

Abstract
Radiotherapy (RT) is commonly employed to treat solid tumors. Immune checkpoint blockade (ICB) of PD-1 and CTLA-4 improves survival in RT patients, yet many fail to respond to combination therapy. NKG2 family receptors, particularly inhibitory NKG2A and activating NKG2D, have emerged as promising therapeutic targets to improve anti-tumor T cell responses, thus we examined how these receptors and their ligands (Qa-1b and Rae-1, respectively) regulate the RT response in C57BL/6 mice bearing syngeneic B16F10 melanoma and MC38 colorectal adenocarcinoma tumors. RT (15Gy) transiently reduced B16F10 tumor burden whereas MC38 tumors exhibited durable response to RT. Intratumoral NK and CD8 T cells expressed NKG2A and NKG2D in both models, which was unaltered by RT. In vitro/in vivo RT increased tumor/stromal cell Qa-1b and Rae-1 expression in both models, especially B16F10 tumors, but IFN-γ stimulation induced both Qa-1b and Rae-1 only in B16F10 tumors. NKG2A/Qa-1b inhibition alone did not improve RT response in either model, but combined RT and NKG2A/PD-1 blockade improved survival in the B16F10 model. Depletion experiments indicate that the triple therapy efficacy is CD8 T cell dependent with negligible NK cell contribution. RNA sequencing of CD8 T cells from triple therapy treated B16F10 tumors showed increased proliferative capacity compared to RT and PD-1 blockade alone. Our work demonstrates that RT modulates NKG2A ligand expression, which inhibits RT-induced T cell responses in tumors that fail to respond to combined RT and PD-1 blockade. These results provide rationale for combining NKG2A blockade with ICB therapies and RT to improve clinical response.
INTRODUCTION
Radiotherapy (RT) is used to treat more than half of tumor malignancies (1). Radiation causes double stranded DNA breaks and eventual cell death to reduce tumor burden (1). We and others have observed that supplementary to direct tumor cytotoxicity, RT limits tumor growth through induction of an anti-tumor immune response. Dying tumor cells release damage associated molecular patterns, cytokines, and antigen (2–4), which activate type one conventional DCs that cross-present to CD8 T cells in the draining lymph node (5). These CD8 T cells then migrate to the tumor microenvironment (TME) to lyse tumor cells and secrete cytokines such as IFN-γ, which is essential to the anti-tumor response (6).
RT elicits a robust anti-tumor response, but many immunosuppressive mechanisms can limit efficacy of therapy. RT promotes recruitment of suppressive myeloid and T regulatory cells (7, 8). Furthermore, heightened IFN-γ secretion can upregulate checkpoint inhibitor ligands, such as PD-L1 (programmed cell death ligand 1), that suppress effector T cell function (9, 10). To combat these regulatory mechanisms, RT has been combined with anti-CTLA-4 or anti-PD-1 (programmed cell death protein 1) antibodies in human patients to improve response (11, 12), yet many patients do not benefit from combination therapy. Even combined inhibition of PD-1 and CTLA-4 with RT does not result in complete cures in mouse models (13, 14). Thus, it is apparent that other immune regulatory mechanisms suppress the intratumoral immune response independent of conventional checkpoint inhibitors.
Recently, the NKG2 (natural killer group 2) family of receptors has attracted interest as a therapeutic option for modulating the anti-tumor immune response. However, analysis of how these molecules contribute to or hinder the anti-tumor immune response is in its infancy, thus little is known about these signaling axes in an RT context. Though typically associated with NK cells, NKG2 receptors are expressed on T cells (15, 16). Upon ligation with HLA-E in humans or Qa-1b in mice, NKG2A suppresses effector activity through recruitment of SHP-1 phosphatase to phosphorylated immunoreceptor tyrosine-based inhibition motifs on the cytosolic tails of the receptor (17). Blocking the NKG2A/Qa-1b axis improves survival in mouse models when combined with immune checkpoint blockade (ICB) or tumor vaccine (18, 19). Additionally, increased intratumoral CD94 expression, which is part of the heterodimeric NKG2A receptor, indicates poor prognosis in human patients with colorectal cancer (20). In contrast, NKG2D binds ligands such as MICA/B in humans and Rae-1 (retinoic acid early inducible 1) in mice to signal through DAP10/12, promoting a cytotoxic response (17). Low expression of NKG2D ligands correlates with worse response in non-small-cell lung carcinoma (NSCLC) (21), and prevention of NKG2D ligand shedding can improve survival in mouse models (22). Importantly, inflammation and cell stress can increase ligand expression for both activating and inhibitory NKG2 receptors (23, 24). As radiation induces cell stress through DNA damage, we hypothesized that RT alters NKG2A and NKG2D ligand expression to modulate the anti-tumor immune response. Using murine models for colorectal adenocarcinoma and melanoma, we observed that RT induces Qa-1b and Rae-1 expression on tumor cells. This response was heightened in the radioresistant melanoma model, resulting in suppression of the anti-tumor immune response that could be alleviated only with combined PD-1 and NKG2A inhibition.
METHODS
Mice and cell lines
C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were treated in accordance with guidelines approved by the University Committee on Animal Resources. MC38 cells were obtained from Dr. Edward Brown. B16F10 Parental and B16F10 Qa-1b CRISPR (clustered regularly interspaced short palindromic repeats) knockout (KO) cell lines were obtained from Dr. Thorbald van Hall from the Leiden University Medical Center. All cell lines were maintained in MAT/P medium containing 100 U/mL penicillin, 100 mg/mL streptomycin, and 2% FBS. Cell lines used for experiments never exceeded 10 consecutive in vitro passages.
Generation of Qa-1b overexpressing and control vector transduced MC38 cell lines
MC38 cells were grown to 50% confluency and media was replaced with 500 μL 2% FBS MAT/P (no antibiotics) containing 8 μg/mL polybrene (Millipore Sigma, St. Louis, MO). Control vector (CV) viral particles (Origene, Rockville, MD) or Qa-1b encoding viral particles (Origene) at a multiplicity of infection of 5 were incubated with cells for 24 hours, after which media was replaced with normal 2% FBS MAT/P media. Three days post transduction, cells were analyzed by flow cytometry for mGFP expression to monitor transduction efficiency and then cultured in selection media containing 1 μg/mL puromycin (GIBCO, Grand Island, NY). Following puromycin selection, CV control transduced cells were sorted on a BD FACSAria II (BD Biosciences, San Jose, CA) for cells with mGFP expression. For Qa-1b vector transduced cells, cells were stimulated with 10 ng/mL murine IFN-γ (Peprotech, Rocky Hill, NJ) or left untreated (UT) for 48 hours and then sorted on a BD FACSaria II (BD Biosciences) for cells with mGFP and Qa-1b expression. Cells were then cloned by limiting dilution (0.5 cells/well) in 5% FBS MAT/P media. After 10 days of incubation, clones were isolated and analyzed for mGFP or mGFP and Qa-1b expression for CV control and Qa-1b transduced clones, respectively, as described above. Three clones with low mGFP expression in CV control transduced cells and three clones with low mGFP but high Qa-1b expression in Qa-1b transduced cells were selected for use in further experiments.
Tumor implantation and monitoring of tumor growth
For primary tumor challenge, 6–8-week-old mice were injected with 1 × 105 MC38/B16F10/B16F10 Qa-1b KO cells intramuscularly in the hind limb. For experiments with CV control or Qa-1b overexpressing MC38 cell lines, three clones of each cell line were combined in equal proportions for a total of 1 × 105 cells per intramuscular injection in the hind limb. Tumors established for seven days then received no treatment or RT (15 Gy) delivered locally to the tumor bearing leg by a cesium source irradiator. Starting on day 7 post implantation and continuing every 2 days until sacrifice, tumor bearing legs were measured in two dimensions by calipers and the geometric mean (GM) was calculated to monitor tumor growth. For tumor rechallenge, surviving mice or age matched controls were injected with 1 × 105 MC38 cells intramuscularly in the contralateral hind limb and growth was monitored as described above. Mice were sacrificed at the indicated time points or when GM of tumor leg burden was greater than or equal to 13 mm.
IFN-γ ELISA of tumor homogenates
MC38 and B16F10 tumors harvested at day nine post tumor implantation were suspended in lysis buffer 11 solution at 400 mg/mL and snap frozen in liquid nitrogen. Samples were then thawed on ice and homogenized for 30 seconds on ice using a Bio-Gen PRO200 Homogenizer (PRO Scientific, Oxford, CT). Tumor homogenate was centrifuged to remove cellular debris, and supernatants were used in bicinchoninic acid assays (BCA, Thermo Scientific, Waltham, MA) and IFN-γ ELISAs (Invitrogen, Waltham, MA) according to the manufacturers’ instructions. Intratumoral IFN-γ concentration was calculated by dividing pg IFN-γ by mg total protein. All BCA and ELISA sample plates were read on a Gen5 plate reader (BioTek, Winooski, VT).
CD8 T cell and NK cell depletion
Tumor bearing mice were injected subcutaneously with control rat IgG antibody (Jackson Immunoresearch, West Grove, PA), 200 μg anti-CD8α (clone 53–6.7, BioXcell) or anti-NK1.1 (clone PK136, BioXcell) (starting on day 3 post tumor implantation and continuing every 3 days until day 12, for a total of four injections). Efficacy of depletion was monitored by analyzing peripheral blood CD8 T cell and NK cell frequency by flow cytometry using anti-CD8β PE (H35-17.2, BD Biosciences) and anti-CD49b (DX5, BD Biosciences) respectively at day 10 post tumor implantation.
