the core fucosylation of 22 N-glycopeptides derived from 17 proteins from 2 μL unenriched serum.
The Goldman group used an exoglycosidase-assisted MRM strategy for the quantitation of isolated haptoglobin T3 glycopeptides among HCC or cirrhosis patients, where neuraminidase or neuraminidase/galactosidase were used to truncate the glycopeptides (Sanda et al., 2013). They used an QSTAR Elite Q-TOF mass spectrometer (SCIEX) for the identification of T3 glycopeptides and used a capillary-LC/4000 Q-TRAP mass analyzer (SCIEX) for the precursor-oxonium ion pair quantitation. The CE for each oxonium ion was found linearly correlated to the precursor m/z after sialic acid removal but each oxonium ion had a different linear equation. T3 glycopeptides with 24 glycoforms were monitored with the intra-class correlation coefficient 0.90 (Sanda et al., 2013).
2). MRM for the Quantitation of Intact Glycopeptide
The Mechref group used the MRM strategy for intact glycopeptide quantitation for the first time in 2012 (Song et al., 2012). They used an LTQ Orbitrap Velos (Thermo Fisher) for the identification of glycopeptides from depleted serum proteins, based on which they obtained fragments profile of glycopeptides and the retention time of glycopeptides for MRM method on a nano-LC/TSQ Vantage (Thermo Fisher). They employed precursor ion scan-based MRM for parent ion selection, where precursor-oxonium ion pairs were used for identification and oxonium ions were used as transitions for quantitation. Due to the lack of glycopeptide standards, they used the same NCE of 40% and the same cone voltage for all rather than a specific NCE for each oxonium ion or an optimal cone voltage for each glycopeptide precursor ion. They also compared MRM method with or without retention time setting, where segmented MRM method with retention time setting enhanced peak intensity and reduced STD. In this pioneer study, they managed to quantitate 11 glycopeptides of 6 glycoproteins from 15 μL top seven high abundance protein depleted serum with STD <1.5% (Song et al., 2012).
The Lebrilla group applied MRM on an Agilent 6520 quadrupole time-of-flight (Q-TOF) and an Agilent 6490 triple quadrupole system on the quantitation of glycopeptides of glycoproteins from serum (Hong et al., 2013; Hong et al., 2015). Oxonium ions were used as transitions for quantitation. Precursor-transition ion pairs, the corresponding optimal CE and retention time information were obtained using digests from standard glycoproteins. The MRM approach was applied directly on 2 μL unenriched serum, where 26 glycopeptides from Ig G1–4 were quantitated in one study (Hong et al., 2013) and 64 glycopeptides of IgG, IgA, and IgM were quantitated in another study with high inter-day repeatability CV<15% and better sensitivity (>two times less amount) compared to immunoprecipitation enrichment method (Hong et al., 2015).
The Pompach group and Goldman group collaborated to further compare the MRM based quantitation with oxonium ions as transitions on a nano LC/6500 Q-TRAP mass spectrometer with MS1 level-based quantitation on a nano LC/12T solariX XR FT-ICR mass spectrometer for albumin depleted serum samples from HCC, colorectal cancer and liver metastasis of colorectal cancer patients (Darebna et al., 2017). They managed to quantitate 23 glycopeptides from 5 high abundance glycoproteins with 8 glycosylation sites. Their results showed that most quantitation results of the two approaches had a similar trend, but MRM quantitation based on oxonium ions resulted in a larger deviation, which is probably due to the non-specificity of oxonium ions.
In a later investigation with the MRM approach, the Goldman and Sanda applied “soft” fragmentation to generate more homogenous Y ions (intact peptide+ partial glycan) and oxonium ions, and used Y ions as transition ions for quantitation (Yuan et al., 2019). In this study, an Orbitrap Fusion Lumos was used for identification and a nano LC/6500 Q-trap (SCIEX) was used for quantitation. Glycoproteins from serum samples were treated with trypsin and neuraminidase digestion without depletion, where 12 fucosylated glycopeptides from 7 glycoprotein with 9 glycosylation sites were quantitated with CV in the range of 1.5%-23%. They showed that using Y ions showed higher specificity and improved the S/N ratio for over 2-times compared with using oxonium ions. Soft fragmentation is more advantageous even with the lower sensitivity (Yuan et al., 2019).
3). Advances to Enhance the Throughput of MRM Quantitation
All the above MRM studies worked on a single glycoprotein or a few high abundance glycoproteins in a mixture. Large scale glycopeptides quantitation is difficult for the MRM approach due to the lack of glycopeptide standards. Without the exact retention time information from glycopeptide standards, all precursor-transition ion pairs should be monitored throughout the entire LC run. More pairs would make the cycle time longer, resulting in fewer data points for peak integration. Ideally, the narrower the monitoring window, the more precursor-transition pairs could be monitored in a LC run. Conventional MRM could separate precursor-transitions into different retention time segments with several minutes per segment to partially alleviate this problem.
The later developed dynamic MRM mode (by Agilent) and scheduled MRM mode (by SCIEX) allow to further narrow down the monitoring window. dMRM (or sMRM) allows one to monitor each precursor-transition pair at a specific retention time with a self-defined delta time, and at the same time it allows to fix the cycle time, where the software can automatically calculate the dwell time for each transition. After a few trials on simple glycoprotein samples (Hong et al., 2013; Hong et al., 2015), the Lebrilla group employed the dMRM for large scale glycopeptide quantitation. Due to the lack of glycopeptide standards and the difference of nano-LC and UPLC chromatograms, it is difficult to know the exact retention time of the targeted glycopeptide. They then predicted the retention time of targeted glycopeptides based on the close correlation of peptide and glycopeptide elution time on the C18 column. They managed to simultaneously monitor the abundances of over 600 glycopeptides from 100 glycosylation sites across 50 serum glycoproteins in a 50 min LC run, some of which were identified by utilizing theoretically predicted ion products and presumed m/z values (Li et al., 2019b). Further development of Scheduled MRM Algorithm Pro (by SCIEX QTRAP 6500) allows the auto assignment of dwell time in sMRM mode to give more time to transitions with weaker signal and give less time to those with stronger signal, which would be very helpful for glycopeptide quantitation.
4.2.2. PRM Approach
In MRM study, although the “soft” fragmentation method improved specificity of glycopeptide quantitation by using Y ions as transitions compared with oxonium ions (Yuan et al., 2019), the lack of fragment information from the peptide chain may lead to potential false identification, especially for glycopeptides which have similar precursor m/z and share many fragment ions. PRM uses one quadruple to select the fixed parent ion with isolation window around 1 Da, another quadruple for collision, and scans all possible daughter transition ions on a high-resolution Orbitrap or TOF mass analyser (Gallien et al., 2012; Peterson et al., 2012; Schilling et al., 2015). Both MRM and PRM perform MS/MS scans on targeted precursors only and quantify based on transition ions (Figure 8). However, PRM is simpler to setup compared with MRM. In PRM, the precursor m/z information is required while neither the transition ion m/z nor the corresponding CE are needed. In PRM, the strongest transition of a precursor at a fixed CE after the data collection would be chosen for quantitation, whereas in MRM, CE should be optimized first for a chosen transition before the data collection starts. However, the maximum ion injection time or the AGC target for Orbitrap mass analyzers or the accumulation time for TOF mass analyzers should also be carefully defined (as discussed in the following subsection 4). The quantitation of glycopeptides using PRM is more complicated than peptides, due to its low ionization efficiency, the biased fragmentation pattern and the shortage of automatic data processing pipeline.
1). PRM for the Quantitation of Truncated Glycopeptide
Early investigations using the PRM approach started with endo H truncated glycopeptides. The Schulz group developed SWAT (Sequential Window Acquisition of Targeted fragment ions) on a TripleTOF 5600, which is very similar to PRM but with wider isolation window (4 Da rather than 1 Da), for the quantitation of endo H truncated glycopeptides from yeast (Yeo et al., 2016). Endo H for yeast is equivalent to Endo F for animal cells which cleaves N-glycans off the glycoprotein, leaving only the innermost GlcNAC with or without fucose. They compared SWAT, SWATH and SRM approaches and found that SWAT showed higher sensitivity and precision than SWATH, and higher specificity than SRM with robust performance (Yeo et al., 2016).
The Aebi group used isotope labeled amino acids to feed yeast and performed PRM quantitation for Endo H truncated glycopeptides on a Q Exactive (Thermo Fisher), where the H/L ratio of glycopeptides with different treatments were quantified using the top 4 transitions and most data showed CV <20% (Poljak et al., 2018).
2). PRM for the Quantitation of Intact Glycopeptide with Manual Data Processing Pipeline
The Goldman group combined “soft” CID with LC-MS PRM on a TripleTOF for the quantitation of glycopeptides of sex-hormone-binding globulin (SHBG) (Yuan et al., 2018). Before that, they identified the glycopeptides using DDA strategy on an Orbitrap Fusion Lumos, based on which they collected the information needed for PRM procedure, the m/z of interested glycopeptide and the corresponding retention time (Yuan et al., 2018). They purified SHBG protein from serum samples of HCC patients with various etiologies, aiming to find specific glycopeptides as marker for HCC with specific etiology. Specific Y-ions were used as transitions and the CE was optimized for each transition. The sum of selected Y-ions was used for quantitation where the glycopeptide intensity was normalized to the total glycopeptide intensity of the glycosylation site. In total, they quantified 15 intact glycopeptides from two glycosylation sites with an average SD around 8%. They found there was difference of N-glycosylation between healthy control and cirrhosis/HCC, but there was no difference between cirrhosis and HCC; one O-glycopeptide was able to distinguish healthy control from cirrhosis and cirrhosis from HCC (Yuan et al., 2018).
