

Abstract
Objective:
Congenital hyperinsulinism (HI) is the most common cause of persistent hypoglycemia in children. In addition to typical focal or diffuse HI, some cases with diazoxide-unresponsive congenital hyperinsulinism (HI) have atypical pancreatic histology termed Localized Islet Nuclear Enlargement (LINE) or mosaic HI, characterized by histologic features similar to diffuse HI, but confined to only a region of pancreas. Our objective was to characterize the phenotype and genotype of children with LINE-HI.
Design:
The phenotype and genotype features of 12 children with pancreatic histology consistent with LINE-HI were examined.
Methods:
We compiled clinical features of 12 children with LINE-HI and performed next-generation sequencing (NGS) on specimens of pancreas from eight of these children to look for mosaic mutations in genes known to be associated with diazoxide-unresponsive HI (ABCC8, KCNJ11, and GCK).
Results:
Children with LINE-HI had lower birth weights and later ages of presentation compared to children with typical focal or diffuse HI. Partial pancreatectomy in LINE-HI cases resulted in euglycemia in 75% of cases; no cases have developed diabetes. Low-level mosaic mutations were identified in the pancreas of six cases with LINE-HI (three in ABCC8, three in GCK). Expression studies confirmed that all novel mutations were pathogenic.
Conclusion:
These results indicate that post-zygotic low-level mosaic mutations of known HI genes are responsible for some cases of LINE-HI which lack an identifiable germline mutation and that partial pancreatectomy may be curative for these cases.
Keywords: beta-cells, genetic diseases, insulin, pancreas, hypoglycemia
Introduction
Congenital hyperinsulinism is the most common cause of persistent hypoglycemia in infants and children and has been associated with mutations in a growing number of genes that play an important role in the regulation of insulin secretion 1. Among children with diazoxide-unresponsive HI, 90% are found by standard testing of peripheral blood DNA to have an identifiable disease-causing mutation involving either ABCC8, KCNJ11, or GCK 30. The most frequently identified mutations occur in the two subunits of the beta-cell ATP-sensitive potassium channel, SUR1 and Kir6.2, encoded by ABCC8 and KCNJ11 on chromosome 11p 30. Mutations in these KATP channel genes can act in either a dominant or recessive fashion and result in one of two histological forms: diffuse-HI which results from biallelic recessive inactivating mutations or from monoallelic dominant-negative mutations; and focal-HI which is a small lesion of islet-cell adenomatosis resulting from a recessive paternally-inherited KATP channel mutation in conjunction with somatic loss of heterozygosity for the maternal chromosome 11p15 region containing both the KATP channel locus and the imprinted growth controlling Beckwith-Wiedemann Syndrome (BWS) region 31 . Children with focal or diffuse HI due to recessive KATP channel mutations are not responsive to treatment with diazoxide, a KATP channel agonist. Children with diffuse HI due to monoallelic dominant-negative KATP channel mutations have channels which are present at the beta-cell surface, but with reduced function 32. Approximately half of the dominant KATP channel mutations reported to date result in diazoxide-unresponsive HI, and all of these have been in ABCC8 33. Children with diffuse HI often require near-total pancreatectomy to control hypoglycemia, while resection of the lesion can be curative in focal HI 34. Dominant activating mutations in GCK are identified in a smaller proportion of diazoxide-unresponsive diffuse cases of hyperinsulinism 35.
In addition to diffuse and focal forms of HI, a third histologic subgroup has been identified in a small number of children that the authors have termed “Localized Islet Nuclear Enlargement” (LINE-HI) and has been termed “mosaic HI” or “atypical HI” by others 36–39. Histologically, LINE-HI, like typical diffuse-HI, is characterized by normal appearing islets and lobular architecture within the pancreas. However, while in diffuse-HI nucleomegaly of individuals islets is observed throughout the pancreas, in LINE-HI, nucleomegaly is constrained to an isolated region of the pancreas, outside of which nucleomegaly is absent. A previous study reported evidence of increased immunohistochemical staining for HK1 in affected areas of the pancreas and mosaic GCK mutations in some cases with atypical/LINE-HI pathology 37. Children with LINE-HI do not have an identifiable mutation in standard sequencing of peripheral blood genomic DNA. Phenotypically, LINE-HI cases identified to date are not responsive to medical therapy with diazoxide and partial pancreatectomy with resection of the abnormal tissue often results in significant improvement or cure of the hypoglycemia 34.
Because 90% of children with diazoxide-unresponsive HI have mutations in ABCC8, KCNJ11, or GCK, we hypothesized that some cases of LINE-HI might have an underlying somatic mutation in one of these genes which could only be detected in the affected pancreatic tissue. To examine this hypothesis, we utilized next-generation sequencing (NGS) in pancreatic tissue and isolated islets to search for mutations in patients with LINE-HI and functionally characterized the found mutations using in vitro assays.
Methods
Probands
The diagnosis of HI was based on previously described criteria 40: fasting hypoglycemia accompanied by inadequate suppression of plasma insulin, inappropriately low plasma free fatty acids and β-hydroxybutyrate concentrations, and inappropriate glycemic response to glucagon stimulation. Patients were defined as being responsive to diazoxide if HI could be completely controlled by treatment with diazoxide at doses ≤15 mg/kg/day, as demonstrated by maintaining plasma glucose concentrations ≥70 mg/dL for 12–18 hr of fasting and/or by developing appropriate fasting hyperketonemia (β-hydroxybutyrate >2 mmol/L before plasma glucose decreased to <50 mg/dl).
Prior to surgery, some cases underwent 18F-6-fluoro-L-dopa PET/CT scan to assess for the presence of a focal lesion 41. LINE cases were characterized histologically as described previously by the presence of islet cell nucleomegaly confined to at least one, but no more than two contiguous, region(s) of the pancreas; nucleomegaly was assessed using the same criteria as diffuse HI as islet cell nuclei having area greater than three times larger than background nuclei 38, 39.
For the purposes of this study, infants were considered to be cured after surgery by demonstration of normal fasting tolerance for age (8-10 hours or greater) without medication. Written informed consent was provided by all subjects or their parents. These studies were approved by the Children’s Hospital of Philadelphia (CHOP) Institution Review Board. All patients undergoing 18F-DOPA PET/CT were enrolled in one of three clinical trials after obtaining informed consent. These studies were performed under an IND with FDA oversight, and approved by the CHOP IRB and the UPENN radiation safety committee.
Genetic Analysis
Standard mutation screening of peripheral blood DNA was performed in a CLIA certified laboratory by NGS for ABCC8, KCNJ11, GCK, GLUD1, UCP2, HNF1A, HNF4A, HADH, and SLC16A1.
Next Generation Sequencing:
DNA and RNA were isolated from peripheral blood and fresh or frozen pancreatic tissue using standard kits following manufacturer’s instructions. Exonic and flanking intronic regions of ABCC8, KCNJ11, and GCK were PCR amplified and combined in equimolar concentrations prior to next-generation sequencing (NGS) (Ion Torrent, Life Technologies, Carlsbad, CA). In order to detect possible low-level mosaic mutations, NGS was performed with a higher depth of coverage than is typically obtained on standard clinical testing. The average coverage for the cases included here was 16,835x compared to ~100x that is typically offered on standard clinical testing 42. Read lengths for KCNJ11 and ABCC8 cDNA was 400bp while ABCC8 gDNA and GCK were 200bp. Analysis was performed using the Torrent Suite Software, starting with signal processing and base calling, followed by alignment with the Torrent Mapping Alignment Program (TMAP). Variant calling and coverage analysis were performed on aligned reads using the Torrent Variant Caller and Coverage Analysis plugins (Life Technologies, Carlsbad, CA). Variant calling included a low stringency filter to identify mosaic variants as low as 2% using the “somatic low stringency” configuration in the Torrent Variant Caller plugin. Resulting variants were visually examined in The Integrated Genomic Viewer (IGV, Broad Institute) to ensure there was no strand bias in variant calls, that nucleotides in adjacent regions were called correctly, and that they were not in homopolymer regions. In addition, nucleotide calls at the position of the resulting variants were examined in the other samples included in each sequencing run (see Miscall Rate column on Table 3). These samples were obtained from blood or pancreas from other children with HI due to germ-line mutations in peripheral blood DNA or children with HI and negative mutation analysis in peripheral blood. These samples underwent the same processing for DNA/RNA isolation, PCR amplification, library preparation and NGS as the cases presented here.
