

Abstract
Mutations in FMR1 are the most common heritable cause of autism spectrum disorder. FMR1 encodes an RNA-binding protein, FMRP, which binds to long, autism-relevant transcripts and is essential for normal neuronal and ovarian development. In contrast to the prevailing model that FMRP acts to block translation elongation, we previously found that FMRP activates the translation initiation of large proteins in Drosophila oocytes. We now provide evidence that FMRP-dependent translation is conserved and occurs in the mammalian brain. Our comparisons of the mammalian cortex and Drosophila oocyte ribosome profiling data show that translation of FMRP-bound mRNAs decreases to a similar magnitude in FMRP-deficient tissues from both species. The steady-state levels of several FMRP targets were reduced in the Fmr1 KO mouse cortex, including a ∼50% reduction of Auts2, a gene implicated in an autosomal dominant autism spectrum disorder. To distinguish between effects on elongation and initiation, we used a novel metric to detect the rate-limiting ribosome stalling. We found no evidence that FMRP target protein production is governed by translation elongation rates. FMRP translational activation of large proteins may be critical for normal human development, as more than 20 FMRP targets including Auts2 are dosage sensitive and are associated with neurodevelopmental disorders caused by haploinsufficiency.
Keywords: Fmr1, FMRP, autism, RNP, translation
Mutations in FMR1, which encodes RNA binding protein FMRP, lead to fragile X syndrome: the most common cause of autism spectrum disorder. Flanagan, Baradaran-Hervari et al. previously found that FMRP promotes translation initiation of large proteins in Drosophila oocytes—in contrast to its previously ascribed function as a repressor of translation elongation. By comparing ribosome profiling data and measuring steady state levels of FMRP targets, they now show that FMRP-dependent large protein translation is highly conserved in the mammalian cortex.
Introduction
Mutations in the FMR1 gene, encoding the RNA-binding protein FMRP, lead to fragile X syndrome (FXS) and fragile X primary ovarian insufficiency, causes of intellectual disability (ID), autism spectrum disorder (ASD), and premature ovarian failure (Sullivan et al. 2011; Hagerman et al. 2017). FMRP binds to hundreds of individual mRNAs, which are longer than average and which are nonrandomly enriched for genes associated with ID and ASD (Darnell et al. 2011; Ascano et al. 2012). FMRP loss leads to a wide array of phenotypes in Fmr1 knockout animals, which extend beyond changes in translation. These include altered RNA transport (Kao et al. 2010; Goering et al. 2020), RNA nuclear export(Hsu et al. 2019), RNA stability (Shu et al. 2020), metabolic signaling (Sharma et al. 2010), ion channel function (Brager et al. 2012), stem cell proliferation (Callan et al. 2010), dendritic spine maturation (Grossman et al. 2006), homeostatic plasticity (Zhang et al. 2018; Bülow et al. 2019), and mGluR5-dependent long-term depression (Osterweil et al. 2010; Hays et al. 2011; Prilutsky et al. 2015). Treatments targeting the primary defects associated with FMRP loss have the potential to alleviate secondary effects as well. However, it has been difficult to determine which phenotypes are a primary result of Fmr1 loss and are a secondary result of the dysregulation of 1 or more of hundreds of potential FMRP target genes. FMRP’s precise molecular function and key targets have remained controversial (Aryal and Klann 2018).
FMRP associates with polysomes, suggesting a role for FMRP in translational regulation. In vitro measurements of translation of neuronal mRNAs performed in reticulocyte extracts showed widespread changes in translation elongation rates in the absence of FMRP (Darnell et al. 2011); however, experiments determining translation elongation rates are performed in the presence of translation initiation inhibitors, confounding their interpretation. Measurements of Drosophila oocyte protein production with ribosome profiling revealed that FMRP preferentially affects large proteins and that it promotes rather than represses translation by promoting the translation initiation (Greenblatt and Spradling 2018). These divergent effects of FMRP in mice and Drosophila could be due to species and/or tissue differences. However, FMRP is highly conserved in structure and function between Drosophila, mouse, and many other species, and the human FMR1 gene rescues neuronal defects in Drosophila Fmr1 mutants (Coffee et al. 2010). Moreover, long protein-coding capacity is emerging as a common property shared by most mRNA targets regulated by FMRP in multiple systems (Greenblatt and Spradling 2018; Das Sharma et al. 2019; Sears et al. 2019; Aryal et al. 2021).
By reanalyzing ribosome profiling data using a common pipeline from experiments conducted with FMRP-deficient Drosophila oocytes (Greenblatt and Spradling 2018) or mouse cortex (Das Sharma et al. 2019), we find that ribosome footprints of FMRP targets encoding large proteins are reduced by comparable magnitudes, suggesting a conserved role of FMRP in translational activation. Consistent with an activator function, large FMRP target proteins examined using capillary electrophoresis immunoassays showed reduced steady-state levels in the cortex of Fmr1 KO mice. These targets include Auts2, which is decreased by ∼50% and which is associated with a human autism disorder caused by Auts2 haploinsufficiency.
The decrease in translation initiation rates of FMRP targets contrasts with previous models suggesting that FMRP acts as a repressor of translation elongation. To determine whether translation of FMRP targets is limited by translation initiation or elongation rates, we developed a bioinformatic method to identify changes in ribosome distribution that would be consistent with the alleviation of rate-limiting stalling in FMRP-deficient tissues. In contrast to the elongation stalling model and consistent with the translation initiation of FMRP targets being rate limiting, we find that the distribution of ribosomes along FMRP target transcripts is nearly identical in control and FMRP-deficient tissues.
Finally, we find that ASD-relevant genes generally encode proteins that are larger than average and that many FMRP ASD target genes are haploinsufficient but not triplosensitive. Our findings are consistent with an ancient and conserved role for FMRP to promote the production of large, dosage-sensitive proteins essential for normal neuronal development. Our data support a model in which FMRP's primary function is to increase the rate of translation initiation of its targets.
Methods
Read alignments and quantification
Raw sequencing data (Woolstenhulme et al. 2015; Fradejas-Villar et al. 2017; Greenblatt and Spradling 2018; Das Sharma et al. 2019; Philippe et al. 2020) from fly, mouse, Escherichiacoli, and human samples were aligned to the Flybase Consortium/Berkeley Drosophila Genome Project (BDGP)/Celera Genomics Drosophila release dm 6.28, Genome Reference Consortium mouse build 38 mm10, University of Wisconsin E.coli strain K-12 sub-strain MG1655 build ASM584v2, and Genome Reference Consortium Human build 38 assemblies, respectively. Alignments were performed with STAR (Dobin et al. 2013). The aligned data from both the mRNA sequencing and ribosome footprinting experiments were imported into Python and quantified using the Plastid (Dunn and Weissman 2016) software package.
Length and expression analyses
SFARI Class I and Class II genes were obtained from the SFARI Gene database (Abrahams et al. 2013). For each gene, the highest expressed transcript isoform was used as the representative form. Transcripts with a transcript per million (TPM) value of <2 were excluded from analyses. Intron, untranslated region (UTR), and coding sequence (CDS) lengths for transcript isoforms were computed from BDGP and refGene gene models for Drosophila and mouse data, respectively. For translation efficiency analyses, fold-change and Padj values were computed using RiboDiff software (Zhong et al. 2017). Stress-granule enrichment data were obtained from Khong et al. (2017).
Ribosome stalling analysis
Ribosome footprints were mapped to their P-sites with 3′ offsets that varied based on the length of the individual ribosome footprints (Supplementary Table 5). These variable 3′ offsets were determined using the psite function from the riboWaltz R package (Lauria et al. 2018). Ribosome footprinting read densities were determined using the Plastid software package. These densities were then smoothed using LOWESS regression and normalized by the total number of reads per transcript. Transcripts were filtered so that only transcripts with a length exceeding 100 codons and an average read density above 0.5 reads/codon were kept. The Kolmogorov–Smirnov (K–S) statistic for all transcripts was calculated as the maximum absolute difference between the control and mutant normalized read densities. All transcripts were grouped into high (>0.3), medium (>0.3 and <0.15), and low (<0.15) K–S statistic groups. The fold enrichment of targets in each group was determined as the fraction of the total targets in that group over the fraction of the total nontargets in that group. The significance of target enrichment was determined using Fisher’s exact test.
Simulations with the inhomogeneous ℓ-Totally Asymmetric Simple Exclusion Process model
Plausible elongation rates for each codon position along simulated transcripts were computed from a gamma distribution to mimic the observed distributions of elongation rates. Elongation rates of control transcripts were greatly reduced at random to simulate a mutation inducing pauses in translation. These elongation rates were inputted into an implementation of the inhomogeneous ℓ-Totally Asymmetric Simple Exclusion Process (TASEP) (Erdmann-Pham et al. 2020; Erdmann-Pham et al. 2021) model in Python. The inhomogeneous ℓ-TASEP model was used to determine if translation was initiation limited, elongation limited, or termination limited. Simulations using the inhomogeneous ℓ-TASEP model were performed to obtain ribosome densities across the simulated transcript. Finally, the transcript was undersampled to simulate the process of obtaining a ribosome footprint. This simulation was repeated many times to obtain many different simulated ribosome footprints for transcripts of varying lengths.