The Yoo group also used CID with LC-MS PRM on a TripleTOF for the quantitation of glycopeptides of α-fetoprotein (AFP) in one early study (Kim et al., 2018) and glycopeptides of α-fetoprotein (AFP), vitronectin (VTN), and α-1-antichymotrypsin (AACT) in another study (Kim et al., 2019). The information about precursor m/z are obtained from previous published studies. The target proteins were purified from serum samples of HCC patients. They further found that the removal of sialic acid enhanced the sensitivity. The sum of selected Y-ions was used for quantification and the CE was fixed for all glycopeptides with the same glycosylation site. The fucosylation ratio was evaluated among HCC patients. The CV of all evaluated 9 fucosylation ratios were around 20%, where the fucosylation ratio of a glycopeptide from AFP showed the biggest fold change among healthy control, cirrhosis and HCC. All these TOF based PRM studies were based on the MS/MS information from Orbitrap for identification and were based on manual identification of TOF results. However, the difference of the two MS systems makes the identification/quantitation inconvenient and error prone.
3). Development of PRM Methods with Simplified and Automatic Analysis Pipeline
The Lubman group recently developed a stepped HCD based LC-HCD-PRM-MS pipeline for glycopeptide quantification directly on an LC-Orbitrap, where an initial run of pooled sample using DDA approach was for identification and a later individual run of each sample using the PRM approach was for glycopeptide quantification, which was based on the precursor m/z and retention time information from the previous DDA run (Yin et al., 2020). All other settings of the two runs, including LC conditions and CE, are exactly the same, ensuring the retention time and the MS/MS fragmentation of targeted precursors are highly consistent to ensure the high specificity of the quantification method. pGlyco software was used for identification and Skyline software was used for quantification. Skyline software was originally designed for automated data processing of peptides quantified by the PRM strategy (MacLean et al., 2010). It was adopted for glycopeptide quantitation in this study by conjugating pGlyco software which can directly export the spectral library from pGlyco analysis of DDA results to Skyline software. This study showed that the quantification using the Y1 ion alone showed lower CV than that of oxonium ions or the sum of all detectable Y ions. The average SD of the integrated transition peak area improved to <5%. The combination of using one LC-MS system for glycopeptide identification and quantitation greatly simplifies the sample analysis process; and the automatic data processing pipeline from pGlyco software to Skyline software also enables the large scale automatic data analysis.
The same strategy was applied by this group to identify a panel of N-glycopeptides for early HCC detection (Lin et al., 2021). The workflow involved high-abundance protein depletion, glycopeptide enrichment, offline fractionation, and LC-stepped HCD-MS/MS analysis for differential determination of changes in glycopeptides in whole serum between patients with HCC versus cirrhosis. Using Byologic software, label-free quantitation was achieved where the relative abundance of a given glycopeptide was quantified at whole serum level. As a result, a panel of site-specific N-glycopeptides were identified as significantly increased in patients with NASH HCC compared to cirrhosis (p<0.05), from which a set of glycopeptide candidates were further selected for validation among 78 patient samples (40 cirrhosis, 28 early HCCs, and 10 late HCCs) using PRM-MS/MS analysis (Lin et al., 2021).
The Mechref group further applied this strategy to investigate the microheterogeneity of haptoglobin extracted from cirrhosis and HCC patient serum (Reyes CDG, 2021). They found many isomeric N-glycopeptides at the glycosylation site Asn207 of haptoglobin, where most of these showed significant differences among cirrhosis and early-stage HCC and especially one of the isomers increased the AUC from 0.85 (AFP alone) to 0.95.
It is essential to use PRM for glycopeptide quantification, as isomers of glycopeptides have been observed using either porous graphitized carbon separations (Zhu et al., 2020b), HILIC separations (Huang et al., 2016; Kozlik et al., 2018), capillary electrophoresis (Kammeijer et al., 2017) or C18 separations (Ji et al., 2019a), which may lead to incomplete quantification of the isomers in DDA strategy. Also, precursors within an isolation window of 1.2 Da may be a mixture of two or more glycopeptides, which may lead to wrong quantification in MRM strategy (Kim et al., 2019; Yin et al., 2020).
4). Orbitrap versus QTOF Instruments for PRM Analysis
Although TOF and Orbitrap are both high resolution mass detectors and both can be used for PRM study, they have some differences. 1. Collision cell. The collision cell of Triple-TOF and Orbitrap are similar, yet the unit and the absolute value of two are different, where Triple-TOF uses eV as the unit and Orbitrap uses % (collision energy eV = NCE (%) ×precursor(m/z) /500 × (charge fact). For peptide fragmentation, a few eV lower in Q-Exactive Focus Orbitrap gives almost exact fragmentation pattern in Q-TOF (Szabo et al., 2021) and yet the exact correlation is unknown. 2. S/N ratio. The Orbitrap has in general a much cleaner background and higher S/N ratio than TOF as in Orbitrap ions are collected in C-trap then injected to the Orbitrap for scanning whereas in the Q-TOF it is beam type scanning which collects all ion signals (Pino et al., 2020) .3. Ion accumulation time and cycle time. TOF accumulates signal at the detector while Orbitrap accumulates signal at the C-trap. For TOF, the cycle time equals to the signal collection time, and can be self-defined; for Orbitrap, the scan time (transient time) and the signal collection time (ion accumulation time) are separated, where the signal collection time is defined by AGC or maximum ion injection time (the larger one) and the scan time is defined by the resolution of the Orbitrap (the ion accumulation time should be similar as the scan time to avoid wasting time in the Orbitrap or during ion accumulation) (Pino et al., 2020).
The main limitation of PRM with the Orbitrap is its scan speed, as shown in Table 2. Both the triple quadrupole and TripleTOF have much higher scan rates than the Orbitrap. However, as glycopeptides normally have much weaker signals than the corresponding peptide, the accumulation time should be much longer to collect enough signal for identification. For example, when the accumulation time exceeds 50 ms, the maximum precursor number at a time of all three mass spectrometers become identical for TOF and Orbitrap analyzers. If the accumulation time needs to be 250 ms for enough signal intensity (which is the case for many low abundance glycopeptides), it will be difficult to monitor more than 8 top abundant glycopeptides at a time for either type of these mass spectrometers.
Table 2.
General comparison of triple quadrupole, TripleTOF and Orbitrap mass spectrometers
| Type | Resolution | Mass accuracy | Max. scan rate | MS/MS accumulation timed (ms) | Max. precursor No.e at a time | |
|---|---|---|---|---|---|---|
| triple quadrupolea | In-beam | 1–2K | low | 10,000 Da/s | 25;50;100;250 | 80;40;20;8 |
| TripleTOFb | In-beam | ≥30K | high | 100 scan/s | 25;50;100;250 | 80;40;20;8 |
| Orbitrapc | Trapping | ≥15K | high | 20 scan/s | 25;50;100;250 | 40;40;20;8 |
Waters Xevo TQD as an example;
Sciex TripleTOF 5600+ as an example;
Thermo Orbitrap Lumos as an example;
for triple quadrupole, accumulation time is dwell time; for TripleTOF, it is the scan time; for orbitrap, it is the ion injection time;
assuming that an elution window is 25.2 s and 12 data points are needed for peak area integration (25.2/12=2.1 s/cycle: 100 ms for precursor scan in each cycle and 2 s for all MS/MS scans in one cycle); for triple quadrupole the number of precursor-transition pairs is used.
4.2.3. Possible Future Developments for MRM and PRM Approaches
1). Optimization of Precursor Selectivity in MRM
For the optimization of precursor selectivity in the MRM approach, the Domon group developed an intelligent MRM for instrument control on a triple quadrupole mass spectrometer (TSQ Vantage, Thermo Scientific) with nanoLC (Kiyonami et al., 2011). They used the specificity of two primary transitions to quantify and intelligently trigger another 6 transitions for confirmation of target peptides, where the detection window is set to 4 min. The detection limit is ten attomole, with the simultaneous qualitative and quantitative analysis of up to 757 peptides in a 60 min LC-MS run. The CV of two primary transitions is mostly <5% whereas the secondary transitions ensure the specificity of precursors (Kiyonami et al., 2011). This feature is MRM triggered MRM and is now also available in Scheduled MRM Algorithm Pro by SCIEX. It can be useful for quantification of glycopeptides which elute at similar retention and share the same peptide backbone.
2). Optimization of PRM Strategy
a. Optimization of Ion Accumulation Time to Enhance the sensitivity
For the optimization of precursor accumulation time in PRM approach, the Domon group further developed an internal standard trigged IS-PRM for peptide quantitation, which can enhance peak integration of transition ions by dynamically assigning the ion fill time for isotope labeled standards and for endogenous peptides, resulting in better quantitation for peptides (Gallien et al., 2015). This strategy should be able to be applied on glycopeptides as well to improve the quantitation of glycopeptides.
To enhance the profile of MS spectrum, the Mann group developed the boxcar MS1 scanning method, which greatly enhances the acquisition of ions with low abundance (Meier et al., 2018). An improved MS1 profile would be also very helpful for selecting glycopeptides for fragmentation. And if this strategy could be also applied on MS2 fragment profiling, it would greatly enhance the identification and quantitation of glycopeptides.
b. Dynamic Control of Orbitrap Scan Speed to Reduce Cycle Time
To enhance the turnover of PRM approach in proteomics study, heavy isotope labeled standard peptides were used for automatic triggering PRM scan, where a low resolution fast pre-scan is used for checking the existence of the target peptides and if detected, a high-resolution scan will be performed, resulting in a narrower retention time window for the target peptide and enhanced overall throughput. The scan mode and the heavy labeled peptide kits are commercially available by SureQuant from Thermo Fisher. This strategy can also be applied for targeted glycopeptides by using the heavy labeled glycopeptides.
V. SPIKE-IN STANDARDS FOR GLYCOPEPTIDE QUANTIFICATION
Due to the shortage of glycopeptide standard, most glycopeptide quantitation studies either use relative labeling approach to compare the abundance of one glycoform with between disease states or normalize the abundance of one glycoform against another glycoform or the sum of all glycoforms of the same sample prior the comparison between disease states. There are a few investigations which developed non-glycosylated peptide as the internal standard for glycopeptide quantitation to reduce the run-to-run variation from sample preparation so that glycopeptide amounts between runs could be compared without labeling.
