

This cohort study analyzes data from 4 independent cohorts to assess the clinical value of proposed biological research criteria intended to identify individuals with preclinical Alzheimer disease.
Key Points
Question
What is the clinical relevance of the amyloid, tau, and neurodegeneration (AT[N]) biological classification of Alzheimer disease in older adults without cognitive impairment?
Findings
In this cohort study of 580 participants without cognitive impairment from 4 independent cohorts, between 43% and 100% of A+T+(N+) participants progressed to mild cognitive impairment within 2 to 3 years after positron emission tomography. The majority of A+T+ nonprogressors also showed cognitive decline.
Meaning
Older adults without cognitive impairment and with biological Alzheimer disease may be at imminent risk of developing mild cognitive impairment and may be ideal candidates for disease-modifying therapies.
Abstract
Importance
National Institute on Aging–Alzheimer’s Association (NIA-AA) workgroups have proposed biological research criteria intended to identify individuals with preclinical Alzheimer disease (AD).
Objective
To assess the clinical value of these biological criteria to identify older individuals without cognitive impairment who are at near-term risk of developing symptomatic AD.
Design, Setting, and Participants
This longitudinal cohort study used data from 4 independent population-based cohorts (PREVENT-AD, HABS, AIBL, and Knight ADRC) collected between 2003 and 2021. Participants were older adults without cognitive impairment with 1 year or more of clinical observation after amyloid β and tau positron emission tomography (PET). Median clinical follow-up after PET ranged from 1.94 to 3.66 years.
Exposures
Based on binary assessment of global amyloid burden (A) and a composite temporal region of tau PET uptake (T), participants were stratified into 4 groups (A+T+, A+T−, A−T+, A−T−). Presence (+) or absence (−) of neurodegeneration (N) was assessed using temporal cortical thickness.
Main Outcomes and Measures
Each cohort was analyzed separately. Primary outcome was clinical progression to mild cognitive impairment (MCI), identified by a Clinical Dementia Rating score of 0.5 or greater in Knight ADRC and by consensus committee review in the other cohorts. Clinical raters were blind to imaging, genetic, and fluid biomarker data. A secondary outcome was cognitive decline, based on a slope greater than 1.5 SD below the mean of an independent subsample of individuals without cognitive impairment. Outcomes were compared across the biomarker groups.
Results
Among 580 participants (PREVENT-AD, 128; HABS, 153; AIBL, 48; Knight ADRC, 251), mean (SD) age ranged from 67 (5) to 76 (6) years across cohorts, with between 55% (137/251) and 74% (95/128) female participants. Across cohorts, 33% to 83% of A+T+ participants progressed to MCI during follow-up (mean progression time, 2-2.72 years), compared with less than 20% of participants in other biomarker groups. Progression further increased to 43% to 100% when restricted to A+T+(N+) individuals. Cox proportional hazard ratios for progression to MCI in the A+T+ group vs other biomarker groups were all 5 or greater. Many A+T+ nonprogressors also showed longitudinal cognitive decline, while cognitive trajectories in other groups remained predominantly stable.
Conclusions and Relevance
The clinical prognostic value of NIA-AA research criteria was confirmed in 4 independent cohorts, with most A+T+(N+) older individuals without cognitive impairment developing AD symptoms within 2 to 3 years.
Introduction
The National Institute on Aging–Alzheimer’s Association (NIA-AA) research criteria for Alzheimer disease (AD) were revised in 2018 to add tau biomarkers. In the resulting AT(N) framework, amyloid β (A) and tau (T) are needed for the diagnosis of AD, while neurodegeneration (N) is used to stage disease severity.1 These biomarkers can be classified as normal (−) and abnormal (+) such that individuals who are A+T+ can be said to have biological AD, even if they do not have cognitive symptoms. The clinical significance of biologically defined AD in individuals without cognitive impairment remains debated,2 given that abnormal levels of amyloid β (Aβ) and tau are apparent in approximately 20% of older adults without cognitive impairment both in vivo3 and at autopsy.4 However, as the cited studies are cross-sectional, it is unclear whether A+T+ individuals are at imminent risk of developing AD-related cognitive impairment. Frequent near-term development of mild cognitive impairment (MCI) in A+T+ individuals without cognitive impairment would provide strong evidence favoring a biological definition of preclinical AD. It would also have important implications both for clinical trial recruitment and prognosis of early clinical disease.
Using positron emission tomographic (PET) signal for Aβ or tau deposition across 4 independent cohorts, we investigated whether elevation of both biomarker signals were associated with near-term progression from cognitively unimpaired to MCI. We also tested whether the evidence of neurodegeneration added clinical prognostic value to the amyloid and tau PET biomarkers. Analyses were performed separately for each cohort to test the robustness of findings across methodologies and samples.
Methods
Participants and Study Design
Participants included 128 individuals from the family history–positive Pre-symptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease (PREVENT-AD) cohort, 153 from the Harvard Aging Brain Study (HABS), 48 from the Australian Imaging, Biomarker & Lifestyle (AIBL) study, and 251 from the Knight Alzheimer Disease Research Center (ADRC) data set (eMethods in Supplement 1). All participants included in this study had at least 1 Aβ and tau PET scan, were cognitively unimpaired at the time of PET, and had at least 12 months of clinical follow-up thereafter. Participants provided written informed consent, and research procedures were approved by the relevant ethics committees. All analyses were performed separately for each cohort.
Full details of all measures, outcomes, their relative timing, and analyses are in the eMethods in Supplement 1. This study follows the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guidelines for cohort studies.
Cognitive Evaluation
Participants completed the Mini-Mental State Examination (MMSE)5 at the time of tau PET and had longitudinal cognitive testing using a composite measure specific to each cohort. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS)6 was used in PREVENT-AD, and the Preclinical Alzheimer Cognitive Composite score (PACC) used in the other cohorts.7,8 Performance was evaluated using cohort-derived z scores. Tau PET was introduced midstudy in all cohorts. All participants were required to be cognitively unimpaired both at cognitive baseline and at the time of PET.
Outcomes
The primary outcome measure was clinical progression to MCI following PET among participants without cognitive impairment. This outcome was adjudicated in all instances by persons masked to PET and MRI data, and to APOE genotype, though not blind to MMSE performance in the PREVENT-AD, HABS, or AIBL cohorts. In PREVENT-AD, HABS, and AIBL, MCI classifications were made by consensus committees comprising expert clinical and research staff. In the Knight ADRC, MCI was defined by a Clinical Dementia Rating (CDR)9 score of 0.5 or greater. Median follow-up after PET ranged from 1.94 to 3.66 years across cohorts. A secondary outcome was cognitive decline, as defined by a longitudinal slope in composite cognitive scores of 1.5 SD or more below the mean of an independent subsample of cognitively normal participants within each cohort.10 For this outcome, we took advantage of the full length of study follow-up (including pre-PET) to create the slopes and characterize participants as “decliners” vs cognitively “stable” (median follow-up across cohorts, 5.10-6.31 years; minimum, 0.90-3.26 years; maximum, 7.26-14.47 years).
