

Abstract
The molecular mechanism by which a mechanical stimulus is translated into a chemical response in biological systems is still unclear. We show that mechanical stretching of single cytoplasmic proteins can activate binding of other molecules. We used magnetic tweezers, total internal reflection fluorescence, and atomic force microscopy to investigate the effect of force on the interaction between talin, a protein that links liganded membrane integrins to the cytoskeleton, and vinculin, a focal adhesion protein that is activated by talin binding, leading to reorganization of the cytoskeleton. Application of physiologically relevant forces caused stretching of single talin rods that exposed cryptic binding sites for vinculin. Thus in the talin-vinculin system, molecular mechanotransduction can occur by protein binding after exposure of buried binding sites in the talin-vinculin system. Such protein stretching may be a more general mechanism for force transduction.
Force sensing by cells is critical for their proper function; however, how cells transform a force stimulus into a chemical response remains unclear. Forces have been shown to open ion channels, activate phosphorylation, and cause catch bond formation. These could all be mechanisms for force transduction [reviewed in (1)]. Another mechanism that has been postulated but never demonstrated experimentally is that stretching of single force-bearing cytoplasmic molecules might expose binding sites for other proteins. We characterized at a single-molecule level the interaction of two cytoskeletal proteins, talin, the adhesion force–bearing protein, and vinculin, its binding partner, in the absence and presence of a physiologically relevant force.
Talin and vinculin are key players in cell signaling, adhesion, and migration (2) and are localized at the sites of cell-matrix adhesion (3). They are ideal candidates for mechanotransduction because the binding of vinculin to focal adhesion complexes was shown to increase upon the application of force (4). Talin couples the cytoskeleton to the extracellular matrix through membrane integrins and can activate signaling through vinculin binding, for assembly and reorganization of the actin cytoskeleton (3, 5). The globular head of talin contains a FERM (4.1/ezrin/radixin/moesin) domain that binds and activates β-integrin cytodomains (6), whereas the talin rod (TR) contains up to 11 vinculin binding sites (VBSs) (7). A portion of the TR spanning residues 482 to 889 (Fig. 1A) contains a five-helix bundle with a cryptic VBS in helix four, followed by a seven-helix bundle containing four additional VBSs (helices 6, 9, 11, and 12). The x-ray structures of VBSs reveal that they are defined by six turns of an α helix and that they are often buried because of extensive hydrophobic interactions with other amphipathic helices (8, 9). VBSs trigger conformational changes in the vinculin head (Vh), in a process of helical bundle conversion: α helices of Vh reorganize to surround the VBS from the TR, burying 50% of its solvent accessible area (5, 10). Figure 1, B and C, shows the proposed sequential unfolding of the helical bundles in the TR and Vh binding to the helix 12, which is exposed in steered molecular dynamic simulations (11).
