

Abstract
Despite potent suppression of HIV-1 viral replication in the CNS by antiretroviral therapy (ART), between 15 and 60% of HIV-1-infected patients receiving ART exhibit neuroinflammation and symptoms of HIV-1-associated neurocognitive disorder (HAND) – a significant unmet challenge. We propose that the emergence of HIV-1 from latency in microglia underlies both neuroinflammation in the CNS and the progression of HAND. Recent molecular studies of cellular silencing mechanisms of HIV-1 in microglia show that HIV-1 latency can be reversed both by pro-inflammatory cytokines and by signals from damaged neurons, potentially creating intermittent cycles of HIV-1 reactivation and silencing in the brain. We posit that anti-inflammatory agents that also block HIV-1 reactivation, such as nuclear receptor agonists, might provide new putative therapeutic avenues for the treatment of HAND.
Keywords: HIV-1-associated neurocognitive disorder (HAND), HIV-1 latency, Microglial cell activation, antiretroviral therapy (ART), anti-inflammatory strategies
The HAND Controversy
Despite the widespread use of antiretroviral therapy (ART), HIV-1-associated neurocognitive disorder (HAND) remains a common cause of cognitive impairment and persists in 15–60% of HIV-1+ individuals [1,2]. An intense debate has arisen about whether HAND results directly from the virus, indirectly from systemic- and neuro-inflammation, or a combination of these and additional mechanisms. One reason why this question is so difficult to resolve is that studies on patients are inherently limited in their ability to monitor virus in the brain. Recently, our understanding of the mechanisms underlying HIV-1 replication in the brain is being driven by the development of both new primary cell models [3] and improved small animal models [4] (Box 1). The new models have greatly improved our molecular understanding of how HIV-1 replicates in microglial cells, and how interactions between microglial and neurons regulates HIV-1 replication. Reasoning from the “bottom up”, we propose that intermittent emergence of HIV-1 from latency may contribute to the development of HAND.
Box 1: New experimental tools to study HAND.
Until recently, there were no adequate cellular models for HIV-1 infection in microglia. Immortalized primary microglia have been instrumental for studying HIV-1 latency and reactivation, but they show proliferative growth in culture [32]. A significant advance is the use of induced pluripotent stem cell (iPSC)-derived microglia (iMG) to study responses to antiretroviral drugs [55], HIV-1 replication and pathogenesis, and microglial activation [34,56]. The culmination of this approach, which may allow the study of microglia in the context of neurons and astrocytes, is the use of three dimensional (3D) cerebral organoids generated from iPSC-derived neural progenitor cells (NPCs) combined with iMG cells [57].
The most commonly used non-human primate model is that of rhesus macaques infected by simian immunodeficient viruses (SIV) [58]. The SIVsm804E strain, which is both T-cell and macrophage tropic, has been shown to induce neuro-AIDS without the need of CD8+ T depletion, and therefore, is considered to be a more reliable model for NeuroHIV pathology [59] than SIV strains such as SIVmac251, which is used primarily to study HIV-1 acute infection in the brain [60]. In general, the CD8+ T cell depletion models show more aggressive neuropathogenesis in SIV infections than in PLWH (with milder forms of HAND, without neuronal loss and only synaptodendritic simplification) [61].
Exciting new developments in small animal models to study HIV-1 infection of microglia in the brain provide complementary experimental approaches. EcoHIV-1 infected mice [62] and rats [63] use a chimeric HIV-1, in which the gp120 in HIV-1 is replaced with gp80 from an amphotropic rodent retrovirus. These animals exhibit HIV-1 mRNA in microglia, synaptic dysfunction, alterations in temporal processing, and altered neurobehavioral mechanisms [64]. Humanized mouse models, created by transplanting human hematopoietic stem cells into immune-deficient mice, allow for reconstitution of the brain with human microglia, and are the only animal models that allow a detailed study of HIV latent reservoirs and their impact on neuroinflammation [65,66]. Genetically engineered versions of NSG mice, such as, NOG-hIL-34 mice, which carry a knock-in gene for hIL-34 [66], and MISTRG-6-15 mice, which carry knock-in genes for 7 human cytokines supporting hematopoiesis [67,68] enhance the numbers of human microglia-like cells in the transplanted animals compared with the original NSG mice. These newer animal models, especially when analyzed with single cell RNA-Seq (scRNA-Seq), hold great promise for transforming NeuroHIV research and supporting pre-clinical drug development.
ART fails to eliminate HAND
Untreated HIV-1 infection of the brain carries a high risk of brain damage, HIV-1-associated encephalitis, dementia (HAD), and eventual death [5]. Longitudinal studies of primary HIV-1 infection of ART-naïve subjects has shown that there is a progressive increase in soluble and cellular markers of inflammation in cerebrospinal fluid (CSF), suggesting an accrual of intrathecal inflammation [6]. Unfortunately, although ART dramatically lowers the amounts of HIV-1 RNA in the central nervous system (CNS) [7,8], ART does not eliminate neuroinflammation and HAND [9].
Studies of HAND have benefited greatly from the establishment of large, extensively characterized, cohorts designed to study NeuroHIV outcomes, such as the CNS HIV-1 Antiretroviral Therapy Effects Research (CHARTER) Study [10,11]. Neurocognitive symptoms seen in well-suppressed people living with HIV-1 (PLWH), range from asymptomatic neurocognitive impairment (ANI) to mild neurocognitive disorder (MND). Epidemiological studies based on neuropsychological testing have shown that there is a change from the prominent slowed motor and speed of processing deficits during pre-ART, to less pronounced learning, memory, and executive functioning impairments during ART [1,12]. With the widespread availability of ART, the prevalence of HAND has dramatically declined to 2 to 8%, but the combined frequencies of MND and ANI have actually increased (to up to 60%) [12]. Unfortunately, even these broad conclusions are not without controversy, since variable results are obtained depending on the methods used to assess cognitive performance [13]. Additionally, many confounding factors in HIV-1-infected patients such as sex, age, comorbidities (including atherosclerotic vascular disease), illicit substance use, and inflammation associated with co-infection by other pathogens, have complicated attempts to correlate ongoing HIV-1 replication in the CNS and HAND in ART-treated patients [14]. Nonetheless, it is clear that early recognition of cognitive impairment, and the development of effective treatments, will be vital to reducing HAND.
The HAND Paradox: Is HIV-1 hiding in the CNS despite ART?