Table 3.
Low-level mosaic mutations detected by next-generation sequencing.
| Case | Gene | Mosaic Mutation | Functional Defect | Minor Allele Frequency (gnomAD) | Mosaicism by Next-Generation Sequencinga (minor/total base calls) | Miscall Rateb | Confirmation by | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||||||
| Peripheral Blood DNA | Whole Pancreas DNA | Isolated Islets DNA | Isolated Islets cDNA | miscall reads/ total reads | Mean % ± SE | Range, % | n | Sanger | Repeat NGS | |||||
| 1 | GCK | c.1340g>t/ p.Arg447Leu | activating | 0 | 0% | 6.8% (1470/21540) | NA | NA | 7/179,016 | 0.0 ± 0.0 | 0.0-0.01 | 8 | pancreas pos | 6.93% (pancreas) |
| 8 | ABCC8 | c.2069c>t/ p.Pro690Leu | trafficks, gating defect | 0 | 0% | 2.5% (69/2738) | NA | NA | 78/106,492 | 0.07 ± 0.01 | 0.06-0.1 | 5 | pancreas neg | - |
| c.2152g>a/ p.Gly718Ser | non-trafficking | 0 | 0% | 2.6% (100/3886) | NA | NA | 24/146,514 | 0.02 ± 0.01 | 0.01-0.03 | 5 | pancreas neg | - | ||
| 9 | ABCC8 | c.3724a>t/ p.Thr1242Ser c | trafficks, normal gating d | 0 | 0% | NA | NA | 5.4% (1557/28907) | 205/161,165 | 0.13 ± 0.03 | 0.08-0.25 | 5 | islets neg | - |
| 10 | GCK | c.190t>c/ p.Ser64Pro | activating | 0 | 0% | 10.3% (4785/46343) | - | - | 567/224,317 | 0.25 ± 0.02 | 0.17-0.29 | 7 | pancreas pos | 10.14% (pancreas) |
| 11 | ABCC8 | c.1600a>g/ p.Arg534Gly | trafficks, gating defect | 0 | 0% | 0% | 0% | 3.4% (832/24202) | 92/99,756 | 0.09 ± 0.02 | 0.03-0.15 | 4 | islets neg | 3.66% (islet cDNA) |
| 12 | GCK | c.271g>t/ p.Val91Leu e | activating | 0 | 0% | NA | 2.2-3.4% (1640/58389) | - | 19/247,053 | 0.01 ± 0.0 | 0.0-0.03 | 7 | islets neg | 2.59-3.49% (islets) |
mosaicism <1% indicated as 0%, see Supplemental Table 1 for more detail;
samples in same NGS run at nucleotide position of the mosaic mutation; NA: not available;
carries intronic variant not predicted to alter splicing (ABCC8:c.4122+15c>t);
possibly pathogenic based on islet functional studies (see text);
previously published mutation (PMID 21831042)
The nucleotides of ABCC8 and corresponding SUR1 amino acids were numbered according to the sequence reported by Nestorowicz, et al 1996 4 that includes the alternatively spliced exon 17 sequence (NCBI accession no. L78224). GCK sequence information is based on GenBank reference sequence NM_000162.3. Novel variants were searched against the gnomAD Browser (v2.1) 43 and functional consequences predicted using SIFT 44, PolyPhen 45 (Table S2). Variants were confirmed by direct sequencing as previously described 30.
Statistics
Statistical analysis was performed using Graph Pad Prism, v8.4.3 (GraphPad Software, San Diego, CA). The Mann-Whitney Test was used to compare disease outcomes in LINE-HI cases vs focal and diffuse-HI cases 46. Fisher’s Exact Test was used to compare NGS miscalls in LINE-HI cases vs control samples.
Functional Characterization of novel ABCC8 Mutations
Functional analyses of KATP channels, including immunoblotting, 86Rb+ efflux, and electrophysiology were as described previously 32, 47. Briefly, for immunoblotting, COSm6 cells were co-transfected with human SUR1 in pCMV6b and human Kir6.2 in pCDNA3 using FuGene6 (Roche Applied Science). SUR1 was probed with rabbit antiserum against the C-terminal 13 amino acids of SUR1 followed by HRP anti-rabbit secondary antibodies (GE Healthcare) and visualized by enhanced chemiluminescence (Super Signal West Femto; Pierce).
For 86Rb+ efflux assays, transfected cells were incubated overnight with 86RbCl (1 μCi/ml medium). The next day, cells were pretreated with 1 mM 2-deoxyglucose and 2.5 μg/ml oligomycin for 30 min to activate the channels. 86Rb+ efflux was followed over 40 minutes and an efflux rate constant was calculated from the rate of initial efflux and fitted to the equation: efflux = 1−e((−k1t)+(−k2t)) ; where k1 is the rate constant calculated from untransfected cells and k2 is the rate constant for the KATP channel mediated efflux 48.
Due to discontinuation of 86Rb+ during the course of the study, the response of the ABCC8: p.Thr1242Ser mutant to metabolic inhibition was assessed by measuring residual currents in 0.1mM ATP or 0.1mM ATP plus 0.5mM MgADP as percent of currents in the absence of ATP or MgADP using inside-out patch-clamp recording as previously described 49.
Biochemical characterization of Novel GCK mutants
Recombinant human islet wild-type glucokinase and novel GCK mutants were generated using methods previously described 50. Briefly, the mutations were cloned for expression as glucokinase fusion proteins containing a COOH-terminal glutathionyl S-transferase (GST). GST-glucokinase was produced in Escherichia coli and then purified from crude extracts to near homogeneity by affinity chromatography using glutathione-agarose (Sigma, St. Louis, Missouri). Kinetic analysis to determine the glucose S0.5 (glucose concentration for half-maximal rate) of the expressed forms of glucokinase was performed using the protocols developed previously 51, 52.
HK1 Expression by Immunoflourescence and cDNA Analysis.
Immunofluorescence.
Paraffin-embedded formalin-fixed tissue obtained at the time of surgery was sectioned and mounted onto glass slides. Paraffin was removed with xylene followed by re-hydration with ethanol. Antigen retrieval was done with Sodium Citrate Buffer. Slides were incubated with primary antibodies (rabbit anti-HK1 1:250, and goat anti-insulin 1:500, Santa Cruz, Dallas, Texas) overnight at 4 degrees Celsius followed by incubation with the secondary antibodies (488 donkey anti-goat 1:500, Cy3 donkey anti-rabbit 1:500 and DAPI 1:1000) for 1 hour in a hydration chamber at room temperature.
cDNA Analysis of HK1 Expression.
RT-PCR of cDNA was performed using custom probes for the ubiquitous HK1 isoform (sequences available upon request); GAPDH was used as the endogenous control. Reactions were prepared in triplicate for each sample using PrimeTime Gene Expression Master Mix (Integrated DNA Technologies, Coralville, IA) and run on a Quant Studio 6 (Thermo Fisher Scientific, Waltham, MA) using standard cycling. Analysis was performed using the delta-delta CT method using a commercially available pancreatic RNA sample as the calibrator (Life Technologies, Carlsbad, CA).
Results
Of 561 children with diazoxide-unresponsive HI seen at CHOP between 1999 and 2019, 12 had histology consistent with LINE-HI (see Figure 1) and had negative mutation analysis in peripheral blood for genes associated with HI. Fresh or frozen pancreatic tissue was available on eight of these 12 to search for mosaic mutations in ABCC8, KCNJ11, and GCK by NGS. In two of the four cases from which pancreas was not available for testing, NGS was performed in peripheral blood DNA.
Figure 1. Histology of LINE-HI.
