

Summary
The RNA editing enzyme ADAR1 is essential for suppression of innate immune activation and pathology caused by aberrant recognition of self-RNA, a role it carries out by disrupting the duplex structure of endogenous double-stranded RNA species1,2. A point mutation in the Z-nucleic-acid binding domain (ZBD) of ADAR1 is associated with severe autoinflammatory disease3–5. ZBP1 is the only other ZBD-containing mammalian protein6 and its activation can trigger both cell death and transcriptional responses via the kinases RIPK1 and RIPK3, and the protease caspase-87–9. Here, we show that the pathology caused by ADAR1 ZBD mutation is driven by activation of ZBP1. We found that ablation of ZBP1 fully rescued the overt pathology caused by ADAR1 mutation, without fully reversing the underlying inflammatory program caused by this mutation. While loss of RIPK3 partially phenocopied the protective effects of ZBP1 ablation, combined deletion of caspase-8 and RIPK3, or of caspase-8 and MLKL, unexpectedly exacerbated the pathogenic effects of ADAR1 mutation. These findings indicate that ADAR1 is a negative regulator of sterile ZBP1 activation, and that ZBP1-dependent signaling underlies the autoinflammatory pathology caused by mutation of ADAR1.
ADAR1 modifies endogenous RNAs to prevent activation of the innate immune RNA sensors MDA5, OAS/RNAseL, and PKR1,2,10,11. The interferon-inducible p150 isoform of ADAR1 includes an N-terminal ZBD, and a naturally occurring point mutation in this region causes a proline-to-alanine substitution at position 193 in human ADAR14 (Fig 1A). This mutation is present at a remarkably high rate (~1/360 in individuals of northern European descent), and if paired with a loss-of-function mutation on the second allele of ADAR1 causes the severe autoinflammatory disease Aicardi-Goutières Syndrome (AGS)12. Recently, a mouse model of this mutation was reported that recapitulates the genetic underpinnings and aspects of the pathology of AGS; mice carrying the P195A mutation (homologous to human P193A) on one or both alleles of ADAR1 are phenotypically normal, but animals with this mutation combined with deletion of the p150 isoform in the second allele of ADAR1 (AdarP195A/p150null) display liver, kidney, and spleen pathology, are runted, and have a median survival of 25 days5.
Figure 1: Immunopathology in ADAR1-mutant mice is driven by ZBP1.

A: Schematic of ADAR1 and ZBP1. B-D: Parental and expected offspring genotypes (B), observed genotypes (C) and percent survival for each genotype produced (D) by the cross of Adarp150null/WT::Zbp1-a+/− mice to AdarP195A/P195A::Zbp1-a−/− mice. E-F: Observed weights for mice of indicated genotypes at 3 (E), 6 or 8 (F) weeks of age. Survival bars represent littermates across 18 litters, AdarP195A / WT:: Zbp1−/− (n=19), AdarP195A / WT:: Zbp1+/− (n=24), AdarP195A/p150null:: Zbp1+/− (n=14), AdarP195A/p150null:: Zbp1−/− (n=27). **** = p ≤ 0.0001 (Log-Rank Mantel Cox). Litter weights: AdarP195A/WT:: Zbp1+/+ (n=17), AdarP195A/p150null:: Zbp1+/+ (n=18), AdarP195A/p150null:: Zbp1+/+ (n=5). **** = p ≤ 0.0001, unpaired t test, two-tailed. 6 week weights, from left to right n = 15, 19, 18, 17, 2, 1. 8 week weights, from left to right n = 10, 18, 17, 15, 2. Whisker bars (1E,F) are presented as mean +/− SD.
ZBP1 loss rescues ADAR1 mutation
The AdarP195A/p150null model is driven by a point mutation in the ZBD of ADAR1 (Fig 1A). Since ZBP1 contains the only other mammalian ZBD, we wondered whether these two proteins might functionally interact. Consistent with this possibility, we found that ADAR1 co-immunoprecipitated with ZBP1, but that this interaction was abrogated when point mutations were introduced to the ZBD of ZBP1 that prevented RNA binding (Extended Data 1A,B). We also observed that the interaction between ZBP1 and ADAR1 was strengthened by UV crosslinking, which covalently links protein to nucleic acid (Extended Data 1C). As these data suggested that ADAR1 and ZBP1 bind a common ligand via their ZBDs, we wondered if the pathology caused by mutation of the ZBD of ADAR1 in the AdarP195A/p150null mouse model was driven by aberrant ZBP1 activation. To test this idea, we crossed AdarP195A/p150null mice to animals lacking ZBP1, using a widely-used ZBP1 knockout mouse strain13, referred to here as ZBP1-a (Fig. 1B). We observed that AdarP195A/p150null:: Zbp1-a−/− mice were born at expected frequencies and appeared phenotypically normal (Fig 1C,D, Extended Data 2A,B). Following birth, AdarP195A/p150null:: Zbp1-a−/− mice gained weight slightly more slowly than AdarP195A/WT littermates (Fig 1E,F), but otherwise displayed normal phenotype, fertility, and survival. The kidney and liver pathology previously reported in AdarP195A/p150null animals5 was also largely normalized by ablation of Zbp1-a (Extended Data 2C).
As we were generating these data, it was reported that the Zbp1-a−/− mouse line was not fully congenic to the C57BL/6 genetic background14, a finding confirmed by our own single nucleotide polymorphism (SNP) analysis (Extended Data 3A,B). We therefore generated a second cross of AdarP195A/p150null mice to a separately derived, fully congenic Zbp1−/− strain15 referred to here as “ZBP1-g” (Extended Data 3C). AdarP195A/p150null::Zbp1-g−/− mice also appeared phenotypically normal (Fig S3D). Notably, we also observed an extension of survival in AdarP195A/p150null::Zbp1-g+/− animals, an effect not observed in AdarP195A/p150null:: Zbp1-a+/− mice (Extended Data 2A). Together, these data confirm that loss of ZBP1 reverses the immunopathology observed in AdarP195A/p150null mice.
Complete ablation of the p150 isoform of ADAR1 causes uniform lethality during embryonic development, but crossing these mice to animals lacking the dsRNA sensor MDA5 (encoded by Ifih1) allows Adarp150null/p150null mice to survive to birth1. Since MDA5 is a potent inducer of type-I interferon (IFN), and since ZBP1 expression is stimulated by IFN (Extended Data 4A), we hypothesized that loss of MDA5 might rescue ADAR1-p150 knockout mice by preventing ZBP1 upregulation. However, in MEF cells, we observed that while loss of MDA5 completely abrogated IFN production induced by ADAR1 depletion, ZBP1 upregulation was only modestly attenuated (Extended Data 4B). This suggested that loss of ADAR1 could lead to ZBP1 upregulation and activation even in the absence of MDA5. Consistent with this finding, we observed that the survival of Adarp150null/p150null::Ifih1−/− mice was modestly but significantly extended by concurrent deletion of ZBP1 (Extended Data 4C). However, deletion of ZBP1 alone did not allow Adarp150null/p150null mice to survive to birth (Extended Data 4D). Together, these data indicate that the developmental lethality induced by ADAR-p150 deletion is mediated by simultaneous activation of ZBP1, MDA5, and additional pathways.
Inflammation in “rescued” mice
AdarP195A/p150null mice display aberrant activation of MDA5, which drives IFN-dependent inflammation. We hypothesized that this IFN-dependent signature would still be present in AdarP195A/p150null:: Zbp1-g−/− mice, despite their normal phenotype. Consistent with this possibility, RNAseq analysis of spleens from 23-day old pups revealed that many aspects of the aberrant inflammatory and interferon stimulated gene (ISG) signature present in AdarP195A/p150null mice was also present in AdarP195A/p150null:: Zbp1−/− animals, despite no alterations in splenic cellularity (Fig 2A-C, Extended Data 5A,B, Extended Data 6). Gene ontology analysis confirmed that the antiviral gene signature induced by ADAR1 mutation was conserved in AdarP195A/p150null:: Zbp1-g−/− animals (Extended Data 5C). This analysis also identified gene signatures present in AdarP195A/p150null mice but absent in AdarP195A/p150null:: Zbp1-g−/− animals (Extended Data 5D). These genes may indicate targets of ZBP1 signaling or may represent pathways upregulated as a result of the immunopathology present in AdarP195A/p150null animals. In carrying out these analyses, we noted that while cells from AdarP195A/p150null:: Zbp1−/− maintain many aspects of the ISG signature observed in AdarP195A/p150null animals, its magnitude is reduced (Fig 2C, Extended Data 5A). This was confirmed by direct comparison of MEF cells isolated from AdarP195A/p150null or AdarP195A/p150null:: Zbp1−/− mice (Fig 2D), and suggested that ZBP1 may play a role in augmenting ISG upregulation, a function previously ascribed to ZBP1 in other contexts16.
Figure 2: The inflammatory program initiated by ADAR1 mutation remains intact upon ZBP1 knockout.
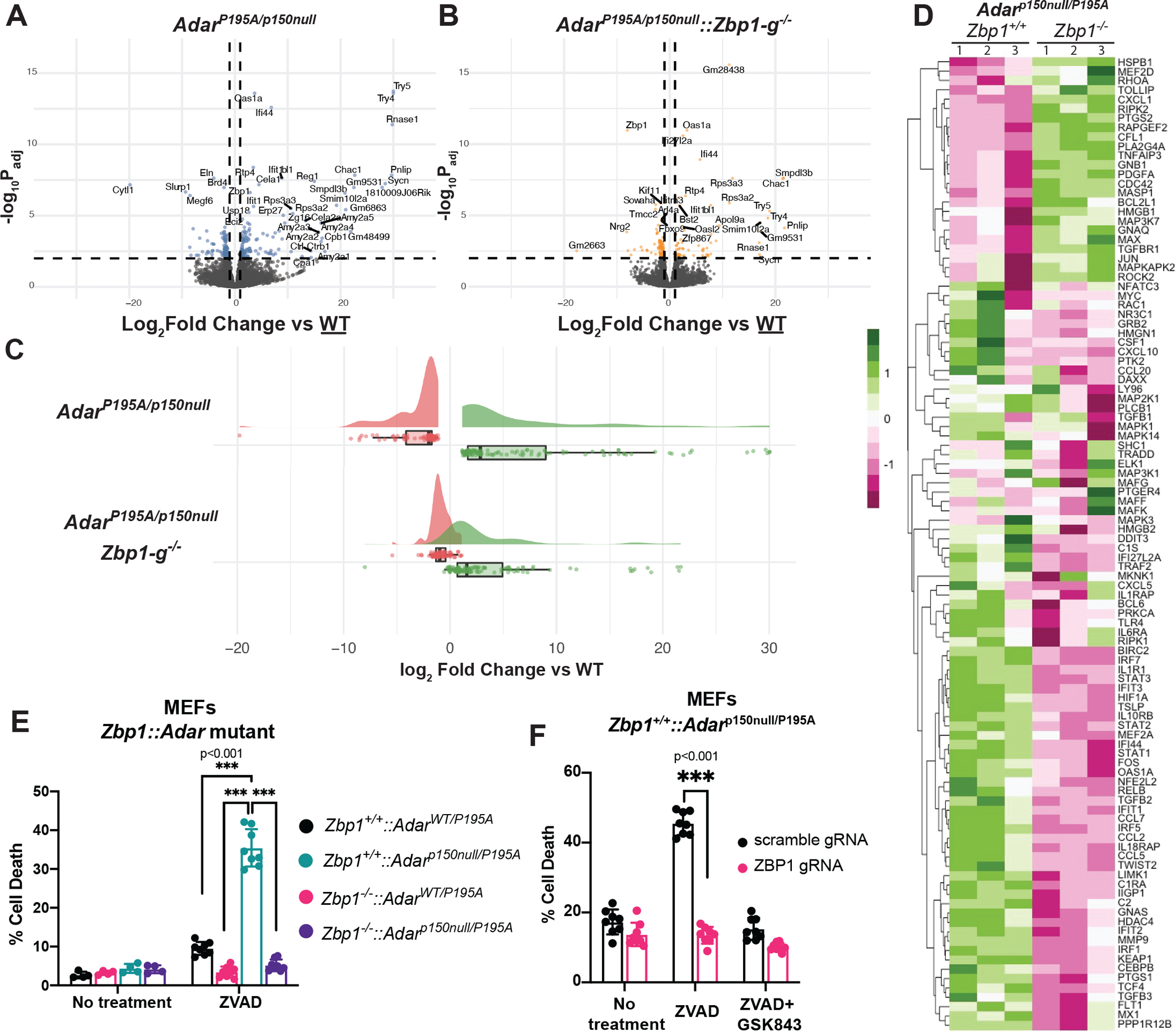
A-B: Volcano plots depicting differential expression of genes detected by RNA sequencing of whole spleen tissue derived from 23 day old mice of the genotypes (A) AdarP195A/p150null (affected) n=4 or (B) AdarP195A/p150null::Zbp1-g−/− (rescued) n=5, in each case compared to spleens derived from wild-type pups (n=4). C: Raincloud plots of affected (n=4) and ZBP1 (n=5) rescued animals depicting the statistically significant up- and down-regulated ADAR signature identified in A. in. ADAR1 signature was identified from the differential analysis of ADAR vs WT from genes with a log2 fold change > 1 and adjusted p-value < 0.01. D. Heatmap analysis of differential gene expression from Nanostring analysis of wild-type vs. Zbp1−/− ADAR mutant MEFs. E-F. Cell death, as measured by loss of plasma membrane integrity using an IncuCyte imager, of Adar mutant MEFs after 8 hour ZVAD treatment on (E) Adar::Zbp1 mutant MEFs (genetic knockout/mutants) or (F) Adar mutant MEFs with CRISPR-Cas9 knockout of ZBP1. Each experimental group (bar) contains n=8 biologic replicates (8 wells). Statistical significance determined by unpaired student t-tests, two-tailed. Incucyte analyses (E and F) are single representatives of independently duplicated experiments. Whisker bars (2E,F) are presented as mean +/− SD.