Flow cytometry
Resected tumors were cut into 1–2 mm pieces using a razor blade and incubated in collagenase solution at 100 mg tissue/1 mL collagenase (2 mg/mL, Sigma) for 30 minutes at 37°C. Digested tumor tissue was passed through a 40 μm filter to obtain a single cell suspension. Spleens were mechanically processed with frosted microscope slides and passed through a 40 μm filter to obtain a single cell suspension. Cells were then incubated with Fc block (24G2) for 10 minutes at 4°C. Cells were then incubated with staining antibodies in PBS solution containing 1% (w/v) bovine serum albumin and 0.1% (w/v) NaN3 for 30 minutes at 4°C and then fixed with BD Cytoperm/CytoFix solution (BD Biosciences) for 20 minutes at 4°C. The following antibodies were used for analysis of cell populations: CD45 BUV395 (30-F11), Ly6C APC-Cy7 (AL-21), Ly6G BV605 (1A8), CD4 APC-Cy7 (GK1.5), CD8β PE (H35-17.2), ΝΚ1.1 PE-Cy7 (PK136), NK1.1 PECF594 (PK136), Qa-1b PE (A8.6F10.1A6), Rae-1 BV605 (186107), and H-2Kb FITC (AF6-88.5) from BD Biosciences as well as CD11b eFluor 450 (M1/70), F4/80 APC (BM8), CD8α eFluor450 (53–6.7), NKG2AB6 PerCP-eFluor 710 (16a11), and NKG2D APC (CX5) from eBioscience (San Diego, CA) and PD-1 BV711 (29F.1A12, Biolegend, San Diego, CA). All samples were run on BD LSR II, BD FACSymphony A1 orBD LSR Fortessa flow cytometers (BD Biosciences) in the University of Rochester flow cytometry core facility.
Anti-PD-1 and anti-NKG2A antibody therapy
For single blockade of NKG2A, B16F10 tumor bearing mice were injected subcutaneously with 200 μg control rat IgG antibody (Jackson Immunoresearch) or 200 μg anti-NKG2A antibody (20D5, BioXcell, Lebanon, NH) starting on day 7 post tumor implantation and continuing every 3 days until day 16 for four total injections. For dual blockade therapy, MC38 and B16F10 tumor bearing mice were injected subcutaneously with the following combinations: 400 μg control rat IgG antibody, 200 μg anti-PD-1 antibody (RMP1-14, BioXcell) and 200 μg control rat IgG antibody, 200 μg anti-NKG2A antibody and 200 μg control rat IgG antibody, or 200 μg anti-NKG2A antibody and 200 μg control anti-PD-1. Importantly, the anti-NKG2A antibody used has been shown to mask NKG2A but not deplete NKG2A expressing cells (19). Injections started on day 7 post tumor implantation and continued every 3 days until day 19 for five total injections. For B16F10 tumor bearing mice, mouse mass was measured concurrently with tumor burden measurements.
Culture of bone marrow derived macrophages
Femoral bone marrow cells isolated from 6–8-week old female C57BL/6 mice were cultured in MAT/P media containing 20 ng/mL murine M-CSF (Shenandoah Biotech, Warwick, PA) for 7 days at 37°C, with murine M-CSF media being replenished on days 3 and 6. On day 7 of in vitro culture, bone marrow derived macrophages were cold shocked by incubation in PBS for 10 minutes at 4°C and then removed from wells with a cell scraper and analyzed by flow cytometry.
In vitro radiation and IFN-γ stimulation of tumor cells
B16F10 Parental, B16F10 Qa-1b KO, and MC38 tumor cells at 50% confluency were treated with a 15 Gy (gray) dose of radiation, 10 ng/mL murine IFN-γ (Peprotech), or left untreated. Forty-eight hours after treatment, cells were harvested and stained for analysis by flow cytometry. For analysis following in vitro irradiation, gMFIs (GM fluorescence intensity) of markers were normalized by subtracting the gMFI of an FMO exposed to the same radiation dose from the sample gMFI to correct for increased autofluorescence due to irradiation.
Cell sorting and RNA sequencing analysis
Mice bearing B16F10 tumors were left untreated or treated with ablative RT combined with subcutaneous injections of control IgG, anti-PD-1 antibody, or anti-PD-1 and anti-NKG2A as described above. On day 9 post tumor implantation, tumors were harvested and cells were stained for surface markers as described above. Cells were sorted on a BD FACSaria II (BD Biosciences) using a 100 μm nozzle into Eppendorf tubes containing 350 μL RLT buffer (Qiagen, Germantown, MD) containing 2-mercaptoethanol (10 μL/mL RLT buffer) at 4°C. To prevent dilution of RLT buffer, a maximum of 4.4 × 104 events was sorted into each tube. Cells were sorted into CD4 T cell, CD8 T cell, NK cell, and macrophage populations and immediately stored at −80°C. CD8 T cell samples were then submitted to the University of Rochester Medical Center Genomics Research Center (URMC GRC) for RNA sequencing (RNASeq) analysis.
RNA isolation, quality analysis, and RNA sequencing and its corresponding analysis was performed by the URMC GRC. RNA quality was assessed using an Agilent Bioanalyzer (Agilent, Santa Clara, CA), and samples with low quality as assessed by the URMC GRC were excluded from further analysis. CDNA libraries were constructed using the SMART-Seq V4 Ultra Low Input RNA Kit (TaKaRa, Mountain View, CA) following the manufacturer’s instructions, and sequencing was performed on an Illumina NovaSeq 6000 Sequencer (Illumina, San Diego, CA).
Log transformed normalized counts from RNASeg data of sorted CD8 T cells were used for gene set enrichment analysis with GSEA 4.1.0 (Broad Institute, UC San Diego, San Diego, CA). The following T cell phenotype datasets from MsigDB (molecular signatures database, Broad Institute) were used for enrichment analysis: GSE9650_1256_200_DN, GSE9650_1256_200_UP, GSE9650_1258_200_DN, GSE9650_1258_200_UP, GSE9650_1254_200_DN, GSE9650_1254_200_UP, GSE41867_1609_200_DN, GSE41867_1609_200_UP, GSE44649_2406_200_DN, GSE44649_2406_200_UP, GSE1000002_1582_200_DN, GSE1000002_1582_200_UP, GSE1000002_1580_200_DN, GSE1000002_1580_200_UP, GSE1000002_1581_200_DN, GSE1000002_1581_200_UP. Enrichment was considered statistically significant if the false discovery rate (FDR) was less than 0.25.
For gene ontology analysis, a gene list consisting of 58 enriched genes shared between GSE1000002_1582_200_UP and GSE1000002_1581_200_DN was run through Gene Ontology (GO) Biological Process 2018 using Enrichr (25, 26).
Flow cytometry and statistical analysis
All flow cytometry data were analyzed using Flowjo (BD Biosciences). All statistical analyses were performed with GraphPad Prism (Graphpad Software Inc., San Diego, CA). For comparisons between two groups, two-tailed student’s T tests or Mann Whitney tests were used, as indicated in figure legends. Survival curves were compared using Log-rank tests and corrected for multiple comparisons using the Bonferroni method.
RESULTS
Radiotherapy induces durable responses in the MC38 model, but only transient responses in the B16F10 model
We compared baseline efficacy of RT in two murine tumor models: MC38, a colorectal adenocarcinoma, and B16F10, a murine metastatic melanoma. Tumors were injected intramuscularly in the leg and allowed to establish for 7 days prior to start of RT. In both models, a single 15 Gy dose reduced tumor burden (Fig. 1A,C), resulting in longer survival for RT treated mice (Fig. 1B,D). RT only induced a transient response in B16F10 tumors, whereas durable responses were observed in the MC38 model (Fig. 1B,D). Our group previously demonstrated that CD8 T cells and IFN-γ are essential for the RT response (6, 27). In both models, RT did not affect intratumoral CD4 T cell, CD8 T cell, or NK cell frequency (Fig. 1E,F). Upregulation of intratumoral IFN-γ was seen 2 days post RT in both the MC38 and B16F10 models. (Fig. 1G,H). Importantly, we and others have observed that RT response is CD8 T cell dependent, as depletion of CD8 T cells abolishes RT efficacy in MC38 and B16F10 tumors treated with ablative RT (6, 28). Altogether, these data demonstrate that RT induces anti-tumor immune responses in both MC38 and B16F10 tumor models, but only results in durable responses in the MC38 model.
Figure 1.