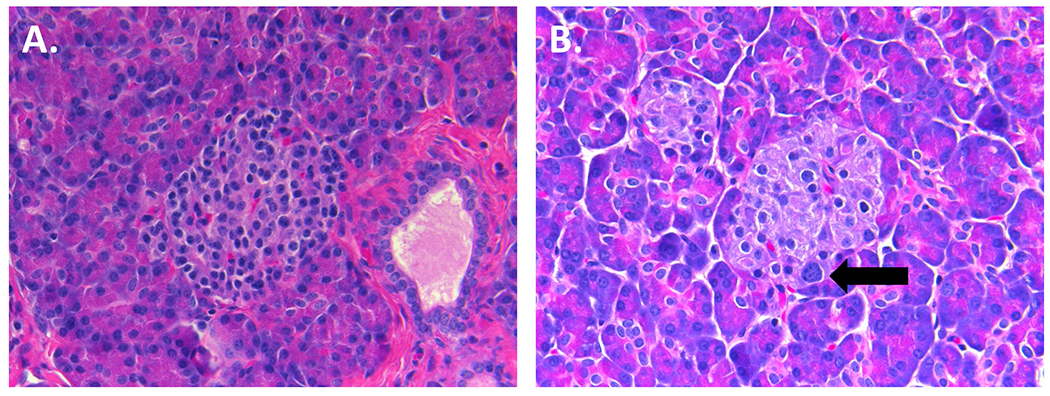
H&E staining in LINE-HI Case 10 (40X). Islets from both unaffected (A) and affected (B) regions of the pancreas in LINE-HI retain normal architecture. Islets from the affected region contain an increased number of islet-cells with nucleomegaly (indicated with an arrow) compared to the unaffected region.
Clinical characteristics of children with LINE-HI
Table 1 shows the clinical characteristics of patients with LINE-HI. All 12 LINE-HI cases presented with a hypoglycemic seizure, were unresponsive to treatment with diazoxide, and required pancreatectomy to control hypoglycemia. Seven of the 12 LINE-HI cases underwent 18F-DOPA PET scan; five of these were interpreted as negative for a focal lesion; one had increased uptake in the pancreatic head, possibly physiologic (Case 9), and another had increased uptake in the body (Case 12). A focal lesion was not identified in either case at surgery though Case 12 had a pancreatic lobule arising from the posterior aspect of the body. In all cases, histology was consistent with LINE-HI. Compared to children with typical diffuse or focal HI, children with LINE-HI had a lower average birth weight (P<0.05) and presented later in infancy (P<0.05) (Table 2). Because the surgical approach for suspected LINE-HI cases is to take biopsies throughout the pancreas and then resect only the regions of pancreas in which nucleomegaly is identified 34, LINE-HI cases had a lesser extent of pancreatic resection at surgery (P<0.05) and had better outcomes after surgery compared to diffuse-HI cases. None of the LINE-HI cases have developed diabetes following surgery over a median follow-up period of 7 years (range 2-15 years), in contrast to cases with diffuse-HI treated with pancreatectomy at our Center, who develop diabetes at a median age of 7.7 years 46. These differences in surgical outcomes and risk of diabetes may reflect both the lesser extent of pancreas resected in most LINE-HI cases and the fact that their residual pancreas may retain predominantly normal islet cells. All 12 LINE-HI cases presented here had distal pancreatectomies with histological evidence of nucleomegaly being present most often in the body and/or tail (8/12, 66%). In three cases, partial pancreatectomy did not result in a cure, possibly due to abnormal cells remaining in the residual pancreas.
Table 1.
Clinical characteristics of children with LINE-HI.
| Case | Gender | Birth Weight (g) | Z-score | Presentation | (18) F-FDOPA PET | Percent Pancreatectomy | Affected region | Outcome | Mosaic Mutation by NGS |
|
|---|---|---|---|---|---|---|---|---|---|---|
| Pancreas | Blood | |||||||||
| 1 | F | 2700 | −0.23 | 4 months; seizure (hypoglycemia at birth) | n/a | 98% | head | control (octreotide) | GCK | Neg |
| 2 | F | 4500 | 2.23 | DOL1; seizure | n/a | 95% | head and body | cure | NA | Neg |
| 3 | M | 3400 | −0.26 | 10 months; seizure | n/a | 95% | head and tail | cure | NA | Neg |
| 4 | M | 3000 | −1.07 | 4 months; seizure (7 months; HI diagnosed) | n/a | 65% | tail | cure | NA | NA |
| 5 | M | 3600 | 0.14 | 7 months; seizure (9 months; HI diagnosed) | n/a | 50% | head | control (octreotide) | NA | NA |
| 6 | F | 2800 | −1.28 | 17 months; seizure | diffuse | 85% | body, tail | cure | neg | Neg |
| 7 | F | 2900 | −0.74 | 4 months; seizure | diffuse | 90% | tail | cure | neg | Neg |
| 8 | F | 3250 | −0.32 | 1 month; seizure | diffuse | 15% | tail | cure | ABCC8 | Neg |
| 9 | F | 3600 | 0.66 | 1.5 months; seizure | increased uptake in head | 60% | body, tail | control (limited fasting) | ABCC8 | Neg |
| 10 | F | 3700 | 2.29 | 2 years; seizure | diffuse | 40% | body, tail | cure | GCK | Neg |
| 11 | F | 2700 | −1.50 | 2 months; seizure | diffuse | 70% | tail | cure | ABCC8 | Neg |
| 12 | F | 3500 | 0.20 | 6 months; seizure | increased uptake in body | 40% | body, tail | cure | GCK | Neg |
NA = material not available
Table 2.
Clinical features of LINE-HI compared to typical Focal or Diffuse-HI.
| n | Birth weight (g) | Z-score | Presentation Age (days) | Pancreatectomy (%) | Outcome a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Mean ± SD | Median (25th,75thile) | Range | Mean ± SD | Median (25th,75thile) | Range | EuG | HypoG | HyperG | ||
| LINE-HI | 12 | 3304 ± 527 | 1.1 ±1.07 | 120 (49, 278) | 1-730 | 67 ± 27 | 68 (43, 94) | 15-98 | 75% | 25% | 0% |
| Diffuse KATP HI b | 43 | 4275 ± 619 | 2.10 ± 1.14 | 0 (0, 0) | 0-63 | 96 ± 5 | 98 (97, 98) | 75-100 | 31% | 49% | 20% |
| Focal KATP HI c | 90 | 3785 ± 573 | 1.47 ± 0.99 | 0.5 (0, 27) | 0-366 | 39 ± 29 | 40 (10, 60) | 2-100 | 97% | ||
Outcomes immediately after surgery for focal and diffuse HI at CHOP in (PMID: 30343978);
Histologically confirmed diffuse HI and compound heterozygous recessive KATP channel mutations (PMID: 26327482);
Histologically confirmed focal HI and paternally inherited recessive KATP channel mutation (PMID: 26327482)
Features of low-level mosaic mutations identified by NGS
Table 3 and Supplemental Table S1 show details of the mosaic mutations identified by NGS in pancreatic DNA or cDNA from the six children with LINE-HI. ABCC8 mutations were identified in three cases of LINE-HI (Table 3) with degree of mosaicism ranging from 2.5-5.4%. Case 8 had two ABCC8 variants, both novel and identified at 2.5% mosaicism; it was not possible to determine if these mutations arose on the same or different alleles. In addition to a mosaic ABCC8 mutation, Case 9 had a maternal intronic ABCC8 variant that was considered to be benign, since it was not predicted to alter splicing and has a high population frequency (1:206 in East Asians). In Case 11, a mosaic ABCC8 mutation was identified in islet cDNA from an affected region of the pancreas, but not in an unaffected region. All of the four ABCC8 mutations were novel missense changes. Expression studies of these ABCC8 mutations are described below.
Three GCK activating mutations were identified in three cases with percent mosaicism ranging from 2.2-10.3% in pancreatic DNA. In one of these (Case 10), the GCK mutation was identified at very low levels of mosaicism in peripheral blood (0.96%) but significantly greater than the background miscall rate at this nucleotide position (0.25%, P<0.05). Two of the three GCK mutations were novel and one was previously reported 53. Functional studies of all three GCK mutations confirmed that they were disease causing (see below).
Validation of mosaic mutations identified by NGS
NGS base calls at the nucleotide position of the mosaic mutations, as well as at the five flanking nucleotides, are shown in Supplemental Table S1. The average sequencing depth at the position of the mosaic mutations in these cases was 22,125x, with a range of 3,886x to 46,343x. The average number of reads that were positive for the mosaic mutation was 1,306, with a range of 69 to 4,785. As shown in Table 3, average background miscall rates in controls from the same sequencing run ranged from 0-0.25% at the nucleotide position where the mosaic mutation was identified and were markedly below the frequency of the mosaic mutation in all cases.