HIV-1 invades the brain during the first two weeks after primary infection [15] and induces high local concentrations of inflammatory cytokines [16] (Figure 1). The early entry of HIV-1 through the blood-brain barrier (BBB) most likely occurs by a Trojan horse mechanism in which HIV-1-infected monocytes, perivascular macrophages, and T-cells, directly cross the BBB and release viral particles into the brain parenchyma [17]. In Tat transgenic mice, HIV-1 Tat exposure is sufficient to destabilize BBB integrity and increase the presence of reactive, phagocytic, perivascular macrophages and microglia in the brain [18]. HIV-1 transcytosis (via the gp120 envelope protein or mannose-6-phosphate receptor in vitro) has also been proposed as a potential additional mechanism permitting HIV-1 entry into the CNS, although this remains to be further investigated [19].
Figure 1. Schematic illustration of the interactions between neurons, astrocytes, and microglia in heathy vs. HAND human brains.
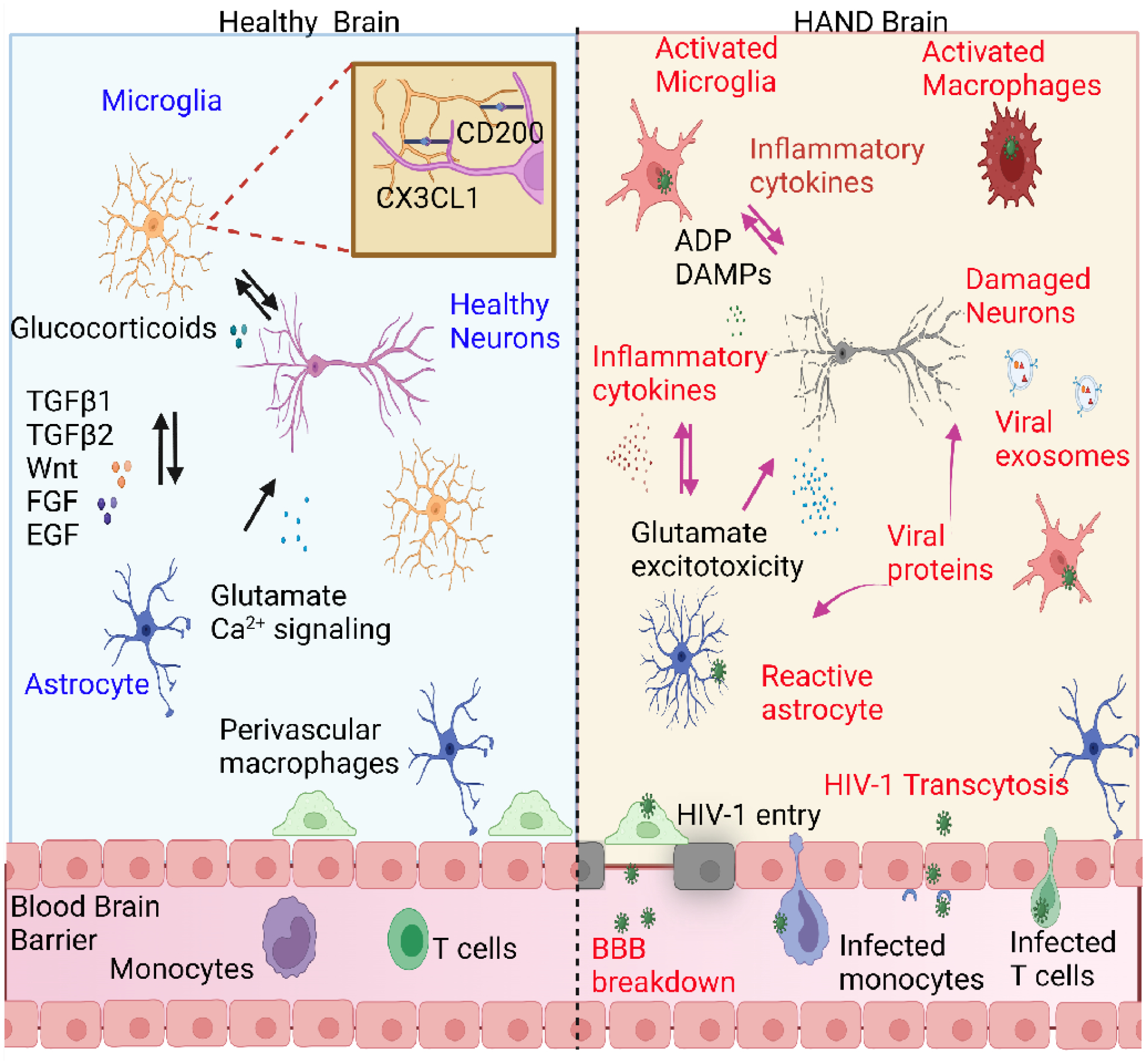
In a healthy brain, neurons and astrocytes induce microglia to attain a surveillance and neuroprotective phenotype by signaling though factors such as fractalkine CX3CL1, CD200, TGFβ1/2, and Wnt [77]. During HIV-1 infections, the “high-jacked” infected monocytes and CD4+ T cells allow viral entry into the brain. Viral particles can also enter the brain by damaging the blood-brain barrier (BBB), altering endothelial cell permeability, or through transcytosis [18]. HIV-1 can establish latent reservoirs in microglia. Reactivation of latent HIV-1 causes a microglial shift to a reactive state, resulting in the release of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, IL-8, CCL2, and CCL5, reactive oxygen species (ROS), and reactive nitrogen species (RNS) [34]. Microglia activation can also cause astrocytes to shift to a reactive state, with secretion of more inflammatory cytokines [72]. Viral proteins such as gp120, Nef, Tat can directly cause neuronal damage and disrupt astrocyte glutamate transporters, resulting in glutamate imbalance in the brain and excitotoxicity of neurons [18,83]. Neural damage-associated molecular pattern (DAMP) molecules and ADP can further cause microglia hyperactivation and HIV-1 reactivation, thus initiating a cycle of progressive neural inflammation. This figure was created using BioRender.com.
Once in the brain, the virus primarily infects perivascular macrophages and microglial cells, but not neurons [20]. Since microglial cells are longer-lived than perivascular macrophages and astrocytes and may support productive HIV-1 replication, they may constitute a main persistent HIV-1 cellular reservoir in the CNS [21]. Although astrocytes can be infected by HIV-1 ex vivo, and in certain humanized mouse models, they do not sustain spreading infections and their role in establishing a viral reservoir and promoting HAND remains uncertain (Box 2).
Box 2. Astrocyte contributions to HAND.