Due to the complication of glycosylation, the relative comparison at the glycopeptide level is not enough to elucidate the real function of glycosylation in the biological and pathological process. For example, the Ruhaak group found the individual difference of relative glycopeptide amount after normalization against the absolute IgG amount was much smaller than the large variation of absolute IgG amounts, indicating that protein concentration contributes considerably to the variation in glycopeptide abundance among individuals (Hong et al., 2013). In another study, the Parker group discovered that in TNF-alpha-induced insulin resistance of adipose there were limited changes in the relative stoichiometry of N-linked glycans after normalizing against the protein level and around 30% of the glycopeptides showed site-specific glycosylation change after normalizing against the protein level (Parker et al., 2016).
These studies suggested that it is essential to evaluate all three levels for glycosylation assessment: protein level, glycoform level and occupancy level. It would be more straightforward if a calibration curve of glycopeptide could be generated so that the absolute quantitation of glycopeptides could be monitored. In combination with the absolute quantitation at peptide level, which is readily available, all three levels could be compared. Recently, there are also some investigations which developed glycosylated peptide standard to generate calibration curves so that the absolute quantity of glycopeptides could be monitored.
5.1. Non-Glycosylated Peptide as the Standard
The internal standard for glycopeptide quantitation was achieved using heavy labeled non-glycosylated peptide. The Zhang group spiked in heavy labeled de-glycosylated peptides into enriched and PNGase F treated serum samples to quantitate the de-glycosylated peptides among prostate cancer patients (Thomas et al., 2015). The coefficient of variation was a bit high around 20%, possibly because the heavy labeled de-glycosylated peptide was spiked in after the glycopeptide enrichment and PNGase F treatment.
The Goldman group spiked the heavy labeled non-glycosylated peptide which elutes close to the glycopeptides as the standard normalization and the matrix effect was minimized by removing the terminal sialic acid to allow the coelution of glycopeptides with the same peptide backbone (so that the same matrix was shared by all glycopeptides), where the intra-class correlation coefficient was 90% (Sanda et al., 2013).
5.2. Glycoprotein Digest as the Standard
The absolute quantitation of glycopeptides was initially achieved using the calibration curve of non-glycosylated peptide. The Lebrilla group (Huang et al., 2017) and the Ruhaak group (Miyamoto et al., 2018) used external standard glycoprotein digests to generate standard curves of glycopeptide and non-glycopeptides, then they quantified the glycopeptide difference from various samples by normalizing ratios relative to the relevant non-glycopeptides, where the absolute quantity of peptides could be measured, and the relative quantity of glycopeptides could be compared. The relative standard deviation was generally below 10%.
5.3. Glycopeptide as the Standard
5.3.1. Glycopeptides with Short Glycan Chain as the Standard
The Yoo group used commercially chemically synthesized glycopeptides with GlcNAc to quantitate immunoprecipitated and endo F2 truncated AFP (endo F2 cuts biantennary N-glycan structures, leaving GlcNAc with or without core-fucose) (Kim et al., 2020). They used a TripleTOF 6600 and nano-ACQUITY for PRM quantitation. The LOD and LOQ can approach 10 and 20 attomole, with a limit of detection (LOD) of <2 ng/ml (the clinical cutoff is 20 ng/ml). The relative percentage of fucosylated AFP has an AUC of 0.962, much higher than the AFP AUC of 0.628.
The Larson group chemoenzymatically synthesized an isotopically labeled glycopeptide Aβ1–15 with Galβ+GalNAc for the quantitation of the same glycopeptide in cerebral spinal fluid after NeuAC removal with the PRM approach using a Q Exactive (Nilsson et al., 2019). They used a wide isolation window to include both spiked glycopeptides and endogenous glycopeptides for quantitation, with CV around 5–11%.
The Liang group used commercially chemoenzymatically synthesized glycopeptides with GlcNAc+dHex to quantitate all glycopeptides with the same glycopeptide backbone and different glycan structures of serum Ig G1 (Cao et al., 2020). They used a Triple Quad™ 5500 and an ACQUITY UPLC with Click maltose HILIC column and Y ions as the transition ions. The drawback of using this glycopeptide as the standard is that the glycopeptides with the same peptide backbone have a wider elution window over several minutes, where the matrix effect may vary.
5.3.2. Glycopeptides with Long Glycan Chain as the Standard
The Nishimura group used yolk glycopeptide to quantitate all mouse serum glycoprotein digested glycopeptides using a Q-trap (Kurogochi et al., 2010). They were able to quantitate about 100 glycoproteins from mouse serum samples with CV<20%. They later chemoenzymatically synthesized one biantennary glycopeptide with 4GlcNAC+5Hex and another bisected glycopeptide with 5GlcNAC+5Hex to generate the calibration curves for the quantitation of the two glycopeptides of IgG1 monoclonal antibody drugs (Hammura et al., 2018). For bisected glycopeptides, oxonium ion was used as a transition ion and for biantennary glycopeptide, Y ion was used as transition ion. The quantitation limit can reach to 200 fmol (Hammura et al., 2018).
The Li group further developed chemoenzymatic method and synthesized 15 isotopic labeled Ig G glycopeptides with 5 glycoforms (4GlcNAC+4Hex+dHex, 4GlcNAC+3Hex+dHex, 4GlcNAC+5Hex+dHex, 4GlcNAC+4Hex+dHex+NeuAC, 4GlcNAC+5Hex+dHex+NeuAC), where Ig G glycopeptides in sera of colon cancer patients and healthy controls were quantified using PRM strategy at 1 nmol/mL level with most RSD <10% (Wang et al., 2021).
The synthesis process of intact glycopeptide standard is still challenging, yet it is the compulsory for the absolute quantitation of glycopeptide with lower detection limit and with better reproducibility.
VI. CONCLUSION AND PERSPECITVE
Recent advances in MS-based quantitative glycoproteomic techniques and strategies have demonstrated significant contributions to glycopeptide analysis, in either intact or truncated forms, and quantitation assessment of glycopeptides in protein level, glycoform level or occupancy level. The MS1 level- and MS/MS level-based quantitative methods exhibit their own strengths in sensitivity, precision, throughput, sample preparation, MS acquisition method, and data interpretation. The strengths and weaknesses of different quantitative methods are summarized in Table 3. Among these methods discussed above, the targeted MRM approach provides the highest sensitivity, high throughput, and simplicity of sample preparation and data interpretation; the targeted PRM approach and non-targeted TMT/iTRAQ label-based approach permit superior precision, however the PRM data bring challenges in the throughput and the labelling step increases the complexity of sample preparation; the DIA approach has the best overall performance in sensitivity, precision, and sample preparation except the challenges in data interpretation; the MS1-based labelling approaches allow simplicity of MS acquisition and moderate precision but low sensitivity; finally, the MS1-based label-free approach, with the advantages in simplicity of sample preparation, MS acquisition and data interpretation, although with low sensitivity and precision in quantitation, is widely used for mapping the landscape of glycopeptides in biological samples, allowing for the largest number of glycopeptides to be identified and semi-quantified.
Table 3.
The strengths and weaknesses of various glycopeptide quantitation methodsa.
| Glycopeptide Quantitation Method | Sensitivity | Precision | Throughput | Sample Prep Simplicity | MS Acquisition Method Simplicity | Data Simplicity | ||
|---|---|---|---|---|---|---|---|---|
| MS1 Level | Label Free | - | ++ | ++ | ++++ | +++++ | +++++ | ++++ |
| Label- Based | Label on the Peptide | ++ | +++ | ++++ | + | +++++ | +++ | |
| SILAC Labeling | ++ | +++ | ++++ | + | +++++ | +++ | ||
| Label on the Glycan | ++ | +++ | ++++ | + | +++++ | +++ | ||
| Isotope-Tagged Cleavable Linker | ++ | +++ | ++++ | + | +++++ | +++ | ||
| MS/MS Level | Non-Targeted | TMT/iTRAQ Label-Based Approach | ++ | +++++ | +++ | + | +++++ | ++++ |
| MSE Approach | + | + | + | +++++ | +++++ | + | ||
| DIA Approach | ++++ | ++++ | +++++ | +++++ | ++++ | + | ||
| Targeted | MRM Approach | +++++ | ++ | +++++ | +++++ | ++ | +++++ | |
| PRM Approach | ++++ | +++++ | +++ | +++++ | +++ | ++ | ||
More “+” marks indicate better performance.
In the future, we expect to see a continuous effort to improve glycopeptide enrichment and LC separation of glycopeptides, to enhance their signal response in the mass spectrometer, to increase the overall throughput for glycopeptide quantitation, and the synthesis of glycopeptide standards for absolute quantification. The enrichment method with high recovery rate and without losing glycan information needs further study. The techniques for developing robust nanoflow or microflow LC columns with various fillings and robust performance is a future direction. Some of the MS analysis methods and data processing algorithms developed for peptide identification and quantification can be applied to glycopeptide analysis, such as the building of a silico-boosted glycopeptide library for the DIA approach, the MRM triggered MRM to increase the specificity in quantitation, internal standard triggered PRM for precise elution window monitoring and for automatic ion filling time adjustment.
With the improvement of in-depth glycoproteomic analysis, researchers will also seek methodologies/assays for accurate quantitation of glycopeptides among small amounts of samples, which could be integrated into clinical translational research to fulfil an unmet clinical need for new biomarkers. These methodologies/assays will be developed to improve both the reproducibility and LOQ for small-volume samples. With efficient glycopeptide enrichment, glycopeptide standards, and/or labeling strategies, quantitative assays are expected to provide sensitive quantitation of even low-abundance glycopeptides to uncover disease-relevant glycopeptides. In the meantime, computational software and tools to facilitate data interpretation and confident glycopeptide assignment will be a continuous need. Quantitative glycoproteomics, with label-free and label-based strategies, will remain a significant area in the next decade, with challenges to be overcome and applications to be explored in biomedical translational research.
ACKNOWLEDGEMENTS
This work was funded by the National Natural Science Foundation of China under grant no. 81601828 (H.Y) and the U.S. National Cancer Institute under grant no. R50 CA221808 (J.Z). We thank Professor David M. Lubman for his supervision and his expert guidance and encouragement in this field.
