

Abstract
Pyruvate dehydrogenase kinase 4 (PDK4) is an important regulator of energy metabolism. Previously, knockdown of PDK4 by specific small interfering RNAs (siRNAs) have been shown to suppress the expression of Kirsten rat sarcoma viral oncogene homolog (KRAS) and the growth of lung and colorectal cancer cells, indicating that PDK4 is an attractive target of cancer therapy by altering energy metabolism. The authors previously reported that a novel small molecule, cryptotanshinone (CPT), which inhibits PDK4 activity, suppresses the in vitro three-dimensional (3D)-spheroid formation and in vivo tumorigenesis of KRAS-activated human pancreatic and colorectal cancer cells. The present study investigated the molecular mechanism of CPT-induced tumor suppression via alteration of glutamine and lipid metabolism in human pancreatic and colon cancer cell lines with mutant and wild-type KRAS. The antitumor effect of CPT was more pronounced in the cancer cells containing mutant KRAS compared with those containing wild-type KRAS. CPT treatment decreased glutamine and lipid metabolism, affected redox regulation and increased reactive oxygen species (ROS) production in the pancreatic cancer cell line MIAPaCa-2 containing mutant KRAS. Suppression of activated KRAS by specific siRNAs decreased 3D-spheroid formation, the expression of acetyl-CoA carboxylase 1 and fatty acid synthase (FASN) and lipid synthesis. The suppression also reduced glutathione-SH/glutathione disulfide and increased the production of ROS. Knockdown of FASN suppressed lipid synthesis in MIAPaCa-2 cells, partially promoted ROS production and mildly suppressed 3D-spheroid formation. These results indicated that CPT reduced tumorigenesis by inhibiting lipid metabolism and promoting ROS production in a mutant KRAS-dependent manner. This PDK4 inhibitor could serve as a novel therapeutic drug for KRAS-driven intractable cancers via alteration of cell metabolism.
Keywords: PDK4 inhibitor, cryptotanshinone, pancreatic neoplasm, KRAS, lipid synthesis, acetyl-CoA carboxylase 1, fatty acid synthase, reactive oxygen species
Introduction
The majority of proliferating tumor cells preferentially metabolize glucose via glycolysis, even under aerobic conditions. This process is known as the Warburg effect and is a primary metabolic hallmark of cancer cells (1-3). Recently, the molecular mechanisms and relationships associated with cancer-related genes, including p53, c-myc and ras, have been investigated (4). The authors reported that the tumor suppressor, drs/SRPX, which is downregulated by oncogenes including v-src and K-ras, was involved in the shift toward glucose metabolism (5). A loss of drs/SRPX induces the Warburg effect by upregulating lactate dehydrogenase (LDH)-B and pyruvate dehydrogenase kinase 4 (PDK4), which suggests that these two enzymes are important in the regulation of glucose metabolism in malignant cancers (5,6). PDK4 is one of four PDK isoforms (PDK1-4) that control the pyruvate dehydrogenase (PDH) complex (7). Phosphorylation of PDH by PDK inhibits its ability to induce a glycolytic shift, thus promoting cytoplasmic glycolysis over mitochondrial oxidative phosphorylation. PDK4 expression is frequently upregulated in cancer tissues and its elevation is important for the induction of the Warburg effect (7,8). Trinidad et al (9) report that PDK4 knockdown by specific small interfering (si)RNAs downregulates mutant Kirsten rat sarcoma viral oncogene homolog (KRAS) expression and consequently suppresses the growth of lung and colorectal cancer (CRC) cells. This finding suggests that PDK4 is an attractive target for cancer therapy by altering energy metabolism (10,11). Recently, glutamine has attracted significant interest for its role in energy metabolism in pancreatic cancer cells (12). The regulation of lipid biosynthesis also serves a central role in energy metabolism and cancer progression (13). Furthermore, lipid metabolism has been found to serve an important role in the development and progression of pancreatic cancer (12,13). Specifically, the key mediators of fatty acid metabolic reprogramming, such as acetyl-CoA carboxylase 1 (ACC1), ATP-citrate lyase (ACLY) and fatty acid synthase (FASN), are abnormally expressed in pancreatic cancer (12,14). These enzymes are considered novel targets for cancer therapy and the drugs targeting lipid metabolism are undergoing clinical trials in various cancers, including pancreatic cancer (12,15). Investigation of the association between this metabolic reprograming and the action of cryptotanshinone (CPT) in pancreatic cancer cells is required to elucidate the molecular mechanism of tumor suppression by CPT.
Tumors driven by mutant KRAS are among the most aggressive and refractory to treatment (16). Unfortunately, drugs targeting activated K-Ras protein, either directly or by acting on downstream signaling molecules, have been largely ineffective (17). However, novel therapeutic opportunities have emerged based on the effect that this oncogene exerts in rewiring cancer cell metabolism (18). Cancer cells that become dependent on KRAS-driven metabolism are sensitive to inhibition via these metabolic routes, which has provided novel opportunities for intervention. KRAS is mutated in 90% of pancreatic cancers and is a major therapeutic target (19). Mutant KRAS promotes the reprogramming of glutamine metabolism, lipid biosynthesis and upregulation of antioxidants, such as GSH, which results in decreased reactive oxygen species (ROS) production (19). The authors previously reported that CPT, a novel PDK4 inhibitor derived from plants, inhibits the formation of 3D spheroids and tumor formation by reducing the expression of mutant K-Ras protein (20). The present study examined the pathway through which CPT suppresses glutamine and lipid metabolism via mutant KRAS to shed light on the mechanism of tumor suppression by CPT.
Materials and methods
Cell culture
Human pancreatic ductal adenocarcinoma (PDAC) cell lines, MIAPaCa-2 (mutant KRAS; cat. no. CRL-1420) and BxPC3 (wild-type KRAS; cat. no. CRL-1687) and human CRC cell line DLD1 (mutant KRAS; cat. no. CCL-221) were obtained from the American Type Culture Collection. The human CRC cell line DKO4 (wild-type KRAS) cell line was established as previously described (21). MIAPaCa-2, DLD1 and DKO4 were cultured in Dulbecco's modified Eagle's medium (DMEM; Nakarai Tesque, Inc.) supplemented with 10% fetal calf serum, penicillin (100 U/ml) and streptomycin (100 µg/ml), whereas BxPC3 was cultured in RPMI1640 (Nakarai Tesque, Inc.) supplemented with 10% fetal calf serum (FCS; Nakarai Tesque, Inc.), penicillin (100 U/ml) and streptomycin (100 µg/ml) at 37°C in a humidified incubator containing 5% CO2. In addition, nonadherent cell culture was performed by coating the dishes with poly-(2-hydroxyethyl methacrylate) reagent (poly-HEMA; MilliporeSigma).
Reagents
CPT (cryptotanshinone) was purchased from Cosmo Bio Co., Ltd. and dissolved in dimethyl sulfoxide (DMSO) to prepare 20 mM stock solutions for the in vitro experiments. The final concentration of DMSO was adjusted to 0.1%.