Astrocytes are the most abundant glial cell type in the human brain and are required to maintain normal neuronal excitations, synaptic plasticity, and neurotransmitter glutamate balance [69]. Although HIV-1 DNA has been detected in a small fraction of human astrocytes, their role as a CNS reservoir is uncertain. The strongest evidence that astrocytes can sustain HIV-1 infections comes from work using a novel humanized mouse model (huAstro/HuPBMCs), which in addition to human stem cells, includes transplanted HIV-1-infected primary human fetal astrocytes (NHAs) [70]. Even under ART, astrocytes can support HIV-1 infection in vivo and exit into peripheral organs, at least partially through the trafficking of infected CD4+ T cells from the brain [70]. But because this represents an artificial model, it is unclear if astrocytes can establish a bona fide latent reservoir. Furthermore, astrocytes lack all HIV-1 receptors (CD4 and CXCR4 or CCR5); therefore, they cannot establish disseminating HIV-1 infections, and infections may only occur via non-specific cell-cell contact mechanisms as evidenced from in vitro experiments using cell lines [71].
Nonetheless, astrocytes probably make important contributions to HAND pathology through indirect mechanisms. Astrocytes become activated during HIV-1 infections, and have disrupted β-catenin signaling, leading to neuronal injury [72]. As seen in the brains of HAND patients and transgenic mice, activated astrocytes can induce migrating monocytes to release inflammatory cytokines that can increase astrocyte-elevated gene-1 (AEG) expression, leading to glutamate imbalance and neuronal injury [35]. Viral proteins can also activate astrocytes directly; Tat can induce the production of chemokines such as CCL2, platelet derived growth factors, and upregulate MMP-9 via the activation of NF-κB in astrocytes in vitro [73]. Similarly, viral gp120 can alter the glutamate transporter in human astrocytes, causing glutamate imbalance, which in turn, caused toxic increases in glutamate signaling in neurons [74]. Thus, it is reasonable to speculate that the crosstalk between microglia and astrocytes as well as the presence of HIV-1 proteins might enhance neuroinflammation and neurodegeneration even if the astrocytes themselves are not infected.
Unfortunately, since access to brain tissue in living patients is impractical, studies of viral replication are mostly limited to measurements of HIV-1 RNA and protein in the CSF that provide only rough estimates of viral titers in the brain [11]. Nonetheless, compartmentalized viral replication has been found in PLWH on ART [22], leading to the development of genetically distinct viruses that are adapted to grow efficiently in microglia and other myeloid cells [23,24].
Virus may persist in the brain, in part, because ART may be less effective in the CNS than in the periphery. Data from patients, humanized mice, and simian immunodeficiency virus (SIV)-infected macaques indicate that the concentrations of ART drugs can be 10 to 100-fold lower in brain tissues compared with plasma and liver concentrations [25]. Furthermore, intracellular ART drug concentrations in microglia are significantly lower than in lymphocytes [26]. Therefore, the presence of HIV-1 in the CNS impacting HAND remains to be fully resolved.
Multiple origins of neuroinflammation during HAND
Despite strong evidence for virus persistence in the CNS, there are also robust findings to support non-viral mechanisms contributing to HAND. Original studies suggested that HAND might correlate more strongly with systemic inflammation and CNS inflammation than with the expression of HIV-1 RNA or viral antigens in the CNS, implying that systemic inflammation, rather than viral persistence in the brain, might be one of the primary drivers of HAND [11,27,28].
In addition, HIV-1-induced neuroinflammation might also contribute to the development of HAND [29]. One compelling direct measure of neuroinflammation in HAND was the observation that antibody titers against myelin oligodendrocyte glycoprotein (MOG) in the CSF were significantly higher in patients with active HAND than in asymptomatic HIV-1-infected patients [29]. Elevated concentrations of neurotoxins were also found in the brains of HIV-1-infected patients [30].
Although the clinical information is incomplete, it is generally accepted that animal models for NeuroHIV have now established a relationship between HIV-1 proteins, microglial activation, and neuronal death. Moreover, exaggerated amounts of neurotoxins produced by reactive microglia can lead to neuronal damage in many viral infections [31]. Importantly, neurotoxin production is strongly and directly enhanced in microglia infected by HIV-1 ex vivo [32–34]. In addition, preclinical findings suggest that HIV-1 proteins including Tat, negative regulatory factor (Nef), envelope glycoprotein gp120 are directly neurotoxic, and can also contribute to CNS inflammation via overactivation of microglia [35].
In a new approach to measure the relationship between HIV-1 replication, inflammation, and neurodegeneration, correlations were established between host gene expression patterns and brain HIV-1 RNA in transcriptional profiles derived from postmortem samples obtained from the National NeuroHIV Tissue Consortium (NNTC) [36]. Viral load directly correlated with increased expression of cytokine and IFN signaling pathways, but was inversely correlated with gene pathways that were relevant to oxidative phosphorylation, electron transfer, and the tricarboxylic acid cycle, suggesting that mitochondrial dysfunction might have also been linked to productive HIV-1 infection [36]. Similarly, histological studies of brain sections of PLWH revealed the elevated expression and functional activation of p53 in microglia, whereas deletion or functional suppression of p53 appeared to suggest reduced inflammation relative to controls, based on transcriptional profiles [37].
Finally, accumulating clinical and ex vivo evidence suggests that at least some anti-retroviral compounds and regimens may also have neurotoxic effects – possibly contributing to the incidence of HAND [38,39]. Therefore, when comparing clinical HAND studies, it is important to consider that ART has the potential for inducing oxidative stress, mitochondrial inhibition, and neuronal damage, possibly masking the benefits of ART-driven reduced HIV-1 gene expression [38,39].
Thus, a combination of factors that include the degree of systemic inflammation, ART toxicities, genetic activation signatures of microglial cells, and abnormal redox environments, might be associated with overall functionality and cognitive impairment in PLWH [40].
HIV-1 latency in microglia and its potential inflammation-mediated reversal
To reconcile the contradictory observations that virus persistence, neuroinflammation, and systemic inflammation are all associated with HAND but do not correlate well with each other, we propose that the central molecular mechanism underlying HAND may be the emergence of HIV-1 from proviral latency due to inflammation and microglial dysregulation. This type of intermittent viral growth at the cellular level can be considered to be a specialized form of arising viral persistence since none of the existing ART drugs target proviral transcription or prevent reactivation of the latent HIV-1 provirus [36].
Central to our hypothesis that neurological complications may result from the periodic emergence of HIV-1 from latency within microglial cells, is the recognition that that the same spectra of inflammatory cytokines (including TNF-α and IL-1β) associated with HAND, can activate microglia and contribute to reactivating dormant proviruses [33,34] (Key Figure, Figure 2). Chronic neuroinflammation and neurodegeneration lead to the abnormal activation of microglial cells and constitute a hallmark not only of HAND, but also of a diverse set of neurological diseases [41] and viral infections [42]. Moreover, activation of microglia occurs in response to both neuronal damage and inflammatory stimuli (Box 3). To prevent a normal pro-inflammatory reaction from causing neuronal damage, healthy neurons can provide signals to microglia via secreted and membrane-bound factors such as CX3CL1, CD200 and neurotransmitters to contribute in restoring microglial homeostasis [44].