AT(N) Classification
Aβ PET imaging was performed using [18F]NAV4694 (NAV) in PREVENT-AD, [11C] Pittsburgh Compound B (PiB) in HABS, [18F]AV45 (florbetapir) and NAV in AIBL, and PiB and florbetapir in Knight ADRC (processing details in the eMethods in Supplement 1). Tau PET was performed using [18F]AV1451 (flortaucipir) in all cohorts.11,12,13,14,15 T1-weighted structural MRI scans were collected on 3-T scanners and segmented with the Freesurfer Desikan-Killiany atlas.16 Preprocessing was performed using cohort-specific pipelines and did not include partial volume correction. Standardized uptake value ratios (SUVRs) (distribution volume ratios [DVRs] for PiB) for each Desikan-Killiany region were computed using the cerebellum gray matter for all scans except for tau PET in PREVENT-AD, which used inferior cerebellar gray matter.17
Participants were allocated to 4 PET biomarker groups (A+T+, A+T−, A−T+, A−T−). Cohort-specific thresholds were used to establish Aβ positivity based on a global amyloid index18 (Centiloid values: PREVENT-AD = 22.32; HABS = 23.9; AIBL = 25; Knight ADRC = 27.1 and 21.9 for PiB and florbetapir, respectively; see the eMethods in Supplement 1 for SUVR/DVR). A temporal meta–region of interest (ROI) was used as the primary measure of tau positivity. This comprised the average SUVR of the bilateral entorhinal cortex, amygdala, parahippocampal gyrus, fusiform gyrus, and inferior and middle temporal gyri.18 Tau positivity was defined as meta-ROI uptake surpassing 2 SD from the mean of A− participants who were cognitively unimpaired (at baseline) in each cohort (SUVR cutoffs: PREVENT-AD = 1.28; HABS = 1.29; AIBL = 1.28; Knight ADRC = 1.26).
In secondary analyses, the presence (+) or absence (−) of neurodegeneration (N) was designated based on average cortical thickness in a bilateral temporal meta-ROI comprising the entorhinal cortex, fusiform, inferior temporal, and middle temporal gyri.17 Participants were classified as neurodegeneration positive if temporal cortical thickness was below the 20th percentile of an independent subsample of cognitively normal participants within the respective cohorts.
Statistical Analysis
Analyses were performed separately for each cohort to assess replicability of results across samples and methodologies. The A−T+ group was excluded from statistical comparisons because of the low number of participants (PREVENT-AD, 0; HABS, 4; AIBL, 1; Knight ADRC, 4), although data from this group are presented visually for completeness. For demographic and clinical variables, we used 1-way analyses of variance with Tukey post hoc tests to compare biomarker groups on continuous variables and Fisher exact tests for categorical variables, including MCI progression status. Cox proportional hazard models tested whether the risk of MCI progression over time was higher in the A+T+ group relative to the other PET biomarker groups, including age, sex, education, and APOE ε4 status as covariates. In follow-up analyses, continuous measures of neurodegeneration (temporal cortical thickness or hippocampal volume) were added to the PET biomarker Cox models to test the additional clinical prognostic value of (N). These were used instead of categorical AT(N) status given the very small sample size of each AT(N) group. Using concordance measures of model fit, we also compared the performance of each of these AT(N) models with clinical models that included MMSE score, age, sex, education, and APOE ε4 status. Furthermore, after confirming linearity of the longitudinal cognitive trajectories across cohorts, we used linear mixed-effects models with random slopes and intercepts to investigate longitudinal cognitive decline across the different AT(N) groups. This secondary outcome was intended to explore whether individuals who had not yet progressed to MCI were nonetheless likely to be on a clinical pathway toward AD symptoms. To do so, participants were further divided into cognitively stable vs decliners based on individual longitudinal cognitive slopes. The proportion of participants who were cognitive decliners vs cognitively stable in each biomarker group was then compared using Fisher exact tests.
We also performed sensitivity analyses in which analyses were repeated using (1) harmonized Aβ and tau thresholds across cohorts, (2) other commonly used regions to define tau PET positivity, and (3) hippocampal volume to define neurodegeneration.
The α was set at P < .05 for all analyses. Analyses were performed using R Studio version 1.1.463.
Results
Demographic and Biological Characteristics Across Biomarker Groups
Among 580 participants (PREVENT-AD, 128; HABS, 153; AIBL, 48; Knight ADRC, 251), mean (SD) age ranged from 67 (5) to 76 (6) years across cohorts, which had between 55% (137/251) and 74% (95/128) female participants. Between 7.17% (18/251) and 12.50% (6/48) of participants were classified as A+T+ using the tau temporal meta-ROI, compared with 20.83% (10/48) to 25.78% (33/128) as A+T−, 0 to 2.61% (4/153) A−T+, and 64.58% (31/48) to 68.13% (171/251) A−T− (eTables 4 and 5 in Supplement 1 show groupings using other regions to define T+ and harmonized Aβ and tau thresholds across cohorts). Characteristics of participants across cohorts and biomarker groups are presented in the Table (the eMethods and eTables 2 and 3 in Supplement 1 contain statistics and characteristics by MCI progression status).
Table. Demographic, Pathological, and Clinical Characteristics of Participants by Biomarker Group Across Cohortsa.