Cell proliferation assays
The three-dimensional (3D) spheroid formation assay was performed using 96-well V plates (PrimeSurface; Sumitomo Bakelite Co., Ltd.) as described previously (20). A total of 1×103 cells were seeded to a 96-well plate and incubated with CPT for 72 h at 37°C in triplicate. To determine the number of viable cells in the 3D spheroid, ATP was quantified using the luminescence-based CellTiter-Glo 3D cell viability assay (Promega Corporation). The viability of the adherent cells was determined using the CellTiter-Glo cell viability assay (Promega Corporation). The morphology of 3D-spheroid was imaged using a light microscope at ×4 magnification (IX73; Olympus Corporation). More than three randomly selected were examined.
Measurement of glutamine, H2O2 and GSH concentration
A total of 5×105 cells were seeded into poly-HEMA-coated 60 mm dishes and cultured with CPT for 48 h at 37°C under nonadherent conditions for metabolite analyses. The culture medium was collected and glutamine and H2O2 concentrations were determined using the Glutamine/Glutamate-Glo Assay kit (Promega Corporation) and Amplite fluorimetric H2O2 kit (ATT Bioquest, Inc.), respectively. The intercellular glutathione-SH/glutathione disulfide (GSH/GSSG) was determined using the GSH/GSSG-Glo Assay kit (Promega Corporation). Glutamine and H2O2 concentrations were normalized to cellular protein content, which was determined using the Bradford method (Bio-Rad protein assay; Bio-Rad Laboratories, Inc.) based on the manufacturer's protocol.
siRNA transfection
For siRNA experiments (GeneDesign, Inc.), 100 nM of siRNA for FASN (siFASN; sense: 5′-AAC CCT GAG ATC CCA GCG CTG-3′, antisense: 5′-CAG CGC TGG GAT CTC AGG GTT-3′) (22) or 10 nM of siRNA for KRAS (siKRAS; sense: 5′-CAG CUA AUU CAG AAU CAU U-3′, antisense: 5′-AAU GAU UCU GAA UUA GCU G-3′) (23) was transfected with Lipofectamine® RNAiMax (Invitrogen; Thermo Fisher Scientific, Inc.) reagent according to the manufacturer's instructions (Lipofectamine® RNAiMax and siRNA were mixed at room temperature for 5 min and then added to MIAPaCa-2 cells). A 100 nM or 10 nM of nonspecific siRNA duplex (sense UUC UCC GAA CGU GUC ACG U, Antisense ACG UGA CAC GUU CGG AGA A; GeneDesign, Inc.) served as the control. A total of 1×105 cells were treated with siRNA for 24 h, followed by spheroid culture in a 96-well V-bottom plate for 72 h in triplicate. For the cell lysate preparation of immunoblot analyses, the cells were cultured under nonadherent conditions for 48 h at 37°C.
Immunoblotting and antibodies
A total of 5×105 cells were seeded into poly-HEMA-coated 60 mm dish and cultured with CPT for 48 h under nonadherent conditions for immunoblot analyses. The cells were lysed in Laemmli sample buffer containing 62.5 mM Tris-HCl [pH 6.8], 10% glycerol, 5% 2-mercaptoethanol, 2% sodium dodecyl sulfate (SDS) and 0.01% bromophenol blue. The total proteins were quantified using Micro BCA Protein Assay Kit (Thermo Fisher Scientific, Inc.). A 20 µg of the cell lysate sample was subjected to 6 or 10% SDS-polyacrylamide gel electrophoresis (PAGE) and the separated proteins were electrotransferred to polyvinylidene difluoride (PVDF) membrane filters (Immobilon-P; MilliporeSigma). After blocking of the filters with TBST [10 mM Tris-HCl (pH 7.6), 150 mM sodium chloride and 0.1% Tween 20] containing 5% bovine serum albumin (BSA; Nakarai Tesque, Inc.) for 1 h at room temperature, the filters were incubated overnight with the indicated primary anti-bodies in TBST (0.1% Tween 20) containing 2% BSA at 4°C. The filters were then washed in TBST and were incubated for 1 h in horseradish peroxidase-conjugated anti-mouse (cat. no. NA931, from sheep) or anti-rabbit (cat. no. NA934, from donkey) immunoglobulin G (IgG; Cytiva) diluted 1:20,000 in TBST containing 2% BSA. After several washes with TBST, the immunoreactivity was detected using the ECL Western Blotting Detection System (Cytiva) according to the manufacturer's instructions. The ECL signal detection was performed using FUSION SOLO. 6S. EDGE (Vilber Lourmat). Protein expression was quantitated by band densitometry using ImageJ v1.51 software (National Institutes of Health). Rabbit monoclonal antibodies for glutamic-oxaloacetic transaminase (GOT)2 (1:3,000; cat. no. ab171739) were purchased from Abcam. Rabbit monoclonal antibodies for GOT1 (1:3,000; cat. no. 14886-1-AP), isocitrate dehydrogenase (IDH)2 (15932-1-AP) and glutamate transport system permease protein (GLUD)1 (1:3,000; cat. no. 14299-1AP) and mouse monoclonal antibodies for GLS (1:3,000; cat. no. 66265-1-Ig) were purchased from ProteinTech Group, Inc. Mouse monoclonal antibodies for GAPDH (1:3,000; cat. no. AC002) and rabbit monoclonal antibodies for KRAS (1:3,000; cat. no. A12704) and IDH1 (1:3,000; cat. no. A13245) were purchased from ABclonal Biotech Co., Ltd. Rabbit monoclonal antibodies for FASN (1:1,000; cat. no. #3189S), anti-ACLY rabbit polyclonal (1:1,000; cat. no. 4332S), anti-phosphorylated (p)ACLY rabbit polyclonal (1:1,000; p-S445; cat. no. 4331S) and anti-ACC1 rabbit polyclonal (1:1,000; cat. no. 3662) antibodies and anti-p-ACC1 rabbit polyclonal (1:1,000; p-S79; cat. no. 3661S) were purchased from Cell Signaling Technology, Inc.
Lipid droplet assay
For lipid droplet staining, the cells were seeded into culture dishes at a density of 1.0×105 cells. When the cell density reached ~60%, CPT (0 or 5 µM) was added and incubated for 24 h. siKRAS and/or siFASN were added and incubated for 72 h at 37°C. The cells were fixed in 4% paraformaldehyde for 10 min at room temperature and then washed twice with phosphate buffered saline (PBS) to remove residual paraformaldehyde. Subsequently, 1 ml of 1 µM LipiDyeII (Funakoshi Co., Ltd.) was added to the cells and incubated at 37°C for 30 min in the dark. The samples were washed three times with PBS, 10 µM DAPI (4′,6-diamidino-2-phenylindole) was added and the cells were imaged immediately using a fluorescent microscope (IX83; Olympus Corporation). Cell fluorescence intensity was quantified using ImageJ v1.51 software (National Institutes of Health). The experiment was repeated three times, with two replicates each time.