Key Figure, Figure 2. Model for the regulation of HIV-1 transcription by inflammation and neuronal interactions.

HIV-1 can infect homeostatic (“ramified”) microglia. However, HIV-1 transcription might become quickly silenced by the recruitment of Nurr1 and the CoREST repressor complex to the long-term repeat regions, based on preliminary findings [48]. In the presence of “Off signals” produced by healthy neurons, HIV-1 infected cells are further suppressed by glucocorticoids (GC) [46] or other anti-inflammatory cytokines such as IL-4, IL-10, and TGF-β, produced by neurons. Latently infected cells can become fully reactive in the presence of exogenous pro-inflammatory activators, including LPS, GM-CSF, TNF-α and IFN-γ [33]. This leads to the activation of NF-κB, which can bind the LTR and activate HIV-1 transcription, releasing viral proteins such as Tat, Env, and Nef -- known neurotoxins. In addition to stimulating HIV-1 expression, NF-κB can activate a variety of cellular inflammatory markers and release additional neurotoxic cytokines including TNF-α, IL-β, and IL-6. This may lead to neuronal damage, further activating infected microglia due to the release of DAMPs [34]. These mechanisms may parallel the sensing behavior of uninfected microglia during a wide variety of neurodegenerative diseases, but microglial activation may become highly exacerbated when HIV-1 is present [40,61,81].
Box 3. Microglial heterogeneity, plasticity, and activation.
Microglia are a heterogenous “residential macrophage-like” population of cells found in the CNS. They not only constitute the first barrier of the innate immune response in the mammalian brain, but are also crucial players in CNS development, homeostasis, immunity, and repair [75]. Microglia are highly plastic cells, displaying a continuum of functional phenotypes ranging from an anti-inflammatory, neuroprotective ramified cell state (performing surveillance and synaptic pruning), to amoeboid forms that are highly pro-inflammatory, antigen-presenting, and neurotoxic [76].
Neurons and astrocytes actively control microglial function and modify CNS inflammatory responses through the release of specific signals, which can be broadly classified as neuron “off” and “on” signals that keep microglia either at a controlled quiescent state or in a reactive phagocytic state [77,78]. Damaged neurons can release purines in the forms of ATP and ADP molecules that could activate microglia purinergic receptors, such as P2RY12, causing microglia to assume an amoeboid state enabling them to phagocytose and pinocytose damaged cells, as well as release pro-inflammatory factors and chemokines to aid in phagocytosis [79]. Therefore, infection and/or neuronal damage in the CNS often results in microglial activation and inflammation.
Recent advances in scRNA-Seq have more precisely defined microglia subpopulations in healthy and neurodegenerative disorders [80,81]. For example, one study reported upregulation of microglia homeostatic genes, and downregulation of interferon genes and TNF-α-responsive genes in microglia-neuron-astrocyte co-cultures, compared to microglial monocultures, showing that healthy astrocytes and neurons could repress microglia inflammatory signatures through TGFβ2 signaling pathways [82], Different microglia phenotypes are also associated with specific transcription factors [37]. Foxp3, Stat6, and Nrf2 signaling in microglia have been reported to direct these cells towards neuroprotective phenotypes via expressional induction of CD200/CD200R and CX3CL1/CX3CR1 whereas, signaling via Msx3, Jmjd3, and nuclear receptors such as Nurr1, estrogen receptors, PPARs, LXRs and RXRs, can result in a neuroprotective and anti-inflammatory phenotype. In contrast, AP1 (FosB), NF-κB (RelA/NF-κB1), and Stat1 signaling yields microglia that contribute to chronic neurodegeneration by inducing the expression of many neurotoxic molecules such as NO, ROS, IL1β, TNF-α, and IL-6 [37]. Consistent with these mechanisms, scRNA-Seq on HIV-1-infected and ART-treated iMG in an iPSC-derived neuron-microglia astrocyte tri-culture model system demonstrated the upregulation of pro-inflammatory pathways associated with IL-8, IL-1β, and TNF-α production during HIV-1 infections [55]. Similarly, studies from our laboratory have shown that HIV-1 transcription can be induced by IL-1β, and TNF-α – the same cytokines that can promote a microglia shift towards a pro-inflammatory state [32,81].
The most direct evidence for HIV-1 latency in patients with cognitive deficits, neurodegenerative alterations, and neuroinflammatory changes, comes from a case-control comparison of 32 HIV-1 seropositive (HIV+) patients [7]. On autopsy, in 10 of these patients, high amounts of viral DNA persisted but there were no detectable or significant amounts of RNA or p24, respectively, suggesting that the majority of HIV-1- infected cells were latently infected [7]. A formal demonstration of latency would require viral reactivation, which is not possible with autopsy samples. Nevertheless, consistent with theidea of CNS HIV-1 latency, latently-infected microglial cells have been isolated from the brains of ART-treated, SIV-infected macaques [43].
Unlike Herpes viruses, HIV-1 does not encode factors that regulate latency. Instead, the establishment of HIV-1 latency and the control of its emergence are inextricably linked to the activation status of the host microglial cell (Figure 2, Key Figure). At a molecular level, there are instructive parallels between HIV-1 transcriptional silencing in CD4+ T-cells [44], and microglial cells [21,45]. When CD4+ T-cells transition into a memory phenotype, they enter a quiescent state that blocks both the initiation of HIV-1 transcription and its elongation [44]. Similarly, HIV-1 latency in microglia is dependent on the cellular activation state [21,45].
HIV-1 readily establishes latency in induced pluripotent stem cell-derived microglial cells (iMG) and immortalized human microglial cells infected ex vivo [32–34,46]. Analogously to CD4+ T-cells, the crucial HIV-1 transcription initiation factor NF-κB is sequestered in the cytoplasm in latently infected microglia, suggesting that the cells reside in their quiescent state, with low or selective cytokine synthesis, and controlled phagocytosis (Key Figure, Figure 2) [32]. Latency reversal in immortalized human microglia by pro-inflammatory cytokines has resulted in the activation and translocation of NF-κB p65/p50 or IRF3 to the nucleus and binding to the HIV-1 promoter regions in the 5’ long terminal repeat (LTR) regions [33]. Furthermore, co-cultures of iMG and neurons shows that exposure to damaged neurons induces HIV-1 transcription whereas exposure to healthy neurons can suppress HIV-1 transcription in infected microglia, potentially generating intermittent cycles of HIV-1 reactivation and silencing in the CNS [47].