We next sought to understand the pathways downstream of ZBP1 that are activated by ADAR1 mutation. We observed that culturing MEF cells from AdarP195A/p150null mice led to the ZBP1-dependent loss of RIPK3 expression (Extended Data 7A), suggesting that the ZBP1-RIPK3 pathway is constitutively active in these cells. Consistent with this, we observed that despite their reduced RIPK3 expression, treatment of these cells with the caspase inhibitor zVAD caused ZBP1-dependent RIPK3 phosphorylation and cell death (Fig 2E,F, Extended Data 7A,B). Together, these results indicated that AdarP195A/p150null cells are sensitized to ZBP1-dependent necroptosis.
Cell death signaling upon ADAR1 mutation
Given this finding, we next sought to address the role of the necroptotic pathway in the pathology of AdarP195A/p150null mice. To do this, we crossed AdarP195A/p150null mice to animals lacking different components of the necroptotic pathway. Ablation of the necroptotic effector MLKL did not alter the phenotype of AdarP195A/p150null mice (Fig 3A, Extended Data 8A), nor did crossing them to mice in which the kinase activity of RIPK1 is absent (Fig 3B, Extended Data 8B), indicating that prevention of canonical necroptosis alone was not sufficient to explain the effect of ZBP1 knockout in these animals.
Figure 3: ZBP1-induced necroptosis does not underlie immunopathology induced by ADAR1 mutation.
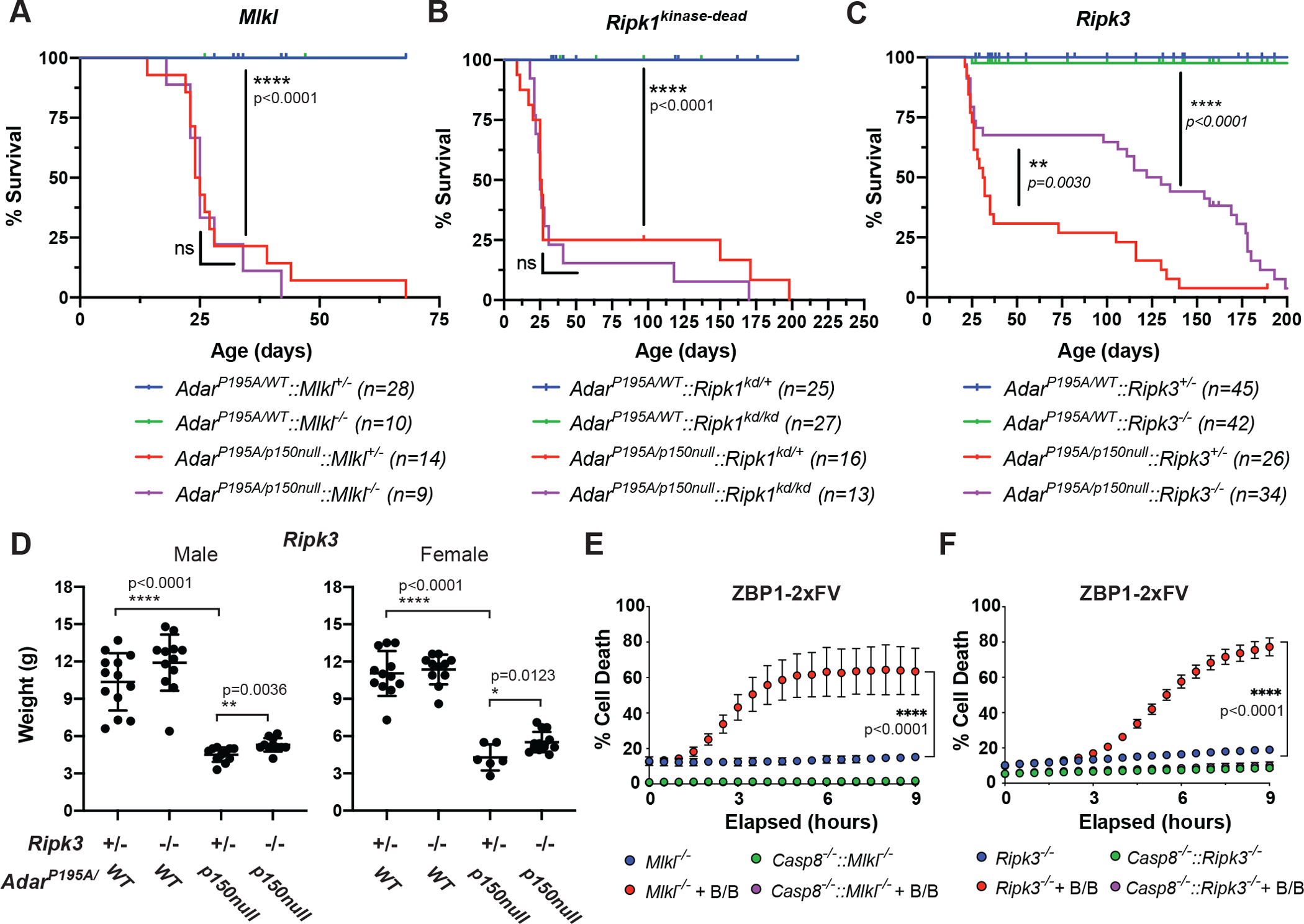
A-C: Survival proportions for AdarP195A/p150null mice crossed to animals lacking MLKL (A), carrying a point mutation abrogating the kinase activity of RIPK1 (Ripk1kd) (B) or lacking RIPK3 (C). In each case, the result of a breeding scheme analogous to that depicted in Fig. 1B is shown. Survival proportions (A-C) represent littermates from 9 (A, Mlkl), 10 (B, Ripk1kd) or 18 (C, Ripk3) litters; AdarP195A / WT:: Mlkl+/− (n=28), AdarP195A / WT:: Mlkl−/− (n=10), AdarP195A/p150null:: Mlkl+/− (n=14), AdarP195A/p150null:: Mlkl−/− (n=9). AdarP195A / WT:: Ripk1kd/+ (n=25), AdarP195A / WT:: Ripk1kd/kd (n=27), AdarP195A/p150null:: Ripk1kd/+ (n=16), AdarP195A/p150null:: Ripk1kd/kd (n=13). AdarP195A / WT:: Ripk3+/− (n=28), AdarP195A / WT:: Ripk3−/− (n=10), AdarP195A/p150null:: Ripk3+/− (n=14), AdarP195A/p150null:: Ripk3−/− (n=9). Survival statistics determined by Log-Rank (Mantel-Cox) test, exact p values indicated on curves. D: Weights of male or female Ripk3−/− mice, observed at 21 days after birth. Litter weights: AdarP195A/WT:: Ripk3+/− (m/f n=13/12), AdarP195A/WT:: Ripk3−/− (m/f n=12/11), AdarP195A/p150null:: Ripk3−/− (m/f n=11/6), AdarP195A/p150null:: Ripk3+/− (m/f n=11/13). Statistical differences determined by individual unpaired t tests (two-tailed), exact p values indicated on plots. E-F: Cell death, as measured by loss of plasma membrane integrity, observed in MEFs from (E) Mlkl−/− and Mlkl−/−::Casp8−/− (n=3 for each group) and (F) Ripk3−/− and Ripk3−/−::Casp8−/− (n=4 for each group) genotypes stably expressing 2xFV-ZBP1, following treatment with the activating drug B/B. E and F are representative of three independently replicated experiments. Whisker bars (3D-F) are presented as mean +/− SD.
In contrast, ablation of RIPK3 led to a significant extension of survival in AdarP195A/p150null mice (Fig 3C). However, this extension of survival was partial, with approximately 1/3rd of AdarP195A/p150null::Ripk3−/− animals succumbing to death within 40 days of birth, followed by slower attrition of mice over the following 200 days. AdarP195A/p150null::Ripk3−/− were severely runted, in contrast to the overtly normal phenotype observed in AdarP195A/p150null:: Zbp1−/− mice (Fig 3D). These findings indicate that ZBP1 can drive pathology in AdarP195A/p150null via signaling that is independent of RIPK3. Following influenza infection, ZBP1 was reported to induce RIPK3 dependent induction of both caspase-8-dependent apoptosis and MLKL-dependent necroptosis17. Nonetheless, our finding that in AdarP195A/p150null mice ablation of RIPK3 failed to fully recapitulate the reversal of pathology observed upon ablation of ZBP1 implies RIPK3-independent functions of ZBP1 when activated downstream of ADAR1 deficiency.