Statistical analyses
All quantitative data were presented as mean ± standard deviation. One-way ANOVA test, followed by Tukey's test, was performed for multiple comparisons and Welch's t-test was used for simple comparisons. All analyses were performed using the R statistical software package, version 2.6.2 (https://www.r-project.org/) and P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of CPT on the 3D-spheroid formation and expression of K-Ras protein in pancreatic and CRC cell lines with or without mutant KRAS
To assess the role of the mutant (activated) KRAS allele on the antitumor activity of CPT, its effects on 3D-spheroid formation in two PDAC cell lines (MIAPaCa-2 and BxPC3) with or without mutant KRAS was determined. The degree of inhibition was compared between the cancer cells containing mutant KRAS and wild-type KRAS. The antioncogenic effect of CPT was assessed by evaluating 3D-spheroid formation, which is frequently used in cancer research to closely mimic the tumor microenvironment (20). 3D-spheroid formation was quantified by using ATP concentration as a surrogate, which is closely associated with the number of viable cancer cells in the spheroids. The 3D-spheroid formation of MIAPaCa-2 cells containing mutant KRAS was strongly inhibited by CPT treatment (3-10 µM) compared with that of the BxPC-3 cells containing wild-type KRAS (Fig. 1A left). The effect of CPT on K-Ras protein was also evaluated in these two cell lines. A 3D-spheroid formation was morphologically altered in these cell lines by CPT treatment (5-10 µM; Fig. S1A). The results indicated that 10-20 µM of CPT effectively suppressed the level of mutant K-Ras protein in MIAPaCa-2 cells (mutant KRAS) as previously described (20), whereas GAPDH an internal control protein remained unaffected by CPT treatment (Figs. 1B and S1B). However, K-Ras protein expression was faintly suppressed by CPT in the BxPC-3 cells (wild-type KRAS). The same experiments were performed in two CRC cell lines (DLD-1 and DKO4), which had the same genetic background with or without mutant KRAS. The 3D-spheroid formation of DLD-1 cells (mutant KRAS) was significantly more inhibited by CPT treatment (3-10 µM) than that of DKO4 cells (Fig. 1A right). CPT more effectively decreased mutant K-Ras protein expression at 10 µM in DLD-1 cells (mutant KRAS) than in DKO4 cells (wild-type KRAS), whereas Although GAPDH expression was barely reduced by CPT treatment, the decrease of K-Ras by CPT was evident (Figs. 1B and S1B). However, K-Ras protein expression was faintly suppressed by CPT in DKO4 cells (wild-type KRAS). These results indicated that CPT treatment specifically decreases K-Ras protein in cancer cells containing mutant KRAS and that the effect of CPT on the suppression of 3D-spheroid formation is dependent on mutant KRAS.
Figure 1.
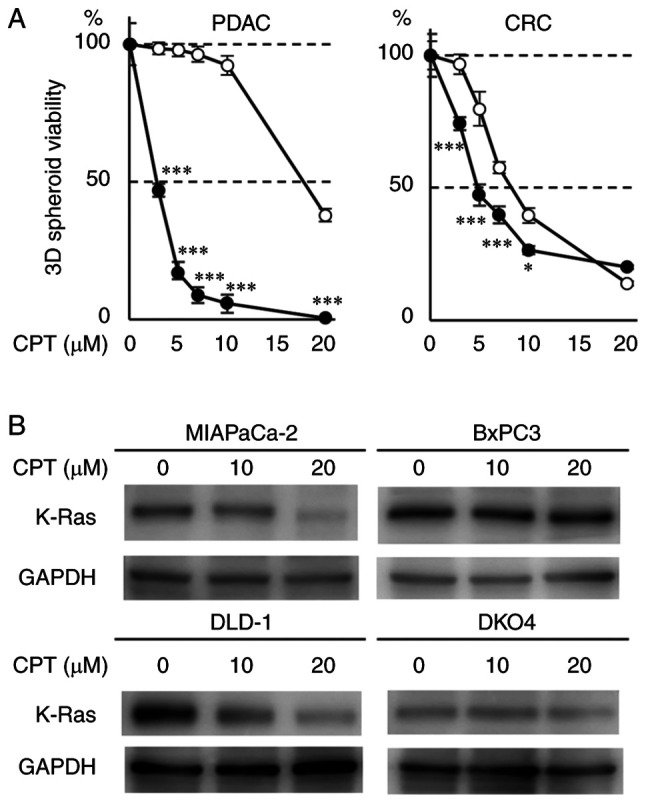
Effects of CPT on human PDAC (MIAPaCa-2 and BxPC3) and CRC (DLD-1 and DKO4) cell lines. (A) Spheroid viability of PDAC (left) and CRC (right) cell lines treated with CPT. A total of 1×103 cells were seeded into a 96-well V-bottom plate and incubated with CPT for 72 h in triplicate. Spheroid viability was determined by measuring the ATP content using the CellTiter-Glo 3D assay. Closed and open circles represent the cell lines with mutant KRAS (MIAPaCa-2 and DLD-1) and wild-type KRAS (BxPC3 and DKO4), respectively. Error bars represent standard deviation. *P<0.05 and ***P<0.001 vs. the wild-type KRAS cells treated with the same CPT concentration. (B) Immunoblot analyses of K-Ras protein in cells treated with CPT. A total of 5×105 cells were seeded into poly-HEMA-coated 60 mm dishes and incubated with CPT at 10 and 20 µM for 48 h. GAPDH was used as the internal control. CPT, cryptotanshinone; PDAC, pancreatic ductal adenocarcinoma; CRC, colorectal cancer; KRAS, Kirsten rat sarcoma viral oncogene homolog.
Effect of CPT on the expression of proteins involved in glutamine and lipid metabolism in pancreatic cancer cell lines containing mutant or wild-type KRAS
Mutant KRAS regulates glutamine and lipid metabolism during the development of malignant tumors (19). The dependency of 3D-spheroid formation on glutamine in PDAC (MIAPaCa-2 and BxPC-3) and CRC (DLD-1 and DKO4) cell lines with or without mutant KRAS was determined. In the PDAC cell lines, spheroid growth of the MIAPaCa-2 cells containing mutant KRAS was more dependent on glutamine concentration compared with BxPC3 cells containing wild-type KRAS (Fig. S2A). As for the CRC cell lines (DLD-1 and DKO4), the dependency of spheroid growth of mutant KRAS on glutamine concentration was not clearly observed. In the mutant KRAS cells (MIAPaCa-2 and DLD-1), the dependency of spheroid growth on glucose concentration was lower compared with that on glutamine concentration, although it was faintly higher than that on glucose concentration in wild-type KRAS cells (Fig. S2A and B). These results indicated that mutant KRAS was associated with the glutamine-dependent cell growth of pancreatic cancer cells. To clarify the molecular mechanisms by which CPT suppressed oncogenesis in a mutant KRAS-dependent manner, alterations in the proteins associated with glutamine and lipid metabolism were evaluated in MIAPaCa-2 (mutant KRAS) and BxPC-3 (wild-type KRAS) cells. As shown in Figs. 2 and S3, CPT concentrations of 10-20 µM decreased the expression of some proteins involved in glutamine metabolism, including GOT1, GLS and IDH1, as well as K-Ras protein in MIAPaCa-2 cells. GOT2, GLUD1 and IDH2 (a protein associated with glutamine metabolism) remained unaffected by CPT treatment. Proteins associated with lipid metabolism, such as FASN, ACC1, P-ACC, ACLY and P-ACLY, were also decreased by CPT treatment in MIAPaCa-2 cells. By contrast, the expression of these proteins and K-Ras protein were faintly or moderately influenced by CPT in BxPC-3. These results suggested that glutamine and lipid metabolism were affected by CPT in MIAPaCa-2 cells containing mutant KRAS.