These molecular mechanisms could also provide a framework for understanding why opportunistic infections and drugs of abuse may exacerbate HAND. For example, drugs of abuse, such as methamphetamine and cocaine, can induce neuronal damage leading to microglial activation and pro-inflammatory cytokine release in HIV-1 infected microglia [35,47].
Cellular mechanisms that can moderate HIV-1 expression and reduce microglial activation
Since ART alone fails to eliminate HAND, there is an urgent demand for developing new pharmacological agents that can inhibit both inflammation and HIV-1 transcription. Functional genomic analyses using genome-wide shRNA screening unveiled an array of factors and pathways that are key to sustaining proviral latency in microglia [46]. This led to preliminary findings from our laboratory that the functional activation of orphan nuclear receptor family member Nurr1 (NR4A2) by specific agonists, or the overexpression of Nurr1, led to the rapid silencing of activated HIV-1 in microglia in biochemical experiments; furthermore, global gene expression analysis indicated that Nurr1 could repress HIV-1 gene expression [48]. We have suggested that Nurr1, and the ligand activated nuclear receptors (LA-NR) (including RXRα and RXRβ, the glucocorticoid receptor (GR, NR3C1)), might play key roles in regulating HIV-1 transcription in microglial cells, although these findings warrant further validation [46,48]. Of note, these results are consistent with the major neuroprotective role of the NR4A/CoREST trans-repression pathway that also acts as a suppressor of inflammatory gene expression in murine microglial cells and astrocytes [49].
Each of these receptors may play a distinct role in regulating HIV-1 transcription. Accordingly, using a set of Nurr1 agonists and genetic manipulation of Nurr1 in immortalized and iMG microglia, we recently reported in the aforementioned preprint article requiring confirmation [48], that Nurr1 could not only potently silence HIV-1 in reactive microglial cells but also drive them towards a quiescent state (Figure 2). Similarly, using shRNA to knockdown (KD) the glucocorticoid receptor (NR3C1, GR), we previously showed that GR binding to the viral promoter induced HIV-1 latency in microglial cells [46]. The production of glucocorticoids by healthy neurons is likely to be one of the major mechanisms used to silence HIV-1, and indeed, activation of the GR by dexamethasone (DEXA), a synthetic glucocorticoid, led to potent repression of HIV-1 transcription, in microglial cell models [34,46]. Of note, although Nurr1 is deemed to be the dominant factor, various nuclear receptor pathways are integrated through protein-protein interactions [50]. For example, NR4A receptors can also interact with GR/NR3C1 and the transcription repressor complex CoREST/HDAC1/2/LSD1 to epigenetically silence inflammatory genes [50]. In our preliminary report, simultaneous activation of these different nuclear receptors led to complete shutdown of HIV-1 expression in microglia [48].
Epigenetic mechanisms governing HIV-1 latency have also been reported in CD4+ T-cells and microglial cells [51]. In both systems, the provirus is silenced epigenetically through the histone lysine methyltransferase (HKMT) machinery [52]. Studies on immortalized human microglial cells have indicated that HIV-1 latency is maintained by multiple HKMTs, including G9a, SUV39H1, and EZH2, which we also reported to be essential for HIV-1 latency in CD4+ T cells [52]. Moreover, in this latter study, histone deacetylases (HDACs) were highly enriched at the long terminal repeat (LTR) of latent HIV-1 in microglial cells and CD4+ T cells [48, 52]. Also, in microglia and CD4+ T cells, latent HIV-1 chromatin was enriched in both H3K4 trimethylation (H3K4me3) and H3K27 trimethylation (H3K27me3), marking it as a bivalent promoter poised for rapid transcription and protected from irreversible silencing by DNA methylation, although some of these findings warrant further confirmation [48,52]. We posit that the targeting of specific epigenetic pathways that regulate HIV-1 latency and microglial activation might provide additional candidate therapeutic opportunities to repress HIV-1 in the CNS.
Concluding remarks
The direct contribution of HIV-1 latency in the CNS to neuroinflammation and development of HAND is increasingly accepted, but many mechanistic questions remain unresolved (see Outstanding Questions). Early ART is crucial for the control of HIV-1 replication and viral load in the CNS and to reduce the incidence of severe HAND. However, since ART unfortunately does not control proviral transcription, it is unable to block reactivation of latent HIV-1 in the CNS. Here, we propose the use of agonists of nuclear receptors NR3C1 (GR), NR4A1(Nur77), NR4A2(Nurr1), NR4A3(Nor1), and RXR-α/RXR-β --- already reported to reduce microglial activation --- as potential agents to presumably block HIV-1 emergence from latency. These drugs, either alone or in combination with other anti-inflammatory agents targeting microglia [53], such as the phosphodiesterase inhibitor Ibudilast [54], could be explored as potential complementary strategies to treat HAND.
Outstanding Questions Box.
How can we accurately distinguish cognitive impairment stemming from HIV-1 infection (or immunosuppression) from cognitive deficits due to age-related disorders and comorbidities?
In HIV-1 infected patients, how do ART regimens, sex differences, inflammation, and immune activation or dysfunction impact the rate of neurocognitive decline and HAND severity?
Are there HIV-1 clade-specific differences that influence HAND in different countries and epidemics?
Does microglia localization in the CNS (e.g. brainstem or frontal cortex) exhibit differential HIV-1 latency? Do these regions selectively respond to HIV-1 reactivation and neuroinflammation?
What proportion of myeloid cells in the CNS (e.g. choroid plexus, perivascular, or meningeal macrophages) harbor latent HIV-1 virus? How do each of these cells respond to HIV-1?
Can latently- or productively-infected microglia be imaged in the brains of HIV-1 infected patients?
Might broadly anti-inflammatory agents targeting microglia also be used as putative HAND therapeutics?
Significance.
Although antiretroviral therapy dramatically lowers HIV-1 RNA in the central nervous system, some patients harbor HIV-1- associated cognitive disorder (HAND). We propose that HIV-1 emergence from latency in microglia in response to inflammatory signals can contribute to this disease.
Highlights.
Antiretroviral therapy (ART) effectively lowers HIV-1 RNA in the central nervous system (CNS) but does not reduce the incidence of HIV-1-associated cognitive disorder (HAND), which occurs in 15 to 60% of people living with HIV-1 (PLWH).
Productive HIV-1 infection of microglia causes neurotoxicity due to enhanced secretion of cytokines and neurotoxins, phagocytic activity, as well as the release of viral proteins.