ZBP1 contains three confirmed RIP homotypic interaction motifs (RHIM), which bind to similar domains present in RIPK1 and RIPK318. We hypothesized that in the absence of RIPK3, ZBP1 may interact with RIPK1 to scaffold and activate caspase-8 dependent apoptosis. To study this, we turned to a reductive system in which ZBP1 could be directly activated. By replacing the ZBD of ZBP1 with tandem inducible dimerization domains derived from the protein FK506, we created a form of ZBP1 that could be activated using the cell permeable small molecule B/B (Extended Data 9A). In wild-type LET1, SVEC or MEF cells this construct, termed “2xFV-ZBP1”, triggered cell death that was blocked only with combined inhibition of RIPK3 and the caspases, consistent with induction of both apoptosis and necroptosis downstream of ZBP1 activation (Extended Data 9B-D). Furthermore, B/B activation of ZBP1 resulted in phosphorylation of RIPK3 and MLKL, and when ZBP1 was directly activated in Mlkl−/− MEF cells it induced robust cell death that was dependent on caspase-8 and involved cleavage of caspase-3, consistent with apoptosis (Fig. 3E, Extended Data 10A, B). ZBP1 activation in Ripk3−/− MEF cells also induced caspase-8-dependent cell death and caspase-3 cleavage, albeit with slower kinetics than those observed in Mlkl−/− cells (Fig. 3E,F). Notably, ZBP1 activation did not trigger detectable cell death in MEF cells lacking both MLKL and caspase-8, or both RIPK3 and caspase-8 (Fig. 3E,F). This finding indicates that while RIPK3 can contribute to ZBP1-dependent apoptosis, ZBP1 can still drive caspase-8-dependent cell death responses in the absence of RIPK3.
Caspase-8 suppresses lethal inflammation
We reasoned that cell death dependent on ZBP1, RIPK1 and caspase-8 could underlie the pathology observed in AdarP195A/p150null::Mlkl−/− and AdarP195A/p150null::Ripk3−/− mice. Deletion of caspase-8 causes embryonic lethality due to unrestrained necroptosis18, but this phenotype is reversed by co-ablation of RIPK3 or MLKL19–21. We therefore generated AdarP195A/p150null::Mlkl−/−::Casp8−/− and AdarP195A/p150null::Ripk3−/−::Casp8−/− mice to test whether ablation of caspase-8 in addition to necroptotic signaling would recapitulate the phenotypic rescue observed upon ablation of ZBP1. Unexpectedly, AdarP195A/p150null::Ripk3−/−::Casp8−/− mice displayed reduced weight and survival compared to AdarP195A/p150null::Ripk3−/−::Casp8+/− littermates (Fig. 4A,B, Extended Data 10C), while AdarP195A/p150null::Mlkl−/−::Casp8−/− mice were born at expected frequencies but uniformly failed to survive to weaning (Fig 4C). Histological analysis of these animals at birth revealed broadly normal development of the liver and kidney (Extended Data 11A-C), but a significant increase in Iba1-positive activated microglia in the brains of AdarP195A/p150null::Mlkl−/−::Casp8−/− neonates, consistent with unrestrained inflammatory signaling at this site (Extended Data 11D,E). These findings imply that caspase-8 suppresses ZBP1 activation in AdarP195A/p150null mice, and that caspase-8 ablation may allow unrestrained ZBP1 transcriptional signaling that is independent of canonical apoptosis or necroptosis. Consistent with this possibility, co-immunoprecipitation experiments revealed recruitment of RIPK1, or of both RIPK1 and RIPK3, upon 2xFV-ZBP1 activation in Ripk3−/−::Casp8−/− or Mlkl−/−::Casp8−/− MEF cells, respectively, despite the lack of cell death responses observed in these conditions (Fig 3E,F Extended Data 12A,B,). We also observed that in AdarP195A/p150null::Mlkl−/− MEF cells, zVAD treatment stabilized the interaction between ZBP1 and RIPK3, again in the absence of cell death responses (Extended Data 12C). These findings indicate that when necroptotic effectors are absent, loss or inhibition of caspase-8 promotes interactions between ZBP1 and the RIP kinases.
Figure 4: Caspase-8 suppresses lethal inflammatory signaling in ADAR1-mutant mice.
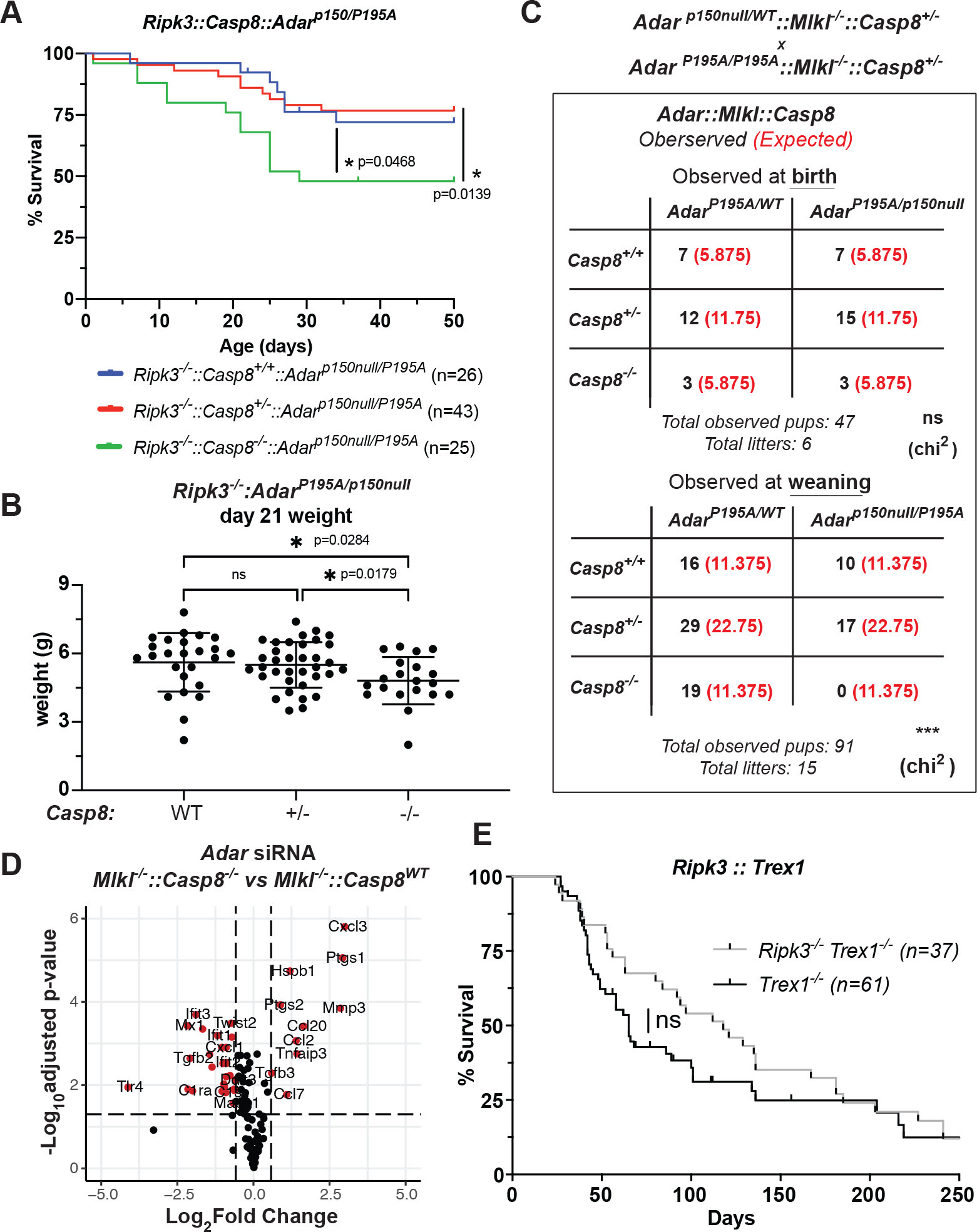
A-B: Ripk3−/−::Casp8−/−::AdarP195A/p150null cross (A) survival and (B) weight at weaning (d.21). A: Log-rank (Mantel-Cox) statistical test performed on survival curves for survival to day 50 between Ripk3−/−::Adarp150nullP195A littermates (AdarP195A/WT mice excluded from comparison) which were Casp8+/+ (n=26), Casp8+/− (n=43) and Casp8−/− (n=25) showed a significant decrease in survival proportion of Casp8−/− mice compared to Casp8+/+ (p=0.0568) and Casp8+/− (p=0.0139) mice. B: Difference between Casp8+/+ and Casp8+/− was not significant. Individual student t-tests performed on affected mouse weights (Adarp150null/P195A) show a statistically significant decrease in Casp8−/− (n=20) compared to Casp8+/+ (n=25), p=0.0284 and Casp8+/− (n=35), p=0.0179 mice. C: Observed (black) and expected (red) mice from Mlkl−/−::Casp8−/−::AdarP195A/p150null cross at birth (day 0) and at weaning (day 21). Chi square power analysis was performed on observed:expected frequencies at birth (not significant) and weaning (p=0.0032**). D: Volcano plots showing differential gene expression from Nanostring analysis of MEF cells derived from Mlkl−/− or Mlkl−/−::Casp8−/− embryos, following siRNA-mediated depletion of ADAR1. E. Survival proportions for Trex1−/− (n=61) or Trex1−/−::Ripk3−/− (n=37) mice. Not significant (Mantel-Cox Log-Rank). Whisker bars (4B) are presented as mean +/− SD.
We next assessed whether loss of caspase-8 potentiated transcriptional signaling induced by ADAR1 insufficiency. We observed that while siRNA-mediated depletion of ADAR1 in Mlkl−/− cells induced the expected upregulation of ISGs, ADAR1 depletion in Mlkl−/−::Casp8−/− MEFs induced a distinct transcriptional response dominated by NF-κB targets (Fig. 4D, Extended Data 13A,B). Since RIPK1 is a key signaling adapter upstream of NF-κB activation22, this finding implied that ZBP1-RIPK1 signaling may underlie these transcriptional effects. Consistent with this, we observed that 2xFV-ZBP1 activation in Mlkl−/− MEF cells induced RIPK1-dependent upregulation of the NF-κB targets CCL2 and CCL7, but not of the canonical ISG IFIT1 (Extended Data 13C). Together, these data indicate that upon ADAR1 mutation or depletion, caspase-8 acts to suppress a ZBP1- and RIPK1-dependent program of inflammatory transcription. The modest extension of survival observed in AdarP195A/p150null::Ripk3−/−::Casp8−/− mice relative to AdarP195A/p150null::Mlkl−/−::Casp8−/− animals suggests that RIPK3 can potentiate, but is not required for ZBP1- and RIPK1-dependent inflammatory signaling.
This study does not address the identity of the ligand(s) responsible for activating ZBP1 in Adar1P195A/p150null mice. Since the P195A mutation in ADAR1 lies in its ZBD, we can speculate that ADAR1P195A may be attenuated in its ability bind ZBD ligands. Interestingly, aligning the ZBD sequences of ADAR1 and ZBP1, along with those present in the fish PKR homologue PKZ23 and the vaccinia virus effector E3L24 reveals that ZBP1 naturally contains an alanine at position 64, the site homologous to ADAR1 P195 (Extended Data 14A). Since substitution of proline with alanine at this site in ADAR1 limits its function, we speculate that the presence of an alanine at the homologous site within ZBP1 may reflect a naturally lower affinity for ligand by ZBP1 relative to other ZBDs. This may represent a means to limit aberrant ZBP1 activation at steady state. We sought to test this idea by creating a mouse line with a “revertant” ZBP1, in which A64 is mutated to proline. However, ZBP1A64P mice did not reveal evidence of increased ZBP1 activation, either when crossed to the Adar1P195A/p150null model or in response to influenza infection, but rather appeared attenuated in their signaling in both settings (Extended Data 14 B,C). While ZBP1A64P protein was properly expressed (Extended Data 14D), this attenuation likely reflects a disruption of protein structure induced by this mutation, and implies that additional mutations or larger domain swaps would be needed to effectively test this hypothesis. Future studies will clarify the identity of ZBP1 ligands that emerge upon ADAR1 mutation.