Mouse brain cortex dissection and capillary electrophoresis western experiments
The animal experiments were carried out in accordance with the protocols of Carnegie Institution of Washington (CIW) IACUC. All mice were kept in specific pathogen-free facilities of CIW. Fmr1 knockout mice (003025) were acquired from the Jackson Laboratory. For brain cortex dissection, 24-day-old male mice were scarified using cervical dislocation. The brains were collected after breaking off the skull and the meninges, followed by freeing the cortical hemisphere from the brain through removing the cerebellum and the pons. The cortex was then dissected, frozen in liquid nitrogen, and stored in −80°C. Samples were lysed in 0.5 ml of cell extraction buffer (FNN0011; Thermo Fisher Scientific) supplemented with 1× complete protease inhibitor cocktail (Roche Molecular Biochemicals) with a Dounce homogenizer on ice. After 30-min incubation on ice, with vortexing at 10-min intervals, lysate was centrifuged at 13,000 rpm for 10 min at 4°C and the supernatant was collected for total protein quantification. The western analysis assays for FMRP, Auts2, Med13L, HUWE1, Arid2, Arid1a, Fat1, Ubr4, and Sptbn2 detection were performed as previously described (Baradaran-Heravi et al. 2016) with minor modifications. In brief, mixtures of cell lysates (0.5 or 1 mg/ml, diluted in 0.1× WES sample buffer) and the fluorescent master mix were heated at 70°C for 5 min. The samples, blocking and chemiluminescent reagents, primary and secondary antibodies, and wash buffer were dispensed into the microplates with 12–230 or 66–440 kDa separation modules and capillary electrophoresis western analysis was carried out with the ProteinSimple WES instrument. Total protein in the lysates was also measured similarly followed by biotin labeling of all proteins. Rabbit anti-FMRP (1:50; Cell Signaling 4317), rabbit anti-Auts2 (1:50; Abcam ab96326), rabbit anti-Med13L (1:50; Bethyl Laboratories A302-420A), rabbit anti-HUWE1 (1:100; Thermo Fisher Scientific A300-486A), rabbit anti-Arid2 (1:50; Abiocode R2380-1), mouse anti-Arid1a (1:50; Santa Cruz sc-32761), rabbit anti-Fat1 (1:50; Abcam ab241372), rabbit anti-Ubr4 antibody (1:100; Abcam ab86738), and rabbit anti-Sptbn2 (1:50; Proteinteck 55107-1-AP) antibodies were used. The data were acquired in electropherograms and analyzed using the inbuilt Compass software (ProteinSimple). The expression level of each target protein was first adjusted using the concentration curve expression generated for 1 wild-type sample and then normalized using the total biotin-labeled protein detected in the same volume of each lysate.
Results
The effect of FMRP deficiency on translation is strikingly similar in Drosophila oocytes and the mouse cortex
To test whether FMRP has a similar function in Drosophila oocytes and in mouse cortex, we compared the effects of FMRP on translation as measured using ribosome profiling in these 2 tissues and organisms (Greenblatt and Spradling 2018; Das Sharma et al. 2019) but re-analyzed here from primary reads with identical bioinformatic pipelines (see Methods and Supplementary Table 1). In this analysis, the translation efficiency is considered to be proportional to the frequency of ribosome footprints mapped onto the mRNA in question by ribosome profiling normalized to mRNA levels as measured by mRNA-seq. Consistent with Greenblatt et al., we found that many transcripts containing long CDS were translationally downregulated in Fmr1 RNAi oocytes as compared to controls (Fig. 1a). The median CDS length of genes whose translation decreased significantly in Fmr1 RNAi oocytes was 3.1 times longer than the median length of all oocyte expressed genes (4,262 vs 1,372 bp, respectively, Fig. 1b). We observed a strikingly similar effect of Fmr1 KO in the mouse cortex. Many genes encoding large proteins were translationally downregulated over a similar range (Fig. 1a), and the affected mRNAs had 3.4 times longer CDS lengths as compared to all cortex-expressed genes (4,797 vs 1,401 bp, respectively, Fig. 1b).
Fig. 1.

FMRP has a highly conserved role to promote translation in Drosophila oocytes and the mouse cortex. a) Volcano and b) box plots showing concordantly diminished translation efficiency of mRNAs with long CDS regions in FMRP-deficient Drosophila oocytes (Greenblatt and Spradling 2018) and mouse cortex (Das Sharma et al. 2019). c) Volcano plots showing concordantly diminished translation efficiency of FMRP-bound mRNAs (McMahon et al. 2016; Liu et al. 2018) in FMRP-deficient Drosophila oocytes and in mouse cortex. d) Scatter plots showing reduced translation efficiency as a function of protein size for the 200 most significantly affected genes in Fmr1 RNAi Drosophila oocytes or FMR1 KO mouse cortex. Histograms of size distributions for all Drosophila or mouse proteins respectively are shown to the right. e) Scatter plot showing reduced translation efficiency as a function of CLIP score (Sawicka et al. 2019) for the 200 most significantly affected genes in the FMR1 KO mouse cortex.
If FMRP acts directly to activate translation, then mRNAs bound by FMRP should be selectively reduced rather than increased in translation in mutant tissues. Consistent with FMRP acting directly, mouse cortex mRNAs bound by FMRP had reduced ribosome footprints in Fmr1-null animals (Das Sharma et al. 2019). Likewise, we found that Drosophila and mouse FMRP direct targets, as determined by TRIBE (McMahon et al. 2016) and in more recent CLIP-seq experiments (Sawicka et al. 2019), were concordantly translationally downregulated. Ninety-eight percentage (43/44) and 100% (50/50) of translationally altered mRNAs bound by FMRP in Drosophila oocytes and the mouse hippocampus, respectively, exhibited reduced translation efficiency in the absence of FMRP (Fig. 1c). FMRP CLIP targets were more than 10 times more likely to be translationally reduced in the Fmr1 mutant mouse cortex compared to all neuronally expressed transcripts irrespective of FMRP binding (25% vs 2.2% respectively, P = 3.8 × 10−107, χ2 test). These data argue that FMRP acts directly to increase the translation of a subset of bound mRNAs.
The magnitude of FMRP translational activation is similar for targets of varying length
FMRP contains multiple RNA-binding domains and studies suggest that it has the potential to bind to widespread tetranucleotide motifs and G-rich sequences (Ascano et al. 2012; Vasilyev et al. 2015; Anderson et al. 2016). FMRP-dependent translational activation might depend on length, with longer mRNAs being more strongly affected on average simply due to a greater number of FMRP-binding sites. An alternative possibility is that FMRP translational activation requires only that a minimum number of binding sites be occupied by FMRP, reflecting a highly cooperative process. This latter model predicts that FMRP translational activation would be independent of mRNA length, at least beyond a threshold (i.e. the length at which the minimum number of binding sites is occupied). To distinguish between these possibilities, we compared the magnitude of downregulation of translation for transcripts of varying length in FMRP-deficient cells. We found that while genes encoding large proteins were more likely to be FMRP targets, the magnitude of the reduction in translation did not increase proportionately with CDS length (Fig. 1d). The magnitude of reduction in translation was similar for FMRP targets of varying length (Fig. 1d) or CLIP score (Fig. 1e), an indicator of the strength of FMRP binding. These data support a threshold model for FMRP-dependent translational activation in both Drosophila oocytes and the mouse cortex.
FMRP targets show decreased expression in the mouse cortex
To further test whether FMRP acts to promote the translation of large proteins in the mouse cortex, we performed western experiments to determine the steady-state levels of FMRP direct targets as determined by CLIP-seq (Darnell et al. 2011; Li et al. 2020) in wild-type or FMRP-deficient tissues. We used automated capillary electrophoresis western analysis, which allows for the efficient detection of large proteins as it avoids a potentially lossy electrophoretic transfer step. The steady-state levels of all FMRP targets tested were reduced in FMRP KO cortex samples between 5% and 50% (Fig. 2, a–c). The largest reduction (49% reduced, P = 0.019, t-test) was observed for Auts2 (Fig. 2, a and a′). Auts2 encodes a PRC1-associated transcriptional activator essential for normal neuronal gene expression (Gao et al. 2014), and haploinsufficiency of Auts2 is associated with ID and autism (Oksenberg and Ahituv 2013). The protein levels of Med13L, HUWE1, and Arid2 were also reduced by 15–17% in the FMRP KO cortex as compared to controls (Fig. 2, a–c) (P = 0.021, P = 0.061, and P = 0.028 respectively). Med13L encodes a subunit of the Mediator complex required for neural crest cell migration (Utami et al. 2014), Arid2 is a core subunit of the PBAF chromatin remodeling complex whose loss results in neuronal hyperconnectivity (Zaslavsky et al. 2019), and HUWE1 encodes a large ubiquitin ligase, which inhibits neuronal progenitor proliferation (Zhao et al. 2008). All 3 of these genes are implicated in ID and autism disorders, and Med13L and Arid2 mutations are associated with dominant disorders caused by Med13L/Arid2 haploinsufficiency (Utami et al. 2014; Bramswig et al. 2017). Consistent with FMRP promoting the translation of a fraction of large proteins in the brain, we observed a small reduction (9%) in the bulk levels of all cellular proteins >300 kDa labeled with biotin (Fig. 2, b′ and b″). The levels of other FMRP CLIP targets, Fat1, Ubr4, and Sptbn2 also exhibited a trend toward reduced rather than increased levels (Fig. 2c). No FMRP target among the 8 tested had increased cortex protein levels in the absence of FMRP. Together, these data indicate that multiple FMRP targets encoding dosage-sensitive genes essential for neurodevelopment are downregulated in the FMRP KO mouse brain.