ABBREVIATIONS
- CE
collision energy
- CID
collision-induced dissociation
- DDA
data-dependent MS/MS acquisition
- dHex
fucose
- DIA
data-independent MS/MS acquisition
- ESI-MS
electrospray ionization mass spectrometry
- ETD
electron transfer dissociation
- EThcD
electron-transfer/higher-energy collision dissociation
- Fuc
fucose
- Gal
galactose
- GalNAc
N-acetylgalactosamine
- GlcNAc
N-acetylglucosamine
- HCD
high-energy collision dissociation
- Hex
hexose
- HexNAc
hexosamine
- HILIC
hydrophilic interaction liquid chromatography
- HPLC
high-performance liquid chromatography
- IgG
immunoglobin
- iTRAQ
isobaric tags for relative and absolute quantitation
- Man
mannose
- MRM
multiple reaction monitoring
- MS
mass spectrometry
- MS1
precursor mass spectrometry scan
- MS2
tandem mass spectrometry scan
- NeuAC
sialic acid
- PRM
parallel reaction monitoring
- QQQ
triple quadrupole mass spectrometer
- TMT
tandem mass tag
- TOF
time of flight mass spectrometer
Biographies

Haidi Yin works in the MS core facility of Shenzhen Bay Laboratory (SZBL). She obtained her Ph.D degree in Peking University and did postdoctoral training with Professor David Lubman in Michigan University. She then worked as a scientific officer in the Hong Kong Polytechnic University from 2015–2021. She maintains mass spectrometers and develops proteomics/metabolomics analysis methods. Her research interests focus on quantitative glycoproteomics and glycomics, especially for biomarker screening and for exploring the regulation mechanism of glycosylation process. She has published 22 peer-reviewed papers and finished one project supported by the Natural Science Foundation of China.

Jianhui Zhu, PhD, is a Lead Research Scientist in the Laboratory of Cancer Proteomics in the Department of Surgery at the University of Michigan. Dr. Zhu obtained her PhD degree from the Nanjing University, China under the direction of Dr. Zijian Guo. She started her postdoc position under the mentorship of Dr. David M. Lubman at the University of Michigan in 2010 and subsequently worked as a senior scientist and a lead research scientist. Her research interest includes the development of mass spectrometry-based methods to identify cancer-related protein markers based on cancer stem cells, exosomes, patient serum and tissues for early cancer detection, monitoring, and prognosis. She has published 55 peer-reviewed research articles. Dr. Zhu received an NCI Research Specialist Award in 2018 to support her work on discovery of new glyco-markers based on the presence of characteristic glycans/glycopeptides in patient serum for early detection of hepatocellular carcinoma.
REFERENCE
- Agrawal P, Fontanals-Cirera B, Sokolova E, Jacob S, Vaiana CA, Argibay D, Davalos V, McDermott M, Nayak S, Darvishian F, et al. (2017). A Systems Biology Approach Identifies FUT8 as a Driver of Melanoma Metastasis. Cancer Cell 31, 804–819 e807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcedo KP, Guerrero A, Basrur V, Fu D, Richardson ML, McLane JS, Tsou CC, Nesvizhskii AI, Welling TH, Lebrilla CB, et al. (2019). Tumor-Selective Altered Glycosylation and Functional Attenuation of CD73 in Human Hepatocellular Carcinoma. Hepatol Commun 3, 1400–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley WR, Mechref Y, and Novotny MV (2009). Use of activated graphitized carbon chips for liquid chromatography/mass spectrometric and tandem mass spectrometric analysis of tryptic glycopeptides. Rapid Commun Mass Sp 23, 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An HJ, Froehlich JW, and Lebrilla CB (2009). Determination of glycosylation sites and site-specific heterogeneity in glycoproteins. Curr Opin Chem Biol 13, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An YM, McCullers JA, Alymova I, Parsons LM, and Cipollo JF (2015). Glycosylation Analysis of Engineered H3N2 Influenza A Virus Hemagglutinins with Sequentially Added Historically Relevant Glycosylation Sites. Journal of Proteome Research 14, 3957–3969. [DOI] [PubMed] [Google Scholar]
- Argade S, Chen T, Shaw T, Berecz Z, Shi W, Choudhury B, Parsons CL, and Sur RL (2015). An evaluation of Tamm-Horsfall protein glycans in kidney stone formers using novel techniques. Urolithiasis 43, 303–312. [DOI] [PubMed] [Google Scholar]
- Ashwood C, Pratt B, MacLean BX, Gundry RL, and Packer NH (2019). Standardization of PGC-LC-MS-based glycomics for sample specific glycotyping. Analyst 144, 3601–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenfaenger M, Moritz M, and Meyer B (2019). Quantitation of Glycopeptides by ESI/MS - size of the peptide part strongly affects the relative proportions and allows discovery of new glycan compositions of Ceruloplasmin. Glycoconj J 36, 13–26. [DOI] [PubMed] [Google Scholar]
- Bahr U, Pfenninger A, Karas M, and Stahl B (1997). High sensitivity analysis of neutral underivatized oligosaccharides by nanoelectrospray mass spectrometry. Analytical Chemistry 69, 4530–4535. [DOI] [PubMed] [Google Scholar]
- Bedair M, and Oleschuk RD (2006). Lectin affinity chromatography using porous polymer monolith assisted nanoelectrospray MS/MS. Analyst 131, 1316–1321. [DOI] [PubMed] [Google Scholar]
- Bern M, Kil YJ, and Becker C (2012). Byonic: advanced peptide and protein identification software. Curr Protoc Bioinformatics Chapter 13, Unit13 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersema PJ, Geiger T, Wisniewski JR, and Mann M (2013). Quantification of the N-glycosylated Secretome by Super-SILAC During Breast Cancer Progression and in Human Blood Samples. Molecular & Cellular Proteomics 12, 158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, and Heck AJ (2009). Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4, 484–494. [DOI] [PubMed] [Google Scholar]
- Breidenbach MA, Gallagher JE, King DS, Smart BP, Wu P, and Bertozzi CR (2010). Targeted metabolic labeling of yeast N-glycans with unnatural sugars. Proc Natl Acad Sci U S A 107, 3988–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camperi J, Combes A, Fournier T, Pichon V, and Delaunay N (2020). Analysis of the human chorionic gonadotropin protein at the intact level by HILIC-MS and comparison with RPLC-MS. Anal Bioanal Chem 412, 4423–4432. [DOI] [PubMed] [Google Scholar]
- Cao C, Yu L, Fu D, Yuan J, and Liang X (2020). Absolute quantitation of high abundant Fc-glycopeptides from human serum IgG-1. Anal Chim Acta 1102, 130–139. [DOI] [PubMed] [Google Scholar]
- Cao W, Liu M, Kong S, Wu M, Zhang Y, and Yang P (2021a). Recent advances in software tools for more generic and precise intact glycopeptide analysis. Mol Cell Proteomics, 100060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Cao Z, Shao Y, Liu C, Yan G, Meng X, Zhang L, Chen C, Huang G, Shu H, et al. (2021b). Analysis of Serum Paraoxonase 1 Using Mass Spectrometry and Lectin Immunoassay in Patients With Alpha-Fetoprotein Negative Hepatocellular Carcinoma. Front Oncol 11, 651421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capelo JL, Carreira RJ, Fernandes L, Lodeiro C, Santos HM, and Simal-Gandara J (2010). Latest developments in sample treatment for 18O-isotopic labeling for proteomics mass spectrometry-based approaches: a critical review. Talanta 80, 1476–1486. [DOI] [PubMed] [Google Scholar]
- Chang D, and Zaia J (2021). Methods to improve quantitative glycoprotein coverage from bottom-up LC-MS data. Mass Spectrom Rev. [DOI] [PubMed] [Google Scholar]
- Chen M, Lu Y, Ma Q, Guo L, and Feng YQ (2009). Boronate affinity monolith for highly selective enrichment of glycopeptides and glycoproteins. Analyst 134, 2158–2164. [DOI] [PubMed] [Google Scholar]
- Chen SY, Dong MM, Yang GL, Zhou YY, Clark DJ, Lih TM, Schnaubelt M, Liu ZC, and Zhang H (2020). Glycans, Glycosite, and Intact Glycopeptide Analysis of N-Linked Glycoproteins Using Liquid Handling Systems. Analytical Chemistry 92, 1680–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Smeekens JM, and Wu R (2014). A universal chemical enrichment method for mapping the yeast N-glycoproteome by mass spectrometry (MS). Mol Cell Proteomics 13, 1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Yu Q, Hao L, Liu F, Johnson J, Tian Z, Kao WJ, Xu W, and Li L (2018). Site-specific characterization and quantitation of N-glycopeptides in PKM2 knockout breast cancer cells using DiLeu isobaric tags enabled by electron-transfer/higher-energy collision dissociation (EThcD). Analyst 143, 2508–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Yu Q, Yu Q, Johnson J, Shipman R, Zhong X, Huang J, Asthana S, Carlsson C, Okonkwo O, et al. (2021). In-depth site-specific analysis of N-glycoproteome in human cerebrospinal fluid (CSF) and glycosylation landscape changes in Alzheimer’s disease (AD). Mol Cell Proteomics, 100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipollo JF, and Parsons LM (2020). Glycomics and glycoproteomics of viruses: Mass spectrometry applications and insights toward structure-function relationships. Mass Spectrom Rev 39, 371–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings RD, and Pierce JM (2014). The challenge and promise of glycomics. Chem Biol 21, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darebna P, Novak P, Kucera R, Topolcan O, Sanda M, Goldman R, and Pompach P (2017). Changes in the expression of N- and O-glycopeptides in patients with colorectal cancer and hepatocellular carcinoma quantified by full-MS scan FT-ICR and multiple reaction monitoring. J Proteomics 153, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delafield DG, and Li L (2021). Recent Advances in Analytical Approaches for Glycan and Glycopeptide Quantitation. Mol Cell Proteomics 20, 100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon B, and Aebersold R (2010). Options and considerations when selecting a quantitative proteomics strategy. Nat Biotechnol 28, 710–721. [DOI] [PubMed] [Google Scholar]