Molecular studies have indicated that HIV-1 can readily enter latency in microglia. Latent HIV-1 can be reactivated by microglia exposure to pro-inflammatory cytokines, toll-like receptor agonists, and damaged neurons. Conversely, healthy neurons produce “off signals” that may drive microglia towards a more resting state and induce HIV-1 latency.
Intermittent reactivation of HIV-1 from latency in microglial cells is a form of viral persistence that is refractory to ART since ART does not inhibit HIV-1 transcription or reactivation of HIV-1 from latency.
We argue that candidate drugs might target both HIV-1 transcription and microglial activation; these might potentially include Nurr1 agonists and other ligand activated and orphan nuclear receptors, which have been reported to play neuroprotective and anti-inflammatory roles in the CNS.
Acknowledgments
Work in the Karn laboratory on NeuroHIV was supported by NIH grants R01 DA043159 and R01 DA049481 to J.K. and R01 MH113457 to Paula Cannon (USC) and JK. We thank Asha Kallianpur, for her helpful comments on the manuscript.
Glossary
- Basal ganglia
(BG) subcortical nuclei; the largest component of the BG, the striatum, receives input from the cerebral cortex, thalamus, and brainstem and is associated with the control of voluntary motor movements, learning, eye movements, cognition, and emotion
- Blood-Brain barrier (BBB)
specialized layer of microvascular endothelial cells, astrocyte end-feet, and pericytes; it restricts the passage of many pathogens
- Anti-Retroviral therapy (ART)
over 30 separate licensed anti-HIV-1 drugs exist that target viral reverse transcriptase, protease, integrase and receptors; used in specific combinations to reduce circulating virus in most HIV-1-infected patients to undetectable titers
- HIV-1-associated neurocognitive disorders (HAND)
People living with HIV-1 (PLWH) can develop a range of neurocognitive, motor, and behavioral dysfunctions, collectively termed HAND
- HIV-1 transcytosis
type of transcellular transport across the interior of a cell; receptor independent transcytosis through endothelial cells is one mechanism believed to enable HIV-1 entry through the blood brain barrier
- Induced pluripotent stem cell (iPSCs)
generated by genetically reprogramming an adult somatic cell (such as a fibroblast or peripheral blood mononuclear cell) using a cocktail of four specific transcription factors (Myc, Oct3/4, Sox2 and Klf4). iPSCs can differentiate into any cell type of the 3 germ layers (ectoderm, mesoderm, and endoderm). In the context of NeuroHIV research, iPSCs provide the only source for obtaining unlimited numbers of donor-matched, microglia, astrocytes, and neurons
- Neurotoxins
Agents that can directly cause neural damage and affect neural communications, and intercellular processes
- Proviral latency
HIV-1 can persist as a transcriptionally silent, or latent, integrated provirus. Key host cell mechanisms that drive HIV-1 into latency are the sequestration of transcription factors and the establishment of epigenetic barriers that inactivate the proviral promoter
- Homeostatic “ramified” microglia
Microglial cells undergo structural changes in response to inflammatory signals, transforming from homeostatic or “ramified” microglia to reactive or “amoeboid” microglia. These transitions allow microglia to defend the CNS on extremely short notice and then revert before causing immunological disturbance
- Toll-like receptors (TLR)
class of pattern recognition receptors that sense pathogen components, such as bacterial surface proteins, viral RNA and DNA. Signaling through TLRs activates transcription factors such as NF-κB or IRF3, mediating the production of inflammatory cytokines, chemokines, and interferons
- EcoHIV
chimeric virus in which the HIV-1 envelope is replaced by an amphotropic retroviral envelope, making it permissive for infection of mice and rats
- NSG mice
The NOD scid gamma (NSG) mouse is an immunodeficient laboratory mouse that lacks mature T cells, B cells, and natural killer (NK) cells. NSG mice are frequently reconstituted by engraftment of primary human hematopoietic stem cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sacktor N (2018) Changing clinical phenotypes of HIV-associated neurocognitive disorders. J Neurovirol 24, 141–145. 10.1007/s13365-017-0556-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Underwood J et al. (2019) Validation of a Novel Multivariate Method of Defining HIV-Associated Cognitive Impairment. Open forum infectious diseases 6, ofz198. 10.1093/ofid/ofz198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gumbs SBH et al. (2022) Human microglial models to study HIV infection and neuropathogenesis: a literature overview and comparative analyses. J Neurovirol. 28(1):64–91 10.1007/s13365-021-01049-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sil S et al. (2021) Advances in the Experimental Models of HIV-Associated Neurological Disorders. Current HIV/AIDS reports 18, 459–474. 10.1007/s11904-021-00570-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Achim CL et al. (1994) Brain viral burden in HIV infection. J Neuropathol Exp Neurol 53, 284–294. 10.1097/00005072-199405000-00010 [DOI] [PubMed] [Google Scholar]
- 6.Suh J et al. (2014) Progressive increase in central nervous system immune activation in untreated primary HIV-1 infection. J Neuroinflammation 11, 199. 10.1186/s12974-014-0199-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desplats P et al. (2013) Molecular and pathologic insights from latent HIV-1 infection in the human brain. Neurology 80, 1415–1423. 10.1212/WNL.0b013e31828c2e9e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson AM et al. (2017) Prevalence and Correlates of Persistent HIV-1 RNA in Cerebrospinal Fluid During Antiretroviral Therapy. J Infect Dis 215, 105–113. 10.1093/infdis/jiw505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alakkas A et al. (2019) White matter damage, neuroinflammation, and neuronal integrity in HAND. J Neurovirol 25, 32–41. 10.1007/s13365-018-0682-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.May PE et al. (2020) Assessing Cognitive Functioning in People Living With HIV (PLWH): Factor Analytic Results From CHARTER and NNTC Cohorts. J Acquir Immune Defic Syndr 83, 251–259. 10.1097/qai.0000000000002252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Livelli A et al. (2019) Correlates of HIV RNA concentrations in cerebrospinal fluid during antiretroviral therapy: a longitudinal cohort study. The lancet. HIV 6, e456–e462. 10.1016/s2352-3018(19)30143-2 [DOI] [PubMed] [Google Scholar]
- 12.Saylor D et al. (2016) HIV-associated neurocognitive disorder--pathogenesis and prospects for treatment. Nat Rev Neurol 12, 234–248. 10.1038/nrneurol.2016.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cysique LA et al. (2021) Reliably Measuring Cognitive Change in the Era of Chronic HIV Infection and Chronic HIV-Associated Neurocognitive Disorders. Curr Top Behav Neurosci 50, 271–298. 10.1007/7854_2019_116 [DOI] [PubMed] [Google Scholar]
- 14.Naveed Z et al. (2022) An assessment of factors associated with neurocognitive decline in people living with HIV. Int J STD AIDS 33, 38–47. 10.1177/09564624211043351 [DOI] [PubMed] [Google Scholar]