Fig. 2.

FMRP targets encoding large proteins are downregulated in the mouse cortex. a, a′) Profiles and quantification of capillary electrophoresis western experiments showing a partial reduction in the levels of FMRP targets Auts2 and Med13L and complete loss of FMRP in the Fmr1 KO mouse cortex as compared to wild-type controls. Insets show standard curves of western signal as a function of total protein concentration. b) Total protein levels of all biotin-labeled proteins in mouse cortex extracts. b′) A zoom-in of (b) showing large proteins >300 kDa. b″) Quantification of (b) showing a decrease in the levels of large proteins but not total proteins in Fmr1 KO mouse cortex samples. c) Quantification of capillary electrophoresis western experiments of indicated proteins performed on mouse cortex extracts from wild-type or Fmr1 KO animals. *P < 0.05, #P < 0.1.
A novel method to detect changes in translation due to ribosomal stalling
Declines in ribosome footprints on target mRNAs in FMRP-deficient tissues could in theory result from reduced translation initiation or increased elongation relative to controls. Similar to our measurements performed in the mouse cortex (Fig. 2, a–c), Drosophila oocytes and neurons are known to contain lower levels of 3 large FMRP target proteins in FMRP-deficient cells, consistent with reduced translation initiation (Greenblatt and Spradling 2018; Sears et al. 2019). However, ribosome density was reported to be more uniform across translated mRNAs in FMRP-deficient neurons (Das Sharma et al. 2019), a finding potentially consistent with a role for FMRP in translation elongation. Translation elongation rates are typically measured in the presence of inhibitors of translation initiation, confounding the interpretation of these experiments (Schmidt et al. 2009; Darnell et al. 2011). Observed changes in translation elongation may not impact overall protein production rates if translation initiation rather than elongation is rate-limiting. While translation of the vast majority of mRNAs is initiation limited (Ingolia et al. 2011; Shah et al. 2013; Erdmann-Pham et al. 2020), elongation limitation has been shown to occur in cells with low levels of tRNAs (Darnell et al. 2018) or in the absence of elongation factors (Woolstenhulme et al. 2015; Schuller et al. 2017).
To disentangle the effects of differential ribosome pausing and reduced translation initiation in FMRP-lacking cells, we sought to develop a quantitative method to detect rate-limiting pause sites using published ribosome profiling data. Rate-limiting pausing during translation elongation leads to the accumulation of stacked ribosomes prior to the pause site (Wolin and Walter 1988). Similar to the distribution of cars before or after a traffic jam, high densities of ribosomes precede rate-limiting pause sites and fewer ribosomes are found downstream of the pause site (Woolstenhulme et al. 2015; Erdmann-Pham et al. 2020). The appearance of “ribosome traffic jams” is therefore indicative of the existence of a rate-limiting ribosomal pause site.
To illustrate this phenomenon, we simulated the effect of ribosomal pauses on ribosome footprinting data from initiation- or elongation-limited genes using a biophysical model of translation (Fig. 3a). This model derives from a stochastic process of interacting particles, called the TASEP, and has been widely used for the past few years to simulate and analyze the determinants of translation speed from ribosome profiles (Zur and Tuller 2016; Dao Duc and Song 2018; Sharma et al. 2019; Szavits-Nossan and Ciandrini 2020). More precisely, one can distinguish under the TASEP model a traffic regime of “low density,” where the flux of ribosome is limited by translation initiation, and leads to a fairly even distribution of ribosomes across the transcript (Fig. 3a). In contrast, a “maximal current” regime is observed when the flux is limited by elongation at the region of minimal elongation rate, with a strong asymmetry in ribosome footprints accumulating upstream in this region (Fig. 3a) as predicted (Erdmann-Pham et al. 2020). Therefore, a slowdown of ribosome traffic within the coding region only results in a transition from initiation- to elongation-limited transition if the corresponding profiles display a discontinuous “phase transition” with asymmetry appearing at the pausing site. For initiation-limited transcripts, even if elongation rates are altered, they will not substantially affect overall translation rates unless they become rate-limiting and vice versa.
Fig. 3.

Elongation limitation leads to a quantifiable change in the distribution of ribosome footprints. a) Simulated ribosome footprinting profiles of initiation-limited and elongation-limited transcript using the inhomogenous ℓ-TASEP model (Erdmann-Pham et al. 2020). b) Cumulative distribution plots showing a large difference in the distribution of ribosome footprints (high K–S scores) in a simulated elongation-limited vs initiation-limited transcript. c) Plot showing high K–S values derived from comparisons (as in b) between elongation-limited but not initiation-limited transcripts. d) Ribosome footprinting profiles of cysQ and Sephs2 showing reduced ribosome densities (Woolstenhulme et al. 2015; Fradejas-Villar et al. 2017b) following stall sites induced in cells lacking efp and Trsp, respectively, whereas no stall is detected for the LARP1 target (Philippe et al. 2020) Rpl13. e) Cumulative distribution plots of (d) showing large differences in the distribution of ribosome footprints for cysQ and Sephs2 but not Rpl13 in mutant vs control cells. f, g) Plots showing that genes with known stall sites, efp targets and selenoproteins, have high K–S scores >0.3 in cells lacking efp and Trsp, respectively. LARP1 targets, which have altered translation initiation, in LARP1 KO cells do not have K–S scores >0.3.
To detect differences in the processes limiting translation (e.g. initiation/elongation) between 2 ribosome footprinting profiles, we introduced using the K–S statistic (Fig. 3b), which measures the maximal difference in the cumulative distributions of 2 samples. We found that low and high K–S statistic values were highly predictive of whether a simulated profile was in the low density or maximal current regimes, respectively (Fig. 3c and Supplementary Fig. 1), with all initiation-limited or elongation-limited transcripts having a K-S statistic value lower than or greater than 0.3, respectively (Fig. 3c).
To validate the utility of this approach, we applied our method to published ribosome footprinting datasets of mutant cells lacking key elongation or translation initiation factors. We analyzed datasets where gene expression was affected by changes in translation elongation: polyproline tract-containing proteins in bacteria lacking efp, a factor required for the efficient incorporation of prolines within polyproline stretches (Doerfel et al. 2013; Ude et al. 2013), and selenoproteins in mouse liver lacking the selenocysteine tRNA gene Trsp (Fradejas-Villar et al. 2017). We also analyzed data from cells lacking LARP1 (Philippe et al. 2020), which acts to repress the translation initiation of ribosomal proteins when mTOR is inhibited. Consistent with published data, the elongation-limited EFP target cysQ and selenoprotein gene Sephs2 had reduced ribosome occupancy downstream of their respective stall sites in mutant cells (Fig. 3d). In contrast, ribosomes were similarly distributed across the length of the initiation-limited LARP1 target Rpl13 (Fig. 3d). Consequently, the K–S scores for cysQ and Sephs2 were substantially higher (0.452 and 0.534, respectively, Fig. 3e) than Rpl13 (0.082, Fig. 3e). We found that among genes with high K–S statistic scores (K–S statistic > 0.3), EFP targets and selenocysteine-containing proteins were strongly enriched, ∼12-16-fold, whereas LARP1 targets were not enriched (Fig. 2, c and d). Thus, the K–S statistic can be used to identify genes whose overall translation is altered due to ribosome pausing.
We performed similar tests to determine whether Drosophila and mouse FMRP targets are limited by translation initiation or elongation. We found that ribosome footprints along Drosophila targets Poe, osa, and HUWE1 and mouse FMRP targets Arid2, HUWE1, and Auts2 were nearly identical in their overall distributions in wild-type vs FMRP-deficient animals, with K–S statistic values varying from 0.027 to 0.105 (Fig. 4, a–d). This trend was true for other FMRP targets. Among genes with high K–S statistic values >0.3, none of the FMRP TRIBE-seq and CLIP-seq targets analyzed passed this threshold (0/111 for Drosophila targets and 0/103 for mouse targets) (Fig. 4, e and f). Together, our analyses do not support a major role for FMRP in controlling the translation elongation of its targets.
Fig. 4.