Aicardi-Goutières Syndrome refers to a family of IFN-driven congenital pathologies driven by mutations in proteins involved in nucleotide sensing and regulation12. Since ZBP1 is strongly induced by IFN, and has been described to bind DNA as well as RNA16, we wondered if ZBP1 and RIPK3 signaling might play a role in AGS-like pathology driven by endogenous DNA ligands. To test this, we assessed mice lacking TREX1, a DNA exonuclease whose ablation causes aberrant activation of the cGAS-STING pathway25,26. However, we did not observe significant amelioration of pathology or extension of survival in Trex1−/− mice when RIPK3 was ablated, unlike the partial rescue observed in Adar1P195A/p150null animals upon RIPK3 knockout (Fig 4F, 3C). This suggests that engagement the ZBP1-RIP kinase pathway is a feature of dysregulated endogenous RNA, but not DNA, sensing.
The unexpected susceptibility of Ripk3−/−::Casp8−/− and Mlkl−/−::Casp8−/− animals to ADAR1 mutation indicates that while these animals develop normally, they are poised for hyperactive inflammatory signaling in response to ZBP1 activation. Indeed, previous studies have found that Fadd−/−::Mlkl−/− mice (comparable to Mlkl−/−::Casp8−/−) are highly susceptible to influenza infection8, a setting in which ZBP1 is strongly activated. While this was interpreted as indicating a requirement for functional cell death pathways for antiviral defense, our data raise the possibility that these mice succumb to overexuberant inflammatory signaling triggered by ZBP1. Notably, we did not observe engagement of pyroptotic cell death upon ZBP1 activation in cells lacking caspase-8 in combination with RIPK3 or MLKL, though our data do not rule out contribution of pyroptotic signaling to the immunopathology we observe.
Our findings identify ZBP1 as a key effector of autoinflammatory pathology induced by mutation of the Z-DNA binding domain of ADAR1. Our data also highlight the pleiotropic nature of ZBP1 signaling; while ZBP1 ablation fully rescued the pathology of the Adar1P195A/p150null model, individual deletion of the necroptotic signaling molecules MLKL or RIPK3 did not, and ablation of caspase-8 unleashed a lethal inflammatory program, apparently in the absence of ZBP1-dependent programmed cell death. These findings are consistent with the dual functions of caspase-8 as both an inducer of cell death and a suppressor of ZBP1-dependent necroptosis and inflammation. Indeed, both the presence and the absence of caspase-8 may drive pathology in ADAR1-mutant mice. Our in vitro data indicate that ZBP1 activation in the absence of RIPK3 can induce caspase-8-dependent cell death, and this pathway may contribute to the pathology of Adar1P195A/p150null::Ripk3−/− mice; conversely, additional ablation of caspase-8 in these animals exacerbates the observed pathology by instead unleashing unrestrained inflammatory signaling. Ultimately, apoptosis, necroptosis and inflammatory transcription may all contribute to the pathology of Adar1P195A/p150null mice, and which of these pathways is engaged upon ADAR1 mutation likely varies between tissues and cell types depending on the abundance of the pathway components as well as of regulatory proteins such as cFLIP and the IAPs. This pleiotropy also suggests that even the combined targeting of apoptosis and necroptosis using small molecule inhibitors is unlikely to reverse AGS pathology driven by ADAR1 mutation.
Materials and Methods
Mice
Mouse strains with modifications to Zbp1-a13, Zbp1-g15, AdarP195A/p150null5, Mlkl27, Ripk1kd28, Ripk1mutRHIM29, Ripk330 and Casp819,31, Trex126 and Ifih1(MDA5)1 have been previously described. Zbp1A64P mice were generated as previously described32, using the sgRNA target sequence CCGCCTATGCTCCATGTTGCAGG and the repair template sequence AAAACCCTCAATCAAGTCCTTTACCGCCTGAAGAAGGAGGACAGAGTGTCCTCCCCA GAGCCTCCAACATGGAGCATAGGCGGGGCTGCTTCTGGAGATGGGGCTCCTGCAATCCCTGAGAACTCCAGT. Briefly, C57BL6/J oocytes were microinjected with Cas9 complexed with sgRNA and ssDNA donor template as described, then implanted into pseudopregnant female mice. Founder pups were screened using Surveyor assay, and resulting mice were bred to homozygosity and genotyped using a Taqman probe system to detect the Zbp1A64P g>c nucleotide change using the primer sequences CCTCAATCAAGTCCTTTACC (Sense) and GACAGATTACCAAGGCTAGG (Antisense), CAGAGCCTGCAACATGGAG (wild type probe), CAGAGCCTCCAACATGGAG (mutant probe). All mice were housed in pathogen-free facilities at the University of Washington under 12-h light– dark cycles with access to food and water ad libitum. Temperatures were set to 74 ± 2 °F with humidity of 30–70%. All animals used were cared for and used in experiments approved by the University of Washington Institutional Animal Care and Use Committee (under protocols 4298–01 and 4190–01) in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facility in accordance with the Guide for the Care and Use of Laboratory Animals and applicable laws and regulations. In breeding experiments, no specific criteria were used to determine final sample size. These experiments were not randomized or blinded.
SNP typing analysis of Zbp1-a and Zbp1-g mouse strains was performed by Taconic Biosciences using the Mouse Genome Scan Panel.
Cell lines
Mouse embryonic fibroblasts were generated from E15 pups and immortalized by retroviral transduction of the SV40 Large T antigen. HEK293T, LET1 (Lung Epithelial Type-1) and MEF (Murine Embryonic Fibroblast) cells were maintained in standard conditions: D-MEM supplemented with 10% fetal bovine serum, glutamine, penicillin and streptomycin. Following isolation and immortalization, MEF lines were tested for cell death competence by stimulation with TNF/ZVAD (RIPK3/RIPK1 dependent cell death) or IAV infection (ZBP1/RIPK3 dependent).
Plasmids and lentiviral vectors and siRNA
2xFV-ZBP1 was generated by replacing the first 146 amino acids of murine ZBP1 (corresponding to the ZBD) with tandem copies of FKBPF36V, available from Clontech. The resulting fusion gene was cloned into the Tet-based inducible expression pSLIK vector33, and this construct was used to create lentiviral particles for transduction of target lines using standard protocols. Expression and activation of this construct was achieved by inducing 2xFV-ZBP1 expression with 1 μg/mL doxycycline for 12 hours, then treating with 100 mM “B/B homodimerizer” (Clontech). The full ADAR1-p150 isoform was cloned from mouse cDNA directly into a ‘pRRL’ lentiviral backbone and subsequently sequenced to confirm identity. Wild-type and mZαβ ZBP1 were subcloned from constructs previously obtained from the lab of Jason Upton into the pRRL backbone, at which point a 3xFLAG tag was added to the C-terminus.
CRISPR-Cas9-mediated deletion was achieved using a lenti-CRISPR construct created by Dr. Daniel Stetson5, into which guide sequences listed below were inserted. These were used to create VSV-G-pseudotyped lentivirus particle, which were used to transduce target cells. Following 10–14 days of antibiotic selection, deletion of target proteins was confirmed by Western blot. The sequences of the guide RNA (gRNA) target sites are as follows, with the protospacer adjacent motif (PAM) sequence underlined: non-targeting control gRNA: Scramble gRNA: GACGGAGGCTAAGCGTCGCAA, Zbp1 gRNA: GAGCCTGCAACATGGAGCAT, Ifih1 (MDA5) gRNA: GTGTGGGTTTGACATAGCGCG.
SiRNA experiments were carried out by transfecting cells with SMARTpool siRNA cocktails (Dharmacon Horizon Discovery) targeting ADAR1 (siGenome mouse Adar, Entrez Gene 56417), RIPK1 (siGenome mouse Ripk1, Entrez Gene 19766), or a non-targeting ‘scramble’ control (siGenome Non-targeting siRNA Pool #1). Transfection of siRNA was performed using the dharmaFECT 1 transfection reagent (Cat. No. T-2001–03, Horizon Discovery) according to the manufacturer’s protocols.
Antibodies and inhibitors
Where indicated, the following drugs were used at the listed concentrations: 50 μM zVAD (SM Biochemicals), 100nM GSK’843 (GlaxoSmithKline).
The following antibodies were used for Western Blots and immunoprecipitations: ADAR1 (15.8.6) SantaCruz, ZBP1 (Zippy-1) AdipoGen, actin (13E5) Cell Signaling Technology, MDA5 (D74E4) Cell Signaling Technologies, p-RIPK3 (GEN135–35-9), Genentech, RIPK3 (1G6.1.4) Genentech, or RIPK3 (2283) ProSci, p-MLKL (D6E3G) Cell Signaling Technologies, MLKL (MABC604) Millipore, Caspase-3 (9662) Cell Signaling Technologies, Cleaved Capsase-3 (9661) Cell Signaling Technologies, RIPK1 (38/RIP) BD Biosciences, anti-FLAG (M2) Sigma, anti-FKBP12, Thermo Fisher (PA1–026A). Iba1 (Cat no. 019–19741) Wako-Chem, Cleaved Caspase 3 (Clone D3E9) Cell Signaling Technologies were used in immunohistochemical analysis.
The following antibodies were used for flow cytometry analysis of splenocytes: FITC anti-CD19 (clone 1D3; BD Biosciences) PerCP-Cy5.5 anti-CD3e (clone 145–2C11; BD Biosciences), PE-Cy7 anti-Ly6C (clone HK1.4; Biolegend), APC anti-F4/80 (clone BM8; eBioscience), AF700 anti-Ly6G (clone 1A8; Biolegend), APC-Cy7 anti-NK1.1 (clone PK136; BD Biosciences), BV510 anti-CD8a (clone 53–6.7; BD Biosciences); BV605 anti-CD4 (clone RM4–5; BD Biosciences), BV650 anti-CD11b (clone M1/70; Biolegend), and BUV395 anti-CD45.2 (clone 104; BD Biosciences).
Western Blots, Immunoprecipitations and IR-CLIP
WT 293T cells or 293T cells expressing FLAG-ZBP1 or FLAG-ZBP1 mZαβ were cultured in 6-well plates and were transfected with pRRL vector expressing ADAR1 p150 or empty pRRL vector using X2 transfection reagent (Mirus). At ~24 hrs post-transfection, cells were washed in PBS and crosslinked with 254 nm UV-C light (0.3 J/cm2) or left un-crosslinked, and lysed in 200 uL IP lysis buffer (25 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5 mM EDTA, 5 mM MgCl2 + 1X protease inhibitor). Lysates were clarified by centrifugation at 5000 xg for 10 mins at 4C, and quantified by Bradford assay. A fraction of each sample was stored for input controls. 200 ug of each lysate was incubated with Protein G Dynabeads (Thermo-Fisher) pre-conjugated with 4 ug anti-FLAG M2 antibody (Sigma) in a final volume of 500 uL IP lysis buffer for 2 hrs at 4C with rotation. Beads were then washed four times with 1 mL IP lysis buffer. Beads were then boiled in 2X Laemmli buffer (Bio-Rad) + 5% beta-mercaptoethanol to eluted protein complexes. Eluates and input controls were resolved on a 4–15% TGX gel (Bio-Rad) for SDS-PAGE, transferred to PVDF membranes. Immunoblotting were performed using HRP-conjugated anti-FLAG and anti-β-Actin antibodies, and with anti-ADAR1 primary antibody and HRP-conjugated anti-mouse secondary antibody (Jackson Immunolabs).