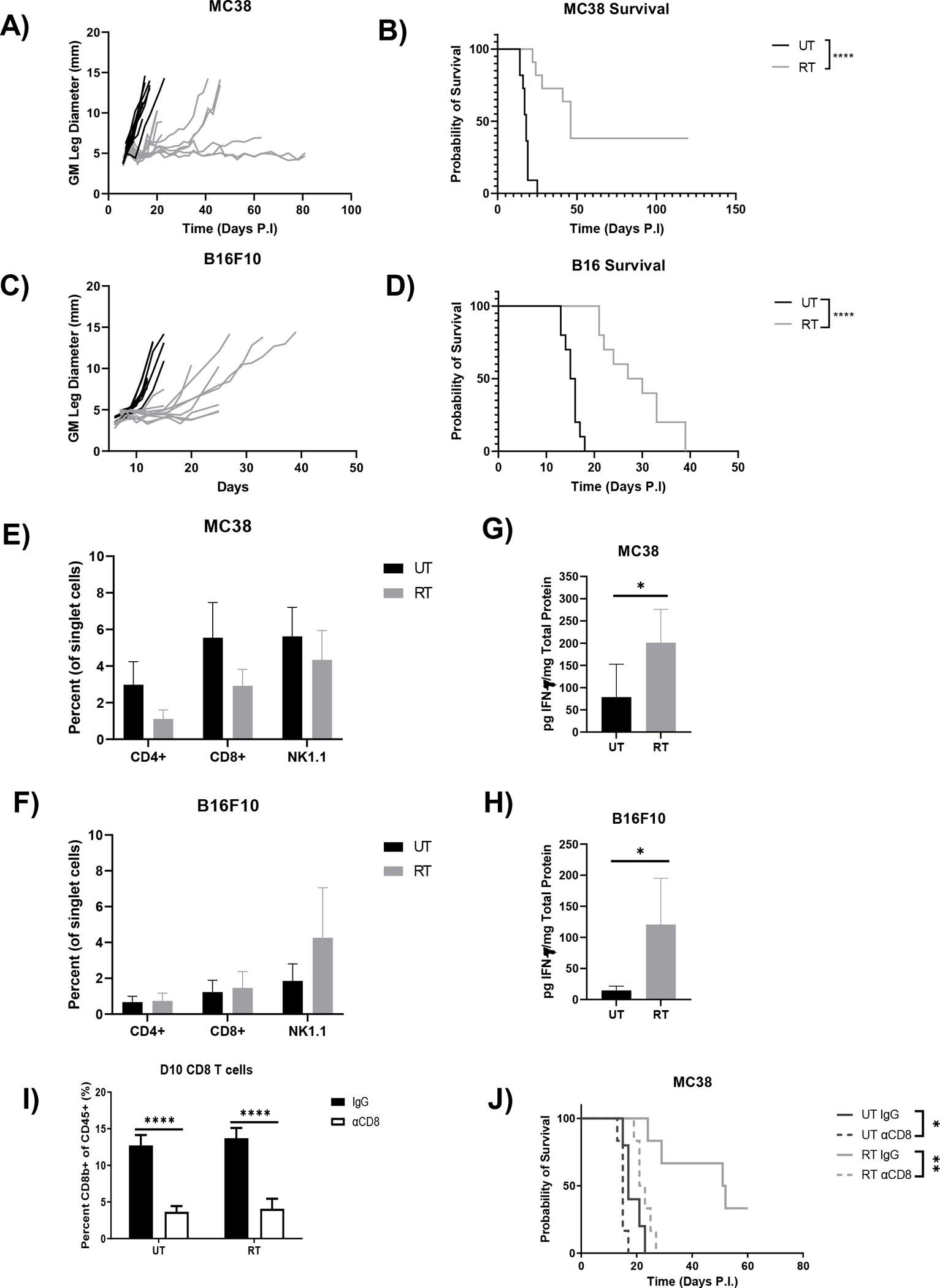
Ablative RT causes durable responses in mice bearing MC38 tumors whereas all mice bearing B16F10 tumors experience tumor rebound. MC38 (A/B) or B16F10 (C/D) tumors were implanted intramuscularly in the leg of 6–8-week-old female C57BL/6 mice, left untreated or treated locally with 15 Gy, and measured every two days until an endpoint of 13 mm was reached to generate growth and survival curves. Frequency of intratumoral CD4 T cells, CD8 T cells, and NK cells in MC38 (E) and B16F10 (F) tumors at day 9 post tumor implantation were analyzed by flow cytometry. Untreated and irradiated MC38 (G) or B16F10 (H) tumors were harvested on day 9 and total intratumoral IFN-γ was measured by ELISA and normalized to total protein determined by BCA. (Survival curves analyzed by Log-rank tests and other comparisons by two-tailed Mann Whitney test). (* p<0.05, ** p<0.01, **** p<0.0001). (n=10–11 mice per group, error bars indicate standard deviation).
NKG2A is expressed by intratumoral NK and CD8 T cells and radiotherapy increases expression of its ligand Qa-1b
Conventional ICB through inhibition of CTLA-4, PD-1, or both has been combined with RT (11, 12, 14), yet non-responders remain with combination therapy, especially in the B16F10 model (13). Thus, we hypothesized that signaling through activating and inhibitory NK receptors, which are expressed by both CD8 and NK cells, may also play a role in inhibiting response following RT. Productive response to RT is evident 2 days post RT (Fig. 1), thus we examined the TME at this timepoint for NK receptor and ligand expression. Gating for NKG2A and NKG2D expression is shown in Supplemental Fig. 1. In the MC38 model, inhibitory and activating receptors NKG2A and NKG2D, respectively, were predominantly expressed on NK and CD8 T cells (Fig. 2A). Importantly, CD8 T cell expression was limited to the TME, as little expression was seen in splenic T cells, whereas NK cells expressed these receptors regardless of tissue compartment. NKG2 receptor expression on NK and CD8 T cells in the B16F10 model largely mirrored observations in the MC38 model (Fig. 2B), though CD4 T cells in this model had modest NKG2A expression. RT had no substantial effect on NK receptor expression on any cell subset in both tumor models (Fig. 2A,B). We also observed NKG2A expression on intratumoral macrophages and patrolling monocytes in the B16F10 model (Supplemental Fig. 2A–C). Macrophage NKG2A expression did not depend on localization to the TME, as bone marrow derived macrophages similarly expressed NKG2A (Supplemental Fig. 2D). In contrast to receptor expression, RT increased expression of the NKG2A and NKG2D ligands Qa-1b and Rae-1, respectively, on CD45− tumor/stromal cells in both models (Fig. 2C,D). Importantly, RT-mediated upregulation of NK receptor ligands was more substantial on tumor/stromal cells in B16F10 tumors compared to MC38 tumors. Qa-1b binds to and is stabilized by class Ia molecule derived peptide (29), but the RT-induced upregulation of Qa-1b surface expression on tumor/stromal cells is likely not due to stabilization from increased MHC class I expression, as H-2Kb levels were unexpectedly not affected by RT in both models at this timepoint (Fig. 2C,D). Basal H-2Kb expression was high in untreated MC38 tumors, therefore it is possible that further increase was not possible with RT, but it is unclear why RT did not increase H-2Kb expression in B16F10 tumors, which have low basal expression in the untreated condition (Fig. 2C,D). Interestingly, Qa-1b expression was not substantially increased by RT on infiltrating immune cells in MC38 and B16F10 tumors (Supplemental Fig. 2E). These data demonstrate that RT increases intratumoral NK receptor ligand expression in vivo on tumor/stromal cells, especially in the B16F10 model, while not affecting NK receptor expression on effector cells.
Figure 2.
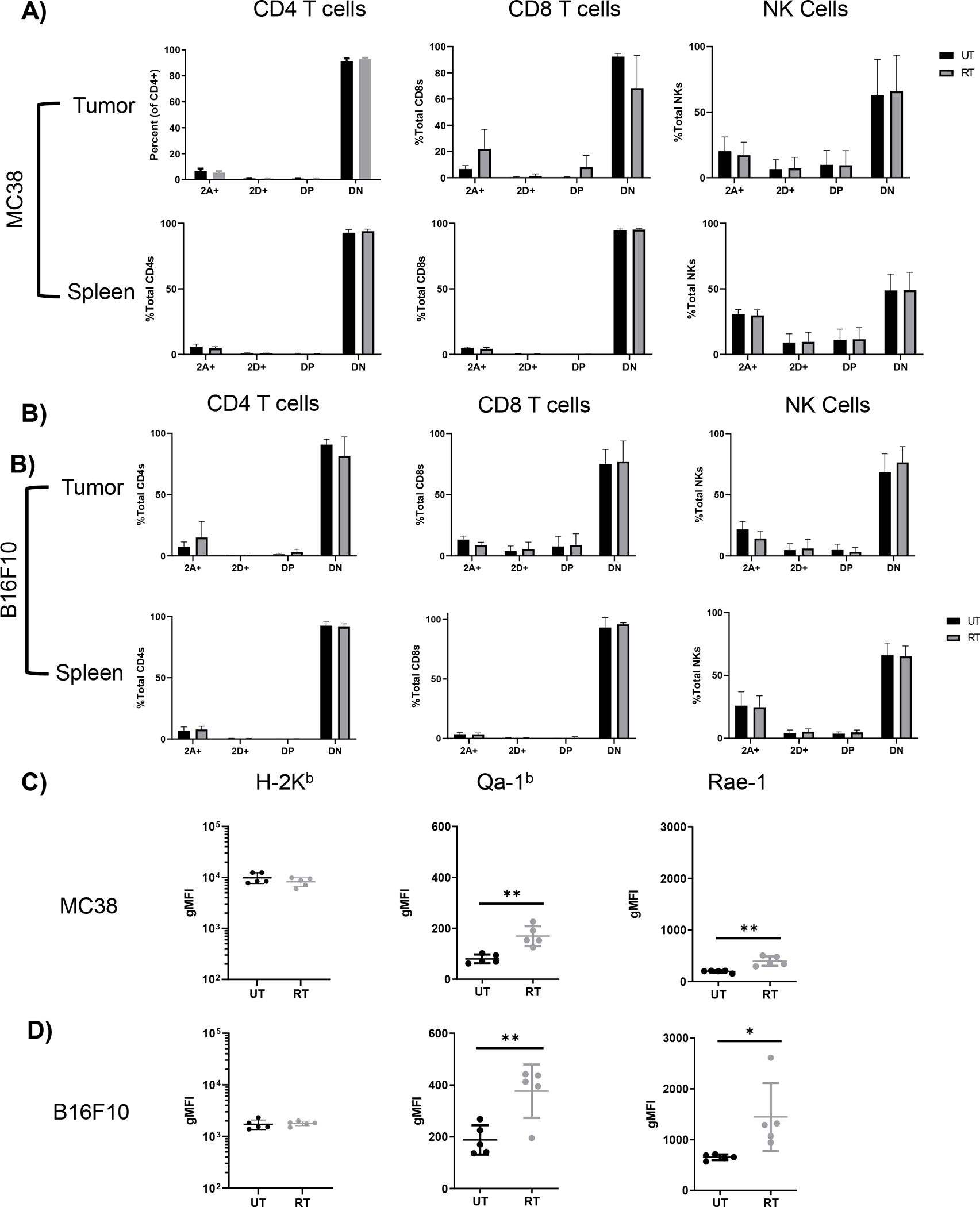
Ablative RT increases in vivo expression of Rae-1 and Qa-1b on tumor cells in both MC38 and B16F10 tumor models but does not affect NKG2A and NKG2D expression on NK and T cells. Tumors and spleens of mice bearing MC38 (A) or B16F10 (B) tumors were harvested on day 9 and analyzed for expression of NKG2 receptors on effector cells (2A = NKG2A, 2D = NKG2D, DP = double positive, DN = double negative). MC38 (C) and B16F10 tumor cells (D) taken from mice at day 9 were analyzed for expression of H-2Kb and the inhibitory and activating ligands Qa-1b and Rae-1, respectively. (Data analyzed by two-tailed Mann Whitney test). (* p<0.05, ** p<0.01). (n=10 mice per group, error bars indicate standard deviation).
RT and IFN-γ both increase Qa-1b expression in vitro, while MC38 only increases expression of Rae-1 in response to RT.