| Characteristic | Mean (SD) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PREVENT-AD | HABS | AIBL | Knight ADRC | |||||||||||||||||
| Full sample (n = 128) | A+T+ (n = 11) | A+T− (n = 33) | A−T+ (n = 0) | A−T− (n = 84) | Full sample (n = 153) | A+T+ (n = 12) | A+T− (n = 35) | A−T+ (n = 4) | A−T− (n = 102) | Full sample (n = 48) | A+T+ (n = 6) | A+T− (n = 10) | A−T+ (n = 1) | A−T− (n = 31) | Full sample (n = 251) | A+T+ (n = 18) | A+T− (n = 58) | A−T+ (n = 4) | A−T− (n = 171) | |
| Demographic | ||||||||||||||||||||
| Age, y | 67.35 (4.87) | 72.17 (5.12) b,c | 66.72 (4.43) | NA | 66.97 (4.71) | 76.11 (6.33) | 78.17 (5.08) | 77.55 (6.24) | 84.06 (3.72) | 75.06 (6.26) | 74.71 (6.87) | 79.17 (6.55) c | 79.50 (7.76) d | 80 | 72.13 (5.40) | 71.97 (5.73) | 74.81 (4.79)c | 72.21 (5.73) | 70.79 (4.74) | 71.62 (5.79) |
| Sex, F:M (% F) | 95:33 (74.22) | 9:2 (81.82) | 26:7 (78.79) | NA | 60:24 (71.43) | 86:67 (56.21) | 9:3 (75) | 18:17 (51.43) | 2:2 (50) | 57:45 (55.88) | 29:19 (60.42) | 5:1 (83.33) | 6:4 (60) | 1:0 (100) | 17:14 (54.84) | 137:114 (54.58) | 14:4 (77.78)c | 37:21 (63.79)d | 3:1 (75) | 83:88 (48.54) |
| Education, y | 15.17 (3.28) | 13.09 (2.81) | 15.21 (2.91) | NA | 15.43 (3.41) | 16.08 (3.06) | 17.00 (2.00) | 16.06 (2.91) | 18 (1.63) | 15.91 (3.23) | 11.79 (2.97) | 9.67 (2.73) | 12.20 (2.86) | 15 | 11.97 (2.96) | 16.33 (2.38) | 15.89 (1.94) | 16.64 (2.34) | 16.00 (1.63) | 16.28 (2.45) |
| APOE ɛ4 carriers, No. (%) | 50 (39.06) | 8 (72.73) c | 19 (57.58) d | NA | 23 (27.38) | 46 (30.07) | 11 (91.67) b,c | 19 (54.29) d | 1 (25) | 15 (14.71) | 12 (25) | 2 (33.33) | 3 (30) | 0 | 7 (22.58) | 76 (30.28) | 13 (72.22)b,c | 26 (44.83)d | 0 | 37 (21.64) |
| Race and ethnicity, No. (%) | ||||||||||||||||||||
| Black and African American | 1 (0.78) | 1 (9.09) | 0 | NA | 0 | 21 (13.73) | 2 (16.67) | 3 (8.57) | 0 | 16 (15.69) | NA | NA | NA | NA | NA | 29 (11.55) | 3 (16.67) | 3 (5.17) | 1 (25) | 22 (12.87) |
| Hispanic | 2 (1.56) | 0 | 0 | NA | 2 (2.38) | 0 | 0 | 0 | 0 | 0 | NA | NA | NA | NA | NA | 0 | 0 | 0 | 0 | 0 |
| White | 125 (97.66) | 10 (90.91) | 33 (100) | NA | 82 (97.62) | 128 (83.66) | 10 (83.33) | 31 (88.57) | 4 (100) | 83 (81.37) | NA | NA | NA | NA | NA | 222 (88.45) | 15 (83.33) | 55 (94.83) | 3 (75) | 149 (87.13) |
| Othere | 0 | 0 | 0 | NA | 0 | 4 (2.61) | 0 | 1 (2.86) | 0 | 3 (2.94) | NA | NA | NA | NA | NA | 0 | 0 | 0 | 0 | 0 |
| PET and MRI | ||||||||||||||||||||
| Global Aβ Centiloid value | 26.59 (28.76) | 73.62 (32.52) b,c | 47.54 (33.57) d | NA | 12.21 (5.13) | 19.00 (20.52) | 50.99 (13.85) c | 43.66 (18.74) d | 10.45 (1.42) | 7.11 (4.05) | 19.35 (38.44) | 72.33 (34.33) b | 64.10 (23.26) d | −16 | −4.19 (10.60) | 21.03 (33.51) | 79.03 (38.64)b,c | 56.12 (30.07)d | 6.67 (7.95) | 3.35 (10.09) |
| Temporal meta-ROI SUVR | 1.17 (0.11) | 1.42 (0.16) b,c | 1.17 (0.06) | NA | 1.14 (0.07) | 1.18 (0.09) | 1.39 (0.06) b,c | 1.19 (0.06) d | 1.31 (0.02) | 1.15 (0.06) | 1.19 (0.16) | 1.51 (0.16) b,c | 1.18 (0.10) | 1.33 | 1.12 (0.09) | 1.14 (0.09) | 1.35 (0.09)b,c | 1.15 (0.07)d | 1.29 (0.02) | 1.11 (0.07) |
| Temporal cortical thickness, mm | 2.89 (0.11) | 2.81 (0.11)b | 2.94 (0.09) d | NA | 2.88 (0.11) | 2.86 (0.16) | 2.71 (0.19) b,c | 2.85 (0.18) | 2.88 (0.13) | 2.88 (0.14) | 2.88 (0.11) | 2.82 (0.09) | 2.86 (0.11) | 3.02 | 2.90 (0.11) | 2.84 (0.14) | 2.80 (0.18) | 2.86 (0.13) | 2.83 (0.13) | 2.83 (0.14) |
| Hippocampal volume, cm3 (% of TIV) | 0.54 (0.06) | 0.51 (0.04)b | 0.56 (0.06) | NA | 0.54 (0.06) | 0.48 (0.06) | 0.44 (0.06) c | 0.47 (0.05) | 0.46 (0.04) | 0.49 (0.06) | 0.51 (0.06) | 0.50 (0.02) | 0.48 (0.04) | 0.57 | 0.52 (0.06) | 0.51 (0.07) | 0.47 (0.07)b | 0.52 (0.07) | 0.50 (0.07) | 0.51 (0.07) |
| Cognition | ||||||||||||||||||||
| MMSE score (/30) | 28.80 (1.26) | 27.73 (1.56) b,c | 29.15 (0.87) | NA | 28.80 (1.29) | 29.26 (0.98) | 28.50 (1.17)b,c | 29.31 (0.87) | 29.75 (0.50) | 29.31 (0.97) | 28.46 (1.61) | 25.83 (2.04)b,c | 28.80 (1.48) | 27 | 28.90 (1.01) | 29.27 (1.08) | 29.17 (1.20) | 29.34 (1.04) | 29.00 (1.41) | 29.27 (1.09) |
| RBANS global cognition score at baseline | −0.09 (0.88) | −0.41 (0.99) | 0.00 (0.89) | NA | −0.09 (0.87) | NA | NA | NA | NA | NA | ||||||||||
| PACC score at baseline | NA | NA | NA | NA | NA | 0.11 (0.61) | 0.18 (0.55) | 0.10 (0.55) | −0.02 (0.25) | 0.11 (0.66) | −0.46 (0.89) | −0.59 (0.58) | −0.71 (0.95) | −1.40 | −0.33 (0.92) | 0.00 (0.69) | −0.14 (0.54) | 0.04 (0.59) | 0.15 (0.35) | 0.00 (0.75) |
Abbreviations: APOE, apolipoprotein E genetic locus; Aβ, amyloid β; DVR, distribution volume ratio; F, female; M, male; MCI, mild cognitive impairment; meta-ROI, meta–region of interest; MMSE, Mini-Mental State Examination; MRI, magnetic resonance imaging; PACC, Preclinical Alzheimer Cognitive Composite; PET, positron emission tomography; RBANS, Repeatable Battery for the Assessment of Neuropsychological Status; SUVR, standardized uptake value ratio; TIV, total intracranial volume.
The A−T+ group is presented for completion but was not included in statistical analysis owing to its small sample size. Age and MMSE performance were calculated at the time of tau PET. Education data were collected in ranges in AIBL, with the lower boundary of the range used in the current analyses. Years of education are therefore likely underestimated in this cohort (further details in the eMethods in Supplement 1). APOE ɛ4 carriers had at least 1 copy of the ɛ4 allele. Cognitive variables (RBANS, PACC) are reported as cohort-derived z scores.
Significant difference between A+T+ and A+T− groups at P < .05.
Significant difference between A+T+ and A−T− groups at P < .05.
Significant difference between A+T− and A−T− groups at P < .05.
Other race and ethnicity included the categories Asian, Native American, or more than 1 race.