Mosaic mutations were confirmed by repeat NGS in all cases from which sufficient pancreatic material was available (4 of 6 cases, Table 3). In two cases with insufficient material for repeat NGS (Cases 8 and 9), the original NGS libraries were re-sequenced and the mutations were confirmed as technical replicates. In one of these (Case 8) pancreatic DNA was available from another region of the pancreas with less characteristic features of LINE, but neither of the two mutations could be confirmed in this sample.
Sanger sequencing confirmed the mutations in pancreas from two of the six LINE-HI cases. In the four cases that were not confirmed by Sanger, the percent mosaicism was below 6% based on NGS.
Functional Characterization of novel ABCC8 Mutations.
To evaluate the effects of the five novel ABCC8 mutations on KATP channel expression and function, we performed Western blotting and Rb efflux assays using COS cells co-transfected with cDNAs encoding human WT Kir6.2 and WT or mutant human SUR1. Complex-glycosylation of SUR1, which shows up in western blot as the “upper” SUR1 band, marks successful assembly of the protein into the channel complex able to traffic to the plasma membrane; it is therefore used to assess channel maturation and surface expression 47. As shown in Figure 2A, three of the SUR1 mutants (p.Arg534Gly, p.Pro690Leu, and p.Thr1242Ser) showed clear complex-glycosylated SUR1 upper band indicating surface expression. In contrast, the other mutant (p.Gly718Ser) showed no upper band indicating this mutant is not expressed at the cell surface. Next, we assessed how the mutations impact the ability of the channel to open in response to metabolic inhibition using 86Rb+ efflux assays. The p.Gly718Ser mutation nearly abolished efflux above that observed in untransfected cells during a 40 min period, consistent with this mutant not being expressed at the plasma membrane (Fig 2B, 2C). The p.Arg534Gly and p.Pro690Leu mutations rendered reduced efflux compared to WT channels, indicating that these mutations compromise channel function. As shown in Fig 2D, patch-clamp recording showed that the p.Thr1242Ser has ATP and MgADP responses similar to WT channels, suggesting that this variant may be benign.
Figure 2. Characterization of mutant KATP channels.
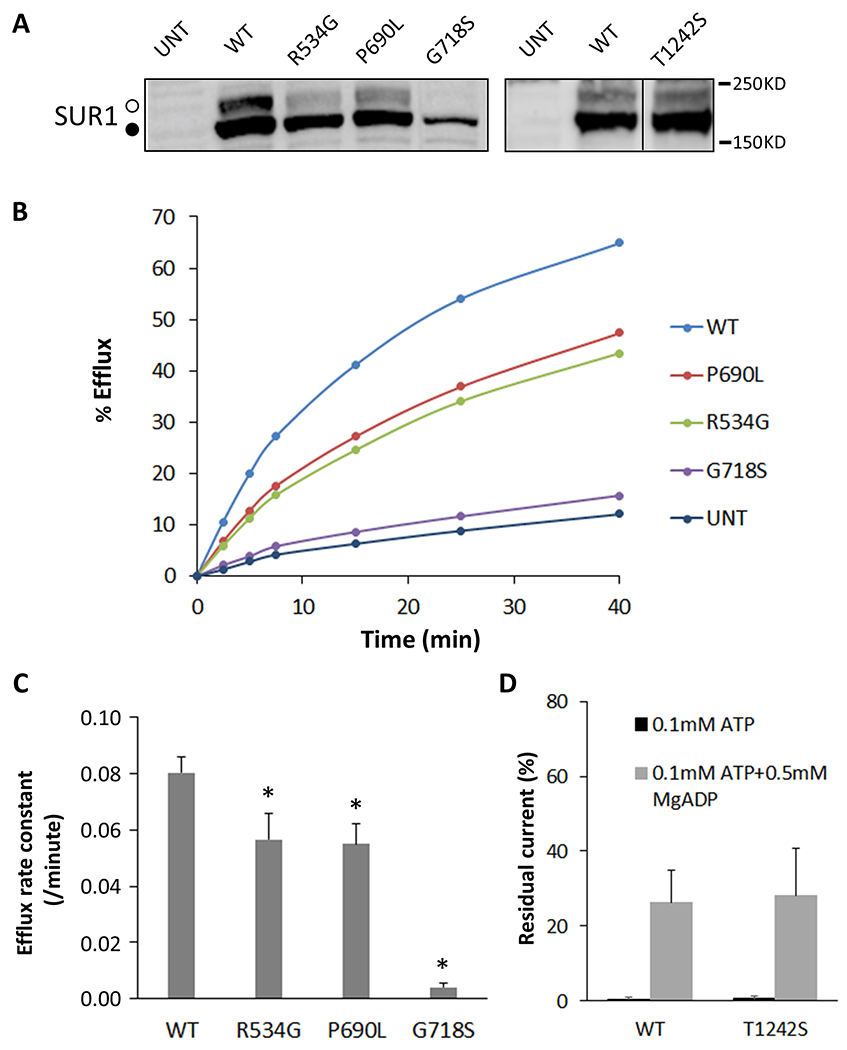
Panel A. A representative western blot of SUR1 in COSm6 cells transiently transfected with WT Kir6.2 together with WT or mutant SUR1 as indicated above the blot. The core-glycosylated immature lower band and the complex-glycosylated mature upper bands of SUR1 are indicated by the solid and open circle, respectively. The vertical line separates different parts of the same blot. The p.Thr1242Ser was analyzed in different experiments and shown in a separate blot. Panel B. Representative results of 86Rb efflux experiments. Similar results were obtained in four independent experiments. Panel C. 86Rb+ efflux rate constant calculated using the equation described in the Methods. Data represent mean ± SEM of three independent experiments. *p<0.05, comparison between WT and the mutant by two-tailed, paired Student’s t-test. Panel D. Patch-clamp recording data showing WT and p.Thr1242Ser mutant channel activity in 0.1mM ATP or 0.1mM ATP+0.5mM MgADP as percent currents in those observed in solutions without added ATP or ADP. Each bar represents the mean ± s.e.m. for four patches.
The above results support a role of three of these mutations in causing hyperinsulinism by impaired KATP channel function either by abolishing expression of the channels on the plasma membrane and/or by reducing channel function. In general, mutations capable of trafficking act in a monoallelic, dominant-negative fashion (p.Arg534Gly and p.Pro690Leu), while mutations that are not capable of trafficking act in bi-allelic, recessive fashion (p.Gly718Ser) 49, 54, 55. In Case 8, with LINE-HI and two mosaic ABCC8 mutations, it appears likely that the HI is due to the first of these mutations, the trafficking-competent p.Pro690Leu compatible with a dominant defect, although the possibility of compound heterozygosity for both a dominant and a recessive mutation (p.Gly718Ser, trafficking defect) cannot be excluded. In Case 9 with LINE-HI, the underlying defect remains unclear since both the maternal intronic variant and the mosaic variant appeared to be non-pathogenic based on in silico analysis and expression studies. The expression studies suggest that the HI in Case 11 can be explained by the mosaic p.Arg534Gly mutation, presuming that it acts in mono-allelic, dominant, fashion.
Functional Characterization of Novel GCK Mutations.
In order to confirm that the two novel GCK mutations as well as the previously reported p.Val91Leu mutation identified in these studies were pathogenic, the corresponding GST-tag mutant glucokinase proteins were produced in E. coli. As shown in Table 4, in all three GCK mutations there was a marked reduction in the glucose concentration for half-maximal enzymatic activity compared to wildtype glucokinase, thus confirming that the mutations caused a pathogenic gain of enzymatic function. This is consistent with other reported activating GCK mutations that result in HI which have an S0.5 range of 0.72-4.5mM 37, 56, 57.
Table 4.
Mutant glucokinase enzyme kinetics.
| GCK Mutant | Glucose S0.5 (mM) |
|---|---|
| p.Ser64Pro | 0.9 |
| p.Val91Leu | 1.2 |
| p.Arg447Leu | 1.1 |
| Normal Control | 7 |
HK1 Expression by Immunoflourescence and cDNA analyses.