The translation of FMRP targets is limited by initiation rather than elongation rates. a) Ribosome footprints of FMRP targets Poe, osa, and HUWE1 in control and Fmr1 RNAi oocytes. b) Cumulative distribution plots showing nearly identical distributions of ribosome footprints for FMRP targets in wild-type and Fmr1 RNAi oocytes. c) Ribosome footprints of mouse FMRP targets Arid2, HUWE1, and Auts2. f) Cumulative distribution plot showing nearly identical distributions of ribosome footprints of Arid2, HUWE1, and Auts2 in wild-type vs FMRP-deficient tissue. e, f) Plots showing that all mouse FMRP targets as analyzed as in (d) have low K–S scores <0.3.
ASD/ID genes encode exceptionally large proteins whose requirement for FMRP is evolutionarily conserved
FMRP is known to associate with mRNAs from many genes associated with ASD and ID (Darnell et al. 2011; Sawicka et al. 2019) and our data indicate a specific function of FMRP RNPs in the translational activation of large proteins. To investigate whether ASD/ID genes, which are known to be exceptionally long (King et al. 2013), also encode proteins larger than average, we compared them with the general population of neuronally expressed genes in the juvenile mouse cortex (Das Sharma et al. 2019). We classified high confidence and strong candidate genes implicated in ASD—SFARI Class I, Class II, and syndromic autism genes (Abrahams et al. 2013; Pereanu et al. 2018)—as “ASD-relevant genes,” and found that they encode proteins averaging 1,234 amino acids, which is more than twice as large as the average neuronally expressed protein, 589 amino acids (Fig. 5a). There was an even greater average difference in the size of these genes (exonic and intronic sequences), 83 vs 23 kb, consistent with prior studies (King et al. 2013; Zhao et al. 2018). The greater gene size was largely because ASD-relevant genes contain larger introns totaling 78.3 kb on average vs 20.6 kb on average for neuronally expressed genes (Fig. 5b). ASD-relevant genes were only slightly larger than the neuronal average with respect to 5′ UTR length (250 vs 160 bp, Fig. 5c) and 3′ UTR length (1,703 vs 1,053 bp, Fig. 5d). ASD-relevant genes were not significantly different from other neuronal genes in mRNA levels (Fig. 5e) or translation levels (Fig. 5f). Not surprisingly given their large average CDS length, ASD-relevant genes as a class were translationally downregulated in the FMRP KO cortex (Fig. 5g). FMRP target ASD-relevant genes encode proteins even larger than the average for ASD-relevant genes generally (2,035 aa vs 1,234 aa).
Fig. 5.
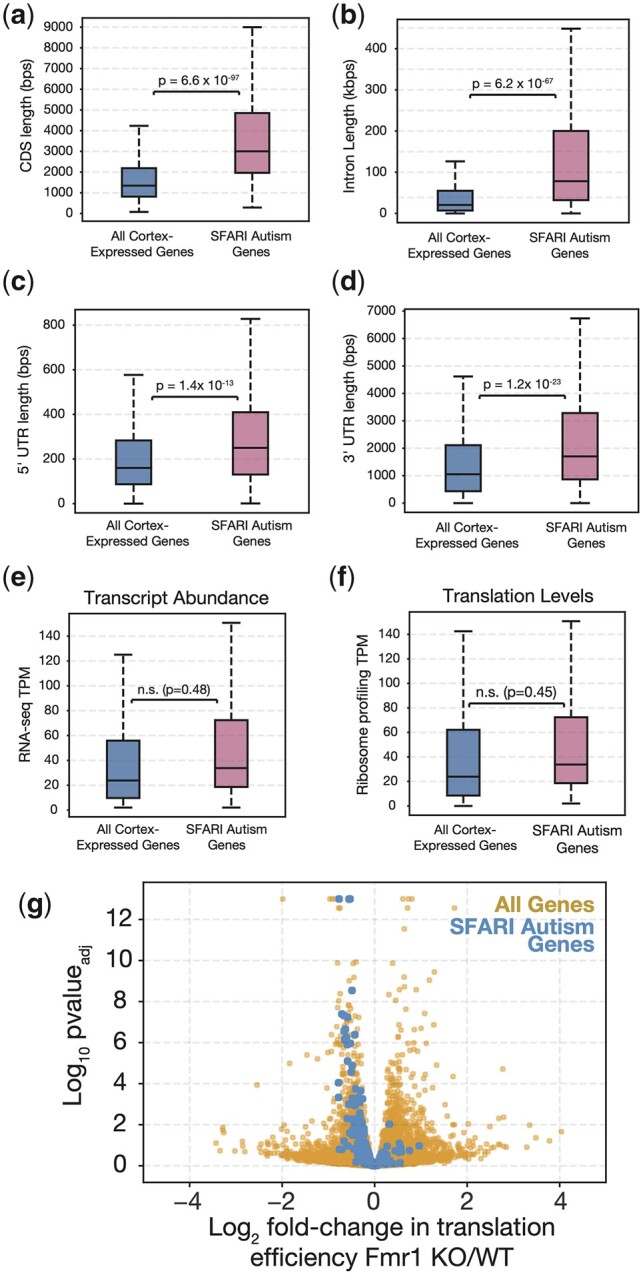
SFARI autism genes have longer open-reading frames, UTRs, and introns than average. Box plots of length distributions showing increased a) coding sequences lengths, b) 5′ UTR lengths, c) intron lengths (summed across all introns), and d) 3′ UTR lengths specifically for SFARI Class I and Class II ASD genes (magenta) as compared to all genes expressed in the juvenile mouse cortex (blue). No significant differences were detected between the distributions of transcript levels (e) or translation levels (f) specifically for ASD-relevant genes vs all cortex-expressed genes as detected by mRNA sequencing and ribosome footprinting, respectively. g) Volcano plot showing a concordant downregulation in the translation efficiency of SFARI Class I and Class II autism genes as compared to all neuronally expressed genes.
No evidence of a role for putative binding motifs in conferring FMRP target specificity
We next tested whether FMRP preferentially promotes the translation of ASD-relevant genes primarily because of their tendency to encode large proteins. If FMRP affects the translation of transcripts from ASD-relevant genes preferentially, then transcripts from these genes would be more likely to be downregulated in the Fmr1 KO cortex as compared to transcripts of similar size from non-ASD-relevant genes. Among the 200 most significantly downregulated genes, we found that ASD-relevant genes encoding long proteins >1,800 amino acids were more enriched than ASD-relevant genes irrespective of protein size, as well as genes generally encoding large proteins (P-value = 0.005 and 0.02, respectively, Chi-squared test, Supplementary Fig. 2a). These data are potentially consistent with FMRP acting preferentially on a subset of large ASD-relevant target mRNAs. However, this result could also be obtained if FMRP acts nonspecifically on mRNAs encoding large proteins, if ASD-relevant genes are more likely than other genes to be expressed in cells in the cortex such as neurons that utilize FMRP-dependent translation.
Previous studies have identified short 3–4 nucleotide motifs in FMRP-bound mRNAs that are slightly enriched in FMRP-bound mRNAs and which have been proposed to contribute to FMRP binding (Ascano et al. 2012; Anderson et al. 2016). To test for a role for these motifs, we compared the frequency of putative FMRP-binding motifs WGGA, ACUK, GAC, and UAY (Anderson et al. 2016) in downregulated genes in the FMR1 KO cortex as compared to nondownregulated genes. While the number of putative FMRP-binding sites was higher in downregulated genes, this was also true for any random 4-mer sequence (Supplementary Fig. 2b). When controlling for transcript length, none of the putative FMRP-binding motifs were enriched in translationally downregulated transcripts as compared to size-matched unaffected transcripts (Supplementary Fig. 2c). Thus, we find no evidence that FMRP acts preferentially on transcripts containing a high density of the putative 3–4 nucleotide-binding motifs. Our data do not exclude a model in which FMRP binds long transcripts via motifs that are present in higher frequency in targets simply because they are longer.
Underproduction rather than overproduction of FMRP ASD target genes may lead to clinical phenotypes
The relatively modest decreases in protein production that result from the loss of FMRP function might be particularly significant in the case of “dosage-sensitive” genes that cause defects when present in only one instead of 2 genomic copies. Dosage-sensitive genes relevant for the FXS phenotype, based on the FMRP activator model, would be “haploinsufficient,” i.e. loss of either the maternal or paternal copy would lead to clinical defects. In contrast, under the repressor model relevant FMRP targets would be “triplosensitive,” where gene duplications leading to the overexpression of FMRP targets result in clinical phenotypes. We identified 61 genes that are translationally downregulated in the Fmr1 KO mouse cortex and that score as SFARI Class I, Class II, or syndromic ASD-relevant genes (Table 1), which we considered to be potentially relevant. From this group, haploinsufficiency and triplosensitivity scores were available in the Clinical Genome Resource (NIH) database from clinical data for 32 FMRP targets (Rehm et al. 2015). Consistent with the activator model, we found that 22 of the 32 downregulated ASD-relevant genes were classified as haploinsufficient at the levels of “emerging evidence” and “sufficient evidence” (score of 2 and 3 respectively, Table 1). In contrast, and in contradiction to the FMRP repressor model, none of these genes (0/32) had emerging or sufficient evidence of triplosensitivity. These data indicate that reduced expression, but not overexpression, of FMRP ASD targets lead to observable clinical phenotypes and further support our model that FMRP acts primarily as an activator and not a repressor of gene expression.