Clinical Progression Rates Across Biomarker Groups
Between 6.54% (10/153) and 16.67% (8/48) of participants across cohorts progressed from cognitively unimpaired to MCI after PET (mean [SD] progression time: PREVENT-AD, 2.00 [1.10] years; HABS, 2.72 [1.49] years; AIBL, 2.55 [1.18] years; Knight ADRC, 2.67 [1.18] years). MCI progression status by biomarker group is displayed in Figure 1 and eTable 4 in Supplement 1. Examples of Aβ and tau PET scans for each biomarker group and cognitive status from PREVENT-AD are presented in Figure 2. Across all cohorts, a greater proportion of A+T+ participants progressed to MCI (ADRC, 33.33%; HABS, 41.67%; PREVENT-AD, 54.55%; AIBL, 83.33%) compared with the other PET biomarker groups (<20%; all P ≤ .004) (Figure 1A-D and eTable 4 in Supplement 1). Compared with other regions (entorhinal, inferior temporal, or any), the meta-ROI for tau positivity detected the highest proportion of MCI progressors in the PREVENT-AD, HABS, and Knight ADRC cohorts, whereas an inferior temporal ROI detected the highest proportion of MCI progressors in AIBL (100% vs 83.33%) (eTable 4 and eFigure 1 in Supplement 1). Harmonized thresholds generally performed worse than cohort-specific thresholds in detecting MCI progressors (eTable 5 and eFigure 2 in Supplement 1). In A+T+ participants, evidence of neurodegeneration (N+), defined using temporal cortical thickness, was associated with a 42.86% to 100% MCI progression rate (Figure 1E-H and the eResults in Supplement 1). Progression rates ranged from 50% to 75% using hippocampal volume to define (N+) (eResults and eFigure 3 in Supplement 1).
Figure 1. Participants Progressing to Mild Cognitive Impairment (MCI) After Positron Emission Tomography (PET) vs Those Remaining Cognitively Unimpaired (CU) in Each Biomarker Group, Across Cohorts .
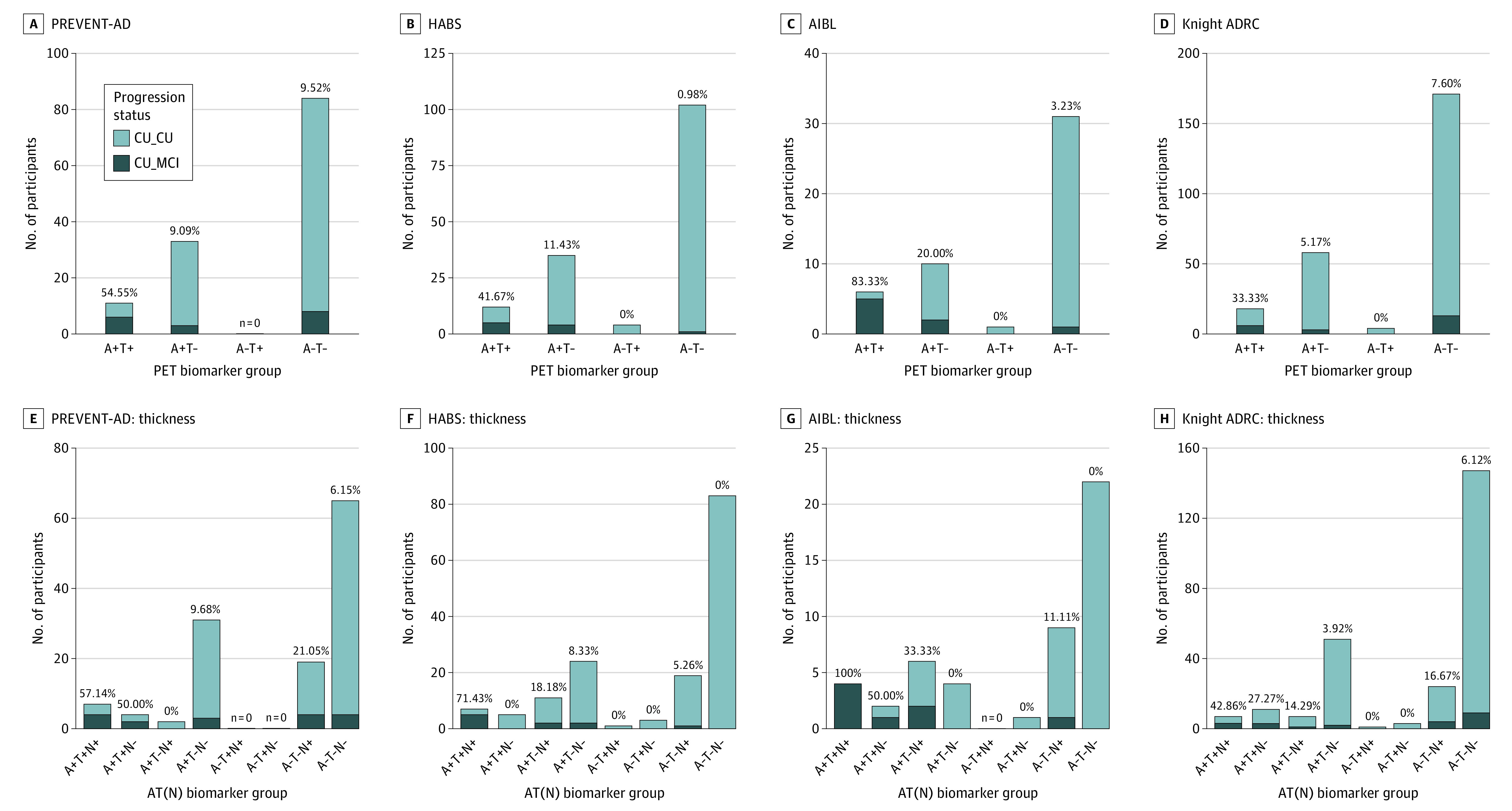
Percentage values represent the proportion of MCI progressors within the group. Note: The amyloid-negative, tau-positive (A−T+) group is displayed for visualization purposes but was not included in statistical analysis because of the small sample size. While the MCI classifications were based on clinical consensus in the PREVENT-AD, HABS, and AIBL cohorts, they were based on a Clinical Dementia Rating of 0.5 or greater for the Knight ADRC cohort. Neurodegeneration (N) was defined by temporal cortical thickness. ADRC indicates Alzheimer Disease Research Center; AIBL, Australian Imaging, Biomarker & Lifestyle study; CU_CU, cognitively unimpaired at time of PET, remaining cognitively unimpaired during follow-up; CU_MCI, cognitively unimpaired at time of PET, progressing to MCI during follow-up; HABS, Harvard Aging Brain Study; PREVENT-AD, Pre-symptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease study.
Figure 2. Sample Amyloid β (A) and Tau (T) Positron Emission Tomography (PET) and Magnetic Resonance Imaging (MRI) Scans From Different Biomarker Groups in the PREVENT-AD Cohort .