Fig. 1.
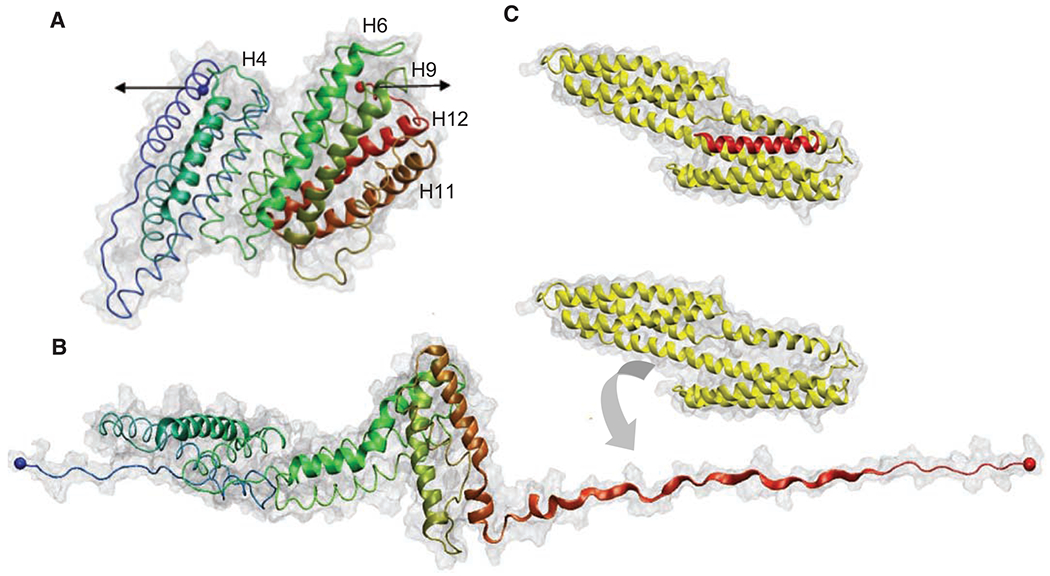
(A) Structure of the 12 helices that form the TR 482 to 889 [Protein Data Bank (PDB) 1xwx]. Color coding: blue, N terminus; green, middle; and red, C terminus. VBS helices are numbered and represented with cartoon models, and the rest of the protein is shown with tube models. (B) Under the application of a force in the direction indicated by the black arrows in (A), TR starts to unfold. Once helix 12 exposed its VBS, Vh (PDB 1u6h), represented in yellow, reorganizes to bind to it. (C) X-ray structure of the complex Vh-VBS-helix 12, PDB 1u6h. Images were generated by using the VMD (Visual Molecular Dynamics) program (www.ks.uiuc.edu/research/vmd/).
Previous studies have shown that four of five VBSs in TR residues 482 to 889 are inactive for binding in the native molecule (8, 12). What activates the VBSs in TR is still unknown. The facts that mechanical force increases vinculin recruitment to focal adhesions (4) and that talin can bind to the actin cytoskeleton suggest that force induced by actomyosin contraction could stretch the TR, exposing the cryptic VBSs to Vh (2). The mechanical stretching of cytoskeletally attached proteins by applied force is documented for the case in which stretch activates tyrosine phosphorylation by Src family kinases (13). Moreover, it has been shown that stretching of DNA leads to changes in the binding of DNA-interacting proteins (14). Molecular dynamics simulations have supported the ability of force-induced conformational changes to expose the cryptic residues of VBSs in the TR (15). In addition, another recent steered molecular dynamic study provided a model in which mechanical force gradually breaks the 12 helices of the TR into three smaller sub-bundles (11).
To experimentally test the hypothesis that force-induced stretching of the TR can open the helical bundles and enable Vh binding, we used a combination of magnetic tweezers to enable stretching of single TR molecules and total internal reflection fluorescence (TIRF) microscopy to observe fluorescent Vh binding (Fig. 2).
Fig. 2.
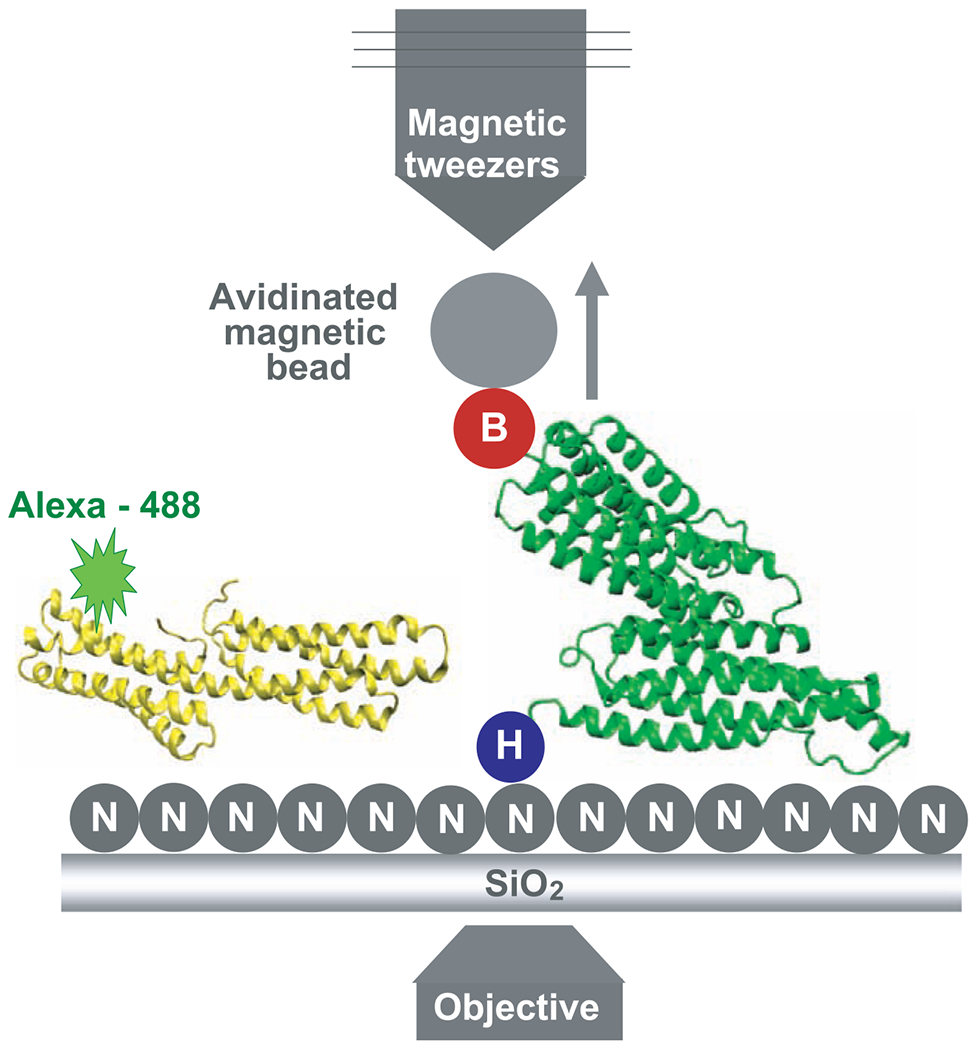
Representation of the device used to measure the binding events. The Ni-NTA (labeled N) grafted slides containing the TR fixed through its 6×His N terminus (labeled H) to the glass and with the avidinated magnetic bead bound to its biotinylated C terminus (labeled B) was placed over the objective. Alexa 488–Vh was added to the slides for the period of the incubation. The TR and Vh structures are represented in green and yellow, respectively. The arrow shows the direction of the movement of the beads when they are pulled using the magnetic tweezers.