Table 1.
SFARI Class I and Class II ASD-relevant genes are translationally reduced in the Fmr1 KO mouse cortex.
| SFARI autism gene | Fold-change TE Fmr1 KO/WT | P-valueadj | # amino acids | Darnell et al. (2011) CLIP target | ClinGen haplo-insufficiency score | ClinGen triplo-sensitivity score |
|---|---|---|---|---|---|---|
| PRR12 | 0.51 | 0.0E+00 | 2,035 | ✓ | n/a | n/a |
| KMT2A | 0.58 | 9.0E−05 | 3,966 | ✓ | 3 | 0 |
| HIVEP3 | 0.58 | 4.7E−04 | 2,348 | ✓ | 1 | 0 |
| SHANK1 | 0.59 | 0.0E+00 | 2,167 | ✓ | 1 | 0 |
| SETD1B | 0.59 | 1.4E−03 | 1,985 | n/a | n/a | |
| KMT2C | 0.61 | 4.0E−08 | 4,904 | ✓ | 3 | 0 |
| SPEN | 0.63 | 2.7E−07 | 3,620 | ✓ | 1 | 0 |
| MED13L | 0.63 | 4.8E−08 | 2,207 | ✓ | 3 | 1 |
| TCF20 | 0.64 | 7.1E−07 | 1,987 | ✓ | 3 | 0 |
| SCAF4 | 0.64 | 6.1E−04 | 1,209 | n/a | n/a | |
| AHDC1 | 0.65 | 2.2E−07 | 1,594 | ✓ | 3 | 0 |
| SRCAP | 0.65 | 6.2E−07 | 3,271 | 1 | 0 | |
| CIC | 0.66 | 5.4E−07 | 1,604 | ✓ | 2 | 0 |
| KDM3B | 0.66 | 1.3E−06 | 1,762 | n/a | n/a | |
| CNOT3 | 0.66 | 5.0E−03 | 751 | 0 | 0 | |
| MAP1A | 0.67 | 0.0E+00 | 2,776 | ✓ | n/a | n/a |
| SHANK2 | 0.67 | 5.9E−08 | 1,472 | ✓ | 2 | 0 |
| RIMS1 | 0.67 | 9.7E−07 | 1,190 | 1 | 0 | |
| AUTS2 | 0.67 | 8.0E−06 | 1,261 | ✓ | 3 | 0 |
| RERE | 0.68 | 1.0E−03 | 1,558 | ✓ | n/a | n/a |
| SYN1 | 0.68 | 1.2E−06 | 670 | ✓ | 2 | 0 |
| HECTD4 | 0.68 | 0.0E+00 | 4,418 | n/a | n/a | |
| LRP1 | 0.69 | 0.0E+00 | 4,545 | ✓ | n/a | n/a |
| CAMK2A | 0.69 | 3.0E−04 | 478 | ✓ | 1 | 0 |
| CREBBP | 0.69 | 1.1E−06 | 2,441 | ✓ | 3 | 0 |
| NOVA2 | 0.70 | 2.6E−03 | 492 | n/a | n/a | |
| CHD8 | 0.70 | 2.7E−05 | 2,582 | ✓ | 3 | 0 |
| SHANK3 | 0.71 | 5.4E−03 | 1,805 | ✓ | 3 | 0 |
| ARID2 | 0.71 | 7.3E−04 | 1,828 | ✓ | 3 | 0 |
| ASH1L | 0.71 | 2.9E−09 | 2,958 | ✓ | 3 | 0 |
| IQSEC2 | 0.71 | 1.3E−05 | 1,479 | ✓ | 3 | 0 |
| ZC3H4 | 0.72 | 3.6E−03 | 1,255 | ✓ | n/a | n/a |
| SON | 0.72 | 5.2E−04 | 2,444 | ✓ | 3 | 0 |
| GRIK5 | 0.72 | 5.6E−03 | 979 | ✓ | n/a | n/a |
| CACNA1A | 0.72 | 1.6E−05 | 2,327 | ✓ | 3 | 0 |
| KIF5C | 0.73 | 1.4E−10 | 956 | ✓ | n/a | n/a |
| HUWE1 | 0.73 | 6.6E−10 | 4,378 | ✓ | 0 | 0 |
| ZMIZ1 | 0.74 | 1.5E−03 | 1,066 | ✓ | n/a | n/a |
| AKAP9 | 0.74 | 1.2E−03 | 3,779 | ✓ | n/a | n/a |
| NCOR1 | 0.74 | 4.1E−07 | 2,454 | ✓ | n/a | n/a |
| ARID1B | 0.75 | 1.8E−04 | 2,244 | ✓ | 3 | 0 |
| AGO2 | 0.76 | 1.2E−03 | 860 | n/a | n/a | |
| MYT1L | 0.77 | 6.6E−03 | 1,185 | ✓ | 3 | 0 |
| FOXG1 | 0.78 | 8.6E−03 | 481 | 3 | 0 | |
| MECP2 | 0.78 | 9.5E−03 | 484 | n/a | n/a | |
| WDFY3 | 0.78 | 6.5E−06 | 3,508 | ✓ | n/a | n/a |
| VAMP2 | 0.78 | 6.7E−05 | 116 | ✓ | n/a | n/a |
| NSD1 | 0.79 | 4.3E−03 | 2,691 | ✓ | 3 | 0 |
| CHD3 | 0.79 | 4.8E−04 | 2,055 | ✓ | n/a | n/a |
| EP400 | 0.80 | 8.2E−04 | 2,999 | ✓ | n/a | n/a |
| INTS1 | 0.80 | 5.2E−03 | 2,222 | ✓ | n/a | n/a |
| PHF3 | 0.80 | 2.7E−03 | 2,025 | 1 | 0 | |
| YWHAG | 0.80 | 5.8E−04 | 247 | ✓ | n/a | n/a |
| SLC12A5 | 0.81 | 5.0E−03 | 1,138 | ✓ | n/a | n/a |
| CTNND2 | 0.81 | 2.3E−04 | 1,221 | ✓ | 2 | 0 |
| SATB1 | 0.82 | 7.4E−03 | 764 | n/a | n/a | |
| TRRAP | 0.82 | 2.0E−04 | 3,847 | ✓ | 1 | 0 |
| NFIX | 0.82 | 5.5E−04 | 433 | ✓ | n/a | n/a |
| SYNE1 | 0.82 | 4.1E−03 | 1,431 | ✓ | n/a | n/a |
| TANC2 | 0.83 | 7.6E−03 | 1,994 | ✓ | n/a | n/a |
| ATP2B2 | 0.83 | 5.5E−04 | 1,198 | ✓ | n/a | n/a |
| ILF2 | 1.25 | 9.5E−03 | 390 | n/a | n/a |
Table showing SFARI Class I and Class II ASD−relevant genes that are significantly altered in their translation efficiency in the Fmr1 KO mouse brain. Ninety-eight percentage (61/62) of altered ASD-relevant genes are translationally reduced. ClinGen Dosage scores (Rehm et al. 2015) (3 = sufficient evidence, 2 = emerging evidence, 1 = little evidence, 0 = no evidence) show that many of these ASD-relevant genes have strong or emerging evidence of human syndromes associated with gene losses (haploinsufficiency) but not gene duplications (triplosensitivity). n/a, not available.
FMRP target ASD-relevant genes were predicted to be translationally downregulated to 51–83% of control levels based on reduced ribosome footprints. On average, FMRP target ASD-relevant genes were translationally reduced to similar levels as compared to generic downregulated genes (72% vs 70%, respectively). FMRP probably acts by directly binding to most of these mRNAs, since 48 of 61 (79%) were cross-linked in FMRP CLIP experiments (Darnell et al. 2011). Thus, at least 22 dosage-sensitive ASD-relevant genes may be translationally downregulated in the Fmr1 KO cortex, likely through the loss of FMRP direct binding, each potentially contributing to defects despite relatively small individual reductions.
Discussion
FMRP has a conserved function to promote the translation of a subset of large proteins
Oocytes and neurons use common mechanisms of RNA transport and translational regulation to control protein production in both time and space. In oocytes, FMRP is required for the constitutive translation of large proteins (Greenblatt and Spradling 2018), and our analyses here suggest a similar role for FMRP in the mammalian brain. Our side-by-side comparison of ribosome profiling data shows that the effect of FMRP loss on translation in Drosophila oocytes and mouse cortex is extremely similar despite the fact that different tissues and organisms are involved. In both cases, the targets are more than 3 times longer than average-sized mRNAs and encode correspondingly larger proteins, including many orthologs. In both cases, the normal stimulatory effects of FMRP are estimated to be 2-fold or less, independent of the length of the mRNA and their predicted FMRP-binding site content. Moreover, based on our analyses, these similar effects do not appear to be to changes in rates of ribosome elongation or stalling, but represent differences in translation initiation and protein output. Consistent with our findings here, western analyses documented the expected reductions in 2 FMRP target proteins in oocytes, the 2,053-amino-acid snRNA 3′ processing factor IntS1 and the 5,322-amino-acid E3 ubiquitin ligase Poe/UBR4 (Greenblatt and Spradling 2018), while the 3,719-amino-acid protein kinase A regulator rugose/NBEA is reduced in Fmr1 mutant Drosophila mushroom body (Sears et al. 2019), as well as mouse cortex FMRP targets in this study which include Auts2, Med13L, HUWE1, and Arid2 (Fig. 2, a–d).