- 15.Resnick L et al. (1988) Early penetration of the blood-brain-barrier by HIV. Neurology 38, 9–14. 10.1212/wnl.38.1.9 [DOI] [PubMed] [Google Scholar]
- 16.Davis LE et al. (1992) Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology 42, 1736–1739. 10.1212/wnl.42.9.1736 [DOI] [PubMed] [Google Scholar]
- 17.Ivey NS et al. (2009) Acquired immunodeficiency syndrome and the blood-brain barrier. J Neurovirol 15, 111–122. 10.1080/13550280902769764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leibrand CR et al. (2017) HIV-1 Tat disrupts blood-brain barrier integrity and increases phagocytic perivascular macrophages and microglia in the dorsal striatum of transgenic mice. Neurosci Lett 640, 136–143. 10.1016/j.neulet.2016.12.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorin V et al. (2020) Antibody Neutralization of HIV-1 Crossing the Blood-Brain Barrier. mBio 11(5):e02424–20. 10.1128/mBio.02424-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castellano P et al. (2017) HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Scientific reports 7, 12866. 10.1038/s41598-017-12758-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallet C et al. (2019) Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front Cell Infect Microbiol 9, 362. 10.3389/fcimb.2019.00362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan P and Spudich S (2022) HIV Compartmentalization in the CNS and Its Impact in Treatment Outcomes and Cure Strategies. Curr HIV/AIDS Rep 19, 207–216. 10.1007/s11904-022-00605-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sturdevant CB et al. (2015) Compartmentalized replication of R5 T cell-tropic HIV-1 in the central nervous system early in the course of infection. PLoS Pathog 11, e1004720. 10.1371/journal.ppat.1004720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunfee RL et al. (2006) The HIV Env variant N283 enhances macrophage tropism and is associated with brain infection and dementia. Proc Natl Acad Sci U S A 103, 15160–15165. 10.1073/pnas.0605513103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osborne O et al. (2020) The Paradox of HIV Blood-Brain Barrier Penetrance and Antiretroviral Drug Delivery Deficiencies. Trends Neurosci 43, 695–708. 10.1016/j.tins.2020.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asahchop EL et al. (2017) Reduced antiretroviral drug efficacy and concentration in HIV-infected microglia contributes to viral persistence in brain. Retrovirology 14, 47. 10.1186/s12977-017-0370-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams ME et al. (2021) Cerebrospinal fluid immune markers and HIV-associated neurocognitive impairments: A systematic review. J Neuroimmunol 358, 577649. 10.1016/j.jneuroim.2021.577649 [DOI] [PubMed] [Google Scholar]
- 28.Swanta N et al. (2020) Blood-based inflammation biomarkers of neurocognitive impairment in people living with HIV. J Neurovirol 26, 358–370. 10.1007/s13365-020-00834-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lackner P et al. (2010) Antibodies to myelin oligodendrocyte glycoprotein in HIV-1 associated neurocognitive disorder: a cross-sectional cohort study. J Neuroinflammation 7, 79. 10.1186/1742-2094-7-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahajan SD et al. (2021) HIV Neuroinflammation: The Role of Exosomes in Cell Signaling, Prognostic and Diagnostic Biomarkers and Drug Delivery. Front Cell Dev Biol 9, 637192. 10.3389/fcell.2021.637192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nosi D et al. (2021) Neuroinflammation: Integrated Nervous Tissue Response through Intercellular Interactions at the “Whole System” Scale. Cells 10(5):1195. 10.3390/cells10051195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia-Mesa Y et al. (2017) Immortalization of primary microglia: a new platform to study HIV regulation in the central nervous system. J Neurovirol 23, 47–66. 10.1007/s13365-016-0499-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alvarez-Carbonell D et al. (2017) Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology 14, 9. 10.1186/s12977-017-0335-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alvarez-Carbonell D et al. (2019) Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog 15, e1008249. 10.1371/journal.ppat.1008249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kraft-Terry SD et al. (2009) A coat of many colors: neuroimmune crosstalk in human immunodeficiency virus infection. Neuron 64, 133–145. 10.1016/j.neuron.2009.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanna PP et al. (2021) Central nervous system (CNS) transcriptomic correlates of human immunodeficiency virus (HIV) brain RNA load in HIV-infected individuals. Sci Rep 11, 12176. 10.1038/s41598-021-88052-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holtman IR et al. (2017) Transcriptional control of microglia phenotypes in health and disease. The Journal of clinical investigation 127, 3220–3229. 10.1172/JCI90604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan NY and Kaul M (2021) Beneficial and Adverse Effects of cART Affect Neurocognitive Function in HIV-1 Infection: Balancing Viral Suppression against Neuronal Stress and Injury. J Neuroimmune Pharmacol 16, 90–112. 10.1007/s11481-019-09868-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lanman T et al. (2021) CNS Neurotoxicity of Antiretrovirals. J Neuroimmune Pharmacol 16, 130–143. 10.1007/s11481-019-09886-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borrajo A et al. (2021) Important role of microglia in HIV-1 associated neurocognitive disorders and the molecular pathways implicated in its pathogenesis. Ann Med 53, 43–69. 10.1080/07853890.2020.1814962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo S et al. (2022) Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front Aging Neurosci 14, 815347. 10.3389/fnagi.2022.815347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Das Sarma J (2014) Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J Neurovirol 20, 122–136. 10.1007/s13365-013-0188-4 [DOI] [PubMed] [Google Scholar]
- 43.Abreu C et al. (2019) Brain macrophages harbor latent, infectious simian immunodeficiency virus. Aids 33 Suppl 2, S181–s188. 10.1097/qad.0000000000002269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mbonye U and Karn J (2017) The Molecular Basis for Human Immunodeficiency Virus Latency. Annual review of virology. 29;4(1):261–285. 10.1146/annurev-virology-101416-041646 [DOI] [PubMed] [Google Scholar]
- 45.Van Lint C et al. (2013) HIV-1 transcription and latency: an update. Retrovirology 10, 67. 10.1186/1742-4690-10-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alvarez-Carbonell D et al. (2018) The Glucocorticoid Receptor Is a Critical Regulator of HIV Latency in Human Microglial Cells. J Neuroimmune Pharmacol. 14(1):94–109. 10.1007/s11481-018-9798-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alvarez-Carbonell D et al. (2020) Cross-talk Between Microglia and Neurons Regulates HIV Latency. PLoS Pathog. 15(12): e1008249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye F et al. (2021) The Nerve Growth Factor IB-like Receptor Nurr1 (NR4A2) Recruits CoREST Transcription Repressor Complexes to Silence HIV Following Proviral Reactivation in Microglial Cells. bioRxiv, 2021.2011.2016.468784. 10.1101/2021.11.16.468784. https://www.biorxiv.org/content/10.1101/2021.11.16.468784v1.full.pdf [DOI] [Google Scholar]