NK receptor ligands have been shown to be regulated by cell stress and inflammatory cytokines (23, 24, 30, 31). To determine which stimuli affected Rae-1 and Qa-1b upregulation following RT in our models, we analyzed ligand expression following RT or IFN-γ stimulation. MC38 and B16F10 cells were treated with 15Gy in vitro and H-2Kb, Rae-1, and Qa-1b expression was analyzed by flow cytometry after 48 hours (Fig. 3A,B). RT increased expression of all ligands on MC38 and B16F10 cells compared to untreated controls, indicating that RT-induced cell stress alone can regulate both activating and inhibitory NK receptor ligands. Interestingly, fold increase of H-2Kb expression in B16F10 cells following RT was substantially lower than that observed in MC38 cells, providing a possible explanation for why increased H-2Kb expression was not observed post RT in vivo. In contrast, direct stimulation of MC38 cells with IFN-γ only increased H-2Kb and inhibitory Qa-1b expression, while Rae-1 expression was unaffected (Fig. 3C). Although IFN-γ stimulation resulted in a statistically significant increase in Qa-1b expression, the magnitude of upregulation was small in MC38 cells (Fig. 3D).
Figure 3.
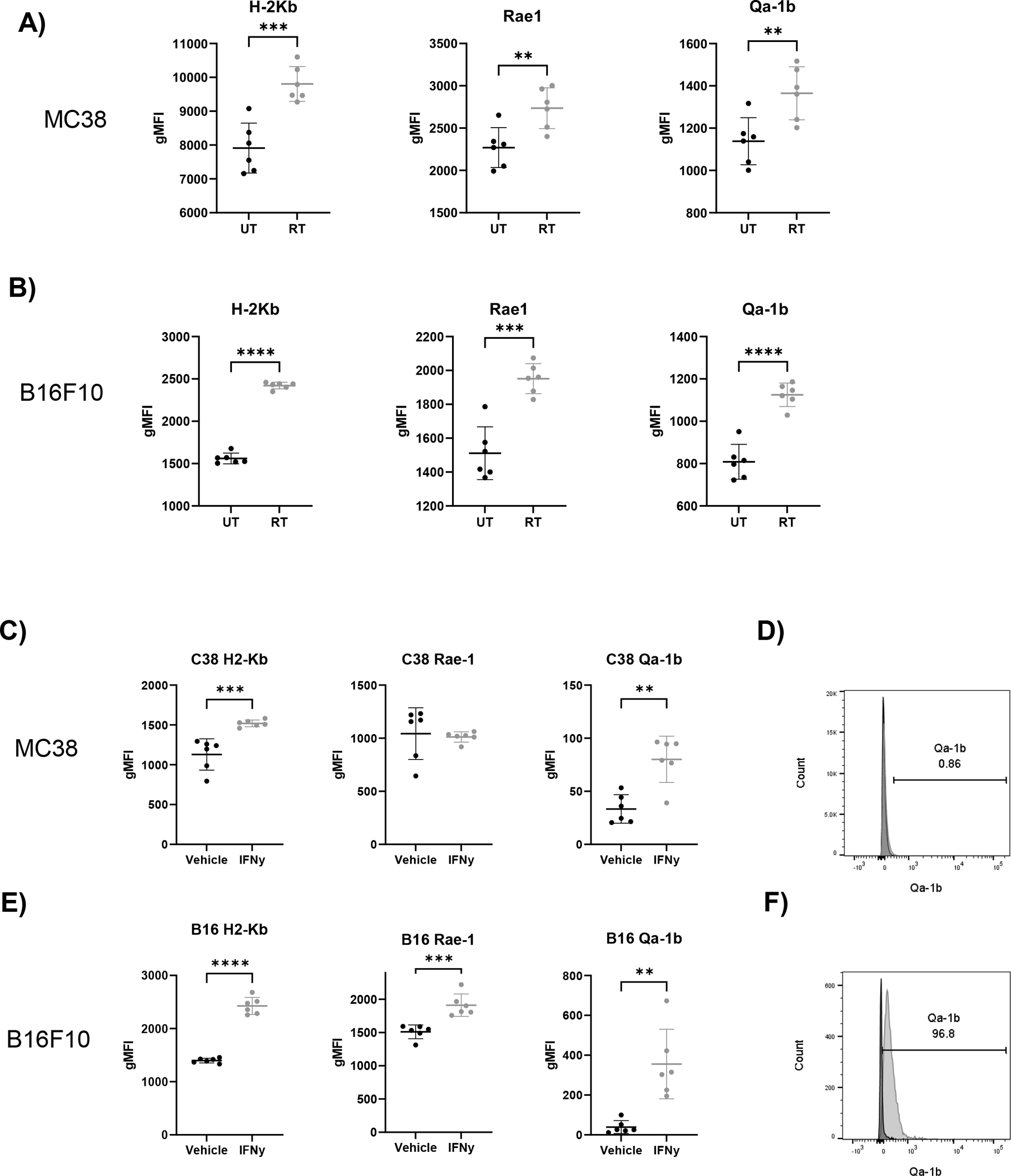
Qa-1b is upregulated by both radiation and IFN-γ whereas Rae-1 is only upregulated by radiation and IFN-γ only in B16F10. MC38 (A) or B16F10 (B) tumor cells left untreated or irradiated with 15 Gy in vitro. After 48 hours, cells were analyzed by flow cytometry for expression of H-2Kb and NKG2 ligands. (C) MC38 tumor cells were incubated with vehicle or 10 ng/mL IFN-γ and then analyzed by flow cytometry for expression of H-2Kb and NKG2 ligands. (D) Representative histogram showing Qa-1b expression on MC38 tumor cells at 48 hours (black = Veh, gray = IFN-γ). (E) B16F10 tumor cells were incubated with vehicle or 10 ng/mL IFN-γ and then analyzed by flow cytometry for expression of H-2Kb and NKG2 ligands. (F) Representative histogram showing Qa-1b expression on B16-F10 tumor cells at 48 hours (black = Veh, gray = IFN-γ). (Data analyzed by two-tailed student’s t-test, normalized gMFI was calculated by subtracting FMO control gMFI from the sample gMFI). (* p<0.05, ** P<0.01, *** p<0.001, **** p<0.0001). (n=6 per group, error bars indicate standard deviation). Data representative of 3 separate experiments.
B16F10 cells responded similarly to MC38 cells as IFN-γ increased H-2Kb and Qa-1b expression, however B16F10 differed from MC38 in that IFN-γ also induced Rae-1 expression mirroring the inductive effect seen in RT (Fig. 3E,F). In summary, IFN-γ selectively induces expression of the inhibitory ligand Qa-1b and the activating ligand Rae-1 in B16F10, while MC38 cells respond by increasing only inhibitory Qa-1b. RT induces activating and inhibitory ligand expression in both B16F10 and MC38.
Blockade of the NKG2A/Qa-1b axis in combination with RT does not improve survival in the B16F10 tumor model
To determine if RT-induced Qa-1b expression suppresses the anti-tumor response elicited by radiation, we combined inhibition of the NKG2A/Qa-1b axis with RT. Due to the more robust upregulation of Qa-1b in the B16F10 model in vitro and in vivo, we focused our initial studies on this model. First, we inoculated wild type (WT) C57BL/6 mice with WT or Qa-1b KO B16F10 cells and treated tumors at day 7 with 15 Gy. B16F10 Qa-1b KO cells failed to upregulate Qa-1b in response to IFN-γ stimulation but retained H-2Kb upregulation after cytokine stimulation in vitro (Supplemental Fig. 3A,B). Qa-1b KO did not affect tumor growth or survival in untreated or irradiated mice (Fig. 4A,B). Other cells in the TME could potentially express Qa-1b, therefore we broadened our inhibition through the use of blocking anti-NKG2A antibodies. NKG2A blocking and control antibodies were administered subcutaneously starting on day 7 prior to irradiation and continuing every 3 days until day 16. Like the Qa-1b tumor cell KO model, combining NKG2A blockade with RT failed to improve response and survival compared to rat IgG controls in both untreated and irradiated mice (Fig. 4C,D). Collectively, these experiments indicate that inhibition of the NKG2A/Qa-1b axis alone does not improve the RT response.
Figure 4.