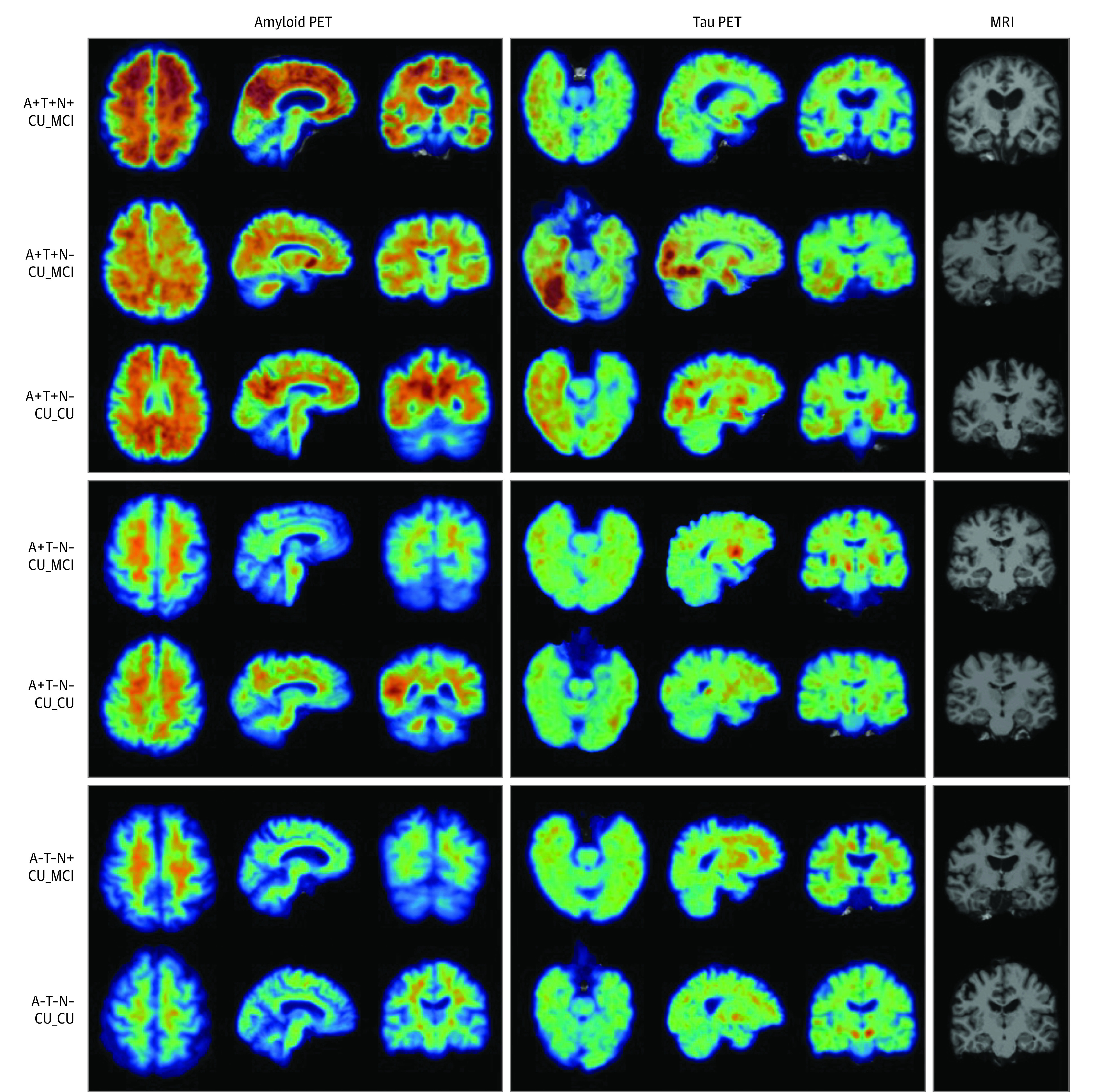
Demographic data in descending order are as follows: 73-year-old female participant; 70-year-old female participant; 65-year-old female participant; 64-year-old female participant; 66-year-old female participant; 61-year-old female participant; 69-year-old female participant. CU_CU indicates cognitively unimpaired at time of PET, remaining cognitively unimpaired during follow-up; CU_MCI, cognitively unimpaired at time of PET, progressing to mild cognitive impairment during follow-up; N, evidence of neurodegeneration; PREVENT-AD, Pre-symptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease study.
Effect of Biomarker Group on Probability of Clinical Progression Across Time
Survival curves representing progression time from cognitive unimpairment to MCI for each AT biomarker group are displayed in Figure 3. Using the meta-ROI to define T+, the A+T+ group demonstrated a greater probability of progression to MCI over time compared with the other groups (hazard ratios >4.75, model concordance values >0.68) (Figure 3 and eTable 6 in Supplement 1). Concordance (ie, model fit) was typically reduced when other regions were used to define tau positivity (eTable 6 and eFigure 4 in Supplement 1) and when models used demographic and clinical information alone without the inclusion of biomarker groups (eTable 7 in Supplement 1). Continuous measurement of neurodegeneration did not add significant prognostic value for MCI progression in the biomarker group models (eTable 8 in Supplement 1).
Figure 3. Survival Curves Reflecting Time From Positron Emission Tomography (PET) Scan to Mild Cognitive Impairment (MCI) Classification for the 4 Biomarker Groups, Across Cohorts.
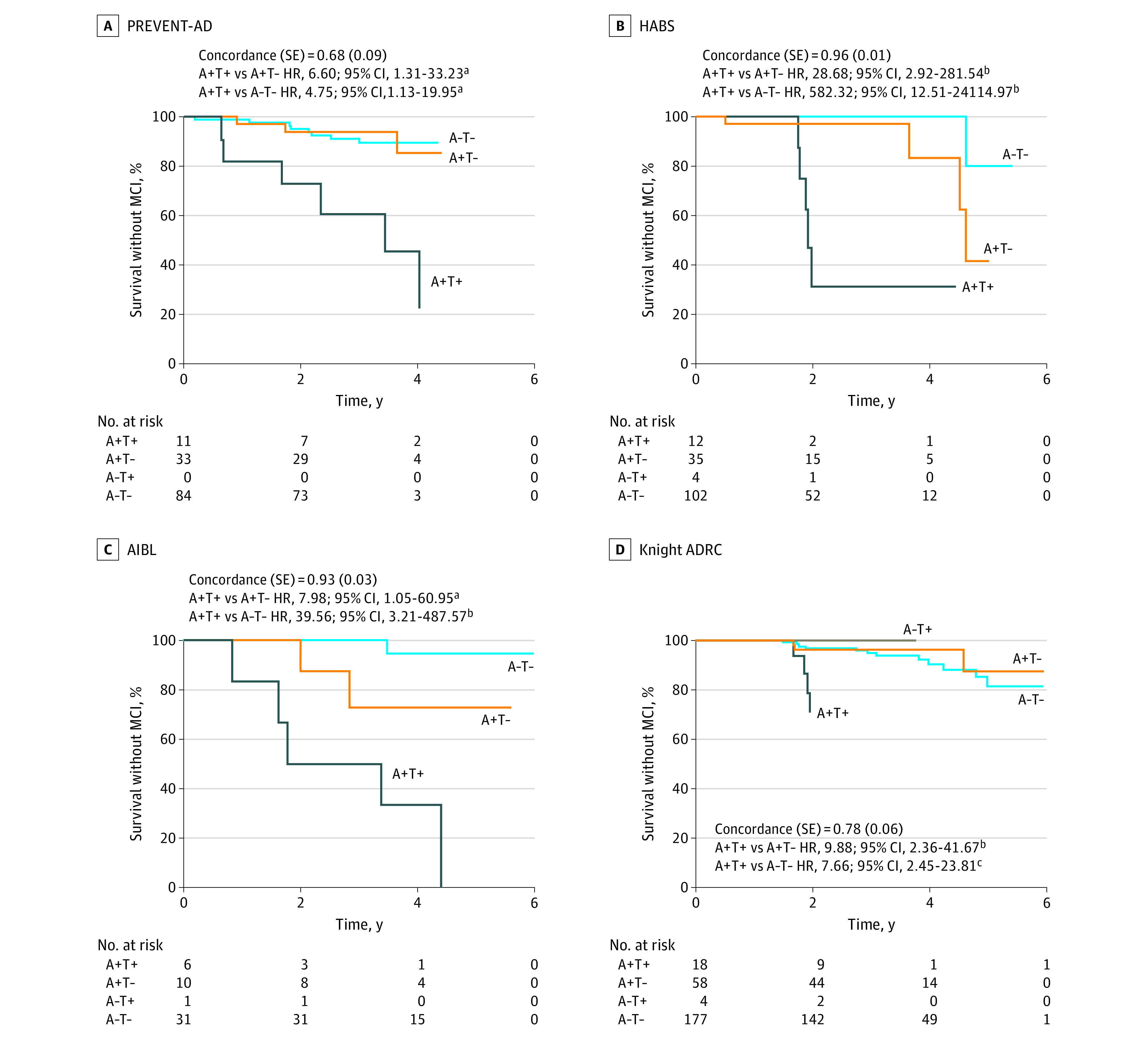
The temporal meta–region of interest was used to define tau positivity (T+). Concordance values and SE are reported for the Cox regression models, which included age at the time of tau PET, sex, education, and APOE e4 status as covariates. Hazard ratios (HRs) for the PET biomarker groups are in reference to the amyloid-positive, tau-positive (A+T+) group. Inverted HRs are reported for ease of interpretation (ie, reflecting risk of progression to MCI in the A+T+ group relative to the other groups). The A−T+ group is displayed for visualization purposes but was not included in statistical analysis because of the small sample size. ADRC indicates Alzheimer Disease Research Center; AIBL, Australian Imaging, Biomarker & Lifestyle study; HABS, Harvard Aging Brain Study; PREVENT-AD, Pre-symptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease study.
aP < .05.
bP < .01.
cP < .001.
Longitudinal Cognition Across Biomarker Groups
In all cohorts, A+T+ participants experienced greater longitudinal cognitive decline compared with the other groups (all β estimates >0.03) (Figure 4 and the eResults in Supplement 1; see eFigures 5-9 in Supplement 1 for performance stratified by MCI progression status and for specific RBANS cognitive indexes in PREVENT-AD). The strength of these associations was reduced when other regions were used to define tau abnormality in PREVENT-AD and HABS, whereas the inferior temporal lobe performed better in AIBL and Knight ADRC (eResults in Supplement 1).
Figure 4. Longitudinal Cognitive Slopes for Each Biomarker Group Across Cohorts .

Models included random slopes and intercepts for each participant and covariates of age, sex, and years of education. For all cohorts, positron emission tomography (PET) was added midstudy and was therefore performed at different cognitive follow-up visits for each participant. Longitudinal cognition analyses included time points both before and after PET scanning. The amyloid-negative, tau-positive (A−T+) group is displayed for visualization purposes but was not included in statistical analyses because of the small sample size. β estimates and SE are for the interaction between biomarker group and visit date, with A+T+ as the reference group. Cognitive scores are reported as cohort-derived z scores. ADRC indicates Alzheimer Disease Research Center; AIBL, Australian Imaging, Biomarker & Lifestyle study; HABS, Harvard Aging Brain Study; PACC, Preclinical Alzheimer Cognitive Composite; PREVENT-AD, Pre-symptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease study; RBANS, Repeatable Battery for the Assessment of Neuropsychological Status.