For RIPK3 and RIPK1 immunoprecipitations, Cells were lysed in ice-cold lysis buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% v/v Triton X-100, 10% v/v glycerol, and 0.01% w/v SDS) supplemented with 1X complete Protease Inhibitor (Roche) for 30 minutes, followed by centrifugation at 1000xg for 10 minutes. Antibodies recognizing ZBP1 or FKBP12 (Invitrogen, PA1–026A) were immobilized to Dynabeads ™ Protein G (Invitrogen) as per the manufacturer’s instruction, and then incubated with total cell lysates overnight at 4°C. Immunoprecipitates were eluted in Laemmli sample buffer (63 mM Tris-HCl, pH 8.0, 10% v/v glycerol, 2% w/v SDS, 0.01% w/v bromophenol blue, 2.5% v/v 2-mercaptoethanol) at 95°C for 10 minutes.
We performed irCLIP as described by Zarnegar et al.34 with slight modifications. Wild-type 293T cells or 293T cells stably expressing FLAG-ZBP1 or FLAG-ZBP1 mZαβ were cultured in 6-well plates. Cells were washed with PBS, crosslinked with 254 nm UV-C light (0.3 J/cm2), and lysed in 200 uL irCLIP lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 1X protease inhibitor). After sonication in ice slurry, lysates were clarified by centrifugation at 5000 × g for 10 mins at 4C, and quantified by Bradford assay (Bio-Rad). A fraction of each sample was stored for input controls. 200 ug of each lysate was incubated with Protein G Dynabeads (Thermo-Fisher) pre-conjugated with 4 ug anti-FLAG M2 antibody (Sigma) in a final volume of 500 uL irCLIP lysis buffer for 2 hrs at 4C with rotation. The beads were then sequentially washed with the following ice-cold buffers: once with 1 mL irCLIP lysis buffer, once with 1 mL high stringency buffer (20 mM Tris pH 7.5, 120 mM NaCl, 25 mM KCl, 5 mM EDTA, 1% Triton X-100, 1% NaDOC, 0.1% SDS), once with 1 mL high salt buffer (20 mM Tris pH 7.5, 500 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1% NaDOC), once with with 1 mL low salt buffer (20 mM Tris pH 7.5, 5 mM NaCl, 5 mM EDTA, 1% Triton X-100), and twice with 0.5 mL NT2 buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40). Beads were then resuspended in 30 uL NT2 buffer containing 25 ng/mL RNase A (Thermo-Fisher) and 15% PEG400 (Sigma) for on-bead RNase digestion at 30C for 15 mins with shaking (1200 rpm) in a Thermomixer. RNase digestion was quenched by the addition of 0.5 mL high stringency buffer. Beads were washed twice with 0.3 mL PNK wash buffer (50 mM Tris pH 7.0, 10 mM MgCl2), and then resuspended in 30 uL PNK dephosphorylation mix (1X PNK buffer (Promega), 0.5 uL RNaseIN (Promega), 1 uL T4 PNK (Promega), 4 uL PEG400). Dephosphorylation reactions were conducted at 37C for 60 mins with shaking (1200 rpm) in a Thermomixer. Dephosphorylation mix was removed and beads were washed with 0.25 mL PNK wash buffer. For ligation of IR-Dye conjugated oligo to RNA crosslinked to protein, beads were resuspended in 30 uL RNA ligation mix (1X RNA ligase I buffer (NEB), 1 uL RNA ligase I (NEB), 1 uL IR-Dye-labeled oligo (cite: PMID: 27111506), 5 uL PEG400, and 0.5 uL RNaseIN) and incubated for 16 hrs at 16C with shaking in Thermomixer (1200 rpm). Ligation mix was then removed and beads were washed twice with 0.25 mL PNK wash buffer, prior to elution of RNA-protein complexes in 20 uL 1X LDS Buffer (Thermo-Fisher) + 10% beta-mercaptoethanol at 80C for 10 mins. 5 uL of eluates, as well as input controls, were then resolved by SDS-PAGE on 4–12% Bis-Tris NuPAGE gels (Thermo-Fisher), and transferred to nitrocellulose membranes. Fluorescent RNA-protein complexes in the eluates were visualized on a LiCOR Odyssey FC imager.
Flow Cytometry
Splenocytes were blocked with Fc block (BD Biosciences) in PBS + 2% heat inactivated FBS for 10 minutes at 4 degrees prior to cell surface staining by subsequent addition of a pre-mixed antibody cocktail. Cells were incubated with fluorescently labeled antibodies, each at a dilution of 1:200 for 30 minutes at 4 degrees, washed, and fixed with 2% paraformaldehyde in PBS for 10 minutes. Data were acquired on a BD FACSymphony A3 Cell Analyzer and using the BD Diva acquisition software (version 9.0) analyzed using FlowJo (Tree Star, version 10.8.1).
Cell death analysis
Cell death was measured using an IncuCyte imaging system, as described previously35. Briefly, cells were imaged in the presence of the cell impermeable DNA intercalator Sytox Green (Thermo Fisher, R37168), and Sytox positive cells quantified at each timepoint using custom processing definitions, available upon request. In parallel, separate cells plated in identical numbers were treated with the cell permeable dye Syto Green (Thermo Fisher, S34854) and quantified using the same approach, and percent cell death was calculated as Sytox+/Syto+ at each timepoint. For siRNA knockdown cell death assays, to avoid excessive non-specific toxicity, cells were transfected with siRNA for 8 hours, at which point cells (adherent) were washed and dye/inhibitors were added at final concentrations just prior to IncuCyte imaging.
Pathology
Pathology analysis for the AdarP195A/p150null:: Zbp1 experiments (Supplemental Figure 2) was performed by the same personnel and using a similar scoring system recently described for analysis of AdarP195A/p150null mice5. Briefly, littermate mice 21 days of age were euthanized via CO2 asphyxiation and livers and kidneys were harvested and washed in PBS and fixed in 10% neutral buffered formalin. Tissues were embedded in paraffin and cut into ~4mm sections for hematoxylin and eosin (HE) staining. Additionally, liver and kidney sections were periodic acid-Schiff (PAS) stained. Slides were evaluated by a board-certified veterinary pathologist, who was blinded to genotype and experimental setup. For kidney, expansion of the glomerular mesangial matrix was scored from 0–4, with 0=normal, 1=minimal, 2=mild, 3=moderate, and 4=severe. For the liver, microvesicular and lesser macrovesicular cytoplasmic vacuolation were scored from 0–5, with 0 = normal; 1 = minimal changes affecting only a small region (< 5%) of the liver; 2 = mild changes throughout the liver but without enlargement of hepatocytes, coalescing lesions, or necrosis; 3 = mild to moderate cytoplasmic vacuolation throughout liver with enlargement of hepatocytes but no necrosis or loss of parenchyma; 4 = moderate, coalescing throughout liver with multifocal mild regions of loss of parenchyma or necrosis; and 5 = severe with moderate multifocal regions of cavitation and necrosis.
Tissues for the AdarP195A/p150null::Mlkl−/−::Casp8−/− experiments (Supplemental Figure 11) were collected from pups euthanized by decapitation on the day of birth, and spleen, kidney, liver, heart, head with brain, and gastrointestinal tract were routinely paraffin embedded and HE stained. PAS stains were also obtained for liver and kidney. These slides were reviewed blindly, with the exception of gastrointestinal tissues, for which the pathologist was not blinded to genotype.
Representative images were captured from scanned slides or from glass slides taken using NIS-Elements BR 3.2 64-bit and plated in Adobe Photoshop. Image white balance, lighting, and contrast were adjusted using auto corrections applied to the entire image. Original magnification is stated.
Iba1 and Cleaved Caspase-3 analysis for Mlkl::Casp8::Adarp150/P195A pups was performed through the University of Washington Histology and Imaging Core (UW-HIC) utilizing the Leica Bond Rx Automated Immunostainer (Leica Microsystems, Buffalo Grove, IL), Slides were deparaffinized with Leica Dewax solution at 72°C for 30 seconds. Antigen retrieval was performed on all slides with EDTA, pH 9, at 100°C for 20 minutes. All subsequent steps were performed at room temperature. Initial blocking consisted of 10% normal goat serum (Jackson ImmunoResearch, cat no. 005–000-121) in tris-buffered saline for 20 minutes and Additional blocking with Leica Bond Peroxide Block for 5 minutes. Slides were incubated with Iba1 (1:1000) or CC3 (1:250) primary antibodies in Leica Primary Antibody Diluent. Slides were scanned in brightfield with a 20X objective using a NanoZoomer Digital Pathology System (Hamamatsu City, Japan). CC3 and Iba1 quantification was performed using the Visiopharm Image Analysis module.
Quantitative PCR analysis
RNA was isolated from primary MEFs or LET1s using Trizol extraction and first strand cDNA synthesis was performed with SuperScript III Reverse Transcriptase (Invitrogen, Cat. No. 18080044). QPCR was performed using a ViiA 7 Real Time PCR System (Thermo Fischer Scientific) using SYBR reagents (Thermo Fisher) The following primers were used for QPCR: Zbp1, S: AAGAGTCCCCTGCGATTATTTG, AS: TCTGGATGGCGTTTGAATTGG, Ripk1, S: GAAGACAGACCTAGACAGCGG, AS: CCAGTAGCTTCACCACTCGAC, Ccl2, S: TGGCTCAGCCAGATGCAGT, AS: TTGGGATCATCTTGCTGGTG, Ccl7, S: CCACATGCTGCTATGTCAAGA, AS: ACACCGACTACTGGTGATCCT, Ifit1, S: GCCATTCAACTGTCTCCTG, AS: GCTCTGTCTGTGTCATATACC, Ifnb, S: CTGGAGCAGCTGAATGGAAAG, AS: CTTCTCCGTCATCTCCATAGGG, Gapdh, S: GGCAAATTCAACGGCACAGT, AS: AGATGGTGATGGGCTTCCC.
RNA-seq and Nanostring analysis
For RNAseq and Nanostring experiments, RNA was isolated from day 23 spleens (RNAseq) or treated cells using Trizol. An on-column DNAse treatment was included for RNAseq experiments. Total RNA was added directly to lysis buffer from the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara), and reverse transcription was performed followed by PCR amplification to generate full-length amplified cDNA. Sequencing libraries were constructed using the NexteraXT DNA sample preparation kit (Illumina) to generate Illumina-compatible barcoded libraries. Libraries were pooled and quantified using a Qubit Fluorometer (Life Technologies). Dual-index, single-read sequencing of pooled libraries was carried out on a HiSeq2500 sequencer (Illumina) with 58-base reads, using HiSeq v4 Cluster and SBS kits (Illumina) with a target depth of 10 million reads per sample.
For Nanostring, two hundred fifty-four transcripts were quantified from total RNA using the mouse nCounter Inflammation V2 panel (Nanostring). The nSolver Analysis Software 4.0 with the nCounter Advanced Analyses package (Version 2.0.134) was used to normalize the data and perform differential gene expression analysis to generate log2-fold-change values and p-values. Data visualizations were carried out in R (version 4.1.1). Differentially expressed genes were visualized as heatmaps and volcano plots using the packages “pheatmap” (version 1.0.12), and “ggplot2” (version 3.3.5).