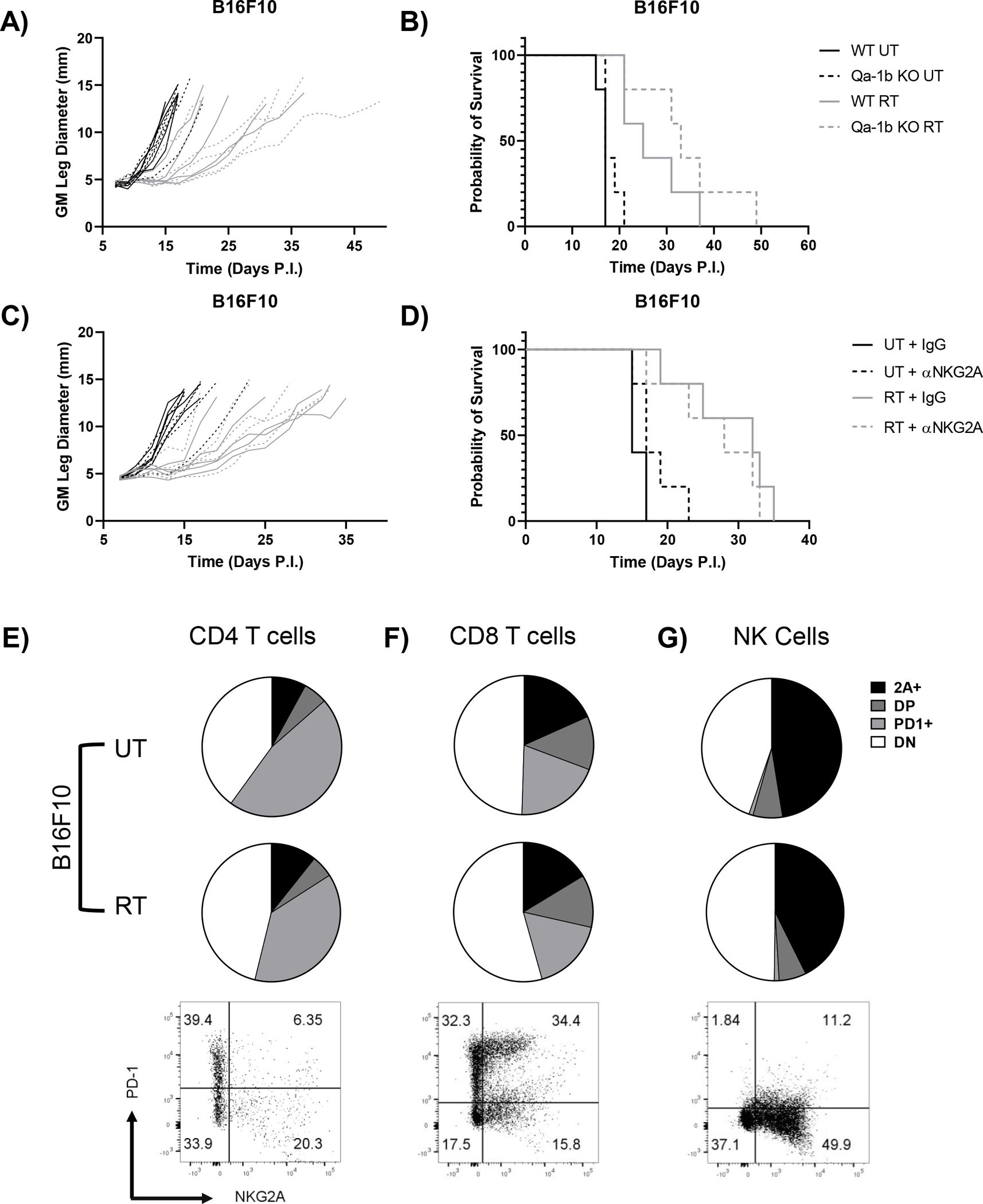
Blockade of NKG2A signaling alone does not improve response to RT in the B16F10 model due to co-expression of NKG2A and PD-1. B16F10 WT and Qa-1b KO tumors were implanted intramuscularly in the leg of 6–8-week-old female C57BL/6 mice, left untreated or treated locally with 15 Gy, and measured every two days until an endpoint of 13 mm was reached to generate growth (A) and survival curves (B). Similarly, B16F10 WT tumors were implanted intramuscularly in the leg of 6–8-week-old female C57BL/6 mice, left untreated or treated locally with 15 Gy, and measured every two days until an endpoint of 13 mm was reached to generate growth and survival curves. On days 7, 10, 13, and 16 untreated and irradiated mice bearing B16F10 tumors were treated with 200 μg of anti-NKG2A antibody (clone 20D5) or rat IgG as a control and monitored for growth (C) and survival (D). CD4 T cells (E), CD8 T cells (F), and NK cells (G) from untreated and irradiated day 9 tumors were analyzed for expression of NKG2A and PD-1 (2A = NKG2A, DP = double positive, DN = double negative). No statistically significant difference in expression between UT and RT groups were observed for these markers in these cell populations. Representative flow cytometry graphs were obtained from an irradiated tumor. (Survival curves analyzed by Log-rank tests, flow cytometry data analyzed by two-tailed student’s t-test). (n=5 mice per group for growth and survival curves, n=3 mice per group for flow cytometry analysis).
These results suggest that NKG2A expression on NK and CD8 T cells alone does not mediate suppression of the immune response after RT, thus we examined intratumoral immune effector cells for expression of the checkpoint inhibitor PD-1. Two days post RT, few CD4 T cells expressed NKG2A (13–20%), but a plurality expressed PD-1 (37–41%) (Fig. 4E). Interestingly, few intratumoral CD8 T cells expressed NKG2A alone (18–20%), but 54–57% expressed PD-1 with about half of those cells co-expressing NKG2A (Fig. 4F). Lastly, approximately half of all NK cells expressed NKG2A, with few expressing PD-1 alone or in combination with NKG2A (Fig. 4G). Similar to NKG2A expression, RT did not alter PD-1 expression in any subset compared to untreated controls (Fig. 4E–F). From these results, we conclude that NKG2A inhibition alone could alleviate suppression in NK cells and a fraction of CD8 T cells following RT, but PD-1 expression on CD4 and CD8 T cells could still limit therapeutic benefit.
Combined NKG2A and PD-1 blockade increases efficacy of RT in the B16F10 tumor model
Phenotyping of immune effector cells in the B16F10 model suggested that combining blockade of NKG2A and PD-1 with radiation could improve response compared to either monotherapy with RT. Thus, NKG2A/PD-1 blocking and control antibodies were administered subcutaneously starting on day 7 prior to irradiation and continuing every 3 days until day 19. Blocking either NKG2A or PD-1 alone failed to significantly improve survival in irradiated mice, but combined NKG2A/PD-1 blockade did improve survival in irradiated mice in an RT dependent manner (Fig. 5A,B). Triple combination resulted in no visible toxicity in mice. Moreover, despite slower weight gain compared to their IgG counterparts, mice in the triple combination group never dropped below their starting weights (supplemental Fig. 3C). Although the MC38 model is more responsive to RT, we applied single NKG2A/PD-1 blockade or combination therapy with RT to determine if we could further improve survival in this model. Both single blockade of NKG2A or PD-1 and combination blockade in conjunction with RT failed to elicit improvement in tumor control and survival (Fig. 5C,D). Furthermore, rechallenge of mice that survived primary MC38 tumor challenge showed no difference between RT treated groups (Supplemental Fig. 3D,E). To determine if low Qa-1b expression in the MC38 model was responsible for lack of therapeutic benefit with triple therapy, we generated MC38 clones transduced with Qa-1b/mGFP fusion protein or control vector (CV) lentivirus (supplementary Fig. 4A,B). The lentiviral vector used only codes for Qa-1b protein and not β2m, which is required for stable surface expression of Qa-1b (29), thus heightened Qa-1b expression was only observed under β2m inducing IFN-γ stimulation (supplementary Fig. 4B). CV and Qa-1b clones with low mGFP expression were utilized for in vivo growth curves to minimize the possibility of response to mGFP as a foreign antigen. Qa-1b overexpression had no effect on survival in untreated or irradiated mice (Supplemental Fig. 4C–E), suggesting low Qa-1b expression on tumor cells alone is not responsible for lack of response to triple therapy in the MC38 model. Overall, these observations indicate that although combined NKG2A/PD-1 blockade has little if any benefit in radioresponsive tumors, this combinatorial approach can significantly improve response to RT in radioresistant tumors.
Figure 5.
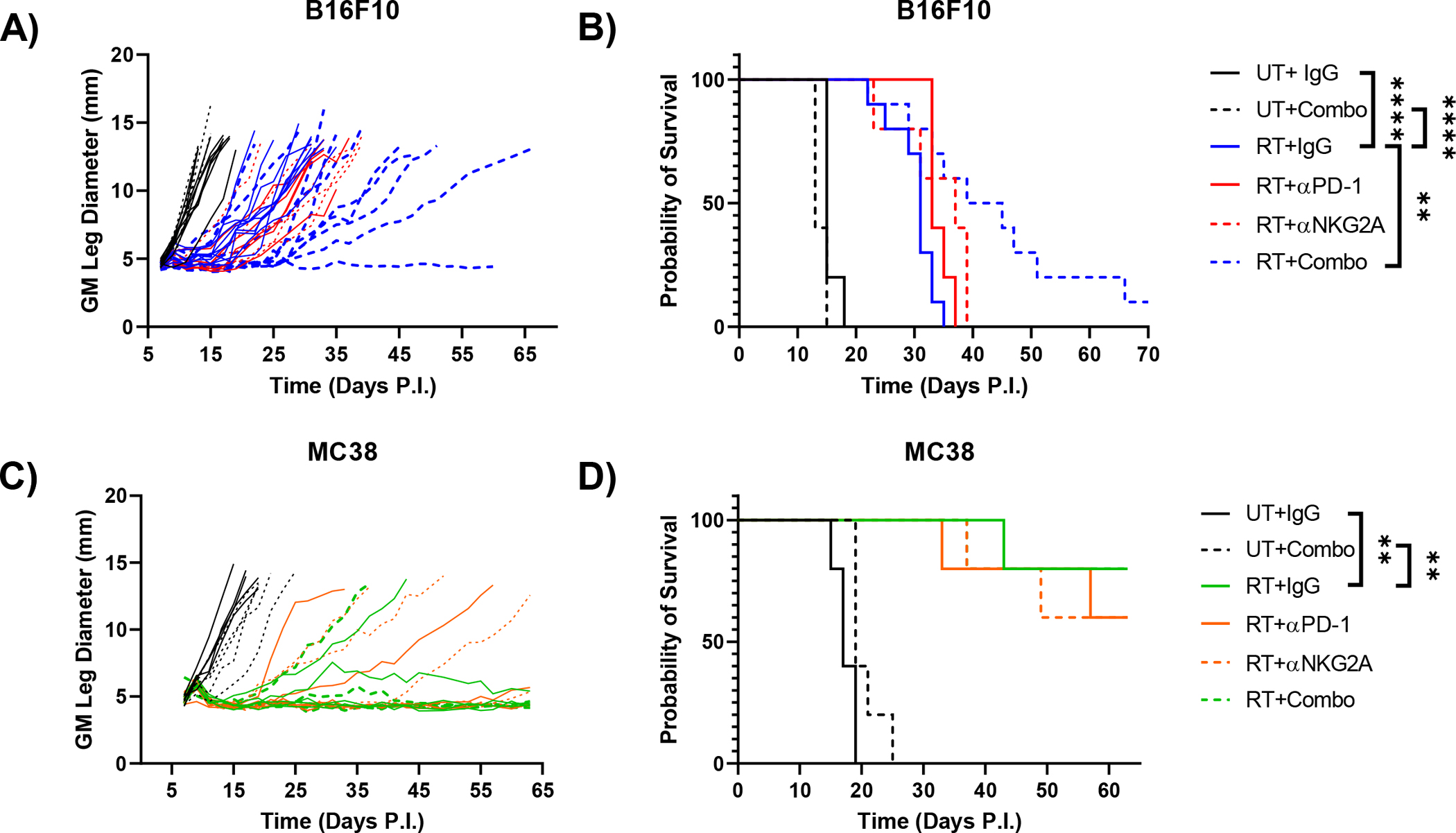
Combined blockade of PD-1 and NKG2A improves RT response only in the B16F10 tumor model. On days 7, 10, 13, 16, and 19 untreated and irradiated mice bearing B16F10 tumors were treated with control rat IgG, 200 μg of anti-NKG2A antibody (clone 20D5) and/or 200 μg of anti-PD-1 antibody (clone RMP1–14) and monitored for growth (A) and survival (B). Untreated and irradiated mice bearing MC38 tumors were treated with NKG2A and/or PD-1 blockade as described above and monitored for growth (C) and survival (D). (Survival curves analyzed by Log-rank tests). (** p<0.01, **** p<0.0001). (n=5–10 mice per group, B16F10 data representative of two individual experiments).