Figure 2.
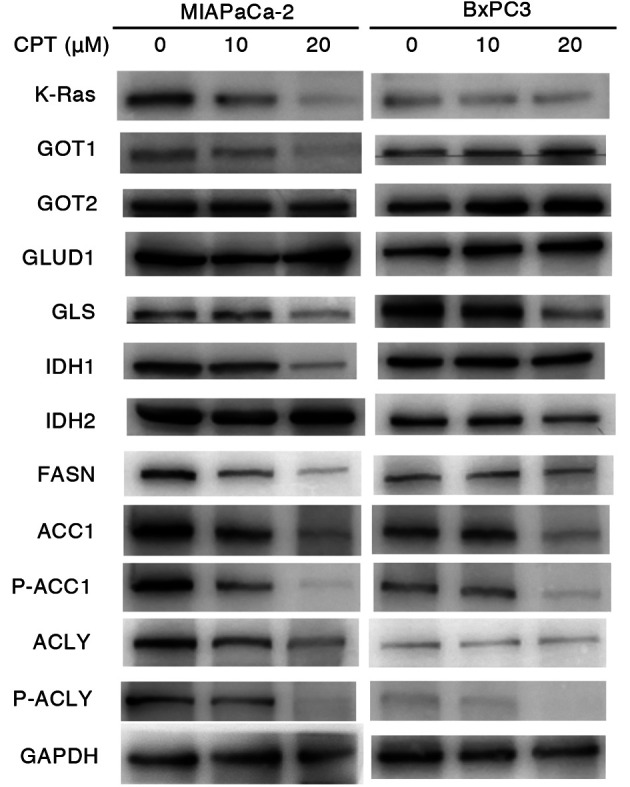
Immunoblot analyses of proteins regulating glutaminolysis and lipogenesis in nonadherent MIAPaCa-2 (mutant KRAS) and BxPC3 (wild-type KRAS) cancer cells treated with CPT. A total of 5×105 cells were seeded into poly-HEMA-coated 60 mm dishes and treated with CPT at 10 and 20 µM for 48 h. GAPDH was used as the internal control. KRAS, Kirsten rat sarcoma viral oncogene homolog; CPT, cryptotanshinone; GOT, glutamic-oxaloacetic transaminase; GLUD, glutamate transport system permease protein; GLS, glutaminase; IDH, isocitrate dehydrogenase; FASN, fatty acid synthase; ACC1, acetyl-CoA carboxylase 1; ACLY, ATP-citrate lyase; p-, phosphorylated.
Effect of CPT on glutamine consumption and lipid synthesis in pancreatic cancer cell lines containing mutant or wild-type KRAS
The effects of CPT on glutamine consumption and lipid synthesis were determined in MIAPaCa-2 and BxPC-3 cells. As shown in Fig. 3A, glutamine concentration in the culture medium was significantly increased by CPT (10 µM), thereby indicating that glutamine consumption was significantly decreased by CPT in MIAPaCa-2 cells. Lipid synthesis was monitored via the formation of lipid droplets and was also decreased by CPT in MIAPaCa-2 cells but not in BxPC-3 cells (Figs. 3B-F and S4). These results indicated that CPT treatment suppresses glutamine and lipid metabolism in MIAPaCa-2 cells containing mutant KRAS.
Figure 3.
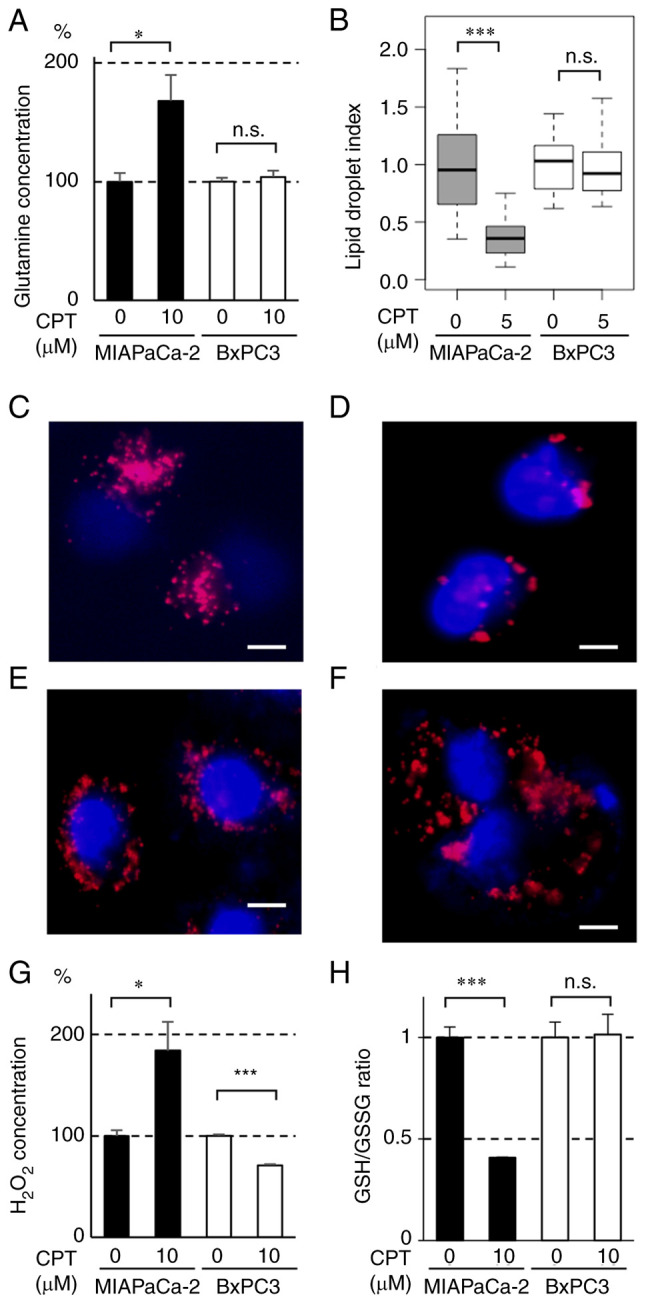
Effect of CPT on (A) glutaminolysis, (B-F) lipogenesis and (G and H) redox regulation in MIAPaCa-2 (mutant KRAS) and BxPC3 (wild-type KRAS) cancer cell lines. (A) Glutamine content in the nonadherent culture medium of MIAPaCa-2 (closed bars) and BxPC3 (open bars) cells treated with CPT. A total of 5×105 cells were seeded into poly-HEMA-coated 60 mm dishes and incubated with 10 µM CPT for 48 h in triplicate. Glutamine content in the culture medium was determined using the Glutamine/Glutamate-Glo Assay kit. The bars indicate mean ± standard deviation. *P<0.05 using Welch's t-test. n.s., not significant. (B) Box plot of the areas of lipid droplets in MIAPaCa-2 (gray boxes) and BxPC3 (white boxes) cells treated with CPT. The cells were seeded in culture dishes at a density of 1.0×105 cells/dish. When the cell density reached ~60%, CPT (0 or 5 µM) was added and incubated for 24 h. The cells were fixed in 4% paraformaldehyde and stained with LipiDyeII and DAPI. The integrated density of LipiDyeII staining (n=20 for each group) was measured using ImageJ software and normalized to the average of each control (0 µM) group. In the plots, the box indicates the lower and upper quartile, the horizontal bar represents the median and the whiskers are the highest and lowest data points that fall within 1.5 times the interquartile range from the lower and upper quartiles. ***P<0.001 using Welch's t-test. n.s., not significant. (C-F) Representative images of lipid droplet formation in (C and D) MIAPaCa-2 and (E and F) BxPC3 cells. The cells were treated or untreated with 5 µM CPT for 24 h and then stained with LipiDyeII and DAPI. The nuclei of the cells were stained with DAPI (blue) and the lipid droplets were stained with LipiDyeII (red). Scale bars=10 µm. (G) Hydrogen peroxide content in the nonadherent culture medium of MIAPaCa-2 (closed bars) and BxPC3 (open bars) cells treated with CPT. A total of 5×105 cells were seeded into poly-HEMA-coated 60 mm dishes and treated with 10 µM CPT for 48 h in triplicate. H2O2 contents in the culture medium were determined using the Amplite fluorimetric H2O2 kit. The bars indicate the mean ± standard deviation. * and ***P<0.05 and P<0.001 using Welch's t-test, respectively. (H) GSH/GSSG in the nonadherent MIAPaCa-2 (closed bars) and BxPC3 (open bars) cells treated with CPT. A total of 5×105 cells were seeded into poly-HEMA-coated 60 mm dishes and treated with 10 µM CPT for 48 h in triplicate. GSH/GSSG was determined by measuring GSH and GSSG using the GSH/GSSG-Glo Assay kit. The bars indicate mean ± standard deviation. ***P<0.001 using Welch's t-test. n.s., not significant; CPT, cryptotanshinone; KRAS, Kirsten rat sarcoma viral oncogene homolog; GSH, glutathione-SH; GSSG, glutathione disulfide.