Three DNA constructs encoding monomeric TR, dimeric tandem TR, and full-length α-actinin were designed with His and Avi tags at their N- and C-termini, respectively. The dimeric tandem TR served as a positive control because it should have twice the number of binding sites that are in a single TR. Because α-actinin has an active binding site for Vh (10), it served as a negative control; we would not expect to see any difference in the number of Vh molecules bound to α-actinin with and without force-induced stretching. All expressed proteins had 6×His and a biotin at their N- and C-termini, respectively (fig. S1). The N-terminal was used to attach the protein to a Ni–nitrilotriacetic acid (NTA) glass surface, whereas avidinated magnetic beads were coupled to the biotinylated C-terminus. The molecule attached to the glass was stretched by applying a magnetic force to the beads in a vertical direction with the magnetic tweezers apparatus described previously (16).
The criterion for deciding that beads were bound to a single talin molecule was to observe a random motion of the bead around a single point close to the surface. The Vh was labeled with the fluorophore Alexa 488 at a molar ratio of 1:1 and further incubated with the TR. This incubation was performed in presence or absence of force depending on whether or not stretching of TR was pursued. Then, all unbound Alexa 488–Vh was washed, and TIRF intensity was measured over time. The number of Alexa 488–Vh molecules bound to the TR was determined by single-molecule fluorescence photobleaching events. The fluorescence intensity abruptly decreased in a standard step if a single fluorophore was bleached. The same protocol was used to measure Vh binding to TR tandem fragment and α-actinin controls.
With monomeric TR in the absence of force, we observed (Fig. 3A) one or no photobleaching events of the Alexa 488–Vh fluorescence for each bead attached to a TR. After the application of 12 pN of force to the beads, as many as three photobleaching events were observed, each the size of a single bleaching event. The histogram next to each graph represents the distribution of photobleaching events for all the analyzed beads. Although fewer than half of the diffusing beads showed binding and individual bleaching events, the percentage of vinculin bound to beads was greater upon application of force. Because each Alexa 488–Vh was labeled with one fluorophore molecule, each photobleaching event was associated with the binding of one Vh molecule to the TR. We suggest that force applied to the beads caused mechanical unfolding of TR, exposing up to two additional VBSs. When 2 pN of force was applied to the beads, the percentage of vinculin bound to beads was larger than in the case of no force but smaller than when 12 pN of force was applied, which supports the hypothesis that the number of vinculin molecules bound to the TR depends on the stretching force applied to the TR.
