

Abstract
Soluble elastin-like peptides (ELPs) can be engineered into a range of physical forms, from hydrogels and scaffolds to fibers and artificial tissues, finding numerous applications in medicine and engineering as “smart polymers”. Elastin-like peptides are attractive candidates as a platform for novel biomaterial design because they exhibit a highly tunable response spectrum, with reversible phase transition capabilities. Here, we report the design of the first virtual library of elastin-like protein models using methods for enhanced sampling to study the effect of peptide chemistry, chain length, and salt concentration on the structural transitions of ELPs, exposing associated molecular mechanisms. We describe the behavior of the local molecular structure under increasing temperatures and the effect of peptide interactions with nearest hydration shell water molecules on peptide mobility and propensity to exhibit structural transitions. Shifts in the magnitude of structural transitions at the single-molecule scale are explained from the perspective of peptide-ion-water interactions in a library of four unique elastin-like peptide systems. Predictions of structural transitions are subsequently validated in experiment. This library is a valuable resource for recombinant protein design and synthesis as it elucidates mechanisms at the single-molecule level, paving a feedback path between simulation and experiment for smart material designs.
Keywords: Elastin-like peptide (ELP), inverse temperature transition, peptide-water interaction, single-molecule, protein simulation library
Introduction
Elastin is an extracellular matrix protein that imparts reversible elastic recoil and resilience to extensible tissues, including the lung, blood vessels, elastic ligaments and skin. Beyond its impressive mechanical elasticity, elastin and artificial elastin-like peptides (ELPs) have been demonstrated, through transmission electron microscopy, circular dichroism and nuclear magnetic resonance [1–5], to exhibit an inverse temperature transition, a reversible propensity to aggregate upon increasing temperature [2, 6–12]. Above a specific temperature, highly dependent on the chemistry and environment of the ELP system, the formation of an aggregating, coacervate phase takes place. This is similar to the natural coacervation process of tropoelastin, the precursor molecule to elastin, in the process of elastic fiber formation [13].
The temperature-responsive feature makes elastin-like peptides versatile and well suited to serve as template materials for tunable design [14, 15]. A number of studies have considered the possibility of designing polymeric ELP materials with introduced temperature-modulated switches. Variability in the temperature transition has been shown experimentally in response to a range of stimuli, including sequence chemistry modification [16–19], changes in salt concentration [17], molecular weight [19–21], pH [20, 22], and light [23].
The stimuli-responsive quality confers controllable functionalities to ELPs, and in conjunction with their biocompatibility and biodegradability, makes them effective candidates as a platform for biomedical materials. ELPs have been designed into a range of material forms, including nanoparticle self-assemblies [24, 25] and hydrogels [1, 26, 27]. The tunability of the ELP phase transitions has been used in biomedical applications from drug delivery [28–30] to tissue engineering [31]. Examples of such applications include elastin-based nanoparticles that have been designed to display cell-penetrating peptide motifs for controlled uptake into cells; thermal targeting of tumors by creation of a diffusion gradient of a drug into the tumor; and sustained release delivery systems for controlled release of peptide drugs to treat type 2 diabetes [32–34].
Inverse-temperature transition studies have preferred to focus on intermolecular or aggregation studies. Here we focus on intramolecular phenomena in order to understand basic events at the single-molecule and peptide domain level. A fundamental understanding at the sub-molecular scale, currently missing, provides a foundation to extend this work for studying intermolecular events. We consider the GVGVP pentapeptide repeat unit, a sequence permutation traditionally used as a representative ELP polymer. Our work considers molecular compactness and secondary structure, hydrogen bonding organization, and distribution and arrangement of water in modulating ELP behavior based on changes on chemistry, salt concentration, and peptide chain length.
To the best of the authors’ knowledge, this is the first study of this type to compare mechanisms involved in the presence and absence of transitions for different ELP sequences with varying conditions at the single-molecule scale. Based on our findings, we propose modeling as a complementary tool to provide fast, reliable predictions based on core molecular mechanisms, because current methods are inadequate and do not consider the contributions of fast and slow molecular dynamics on the one hand; and the balancing of interactions between peptide, ions and water on the other. Furthermore, understanding single molecule behavior mechanisms is a necessary prelude to studying inter-molecular events such as coacervation. This approach can be used to quickly screen the sequence space from a library of candidate sequences, in order to define sequences that might be useful experimentally (Figure 1).
Figure 1 |.
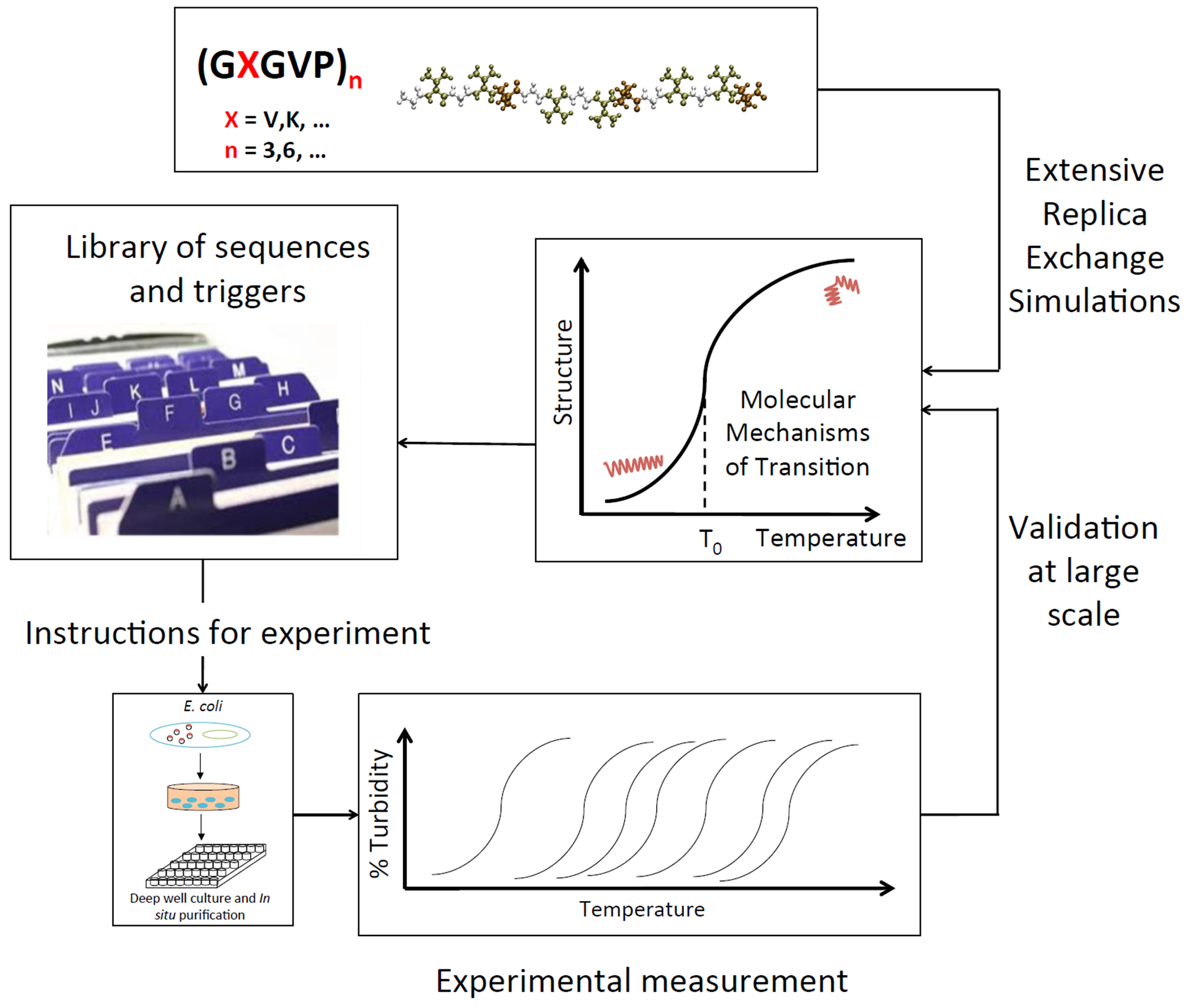
Simulation-experiment material design paradigm flow-chart. Sequences of the form [(GVGVP)(GXGVP)(GVGVP)]n are simulated with extensive Replica Exchange Molecular Dynamics simulations to identify molecular mechanisms of structural transitions. This yields a basis for a library of available sequences to instruct experimental design. Recombinant DNA technology is used to synthesize proteins in vitro, feeding back into the simulation-experiment design loop.