For RNA seq analysis of day 23 spleens, reads were aligned using kallisto36 to the mouse reference genome (GRCm39) using default parameters. Quality control was performed on raw reads using fastqc and then combined with aligned reads using multiqc37, with no samples removed from the final dataset due to QC checks. Analysis of aligned read was performed with R using DESeq238 using standard parameters to generate differential gene expressions for each of the conditions against wild type, and significant differential expression was defined by adjust P-value < 0.01 and absolute log2 fold change > 1. The differential expression data was annotated using the bioMaRt package39,40. Fold changes against wild-type between ADAR1 deficient (AdarP195A/p150null), with and without Zbp1-a knockout were compared, defining high recovery genes as those with a >50% regression to a fold change of 0 after Zbp1-a knockout and as low recovery otherwise. Gene ontology analysis was performed using the clusterProfiler package41 using standard parameters comparing both high and low recovery genes against background separately.
Statistical analysis
Comparison of survival cures was done using a Log-rank (Mantel-Cox) test. P values less than 0.0001 were a result of the statistical analysis package and are represented as “>0.0001”. Data shown in graphs are mean or mean ± SD. If the data fulfilled the criteria for Gaussian distribution tested by column statistics, an unpaired parametric t-test with Welch’s correction was performed for statistical analysis. All statistical tests listed in the figure legends were two-sided and were performed using Graphpad Prism, or Microsoft Excel (Chi-Square Power values for mendelian distributions). P values are presented in the figure or figure legends. All in vitro experiments, unless otherwise stated were independently replicated a minimum of two times, and details on replication of displayed data is stated in the figure legends.
Data and code availability
The R analysis was performed using publicly available code, described above; Custom R scripts are available on https://github.com/OberstLab/Hubbard-et-al-2022-Nature. RNAseq and Nanostring data are available via the NIH Gene Expression Omnibus, accession numbers GSE200854 (RNAseq) and GSE200985 and GSE200986 (NanoString).
Extended Data
Extended Data 1: Interaction of ZBP1 with RNA and ADAR1.
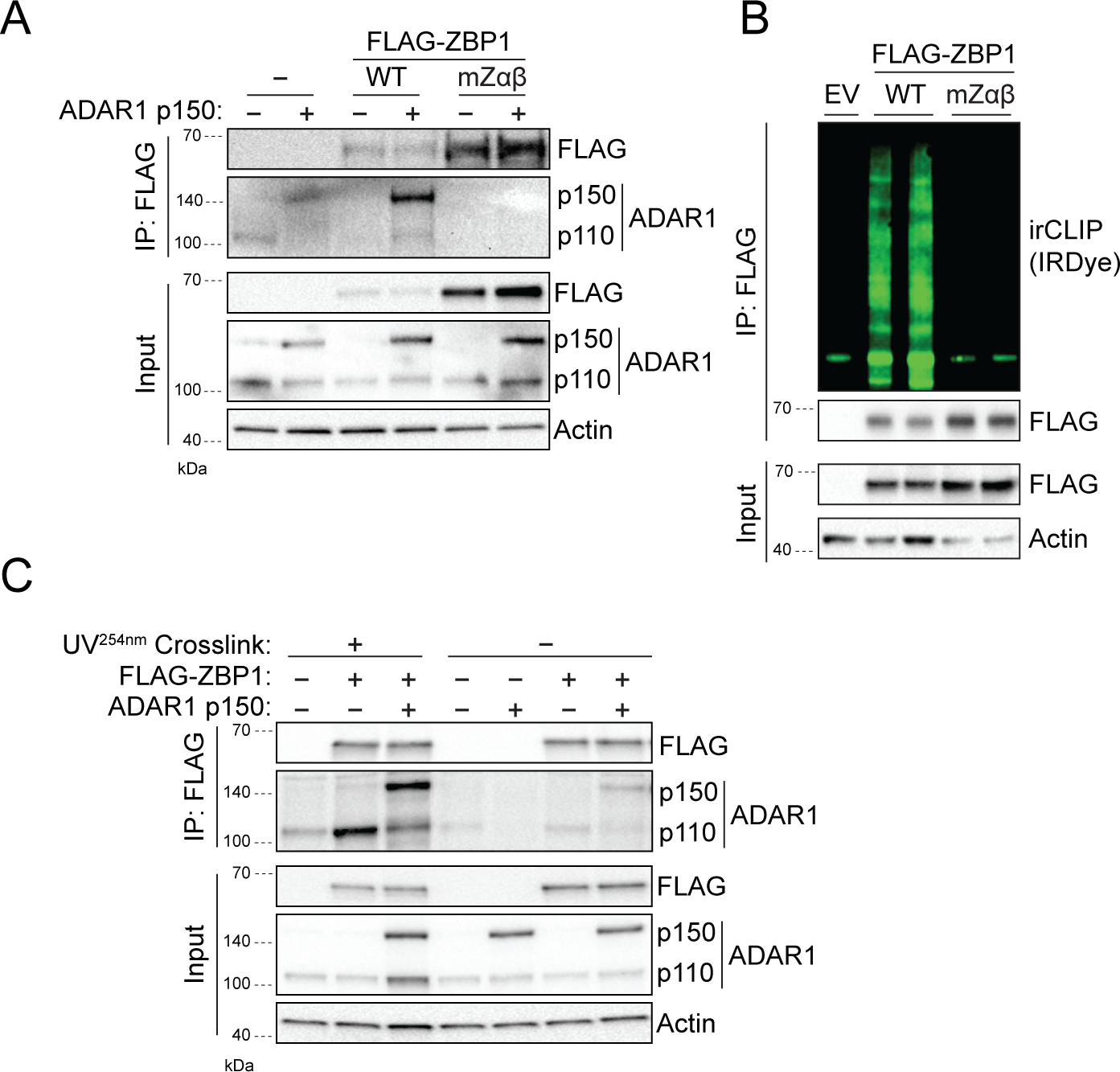
A. Co-precipitation ADAR1 with WT or mutant Zαβ (mZαβ) Flag-tagged ZBP1 (FLAG-ZBP1) after FLAG immunoprecipitation. Β. Immunoprecipitation and IR-CLIP analysis for RNA binding by of WT or mZαβ FLAG-ZBP1. C. Co-precipitation of ADAR1 and FLAG-tagged ZBP1 after UV-crosslinking. These experiments were performed in HEK293T cells.
Extended Data 2: Immunopathology in ADAR-mutant mice is ZBP1 dependent.

Survival proportions observed upon cross of Adarp150null/WT::Zbp1-a+/− mice to AdarP195A/P195A::Zbp1-+/−, **** p<0.0001 (Mantel-Cox Log-Rank test) (A) or Adarp150null/WT::Zbp1-a−/− mice to AdarP195A/P195A::Zbp1-a−/−, not significant (Mantel-Cox Log-Rank test) (B). (C) Histopathological analysis of liver and kidney from affected, rescued and unaffected mice (genotypes indicated.) For liver samples, regions of cytoplasmic vacuolation indicated with asterisk. Original magnification 20x, HE staining. For kidney, glomeruli are indicated with arrows, from original magnification 40x HE staining.
Extended Data 3: Cross of AdarP195A/p150null mice to a separately derived, fully congenic Zbp1−/−-g strain.

A-B. SNP typing analysis of ZBP1-g (A) and ZBP1-a (B) mice. C-D. Zbp1−/−-g::AdarP195A/p150null survival proportions(C) and observed weight (D) at 21 days (weaning). Combined male & female, Zbp1+/+::AdarP195A/WT (n=17), Zbp1+/−::AdarP195A/WT (n=21), Zbp1−/−::AdarP195A/WT (n=10), Zbp1+/+::AdarP195A/p150null (n=9), Zbp1+/−::AdarP195A/WT (n=25), Zbp1−/−::AdarP195A/WT (n=7).
Extended Data 4: ZBP1 is IFN dependent and partially dependent on MDA5.

A. Twelve-hour stimulation of LET1 and SVEC cells with varying concentrations of IFN-β followed by Western Blot analysis for ZBP1 protein. B. Quantitative PCR analysis for Zbp1 and Ifnb after ADAR1 depletion in wild-type (scramble gRNA) or MDA5 knockout LET1. Gene was normalized against Gapdh. Significance determined by individual student t-tests. Each group (Zbp and Ifnb) contains 3 biologic replicates, each comprising the average of 4 technical replicates). This experiment is representative of two independent repeats. Whisker bars are presented as mean +/− SD. C. Survival of Zbp1−/−::MDA5−/−::ADARp150null/p150null mice. Zbp1-a+/+::Ifih1−/−::Adarp150null/+ n=6, Zbp1-a+/+::Ifih1−/−::Adarp150null/p150null n=4, Zbp1-a−/−::Ifih1−/−::Adarp150null/+ n=5, Zbp1-a−/−::Ifih1−/−::Adarp150null/p150null n=3. Statistical significance determined by Mantel-Cox (Log-Rank) test. D. Survival proportions of Zbp1−/−-a::Adarp150/WT intercross. Chi square power analysis performed, indicating significance at p=1.82×10−5.
Extended Data 5: Identification of ZBP1 dependent and independent aspects of the ADAR1 inflammatory signature.
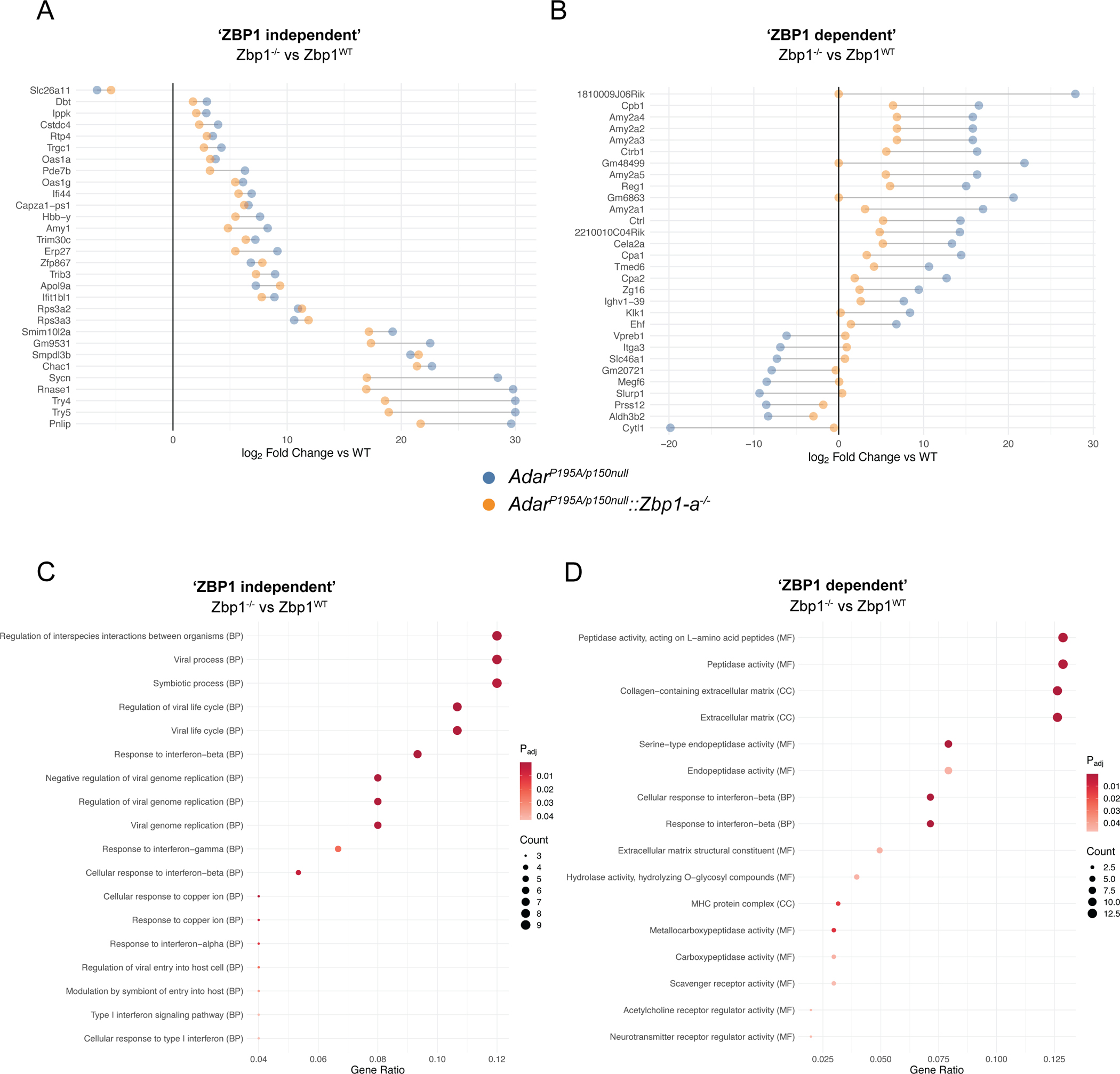
A-B: Cleveland plots indicating changes in the ADAR1 dependent gene signature observed in the spleens of 23 day old mice, indicating most- (A) and least- (B) changed genes upon ZBP1 ablation in AdarP195A/p150null mice. Gene selection is the top 30 largest contributors to the ZBP1 dependent (A) and independent (B) signature from ADARP195A/p150null mice, in comparison to WT mice. Gene Ontology analysis was performed on the signatures from A and B. C-D: GO-terms analysis for ZBP1-independent signature (C), and the ZBP1 dependent signature analysis (D).