These findings are consistent with the important and ancient role FMRP plays in neurons, oocytes, and spermatocytes from diverse animals ranging from Drosophila to humans (Hagerman et al. 2017; Drozd et al. 2018). Mammalian and Drosophila FMRP contain the same protein domains and the expression of human FMRP rescues neural defects in Drosophila Fmr1 mutants (Coffee et al. 2010). FMRP had long been considered primarily a general translational repressor (Laggerbauer et al. 2001; Li et al. 2001; Darnell et al. 2011); however, bulk translation in the brain has been observed to be unaffected in the brains of FXS patients (Schmidt et al. 2020). Our data indicate that FMRP likely acts directly on its targets to increase translation. FMRP was found to bind to Poe mRNA in neurons and glia in vivo as well as in cultured Schneider 2 cells (McMahon et al. 2016) as well as to the mRNA of mouse ortholog Ubr4 in hippocampal neurons (Sawicka et al. 2019), and FMRP direct targets are concordantly reduced in translation in both Drosophila ovaries (Fig. 1c) as well as the mouse cortex (Fig. 1c). These data are corroborated by previous Ribo-tag studies of Fmr1 KO hippocampal cells, which similarly showed that 11/12 differentially expressed FMRP targets are reduced rather than increased in expression (Ceolin et al. 2017). Our findings here are similarly consistent with observations that FMRP CLIP targets have concordantly reduced ribosome footprints (Das Sharma et al. 2019), that FMRP preferentially binds to long transcripts (Sawicka et al. 2019; Li et al. 2020), and that the protein levels of large proteins are reduced rather than increased in the Fmr1 KO hippocampal proteome (Seo et al. 2022). Our data are also consistent with other prior reports that FMRP activates the translation of its targets (Bechara et al. 2009; Tabet et al. 2016). Indeed, none of the mRNAs with increased translation in Fmr1-null animals were among the top 200 CLIP targets (Darnell et al. 2011; Das Sharma et al. 2019). The increased translation of some genes in Fmr1 mutant animals is likely due to secondary, indirect effects of FMRP loss. FMRP targets include transcription repressors, and E3 ubiquitin ligases that stimulate protein degradation via the proteasome, while some proteins increase in translation due to the dysregulation of mTOR signaling (Sharma et al. 2010; Das Sharma et al. 2019). These data help explain the failure of the mTOR inhibitor rapamycin to reverse behavior deficits in Fmr1-null mice (Saré et al. 2017), since treating the increased translation levels of secondary FMRP targets would not rescue the primary translational defect in these animals—the underproduction of dozens of ASD/ID-associated large proteins. These data are also consistent with observations that underproduction, but not overproduction, of FMRP ASD targets leads to clinical phenotypes (Table 1).
FMRP is used to support translation in neurons and other cells that depend on stored mRNAs
Large cells such as neurons and oocytes rely heavily on the transport and storage of stable, untranslated mRNAs. For example, a large fraction of oocyte mRNAs are translationally repressed for later use during early embryonic development, including CNS development (Kronja et al. 2014; Greenblatt et al. 2019). Neurons transport translationally regulated synaptic mRNAs up to a meter or more down long axonal projections. Such transport requires cells to partition mRNAs into repressed or actively translated states through the use of RNP particles, including P bodies and neuronal particles, that are structurally related to stress granules. These complexes often associate binding partners together through phase separation, which is sensitive to protein concentration (Molliex et al. 2015). Thus, there are periods when mRNAs undergoing transport from the cell body along an axon, or while stored in the vicinity of a synapse, must remain functional while in an inactive state. The rate of growth of Drosophila oocytes varies strongly in response to available nutrition, and both oocytes and neurons grow at very different rates at different stages of development. P bodies and stress granules can be seen to form and disperse within minutes when growth conditions vary sharply (Shimada et al. 2011; Wheeler et al. 2016). FMRP function is particularly required under conditions where development is slowed, for example after mature oocytes enter quiescence (Greenblatt and Spradling 2018). In such cells, long mRNAs are inefficiently translated as compared to shorter mRNAs (Greenblatt and Spradling 2018). Fmr1 may have evolved during animal evolution to support the production of large proteins which play critical roles in the reproductive and nervous systems. This model is consistent with FMRP′s preferential binding to mRNA CDS regions (Darnell et al. 2011; Maurin et al. 2018). Proteins do not have to bind to the 5′ UTR or mRNA cap to affect translation initiation—for example the repressive cap-binding complex 4EHP-GYF2 is recruited to the 3′ UTR of AU-rich element-containing mRNAs through tristetraprolin to compete with eIF4E-dependent translation initiation (Fu et al. 2016). Another example is poly(A)-binding protein, which stimulates translation initiation through the binding of the poly(A) tail of mRNAs (Sachs and Davis 1989).
Previous observations showed that genes expressed during neural development on average encode larger proteins than in many other tissues (Gabel et al. 2015). Proteins regulated by FMRP are more than 3 times larger still and include many genes associated with autism and ID syndromes. There is currently no well-understood functional explanation for what exceptionally large protein size contributes to higher neural functions. Neural development likely involves complex tasks such as integrating sensory inputs and outputs in ways that are vital to survival. It may be advantageous to place multiple protein domains along a single polypeptide chain to ensure they are present at equimolar levels to help guarantee the proper stoichiometries of protein domains that function together in a complex process. Another possibility is that large neural proteins function as scaffolds for exceptionally large and specific protein complexes that play critical functions essential for neural processing. Such proteins might require many critical binding sites for additional binding partners spaced substantial distances apart along the primary sequence. If processes associated with the most complex neuronal-based decision-making are especially dependent on complexes catalyzed by enormous proteins, this might explain the enrichment of the ASD/ID gene products among the largest cohorts of FMRP targets.
Data availability
All sequencing data were obtained from public NCBI GEO and BioProject databases with the following accessions: GSE64488 (Woolstenhulme et al. 2015), GSE114064 (Das Sharma et al. 2019), GSE132703 (Philippe et al. 2020), GSE84112 (Fradejas-Villar et al. 2017), and PRJNA466150 (Greenblatt and Spradling 2018). The compiled translation efficiency data for the Fmr1 RNAi Drosophila ovary and Fmr1 KO mouse cortex analyses are available in Supplementary Table 1. A compiled table showing the effect of Fmr1 RNAi on the translation of Drosophila orthologs of ASD-relevant genes is available in Supplementary Table 2. K–S statistic values computed for Drosophila and mouse transcripts in comparisons of control and Fmr1-deficient tissues are available in Supplementary Table 3. Unprocessed RNA sequencing and ribosome profiling TPM values are available in Supplementary Table 4.
Supplemental material is available at GENETICS online.
Funding
This work was supported by funding through the Howard Hughes Medical Institute (Q.Y., A.C.S., and E.G.), the Simons Foundation Autism Research Initiative (K.F., A.B.H., and E.G.), and Michael Smith Health Research BC (E.G.).
Conflicts of interest
None declared.
Supplementary Material
Contributor Information
Keegan Flanagan, Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, BC V6T 1Z3, Canada; Department of Mathematics, University of British Columbia, Vancouver, BC V6T 1Z2, Canada.
Alireza Baradaran-Heravi, Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, BC V6T 1Z3, Canada.
Qi Yin, Howard Hughes Medical Institute Research Laboratories, Department of Embryology, Carnegie Institution for Science, Baltimore, MD 21218 USA.
Khanh Dao Duc, Department of Mathematics, University of British Columbia, Vancouver, BC V6T 1Z2, Canada.
Allan C Spradling, Howard Hughes Medical Institute Research Laboratories, Department of Embryology, Carnegie Institution for Science, Baltimore, MD 21218 USA.
Ethan J Greenblatt, Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, BC V6T 1Z3, Canada; Howard Hughes Medical Institute Research Laboratories, Department of Embryology, Carnegie Institution for Science, Baltimore, MD 21218 USA.