Fig. 3.
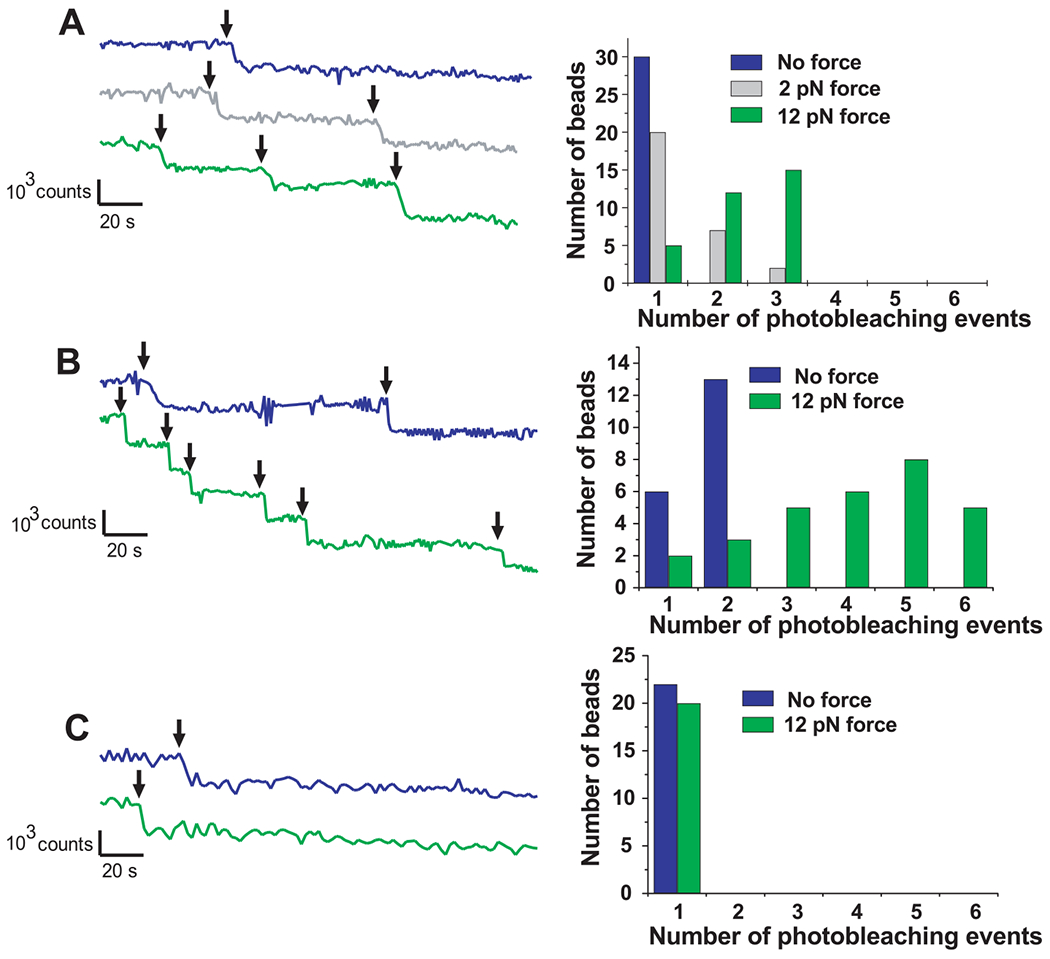
Diagram of photobleaching events of Alexa 488–Vh bound to (A) TR, (B) dimeric tandem TR, and (C) α-actinin. Histograms show the number of beads per photobleaching event. In all cases, blue, gray, and green colors correspond with no force, 2-pN force, and 12-pN force applied, respectively. The TR, talin dimeric tandem (positive control), and α-actinin (negative control) showed maximally 1 and 3, 2 and 6, and 1 and 1 photobleaching events (black arrows) when no force and 12 pN force, respectively, was applied.