A study from Professor Henquin’s lab on infants with congenital HI and a histologic appearance of “atypical hyperinsulinism” (similar to our cases with LINE-HI) found evidence in some cases of increased HK1 immunofluorescent staining in affected islets and suggested that their hyperinsulinism might be explained by abnormal expression of HK1 enzymatic activity 37. To evaluate this observation in our series of LINE-HI cases, we performed immunofluorescent staining for HK1 in pancreas from our cases of LINE-HI, as well as in controls with typical diffuse or focal HI due to KATP channel mutations or GCK mutations. As shown in Figure 3A, increased HK1 staining in islets within regions of pancreas showing islet cell nucleomegaly was observed only in two cases of LINE-HI in our series (Cases 3 and 6); increased HK1 staining was not present in islets from the normal regions of pancreas. Fresh or frozen pancreas tissue was not available for NGS in Case 3, but NGS in peripheral blood DNA did not reveal a mutation in the genes tested. NGS of whole pancreas DNA in Case 6 did not reveal a mutation in ABCC8, KCNJ11 or GCK. HK1 staining was negative in the other 10 LINE-HI cases (illustrated by LINE-HI cases 7 and 8 in Fig 3A), as well as in pancreas from cases with focal-HI and diffuse-HI due to KATP or GCK mutations.
Figure 3. HK1 Expression by immunofluorescence staining and RT-PCR.
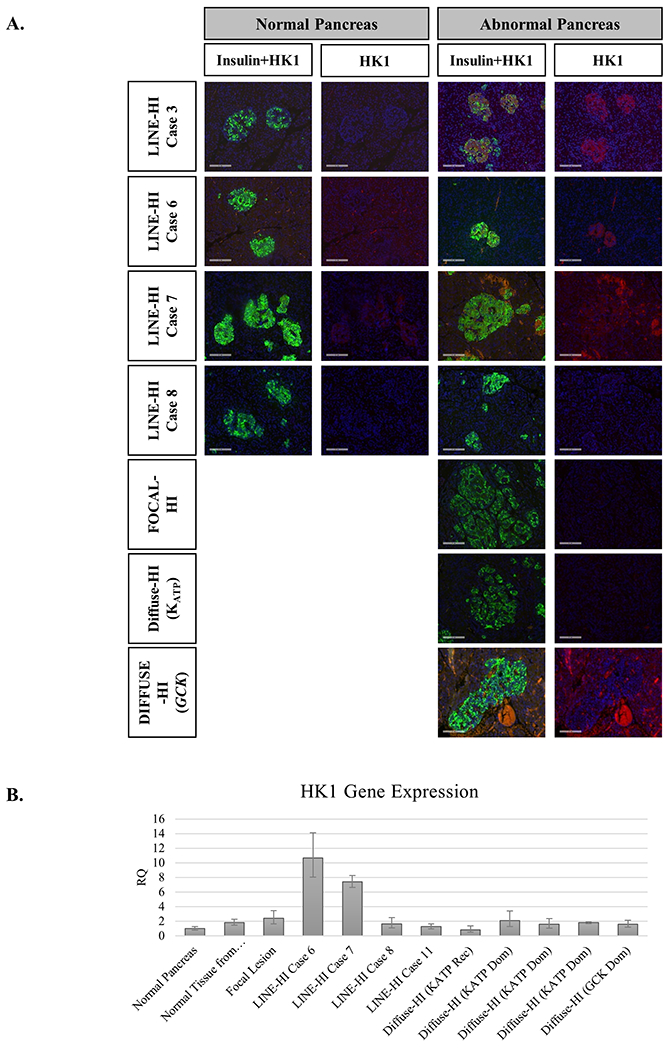
A. HK1 Expression by immunofluorescence staining. Formalin fixed paraffin embedded tissue sections from normal and abnormal regions of LINE-HI cases and controls (Diffuse-HI due to KATP channel or GCK mutations) from resected pancreas were stained for insulin (green) and HK1 (red). Islets from the abnormal region of the pancreas in LINE-HI cases 3 and 6 showed increased HK1 staining compared to normal pancreas. Although some HK1 staining appears to be present in the abnormal region in LINE-HI case 7, it does not appear to be localized to the islets. HK1 staining was absent in normal and abnormal pancreatic regions for the nine other LINE-HI patients, as demonstrated by LINE-HI case 8) . HK1 staining was also absent in pancreas from focal-HI and diffuse-HI due to either KATP or GCK mutations. B. Quantitation of HK1 Expression by RT-PCR. HK1 expression was assessed by RT-PCR in four cases of LINE-HI compared to normal pancreas, a focal lesion, and typical diffuse-HI cases due to either KATP or GCK mutation. Two cases of LINE-HI (Cases 6 and 7 showed an increase in HK1 staining compared to control samples (10 and 7 fold increase, respectively).
In order to determine whether the increase in HK1 immunofluoresence reflected increased HK1 gene expression, we measured pancreatic tissue levels of HK1 mRNA by synthesizing cDNA by RT-PCR in those cases from which fresh or frozen whole pancreas or isolated islets were available. As shown in Figure 3B, Case 6 showed increased HK1 gene expression, confirming the increased HK1 immunofluorescent staining. Case 7 also showed increased HK1 gene expression, although increased HK1 immunofluorescent staining was not observed in this case (Fig 3A). The other LINE-HI cases, as well as the control cases with focal or diffuse HI due to KATP channel mutations, and the one case with diffuse HI due to GCK mutation, did not show increased HK1 gene expression compared to pancreas from a normal control.
Discussion
The results of these studies support our hypothesis that post-zygotic mosaic mutations of known HI genes may be the underlying genetic defect in some children with LINE-HI. Mosaic mutations were identified in two of the genes that are most commonly involved in congenital hyperinsulinism: GCK and the ABCC8 subunit of the KATP channel. These mutations were identified at low levels of mosaicism in pancreatic tissue by NGS.
We and others have previously reported somatic post-zygotic mutations in genes associated with HI identified in pancreatic DNA. We have previously reported post-zygotic mutations in GLUD1 and GCK identified in pancreas from two diffuse cases 30. The case with mosaic GLUD1 mutation (c.1493c>t/p.Ser498Leu) elected for pancreatectomy despite being diazoxide responsive and the GLUD1 mutation was subsequently identified at low levels of mosaicism in peripheral blood DNA (data unpublished). In the second case with a somatic GCK mutation (c.1361_1363dupCGG/p.Ala454dup) identified in pancreatic DNA, the mutation was not reported on clinical genetic testing of peripheral blood DNA, done by Sanger Sequencing. Because peripheral blood DNA was not available for sequencing by NGS, it is unknown whether or not the GCK mutation in this case is present in peripheral blood at levels below the limit of detection of the clinical genetic test. In addition, Houghton, et al. reported a case with HI associated with 11pUPD and a somatic mutation in ABCC8 (p.Glu1507Lys) that was detected in lesion tissue by NGS at a level of mosaicism (28% of 419 reads) high enough to be found by Sanger sequencing, but was not found in normal regions of pancreas or in peripheral blood DNA 58. Henquin, et al. reported a patient with histology consistent with LINE-HI and a mosaic GCK (p.Ile211Phe) mutation identified in affected pancreatic tissue; blood and unaffected pancreas were negative for the mutation (see below) 37.
Two series of children with histology consistent with LINE-HI (but termed “mosaic” or “atypical” by the authors of the respective series) have previously been reported in the literature. Sempoux, et al. described a series of 16 children with negative mutation analysis for ABCC8, KCNJ11, and GCK who had normal birth weights and late onset of hypoglycemia. Ten of the 16 children were cured after partial pancreatectomy 36. Henquin, et al. described a series of six children with negative mutation analysis in peripheral blood DNA for ABCC8, KCNJ11, and GCK who underwent surgery. In one of these cases, functional studies of isolated islets from an affected region of pancreas showed an abnormally low glucose threshold for insulin release. Sanger sequencing of cDNA from the affected tissue detected an activating mutation of GCK (p.Ile211Phe) that was not detected in blood or in unaffected regions of pancreas 37.