- Fang J, Sheng X, Bao H, Zhang Y, and Lu H (2021). Comparative analysis of intact glycopeptides from mannose receptor among different breast cancer subtypes using mass spectrometry. Talanta 223, 121676. [DOI] [PubMed] [Google Scholar]
- Fang P, Ji YL, Silbern I, Doebele C, Ninov M, Lenz C, Oellerich T, Pan KT, and Urlaub H (2020). A streamlined pipeline for multiplexed quantitative site-specific N-glycoproteomics. Nature Communications 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Yang N, Pennathur S, Goodison S, and Lubman DM (2009). Enrichment of Glycoproteins Using Nanoscale Chelating Concanavalin A Monolithic Capillary Chromatography (vol 81, pg 3776, 2009). Analytical Chemistry 81, 8654–8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuki K, and Toyo’oka T (2017). Retention of glycopeptides analyzed using hydrophilic interaction chromatography is influenced by charge and carbon chain length of ion-pairing reagent for mobile phase. Biomed Chromatogr 31. [DOI] [PubMed] [Google Scholar]
- Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, and Domon B (2012). Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics 11, 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallien S, Kim SY, and Domon B (2015). Large-Scale Targeted Proteomics Using Internal Standard Triggered-Parallel Reaction Monitoring (IS-PRM). Mol Cell Proteomics 14, 1630–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam S, Banazadeh A, Cho BG, Goli M, Zhong J, and Mechref Y (2021). Mesoporous Graphitized Carbon Column for Efficient Isomeric Separation of Permethylated Glycans. Anal Chem 93, 5061–5070. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, and Aebersold R (2012). Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Molecular & Cellular Proteomics 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover MS, Yu Q, Chen Z, Shi X, Kent KC, and Li L (2018). Characterization of intact sialylated glycopeptides and phosphorylated glycopeptides from IMAC enriched samples by EThcD fragmentation: Toward combining phosphoproteomics and glycoproteomics. International Journal of Mass Spectrometry 427, 35–42. [Google Scholar]
- Hammura K, Ishikawa A, Kumar HVR, Miyoshi R, Yokoi Y, Tanaka M, Hinou H, and Nishimura SI (2018). Synthetic Glycopeptides Allow for the Quantitation of Scarce Nonfucosylated IgG Fc N-Glycans of Therapeutic Antibody. Acs Med Chem Lett 9, 889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong QT, Lebrilla CB, Miyamoto S, and Ruhaak LR (2013). Absolute Quantitation of Immunoglobulin G and Its Glycoforms Using Multiple Reaction Monitoring. Analytical Chemistry 85, 8585–8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong QT, Ruhaak LR, Stroble C, Parker E, Huang JC, Maverakis E, and Lebrilla CB (2015). A Method for Comprehensive Glycosite-Mapping and Direct Quantitation of Serum Glycoproteins. Journal of Proteome Research 14, 5179–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang BY, Yang CK, Liu CP, and Liu CY (2014). Stationary phases for the enrichment of glycoproteins and glycopeptides. Electrophoresis 35, 2091–2107. [DOI] [PubMed] [Google Scholar]
- Huang JC, Kailemia MJ, Goonatilleke E, Parker EA, Hong QT, Sabia R, Smilowitz JT, German JB, and Lebrilla CB (2017). Quantitation of human milk proteins and their glycoforms using multiple reaction monitoring (MRM). Anal Bioanal Chem 409, 589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Nie Y, Boyes B, and Orlando R (2016). Resolving Isomeric Glycopeptide Glycoforms with Hydrophilic Interaction Chromatography (HILIC). J Biomol Tech 27, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen BC, Falck D, de Haan N, Hipgrave Ederveen AL, Razdorov G, Lauc G, and Wuhrer M (2016). LaCyTools: A Targeted Liquid Chromatography-Mass Spectrometry Data Processing Package for Relative Quantitation of Glycopeptides. J Proteome Res 15, 2198–2210. [DOI] [PubMed] [Google Scholar]
- Ji ES, Lee HK, Park GW, Kim KH, Kim JY, and Yoo JS (2019a). Isomer separation of sialylated O- and N-linked glycopeptides using reversed-phase LC-MS/MS at high temperature. J Chromatogr B 1110, 101–107. [DOI] [PubMed] [Google Scholar]
- Ji ES, Lee HK, Park GW, Kim KH, Kim JY, and Yoo JS (2019b). Isomer separation of sialylated O- and N-linked glycopeptides using reversed-phase LC-MS/MS at high temperature. J Chromatogr B Analyt Technol Biomed Life Sci 1110–1111, 101–107. [DOI] [PubMed] [Google Scholar]
- Jiang H, Yuan HM, Qu YY, Liang Y, Jiang B, Wu Q, Deng N, Liang Z, Zhang LH, and Zhang YK (2016). Preparation of hydrophilic monolithic capillary column by in situ photo-polymerization of N-vinyl-2-pyrrolidinone and acrylamide for highly selective and sensitive enrichment of N-linked glycopeptides. Talanta 146, 225–230. [DOI] [PubMed] [Google Scholar]
- Kaji H, Ocho M, Togayachi A, Kuno A, Sogabe M, Ohkura T, Nozaki H, Angata T, Chiba Y, Ozaki H, et al. (2013). Glycoproteomic discovery of serological biomarker candidates for HCV/HBV infection-associated liver fibrosis and hepatocellular carcinoma. J Proteome Res 12, 2630–2640. [DOI] [PubMed] [Google Scholar]
- Kammeijer GSM, Jansen BC, Kohler I, Heemskerk AAM, Mayboroda OA, Hensbergen PJ, Schappler J, and Wuhrer M (2017). Sialic acid linkage differentiation of glycopeptides using capillary electrophoresis - electrospray ionization - mass spectrometry. Sci Rep-Uk 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Lee SY, Hwang H, Lee JY, Ji ES, An HJ, Kim JY, and Yoo JS (2018). Direct Monitoring of Fucosylated Glycopeptides of Alpha-Fetoprotein in Human Serum for Early Hepatocellular Carcinoma by Liquid Chromatography-Tandem Mass Spectrometry with Immunoprecipitation. Proteom Clin Appl 12. [DOI] [PubMed] [Google Scholar]
- Kim KH, Lee SY, Kim DG, Lee SY, Kim JY, and Yoo JS (2020). Absolute Quantification of N-Glycosylation of Alpha-Fetoprotein Using Parallel Reaction Monitoring with Stable Isotope-Labeled N-Glycopeptide as an Internal Standard. Analytical Chemistry 92, 12588–12595. [DOI] [PubMed] [Google Scholar]
- Kim KH, Park GW, Jeong JE, Ji ES, An HJ, Kim JY, and Yoo JS (2019). Parallel reaction monitoring with multiplex immunoprecipitation of N-glycoproteins in human serum for detection of hepatocellular carcinoma. Anal Bioanal Chem 411, 3009–3019. [DOI] [PubMed] [Google Scholar]
- Kiyonami R, Schoen A, Prakash A, Peterman S, Zabrouskov V, Picotti P, Aebersold R, Huhmer A, and Domon B (2011). Increased selectivity, analytical precision, and throughput in targeted proteomics. Mol Cell Proteomics 10, M110 002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JA, and Zaia J (2020). A Perspective on the Confident Comparison of Glycoprotein Site-Specific Glycosylation in Sample Cohorts. Biochemistry-Us 59, 3089–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlik P, Goldman R, and Sanda M (2018). Hydrophilic interaction liquid chromatography in the separation of glycopeptides and their isomers. Anal Bioanal Chem 410, 5001–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlik P, Sanda M, and Goldman R (2017). Nano reversed phase versus nano hydrophilic interaction liquid chromatography on a chip in the analysis of hemopexin glycopeptides. J Chromatogr A 1519, 152–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurogochi M, and Amano J (2014). Relative quantitation of glycopeptides based on stable isotope labeling using MALDI-TOF MS. Molecules 19, 9944–9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurogochi M, Matsushista T, Amano M, Furukawa J, Shinohara Y, Aoshima M, and Nishimura S (2010). Sialic acid-focused quantitative mouse serum glycoproteomics by multiple reaction monitoring assay. Mol Cell Proteomics 9, 2354–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Cha HJ, Lim JS, Lee SH, Song SY, Kim H, Hancock WS, Yoo JS, and Paik YK (2014). Abundance-Ratio-Based Semiquantitative Analysis of Site-Specific N-Linked Glycopeptides Present in the Plasma of Hepatocellular Carcinoma Patients. Journal of Proteome Research 13, 2328–2338. [DOI] [PubMed] [Google Scholar]
- Lee JY, Lee HK, Park GW, Hwang H, Jeong HK, Yun KN, Ji ES, Kim KH, Kim JS, Kim JW, et al. (2016). Characterization of Site-Specific N-Glycopeptide Isoforms of alpha-1-Acid Glycoprotein from an Interlaboratory Study Using LC-MS/MS. J Proteome Res 15, 4146–4164. [DOI] [PubMed] [Google Scholar]
- Li J, Li Z, Duan X, Qin K, Dang L, Sun S, Cai L, Hsieh-Wilson LC, Wu L, and Yi W (2019a). An Isotope-Coded Photocleavable Probe for Quantitative Profiling of Protein O-GlcNAcylation. ACS Chem Biol 14, 4–10. [DOI] [PubMed] [Google Scholar]
- Li Q, Kailemia MJ, Merleev AA, Xu G, Serie D, Danan LM, Haj FG, Maverakis E, and Lebrilla CB (2019b). Site-Specific Glycosylation Quantitation of 50 Serum Glycoproteins Enhanced by Predictive Glycopeptidomics for Improved Disease Biomarker Discovery. Anal Chem 91, 5433–5445. [DOI] [PubMed] [Google Scholar]
- Li Q, Xie Y, Wong M, Barboza M, and Lebrilla CB (2020). Comprehensive structural glycomic characterization of the glycocalyxes of cells and tissues. Nat Protoc 15, 2668–2704. [DOI] [PubMed] [Google Scholar]
- Lin CH, Krisp C, Packer NH, and Molloy MP (2018). Development of a data independent acquisition mass spectrometry workflow to enable glycopeptide analysis without predefined glycan compositional knowledge. J Proteomics 172, 68–75. [DOI] [PubMed] [Google Scholar]