Methods
Molecular Simulation Setup
Input structures for simulation are created based on elastin-like protein polymer (ELP) sequences of the form [(GVGVP)(GXGVP)(GVGVP)]n in single amino acid letter code (SI Figure 1a). X is an interchangeable amino acid considered for shifting and modifying transition temperatures and determines sequence length. The systems considered in this study are described in SI Table 1. X-residue identity, chain length, and NaCl concentration are considered.
Extended conformations of the sequences are built using CHARMM version 24b1 [35]. Following sequence construction, all subsequent system modifications and simulations are carried out using GROMACS version 5.01 [36]. Each system includes the protein sequence [(GVGVP)(GXGVP)(GVGVP)]n in a rectangular water box in 1 M or 100 mM NaCl, with periodic boundary conditions (SI Figure 1b). The number of atoms in each system studied here is indicated in SI Table 1. The CHARMM27 force field is used, which includes CHARMM22 and CMAP for proteins [37].
First, the structure is relaxed to ensure no steric clashes using energy minimization through the steepest descent algorithm. Then, solvent and ions are equilibrated around the protein, where the protein is fixed, through two equilibration stages, each 100 ps in length. The first phase is equilibration in an NVT ensemble to stabilize temperature, followed by a second stage in an NPT ensemble to stabilize system pressure. After the solvent is equilibrated, the protein restraint is removed and the protein and solvent are equilibrated in an NPT ensemble for an additional 100 ps. After this stage, final structures are input into Replica Exchange Molecular Dynamics (REMD) simulations [38] (SI Figure 1c). The Berendsen thermostat [39] is used for temperature coupling and the Parrinello-Rahman barostat [40] is used for pressure coupling. The short range electrostatic interactions and Lennard-Jones interactions were evaluated with a cutoff of 10 Å. Particle-mesh Ewald summation [41] was used to calculate long range electrostatic interactions with a grid spacing of 1.6 Å and a fourth order interpolation.
Replica Exchange Molecular Dynamics
We build on existing work by using accelerated molecular modeling methods, specifically Replica Exchange Molecular Dynamics (REMD), to introduce better sampling of the system, identifying smooth-transition temperature regions [42–45]. Using REMD enhances the number of temperatures that can be studied simultaneously, ensuring better ensemble sampling at each temperature. A key challenge in studying elastin-like sequences lies in accurate characterization of a highly dynamic, multi-state system. By performing intensive REMD simulations with read-outs for a large number of temperature data points, we are able to show, for the first time, that the molecular collapse previously described happens as a gradual transition in a well-defined temperature region at the single-molecule scale.
The ELP systems are highly dynamic; therefore, effective sampling, ensured by Replica Exchange simulations, are needed to capture the full range of accessible molecular structures at each temperature. A large spread is observed in molecular size across all temperatures, illustrated by the distribution of radii of gyration across temperatures in SI Figure 11. At high temperatures, the range of accessible structures shrinks by over 15%, with structures more densely concentrated around a smaller radius of gyration, manifested by a narrower distribution peak.
As a result of a flat energy landscape traversed by the system even at high temperature, open structures populate the ensemble, though they compose a smaller fraction than at low temperature, evident in the wide distributions of radii of gyration across temperatures (SI Figure 11). These tools permit the identification of molecular-level detail comparisons across sequences and environmental conditions to shed light on the transition behavior of single-molecule ELP systems, and on ways of enhancing, reducing and suppressing such transitions.
After initial equilibration, subsequent Replica Exchange Molecular Dynamics simulations are carried out in the canonical ensemble. REMD improves sampling compared to classical molecular dynamics (MD) simulations by integrating Monte Carlo exchanges into a classical MD simulation scheme. High temperatures allow for wide conformational space sampling, avoiding protein entrapment in local-minima free energy states via thermal stimulation. Frequent exchanges ensure wide sampling across temperature replicas.
For each system, 60 temperature replicas are used, exponentially distributed from 280 K to 400 K [46]. Each replica is simulated for 10 ns, for a total simulation time of 600 ns across all temperatures. Exchanges are attempted after 2 ps equilibration runs, and are accepted according to the Metropolis criterion. Exchange acceptance ratios are between 30-40%, signifying sufficient sampling. Representative structures are determined by analyzing the ensemble of structures in the final 2 ns of each replica. K-means clustering is used to group structures into clusters according to root mean square deviation of 4 Å for the low-temperature replica at 280 K and 3.5 Å for the high-temperature replica at 340 K. Representative structures with lowest potential energy are chosen from most populated clusters, represented by significance factor p in SI Figures 2–5. Analysis of representative structures is carried out using the MMTSB script package [47]. All simulations are completed using the Extreme Science and Engineering Discovery Environment (XSEDE) [48].
Analysis of Molecular Structures
We use the last 8 ns of each REMD run to analyze the molecular structures of ELP sequences. We identify molecular transitions characterized by changes in radius of gyration, hydrogen bonding, solvent accessible surface area, secondary structure and behavior of solvation waters. Radius of gyration gives a measure of the compactness of the structure, defined as
| (1) |
where is the mass of atom and is the position of atom with respect to the center of mass of the molecule. Protein secondary structure is computed using the DSSP algorithm [49, 50]. Hydrogen bonds are determined using a geometric definition, with the donor–hydrogen–acceptor angle of 30 degrees and a cutoff distance of 0.35 nm between the donor and the acceptor. To measure hydrogen bond distributions within peptides, we define , the hydrogen bond scaled occupancy spread as
| (2) |
where is the fraction of time a hydrogen bond exists in the ensemble between donor and acceptor and are the donor and acceptor residue indices and is the total number of donor-acceptor pairs where a hydrogen bond appears.
Solvent accessible surface area is computed using the double cubic lattice method [51] implemented in Gromacs [36]. Radius of gyration, secondary structure and solvent accessible surface area analysis is done using Gromacs analysis tools [36] and in-house scripts. Hydrogen bond analysis and visualization of molecular models is performed using VMD 1.9.1 [52] and in-house scripts. Average errors are calculated from 400 ps block averages.
DSSP
DSSP [49, 50] calculates hydrogen bond energies based on 3D protein structure, and assigns the most likely class of secondary structure based on hydrogen bond distributions, identifying helix, beta sheet, hydrogen-bonded turn, bend and random coil structure. Coil structures are defined as regions of low curvature, compared to the high curvature of at least 70° in bend structures. For the sequences studied here, beta sheet and helix structure content is less than 1%; bend structure represents 15-30% of the secondary structure content; coil structure 48-61%; and turn structure 14-22%.
UV–Vis Spectrophotometry
Lyophilized elastin-like peptides are purchased from Biomatik (Wilmington, Delaware, USA). The UV-Vis spectra of ELP solutions are obtained by a 14DS spectrophotometer equipped with a Peltier temperature controller (Aviv Biomedical, Lakewood, NJ). The 40 mg/ml ELP solution is well-mixed and pipetted into 1 mm path length absorption cuvettes (Hellma USA, Plainview, NY) and allowed to equilibrate at 4°C (277 K) before measurements. To characterize the inverse temperature transition of ELPs, temperature scans are performed at 350 nm from 3 to 100°C (276 K to 373 K) at a rate of 2 degrees/min and then cooled to 0°C (273 K) at the same rate. Absorbance readings are taken after equilibrating ELP solutions at each desired temperature for 30 s. The averaging time of each measurement is 10 s per step. The baseline scans are taken with the solvent and cuvette under the same conditions and subtracted from the sample scans.