aP < .05.
bP < .01.
cP < .001.
Cognitive Decline Status of Nonprogressors Across Biomarker Groups
Cognitive status (declining vs stable) for nonprogressors by biomarker group is displayed in eTable 9 and eFigure 10 in Supplement 1. A greater proportion of A+T+ nonprogressors showed cognitive decline compared with the A−T− group in PREVENT-AD (4/5 participants declining [80%] vs 21/76 declining [27.63%] P = .03) and HABS (4/7 participants declining [57.14%] vs 14/101 declining [13.86%]; P = .01). No group difference reached significance in the other cohorts, but adding the decliners to the progressors in AIBL captured 100% of the A+T+ participants. Using different regions to classify tau positivity produced varying results across cohorts, with regions other than the temporal meta-ROI performing better at capturing decliners in some cases (eTable 9 and eFigure 10 in Supplement 1).
Discussion
The AT(N) biological framework for AD has been proposed for research purposes,1 but its clinical significance for individuals without cognitive impairment is unclear. We examined the implications of Aβ-positive and tau-positive PET signals for clinical progression from cognitively unimpaired to MCI over short-term intervals. Across 4 independent cohorts, 33% to 83% of individuals without cognitive impairment and with abnormal elevation of both Aβ and tau progressed to MCI within a mean (SD) of 2.00 (1.10) to 2.72 (1.49) years after PET scanning. These numbers increased across all cohorts when restricted to individuals who were N+, reaching a progression rate of 43% to 100%. Most of the remaining A+T+ participants also experienced cognitive decline, suggesting that they too are on a pathway toward AD symptoms.
Clinical trials for AD often require an abnormal amyloid biomarker for inclusion.19,20 Here, positivity on both Aβ and tau PET was associated with a 7 to 29 times greater hazard of progression from cognitively unimpaired to MCI, as compared with a positive Aβ scan in the absence of tau positivity. The A−T+ group was not considered in analyses because it represented less than 2% of all participants. These results suggest (1) that the presence of Aβ is typically needed as a precondition to tau-PET tracer binding detection and (2) that tau pathology is critical for imminent decline. Models based on A+T+ PET biomarkers outperformed models based on demographic and clinical data alone in identifying risk of progression to MCI. Therefore, combining both tau and Aβ PET greatly boosts associations with near-term clinical progression in preclinical disease stages, a finding that is highly relevant for future clinical trials. Examination of longitudinal cognitive trajectories further indicated that 25% to 100% of A+T+ participants who remained “cognitively unimpaired” nonetheless demonstrated cognitive decline.
The research framework for the biological definition of AD uses dichotomous categories to define biomarker abnormality, ie, positive or negative.1 One challenge for this framework in PET studies is choice of anatomical region from which to define tau positivity.21 While the entorhinal cortex is often the site of earliest tau deposition in AD,22 tau in this region is not necessarily specific to AD and may also occur with increasing age, independent of Aβ.23,24 Accordingly, a temporal meta-ROI, comprising both medial and neocortical temporal regions, has been proposed as an alternative to the entorhinal cortex ROI for detecting AD-specific early tau deposition.18 Here, use of a temporal meta-ROI to define T+ typically identified a larger percentage of MCI progressors in the A+T+ group and showed stronger associations with longitudinal cognitive decline when compared with entorhinal cortex and inferior temporal ROIs.