Henquin et al. reported evidence of increased immunohistochemical staining for HK1 in affected areas of pancreas from four cases with “atypical”/LINE-HI pathology 37. Isolated islets from these cases demonstrated low glucose thresholds for insulin secretion, suggesting that increased HK1 enzymatic activity could be responsible, at least in part, for hyperinsulinism due to the very low Km of this enzyme for glucose. Our results in two cases with LINE-HI confirmed their observation of increased HK1 immunostaining and also demonstrated that increased HK1 immunostaining in at least one case was associated with increased levels of HK1 mRNA. It is unclear whether increased HK1 expression, or other factors, such as altered expression of additional genes controlling insulin release, are contributing to the HI in these cases. Interestingly, non-coding regulatory mutations resulting in aberrant HK1 expression in beta cells have been recently identified as a cause of hyperinsulinism 59.
Because the diagnosis of LINE-HI is based on histological findings in resected pancreas, the cases presented here and in other series have been diazoxide-unresponsive 36, 37. However, it is reasonable to predict that some children with diazoxide-responsive HI and negative genetic analysis in peripheral blood may have mosaic mutations in known HI genes within the pancreas and also have histology consistent with LINE-HI. Over half of the children with diazoxide-responsive HI do not have an identifiable mutation in known HI genes 30. While some dominant mutations in ABCC8 result in diazoxide-unresponsive HI, others result in HI that is responsive to diazoxide 33.
The very low levels of mosaicism for some of the mutations in this study raises possible concern whether they can reasonably explain HI severe enough to require pancreatectomy. To address this potential limitation, efforts were made to validate mutations in multiple ways, including repeated NGS and expression studies to confirm that novel mutations were pathogenic. With regard to the likelihood that severe HI can result from mutations affecting only a very small proportion of pancreatic beta-cells, there is a rough correlation in patients with HI due to inactivating KATP channel mutations between the severity of hyperinsulinism and the proportion of pancreatic beta-cells affected: the most severe disease occurs in patients with paternal 11p uniparental isodisomy and BWS that results in overgrowth of islets expressing isodisomic KATP mutations 60, followed by patients with diffuse KATP-HI affecting all beta-cells, and then patients with focal-HI lesions 46. In some of the latter, the percentage of total pancreatic beta-cells affected may be very small with lesions measuring half a centimeter or less. In children with diffuse KATP-HI, following near-total pancreatectomy, 2-5% residual pancreas is often associated with continuing severe hypoglycemia 34. Thus, in our patients with LINE-HI and mosaic ABCC8 mutations, their relatively mild HI phenotype and later age at presentation compared to patients with focal or diffuse HI appears to be consistent with their mutations affecting only a small proportion of beta-cells.
A limitation of the present study was the fact that the search for mosaic mutations was retrospective and dependent on availability of appropriate specimens of pancreas and other tissues. In two cases, repeat NGS was not feasible due to inadequate pancreatic DNA (Cases 8 and 9). Although these mutations were validated by technical replicate, we cannot fully exclude the possibility that the mutations identified in these two cases are false positive calls. In the remaining four cases, repeat NGS validated the mosaic mutations and Sanger sequencing also confirmed the mutations in two of these cases (Cases 1 and 10). In the two cases that were not validated by Sanger (Cases 11 and 12), the mosaicism was less than 6% and likely represents the limit of detection of the methodology. In both cases, there is evidence for the presence of the mosaic mutation in multiple tissues. In Case 11, the mosaic ABCC8 mutation identified in islet cDNA at 3.4% was present at low levels in DNA from whole pancreas (0.2-0.34%) and blood (0.27%). Though these levels of mosaicism are low, they are significantly greater than the miscall rate at this nucleotide position in controls (P<0.05). In Case 12, the mosaic GCK mutation was identified in two separate pieces of pancreas at levels of mosaicism of 2.2 and 3.3% (Table S1). The present findings may aid in designing improved approaches to detecting and validating low level mosaic mutations.
In summary, LINE-HI histology should be considered in children with diazoxide-unresponsive HI who present later in infancy. These children are negative for known HI mutations in peripheral blood DNA and partial pancreatectomy in these cases may be curative. Our results demonstrate that mosaic mutations in ABCC8 or GCK should be considered in these children and may only be detectable using highly sensitive NGS methods on samples from affected pancreatic regions. We speculate that low-level mosaic mutations of dominant HI genes, such as ABCC8 and GCK, could be present in other HI cases with negative genetic testing.
Supplementary Material
Acknowledgements and Funding
This work was supported by National Institutes of Health grants R01-DK056268 (C.A.S., A.G., D.D.D.L.), R01-DK098517 (D.D.D.L.), and R01-DK057699 and R01-DK066485 (S.L.S.). Additional support was provided by the CHOP Center for Human Phenomic Science supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1TR001878. Next generation sequencing and mosaic variant calling was performed in the Penn Genomic Analysis Core at the University of Pennsylvania Perelman School of Medicine. The authors would like to thank Erik Toorens from the Penn Genomic Analysis core for his help in the analysis of the mosaic mutations included in this manuscript. The authors would like to thank the Genome Aggregation Database (gnomAD) and the groups that provided exome and genome variant data to this resource. A full list of contributing groups can be found at https://gnomad.broadinstitute.org/about.
Declaration of interest
DDDL owns stock/stock options from Merck & Co. DDDL has received research funding from Zealand Pharma, Tiburio Therapeutics and Crinetics Pharmaceuticals for studies not included in this manuscript. DDDL has received consulting fees from Zealand Pharma, Crinetics Pharmaceuticals, Hanmi Pharmaceutical, Poxel SA, and Twist Pharma not related to this manuscript. CAS and DDDL are named inventors in these patents # USA Patent Number 9,616,108, 2017, USA Patent Number 9,821,031, 2017, Europe Patent Number EP 2120994, 2018, Europe Patent Number EP2818181, 2019. These patents are not related to the work included in this manuscript.
Funding:
R01-DK056268, R01-DK098517, R01-DK057699, R01-DK066485, UL1TR001878
Disclosure Statement:
Ownership: DDDL owns stock/stock options from Merck & Co
Research support: DDDL has received research funding from Zealand Pharma, Tiburio Therapeutics, Twist Pharma, and Crinetics Pharmaceuticals for studies not included in this manuscript
Consulting: DDDL has received consulting fees from Zealand Pharma, Crinetics Pharmaceuticals, Hanmi Pharmaceutical, Poxel SA, and Heptares Therapeutics not related to this manuscript.
Intellectual property: CAS and DDDL are named inventors in these patents # USA Patent Number 9,616,108, 2017, USA Patent Number 9,821,031, 2017, Europe Patent Number EP 2120994, 2018, Europe Patent Number EP2818181, 2019. These patents are not related to the work included in this manuscript.
Data Availability
Some or all datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.