Results
We compare the effect of temperature for each system by considering the change in radius of gyration (Figure 2a–d). (GVGVP)3 and (GVGVP)6 sequences in 1 M NaCl show a distinct, gradual transition to a reduced radius of gyration, indicating a shift from extended to contracted structures with increasing temperature (Figure 2a,c). This is consistent with observations of molecular folding upon increasing temperature, representative of single-molecule inverse temperature transitions [42–44]. For the (GVGVP)3 sequence the structural transition occurs gradually over a 35 degree temperature range (Figure 2a), between 294 K and 329 K, consistent with experimental studies of similar sequences, where a transition is identified at 24°C (i.e. 297 K) [16]. The transition region is defined by the intersection of linear fits to regions below, at, and above transition temperatures. Peptide structures for System 1 are shown at 280 K (Figure 2e) and 340 K (Figure 2f), and display molecular collapse accompanied by a reorientation of water molecules and restructuring of hydrogen bond networks.
Figure 2 |.
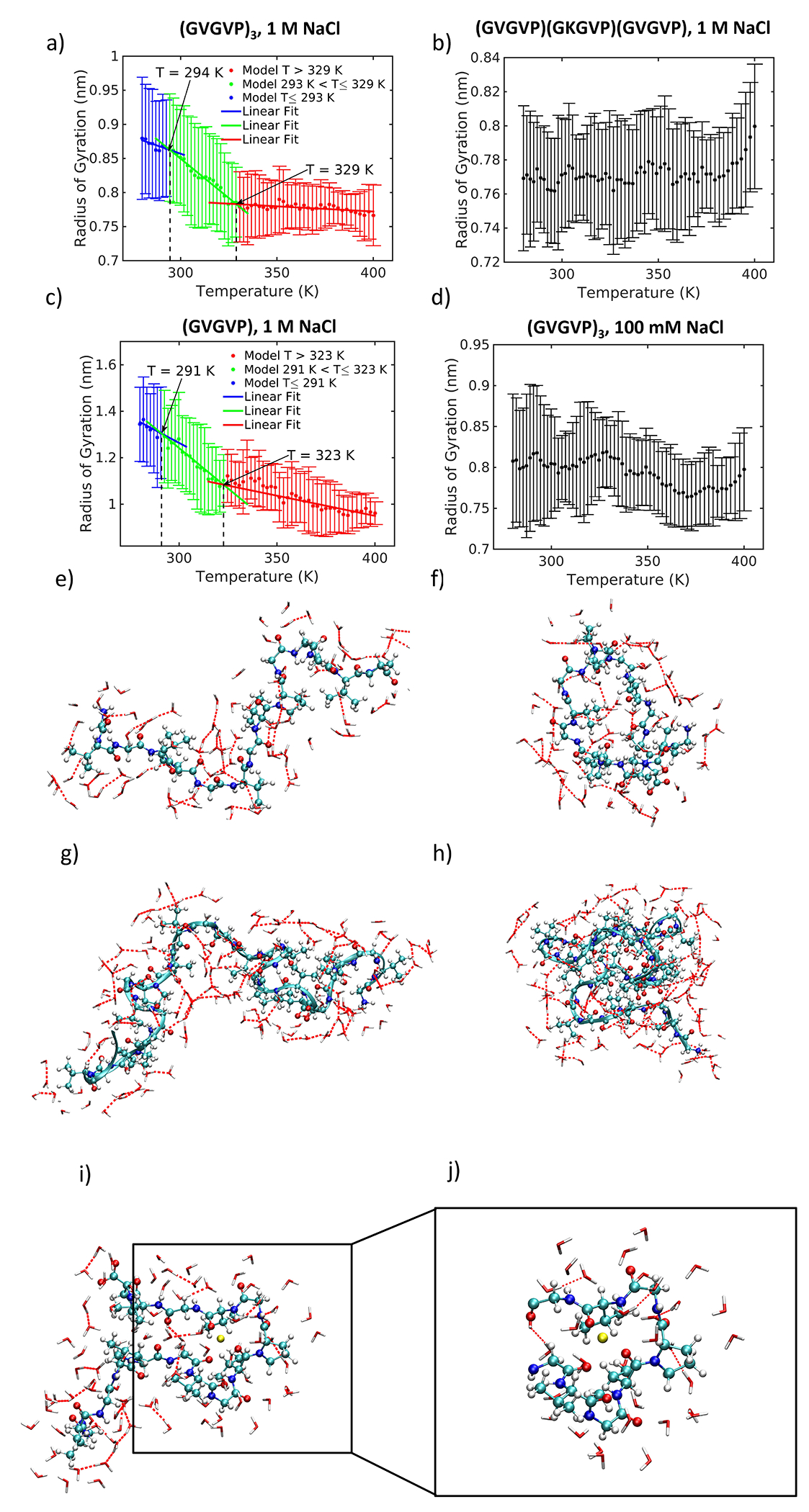
Radius of gyration change with temperature. Green represents transition region; blue represents structures below transition range; red represents structures above transition range. a) (GVGVP)3 in 1 M NaCl with snapshots at low and high temperature; b) (GVGVP)(GKGVP)(GVGVP) in 1 M NaCl; c) (GVGVP)6 in 1 M NaCl; d) (GVGVP)3 in 100 mM NaCl; e) System 1 at 280 K; f) System 1 at 340 K; g) System 3 at 280 K; h) System 3 at 340 K; i) System 1 at 340 K showing peptide, hydrogen-bonded water molecules and a single sodium ion (in yellow) within a 3 Å solvation shell; j) Expanded image showing sodium ion (in yellow) interacting with electronegative atoms of the peptide. The sodium ion is hinged on the interaction with three oxygen atoms (in red).
Doubling the chain length of the peptide almost doubles the change in the radius of gyration across the transition range (Figure 2c,g,h). The temperature transition range is marginally lower than that of the shorter sequence; between 291 K and 323 K (Figure 2c). These findings are consistent with experimental work [17, 53] where transitions occur at lower temperatures as the chain length increases. We note that at the single-molecule scale, the main difference between shorter and longer chain length sequences is the magnitude of the transition, or the degree of molecular collapse, rather than a significant shift in the temperature transition region. A potential discrepancy with experimental findings where longer chain lengths introduce a pronounced shift to lower transition temperatures, appreciates that at the single-molecule scale, the molecular collapse occurs gradually. Thus, we propose that because the magnitude of this transition is more significant for longer chain lengths, this system reaches a saturated state inducing a macroscale phase transition at a lower temperature.
In contrast, no significant transition is exhibited by (GVGVP)(GKGVP)(GVGVP) in 1 M NaCl. A single residue substitution from valine (V) to lysine (K) in the seventh position of the (GVGVP)3 sequence suppresses a structural transition with temperature (Figure 2b). The difference between low- and high- temperature values is not significant: for 280 K and 340 K ( for and for 280 K and 380 K (for . The molecular model agrees with experimental findings [16] where a lysine substitution leads to an upshift in the transition temperature from 24°C to 120°C (297 K to 393 K), albeit beyond the temperature range we consider here. Our results match predictions that ELPs containing charged residues would have high transition temperatures inaccessible in aqueous solutions [16, 54].
As sequence modification serves as a trigger for modulating transition temperature, we next turn to considering the effect of changing salt concentration on the molecule’s structural integrity. On this basis, a reduction in salt concentration from 1 M to 100 mM NaCl decreases the magnitude of the structural transition of (GVGVP)3 (Figure 2d). The difference between low- and high-temperatures is not significant: for 280 K and 340 K for and for 280 K and 380 K (for . We find that higher salt concentration results in ion penetration into the protein core, which likely facilitates the transition observed (Figure 2i,j). Our results are consistent with experimental work showing that higher salt concentration can induce transitions by lowering the transition temperature [17].
To validate our predictions, we conduct a series of experiments and consider the effect of increasing salt concentration to induce a transition in the lysine-containing sequence. First, we confirm with UV spectroscopy that a lysine substitution effectively eliminates a temperature transition (Figure 3a). The turbidity profile of 40 mg/ml (GVGVP)(GKGVP)(GVGVP) in deionized water at 350 nm shows no significant change in the temperature range from 293 to 353 K, consistent with a model where the structural transition is suppressed by inserting a charged residue into the elastin sequence. To demonstrate the predictive nature of the model system, we increase the NaCl concentration of (GVGVP)(GKGVP)(GVGVP) from 0.1M NaCl, where no transition is observed, to 5M NaCl, finding a structural transition at 333 K, as predicted by the modeling (Figure 3b).