The efficacy of combined RT + NKG2A/PD1 blockade is dependent on CD8 T cells that exhibit enhanced transcriptomic markers of effector function.
Given the increased therapeutic benefit of RT + combination NKG2A/PD-1 blockade in the B16F10 model (Fig. 5) and that both CD8 T cells and NK cells express NKG2A (Fig. 2), we hypothesized that both NKG2A expressing cell types were required for the increased efficacy of triple combination therapy To test this hypothesis, we administered depleting antibodies for both CD8 T cells and NK cells as described in the materials and methods. Mice depleted of NK cells demonstrated similar tumor control and survival as control isotype-treated mice, however CD8 depletion completely abrogated treatment efficacy of triple combination therapy (Fig 6A,B). These results indicate that the efficacy of this therapy is dependent on CD8 T cells with negligible contribution from NK cells.
Figure 6.
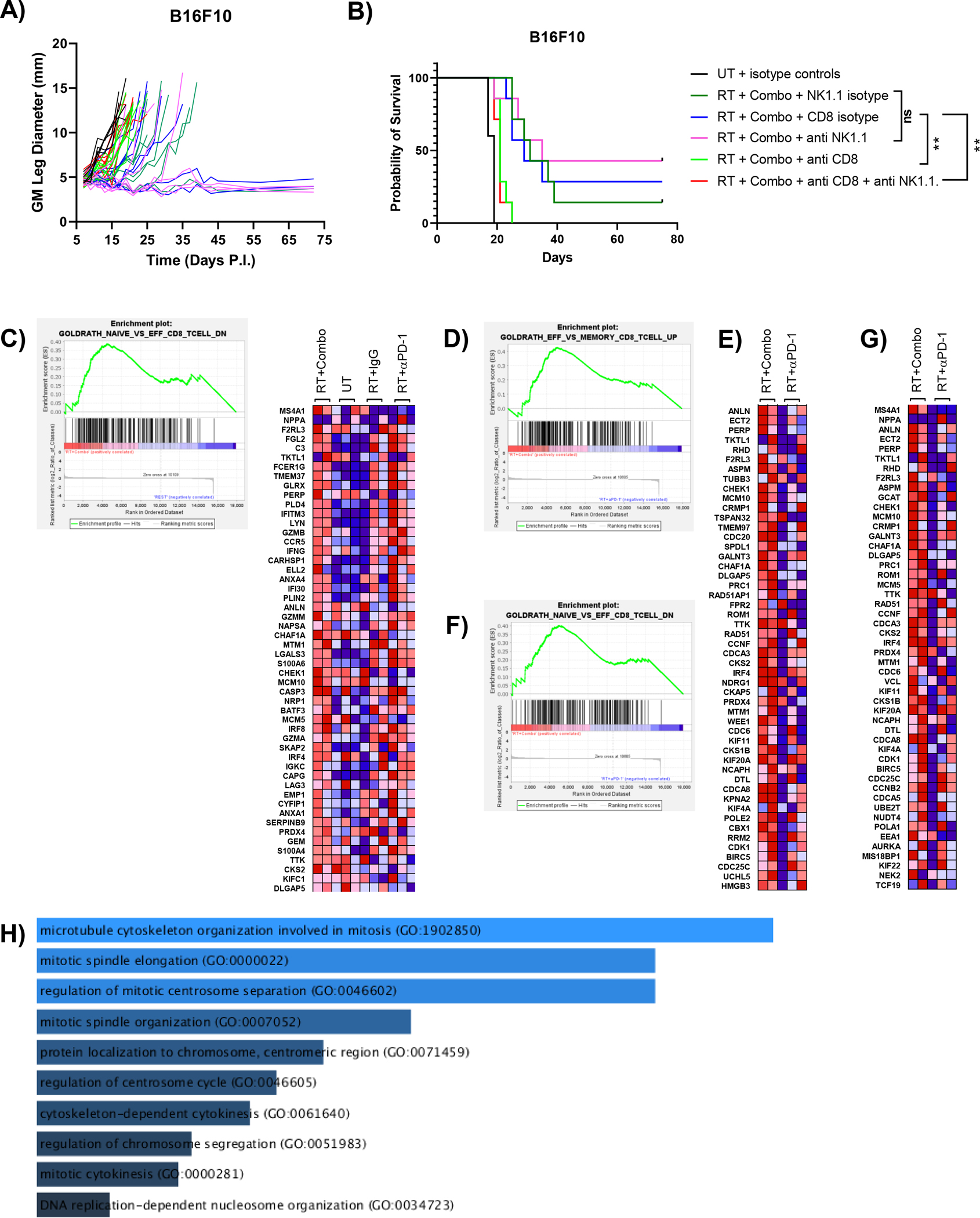
The efficacy of triple therapy is due to enhanced CD8 T cell effector function. Antibody mediated depletion indicates that the efficacy of triple combination therapy is dependent on CD8 T cells. (A) Growth curve illustrating loss of tumor control only in mice treated with αCD8 antibody. Tumor-bearing mice treated with anti-NK1.1 demonstrated similar growth curves to control. Control groups were treated with isotype rat IgG or mouse IgG2a or both. (B) Survival curve of each group showing the reduced efficacy of mice treated with triple combination alongside αCD8 antibody (Survival curves analyzed by log rank test, ** p<0.01). Log transformed normalized counts from RNA sequencing data of sorted CD8 T cells from Supplemental Fig. 4 were used for gene set enrichment analysis using 16 T cell phenotype datasets from MsigDB. Enrichment was considered statistically significant if false discovery rate (FDR) was less than 0.25. Only significantly enriched data sets are shown. (C) Enrichment plot for genes downregulated in naïve versus effector T cells comparing RT+Combo CD8 T cells to T cells from all other groups (FDR = 0.231, nominal p-value = 0.069). (D) Heat map showing the top 50 ranked genes from the comparison in (A) that were enriched in RT+Combo CD8 T cells compared to all other groups. (E) Enrichment plot for genes upregulated in effector versus memory T cells comparing RT+Combo CD8 T cells to RT+αPD-1 CD8 T cells (FDR = 0.112, nominal p-value < 0.01). (F) Heat map showing the top 50 ranked genes from the comparison in (C) that were enriched in RT+Combo CD8 T cells compared to RT+αPD-1 CD8 T cells. (G) Enrichment plot for genes downregulated in naïve versus effector T cells comparing RT+Combo CD8 T cells to RT+αPD-1 CD8 T cells (FDR = 0.147, nominal p-value < 0.01). (H) Heat map showing the top 50 ranked genes from the comparison in (E) that were enriched in RT+Combo CD8 T cells compared to RT+αPD-1 CD8 T cells. (G) Gene ontology analysis of enriched genes that were shared between the data sets used in (C) and (D). Increased length of bars (and increasing brightness of blue) indicates increased significance as determined by a combined ranking metric that considers p-value and z-scores. (n=2–3 mice per group).
To further investigate this essential immune subset, we performed gene set enrichment analysis (GSEA) of CD8 T cells isolated from B16F10 tumors to determine how they differed phenotypically in multiple treatment conditions. Singlet CD8 T cells were sorted from UT, RT+IgG, RT+αPD-1, and RT+Combination treated tumors at day 9 post tumor implantation according to the gating scheme in Supplemental Fig. 1. Sorted CD8 T cells were then used for RNAseq, followed by GSEA using gene sets comparing naïve, effector, memory, and exhausted T cells. GSEA comparing the RT+Combination group to all other groups using 16 CD8 T cell gene sets from MSigDB showed that triple therapy CD8 T cells were enriched in genes commonly associated with enhanced effector function (Fig. 6C). Enriched genes contained effector molecules such as granzyme A, granzyme B, granzyme M, and IFN-γ, as well as transcription factors that support T cell responses, such as IRF4 and IRF8 (interferon regulatory factor 4/8, Fig. 6D). This comparison indicated that CD8 T cells in the triple therapy group had a heightened effector function compared to all other groups combined, but expression of many molecules appeared similarly enriched in T cells from the RT+αPD-1 group. When specifically comparing RT+Combination CD8 T cells to RT+αPD-1 CD8 T cells, two gene sets showed significant enrichment in the former compared to the latter: genes upregulated in effector versus memory CD8 T cells and genes downregulated in naïve versus effector CD8 T cells (Fig. 6C–F). Although some genes associated with effector response, such as IFN-γ and IRF4, were enriched in the combination therapy group, the majority of the 58 enriched genes that are shared between these two data sets function in DNA replication and cell cycling (Fig. 6G). Altogether, GSEA analysis suggests that combination blockade induces more robust effector function than PD-1 blockade alone. Additionally, proliferative function of CD8 T cells, in addition to effector molecule production, are important contributors to the enhanced response observed with triple therapy.