Effect of CPT on the redox regulation and production of ROS in pancreatic cancer cells containing mutant or wild-type KRAS
ROS exhibits potent antitumor activity by reacting with DNA, proteins, lipids and enzymes (24). Redox molecules, such as GSH and GSSG, serve key roles in ROS production (25). Furthermore, glutamine and lipid metabolism have been shown to be closely associated with redox regulation (19,26). To determine whether CPT also affects redox regulation, the effect of CPT treatment on GSH/GSSG and ROS production was examined in MIAPaCa-2 and BxPC-3 cells. As shown in Fig. 3G, CPT treatment (10 µM) significantly increased H2O2 (ROS) production in MIAPaCa-2 cells but not in BxPC-3 cells. GSH is known to react with ROS to suppress its affects (25). GSH/GSSG was decreased by CPT treatment (10 µM) in MIAPaCa-2 cells but not in BxPC-3 cells (Fig. 3H). These results signified that CPT also affects redox regulation (GSH/GSSG) and ROS production in MIAPaCa-2 cells containing mutant KRAS, which suggests that these changes contribute to the suppression of tumorigenesis by CPT.
Effect of KRAS knockdown on the expression of proteins involved in glutamine and lipid metabolism, redox regulation and ROS production in MIAPaCa-2 pancreatic cancer cells containing mutant KRAS
To clarify the role of K-Ras protein in the suppression of glutamine and lipid metabolism, KRAS was knocked down in MIAPaCa-2 cells using specific siRNAs and the changes in related proteins examined. Introduction of siRNA against KRAS decreased the expression of FASN, P-ACC1 and ACC1 as well as K-Ras protein (Figs. 4A and B, 5A and S5A and B). Expression of GAPDH was unaffected by KRAS siRNA. The expressions of GOT1, GOT2, IDH1, IDH2, GLUD1, GLS and ACLY were minimally affected by KRAS siRNA (Figs. 4A and S5A). These results indicated that the suppression of proteins involved in lipid synthesis, such as FASN and ACC1, resulted from the downregulation of mutant K-Ras protein. Knockdown of KRAS also markedly decreased 3D spheroid formation (~90% decrease; Fig. 5B). The effect of KRAS siRNA on lipid synthesis, ROS production and GSH/GSSG in MIAPaCa-2 cells was also determined. KRAS knockdown decreased the formation of lipid droplets (Figs. 5D and E and S6A and B). It also increased ROS production (Fig. 5G) and reduced the GSH/GSSG (Fig. 5H) similar to CPT treatment (Figs. 3 and S4). To further investigate the relationship among lipid synthesis, ROS production and 3D-spheroid formation, knockdown experiments using siRNA against FASN, a key enzyme in lipid synthesis, were also performed in MIAPaCa-2 cells. As shown in Figs. 5D and F and S6A and C, the formation of lipid droplets was significantly decreased by FASN knockdown. 3D-spheroid formation was also decreased by FASN siRNA treatment, although the degree of suppression (~50%) was less than that observed with KRAS siRNA (~90%; Fig. 5B). Treatment with siRNA against FASN also partially increased ROS production (Fig. 5G) but did not decrease GSH/GSSG (Fig. 5H). FASN knockdown barely altered the level of K-Ras protein (Figs. 5A and S5B). These results indicated that the suppression of lipid synthesis mediated by ACC1 and FASN served an important role in the antioncogenic activity of CPT via the downregulation of K-Ras protein, although pathways other than lipid synthesis may also contribute to this suppression. Collectively, these results demonstrated that the antioncogenic activity of CPT via the downregulation of the K-Ras protein is primarily mediated by the regulation of lipid metabolism. In addition, the enhancement of ROS production is important for this activity (Fig. 6).
Figure 4.
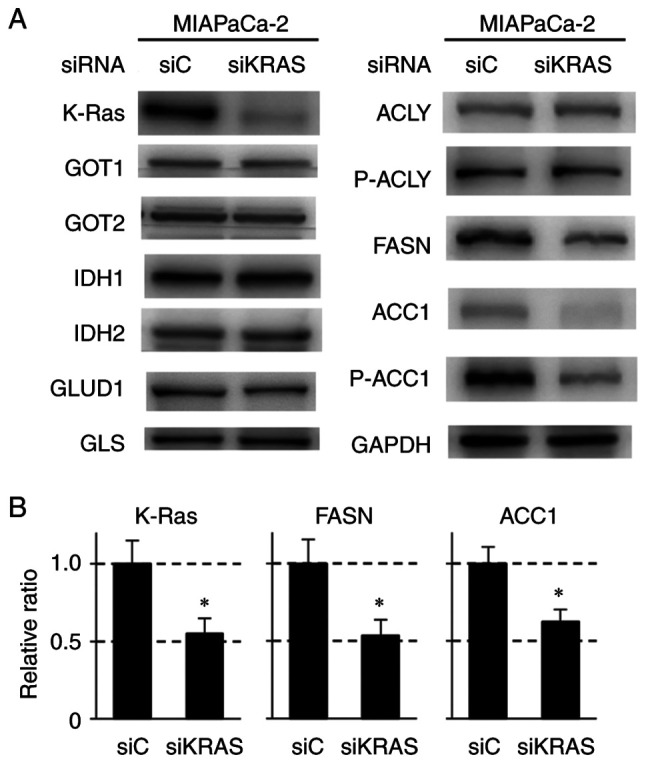
Immunoblot analyses of proteins regulating glutaminolysis and lipogenesis in MIAPaCa-2 cells treated with control (siC) and KRAS siRNA (siKRAS). (A) A total of 1×105 cells were treated with siRNA for 24 h, followed by nonadherent culture (48 h). GAPDH was used as the internal control. (B) Quantitative analysis of protein expression levels of K-Ras, FASN and ACC1 after siKRAS treatment in MIAPaCa-2 cells. The bars indicate the relative expression value normalized to those of GAPDH and are presented as mean ± standard deviation of three independent assays. *P<0.05 compared with siC using Welch's t-test. KRAS, Kirsten rat sarcoma viral oncogene homolog; si, short interfering; FASN, fatty acid synthase; ACC1, acetyl-CoA carboxylase 1; ACLY, ATP-citrate lyase; GOT, glutamic-oxaloacetic transaminase; IDH, isocitrate dehydrogenase; GLUD, glutamate transport system permease protein; GLS, glutaminase; p-, phosphorylated.