Evidence of neurodegeneration is not required for a diagnosis of biological AD but is thought instead to reflect a nonspecific marker of disease severity typical of more advanced stages. In the HABS cohort, A+T+ individuals with thinner temporal cortices had increased MCI progression rates. In the other cohorts, the progression rate was numerically but not significantly higher in the A+T+(N+) than the A+T+(N−) group when cortical thickness was used to define presence of neurodegeneration, though it is notable that 100% of the A+T+(N+) AIBL participants progressed to MCI. The percentage of MCI progression was also numerically higher in the A+T+(N+) than A+T+(N−) groups in PREVENT-AD, HABS, and Knight ADRC cohorts when neurodegeneration was defined based on the hippocampal volume. The absence of a significant difference in AIBL, PREVENT-AD, and Knight ADRC may be attributable to the very high percentage of A+T+ progressors in AIBL (83%) and low statistical power in PREVENT-AD and Knight ADRC.
Strengths and Limitations
While the magnitude of the associations varied, the results were replicated across 4 independent cohorts using related but different methods. This replicability across methodologies and samples represents a key strength of our study, as does the robustness of the reported findings in multiple sensitivity analyses. Of note, the lowest progression rate for the A+T+ group was found in the Knight ADRC, the only cohort for which MCI was not defined on a consensus committee review but based on a CDR of 0.5 or greater. We also found that cohort-specific biomarker thresholds performed better at detecting MCI progressors than harmonized cutoffs, likely due to between-sample differences such as participant age. The development of demographically adjusted thresholds will be important for future clinical applicability of our findings. Study limitations include the modest sample sizes of the A+T+ groups, though the proportion of participants assigned to this biomarker group was similar to previous studies.25,26,27 Given the majority of our participants were non-Hispanic White individuals, further studies are required to determine the applicability of our findings in more diverse and representative samples.
Conclusions
In 4 independent cohorts, we demonstrate that Aβ and tau PET positivity in individuals without cognitive impairment is associated both with near-term progression to MCI and, among those who do not show such categorical change, longitudinal cognitive decline. Additional evidence of neurodegeneration (N) implies substantial additional probability of clinical progression, reaching a 100% progression rate over an approximately 3-year follow-up in 1 of the cohorts. Crucially, abnormality in both Aβ and tau PET was associated with a considerably greater risk of near-term clinical progression than abnormality of Aβ PET alone. These findings support the clinical validity of a biological definition of AD in participants without cognitive impairment that is based on the presence of both Aβ and tau. When preventive treatments become available, the use of such a biological definition of AD to identify persons with probable preclinical AD could substantially mitigate the AD epidemic. Until then, elevations in both Aβ and tau PET indicate imminent clinical progression in most individuals without cognitive impairment.
eMethods
eResults
eTable 1. Demographic characteristics of independent subsamples used to define cognitive decliners and neurodegeneration positivity, in each cohort
eTable 2. Demographic, pathological and clinical characteristics of participants by MCI progression status and biomarker group in PREVENT-AD and HABS
eTable 3. Demographic, pathological and clinical characteristics of participants by MCI progression status and biomarker group in AIBL and Knight ADRC
eTable 4. MCI progression status within biomarker groups across cohorts, using different tau regions to define positivity
eFigure 1. Number of participants progressing to MCI after PET versus those remaining cognitively unimpaired in each AT biomarker group, across cohorts, using different regions to define tau positivity
eTable 5. MCI progression status within biomarker groups across cohorts, using harmonised Aβ and tau thresholds
eFigure 2. Number of participants progressing to MCI after PET versus those remaining cognitively unimpaired in each AT biomarker group, across cohorts, defined using harmonised A and T thresholds
eFigure 3. Number of participants progressing to MCI after PET versus those remaining cognitively unimpaired in each AT(N) biomarker group, using hippocampal volume to define N+, across cohorts
eTable 6. Cox proportional hazard models determining effect of PET-biomarker group on time to incident MCI classification
eFigure 4. Survival curves reflecting time from PET scan to MCI for the four PET biomarker groups, across cohorts, using different regions to define tau positivity
eTable 7. Cox proportional hazard models assessing the effect of demographic/clinical information on time to incident MCI classification
eTable 8. Cox proportional hazard models incorporating the effect of continuous measures of neurodegeneration on time to incident MCI classification
eFigure 5. Group mean and individual longitudinal cognitive slopes for participants who progressed to MCI after PET and those who remained cognitively unimpaired across each biomarker group across cohorts, using temporal meta-ROI to define tau positivity
eFigure 6. Group mean and individual longitudinal cognitive slopes for participants who progressed to MCI after PET and those who remained cognitively unimpaired across each biomarker group across cohorts, using entorhinal cortex to define tau positivity
eFigure 7. Group mean and individual longitudinal cognitive slopes for participants who progressed to MCI after PET and those who remained cognitively unimpaired across each biomarker group across cohorts, using inferior temporal cortex to define tau positivity
eFigure 8. Group mean and individual longitudinal cognitive slopes for participants who progressed to MCI after PET and those who remained cognitively unimpaired across each biomarker group across cohorts, using any of temporal meta-ROI, entorhinal cortex, and inferior temporal cortex to define tau positivity
eFigure 9. Group mean and individual longitudinal cognitive slopes for each of the RBANS index scores for participants who progressed to MCI after PET and those who remained cognitively unimpaired across each biomarker group in Prevent-AD, using temporal meta-ROI to define tau positivity
eTable 9. Cognitive decline status amongst nonprogressors participants within each PET-biomarker group across cohorts and tau positivity regions
eFigure 10. Percentage of cognitively unimpaired participants, who did not progress to MCI, but who showed longitudinal cognitive decline versus those who remained cognitively stable in each AT biomarker group, using different regions to define tau positivity, across cohorts.