DISCUSSION
Despite the success of PD-1 and CTLA-4 checkpoint blockade therapy, many patients fail to respond to therapy, even when combined with RT. We compared a radioresponsive (MC38) tumor model to a radioresistant (B16F10) model to determine if NKG2 receptors regulate the RT-induced immune response. In our analysis, we observed increased IFN-γ following RT in both models, as well as increased expression of the NKG2D and NKG2A ligands Rae-1 and Qa-1b, respectively, on tumor/stromal cells. Immunostimulatory Rae-1 upregulation was primarily dependent on exposure to radiation in MC38 whereas B16F10 expressed increased Rae-1 when exposed to both RT and IFN-γ. This result is consistent with previous work showing that DNA damage response upregulates Rae-1 (23). Inhibitory Qa-1b was increased by both radiation and IFN-γ, both of which have previously been noted to upregulate Qa-1b or its human homolog HLA-E (19, 30). Additionally, CD8 T cells and NK cells expressed NKG2A and NKG2D receptors in both models. This expression was limited to the TME for CD8 T cells, whereas NK cells expressed receptor regardless of anatomical location, as described by others (19, 32). Furthermore, intratumoral macrophages uniformly expressed NKG2A, confirming previous work by Maeda et al. that showed NKG2A expression on human macrophages (33). Combined, these data suggest that RT has the potential to both positively and negatively regulate the RT-induced anti-tumor immune response.
To determine if NKG2A signaling inhibited RT response, we blocked this pathway using Qa-1b KO B16F10 cells and NKG2A blocking antibodies. Single blockade of NKG2A signaling with either method combined with RT did not improve survival compared to RT alone in both tumor models. Upon further analysis of intratumoral lymphocytes, we observed PD-1 and NKG2A co-expression on CD8 T cells in the B16F10 model. Supporting this data, NKG2A and PD-1 co-expression has previously been observed in both mouse and human intratumoral T cells (19, 32). In our observations, approximately 30% of CD8 T cells in the B16F10 TME were single positive for PD-1 and approximately 15% were single positive for NKG2A. Approximately 30% of CD8 T cells expressed both PD-1 and NKG2A, thus single blockade of only PD-1 or NKG2A alone would only effectively block inhibitory signals in a minority of intratumoral CD8 T cells. By combining RT with NKG2A and PD-1 blockade, inhibitory signals could be decreased for NKG2A single positive, PD-1 single positive, and NKG2A/PD-1 double positive cells, which altogether comprise approximately 80% of intratumoral CD8 T cells. Thus, we treated irradiated mice with combined NKG2A and PD-1 blockade. Triple therapy or dual therapy with RT and either NKG2A or PD-1 blockade alone failed to improve primary and rechallenge response in MC38 tumors. In contrast, triple therapy improved survival in B16F10 mice compared to mice receiving RT alone, whereas either dual therapy combination did not. Importantly, this therapy had negligible toxicity that resolved upon completion of blockade therapy. Additionally, this survival benefit was dependent on the presence of CD8 T cells and not NK cells. RNAseq GSEA of T cells in the B16F10 model revealed that triple combination therapy primarily increased genes associated with cell cycle, as well as some effector molecules, such as IFN-γ, compared to all other groups and specifically RT combined with PD-1 blockade alone suggesting more robust effector function with triple combination therapy. Previous analysis of dual NKG2A and PD-1 blockade has shown increased IFN-γ expression in CD8 T cells compared to either therapy alone (19), although no previous studies have described increased proliferative capacity of CD8 T cells following combined therapy to our knowledge. Altogether, our data suggest that combining NKG2A blockade with existing immunotherapies and RT may improve responses in patients who fail to respond to dual therapy.
Triple combination therapy did not noticeably improve response in the MC38 model, despite increasing survival in the B16F10 model. Our results suggest the increased survival in the B16F10 model is dependent on CD8 T cells and not NK cells. This is in line with studies demonstrating responses to NKG2A blockade to be independent of NK cells (19). Alternative studies have shown an NK dependence for RT responses in B16F10 tumors (34) but this was not the case in our novel combinatorial regimen.
Higher Qa-1b expression in the B16F10 model may also explain enhanced efficacy of triple therapy in this model compared to the MC38 model and therefore act as a predictive marker for therapy response. To test this hypothesis, we generated MC38 clones transduced that overexpress Qa-1b, but did not see any inhibition of the RT response in vivo using these cells. Thus, lower MC38 tumor cell Qa-1b expression alone does not explain lack of response to triple therapy in this model. As our in vivo analysis of Qa-1b expression monitored CD45− cells, which include tumor and stromal cells, it is possible that stromal cells significantly contribute to Qa-1b mediated inhibition of NKG2A-expressing cells in the B16F10 model. Therefore, to observe NKG2A mediated inhibition in the MC38 model Qa-1b would have to be overexpressed on both tumor and stromal cells. Additionally, expression of HLA-E, the human homolog of Qa-1b, did not correlate with response in a clinical trial using monalizumab (anti-NKG2A) as a monotherapy in gynecologic cancers, supporting our observations (35). It is possible that expression of additional markers in conjunction with Qa-1b/HLA-E is necessary to determine if a given tumor will respond to combined NKG2A/PD-1 blockade. For example, in ovarian cancer patients intratumoral HLA-E alone did not act as a prognostic marker, but combined high intratumoral CD8 T cell count and low HLA-E expression did (36). Increased NKG2D ligand expression has been shown to act as a prognostic marker in NSCLC patients (21). We see elevated basal Rae-1 expression in B16F10 tumors compared to MC38 tumors (Fig. 2), thus, using combined NKG2D and NKG2A ligand expression as a predictive indicator may be prudent in the consideration of NKG2A/PD-1 blockade with RT. Further work analyzing the predictive capability of these markers in multiple tumor models as well as human patients is needed. NKG2A blockade could also be combined with antibodies that stabilize NKG2D ligand expression, which have been shown to improve the anti-tumor immune response (22), regardless of the predictive value of these molecules.
Regulatory CD8 T cells that recognize Qa-1b have been described in mouse models (37). As our triple therapy blocks NKG2A receptor rather than its ligand, these regulatory T cells may still exert suppressive function in our models. Qa-1b blockade may remedy this deficiency, but this strategy also eliminates any contribution of tumor specific Qa-1b restricted CD8 T cells (38).
Expression of NKG2A and its ligand Qa-1b/HLA-E has been observed in multiple human and murine cancers (18, 19, 32). Additionally, NKG2A inhibition has been demonstrated to improve the anti-tumor response when combined with existing checkpoint blockade and tumor vaccination strategies (18, 19). In this work, we have demonstrated that NKG2 ligands can be regulated to both benefit and inhibit the anti-tumor immune response induced by RT. Notably, combining NKG2A and PD-1 blockade with RT significantly improves survival in mice compared to either therapy alone with RT in a radioresistant tumor model. Recent clinical studies have indicated that single agent NKG2A blockade with monalizumab in gynecological cancer patients had minimal toxicity but also showed minimal therapeutic benefit (35). Our work provides rationale for combining monalizumab with existing ICB therapies and RT in the clinic.
Supplementary Material
KEY POINTS.
The NKG2A/Qa1b axis inhibits RT induced T cell responses.
Combined RT plus NKG2A/PD1 blockade improved survival in B16F10 tumors.
ACKNOWLEDGEMENTS
We would like to thank Dr. Thorbald van Hall (Leiden University Medical Center, Leiden, The Netherlands) for providing wild type and Qa-1b knockout B16F10 cell lines. We would also like to thank Dr. John Frelinger (University of Rochester Medical Center, Rochester, NY, USA) for recommendations on study design and directions.
This work was supported by the University of Rochester Sproull Fellowship, National Cancer Institute Grants 5R01CA028332 and R01CA230277, and the University of Rochester Immunology training grant (T32AI007285) from the National Institute of Allergy and Infectious Diseases.