- Lin Y, Zhu J, Pan L, Zhang J, Tan Z, Olivares J, Singal AG, Parikh ND, and Lubman DM (2021). A Panel of Glycopeptides as Candidate Biomarkers for Early Diagnosis of NASH Hepatocellular Carcinoma Using a Stepped HCD Method and PRM Evaluation. J Proteome Res 20, 3278–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Finch JW, Lavallee MJ, Collamati RA, Benevides CC, and Gebler JC (2007). Effects of column length, particle size, gradient length and flow rate on peak capacity of nano-scale liquid chromatography for peptide separations. J Chromatogr A 1147, 30–36. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhu B, Fang Z, Zhang N, Qin H, Guo Z, Liang X, Yao Z, and Ye M (2021). Automated Intact Glycopeptide Enrichment Method Facilitating Highly Reproducible Analysis of Serum Site-Specific N-Glycoproteome. Anal Chem 93, 7473–7480. [DOI] [PubMed] [Google Scholar]
- Liu MQ, Zeng WF, Fang P, Cao WQ, Liu C, Yan GQ, Zhang Y, Peng C, Wu JQ, Zhang XJ, et al. (2017a). pGlyco 2.0 enables precision N-glycoproteomics with comprehensive quality control and one-step mass spectrometry for intact glycopeptide identification. Nat Commun 8, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Shang S, Li W, Qin X, Sun L, Zhang S, and Liu Y (2017b). Assessment of Hepatocellular Carcinoma Metastasis Glycobiomarkers Using Advanced Quantitative N-glycoproteome Analysis. Front Physiol 8, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, He J, Li C, Benitez R, Fu S, Marrero J, and Lubman DM (2010). Identification and confirmation of biomarkers using an integrated platform for quantitative analysis of glycoproteins and their glycosylations. J Proteome Res 9, 798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Huttenhain R, Surinova S, Gillet LC, Mouritsen J, Brunner R, Navarro P, and Aebersold R (2013). Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 13, 1247–1256. [DOI] [PubMed] [Google Scholar]
- Lu J, Fu DM, Yu L, Cao CY, Zou LJ, and Liang XM (2017). Determination of N-Glycopeptides by Hydrophilic Interaction Liquid Chromatography and Porous Graphitized Carbon Chromatography with Mass Spectrometry Detection. Anal Lett 50, 315–324. [Google Scholar]
- Ma J, Sanda M, Wei R, Zhang L, and Goldman R (2018). Quantitative analysis of core fucosylation of serum proteins in liver diseases by LC-MS-MRM. J Proteomics 189, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes A, Duarte HO, and Reis CA (2017). Aberrant Glycosylation in Cancer: A Novel Molecular Mechanism Controlling Metastasis. Cancer Cell 31, 733–735. [DOI] [PubMed] [Google Scholar]
- Magalhaes A, Duarte HO, and Reis CA (2021). The role of O-glycosylation in human disease. Mol Aspects Med 79, 100964. [DOI] [PubMed] [Google Scholar]
- Mechref Y (2012). Use of CID/ETD mass spectrometry to analyze glycopeptides. Curr Protoc Protein Sci Chapter 12, Unit 12 11 11–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier F, Geyer PE, Winter SV, Cox J, and Mann M (2018). BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nature Methods 15, 440–+. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Ruhaak LR, Stroble C, Salemi MR, Phinney B, Lebrilla CB, and Leiserowitz GS (2016). Glycoproteomic Analysis of Malignant Ovarian Cancer Ascites Fluid Identifies Unusual Glycopeptides. J Proteome Res 15, 3358–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Stroble CD, Taylor S, Hong QT, Lebrilla CB, Leiserowitz GS, Kim K, and Ruhaak LR (2018). Multiple Reaction Monitoring for the Quantitation of Serum Protein Glycosylation Profiles: Application to Ovarian Cancer. Journal of Proteome Research 17, 222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnarova K, and Kozlik P (2020). Comparison of Different HILIC Stationary Phases in the Separation of Hemopexin and Immunoglobulin G Glycopeptides and Their Isomers. Molecules 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montacir O, Montacir H, Springer A, Hinderlich S, Mahboudi F, Saadati A, and Parr MK (2018). Physicochemical Characterization, Glycosylation Pattern and Biosimilarity Assessment of the Fusion Protein Etanercept. Protein J 37, 164–179. [DOI] [PubMed] [Google Scholar]
- Nasir W, Toledo AG, Noborn F, Nilsson J, Wang M, Bandeira N, and Larson G (2016). SweetNET: A Bioinformatics Workflow for Glycopeptide MS/MS Spectral Analysis. J Proteome Res 15, 2826–2840. [DOI] [PubMed] [Google Scholar]
- Nie S, Lo A, Wu J, Zhu J, Tan Z, Simeone DM, Anderson MA, Shedden KA, Ruffin MT, and Lubman DM (2014). Glycoprotein biomarker panel for pancreatic cancer discovered by quantitative proteomics analysis. J Proteome Res 13, 1873–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie S, Lo A, Zhu JH, Wu J, Ruffin MT, and Lubman DM (2013). Isobaric Protein-Level Labeling Strategy for Serum Glycoprotein Quantification Analysis by Liquid Chromatography-Tandem Mass Spectrometry. Analytical Chemistry 85, 5353–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson J, Brinkmalm G, Ramadan S, Gilborne L, Noborn F, Blennow K, Wallin A, Svensson J, Abo-Riya MA, Huang X, et al. (2019). Synthetic standard aided quantification and structural characterization of amyloid-beta glycopeptides enriched from cerebrospinal fluid of Alzheimer’s disease patients. Sci Rep 9, 5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan KT, Chen CC, Urlaub H, and Khoo KH (2017). Adapting Data-Independent Acquisition for Mass Spectrometry-Based Protein Site-Specific N-Glycosylation Analysis. Anal Chem 89, 4532–4539. [DOI] [PubMed] [Google Scholar]
- Pap A, Klement E, Hunyadi-Gulyas E, Darula Z, and Medzihradszky KF (2018). Status Report on the High-Throughput Characterization of Complex Intact O-Glycopeptide Mixtures. J Am Soc Mass Spectrom 29, 1210–1220. [DOI] [PubMed] [Google Scholar]
- Park GW, Kim JY, Hwang H, Lee JY, Ahn YH, Lee HK, Ji ES, Kim KH, Jeong HK, Yun KN, et al. (2016). Integrated GlycoProteome Analyzer (I-GPA) for Automated Identification and Quantitation of Site-Specific N-Glycosylation. Sci Rep 6, 21175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BL, Thaysen-Andersen M, Fazakerley DJ, Holliday M, Packer NH, and James DE (2016). Terminal Galactosylation and Sialylation Switching on Membrane Glycoproteins upon TNF-Alpha-Induced Insulin Resistance in Adipocytes. Molecular & Cellular Proteomics 15, 141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons LM, An Y, Qi L, White MR, van der Woude R, Hartshorn KL, Taubenberger JK, de Vries RP, and Cipollo JF (2020). Influenza Virus Hemagglutinins H2, H5, H6, and H11 Are Not Targets of Pulmonary Surfactant Protein D: N-Glycan Subtypes in Host-Pathogen Interactions. J Virol 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patabandige MW, Pfeifer LD, Nguyen HT, and Desaire H (2021). Quantitative clinical glycomics strategies: A guide for selecting the best analysis approach. Mass Spectrom Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwa T, Li C, Simeone DM, and Lubman DM (2010). Glycoprotein Analysis Using Protein Microarrays and Mass Spectrometry. Mass Spectrometry Reviews 29, 830–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Gutierrez Reyes CD, Gautam S, Yu A, Cho BG, Goli M, Donohoo K, Mondello S, Kobeissy F, and Mechref Y (2021). MS-based glycomics and glycoproteomics methods enabling isomeric characterization. Mass Spectrom Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson AC, Russell JD, Bailey DJ, Westphall MS, and Coon JJ (2012). Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics 11, 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phung TK, Zacchi LF, and Schulz BL (2020). DIALib: an automated ion library generator for data independent acquisition mass spectrometry analysis of peptides and glycopeptides. Mol Omics 16, 100–112. [DOI] [PubMed] [Google Scholar]
- Pinho SS, and Reis CA (2015). Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 15, 540–555. [DOI] [PubMed] [Google Scholar]
- Pino LK, Just SC, MacCoss MJ, and Searle BC (2020). Acquiring and Analyzing Data Independent Acquisition Proteomics Experiments without Spectrum Libraries. Molecular & Cellular Proteomics 19, 1088–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polasky DA, Yu F, Teo GC, and Nesvizhskii AI (2020). Fast and comprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco. Nat Methods 17, 1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poljak K, Selevsek N, Ngwa E, Grossmann J, Losfeld ME, and Aebi M (2018). Quantitative Profiling of N-linked Glycosylation Machinery in Yeast Saccharomyces cerevisiae. Mol Cell Proteomics 17, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujic I, and Perreault H (2021). Recent advancements in glycoproteomic studies: Glycopeptide enrichment and derivatization, characterization of glycosylation in SARS CoV2, and interacting glycoproteins. Mass Spectrom Rev. [DOI] [PubMed] [Google Scholar]
- Qin K, Zhu Y, Qin W, Gao J, Shao X, Wang YL, Zhou W, Wang C, and Chen X (2018). Quantitative Profiling of Protein O-GlcNAcylation Sites by an Isotope-Tagged Cleavable Linker. ACS Chem Biol 13, 1983–1989. [DOI] [PubMed] [Google Scholar]
- Qing G, Yan J, He X, Li X, and Liang X (2020). Recent advances in hydrophilic interaction liquid interactionchromatography materials for glycopeptide enrichment and glycanseparation. Trends in Analytical Chemistry 124, 115570. [Google Scholar]
- Rebecchi KR, Wenke JL, Go EP, and Desaire H (2009). Label-free quantitation: a new glycoproteomics approach. J Am Soc Mass Spectrom 20, 1048–1059. [DOI] [PubMed] [Google Scholar]