Extended Data 6: Flow cytometry analysis of splenic cellular subsets.

B cell, T cell, monocyte, macrophage percentages from day 23 spleens of Zbp1::Adarp150/P195A mice.
Extended Data 7: ZVAD treatment induces phosphorylation of RIPK3 in ADAR mutant MEFs in a ZBP1 dependent fashion.
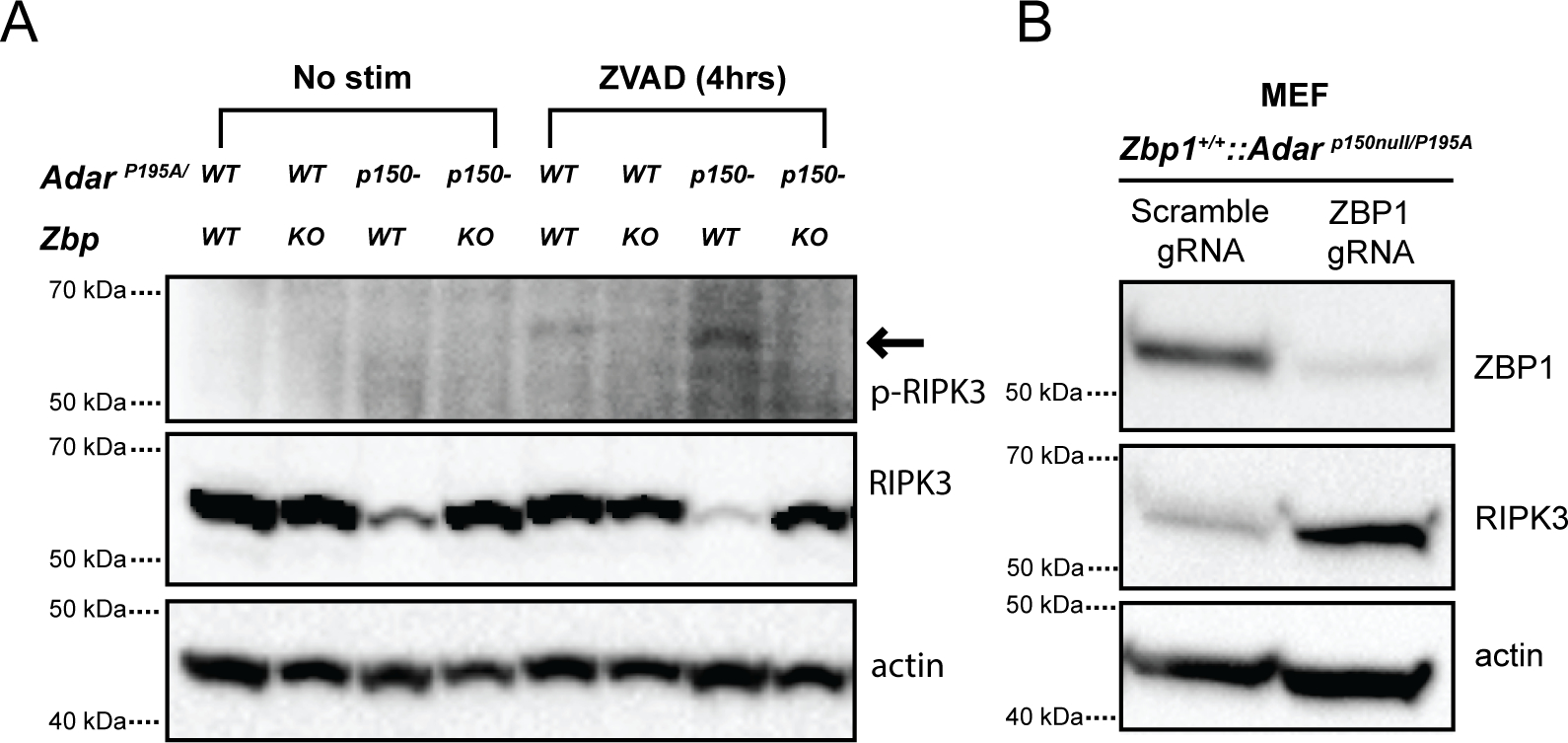
A. Analysis of phospho-RIPK3 in ADAR1 mutant MEFs after 4 hour ZVAD treatment. B. Confirmation of ZBP1 gRNA knockout in ADAR1 mutant MEFs.
Extended Data 8: MLKL or RIPK1kinase dead mutations do not rescue ADARP195A/p150null mutation.

Three-week-old weights (weaning) of male or female AdarP195A/p150null mice crossed to animals lacking, A: MLKL. Mlkl+/−::AdarP195A/WT (m/f n=9/7), Mlkl+/−::AdarP195A/p150null (m/f n=8/2), Mlkl−/−::AdarP195A/WT (m/f n=8/5), Mlkl−/−::AdarP195A/p150null (m/f n=6/3). or B: carrying a point mutation abrogating the kinase activity of RIPK1 (Ripk1kd). Ripk1kd/+::AdarP195A/WT (m/f n=3/11), Ripk1kd/+::AdarP195A/p150null (m/f n=7/5), Ripk1kd/kd::AdarP195A/WT (m/f n=11/8), Ripk1kd/kd::AdarP195A/p150null (m/f n=5/5). Statistical differences determined by individual student t-tests (two tailed). All genotypes are littermates from mixed litters.
Extended Data 9: Oligomerization of ZBP1 triggers necroptotic cell death.
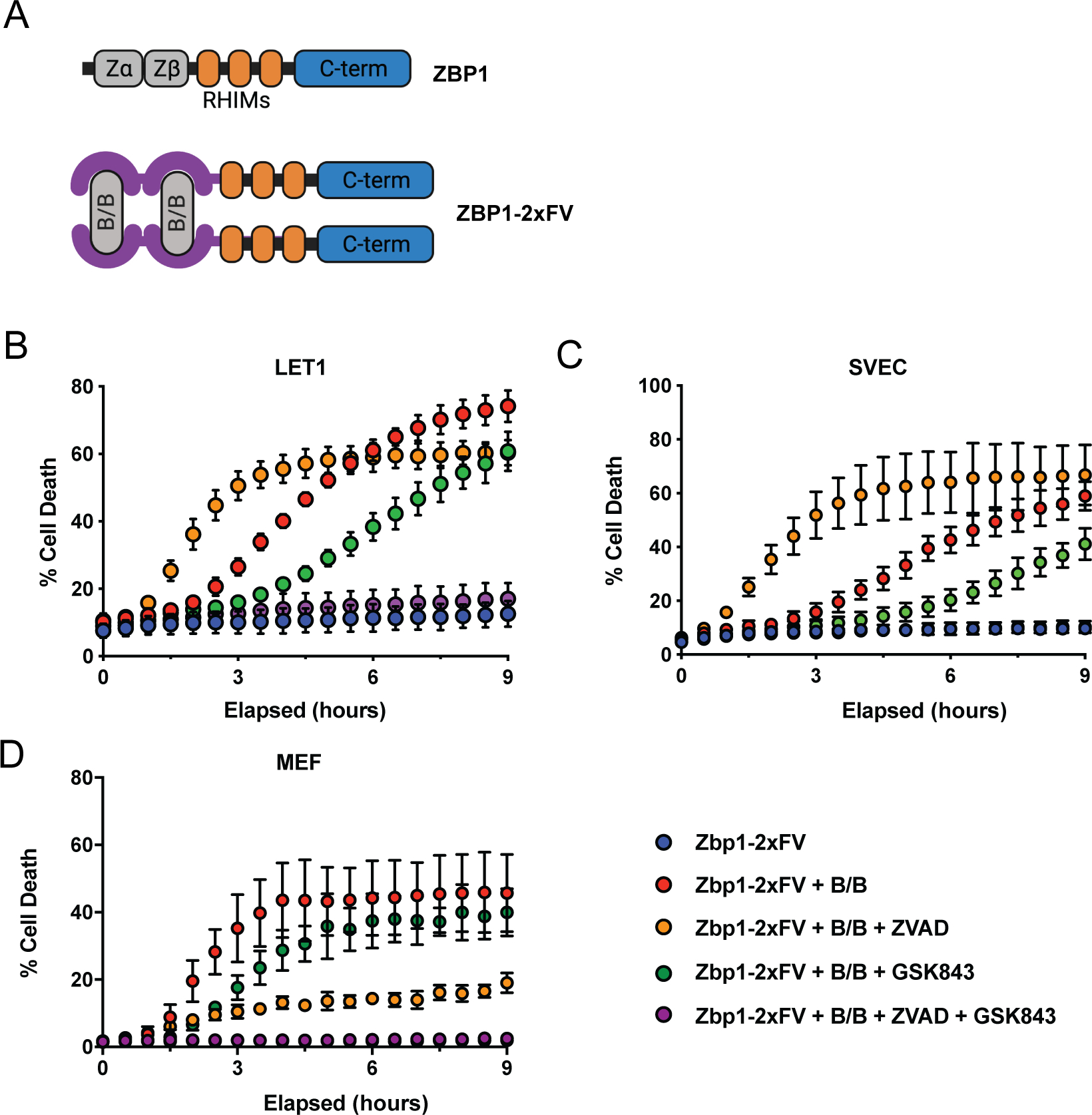
A. Schematic indicating the replacement of ZBP1’s Z-DNA binding domain with a tandem FKBP domain. B-D: Cell death following addition of B/B homodimerizer with indicated combinations of ZVAD and GSK843 in LET1s (each group, n=4 biologic replicates) (B), SVECs (each group n=4 biologic replicates) (C) or MEFs (each group n=3 biologic replicates) (D). Statistical significance was determined by unpaired t tests (two-tailed). Experiments B and C are representative of two independent experiments, and D is representative of 3 independent experiments. All whisker bars are presented as mean +/− SD.
Extended Data 10: Molecular analysis of cell death induced by ZBP1–2xFV homodimerization.