Figure 3 |.
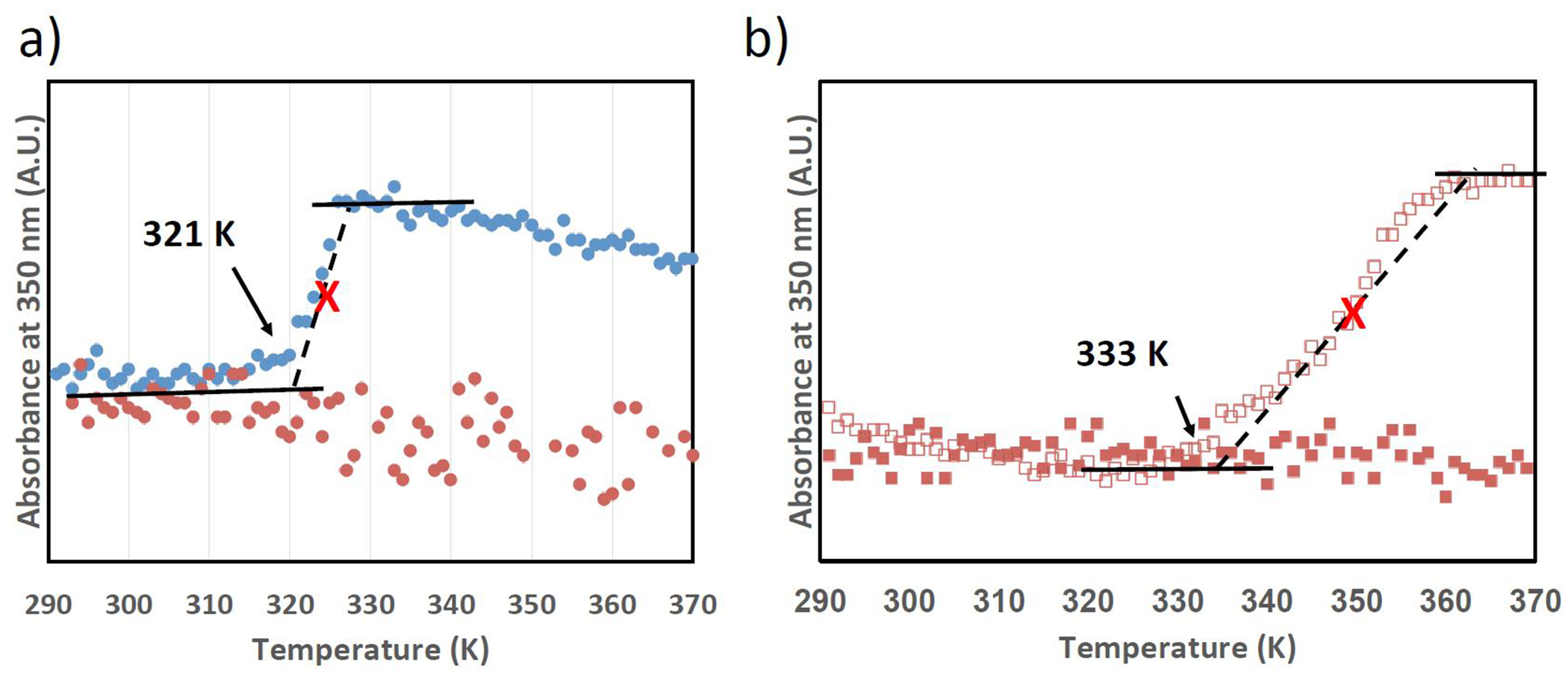
(a) Turbidity profiles of (GVGVP)(GVGVP)(GVGVP) (blue dots) and (GVGVP)(GKGVP)(GVGVP) (orange dots) in deionized water, 60mg/ml, suggesting the transition temperature can be suppressed by switching V to K, a charged residue. (b) Turbidity profiles of (GVGVP)(GKGVP)(GVGVP) in 0.1M NaCl (orange squares) and 5M NaCl (open orange squares), 60mg/ml, suggesting the transition temperature can be lowered by increasing the ionic strength. The black arrows represent the onsets of transition, and red crosses represent the inflection point.
The number of hydrogen bonds (Figure 4a–d) formed within the protein for Systems 1 and 3 increases with temperature, corresponding to the local structural ordering of the peptides (Figure 4a,c). Peptide-peptide hydrogen bonds replace water-water hydrogen bonds in the nearest solvation shells and peptide-water hydrogen bonds as the structure collapses (SI Figure 6a,c, SI Figure 7a,c). The increased formation of hydrogen bonds within the peptides at higher temperatures is counterintuitive from a kinetic standpoint, and drives structural stabilization. This agrees with findings that suggest the presence of dynamic beta turns that oscillate between disordered and hydrogen bonded states [55], and with a general absence of ordered secondary structure identified through nuclear magnetic resonance [56–58]. Earlier studies additionally point to the joint contributions of glycine (G) and proline (P) to the solvation of the peptide backbone and resultant disordered nature of the peptide, where glycine adds a high degree of chain mobility due to its small size and proline is a rigidity promoter that prohibits the formation of structure-stabilizing hydrogen bonds [59].
Figure 4 |.
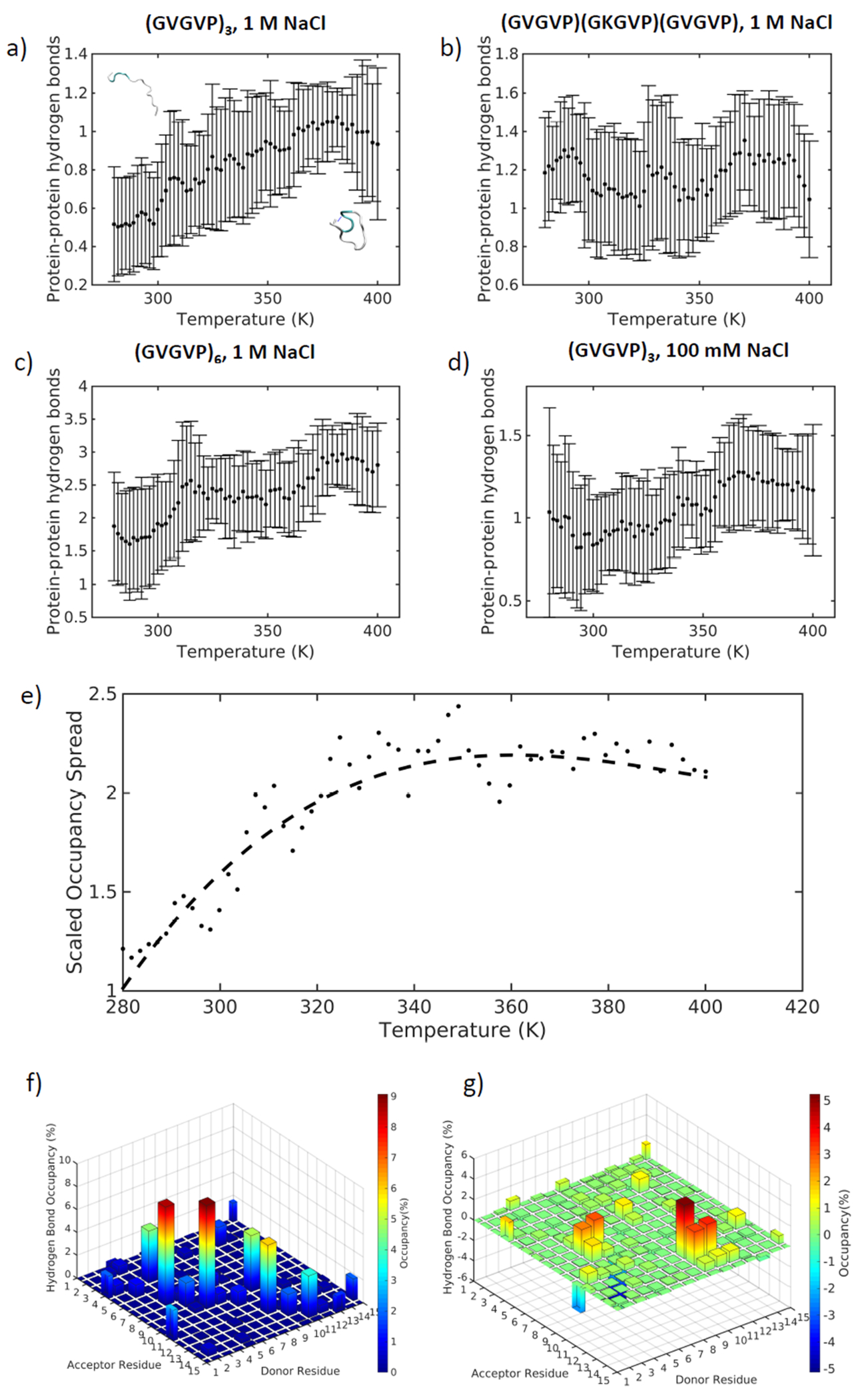
Hydrogen bonds between protein residues change with temperature for a) (GVGVP)3 in 1 M NaCl, b) (GVGVP)(GKGVP)(GVGVP) in 1 M NaCl, c) (GVGVP)6 in 1 M NaCl, d) (GVGVP)3 in 100 mM NaCl; e) Occupancy of hydrogen bonds between donor and acceptor residues as a function of temperature as defined by SI Equation 2; f) Hydrogen bond occupancy at 280 K; g) Hydrogen bond occupancy difference between 340 K and 280 K.