Literature cited
- Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, Menashe I, Wadkins T, Banerjee-Basu S, Packer A.. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism. 2013;4(1):36. doi: 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson BR, Chopra P, Suhl JA, Warren ST, Bassell GJ.. Identification of consensus binding sites clarifies FMRP binding determinants. Nucleic Acids Res. 2016;44(14):6649–6659. doi: 10.1093/nar/gkw593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryal S, Klann E.. Turning up translation in fragile X syndrome. Science. 2018;361(6403):648–649. doi: 10.1126/science.aau6450. [DOI] [PubMed] [Google Scholar]
- Aryal S, Longo F, Klann E.. Genetic removal of p70 S6K1 corrects coding sequence length-dependent alterations in mRNA translation in fragile X syndrome mice. Proc Natl Acad Sci USA. 2021;118(18). doi: 10.1073/pnas.2001681118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492(7429):382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baradaran-Heravi A, Balgi AD, Zimmerman C, Choi K, Shidmoossavee FS, Tan JS, Bergeaud C, Krause A, Flibotte S, Shimizu Y, et al. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016;44(14):6583–6598. doi: 10.1093/nar/gkw638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara EG, Didiot MC, Melko M, Davidovic L, Bensaid M, Martin P, Castets M, Pognonec P, Khandjian EW, Moine H, et al. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 2009;7(1):e16. doi: 10.1371/journal.pbio.1000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH, Akhavan AR, Johnston D.. Impaired dendritic expression and plasticity of h-channels in the fmr1(-/y) mouse model of fragile X syndrome. Cell Rep. 2012;1(3):225–233. doi: 10.1016/j.celrep.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramswig NC, Caluseriu O, Lüdecke H-J, Bolduc FV, Noel NCL, Wieland T, Surowy HM, Christen H-J, Engels H, Strom TM, et al. Heterozygosity for ARID2 loss-of-function mutations in individuals with a Coffin-Siris syndrome-like phenotype. Hum Genet. 2017;136(3):297–305. doi: 10.1007/s00439-017-1757-z. [DOI] [PubMed] [Google Scholar]
- Bülow P, Murphy TJ, Bassell GJ, Wenner P.. Homeostatic intrinsic plasticity is functionally altered in fmr1 KO cortical neurons. Cell Rep. 2019;26(6):1378–1388.e3. doi: 10.1016/j.celrep.2019.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callan MA, Cabernard C, Heck J, Luois S, Doe CQ, Zarnescu DC.. Fragile X protein controls neural stem cell proliferation in the Drosophila brain. Hum Mol Genet. 2010;19(15):3068–3079. doi: 10.1093/hmg/ddq213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceolin L, Bouquier N, Vitre-Boubaker J, Rialle S, Severac D, Valjent E, Perroy J, Puighermanal E.. Cell type-specific mRNA dysregulation in hippocampal CA1 pyramidal neurons of the fragile X syndrome mouse model. Front Mol Neurosci. 2017;10:340. doi: 10.3389/fnmol.2017.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee RL, Tessier CR, Woodruff EA, Broadie K.. Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis Models Mech. 2010;3(7–8):471–485. doi: 10.1242/dmm.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao Duc K, Song YS.. The impact of ribosomal interference, codon usage, and exit tunnel interactions on translation elongation rate variation. PLoS Genet. 2018;14(1):e1007166. doi: 10.1371/journal.pgen.1007166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell AM, Subramaniam AR, O'Shea EK.. Translational control through differential ribosome pausing during amino acid limitation in mammalian cells. Mol Cell. 2018;71(2):229–243.e11. doi: 10.1016/j.molcel.2018.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KYS, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Sharma S, Metz JB, Li H, Hobson BD, Hornstein N, Sulzer D, Tang G, Sims PA.. Widespread alterations in translation elongation in the brain of juvenile fmr1 knockout mice. Cell Rep. 2019;26(12):3313–3322.e5. doi: 10.1016/j.celrep.2019.02.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV.. EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science. 2013;339(6115):85–88. doi: 10.1126/science.1229017. [DOI] [PubMed] [Google Scholar]
- Drozd M, Bardoni B, Capovilla M.. Modeling fragile X syndrome in Drosophila. Front Mol Neurosci. 2018;11:124. doi: 10.3389/fnmol.2018.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, Weissman JS.. Plastid: nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genomics. 2016;17(1):958. doi: 10.1186/s12864-016-3278-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann-Pham DD, Dao Duc K, Song YS.. The key parameters that govern translation efficiency. Cell Syst. 2020;10(2):183–192.e6. doi: 10.1016/j.cels.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann-Pham DD, Son W, Dao Duc K, Song YS.. EGGTART: a tool to visualize the dynamics of biophysical transport under the inhomogeneous l-TASEP. Biophys J. 2021;120(8):1309–1313. doi: 10.1016/j.bpj.2021.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradejas-Villar N, Seeher S, Anderson CB, Doengi M, Carlson BA, Hatfield DL, Schweizer U, Howard MT.. The RNA-binding protein Secisbp2 differentially modulates UGA codon reassignment and RNA decay. Nucleic Acids Res. 2017;45(7):4094–4107. doi: 10.1093/nar/gkw1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R, Olsen MT, Webb K, Bennett EJ, Lykke-Andersen J.. Recruitment of the 4EHP-GYF2 cap-binding complex to tetraproline motifs of tristetraprolin promotes repression and degradation of mRNAs with AU-rich elements. RNA. 2016;22(3):373–382. doi: 10.1261/rna.054833.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, Greenberg ME.. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015;522(7554):89–93. doi: 10.1038/nature14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Lee P, Stafford JM, von Schimmelmann M, Schaefer A, Reinberg D.. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature. 2014;516(7531):349–354. doi: 10.1038/nature13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goering R, Hudish LI, Guzman BB, Raj N, Bassell GJ, Russ HA, Dominguez D, Taliaferro JM.. FMRP promotes RNA localization to neuronal projections through interactions between its RGG domain and G-quadruplex RNA sequences. eLife. 2020;9. doi: 10.7554/eLife.52621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt EJ, Obniski R, Mical C, Spradling AC.. Prolonged ovarian storage of mature Drosophila oocytes dramatically increases meiotic spindle instability. eLife. 2019;8. doi: 10.7554/eLife.49455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt EJ, Spradling AC.. Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science. 2018;361(6403):709–712. doi: 10.1126/science.aas9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AW, Elisseou NM, McKinney BC, Greenough WT.. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006;1084(1):158–164. doi: 10.1016/j.brainres.2006.02.044. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB, Moine H, Kooy RF, Tassone F, Gantois I, Sonenberg N, Mandel JL, et al. Fragile X syndrome. Nat Rev Dis Primers. 2017;3:17065. doi: 10.1038/nrdp.2017.65. [DOI] [PubMed] [Google Scholar]
- Hays SA, Huber KM, Gibson JR.. Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J Neurosci. 2011;31(40):14223–14234. doi: 10.1523/JNEUROSCI.3157-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PJ, Shi H, Zhu AC, Lu Z, Miller N, Edens BM, Ma YC, He C.. The RNA-binding protein FMRP facilitates the nuclear export of N6-methyladenosine-containing mRNAs. J Biol Chem. 2019;294(52):19889–19895. doi: 10.1074/jbc.AC119.010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS.. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147(4):789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao D-I, Aldridge GM, Weiler IJ, Greenough WT.. Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc Natl Acad Sci U S A. 2010;107(35):15601–15606. doi: 10.1073/pnas.1010564107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, Parker R.. The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol Cell. 2017;68(4):808–820.e5. doi: 10.1016/j.molcel.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IF, Yandava CN, Mabb AM, Hsiao JS, Huang H-S, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501(7465):58–62. doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronja I, Yuan B, Eichhorn SW, Dzeyk K, Krijgsveld J, Bartel DP, Orr-Weaver TL.. Widespread changes in the posttranscriptional landscape at the Drosophila oocyte-to-embryo transition. Cell Rep. 2014;7(5):1495–1508. doi: 10.1016/j.celrep.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U.. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10(4):329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- Lauria F, Tebaldi T, Bernabò P, Groen EJN, Gillingwater TH, Viero G.. riboWaltz: optimization of ribosome P-site positioning in ribosome profiling data. PLoS Comput Biol. 2018;14(8):e1006169. doi: 10.1371/journal.pcbi.1006169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Shin J, Risgaard RD, Parries MJ, Wang J, Chasman D, Liu S, Roy S, Bhattacharyya A, Zhao X.. Identification of FMR1-regulated molecular networks in human neurodevelopment. Genome Res. 2020;30(3):361–374. doi: 10.1101/gr.251405.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y.. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29(11):2276–2283. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Li Y, Stackpole EE, Novak A, Gao Y, Zhao Y, Zhao X, Richter JD.. Regulatory discrimination of mRNAs by FMRP controls mouse adult neural stem cell differentiation. Proc Natl Acad Sci U S A. 2018;115(48):E11397–E11405. doi: 10.1073/pnas.1809588115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin T, Lebrigand K, Castagnola S, Paquet A, Jarjat M, Popa A, Grossi M, Rage F, Bardoni B.. HITS-CLIP in various brain areas reveals new targets and new modalities of RNA binding by fragile X mental retardation protein. Nucleic Acids Res. 2018;46(12):6344–6355. doi: 10.1093/nar/gky267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AC, Rahman R, Jin H, Shen JL, Fieldsend A, Luo W, Rosbash M.. TRIBE: hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell. 2016;165(3):742–753. doi: 10.1016/j.cell.