- Reyes CDG, H. Y, Atashi M, Zhang J, Zhu J, Liu S, Parikh ND, Singal AG, Dai J, Lubman DM, Mechref Y (2021). PRM-MS Quantitative Analysis of Isomeric N-Glycopeptides Derived From Human Serum Haptoglobin of Patients With Cirrhosis and Hepatocellular Carcinoma. preprint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley NM, Bertozzi CR, and Pitteri SJ (2020a). A Pragmatic Guide to Enrichment Strategies for Mass Spectrometry-based Glycoproteomics. Mol Cell Proteomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley NM, Hebert AS, Westphall MS, and Coon JJ (2019). Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat Commun 10, 1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley NM, Malaker SA, Driessen MD, and Bertozzi CR (2020b). Optimal Dissociation Methods Differ for N- and O-Glycopeptides. J Proteome Res 19, 3286–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost HL, Rosenberger G, Navarro P, Gillet L, Miladinovic SM, Schubert OT, Wolski W, Collins BC, Malmstrom J, Malmstrom L, et al. (2014). OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol 32, 219–223. [DOI] [PubMed] [Google Scholar]
- Ruhaak LR, Taylor SL, Miyamoto S, Kelly K, Leiserowitz GS, Gandara D, Lebrilla CB, and Kim K (2013). Chip-based nLC-TOF-MS is a highly stable technology for large-scale high-throughput analyses. Anal Bioanal Chem 405, 4953–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhaak LR, Xu G, Li Q, Goonatilleke E, and Lebrilla CB (2018). Mass Spectrometry Approaches to Glycomic and Glycoproteomic Analyses. Chem Rev 118, 7886–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda M, and Goldman R (2016). Data Independent Analysis of IgG Glycoforms in Samples of Unfractionated Human Plasma. Analytical Chemistry 88, 10118–10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda M, Pompach P, Brnakova Z, Wu J, Makambi K, and Goldman R (2013). Quantitative liquid chromatography-mass spectrometry-multiple reaction monitoring (LC-MS-MRM) analysis of site-specific glycoforms of haptoglobin in liver disease. Mol Cell Proteomics 12, 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling B, MacLean B, Held JM, Sahu AK, Rardin MJ, Sorensen DJ, Peters T, Wolfe AJ, Hunter CL, MacCoss MJ, et al. (2015). Multiplexed, Scheduled, High-Resolution Parallel Reaction Monitoring on a Full Scan QqTOF Instrument with Integrated Data-Dependent and Targeted Mass Spectrometric Workflows. Anal Chem 87, 10222–10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schjoldager KT, Narimatsu Y, Joshi HJ, and Clausen H (2020). Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol 21, 729–749. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Karas M, and Dulcks T (2003). Effect of different solution flow rates on analyte ion signals in nano-ESI MS, or: When does ESI turn into nano-ESI? J Am Soc Mass Spectr 14, 492–500. [DOI] [PubMed] [Google Scholar]
- Searle BC, Pino LK, Egertson JD, Ting YS, Lawrence RT, MacLean BX, Villen J, and MacCoss MJ (2018). Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat Commun 9, 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman MH, Derks RJ, Bondt A, Palmblad M, Schoenmaker B, Koeleman CA, van de Geijn FE, Dolhain RJ, Deelder AM, and Wuhrer M (2012). Fc specific IgG glycosylation profiling by robust nano-reverse phase HPLC-MS using a sheath-flow ESI sprayer interface. J Proteomics 75, 1318–1329. [DOI] [PubMed] [Google Scholar]
- Seo Y, Oh MJ, Park JY, Ko JK, Kim JY, and An HJ (2019). Comprehensive Characterization of Biotherapeutics by Selective Capturing of Highly Acidic Glycans Using Stepwise PGC-SPE and LC/MS/MS. Analytical Chemistry 91, 6064–6071. [DOI] [PubMed] [Google Scholar]
- Shu H, Zhang L, Chen Y, Guo Y, Li L, Chen F, Cao Z, Yan G, Lu C, Liu C, et al. (2021). Quantification of Intact O-Glycopeptides on Haptoglobin in Sera of Patients With Hepatocellular Carcinoma and Liver Cirrhosis. Front Chem 9, 705341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Q, Li M, Shu L, An Z, Wang J, Lv H, Yang M, Cai T, Hu T, Fu Y, et al. (2020). Large-scale Identification of N-linked Intact Glycopeptides in Human Serum using HILIC Enrichment and Spectral Library Search. Mol Cell Proteomics 19, 672–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh C, Zampronio CG, Creese AJ, and Cooper HJ (2012). Higher energy collision dissociation (HCD) product ion-triggered electron transfer dissociation (ETD) mass spectrometry for the analysis of N-linked glycoproteins. J Proteome Res 11, 4517–4525. [DOI] [PubMed] [Google Scholar]
- Song E, Pyreddy S, and Mechref Y (2012). Quantification of glycopeptides by multiple reaction monitoring liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 26, 1941–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavenhagen K, Hinneburg H, Kolarich D, and Wuhrer M (2017). Site-Specific N- and O-Glycopeptide Analysis Using an Integrated C18-PGC-LC-ESI-QTOF-MS/MS Approach. Methods Mol Biol 1503, 109–119. [DOI] [PubMed] [Google Scholar]
- Stavenhagen K, Hinneburg H, Thaysen-Andersen M, Hartmann L, Silva DV, Fuchser J, Kaspar S, Rapp E, Seeberger PH, and Kolarich D (2013). Quantitative mapping of glycoprotein micro-heterogeneity and macro-heterogeneity: an evaluation of mass spectrometry signal strengths using synthetic peptides and glycopeptides. J Mass Spectrom 48, 627–639. [DOI] [PubMed] [Google Scholar]
- Strum JS, Nwosu CC, Hua S, Kronewitter SR, Seipert RR, Bachelor RJ, An HJ, and Lebrilla CB (2013). Automated Assignments of N- and O-Site Specific Glycosylation with Extensive Glycan Heterogeneity of Glycoprotein Mixtures. Analytical Chemistry 85, 5666–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Qin H, Wang F, Cheng K, Dong M, Ye M, and Zou H (2012). Capture and dimethyl labeling of glycopeptides on hydrazide beads for quantitative glycoproteomics analysis. Anal Chem 84, 8452–8456. [DOI] [PubMed] [Google Scholar]
- Suttapitugsakul S, Sun F, and Wu R (2020). Recent Advances in Glycoproteomic Analysis by Mass Spectrometry. Anal Chem 92, 267–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo D, Schlosser G, Vekey K, Drahos L, and Revesz A (2021). Collision energies on QTof and Orbitrap instruments: How to make proteomics measurements comparable? J Mass Spectrom 56, e4693. [DOI] [PubMed] [Google Scholar]
- Taga Y, Kusubata M, Ogawa-Goto K, and Hattori S (2013). Site-specific Quantitative Analysis of Overglycosylation of Collagen in Osteogenesis Imperfecta Using Hydrazide Chemistry and SILAC. Journal of Proteome Research 12, 2225–2232. [DOI] [PubMed] [Google Scholar]
- Tan ZJ, Yin HD, Nie S, Lin ZX, Zhu JH, Ruffin MT, Anderson MA, Simone DM, and Lubman DM (2015). c. Journal of Proteome Research 14, 1968–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Kitagawa K, Kojima N, and Iijima S (2016). Multifucosylated Alpha-1-acid Glycoprotein as a Novel Marker for Hepatocellular Carcinoma. J Proteome Res 15, 2935–2944. [DOI] [PubMed] [Google Scholar]
- Thomas SN, Harlan R, Chen J, Aiyetan P, Liu YS, Sokoll LJ, Aebersold R, Chan DW, and Zhang H (2015). Multiplexed Targeted Mass Spectrometry-Based Assays for the Quantification of N-Linked Glycosite-Containing Peptides in Serum. Analytical Chemistry 87, 10830–10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toernkvist A (2003). Aspects of Porous Graphitic Carbon as Packing Material in Capillary Liquid Chromatography (Uppsala: Acta Universitatis Upsaliensis; ). [Google Scholar]
- Toghi Eshghi S, Shah P, Yang W, Li X, and Zhang H (2015). GPQuest: A Spectral Library Matching Algorithm for Site-Specific Assignment of Tandem Mass Spectra to Intact N-glycopeptides. Anal Chem 87, 5181–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, and Nesvizhskii AI (2015). DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods 12, 258–264, 257 p following 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A, Cummings R, Esko J, Stanley P, Hart G, Aebi M,Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, and Seeberger PH (2017). Essentials of Glycobiology (Cold Spring Harbor Laboratory Press, Plainview, NY.). [PubMed] [Google Scholar]
- Veillon L, Fakih C, Abou-El-Hassan H, Kobeissy F, and Mechref Y (2018). Glycosylation Changes in Brain Cancer. ACS Chem Neurosci 9, 51–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Liu D, Qu J, Zhu H, Chen C, Gibbons C, Greenway H, Wang P, Bollag RJ, Liu K, et al. (2021). Streamlined Subclass-Specific Absolute Quantification of Serum IgG Glycopeptides Using Synthetic Isotope-Labeled Standards. Anal Chem 93, 4449–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xia N, and Liu L (2013). Boronic Acid-based approach for separation and immobilization of glycoproteins and its application in sensing. Int J Mol Sci 14, 20890–20912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xu F, Chen Y, and Tian Z (2020). A quantitative N-glycoproteomics study of cell-surface N-glycoprotein markers of MCF-7/ADR cancer stem cells. Anal Bioanal Chem 412, 2423–2432. [DOI] [PubMed] [Google Scholar]
- Wilm M, and Mann M (1996). Analytical properties of the nanoelectrospray ion source. Anal Chem 68, 1–8. [DOI] [PubMed] [Google Scholar]
- Wohlgemuth J, Karas M, Eichhorn T, Hendriks R, and Andrecht S (2009). Quantitative site-specific analysis of protein glycosylation by LC-MS using different glycopeptide-enrichment strategies. Anal Biochem 395, 178–188. [DOI] [PubMed] [Google Scholar]