To determine the relative locations of the hydrogen bonds along the length of the protein, we define η, or scaled occupancy spread (SI Equation 2). This measure captures the relative distance between donor and acceptor residues where a hydrogen bond may exist. The scaled occupancy spread almost doubles across the temperature transition range for Systems 1 and 3, indicating that close-neighbor hydrogen bonds are replaced by hydrogen bonds connecting distant residues as temperature increases (Figure 4e, SI Figure 8a). This is consistent with the observation that the peptide folds up and is then stabilized by forming a hydrogen bond between opposite ends of the molecule. Hydrogen bond occupancy at low temperature (280 K) for sequence (GVGVP)3 is shown in Figure 4f. The occupancy indicates the fraction of time that a particular hydrogen bond exists between a donor-acceptor pair. The highest occurrence of hydrogen bonds appears along the diagonal of donor-acceptor residue pairs at low temperature, implying that the majority of hydrogen bonds that can form are between nearest neighbor residues. At high temperature (340 K), the distribution of hydrogen bond occupancy shifts away from the diagonal, as illustrated by the difference in occupancy between high and low temperatures in Figure 4g. This trend is not observed for Systems 2 and 4 (SI Figure 8b,c). The shift in hydrogen bond position for Systems 1 and 3 may be responsible for stabilizing structures at higher temperatures. Indeed, we find that the standard deviation of the radius of gyration is lower at high temperatures for Systems 1 and 3, and to a lower extent for System 4, indicating less variance in structure and greater stability at increased temperatures (SI Figure 9). This counterintuitive result from a kinetics standpoint highlights the unique inverse nature of ELP temperature transitions.
Trends in the amount of solvent accessible surface area across the temperature range (Figure 5a–d) are consistent with patterns in radius of gyration: when structural folding occurs, it is accompanied by a reduced solvent accessible surface area. This is clearly visible for Systems 1 and 3 (Figure 5a,c), whereas no shift is detected for System 2 (Figure 5b). For System 4, a mild shift towards a reduced solvent accessible surface area is observed at a higher temperature than for System 1 (Figure 5d). The patterns observed in solvation water molecules around the protein are similarly consistent (Figure 5e–h). The first hydration shell in each system occurs approximately within 2.1 Å of the protein, indicated by the first local minimum in the protein-water radial distribution functions (SI Figure 10). After the first local minima, a mild second local minima is discerned at approximately 3 Å. Beyond the first two hydration shells, water molecules are arranged as in bulk, as indicated by the flattening of the radial distribution functions, as a result of the small size of the protein that does not significantly disturb the bulk water. Because there is a constant transfer of molecules across the boundary of the first solvation shell, we consider a shell of 3 Å for solvation water calculations to capture the behavior of water molecules at the protein’s surface. We observe that the radial distribution function peaks flatten with temperature, most significantly for Systems 1 and 3 that display transitions, indicating that water molecules in the local hydration shells are replaced by bulk water as temperature increases.
Figure 5 |.
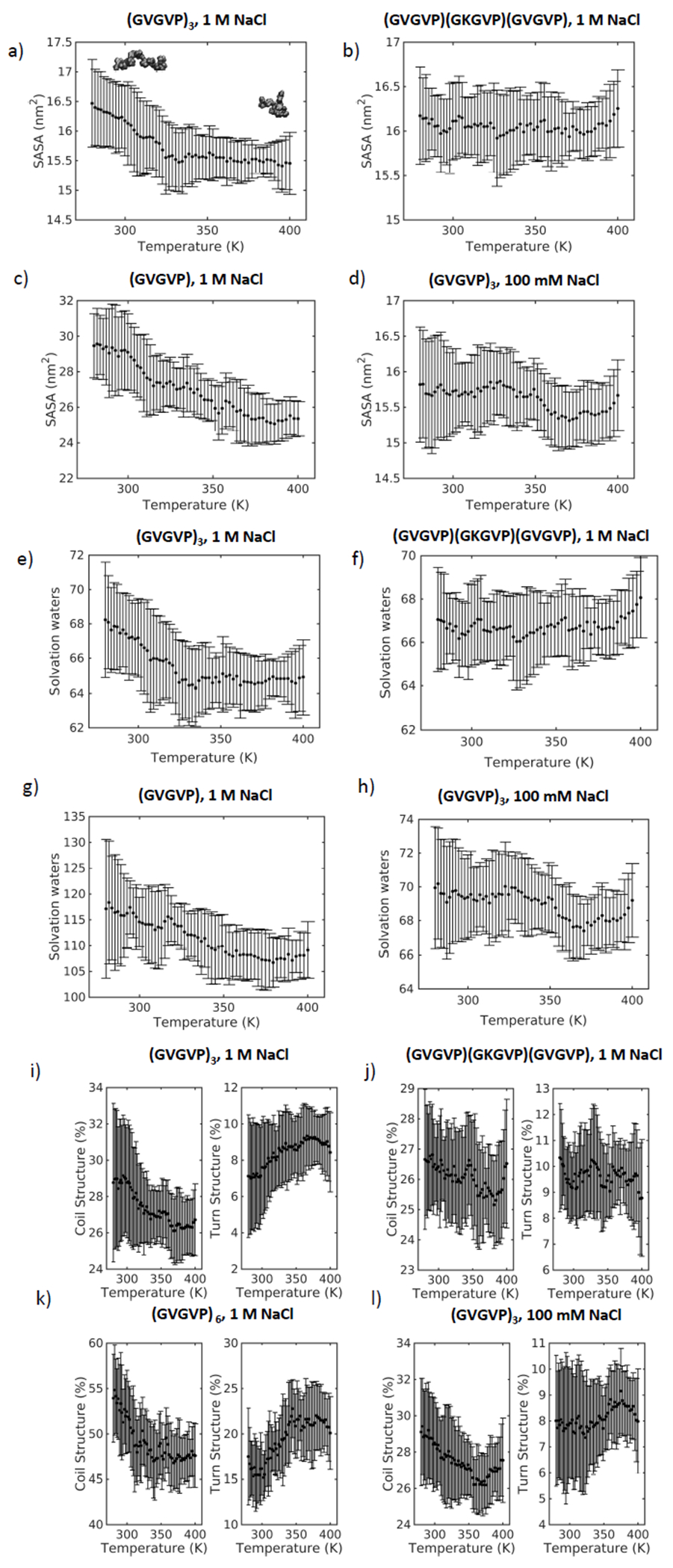
Solvent accessible surface area change with temperature for a) (GVGVP)3 in 1 M NaCl with snapshots at low and high temperature, b) (GVGVP)(GKGVP)(GVGVP) in 1 M NaCl, c) (GVGVP)6 in 1 M NaCl, d) (GVGVP)3 in 100 mM NaCl; e) Change with temperature of the number of water molecules within a 3 Å solvation shell for (GVGVP)3 in 1 M NaCl, f) (GVGVP)(GKGVP)(GVGVP) in 1 M NaCl, g) (GVGVP)6 in 1 M NaCl, h) (GVGVP)3 in 100 mM NaCl; i) Coil and turn secondary structure changes with temperature for (GVGVP)3 in 1 M NaCl, j) (GVGVP)(GKGVP)(GVGVP) in 1 M NaCl, k) (GVGVP)6 in 1 M NaCl, l) (GVGVP)3 in 100 mM NaCl.