The analysis of the photobleaching events for the experiments performed using the tandem TR as the positive control (Fig. 3B) revealed up to two Vh binding events to the tandem TR in the absence of force. When 12 pN of force was applied to the beads, up to six binding events were observed. In all experiments performed using α-actinin as the negative control (Fig. 3C), one or no binding event was observed with and without the application of 12 pN of force.
In order to measure the pattern of force-dependent stretching of TR, we designed a DNA construct encoding for I272-TR-I272 [TR flanked by two molecules of the I27 (27th immunoglobular domain of human cardiac titin), which has well-characterized mechanical properties (17) (Fig. 4A)]. The protein I272-TR-I272 was manipulated by using single-molecule force extension spectroscopy (18). Figure 4B shows a characteristic force extension curve corresponding to the unfolding of I272-TR-I272. In this trace, the four equally spaced peaks at ~200 pN with contour length increments of ~28.5 nm correspond to the unfolding of the four flanking I27 handles (17). The largest peak at the right of Fig. 4B represents the detachment of the cantilever from the polyprotein. Before the four force peaks for the I27 domains, there are several smaller peaks over a distance of about 160 nm, which corresponds closely to the expected length of the fully stretched 407-residue TR domain. Distances between the unfolding peaks were determined by using the contour length measured by fits of the wormlike chain model (green lines in Fig. 4B). [The contour length of a fully stretched protein is about 0.4 nm per amino acid (19).] A histogram of positions for each of the five TR force peaks (Fig. 4C, n = 120 traces) with the resting length of the protein as reference (163 nm minus the measured distance to the first I27 module) showed that the unfolding pattern was very regular. Five unfolding peaks with average forces of 29 ± 19, 31 ± 19, 36 ± 22, 42 ± 22, and 51 ± 26 pN were identified at 24 ± 7, 81 ± 9, 107 ± 3, 129 ± 6, and 152 ± 5 nm, respectively. Histograms showing the force distribution for each peak can be found in fig. S2.
Fig. 4.
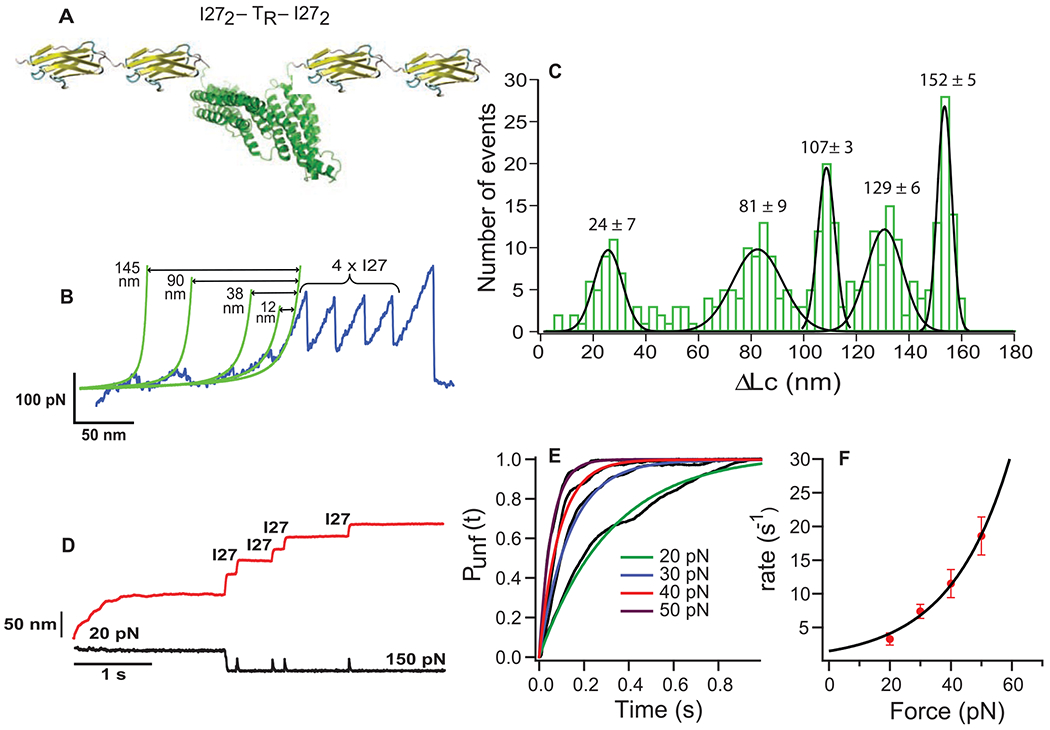
AFM experiments. (A) Diagram of the polyprotein designed for AFM, I272–TR–I272. (B) Force extension trace for I272-TR-I272. (C) Histogram of the contour length increments (ΔLc) for the unfolding peaks of the TR (120 traces). (D) Force-clamp trace obtained by stretching I272-TR-I272 at 20 and 150 pN of force. The red and black lines represent extension and force, respectively. (E) Probability of unfolding versus time for TR. Average recordings of TR unfolding at 20 pN (15 traces), 30 pN (14 traces), 40 pN (17 traces), and 50 pN (22 traces). A single exponential (colored smooth lines) is fitted to each average trace (in black). (F) The rate constant of unfolding as a function of force. Data are represented as mean ± SD; error bars were calculated by using the bootstrapping method (26).
The force extension experiment provides a mechanical description for TR, that is, unfolding forces and contour lengths. To study the unfolding rate of TR, we used force-clamp spectroscopy, a technique that allows us to apply a constant calibrated force to a single TR for determining the unfolding trajectory as a function of time (20). A double pulse protocol was used in order to separate the unfolding of TR from those of I27 modules. The first force pulse was set up at forces ranging from 20 to 50 pN to allow for the unfolding of TR. A second pulse