References
- 1.Galcheva S, Demirbilek H, Al-Khawaga S & Hussain K. The Genetic and Molecular Mechanisms of Congenital Hyperinsulinism. Front Endocrinol (Lausanne) 2019. 10 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JPt, Boyd AE 3rd, Gonzalez G, Herrera-Sosa H, Nguy K, Bryan J & Nelson DA. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science 1995. 268 423–426. [DOI] [PubMed] [Google Scholar]
- 3.Nestorowicz A, Inagaki N, Gonoi T, Schoor KP, Wilson BA, Glaser B, Landau H, Stanley CA, Thornton PS, Seino S & Permutt MA. A nonsense mutation in the inward rectifier potassium channel gene, Kir6.2, is associated with familial hyperinsulinism. Diabetes 1997. 46 1743–1748. [DOI] [PubMed] [Google Scholar]
- 4.Nestorowicz A, Wilson BA, Schoor KP, Inoue H, Glaser B, Landau H, Stanley CA, Thornton PS, Clement JPt, Bryan J, Aguilar-Bryan L & Permutt MA. Mutations in the sulonylurea receptor gene are associated with familial hyperinsulinism in Ashkenazi Jews. Hum Mol Genet 1996. 5 1813–1822. [DOI] [PubMed] [Google Scholar]
- 5.Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, Aguilar-Bryan L, Gagel RF & Bryan J. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 1995. 268 426–429. [DOI] [PubMed] [Google Scholar]
- 6.Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, Stanley CA, Thornton PS, Permutt MA, Matschinsky FM & Herold KC. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med 1998. 338 226–230. [DOI] [PubMed] [Google Scholar]
- 7.Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, Perlman K, Rich BH, Zammarchi E & Poncz M. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med 1998. 338 1352–1357. [DOI] [PubMed] [Google Scholar]
- 8.Dusatkova P, Pruhova S, Sumnik Z, Kolouskova S, Obermannova B, Cinek O & Lebl J. HNF1A mutation presenting with fetal macrosomia and hypoglycemia in childhood prior to onset of overt diabetes. J Pediatr Endocrinol Metab 2011. 24 187–189. [DOI] [PubMed] [Google Scholar]
- 9.Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, Siahanidou T, Banerjee I, Akcay T, Rubio-Cabezas O, Shield JP, Hussain K & Ellard S. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol 2010. 162 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, Ellard S, Ferrer J & Hattersley AT. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med 2007. 4 e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pingul MM, Hughes N, Wu A, Stanley CA & Gruppuso PA. Hepatocyte nuclear factor 4alpha gene mutation associated with familial neonatal hyperinsulinism and maturity-onset diabetes of the young. J Pediatr 2011. 158 852–854. [DOI] [PubMed] [Google Scholar]
- 12.Stanescu DE, Hughes N, Kaplan B, Stanley CA & De Leon DD. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. J Clin Endocrinol Metab 2012. 97 E2026–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tung JY, Boodhansingh K, Stanley CA & De Leon DD. Clinical heterogeneity of hyperinsulinism due to HNF1A and HNF4A mutations. Pediatr Diabetes 2018. 19 910–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrara CT, Boodhansingh KE, Paradies E, Giuseppe F, Steinkrauss LJ, Topor LS, Quintos JB, Ganguly A, De Leon DD, Palmieri F & Stanley CA. Novel Hypoglycemia Phenotype in Congenital Hyperinsulinism Due to Dominant Mutations of Uncoupling Protein 2. J Clin Endocrinol Metab 2017. 102 942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez-Barroso MM, Giurgea I, Bouillaud F, Anedda A, Bellanne-Chantelot C, Hubert L, de Keyzer Y, de Lonlay P & Ricquier D. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS One 2008. 3 e3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clayton PT, Eaton S, Aynsley-Green A, Edginton M, Hussain K, Krywawych S, Datta V, Malingre HE, Berger R & van den Berg IE. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J Clin Invest 2001. 108 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molven A, Matre GE, Duran M, Wanders RJ, Rishaug U, Njolstad PR, Jellum E & Sovik O. Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes 2004. 53 221–227. [DOI] [PubMed] [Google Scholar]
- 18.Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, Kere J & Sipila I. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes 2003. 52 199–204. [DOI] [PubMed] [Google Scholar]
- 19.Grand K, Gonzalez-Gandolfi C, Ackermann AM, Aljeaid D, Bedoukian E, Bird LM, De Leon DD, Diaz J, Hopkin RJ, Kadakia SP, Keena B, Klein KO, Krantz I, Leon E, Lord K, McDougall C, Medne L, Skraban CM, Stanley CA, Tarpinian J, Zackai E, Deardorff MA & Kalish JM. Hyperinsulinemic hypoglycemia in seven patients with de novo NSD1 mutations. Am J Med Genet A 2019. 179 542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flanagan SE, Vairo F, Johnson MB, Caswell R, Laver TW, Lango Allen H, Hussain K & Ellard S. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr Diabetes 2017. 18 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giri D, Vignola ML, Gualtieri A, Scagliotti V, McNamara P, Peak M, Didi M, Gaston-Massuet C & Senniappan S. Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm-derived organ abnormalities. Hum Mol Genet 2017. 26 4315–4326. [DOI] [PubMed] [Google Scholar]
- 22.Yap KL, Johnson AEK, Fischer D, Kandikatla P, Deml J, Nelakuditi V, Halbach S, Jeha GS, Burrage LC, Bodamer O, Benavides VC, Lewis AM, Ellard S, Shah P, Cody D, Diaz A, Devarajan A, Truong L, Greeley SAW, De Leo-Crutchlow DD, Edmondson AC, Das S, Thornton P, Waggoner D & Del Gaudio D. Congenital hyperinsulinism as the presenting feature of Kabuki syndrome: clinical and molecular characterization of 9 affected individuals. Genet Med 2019. 21 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabezas OR, Flanagan SE, Stanescu H, Garcia-Martinez E, Caswell R, Lango-Allen H, Anton-Gamero M, Argente J, Bussell AM, Brandli A, Cheshire C, Crowne E, Dumitriu S, Drynda R, Hamilton-Shield JP, Hayes W, Hofherr A, Iancu D, Issler N, Jefferies C, Jones P, Johnson M, Kesselheim A, Klootwijk E, Koettgen M, Lewis W, Martos JM, Mozere M, Norman J, Patel V, Parrish A, Perez-Cerda C, Pozo J, Rahman SA, Sebire N, Tekman M, Turnpenny PD, Hoff WV, Viering D, Weedon MN, Wilson P, Guay-Woodford L, Kleta R, Hussain K, Ellard S & Bockenhauer D. Polycystic Kidney Disease with Hyperinsulinemic Hypoglycemia Caused by a Promoter Mutation in Phosphomannomutase 2. J Am Soc Nephrol 2017. 28 2529–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld ME, Timal S, Raymond K, He P, Ichikawa M, Veltman J, Huijben K, Shin YS, Sharma V, Adamowicz M, Lammens M, Reunert J, Witten A, Schrapers E, Matthijs G, Jaeken J, Rymen D, Stojkovic T, Laforet P, Petit F, Aumaitre O, Czarnowska E, Piraud M, Podskarbi T, Stanley CA, Matalon R, Burda P, Seyyedi S, Debus V, Socha P, Sykut-Cegielska J, van Spronsen F, de Meirleir L, Vajro P, DeClue T, Ficicioglu C, Wada Y, Wevers RA, Vanderschaeghe D, Callewaert N, Fingerhut R, van Schaftingen E, Freeze HH, Morava E, Lefeber DJ & Marquardt T. Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med 2014. 370 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sethi A, Foulds N, Ehtisham S, Ahmed SH, Houghton J, Colclough K, Didi M, Flanagan SE & Senniappan S. Heterozygous Insulin Receptor (INSR) Mutation Associated with Neonatal Hyperinsulinemic Hypoglycaemia and Familial Diabetes Mellitus: Case Series. J Clin Res Pediatr Endocrinol 2020. 12 420–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Welters A, El-Khairi R, Dastamani A, Bachmann N, Bergmann C, Gilbert C, Clement E, Hurst JA, Quercia N, Wasserman JD, Meissner T, Shah P & Kummer S. Persistent hyperinsulinaemic hypoglycaemia in children with Rubinstein-Taybi syndrome. Eur J Endocrinol 2019. 181 121–128. [DOI] [PubMed] [Google Scholar]
- 27.Torekov SS, Iepsen E, Christiansen M, Linneberg A, Pedersen O, Holst JJ, Kanters JK & Hansen T. KCNQ1 long QT syndrome patients have hyperinsulinemia and symptomatic hypoglycemia. Diabetes 2014. 63 1315–1325. [DOI] [PubMed] [Google Scholar]