- Woo CM, Felix A, Byrd WE, Zuegel DK, Ishihara M, Azadi P, Iavarone AT, Pitteri SJ, and Bertozzi CR (2017). Development ofc. J Proteome Res 16, 1706–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo CM, Iavarone AT, Spiciarich DR, Palaniappan KK, and Bertozzi CR (2015). Isotope-targeted glycoproteomics (IsoTaG): a mass-independent platform for intact N- and O-glycopeptide discovery and analysis. Nat Methods 12, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Chen W, Smeekens JM, and Wu R (2018). An enrichment method based on synergistic and reversible covalent interactions for large-scale analysis of glycoproteins. Nat Commun 9, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Sun F, Suttapitugsakul S, and Wu R (2019). Global and site-specific analysis of protein glycosylation in complex biological systems with Mass Spectrometry. Mass Spectrom Rev 38, 356–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, and Tian Z (2019). GPSeeker Enables Quantitative Structural N-Glycoproteomics for Site- and Structure-Specific Characterization of Differentially Expressed N-Glycosylation in Hepatocellular Carcinoma. J Proteome Res 18, 2885–2895. [DOI] [PubMed] [Google Scholar]
- Xu Y, Bailey UM, and Schulz BL (2015). Automated measurement of site-specific N-glycosylation occupancy with SWATH-MS. Proteomics 15, 2177–2186. [DOI] [PubMed] [Google Scholar]
- Yang GL, Hoti N, Chen SY, Zhou YY, Wang Q, Betenbaugh M, and Zhang H (2020a). One-Step Enrichment of Intact Glycopeptides From Glycoengineered Chinese Hamster Ovary Cells. Frontiers in Chemistry 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Chen L, Chan DW, Li QK, and Zhang H (2017a). Protein signatures of molecular pathways in non-small cell lung carcinoma (NSCLC): comparison of glycoproteomics and global proteomics. Clin Proteomics 14, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WM, Ao MG, Song A, Xu YW, Sokoll L, and Zhang H (2020b). Mass Spectrometric Mapping of Glycoproteins Modified by Tn-Antigen Using Solid-Phase Capture and Enzymatic Release. Analytical Chemistry 92, 9230–9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WM, Shah P, Hu YW, Eshghi ST, Sun SS, Liu Y, and Zhang H (2017b). Comparison of Enrichment Methods for Intact N- and O-Linked Glycopeptides Using Strong Anion Exchange and Hydrophilic Interaction Liquid Chromatography. Analytical Chemistry 89, 11193–11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WM, Song A, Ao MH, Xu YW, and Zhang H (2020c). Large-scale site-specific mapping of the O-GalNAc glycoproteome. Nat Protoc 15, 2589–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Mao Y, Clausen H, and Vakhrushev SY (2019). Glyco-DIA: a method for quantitative O-glycoproteomics with in silico-boosted glycopeptide libraries. Nat Methods 16, 902–910. [DOI] [PubMed] [Google Scholar]
- Yeo KYB, Chrysanthopoulos PK, Nouwens AS, Marcellin E, and Schulz BL (2016). High-performance targeted mass spectrometry with precision data-independent acquisition reveals site-specific glycosylation macroheterogeneity. Anal Biochem 510, 106–113. [DOI] [PubMed] [Google Scholar]
- Yin H, An M, So PK, Wong MY, Lubman DM, and Yao Z (2018). The analysis of alpha-1-antitrypsin glycosylation with direct LC-MS/MS. Electrophoresis 39, 2351–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Tan Z, Wu J, Zhu J, Shedden KA, Marrero J, and Lubman DM (2015a). Mass-Selected Site-Specific Core-Fucosylation of Serum Proteins in Hepatocellular Carcinoma. J Proteome Res 14, 4876–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Zhu J, Wang M, Yao ZP, and Lubman DM (2020). Quantitative Analysis of alpha-1-Antitrypsin Glycosylation Isoforms in HCC Patients Using LC-HCD-PRM-MS. Anal Chem 92, 8201–8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HD, Tan ZJ, Wu J, Zhu JH, Shedden KA, Marrero J, and Lubman DM (2015b). Mass-Selected Site-Specific Core-Fucosylation of Serum Proteins in Hepatocellular Carcinoma. Journal of Proteome Research 14, 4876–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You X, Yao Y, Mao J, Qin H, Liang X, Wang L, and Ye M (2018). Chemoenzymatic Approach for the Proteomics Analysis of Mucin-Type Core-1 O-Glycosylation in Human Serum. Anal Chem 90, 12714–12722. [DOI] [PubMed] [Google Scholar]
- Yu A, Zhao J, Peng W, Banazadeh A, Williamson SD, Goli M, Huang Y, and Mechref Y (2018). Advances in mass spectrometry-based glycoproteomics. Electrophoresis 39, 3104–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Wang B, Chen Z, Urabe G, Glover MS, Shi X, Guo LW, Kent KC, and Li L (2017a). Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J Am Soc Mass Spectrom 28, 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Wang BW, Chen ZW, Urabe G, Glover MS, Shi XD, Guo LW, Kent KC, and Li LJ (2017b). Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J Am Soc Mass Spectr 28, 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Benicky J, Wei R, Goldman R, and Sanda M (2018). Quantitative Analysis of Sex-Hormone-Binding Globulin Glycosylation in Liver Diseases by Liquid Chromatography-Mass Spectrometry Parallel Reaction Monitoring. Journal of Proteome Research 17, 2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Wei RHZ, Goldman R, and Sanda M (2019). Optimized Fragmentation for Quantitative Analysis of Fucosylated N-Glycoproteins by LC-MS-MRM. Analytical Chemistry 91, 9206–9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias LG, Hartmann AK, Song E, Zhao J, Zhu R, Mirzaei P, and Mechref Y (2016). HILIC and ERLIC Enrichment of Glycopeptides Derived from Breast and Brain Cancer Cells. J Proteome Res 15, 3624–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li XJ, Martin DB, and Aebersold R (2003). Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol 21, 660–666. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Ma C, Chin LS, and Li L (2020a). Integrative glycoproteomics reveals protein N-glycosylation aberrations and glycoproteomic network alterations in Alzheimer’s disease. Sci Adv 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Cao X, Liu C, Li W, Zeng W, Li B, Chi H, Liu M, Qin X, Tang L, et al. (2019). N-glycopeptide Signatures of IgA2 in Serum from Patients with Hepatitis B Virus-related Liver Diseases. Mol Cell Proteomics 18, 2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Madunic K, Holst S, Zhang J, Jin C, Ten Dijke P, Karlsson NG, Stavenhagen K, and Wuhrer M (2020b). Development of a 96-well plate sample preparation method for integrated N- and O-glycomics using porous graphitized carbon liquid chromatography-mass spectrometry. Mol Omics 16, 355–363. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lin T, Zhao Y, Mao Y, Tao Y, Huang Y, Wang S, Hu L, Cheng J, and Yang H (2020c). Characterization of N-linked intact glycopeptide signatures of plasma IgGs from patients with prostate carcinoma and benign prostatic hyperplasia for diagnosis pre-stratification. Analyst 145, 5353–5362. [DOI] [PubMed] [Google Scholar]
- Zhao J, Qiu W, Simeone DM, and Lubman DM (2007). N-linked glycosylation profiling of pancreatic cancer serum using capillary liquid phase separation coupled with mass spectrometric analysis. J Proteome Res 6, 1126–1138. [DOI] [PubMed] [Google Scholar]
- Zhao J, Simeone DM, Heidt D, Anderson MA, and Lubman DM (2006). Comparative serum glycoproteomics using lectin selected sialic acid glycoproteins with mass spectrometric analysis: application to pancreatic cancer serum. J Proteome Res 5, 1792–1802. [DOI] [PubMed] [Google Scholar]
- Zhao X, Zheng S, Li Y, Huang J, Zhang W, Xie Y, Qin W, and Qian X (2020). An Integrated Mass Spectroscopy Data Processing Strategy for Fast Identification, In-Depth, and Reproducible Quantification of Protein O-Glycosylation in a Large Cohort of Human Urine Samples. Anal Chem 92, 690–698. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Jia W, Wang J, Ying W, Zhang Y, and Qian X (2011). Fragmentation and site-specific quantification of core fucosylated glycoprotein by multiple reaction monitoring-mass spectrometry. Anal Chem 83, 8802–8809. [DOI] [PubMed] [Google Scholar]
- Zhou C, and Schulz BL (2020). Glycopeptide variable window SWATH for improved data independent acquisition glycoprotein analysis. Anal Biochem 597, 113667. [DOI] [PubMed] [Google Scholar]
- Zhou S, Huang Y, Dong X, Peng W, Veillon L, Kitagawa DAS, Aquino AJA, and Mechref Y (2017). Isomeric Separation of Permethylated Glycans by Porous Graphitic Carbon (PGC)-LC-MS/MS at High Temperatures. Anal Chem 89, 6590–6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Chen Z, Zhang J, An M, Wu J, Yu Q, Skilton SJ, Bern M, Ilker Sen K, Li L, et al. (2019a). Differential Quantitative Determination of Site-Specific Intact N-Glycopeptides in Serum Haptoglobin between Hepatocellular Carcinoma and Cirrhosis Using LC-EThcD-MS/MS. J Proteome Res 18, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, He J, Liu Y, Simeone DM, and Lubman DM (2012). Identification of glycoprotein markers for pancreatic cancer CD24+CD44+ stem-like cells using nano-LC-MS/MS and tissue microarray. J Proteome Res 11, 2272–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Huang J, Zhang J, Chen Z, Lin Y, Grigorean G, Li L, Liu S, Singal AG, Parikh ND, et al. (2020a). Glycopeptide Biomarkers in Serum Haptoglobin for Hepatocellular Carcinoma Detection in Patients with Nonalcoholic Steatohepatitis. J Proteome Res 19, 3452–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Warner E, Parikh ND, and Lubman DM (2019b). Glycoproteomic markers of hepatocellular carcinoma-mass spectrometry based approaches. Mass Spectrom Rev 38, 265–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R, Huang Y, Zhao J, Zhong J, and Mechref Y (2020b). Isomeric Separation of N-Glycopeptides Derived from Glycoproteins by Porous Graphitic Carbon (PGC) LC-MS/MS. Anal Chem 92, 9556–9565. [DOI] [PMC free article] [PubMed] [Google Scholar]