of force of 150 pN was applied to capture the unfolding of the I27 modules, represented with the four identical steps of 24 nm (Fig. 4D). Only those recordings showing at least three I27 unfolding events were analyzed. The portion of the traces containing the TR unfolding was extracted, averaged, and fitted to a single exponential with a time constant, τ, which provides the unfolding rate, r = 1/τ (Fig. 4E). Considering the number of traces collected per force (~20 traces), the exponential fitting provides a good approximation for the unfolding rate that adequately describes the process over the entire range of forces. The unfolding rate versus the applied force is presented in Fig. 4F. The extrapolation at 12 pN, the unfolding force value used in the binding experiments, yields a constant rate of 2.7 s−1.
Taking together the results of the atomic force spectroscopy and the binding experiments of Vh to the TR, it is possible to make some conclusions. First, the fact that only one binding event was observed in the absence of force-induced stretching indicates that, in its intact state, TR contains one active VBS, as was predicted in previous studies using several biophysical techniques (9, 12). This active VBS could be located in the helix 6, 9, 11, or 12, most likely helix 9, where the trypsin cleavage site is found (12) and which is likely the initial vinculin interaction site in cells. Second, stretching the TR with 12 pN of force can increase the Vh binding events from one to three. Thus, force-induced stretching of the TR caused conformational changes in its helical bundle, exposing two of the four remaining buried VBSs, which become available to bind Vh.
Previous steered molecular dynamic calculations potentially provide insights into the number of VBSs activated by force-induced stretching (11). Because the mechanical stabilities of the domains that form the TR are different (8, 12), force applied across the H1-H12 fragment will sequentially break the rod into smaller helix bundles. In the calculations, force first splits the fragments H1-H8 from H9-H12, and then H1-H8 breaks into the sub-bundles H1-H5 and H6-H8. The formation of these two intermediate states is in agreement with the unfolding pattern that we obtained by using force extension spectroscopy, because the subsequent unfolding of the three domains would give five mechanical transition states as observed. The predicted fragmentation pattern involves the sequential exposure of VBSs in TR, and this could explain why we observed up to three instead of five Vh binding events when force is applied. Although the 12-pN force used in the binding experiments is lower than the average unfolding forces in the atomic force microscopy (AFM) experiments, it is applied for a much longer time and could be as effective. More experimentation is needed to define the partial unfolding of the TR to intermediate states, in which the cryptic VBSs become exposed but the secondary structure of helices enables recognition by Vh. Similarly, lower unfolding forces have been used in AFM force-clamp unfolding of polyubiquitins (21) as compared with AFM force extension experiments (22). Furthermore, it is known that at constant low force (i.e., 12 pN), the unfolding and refolding processes may compete with each other, leading to situations such as that reported by Cecconi et al. (23) in which only up to three VBSs are available. Higher forces might stretch the VBS helices and reduce the binding probability; however, a new experimental setup is needed to generate the higher forces.