The structural collapse and associated reduction in the solvent accessible surface area is driven by changes in local secondary structure (Figure 5i–l). These changes are characterized by a distinct transition from coil to turn structures, defined by the DSSP (Define Secondary Structure of Proteins) algorithm [49, 50]. In systems 1 and 3, with increasing temperature, coils are replaced by ordered secondary structure, including a very low percentage (< 1%) of sheet and helix, but primarily hydrogen-bonded turns. We note that mild changes in secondary structure are observed for Systems 2 and 4. This observation indicates that at the local level, these sequences exhibit some structural folding, though a full molecular collapse is not observed. In System 4, the transition shifts to higher temperatures, consistent with experimental findings in this study and earlier work [17, 60].
Salt Concentration Effects
The evident transition at higher salt concentration of System 1 compared to System 4, is attributed to an increased interaction between ions, protein and water molecules in the nearest hydration shells. The presence of ions in the first two nearest hydration shells increases noticeably at higher NaCl concentration. On average, 1.8 sodium and chloride ions exist in a 3 Å solvation shell around the protein in 1 M NaCl, in contrast to 0.5 ions in 100 mM NaCl. This is consistent across all temperatures. The ions that diffuse into the nearest hydration shell of the protein can form strong interactions with electronegative atoms in the peptide chain, facilitating structural folding (Figure 2i,j). Moreover, ions act to disturb the hydrogen-bonded water network surrounding the protein, promoting the formation of hydrogen bonds within the peptide as solvation waters are expelled from the nearest hydration shell.
Chain Length Effects
As the chain length is increased, the expanded form of the molecule expels an increased number of solvation waters upon folding. Water-water and peptide-water hydrogen bonds break, ejecting water molecules trapped at the surface of the protein out of the solvation shell into the bulk. As the water molecules shift in the longer-length peptide, it folds over itself, forming turn secondary structure stabilized by hydrogen bonds.
Chemistry Effects
Amino acid substitutions can substantially shift the transition temperatures of ELPs [16, 17]. At the molecular level, we find that transitions in local hydrogen bonding and secondary structure, as well as any structural peptide collapse are absent for the lysine-containing sequence. To evaluate these effects we consider the local environment in a 3 Å radius solvation shell around the seventh amino acid, valine or lysine, in ELP Systems 1 and 4, respectively. Notably, the valine in the seventh-position in System 1 contains approximately 6 water molecules in a 3 Å radius solvation shell, compared to approximately 10 water molecules surrounding the lysine residue in System 4 (Figure 6a,b). Reduction in the number of water molecules with increasing temperature is more prominent around the seventh position valine, than for lysine, yet in both systems the change in solvation waters is minimal – a difference of < 1 solvation water molecule between high and low temperatures – compared to the raw difference in the number of water molecules surrounding the seventh position residue.
Figure 6 |.
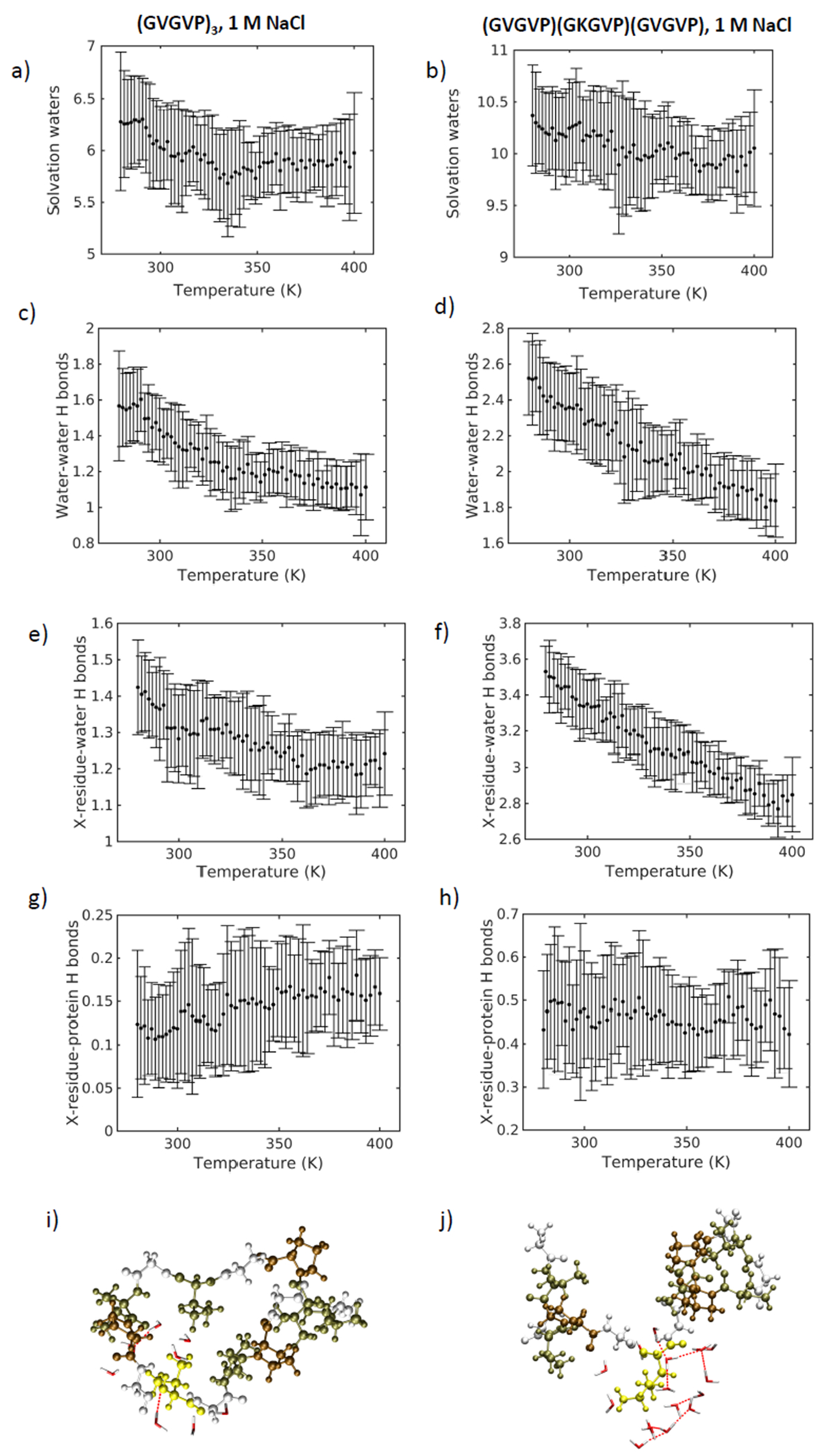
a) Solvation waters within a 3 Å solvation shell of the seventh residue valine for System 1 and b) lysine for System 4. Water-water hydrogen bonds around the seventh residue of c) System1 and d) System 4. Hydrogen bonds between water molecules and the seventh residue for e) System 1 and f) System 4. Hydrogen bonds formed between the peptide and the seventh residue of g) System 1 and h) System 4. Hydrogen bonded water molecules around the seventh residue (in yellow) of i) System 1 and j) System 4.