A. Phospho-MLKL analysis of ZBP1–2xFV MEFs (wild-type, Ripk3−/−, MLKL−/−) after 1 hour stimulation with B/B homodimerizer. B. Cleaved-caspase 3 analysis of ZBP1–2xFV MEFs (wild-type, Ripk3−/−, MLKL−/−) after 3 hour stimulation with B/B homodimerizer. C. Representative image depicting Caspase-8 deficiency exacerbation of disease phenotype in ADARP195A/p150null::RIPK3−/− mice. Image of 20-day old littermates from the cross depicted in Fig. 4B.
Extended Data 11: Pathologic analysis of Mlkl−/−::Casp8−/−::Adarp150null/P195A mice.

A-B. Immunohistochemical staining (A) and quantification (B) in liver for cleaved-caspase 3. (n=3 d.0 pups, each group) Original magnification 10x. C. Additional histological images of other tissue sites (kidney, liver and small intestine). Kidney (20x) and liver (10x) PAS staining, small intestine HE staining (original magnification as stated). D-E. Immunohistochemical staining, with 2.5x and 10x images (D) and quantification (E) in brain for Iba1 (n=3 d.0 pups, each group). B & E use tissues from matched animals.
Extended Data 12: Immunoprecipitation of RIPK3 and RIPK1 by ZBP1 is enhanced by Casp8 deficiency.

A. Pulldown of ZBP1–2xFV (FKBP) and co-precipitation of RIPK1 in Ripk3−/−::Casp8−/− MEFs. B. Pulldown of ZBP1–2xFV (FKBP) and co-precipitation of RIPK1 and RIPK3 in Mkl−/−::Casp8−/− MEFs. C. Pulldown of ZBP1 and co-precipitation of RIPK3 and in Mlkl−/−::Adarp150null/P195A MEFs.
Extended Data 13: ZBP1 activation results in RIPK1 dependent, RIPK3 independent gene transcription which is enhanced by knockout of Casp8.
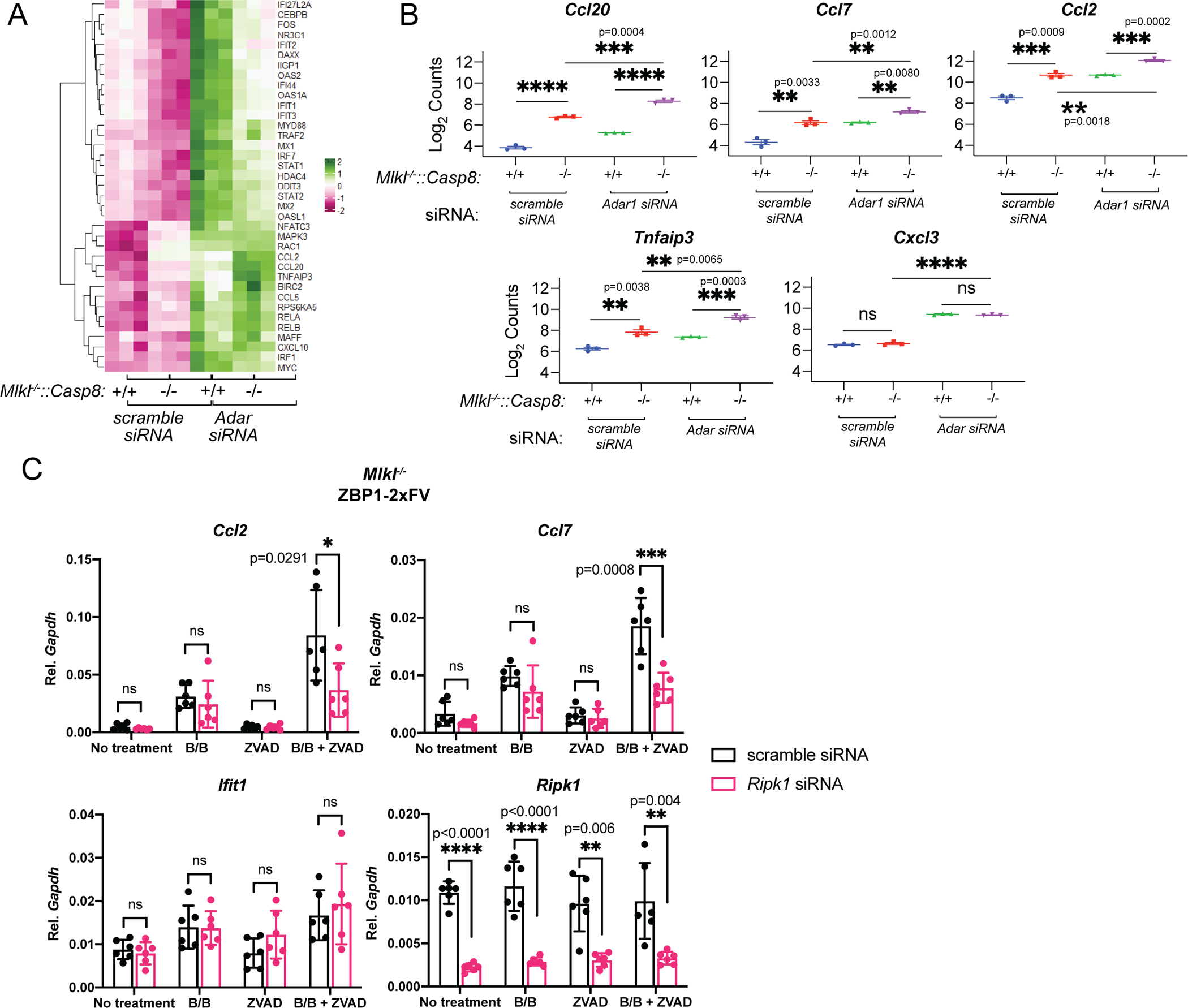
A-B Differential transcript analysis of Mlkl−/−::Casp8+/+ and Mlkl−/−::Casp8−/− MEFs (n=3) following ADAR depletion, by (A) Heatmap analysis and (B) individual quantification of the top 5 differentially expressed genes. Statistical significance determined by individual unpaired t tests (two-tailed). Where not indicated: **** p<0.0001. C. QPCR analysis of top 5 differentially expressed genes identified (B) in MEFs expressing ZBP1–2xFV after B/B activation after depletion of RIPK1. n=6 biologic replicates for each treatment group, each representing the average of three technical replicates. Data is compiled from two independent experiments. Statistical significance determined by individual unpaired t tests (two-tailed). Where indicated: **** p<0.0001. Whisker bars represent the mean +/− SD.
Extended Data 14: ZBP1A64P mutation attenuates ZBP1 activity in ADAR deficiency model and during influenza infection.

A. Previously-reported structures of the ZBDs of ZBP1 (left) and ADAR1 (right), with A64 and P195A (respectively) highlighted in green. PDB accession #s: 1J75 and 3F21. B. Survival proportions of Adar1P195A/p150null::ZBP1A64P mice (n=9 animals) compared with previous survival statistics of AdarP195A/p150null::ZBP1-a+/− animals. C. Cell death analysis following influenza (X31, MOI 2) infection of primary MEFs derived from Wild Type (B6/J), Zbp1−/− or ZBP1A64P/A64P mice. Each group, n=2 biologic replicates. Whisker bars represent the mean +/− SD. Experiment was independently replicated using MEFs derived from a different embryo. D. Expression of ZBP1 in wild-type, Zbp1−/−, and Zbp1A64P/A64P MEFs following 24 stimulation with 1000 IU/mL murine IFN-β.
Supplementary Material
Acknowledgements:
This work is supported by grants R01 AI153246 (to AO and DBS), R01 CA228098 (to AO), R01 AI084914 (to DBS), R01 AI143227 and R01 AI147177 (to RS), by the Titus Fellowship (to NWH), T32 T32AR7108-41 (to JMA), and by the Helen Hay Whitney Foundation award (to NSG). Extended Data figure 9A was created using Biorender.com. The authors thank Dr. Pooja Jain, Isabel Silva and Ricky Lee for technical assistance, and Dr. Annelise Snyder for the mouse drawing used in Fig. 1B.
Footnotes
Competing interests
DBS is a co-founder and shareholder of Danger Bio, LLC, and a scientific advisor for Related Sciences LLC. AO is a co-founder and shareholder of Walking Fish Therapeutics.
Additional Information
Requests for reprints, permissions, materials or additional information may be directed to Dr. Andrew Oberst: oberst@uw.edu
Main text references
- 1.Pestal K et al. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 43, 933–944 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad S et al. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 172, 797–810.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbert A et al. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc National Acad Sci 94, 8421–8426 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rice GI et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet 44, 1243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maurano M et al. Protein kinase R and the integrated stress response drive immunopathology caused by mutations in the RNA deaminase ADAR1. Immunity (2021) doi: 10.1016/j.immuni.2021.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz T, Behlke J, Lowenhaupt K, Heinemann U & Rich A Structure of the DLM-1–Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat Struct Mol Biol 8, 761–765 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Rebsamen M et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. Embo Rep 10, 916–922 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thapa RJ et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 20, 674–681 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Upton JW, Kaiser WJ & Mocarski ES DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung H et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 172, 811–824.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y et al. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife 6, e25687 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crow YJ et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 167, 296–312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishii KJ et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451, 725–729 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Koehler HS, Feng Y, Mandal P & Mocarski ES Recognizing limits of Z-nucleic acid binding protein (ZBP1/DAI/DLM1) function. Febs J 287, 4362–4369 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Newton K et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540, (2016). [DOI] [PubMed] [Google Scholar]
- 16.Takaoka A et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Nogusa S et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 20, 13–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varfolomeev EE et al. Targeted Disruption of the Mouse Caspase 8 Gene Ablates Cell Death Induction by the TNF Receptors, Fas/Apo1, and DR3 and Is Lethal Prenatally. Immunity 9, 267–276 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Oberst A et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaiser WJ et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alvarez-Diaz S et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 45, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Najjar M et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity 45, 46–59 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothenburg S et al. A PKR-like eukaryotic initiation factor 2α kinase from zebrafish contains Z-DNA binding domains instead of dsRNA binding domains. P Natl Acad Sci Usa 102, 1602–1607 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koehler H et al. Vaccinia virus E3 prevents sensing of Z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe 29, 1266–1276.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray EE, Treuting PM, Woodward JJ & Stetson DB Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutières Syndrome. J Immunol 195, 1939–1943 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gall A et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 27.Murphy JM et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Kasparcova V et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol 192, 5476–5480 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin J et al. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540, 124–128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newton K, Sun X & Dixit VM Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Molecular and Cellular Biology 24, 1464–1469 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salmena L et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Gene Dev 17, 883–895 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henao-Mejia J et al. Generation of Genetically Modified Mice Using the CRISPR–Cas9 Genome-Editing System. Cold Spring Harb Protoc 2016, pdb.prot090704 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin K-J et al. A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc National Acad Sci 103, 13759–13764 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zarnegar BJ et al. irCLIP platform for efficient characterization of protein–RNA interactions. Nat Methods 13, 489–492 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orozco S et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ 21, 1511–1521 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bray NL, Pimentel H, Melsted P & Pachter L Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Ewels P, Magnusson M, Lundin S & Käller M MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Durinck S et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Durinck S, Spellman PT, Birney E & Huber W Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu G, Wang L-G, Han Y & He Q-Y clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. Omics J Integr Biology 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R analysis was performed using publicly available code, described above; Custom R scripts are available on https://github.com/OberstLab/Hubbard-et-al-2022-Nature. RNAseq and Nanostring data are available via the NIH Gene Expression Omnibus, accession numbers GSE200854 (RNAseq) and GSE200985 and GSE200986 (NanoString).