- 49.Saijo K et al. (2009) A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137, 47–59. 10.1016/j.cell.2009.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurakula K et al. (2014) NR4A nuclear receptors are orphans but not lonesome. Biochimica et biophysica acta 1843, 2543–2555. 10.1016/j.bbamcr.2014.06.010 [DOI] [PubMed] [Google Scholar]
- 51.Kumar A et al. (2015) Epigenetic control of HIV-1 post integration latency: implications for therapy. Clinical epigenetics 7, 103. 10.1186/s13148-015-0137-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nguyen K et al. (2021) Inhibition of the H3K27 demethylase UTX enhances the epigenetic silencing of HIV proviruses and induces HIV-1 DNA hypermethylation but fails to permanently block HIV reactivation. PLoS Pathog 17, e1010014. 10.1371/journal.ppat.1010014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ambrosius B et al. (2019) Antineuroinflammatory drugs in HIV-associated neurocognitive disorders as potential therapy. Neurol Neuroimmunol Neuroinflamm 6, e551. 10.1212/nxi.0000000000000551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oliveros G et al. (2022) Repurposing ibudilast to mitigate Alzheimer’s disease by targeting inflammation. Brain. 12, awac136. 10.1093/brain/awac136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ryan SK et al. (2020) Neuroinflammation and EIF2 Signaling Persist despite Antiretroviral Treatment in an hiPSC Tri-culture Model of HIV Infection. Stem Cell Reports 14, 703–716. 10.1016/j.stemcr.2020.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rai MA et al. (2020) Comparative analysis of human microglial models for studies of HIV replication and pathogenesis. Retrovirology 17, 35. 10.1186/s12977-020-00544-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gumbs SBH et al. (2022) Characterization of HIV-1 Infection in Microglia-Containing Human Cerebral Organoids. Viruses 14(4):829. 10.3390/v14040829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moretti S et al. (2021) Advances in SIV/SHIV Non-Human Primate Models of NeuroAIDS. Pathogens 10(8):1018. 10.3390/pathogens10081018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martinez-Navio JM (2021) Neurological complications during HIV infection. Explor Neuroprot Ther. 1, 19–32. 10.37349/ent.2021.00004 [DOI] [Google Scholar]
- 60.Garcia-Mesa Y et al. (2020) Regional Brain Recovery from Acute Synaptic Injury in Simian Immunodeficiency Virus-Infected Rhesus Macaques Associates with Heme Oxygenase Isoform Expression. J Virol 94. 10.1128/jvi.01102-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Irollo E et al. (2021) Mechanisms of neuronal dysfunction in HIV-associated neurocognitive disorders. Cell Mol Life Sci 78, 4283–4303. 10.1007/s00018-021-03785-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gu CJ et al. (2018) EcoHIV infection of mice establishes latent viral reservoirs in T cells and active viral reservoirs in macrophages that are sufficient for induction of neurocognitive impairment. PLoS Pathog 14, e1007061. 10.1371/journal.ppat.1007061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H et al. (2021) Microglial HIV-1 Expression: Role in HIV-1 Associated Neurocognitive Disorders. Viruses 13(5):924. 10.3390/v13050924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H et al. (2021) A Rat Model of EcoHIV Brain Infection. J Vis Exp. Jan 21;(167):10.3791/62137 10.3791/62137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Llewellyn GN et al. (2018) HIV-1 infection of microglial cells in a reconstituted humanized mouse model and identification of compounds that selectively reverse HIV latency. J Neurovirol 24, 192–203. 10.1007/s13365-017-0604-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mathews S et al. (2019) Human Interleukin-34 facilitates microglia-like cell differentiation and persistent HIV-1 infection in humanized mice. Mol Neurodegener 14, 12. 10.1186/s13024-019-0311-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shan L et al. (2022) Development of Humanized Mouse Models for Studying Human NK Cells in Health and Disease. Methods Mol Biol 2463, 53–66. 10.1007/978-1-0716-2160-8_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ivic S et al. (2017) Differential Dynamics of HIV Infection in Humanized MISTRG versus MITRG Mice. ImmunoHorizons 1, 162. 10.4049/immunohorizons.1700042 [DOI] [Google Scholar]
- 69.Kimelberg HK and Nedergaard M (2010) Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics 7, 338–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lutgen V et al. (2020) HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLoS Pathog 16, e1008381. 10.1371/journal.ppat.1008381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo X and He JJ (2015) Cell-cell contact viral transfer contributes to HIV infection and persistence in astrocytes. J Neurovirol 21, 66–80. 10.1007/s13365-014-0304-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garland EF et al. (2022) Microglia and Astrocyte Function and Communication: What Do We Know in Humans? Front Neurosci 16, 824888. 10.3389/fnins.2022.824888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bozzelli PL et al. (2019) HIV-1 Tat promotes astrocytic release of CCL2 through MMP/PAR-1 signaling. Glia 67, 1719–1729. 10.1002/glia.23642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rao VR et al. (2014) Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND). AIDS Res Ther 11, 13. 10.1186/1742-6405-11-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nimmerjahn A et al. (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- 76.Stratoulias V et al. (2019) Microglial subtypes: diversity within the microglial community. Embo J 38, e101997. 10.15252/embj.2019101997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Szepesi Z et al. (2018) Bidirectional Microglia-Neuron Communication in Health and Disease. Front Cell Neurosci 12, 323. 10.3389/fncel.2018.00323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Biber K et al. (2007) Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci 30, 596–602. 10.1016/j.tins.2007.08.007 [DOI] [PubMed] [Google Scholar]
- 79.Illes P et al. (2020) Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells 9(5): 1108. 10.3390/cells9051108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mathys H et al. (2017) Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep 21, 366–380. 10.1016/j.celrep.2017.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Q et al. (2019) Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 101, 207–223.e210. 10.1016/j.neuron.2018.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baxter PS et al. (2021) Microglial identity and inflammatory responses are controlled by the combined effects of neurons and astrocytes. Cell Rep 34, 108882. 10.1016/j.celrep.2021.108882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gorska AM and Eugenin EA (2020) The Glutamate System as a Crucial Regulator of CNS Toxicity and Survival of HIV Reservoirs. Front Cell Infect Microbiol 10, 261. 10.3389/fcimb.2020.00261 [DOI] [PMC free article] [PubMed] [Google Scholar]