We can understand this result by considering the longer side chain of lysine compared to that of valine, a correspondingly larger exposed surface area to water and exposed positive charge. At high temperatures, both sequences exhibit a loss in water – water hydrogen bonds in the 3 Å radius solvation shell around the seventh residue (Figure 6c,d), as well as a decrease in seventh residue – water hydrogen bonds (Figure 6e,f). We see a greater number of water-water and residue-water hydrogen bonds in the lysine-containing sequence. Indeed at higher temperature where the network is disrupted, the number of hydrogen bonds associated with lysine exceeds the number of hydrogen bonds surrounding the seventh valine in System 1 - even at low temperature. This suggests that the lysine residue acts as a stiffened hinge that is densely hydrogen-bonded to an interconnected network of water molecules. The presence of a larger number of interconnected waters enveloping the center of the peptide chain prevents a structural collapse representative of the transition in System 1. This is supported by the fact that the number of hydrogen bonds between the peptide and the seventh valine in System 1 increases modestly, while no increase is observed for lysine (Figure 6g,h).
Remarkably, the local effects around the guest residue in the seventh position explain the formation of hydrogen bonds between distant residues in the peptide chain (GVGVP)3 (Figure 6i,j). The small, flexible valine hinge allows for structural collapse of the molecule and consequent system stabilization, in contrast to the lysine-containing sequence. We recognize two disparate roles for water: it acts as a plasticizer, enhancing the conformational mobility of the peptide [55]; yet, simultaneously, it provides an obstacle to structural flexibility based on its interaction with local components of the peptide.
Conclusion
In summary, in this study the effects of ionic concentration, chain length, and sequence chemistry on temperature-induced structural transitions in elastin-like peptides are presented. We consider these systems at the single-molecule scale finding that structural transitions occur as smooth shifts within defined transition regions. The behavior of water networks around the peptide is a driving factor in structural transitions. In the presence of higher salt, we find that ions can more easily diffuse into the nearest hydration shell of the protein, forming strong interactions with electronegative atoms in the peptide chain, which facilitates structural folding. Ions act to disturb the hydrogen-bonded water network surrounding the protein, promoting the formation of hydrogen bonds within the peptide as solvation waters are expelled from the nearest hydration shell. Longer chain length drives the expulsion of solvation waters as temperature is increased. As water-water and peptide-water hydrogen bonds brake, water molecules trapped at the surface of the protein are pushed out of the solvation shell into the bulk. This introduces the formation of turns stabilized by hydrogen bonds and an overall shift to compact molecular structure. Finally, sequence chemistry changes influence the local interactions with water. We compare a lysine-for-valine substitution and find that the lysine residue stiffens the hinge by hydrogen-bonding to an interconnected network of water molecules, and so prevents a structural collapse. We find that water acts as a plasticizer, enhancing the conformational mobility of the peptide [55] and simultaneously reducing structural flexibility based on its interactions with local components of the peptide.
Supplementary Material
Acknowledgements
We thank Zhao Qin and Davoud Ebrahimi for fruitful discussions. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), supported by the National Science Foundation grant number ACI-1053575. The authors acknowledge support from the NIH (5U01EB014976 and EB014283), ONR (N000141612333) and AFOSR (FA9550-11-1-0199).
References
- 1.Lee J, Macosko CW, and Urry DW, Phase transition and elasticity of protein-based hydrogels. Journal Of Biomaterials Science. Polymer Edition, 2001. 12(2): p. 229–242. [DOI] [PubMed] [Google Scholar]
- 2.Urry DW, Entropic elastic processes in protein mechanisms. I. Elastic structure due to an inverse temperature transition and elasticity due to internal chain dynamics. J Protein Chem, 1988. 7(1): p. 1–34. [DOI] [PubMed] [Google Scholar]
- 3.Urry DW, Shaw RG, and Prasad KU, Polypentapeptide of elastin: temperature dependence of ellipticity and correlation with elastomeric force. Biochemical And Biophysical Research Communications, 1985. 130(1): p. 50–57. [DOI] [PubMed] [Google Scholar]
- 4.Urry DW, et al. , Carbon-13 NMR relaxation studies demonstrate an inverse temperature transition in the elastin polypentapeptide. Biochemistry, 1985. 24(19): p. 5182–5189. [DOI] [PubMed] [Google Scholar]
- 5.Urry DW, Trapane TL, and Prasad KU, Phase-structure transitions of the elastin polypentapeptide-water system within the framework of composition-temperature studies. Biopolymers, 1985. 24(12): p. 2345. [DOI] [PubMed] [Google Scholar]
- 6.Urry DW, Entropic elastic processes in protein mechanisms. II. Simple (passive) and coupled (active) development of elastic forces. J Protein Chem, 1988. 7(2): p. 81–114. [DOI] [PubMed] [Google Scholar]
- 7.Urry DW, Free energy transduction in polypeptides and proteins based on inverse temperature transitions. Progress In Biophysics And Molecular Biology, 1992. 57(1): p. 23–57. [DOI] [PubMed] [Google Scholar]
- 8.Urry DW, MOLECULAR MACHINES - HOW MOTION AND OTHER FUNCTIONS OF LIVING ORGANISMS CAN RESULT FROM REVERSIBLE CHEMICAL-CHANGES. ANGEWANDTE CHEMIE-INTERNATIONAL EDITION, 1993. 32(6): p. 819–841. [Google Scholar]
- 9.Urry DW, What sustains life? [electronic resource] : consilient mechanisms for protein-based machines and materials. 2006: New York, N.Y. : Springer, c2006. [Google Scholar]
- 10.Roberts S, Dzuricky M, and Chilkoti A, Review: Elastin-like polypeptides as models of intrinsically disordered proteins. FEBS Letters, 2015. 589(Part A): p. 2477–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDaniel JR, Radford DC, and Chilkoti A, A Unified Model for De Novo Design of Elastin-like Polypeptides with Tunable Inverse Transition Temperatures. BIOMACROMOLECULES, 2013. 14(8): p. 2866–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurzbach D, et al. , Hydration layer coupling and cooperativity in phase behavior of stimulus responsive peptide polymers. J Am Chem Soc, 2013. 135(30): p. 11299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeo GC, Keeley FW, and Weiss AS, Coacervation of tropoelastin. Advances in Colloid and Interface Science, 2011. 167: p. 94–103. [DOI] [PubMed] [Google Scholar]
- 14.Urry DW, Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers. JOURNAL OF PHYSICAL CHEMISTRY B, 1997. 101(51): p. 11007–11028. [Google Scholar]
- 15.Urry DW, Five axioms for the functional design of peptide-based polymers as molecular machines and materials: Principle for macromolecular assemblies. BIOPOLYMERS, 1998. 47(2): p. 167–178. [Google Scholar]
- 16.Urry DW, et al. , Hydrophobicity scale for proteins based on inverse temperature transitions. Biopolymers, 1992. 32(9): p. 1243–1250. [DOI] [PubMed] [Google Scholar]
- 17.Nuhn H and Klok HA, Secondary Structure Formation and LCST Behavior of Short Elastin-Like Peptides. BIOMACROMOLECULES, 2008. 9(10): p. 2755–2763. [DOI] [PubMed] [Google Scholar]
- 18.Ribeiro A, et al. , Article: Influence of the Amino-Acid Sequence on the Inverse Temperature Transition of Elastin-Like Polymers. Biophysical Journal, 2009. 97: p. 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer DE and Chilkoti A, Genetically encoded synthesis of protein-based polymers with precisely specified molecular weight and sequence by recursive directional ligation: Examples from the elastin-like polypeptide system. BIOMACROMOLECULES, 2002. 3(2): p. 357–367. [DOI] [PubMed] [Google Scholar]