These findings establish that force-induced stretching can expose previously buried binding sites for vinculin in the TR. The consensus sequence for the 11 VBSs in the TR indicates that they have extensive hydrophobic interactions with adjacent amphipathic helices (7). If this scenario is correct, the proposed mechanotransduction mechanism for the VBSs in TR may be a general pathway used by the talin molecule to amplify biochemical signals upon activation by force. Indeed, talin head interacts with membrane integrins while its tail binds actin filaments, so that force transmitted to the matrix from the actin cytoskeleton would logically be borne by talin. Actomyosin contraction could develop force-dependent talin unfolding that may cause vinculin binding and activation, leading to several intracellular responses. Although the force per integrin is 1 to 2 pN on average, the bond between integrin and fibronectin is able to withstand a force of about 20 pN before breaking (24). Thus, forces used in this study are in a physiological range.
Talin stretching may provide a prototype for a wide range of signaling events in cells because there are many multidomain proteins that link cytoskeleton with membrane sites (25). Learning the mechanism by which stretching forces activate binding may broaden our understanding of the fundamental workings of mechano-sensory systems in cells.
Supplementary Material
Acknowledgments
We thank D. Critchley and N. Bate for Vh and TR monomer constructs, C. Badilla-Fernandez for advisement with constructs, and V. Hytonen for contribution to Fig. 1. This work was supported by NIH grants to J.M.F. and M.P.S., an NIH award to A.d.R., a grant from the Spanish Ministry of Science and Education to R.P.-J., and a Marie Curie international fellowship to P.R.-C.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/323/5914/638/DC1
Materials and Methods
References
References and Notes
- 1.Vogel V, Sheetz M, Nat. Rev. Mol. Cell Biol 7, 265 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Critchley DR, Biochem. Soc. Trans 33, 1308 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Critchley DR, Curr. Opin. Cell Biol 12, 133 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Galbraith CG, Yamada KM, Sheetz MP, J. Cell Biol 159, 695 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Izard T, Vonrhein C, J. Biol. Chem 279, 27667 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Calderwood DA, Tai V, Di Paolo G, De Camilli P, Ginsberg MH, J. Biol. Chem 279, 28889 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Gingras AR et al. , J. Biol. Chem 280, 37217 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Papagrigoriou E et al. , EMBO J. 23, 2942 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fillingham I et al. , Structure 13, 65 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Izard T et al. , Nature 427, 171 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Hytonen VP, Vogel V, PLOS Comput. Biol 4, e24 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel B et al. , J. Biol. Chem 281, 7458 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Sawada Y et al. , Cell 127, 1015 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harada Y et al. , Biophys. J 76, 709 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SE, Kamm RD, Mofrad MR, J. Biomech 40, 2096 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Tanase M, Biais N, Sheetz M, Methods Cell Biol. 83, 473 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Carrion-Vazquez M et al. , Proc. Natl. Acad. Sci. U.S.A 96, 3694 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oberhauser AF, Marszalek PE, Erickson HP, Fernandez JM, Nature 393, 181 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Ainavarapu SR et al. , Biophys. J 92, 225 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez JM, Li H, Science 303, 1674 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Schlierf M, Li HB, Fernandez JM, Proc. Natl. Acad. Sci. U.S.A 101, 7299 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrion-Vazquez M et al. , Nat. Struct. Biol 10, 738 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Cecconi C, Shank EA, Bustamante C, Marqusee S, Science 309, 2057 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Jiang G, Giannone G, Critchley DR, Fukumoto E, Sheetz MP, Nature 424, 334 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Calderwood DA, Ginsberg MH, Nat. Cell Biol 5, 694 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Materials and methods are available as supporting material on Science Online.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.