Figure 5.
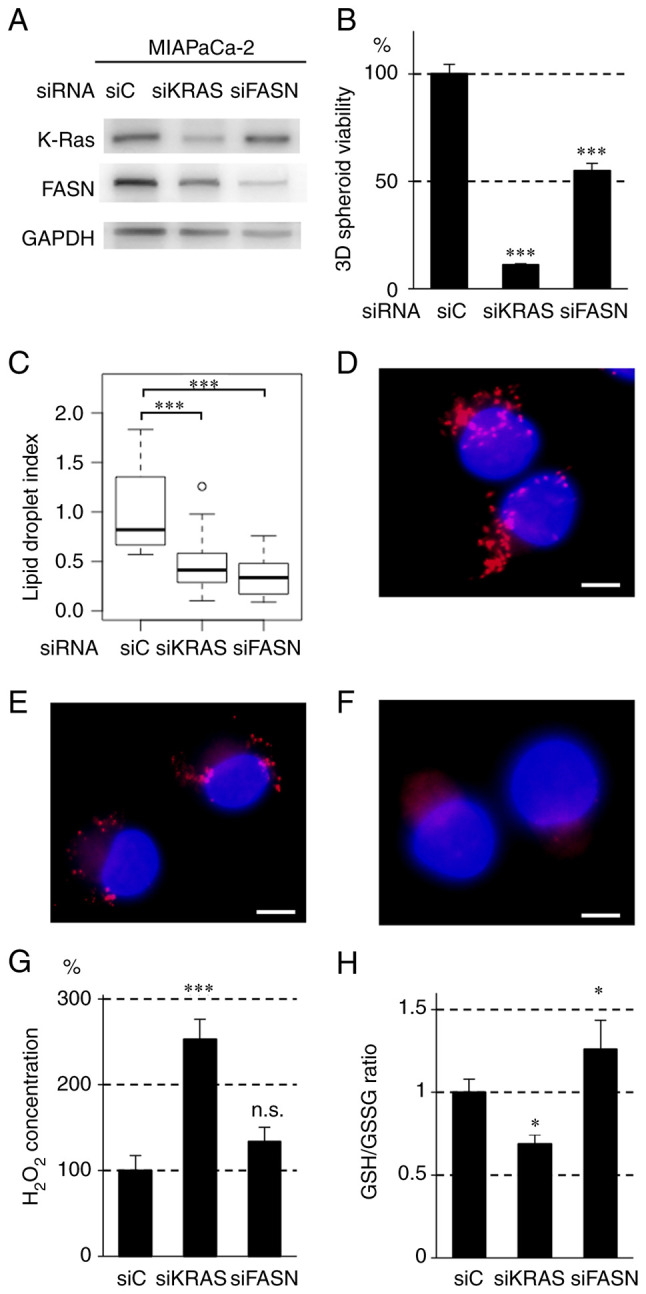
Effect of siRNA against KRAS (siKRAS) and FASN (siFASN) on (B) 3D-spheroid formation, (C-F) lipogenesis and (G and H) redox regulation in MIAPaCa-2 cells. (A) Immunoblot analysis of MIAPaCa-2 cells treated with control (siC), siKRAS and siFASN. A total of 1×105 cells were treated with the siRNAs for 24 h, followed by nonadherent culture for 48 h. GAPDH was used as the internal control. (B) 3D-spheroid viability of cell lines treated with siC, siKRAS and siFASN. A total of 1×105 cells were treated with siRNA for 24 h, followed by spheroid culture in a 96-well V-bottom plate for 72 h in triplicate. 3D-spheroid viability was determined by measuring adenosine triphosphate (ATP) content using the CellTiter-Glo® 3D assay. Error bars represent standard deviation. ***P<0.001 vs. siC using Tukey's test. (C) Box plot of the areas of lipid droplets in MIAPaCa-2 cells treated with siC, siKRAS and siFASN. The cells were seeded into culture dishes at a density of 1.0×105 cells/dish. When the cell density reached ~60%, siKRAS and siFASN were added and incubated for 72 h. The cells were fixed in 4% paraformaldehyde and stained with LipiDyeII and DAPI. The integrated density of LipiDyeII staining (n=20 for each group) was measured using ImageJ software and normalized to the average of the control (siC) group. In the plots, the box indicates the lower and upper quartiles, the horizontal bar represents the median, the whiskers are the highest and lowest data points that fall within 1.5 times the interquartile range from the lower and upper quartiles and the dots are the outliers. ***P<0.001 compared with siC using Tukey's test. Representative images of lipid droplet formation treated with (D) siC, (E) siKRAS and (F) siFASN for 72 h in MIAPaCa-2 cells. The nuclei of the cells were stained with DAPI (blue) and the lipid droplets were stained with LipiDyeII (red). Scale bars=10 µm. (G) H2O2 content in MIAPaCa-2 cells treated with siC, siKRAS and siFASN. A total of 1×105 cells were treated with siKRAS and siFASN for 24 h, followed by nonadherent culture for 48 h. H2O2 concentration in the culture medium was determined using the Amplite fluorimetric H2O2 kit and normalized to cellular protein content. The bars indicate the mean ± standard deviation. ***P<0.001 compared with siC using Tukey's test. n.s., not significant. (H) GSH/GSSG in MIAPaCa-2 cells treated with siC, siKRAS and siFASN. A total of 1×105 cells were treated with siKRAS and siFASN for 24 h, followed by nonadherent culture for 48 h. GSH/GSSG was determined by measuring GSH and GSSG using the GSH/GSSG-Glo Assay kit. The bars indicate mean ± standard deviation. *P<0.05 compared with siC using Tukey's test. si, short interfering; KRAS, Kirsten rat sarcoma viral oncogene homolog; FASN, fatty acid synthase; GSH, glutathione-SH; GSSG, glutathione disulfide.
Figure 6.