- 20.Girotti A, et al. , Influence of the molecular weight on the inverse temperature transition of a model genetically engineered elastin-like pH-responsive polymer. MACROMOLECULES, 2004. 37(9): p. 3396–3400. [Google Scholar]
- 21.Reguera J, et al. , Effect of NaCl on the exothermic and endothermic components of the inverse temperature transition of a model elastin-like polymer. BIOMACROMOLECULES, 2007. 8(2): p. 354–358. [DOI] [PubMed] [Google Scholar]
- 22.MacKay JA, et al. , Quantitative Model of the Phase Behavior of Recombinant pH-Responsive Elastin-Like Polypeptides. BIOMACROMOLECULES, 2010. 11(11): p. 2873–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strzegowski LA and Martinez MB, Photomodulation of the inverse temperature transition of a modified elastin poly(pentapeptide). Journal of the American Chemical Society, 1994. 116(2): p. 813. [Google Scholar]
- 24.Dreher MR, et al. , Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, 2008. 130(2): p. 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDaniel JR, et al. , Noncanonical self-assembly of highly asymmetric genetically encoded polypeptide amphiphiles into cylindrical micelles. Nano Letters, 2014. 14(11): p. 6590–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trabbic-Carlson K, Setton LA, and Chilkoti A, Swelling and mechanical behaviors of chemically cross-linked hydrogels of elastin-like polypeptides. BIOMACROMOLECULES, 2003. 4(3): p. 572–580. [DOI] [PubMed] [Google Scholar]
- 27.Martín L, et al. , Synthesis and characterization of macroporous thermosensitive hydrogels from recombinant elastin-like polymers. Biomacromolecules, 2009. 10(11): p. 3015–3022. [DOI] [PubMed] [Google Scholar]
- 28.MacEwan SR and Chilkoti A, Review: Applications of elastin-like polypeptides in drug delivery. Journal of Controlled Release, 2014. 190: p. 314–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDaniel JR, Callahan DJ, and Chilkoti A, Drug delivery to solid tumors by elastin-like polypeptides. Advanced Drug Delivery Reviews, 2010. 62: p. 1456–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herrero-Vanrell R, et al. , Self-assembled particles of an elastin-like polymer as vehicles for controlled drug release. Journal of Controlled Release, 2005. 102: p. 113–122. [DOI] [PubMed] [Google Scholar]
- 31.Nettles DL, Chilkoti A, and Setton LA, Applications of elastin-like polypeptides in tissue engineering. Advanced Drug Delivery Reviews, 2010. 62: p. 1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacEwan SR and Chilkoti A, Digital Switching of Local Arginine Density in a Genetically Encoded Self-Assembled Polypeptide Nanoparticle Controls Cellular Uptake. NANO LETTERS, 2012. 12(6): p. 3322–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dreher MR, et al. , Thermal cycling enhances the accumulation of a temperature-sensitive biopolymer in solid tumors. CANCER RESEARCH, 2007. 67(9): p. 4418–4424. [DOI] [PubMed] [Google Scholar]
- 34.Amiram M, et al. , Injectable protease-operated depots of glucagon-like peptide-1 provide extended and tunable glucose control. PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA, 2013. 110(8): p. 2792–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brooks BR, et al. , CHARMM: a program for macromolecular energy, minimisation, and dynamics calculations. Journal of Computational Chemistry, 1983. 4(2): p. 187–217. [Google Scholar]
- 36.Van Der Spoel D, et al. , GROMACS: fast, flexible, and free. J Comput Chem, 2005. 26(16): p. 1701–18. [DOI] [PubMed] [Google Scholar]
- 37.MacKerell AD, et al. , All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. The Journal of Physical Chemistry B, 1998. 102(18): p. 3586–3616. [DOI] [PubMed] [Google Scholar]
- 38.Sugita Y and Okamoto Y, Replica-exchange molecular dynamics method for protein folding. Chemical Physics Letters, 1999. 314(1–2): p. 141–151. [Google Scholar]
- 39.Berendsen HJC, et al. , Molecular dynamics with coupling to an external bath. Journal of Chemical Physics, 1984. 81(8): p. 3684. [Google Scholar]
- 40.Parrinello M and Rahman A, Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics, 1981. 52(12): p. 7182. [Google Scholar]
- 41.Essmann U, et al. , A smooth particle mesh Ewald method. Journal of Chemical Physics, 1995. 103(19): p. 8577. [Google Scholar]
- 42.Rousseau R, et al. , Temperature-dependent conformational transitions and hydrogen-bond dynamics of the elastin-like octapeptide GVG(VPGVG): a molecular-dynamics study. Biophysical Journal, 2004. 86(3): p. 1393–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schreiner E, et al. , Folding and unfolding of an elastinlike oligopeptide: “Inverse temperature Transition,” reentrance, and hydrogen-bond dynamics. Physical Review Letters, 2004. 92(14): p. 148101/1-4. [DOI] [PubMed] [Google Scholar]
- 44.Li B, Alonso DOV, and Daggett V, The molecular basis for the inverse temperature transition of elastin. JOURNAL OF MOLECULAR BIOLOGY, 2001. 305(3): p. 581–592. [DOI] [PubMed] [Google Scholar]
- 45.Li NK, et al. , Molecular description of the LCST behavior of an elastin-like polypeptide. Biomacromolecules, 2014. 15(10): p. 3522–3530. [DOI] [PubMed] [Google Scholar]
- 46.Patriksson A and van der Spoel D, A temperature predictor for parallel tempering simulations. Physical Chemistry Chemical Physics, 2008. 10(15): p. 2073–2077. [DOI] [PubMed] [Google Scholar]
- 47.Feig M, Karanicolas J, and Brooks IIICL, MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. Journal of Molecular Graphics and Modelling, 2004. 22: p. 377–395. [DOI] [PubMed] [Google Scholar]
- 48.Towns J, et al. , XSEDE: Accelerating Scientific Discovery. Computing in Science & Engineering, 2014. 16(5): p. 62–74. [Google Scholar]
- 49.Joosten RP, et al. , A series of PDB related databases for everyday needs. Nucleic Acids Research, 2011. 39(Database issue): p. D411–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kabsch W and Sander C, Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 1983. 22(12): p. 2577–2637. [DOI] [PubMed] [Google Scholar]
- 51.Eisenhaber F, et al. , The double cubic lattice method: Efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. Journal of Computational Chemistry, 1995. 16(3): p. 273–284. [Google Scholar]
- 52.Humphrey W, Dalke A, and Schulten K, VMD: Visual molecular dynamics. Journal of Molecular Graphics, 1996. 14(33). [DOI] [PubMed] [Google Scholar]
- 53.Meyer DE and Chilkoti A, Quantification of the effects of chain length and concentration on the thermal behavior of elastin-like polypeptides. BIOMACROMOLECULES, 2004. 5(3): p. 846–851. [DOI] [PubMed] [Google Scholar]
- 54.Christensen T, et al. , Predicting transition temperatures of elastin-like polypeptide fusion proteins. Biomacromolecules, 2013. 14(5): p. 1514–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Debelle L and Tamburro AM, Elastin: molecular description and function. INTERNATIONAL JOURNAL OF BIOCHEMISTRY & CELL BIOLOGY, 1999. 31(2): p. 261–272. [DOI] [PubMed] [Google Scholar]
- 56.Hong M, et al. , Structure of an elastin-mimetic polypeptide by solid-state NMR chemical shift analysis. BIOPOLYMERS, 2003. 70(2): p. 158–168. [DOI] [PubMed] [Google Scholar]
- 57.Kurkova D, et al. , Structure and dynamics of two elastin-like polypentapeptides studied by NMR spectroscopy. BIOMACROMOLECULES, 2003. 4(3): p. 589–601. [DOI] [PubMed] [Google Scholar]
- 58.Pometun MS, Chekmenev EY, and Wittebort RJ, Quantitative observation of backbone disorder in native elastin. JOURNAL OF BIOLOGICAL CHEMISTRY, 2004. 279(9): p. 7982–7987. [DOI] [PubMed] [Google Scholar]
- 59.Rauscher S, et al. , Proline and glycine control protein self-organization into elastomeric or amyloid fibrils. STRUCTURE, 2006. 14(11): p. 1667–1676. [DOI] [PubMed] [Google Scholar]
- 60.Luan CH, et al. , Differential scanning calorimetry studies of NaCl effect on the inverse temperature transition of some elastin-based polytetra-, polypenta-, and polynonapeptides. Biopolymers, 1991. 31(5): p. 465–475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.