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP.. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163(1):123–133. doi: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenberg N, Ahituv N.. The role of AUTS2 in neurodevelopment and human evolution. Trends Genet. 2013;29(10):600–608. doi: 10.1016/j.tig.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF.. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30(46):15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereanu W, Larsen EC, Das I, Estévez MA, Sarkar AA, Spring-Pearson S, Kollu R, Basu SN, Banerjee-Basu S.. AutDB: a platform to decode the genetic architecture of autism. Nucleic Acids Res. 2018;46(D1):D1049–D1054. doi: 10.1093/nar/gkx1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe L, G van den Elzen AM, Watson MJ, Thoreen CC.. Global analysis of LARP1 translation targets reveals tunable and dynamic features of 5’ TOP motifs. Proc Natl Acad Sci U S A. 2020;117(10):5319–5328. doi: 10.1073/pnas.1912864117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prilutsky D, Kho AT, Palmer NP, Bhakar AL, Smedemark-Margulies N, Kong SW, Margulies DM, Bear MF, Kohane IS.. Gene expression analysis in Fmr1KO mice identifies an immunological signature in brain tissue and mGluR5-related signaling in primary neuronal cultures. Mol Autism. 2015;6:66. doi: 10.1186/s13229-015-0061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, et al. ClinGen—the clinical genome resource. N Engl J Med. 2015;372(23):2235–2242. – doi:10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs AB, Davis RW.. The poly(A) binding protein is required for poly(A) shortening and 60S ribosomal subunit-dependent translation initiation. Cell. 1989;58(5):857–867. doi: 10.1016/0092-8674(89)90938-0. [DOI] [PubMed] [Google Scholar]
- Saré RM, Song A, Loutaev I, Cook A, Maita I, Lemons A, Sheeler C, Smith CB.. Negative effects of chronic rapamycin treatment on behavior in a mouse model of fragile X syndrome. Front Mol Neurosci. 2017;10:452. doi: 10.3389/fnmol.2017.00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka K, Hale CR, Park CY, Fak JJ, Gresack JE, Van Driesche SJ, Kang JJ, Darnell JC, Darnell RB.. FMRP has a cell-type-specific role in CA1 pyramidal neurons to regulate autism-related transcripts and circadian memory. eLife 8. 2019;8. doi: 10.7554/eLife.46919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P.. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6(4):275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Schmidt KC, Loutaev I, Quezado Z, Sheeler C, Smith CB.. Regional rates of brain protein synthesis are unaltered in dexmedetomidine sedated young men with fragile X syndrome: a L-[1-11C]leucine PET study. Neurobiol Dis. 2020;143:104978. doi: 10.1016/j.nbd.2020.104978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller AP, Wu CC-C, Dever TE, Buskirk AR, Green R.. eIF5A functions globally in translation elongation and termination. Mol Cell. 2017;66(2):194–205.e5. doi: 10.1016/j.molcel.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears JC, Choi WJ, Broadie K.. Fragile X Mental Retardation Protein positively regulates PKA anchor Rugose and PKA activity to control actin assembly in learning/memory circuitry. Neurobiol Dis. 2019;127:53–64. doi: 10.1016/j.nbd.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SS, Louros SR, Anstey N, Gonzalez-Lozano MA, Harper CB, Verity NC, Dando O, Thomson SR, Darnell JC, Kind PC, et al. Excess ribosomal protein production unbalances translation in a model of fragile X syndrome. Nat Commun. 2022;13(1):3236. doi: 10.1038/s41467-022-30979-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB.. Rate-limiting steps in yeast protein translation. Cell. 2013;153(7):1589–1601. doi: 10.1016/j.cell.2013.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AK, Sormanni P, Ahmed N, Ciryam P, Friedrich UA, Kramer G, O'Brien EP.. A chemical kinetic basis for measuring translation initiation and elongation rates from ribosome profiling data. PLoS Comput Biol. 2019;15(5):e1007070. doi: 10.1371/journal.pcbi.1007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS.. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30(2):694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada Y, Burn KM, Niwa R, Cooley L.. Reversible response of protein localization and microtubule organization to nutrient stress during Drosophila early oogenesis. Dev Biol. 2011;355(2):250–262. doi: 10.1016/j.ydbio.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu H, Donnard E, Liu B, Jung S, Wang RRichter JD. FMRP links optimal codons to mRNA stability in neurons. Proceedings of the National Academy of Sciences of the United States of America 117:30400–30411. doi: 10.1073/pnas.2009161117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan SD, Welt C, Sherman S.. FMR1 and the continuum of primary ovarian insufficiency. Semin Reprod Med. 2011;29(4):299–307. doi: 10.1055/s-0031-1280915. [DOI] [PubMed] [Google Scholar]
- Szavits-Nossan J, Ciandrini L.. Inferring efficiency of translation initiation and elongation from ribosome profiling. Nucleic Acids Res. 2020;48(17):9478–9490. doi: 10.1093/nar/gkaa678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabet R, Moutin E, Becker JAJ, Heintz D, Fouillen L, Flatter E, Krężel W, Alunni V, Koebel P, Dembélé D, et al. Fragile X Mental Retardation Protein (FMRP) controls diacylglycerol kinase activity in neurons. Proc Natl Acad Sci U S A. 2016;113(26):E3619–28. doi: 10.1073/pnas.1522631113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, Jung K.. Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science. 2013;339(6115):82–85. doi: 10.1126/science.1228985. [DOI] [PubMed] [Google Scholar]
- Utami KH, Winata CL, Hillmer AM, Aksoy I, Long HT, Liany H, Chew EGY, Mathavan S, Tay SKH, Korzh V, et al. Impaired development of neural-crest cell-derived organs and intellectual disability caused by MED13L haploinsufficiency. Hum Mutat. 2014;35(11):1311–1320. doi: 10.1002/humu.22636. [DOI] [PubMed] [Google Scholar]
- Vasilyev N, Polonskaia A, Darnell JC, Darnell RB, Patel DJ, Serganov A.. Crystal structure reveals specific recognition of a G-quadruplex RNA by a β-turn in the RGG motif of FMRP. Proc Natl Acad Sci U S A. 2015;112(39):E5391–400. doi: 10.1073/pnas.1515737112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JR, Matheny T, Jain S, Abrisch R, Parker R.. Distinct stages in stress granule assembly and disassembly. eLife. 2016;5. doi: 10.7554/eLife.18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin SL, Walter P.. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 1988;7(11):3559–3569. doi: 10.1002/j.1460-2075.1988.tb03233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolstenhulme CJ, Guydosh NR, Green R, Buskirk AR.. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 2015;11(1):13–21. doi: 10.1016/j.celrep.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaslavsky K, Zhang W-B, McCready FP, Rodrigues DC, Deneault E, Loo C, Zhao M, Ross PJ, El Hajjar J, Romm A, et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat Neurosci. 2019;22(4):556–564. doi: 10.1038/s41593-019-0365-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Marro SG, Zhang Y, Arendt KL, Patzke C, Zhou B, Fair T, Yang N, Südhof TC, Wernig M, et al. The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci Transl Med. 2018;10(452). doi: 10.1126/scitranslmed.aar4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Heng JI-T, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, Iavarone A, Lasorella A.. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat Cell Biol. 2008;10(6):643–653. doi: 10.1038/ncb1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y-T, Kwon DY, Johnson BS, Fasolino M, Lamonica JM, Kim YJ, Zhao BS, He C, Vahedi G, Kim TH, et al. Long genes linked to autism spectrum disorders harbor broad enhancer-like chromatin domains. Genome Res. 2018;28(7):933–942. doi:10.1101/gr.233775.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Karaletsos T, Drewe P, Sreedharan VT, Kuo D, Singh K, Wendel H-G, Rätsch G.. RiboDiff: detecting changes of mRNA translation efficiency from ribosome footprints. Bioinformatics. 2017;33(1):139–141. doi: 10.1093/bioinformatics/btw585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur H, Tuller T.. Predictive biophysical modeling and understanding of the dynamics of mRNA translation and its evolution. Nucleic Acids Res. 2016;44(19):9031–9049. doi: 10.1093/nar/gkw764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data were obtained from public NCBI GEO and BioProject databases with the following accessions: GSE64488 (Woolstenhulme et al. 2015), GSE114064 (Das Sharma et al. 2019), GSE132703 (Philippe et al. 2020), GSE84112 (Fradejas-Villar et al. 2017), and PRJNA466150 (Greenblatt and Spradling 2018). The compiled translation efficiency data for the Fmr1 RNAi Drosophila ovary and Fmr1 KO mouse cortex analyses are available in Supplementary Table 1. A compiled table showing the effect of Fmr1 RNAi on the translation of Drosophila orthologs of ASD-relevant genes is available in Supplementary Table 2. K–S statistic values computed for Drosophila and mouse transcripts in comparisons of control and Fmr1-deficient tissues are available in Supplementary Table 3. Unprocessed RNA sequencing and ribosome profiling TPM values are available in Supplementary Table 4.
Supplemental material is available at GENETICS online.