eReferences
Nonauthor collaborators
References
- 1.Jack CR Jr, Bennett DA, Blennow K, et al. ; Contributors . NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535-562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dubois B, Villain N, Frisoni GB, et al. Clinical diagnosis of Alzheimer’s disease: recommendations of the International Working Group. Lancet Neurol. 2021;20(6):484-496. doi: 10.1016/S1474-4422(21)00066-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lowe VJ, Bruinsma TJ, Min HK, et al. Elevated medial temporal lobe and pervasive brain tau-PET signal in normal participants. Alzheimers Dement (Amst). 2018;10:210-216. doi: 10.1016/j.dadm.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Driscoll I, Troncoso J. Asymptomatic Alzheimer’s disease: a prodrome or a state of resilience? Curr Alzheimer Res. 2011;8(4):330-335. doi: 10.2174/156720511795745348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198. doi: 10.1016/0022-3956(75)90026-6 [DOI] [PubMed] [Google Scholar]
- 6.Duff K, Humphreys Clark JD, O’Bryant SE, Mold JW, Schiffer RB, Sutker PB. Utility of the RBANS in detecting cognitive impairment associated with Alzheimer’s disease: sensitivity, specificity, and positive and negative predictive powers. Arch Clin Neuropsychol. 2008;23(5):603-612. doi: 10.1016/j.acn.2008.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papp KV, Rentz DM, Orlovsky I, Sperling RA, Mormino EC. Optimizing the preclinical Alzheimer’s cognitive composite with semantic processing: the PACC5. Alzheimers Dement (N Y). 2017;3(4):668-677. doi: 10.1016/j.trci.2017.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donohue MC, Sperling RA, Salmon DP, et al. ; Australian Imaging, Biomarkers, and Lifestyle Flagship Study of Ageing; Alzheimer’s Disease Neuroimaging Initiative; Alzheimer’s Disease Cooperative Study . The preclinical Alzheimer cognitive composite: measuring amyloid-related decline. JAMA Neurol. 2014;71(8):961-970. doi: 10.1001/jamaneurol.2014.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412-2414. doi: 10.1212/wnl.43.11.2412-a [DOI] [PubMed] [Google Scholar]
- 10.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270-279. doi: 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson KA, Gregas M, Becker JA, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62(3):229-234. doi: 10.1002/ana.21164 [DOI] [PubMed] [Google Scholar]
- 12.Gordon BA, Friedrichsen K, Brier M, et al. The relationship between cerebrospinal fluid markers of Alzheimer pathology and positron emission tomography tau imaging. Brain. 2016;139(pt 8):2249-2260. doi: 10.1093/brain/aww139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McSweeney M, Pichet Binette A, Meyer P-F, et al. ; PREVENT-AD Research Group . Intermediate flortaucipir uptake is associated with Aβ-PET and CSF tau in asymptomatic adults. Neurology. 2020;94(11):e1190-e1200. doi: 10.1212/WNL.0000000000008905 [DOI] [PubMed] [Google Scholar]
- 14.Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79(1):110-119. doi: 10.1002/ana.24546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fowler C, Rainey-Smith SR, Bird S, et al. ; the AIBL investigators . Fifteen years of the Australian Imaging, Biomarkers and Lifestyle (AIBL) study: progress and observations from 2,359 older adults spanning the spectrum from cognitive normality to Alzheimer’s disease. J Alzheimers Dis Rep. 2021;5(1):443-468. doi: 10.3233/ADR-210005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968-980. doi: 10.1016/j.neuroimage.2006.01.021 [DOI] [PubMed] [Google Scholar]
- 17.Baker SL, Maass A, Jagust WJ. Considerations and code for partial volume correcting [18F]-AV-1451 tau PET data. Data Brief. 2017;15:648-657. doi: 10.1016/j.dib.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jack CR Jr, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement. 2017;13(3):205-216. doi: 10.1016/j.jalz.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6(228):228fs13. doi: 10.1126/scitranslmed.3007941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537(7618):50-56. doi: 10.1038/nature19323 [DOI] [PubMed] [Google Scholar]
- 21.Villemagne VL, Lopresti BJ, Doré V, et al. What is T+? a Gordian knot of tracers, thresholds, and topographies. J Nucl Med. 2021;62(5):614-619. doi: 10.2967/jnumed.120.245423 [DOI] [PubMed] [Google Scholar]
- 22.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-259. doi: 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- 23.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128(6):755-766. doi: 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoon B, Guo T, Provost K, et al. ; Alzheimer’s Disease Neuroimaging Initiative . Abnormal tau in amyloid PET negative individuals. Neurobiol Aging. 2022;109:125-134. doi: 10.1016/j.neurobiolaging.2021.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Therriault J, Pascoal TA, Benedet AL, et al. Frequency of biologically defined Alzheimer disease in relation to age, sex, APOE ε4, and cognitive impairment. Neurology. 2021;96(7):e975-e985. doi: 10.1212/WNL.0000000000012586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Betthauser TJ, Koscik RL, Jonaitis EM, et al. Amyloid and tau imaging biomarkers explain cognitive decline from late middle-age. Brain. 2020;143(1):320-335. doi: 10.1093/brain/awz378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jack CR Jr, Therneau TM, Weigand SD, et al. Prevalence of biologically vs clinically defined Alzheimer spectrum entities using the National Institute on Aging-Alzheimer’s Association research framework. JAMA Neurol. 2019;76(10):1174-1183. doi: 10.1001/jamaneurol.2019.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods
eResults
eTable 1. Demographic characteristics of independent subsamples used to define cognitive decliners and neurodegeneration positivity, in each cohort
eTable 2. Demographic, pathological and clinical characteristics of participants by MCI progression status and biomarker group in PREVENT-AD and HABS
eTable 3. Demographic, pathological and clinical characteristics of participants by MCI progression status and biomarker group in AIBL and Knight ADRC
eTable 4. MCI progression status within biomarker groups across cohorts, using different tau regions to define positivity
eFigure 1. Number of participants progressing to MCI after PET versus those remaining cognitively unimpaired in each AT biomarker group, across cohorts, using different regions to define tau positivity
eTable 5. MCI progression status within biomarker groups across cohorts, using harmonised Aβ and tau thresholds
eFigure 2. Number of participants progressing to MCI after PET versus those remaining cognitively unimpaired in each AT biomarker group, across cohorts, defined using harmonised A and T thresholds
eFigure 3. Number of participants progressing to MCI after PET versus those remaining cognitively unimpaired in each AT(N) biomarker group, using hippocampal volume to define N+, across cohorts
eTable 6. Cox proportional hazard models determining effect of PET-biomarker group on time to incident MCI classification
eFigure 4. Survival curves reflecting time from PET scan to MCI for the four PET biomarker groups, across cohorts, using different regions to define tau positivity
eTable 7. Cox proportional hazard models assessing the effect of demographic/clinical information on time to incident MCI classification
eTable 8. Cox proportional hazard models incorporating the effect of continuous measures of neurodegeneration on time to incident MCI classification
eFigure 5. Group mean and individual longitudinal cognitive slopes for participants who progressed to MCI after PET and those who remained cognitively unimpaired across each biomarker group across cohorts, using temporal meta-ROI to define tau positivity
eFigure 6. Group mean and individual longitudinal cognitive slopes for participants who progressed to MCI after PET and those who remained cognitively unimpaired across each biomarker group across cohorts, using entorhinal cortex to define tau positivity
eFigure 7. Group mean and individual longitudinal cognitive slopes for participants who progressed to MCI after PET and those who remained cognitively unimpaired across each biomarker group across cohorts, using inferior temporal cortex to define tau positivity
eFigure 8. Group mean and individual longitudinal cognitive slopes for participants who progressed to MCI after PET and those who remained cognitively unimpaired across each biomarker group across cohorts, using any of temporal meta-ROI, entorhinal cortex, and inferior temporal cortex to define tau positivity
eFigure 9. Group mean and individual longitudinal cognitive slopes for each of the RBANS index scores for participants who progressed to MCI after PET and those who remained cognitively unimpaired across each biomarker group in Prevent-AD, using temporal meta-ROI to define tau positivity
eTable 9. Cognitive decline status amongst nonprogressors participants within each PET-biomarker group across cohorts and tau positivity regions
eFigure 10. Percentage of cognitively unimpaired participants, who did not progress to MCI, but who showed longitudinal cognitive decline versus those who remained cognitively stable in each AT biomarker group, using different regions to define tau positivity, across cohorts.
eReferences
Nonauthor collaborators