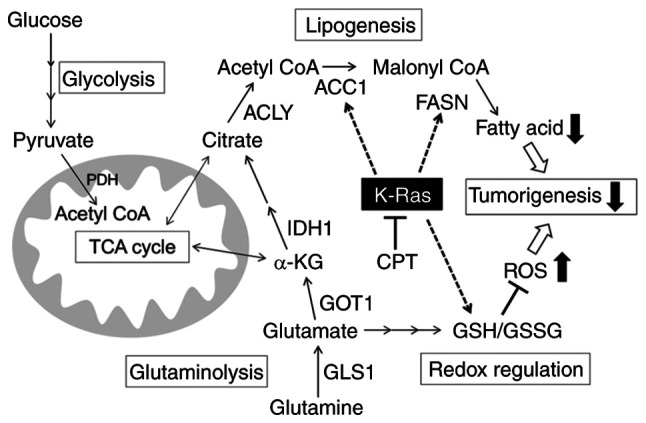
Schematic illustration of the antioncogenic effect of CPT mediated by mutant K-Ras protein. KRAS, Kirsten rat sarcoma viral oncogene homolog; CPT, cryptotanshinone; GOT, glutamic-oxaloacetic transaminase; GLUD, glutamate transport system permease protein; GLS, glutaminase; IDH, isocitrate dehydrogenase; FASN, fatty acid synthase; ACC1, acetyl-CoA carboxylase 1; ACLY, ATP-citrate lyase; ROS, oxygen species.
Discussion
The present study initially demonstrated that the antioncogenic effects of CPT were more pronounced in pancreatic and CRC cells containing mutant KRAS compared with those harboring wild-type KRAS. The 3D-spheroid growth of the cells containing mutant KRAS was more dependent on glutamine than glucose. Moreover, CPT treatment reduced glutamine and lipid metabolism in the MIAPaCa-2 pancreatic cancer cell line containing mutant KRAS. Furthermore, CPT affected redox regulation and increased ROS production. In MIAPaCa-2 cells, CPT treatment decreased the levels of enzymes involved in glutamine and lipid metabolism, including GOT1, GLS, IDH1, ACLY, ACC1 and FASN, as well as K-Ras protein. However, knockdown experiments using siRNA specific for KRAS revealed that the downregulation of the proteins involved in lipid metabolism, ACC1 and FASN and enhancement of ROS production was dependent on the decreased expression of mutant K-Ras protein by CPT. Based on these results, it was concluded that CPT is able to suppress oncogenic growth by inhibiting lipid metabolism and promoting ROS production in a mutant KRAS-dependent manner (Fig. 6).
Although glucose and glutamine are important substrates for the major anabolic pathways, glutamine was predominantly used for cell growth in pancreatic and CRC cells containing mutant KRAS (Fig. S2) and the suppressive effect of CPT was also dependent on mutant KRAS (Fig. 1). The authors previously demonstrated that the downregulation of K-Ras protein was induced by CPT treatment at the post-transcriptional level in pancreatic cells (20). Mutant KRAS exhibits downstream effects by regulating glucose, glutamine and lipid metabolism (19). Indeed, activated mutant KRAS is reported to upregulate GOT1 and downregulate GLUD1 to shift glutamine metabolism from the mitochondrial canonical pathway to the noncanonical pathway for nicotinamide adenine dinucleotide phosphate-reduced (NADPH) production (25). Lipogenic enzymes, including ACLY, ACC1 and FASN, are also increased by mutant KRAS (19), although the underlying molecular mechanisms have not been elucidated. In the present study, the expressions of the enzymes involved in glutamine and lipid metabolism were decreased by CPT treatment in MIAPaCa-2 cells. However, siRNA specific for KRAS suppressed the levels of only ACC1 and FASN, which are involved in fatty acid biosynthesis (Figs. 4A and B and S5A). FASN knockdown did not affect the expression of the K-Ras protein (Figs. 5A and S5B), which indicated that KRAS regulates FASN expression upstream but that its expression is not affected by FASN. ACC1 and FASN are involved in fatty acid synthesis and were attenuated by mutant K-Ras protein in this pancreatic cancer cell line. Other proteins suppressed by CPT treatment are also regulated in a KRAS-independent manner in MIAPacCa-2 cells, which alludes that both KRAS-dependent and -independent pathways regulate glutamine and lipid metabolism via CPT exposure. Although KRAS siRNA markedly suppressed (90%) 3D-spheroid formation, the degree of suppression for FASN was lower (50%) compared with that by KRAS siRNA (Fig. 5B). With respect to ROS production, the effect of KRAS knockdown was significantly higher than that of FASN. Although the KRAS-dependent suppression of lipogenesis contributes to antitumor activity and increased ROS production, its effect is limited. In addition to K-Ras protein activity, other KRAS-independent function(s) of CPT is considered to be required for the complete suppression of tumorigenicity. The upregulation of fatty acid synthesis is a classically described metabolic alteration in cancer (15). This process occurs via the upregulation of various lipogenic enzymes, such as ACLY, ACC1 and FASN. FASN is a multienzyme protein that catalyzes the synthesis of fatty acids from acetyl-CoA and malonyl-CoA in an NADPH-dependent reaction. Increased expression of FASN has been associated with a variety of cancers and an association exists between higher levels of FASN and later stages of disease as well as prognosis (25). FASN inhibitors have attracted interest as a novel therapeutic target for cancer treatment (15). Although a number of FASN inhibitors have been preclinically tested, the antitumor effects of most are pharmacologically limited and exhibit some side effects, with only TVB-2640 entering clinical study (15). As CPT inhibits the expression of FASN and exhibits additional antitumor effects in mouse models (5), favorable clinical activity is expected. CPT treatment also suppressed the expression of IDH1 and other enzymes that regulate glutamine metabolism (Figs. 2 and S3), although downregulation was independent of K-Ras protein suppression (Figs. 4A and S5A). IDH1 is mutated or highly expressed in a number of cancers (27). Diminished IDH1 activity results in reduced α-ketoglutarate (αKG) and NADPH production (25), which results in the exhaustion of reduced glutathione and increased ROS levels (25). Pharmacological inhibition of IDH1 has also been reported to be effective against certain cancers, such as glioblastoma (27). IDH1 may be a target for the KRAS-independent antitumor effect of CPT.
The reduced form of glutathione (GSH) inhibits ROS production (24). GSH/GSSG was reduced and ROS production was increased following CPT treatment and knockdown of KRAS in MIAPaCa-2 cells, which indicated that redox and the ROS axis contributed to the mutant KRAS-dependent antioncogenic effects of CPT (Fig. 6). FASN knockdown resulted in the suppression of lipogenesis but did not reduce GSH/GSSG and minimally increased ROS production (Fig. 5G and H). This finding suggested that the KRAS-dependent suppression of lipid synthesis by CPT was not directly involved in the promotion of ROS production in MIAPaCa-2 cells, although reduced lipogenesis is important for the KRAS-dependent antioncogenic effects of CPT. The KRAS-independent function of CPT, such as the suppression of glutamine metabolism, may serve an additional role in enhanced ROS production by decreasing GSH/GSSG. Metabolic reprogramming and the regulation of ROS production are closely correlated with the malignant progression of cancer (26). To prevent oxidative damage, cancer cells modulate metabolic adaptation to maintain redox balance (24). The present study demonstrated that CPT can alter the reprogrammed metabolic adaptation, including glutamine and lipid metabolisms, to decrease GSH/GSSG and promote ROS production. This process is detrimental to cancer cells and suppresses 3D-spheroid formation in malignant pancreatic cancer cells containing mutant KRAS. Previously, we showed that CPT suppresses cancer cell invasiveness and metastasis in a mouse model (28). Thus, CPT is expected to be a potent therapeutic drug for KRAS-driven intractable cancers.