Abbreviations used in this article:
- BCA
bicinchoninic acid assay
- BMDM
bone marrow derived macrophages
- CRISPR
clustered regularly interspaced short palindromic repeats
- CV
control vector
- GM
geometric mean
- gMFI
geometric mean fluorescence intensity
- GSEA
gene set enrichment analysis
- Gy
Gray
- ICB
immune checkpoint blockade
- IRF
interferon regulatory factor
- KIR
killer-cell immunoglobulin-like receptor
- KO
knockout
- MsigDB
molecular signatures database
- NKG2
natural killer group 2
- NSCLC
non-small-cell lung carcinoma
- PD-1
programmed cell death protein 1
- PD-L1
programmed cell death ligand 1
- Rae-1
retinoic acid early inducible 1
- RNAseq
RNA sequencing
- RT
radiotherapy
- TME
tumor microenvironment
- URMC
University of Rochester Medical Center
- GRC
genomics research center
- UT
untreated
- WT
wild type
REFERENCES
- 1.Ahmed MM, Hodge JW, Guha C, Bernhard EJ, Vikram B, and Coleman CN. 2013. Harnessing the Potential of Radiation-Induced Immune Modulation for Cancer Therapy. Cancer Immunol Res 1: 280–4. [DOI] [PubMed] [Google Scholar]
- 2.Murray D, McBride WH, and Schwartz JL. 2014. Radiation biology in the context of changing patterns of radiotherapy. Radiat. Res 182: 259–72. [DOI] [PubMed] [Google Scholar]
- 3.Lim JYH, Gerber SA, Murphy SP, and Lord EM. 2014. Type I Interferons Induced by Radiation Therapy Mediate Recruitment and Effector Function of CD8+ T cells. Cancer Immunol. Immunother 63: 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, and Lord EM. 2005. Local Radiation Therapy of B16 Melanoma Tumors Increases the Generation of Tumor Antigen-Specific Effector Cells That Traffic to the Tumor. J. Immunol 174: 7516–7523. [DOI] [PubMed] [Google Scholar]
- 5.Filatenkov A, Baker J, Mueller AMS, Kenkel J, Ahn GO, Dutt S, Zhang N, Kohrt H, Jensen K, Dejbakhsh-Jones S, Shizuru JA, Negrin RN, Engleman EG, and Strober S. 2015. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin. Cancer Res 21: 3727–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerber SA, Sedlacek AL, Cron KR, Murphy SP, Frelinger JG, and Lord EM. 2013. IFN-γ mediates the antitumor effects of radiation therapy in a murine colon tumor. Am. J. Pathol 182: 2345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connolly KA, Belt BA, Figueroa NM, Murthy A, Patel A, Kim M, Lord EM, Linehan DC, and Gerber SA. 2016. Increasing the efficacy of radiotherapy by modulating the CCR2/CCR5 chemokine axes. Oncotarget 7: 86522–86535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Price JG, Idoyaga J, Salmon H, Hogstad B, Bigarella CL, Ghaffari S, Leboeuf M, and Merad M. 2015. CDKN1A regulates Langerhans cell survival and promotes Treg cell generation upon exposure to ionizing irradiation. Nat. Immunol 16: 1060–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, and Gajewski TF. 2013. Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells. Sci. Transl. Med 5: 200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, Gangadhar TC, Amaravadi RK, Schuchter LM, Feldman MD, Ishwaran H, Vonderheide RH, Maity A, Wherry EJ, and Minn AJ. 2016. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167: 1540–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiess AP, Wolchok JD, Barker CA, Postow MA, Tabar V, Huse JT, Chan TA, Yamada Y, and Beal K. 2015. Stereotactic radiosurgery for melanoma brain metastases in patients receiving ipilimumab: Safety profile and efficacy of combined treatment. Int. J. Radiat. Oncol. Biol. Phys 92: 368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luke JJ, Lemons JM, Karrison TG, Pitroda SP, Melotek JM, Zha Y, Al-Hallaq HA, Arina A, Khodarev NN, Janisch L, Chang P, Patel JD, Fleming GF, Moroney J, Sharma MR, White JR, Ratain MJ, Gajewski TF, Weichselbaum RR, and Chmura SJ. 2018. Safety and clinical activity of pembrolizumab and multisite stereotactic body radiotherapy in patients with advanced solid tumors. J. Clin. Oncol 36: 1611–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Herati RS, Mansfield KD, Patsch D, Amaravadi RK, Schuchter LM, Ishwaran H, Mick R, Pryma DA, Xu X, Feldman MD, Gangadhar TC, Hahn SM, Wherry EJ, Vonderheide RH, and Minn AJ. 2015. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520: 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kroon P, Gadiot J, Peeters M, Gasparini A, Deken MA, Yagita H, Verheij M, Borst J, Blank CU, and Verbrugge I. 2016. Concomitant targeting of programmed death-1 (PD-1) and CD137 improves the efficacy of radiotherapy in a mouse model of human BRAFV600-mutant melanoma. Cancer Immunol. Immunother 65: 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho J, Kim H, Webster K, Palendira M, Hahm B, Kim K, King C, Tangye SG, and Sprent J. 2019. Calcineurin-dependent negative regulation of CD94 / NKG2A expression on naive CD8+ T cells. 118: 116–129. [DOI] [PubMed] [Google Scholar]
- 16.Sasawatari S, Okamoto Y, Kumanogoh A, and Toyofuku T. 2020. Blockade of N -Glycosylation Promotes Antitumor Immune Response of T Cells. J. Immunol 204: 1373–1385. [DOI] [PubMed] [Google Scholar]
- 17.Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, and Kershaw MH. 2011. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol 89: 216–224. [DOI] [PubMed] [Google Scholar]
- 18.André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, Bléry M, Bonnafous C, Gauthier L, Morel A, Rossi B, Remark R, Breso V, Bonnet E, Habif G, Guia S, Lalanne AI, Hoffmann C, Lantz O, Fayette J, Boyer-Chammard A, Zerbib R, Dodion P, Ghadially H, Jure-Kunkel M, Morel Y, Herbst R, Narni-Mancinelli E, Cohen RB, and Vivier E. 2018. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 175: 1731–1743.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marijt KA, Santegoets SJ, van Ham VJ, Ehsan I, Charoentong P, André P, Wagtmann N, Welters MJP, Kim YJ, Piersma SJ, van der Burg SH, and van Hall T. 2018. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 175: 1744–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eugène J, Jouand N, Ducoin K, Dansette D, Oger R, Deleine C, Leveque E, Meurette G, Podevin J, Matysiak T, Bennouna J, Bezieau S, Volteau C, Thomas WEA, Chetritt J, Kerdraon O, Fourquier P, Thibaudeau E, Dumont F, Mosnier J-FF, Toquet C, Jarry A, Gervois N, and Bossard C. 2020. The inhibitory receptor CD94/NKG2A on CD8+ tumor-infiltrating lymphocytes in colorectal cancer: a promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Mod. Pathol 33: 468–482. [DOI] [PubMed] [Google Scholar]
- 21.Okita R, Maeda A, Shimizu K, Nojima Y, Saisho S, and Nakata M. 2019. Clinicopathological relevance of tumor expression of NK group 2 member D ligands in resected non-small cell lung cancer. Oncotarget 10: 6805–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrari de Andrade L, Kumar S, Luoma AM, Ito Y, Alves da Silva PH, Pan D, Pyrdol JW, Yoon CH, and Wucherpfennig KW. 2020. Inhibition of MICA and MICB Shedding Elicits NK-Cell-Mediated Immunity against Tumors Resistant to Cytotoxic T Cells. Cancer Immunol. Res 8: 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raulet David H., Gassr Stephan, Gown Benjamin G., Deng Weiwen, and H. J. 2013. Regulation of Ligands for the NKG2D activating receptor,. Annu Rev Immunol. 31: 413–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki T, Kanaseki T, Shionoya Y, Tokita S, Miyamoto S, Saka E, Kochin V, Takasawa A, Hirohashi Y, Tamura Y, Miyazaki A, Torigoe T, Hiratsuka H, and Sato N. 2016. Microenvironmental stresses induce HLA-E/Qa-1 surface expression and thereby reduce CD8+ T-cell recognition of stressed cells. Eur. J. Immunol 46: 929–940. [DOI] [PubMed] [Google Scholar]
- 25.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, and Ma’ayan A. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44: W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A. 2013. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lugade AA, Sorensen EW, Gerber SA, Moran JP, Frelinger JG, and Lord EM. 2008. Radiation-Induced IFN-g Production within the Tumor Microenvironment Influences Antitumor Immunity. J. Immunol 180: 3132–3139. [DOI] [PubMed] [Google Scholar]
- 28.Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, Schwendener R, Pruschy M, Knuth A, and van den Broek M. 2012. Radiotherapy Promotes Tumor-Specific Effector CD8 + T Cells via Dendritic Cell Activation. J. Immunol 189: 558–566. [DOI] [PubMed] [Google Scholar]
- 29.Kraft JR, Vance RE, Pohl J, Martin AM, Raulet DH, and Jensen PE. 2000. Analysis of Qa-1(b) peptide binding specificity and the capacity of CD94/NKG2A to discriminate between Qa-1-peptide complexes. J. Exp. Med 192: 613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riederer I, Sievert W, Eissner G, Molls M, and Multhoff G. 2010. Irradiation-Induced Up-Regulation of HLA-E on Macrovascular Endothelial Cells Confers Protection against Killing by Activated Natural Killer Cells. PLoS One 5: e15339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, Von Laer D, Kalinke U, Vivier E, Jonjic S, and Oxenius A. 2014. Type I Interferons Protect T Cells against NK Cell Attack Mediated by the Activating Receptor NCR1. Immunity 40: 961–973. [DOI] [PubMed] [Google Scholar]
- 32.Abd Hamid M, Wang R-ZZ, Yao X, Fan P, Li X, Chang X-MM, Feng Y, Jones S, Maldonado-Perez D, Waugh C, Verrill C, Simmons A, Cerundolo V, McMichael A, Conlon C, Wang X, Peng Y, Dong T, Hamid MA, Wang R-ZZ, Yao X, Fan P, Li X, Chang X-MM, Feng Y, Jones S, Maldonado-Perez D, Waugh C, Verrill C, Simmons A, Cerundolo V, McMichael A, Conlon C, Wang X, Peng Y, and Dong T. 2019. Enriched HLA-E and CD94/NKG2A Interaction Limits Antitumor CD8 + Tumor-Infiltrating T Lymphocyte Responses. Cancer Immunol. Res 7: 1293–1306. [DOI] [PubMed] [Google Scholar]
- 33.Maeda A, Kawamura T, Ueno T, Usui N, Eguchi H, and Miyagawa S. 2013. The suppression of inflammatory macrophage-mediated cytotoxicity and proinflammatory cytokine production by transgenic expression of HLA-E. Transpl. Immunol 29: 76–81. [DOI] [PubMed] [Google Scholar]
- 34.Finkel P, Frey B, Mayer F, Bösl K, Werthmöller N, Mackensen A, Gaipl US, and Ullrich E. 2016. The dual role of NK cells in antitumor reactions triggered by ionizing radiation in combination with hyperthermia. Oncoimmunology 5: e1101206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tinker AV, Hirte HWHWHWHW, Provencher DM, Butler MO, Ritter H, Tu D, Azim HA, Paralejas P, Grenier N, Hahn S-AA, Ramsahai J, and Seymour L. 2019. Dose-ranging and cohort-expansion study of monalizumab (IPH2201) in patients with advanced gynecologic malignancies: A trial of the Canadian Cancer Trials Group (CCTG): IND221. Clin. Cancer Res 25: 6052–6060. [DOI] [PubMed] [Google Scholar]
- 36.Gooden M, Lampen M, Jordanova ES, Leffers N, Trimbos JB, Van Der Burg SH, Nijman H, and Van Hall T. 2011. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8 + T lymphocytes. Proc. Natl. Acad. Sci. U. S. A 108: 10656–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi JY, Eskandari SK, Cai S, Sulkaj I, Assaker JP, Allos H, AlHaddad J, Muhsin SA, Alhussain E, Mansouri A, Yeung MY, Seelen MAJ, Kim HJ, Cantor H, and Azzi JR. 2020. Regulatory CD8 T cells that recognize Qa-1 expressed by CD4 T-helper cells inhibit rejection of heart allografts. Proc. Natl. Acad. Sci. U. S. A 117: 6042–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oliveira CC, Van Veelen PA, Querido B, De Ru A, Sluijter M, Laban S, Van Der Burg SH, Offringa R, and Van Hall T. 2010. The nonpolymorphic MHC Qa-1b mediates CD8+ T cell surveillance of antigen-processing defects. J. Exp. Med 207: 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.