Supplementary Data
Acknowledgments
Not applicable.
Abbreviations
- CPT
cryptotanshinone
- CRC
colorectal cancer
- PDAC
pancreatic ductal adenocarcinoma
- PDH
pyruvate dehydrogenase
- PDK
pyruvate dehydrogenase kinase
- siRNA
short interfering RNA
- ROS
reactive oxygen species
- ACC1
acetyl-CoA carboxylase 1
- FASN
fatty acid synthase
- ACLY
ATP-citrate lyase
- GSH
glutathione-SH
- GSSG
glutathione disulfide
- NADH
nicotinamide adenine dinucleotide phosphate
- NADPH
nicotinamide adenine dinucleotide phosphate-reduced
Funding Statement
This study was supported by the Japan Society for the Promotion of Scientific KAKENHI (Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science; grant nos. 19K09687, 19K07480 and 19K07663).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
TokT, HT and HI designed the study. TokT, CJK, KM, AU, YT and HI performed the experiments. TokT, YT and HI analyzed and interpreted of data and also confirm the authenticity of all the raw data. YT and HI were responsible for the statistical analyses. TosT and SS provided the colorectal cancer cell lines. TosT, SS, SK and AK were involved in conceiving the project and provided several important suggestions for the research plan. TokT, HT, YT and HI wrote the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sugden MC, Holness MJ. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem. 2006;112:139–149. doi: 10.1080/13813450600935263. [DOI] [PubMed] [Google Scholar]
- 5.Tambe Y, Hasebe M, Kim CJ, Yamamoto A, Inoue H. The drs tumor suppressor regulates glucose metabolism via lactate dehydrogenase-B. Mol Carcinog. 2016;55:52–63. doi: 10.1002/mc.22258. [DOI] [PubMed] [Google Scholar]
- 6.Kinnaird A, Dromparis P, Saleme B, Gurtu V, Watson K, Paulin R, Zervopoulos S, Stenson T, Sutendra G, Pink DB, et al. Metabolic modulation of clear-cell renal cell carcinoma with dichloroacetate, an inhibitor of pyruvate dehydrogenase kinase. Eur Urol. 2016;69:734–744. doi: 10.1016/j.eururo.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 7.Saunier E, Benelli C, Bortoli S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J Cancer. 2016;138:809–817. doi: 10.1002/ijc.29564. [DOI] [PubMed] [Google Scholar]
- 8.Leclerc D, Pham DN, Lévesque N, Truongcao M, Foulkes WD, Sapienza C, Rozen R. Oncogenic role of PDK4 in human colon cancer cells. Br J Cancer. 2017;116:930–936. doi: 10.1038/bjc.2017.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trinidad AG, Whalley N, Rowlinson R, Delpuech O, Dudley P, Rooney C, Critchlow SE. Pyruvate dehydrogenase kinase 4 exhibits a novel role in the activation of mutant KRAS, regulating cell growth in lung and colorectal tumour cells. Oncogene. 2017;36:6164–6176. doi: 10.1038/onc.2017.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stacpoole PW. Therapeutic targeting of the pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase (PDC/PDK) axis in cancer. J Natl Cancer Inst. 2017;109 doi: 10.1093/jnci/djx071. [DOI] [PubMed] [Google Scholar]
- 11.Sutendra G, Michelakis ED. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol. 2013;3:38. doi: 10.3389/fonc.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin C, Yang G, Yang J, Ren B, Wang H, Chen G, Zhao F, You L, Wang W, Zhao Y. Metabolism of pancreatic cancer: Paving the way to better anticancer strategies. Mol Cancer. 2020;19:50. doi: 10.1186/s12943-020-01169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368:eeaw5473. doi: 10.1126/science.aaw5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen M, Hang J. The expanded role of fatty acid metabolism in cancer: New aspects and targets. Precis Clin Med. 2019;2:183–191. doi: 10.1093/pcmedi/pbz017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W, Bai L, Li W, Cui J. The lipid metabolic landscape of cancers and new therapeutic perspectives. Front Oncol. 2020;10:605154. doi: 10.3389/fonc.2020.605154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mustachio LM, Chelariu-Raicu A, Szekvolgyi L, Roszik J. Targeting KRAS in cancer: Promising therapeutic strategies. Cancers (Basel) 2021;13:1024. doi: 10.3390/cancers13061204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han CW, Jeong MS, Jang SB. Understand KRAS and the quest for anti-cancer drugs. Cells. 2021;10:842. doi: 10.3390/cells10040842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaek SA, Papagiannakopoulos T, Shah YM, Lyssiotis CA. Metabolic networks in mutant KRAS-driven tumours: Tissue specificities and the microenvironment. Nat Rev Cancer. 2021;21:510–525. doi: 10.1038/s41568-021-00375-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muyinda IJ, Park JG, Jang EJ, Yoo BC. KRAS, A prime mediator in pancreatic lipid synthesis through extra mitochondrial glutamine and citrate metabolism. Int J Mol Sci. 2021;22:5070. doi: 10.3390/ijms22105070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tambe Y, Terado T, Kim CJ, Mukaisho K, Yoshida S, Sugihara H, Tanaka H, Chida J, Kido H, Yamaji K, et al. Antitumor activity of potent pyruvate dehydrogenase kinase 4 inhibitors from plants in pancreatic cancer. Mol Carcinog. 2019;58:1726–1737. doi: 10.1002/mc.23045. [DOI] [PubMed] [Google Scholar]
- 21.Shirasawa S, Furuse M, Yokoyama A, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–88. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- 22.Chang L, Fang S, Chen Y, Yang Z, Yuan Y, Zhang J, Ye L, Gu W. Inhibition of FASN suppresses the malignant biological behavior of non-small cell lung cancer cells via deregulating glucose metabolism and AKT/ERK pathway. Lipids Health Dis. 2019;18:118. doi: 10.1186/s12944-019-1058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pecot CV, Wu SY, Bellister S, Filant J, Rupaimoole R, Hisamatsu T, Bhattacharya R, Maharaj A, Azam S, Rodriguez-Aguayo C, et al. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol Cancer Ther. 2014;13:2876–2885. doi: 10.1158/1535-7163.MCT-14-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative stress in cancer. Cancer Cell. 2020;38:167–197. doi: 10.1016/j.ccell.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sang-Min J, Navdeep SC, Nissim H. AMPK regulates NADPH homeostasiss to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bueno MJ, Quintela-Fandino M. Emerging role of fatty acid synthase in tumor initiation: Implications for cancer prevention. Mol Cell Oncol. 2020;7:e1709389. doi: 10.1080/23723556.2019.1709389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJ. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene. 2018;37:1949–1960. doi: 10.1038/s41388-017-0077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim CJ, Terado T, Tambe Y, Mukaisho K, Kageyama S, Kawauchi A, Inoue H. Cryptotanshinone, a novel PDK4 inhibitor, suppresses bladder cancer cell invasiveness via the mTOR/β-catenin/N-cadherin axis. Int J Oncol. 2021;59:40. doi: 10.3892/ijo.2021.5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article.