- 28.Pinney SE, Ganapathy K, Bradfield J, Stokes D, Sasson A, Mackiewicz K, Boodhansingh K, Hughes N, Becker S, Givler S, Macmullen C, Monos D, Ganguly A, Hakonarson H & Stanley CA. Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Horm Res Paediatr 2013. 80 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregory LC, Ferreira CB, Young-Baird SK, Williams HJ, Harakalova M, van Haaften G, Rahman SA, Gaston-Massuet C, Kelberman D, Gosgene, Qasim W, Camper SA, Dever TE, Shah P, Robinson I & Dattani MT. Impaired EIF2S3 function associated with a novel phenotype of X-linked hypopituitarism with glucose dysregulation. EBioMedicine 2019. 42 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, Ganapathy K, Bhatti T, Stanley CA & Ganguly A. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab 2013. 98 E355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Lonlay P, Fournet JC, Rahier J, Gross-Morand MS, Poggi-Travert F, Foussier V, Bonnefont JP, Brusset MC, Brunelle F, Robert JJ, Nihoul-Fekete C, Saudubray JM & Junien C. Somatic deletion of the imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous hyperplasia and endorses partial pancreatectomy. J Clin Invest 1997. 100 802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macmullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, Ganguly A, Shyng SL & Stanley CA. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the beta-cell sulfonylurea receptor SUR1. Diabetes 2011. 60 1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boodhansingh KE, Kandasamy B, Mitteer L, Givler S, De Leon DD, Shyng SL, Ganguly A & Stanley CA. Novel dominant KATP channel mutations in infants with congenital hyperinsulinism: Validation by in vitro expression studies and in vivo carrier phenotyping. Am J Med Genet A 2019. 179 2214–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adzick NS, De Leon DD, States LJ, Lord K, Bhatti TR, Becker SA & Stanley CA. Surgical treatment of congenital hyperinsulinism: Results from 500 pancreatectomies in neonates and children. J Pediatr Surg 2019. 54 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sayed S, Langdon DR, Odili S, Chen P, Buettger C, Schiffman AB, Suchi M, Taub R, Grimsby J, Matschinsky FM & Stanley CA. Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes 2009. 58 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sempoux C, Capito C, Bellanne-Chantelot C, Verkarre V, de Lonlay P, Aigrain Y, Fekete C, Guiot Y & Rahier J. Morphological mosaicism of the pancreatic islets: a novel anatomopathological form of persistent hyperinsulinemic hypoglycemia of infancy. J Clin Endocrinol Metab 2011. 96 3785–3793. [DOI] [PubMed] [Google Scholar]
- 37.Henquin JC, Sempoux C, Marchandise J, Godecharles S, Guiot Y, Nenquin M & Rahier J. Congenital hyperinsulinism caused by hexokinase I expression or glucokinase-activating mutation in a subset of beta-cells. Diabetes 2013. 62 1689–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suchi M, MacMullen C, Thornton PS, Ganguly A, Stanley CA & Ruchelli ED. Histopathology of congenital hyperinsulinism: retrospective study with genotype correlations. Pediatr Dev Pathol 2003. 6 322–333. [DOI] [PubMed] [Google Scholar]
- 39.Ernst LE, Suchi M, SC A, Adzick NS, MacMullen C & Ruchelli E. Localized islet cell nuclear enlargement in congenital hyperinsulinism: a distinct clinicopathologic entity. Mod Pathol 2007. 287. [Google Scholar]
- 40.Ferrara C, Patel P, Becker S, Stanley CA & Kelly A. Biomarkers of Insulin for the Diagnosis of Hyperinsulinemic Hypoglycemia in Infants and Children. J Pediatr 2016. 168 212–219. [DOI] [PubMed] [Google Scholar]
- 41.States LJ, Davis JC, Hamel SM, Becker SA & Zhuang H. (18)F-6-Fluoro-l-Dopa PET/CT Imaging of Congenital Hyperinsulinism. J Nucl Med 2021. 62 51S–56S. [DOI] [PubMed] [Google Scholar]
- 42.Koboldt DC. Best practices for variant calling in clinical sequencing. Genome Med 2020. 12 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O’Donnell-Luria A, Ware J, Hill A, Cummings B, Tukiainen T, Birnbaum D, Kosmicki J, Duncan L, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Cooper D, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki M, Levy Moonshine A, Natarajan P, Orozco L, Peloso G, Poplin R, Rivas M, Ruano-Rubio V, Ruderfer D, Shakir K, Stenson P, Stevens C, Thomas B, Tiao G, Tusie-Luna M, Weisburd B, Won H-H, Yu D, Altshuler D, Ardissino D, Boehnke M, Danesh J, Roberto E, Florez J, Gabriel S, Getz G, Hultman C, Kathiresan S, Laakso M, McCarroll S, McCarthy M, McGovern D, McPherson R, Neale B, Palotie A, Purcell S, Saleheen D, Scharf J, Sklar P, Patrick S, Tuomilehto J, Watkins H, Wilson J, Daly M & MacArthur D. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G & Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012. 40 W452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS & Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods 2010. 7 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lord K, Radcliffe J, Gallagher PR, Adzick NS, Stanley CA & De Leon DD. High Risk of Diabetes and Neurobehavioral Deficits in Individuals With Surgically Treated Hyperinsulinism. J Clin Endocrinol Metab 2015. 100 4133–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan FF, Lin YW, MacMullen C, Ganguly A, Stanley CA & Shyng SL. Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+ channels: identification and rescue. Diabetes 2007. 56 2339–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper PE, Sala-Rabanal M, Lee SJ & Nichols CG. Differential mechanisms of Cantu syndrome-associated gain of function mutations in the ABCC9 (SUR2) subunit of the KATP channel. J Gen Physiol 2015. 146 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, Ganguly A, Shyng SL & Stanley CA. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest 2008. 118 2877–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang Y, Kesavan P, Wang LQ, Niswender K, Tanizawa Y, Permutt MA, Magnuson MA & Matschinsky FM. Variable effects of maturity-onset-diabetes-of-youth (MODY)-associated glucokinase mutations on substrate interactions and stability of the enzyme. Biochem J 1995. 309 ( Pt 1) 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Christesen HB, Jacobsen BB, Odili S, Buettger C, Cuesta-Munoz A, Hansen T, Brusgaard K, Massa O, Magnuson MA, Shiota C, Matschinsky FM & Barbetti F. The second activating glucokinase mutation (A456V): implications for glucose homeostasis and diabetes therapy. Diabetes 2002. 51 1240–1246. [DOI] [PubMed] [Google Scholar]
- 52.Gloyn AL, Noordam K, Willemsen MA, Ellard S, Lam WW, Campbell IW, Midgley P, Shiota C, Buettger C, Magnuson MA, Matschinsky FM & Hattersley AT. Insights into the biochemical and genetic basis of glucokinase activation from naturally occurring hypoglycemia mutations. Diabetes 2003. 52 2433–2440. [DOI] [PubMed] [Google Scholar]
- 53.Zelent B, Odili S, Buettger C, Zelent DK, Chen P, Fenner D, Bass J, Stanley C, Laberge M, Vanderkooi JM, Sarabu R, Grimsby J & Matschinsky FM. Mutational analysis of allosteric activation and inhibition of glucokinase. Biochem J 2011. 440 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shyng SL, Bushman JD, Pratt EB & Zhou Q. Molecular Defects of ATP-Sensitive Potassium Channels in Congenital Hyperinsulinism. In Monogenic Hyperinsulinemic Hypoglycemia Disorders. Front Diabetes., pp 30–42. Eds Stanley CA & De Leon DD. Basel: Karger, 2012. [Google Scholar]
- 55.Crane A & Aguilar-Bryan L. Assembly, maturation, and turnover of K(ATP) channel subunits. J Biol Chem 2004. 279 9080–9090. [DOI] [PubMed] [Google Scholar]
- 56.Sayed S, Matschinsky FM & Stanley CA. Hyperinsulinism Due to Activating Mutations of Glucokinase. In Monogenic Hyperinsulinemic Hypoglycemia Disorders, pp 146–157. Eds Stanley CA & DeLeon DD. Basel: Karger, 2012. [Google Scholar]
- 57.Pal P & Miller BG. Activating mutations in the human glucokinase gene revealed by genetic selection. Biochemistry 2009. 48 814–816. [DOI] [PubMed] [Google Scholar]
- 58.Houghton JA, Banerjee I, Shaikh G, Jabbar S, Laver TW, Cheesman E, Chinnoy A, Yau D, Salomon-Estebanez M, Dunne MJ & Flanagan SE. Unravelling the genetic causes of mosaic islet morphology in congenital hyperinsulinism. J Pathol Clin Res 2020. 6 12–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wakeling MN, Owens NDL, Hopkinson JR, Johnson MB, Houghton JAL, Dastamani A, Flaxman CS, Wyatt RC, Hewat TI, Hopkins JJ, Laver TW, Van Heugten R, Weedon MN, De Franco E, Patel KA, Ellard S, Morgan NG, Cheesman E, Banerjee I, Hattersley AT, Dunne MJ, Consortium ICH, Richardson SJ & Flanagan S. A novel disease mechanism leading to the expression of a disallowed gene in the pancreatic beta-cell identified by non-codign, regulatory mutations controlling HK1. medRxiv 2022. doi: 10.1101/2021.12.03.21267240. [DOI] [Google Scholar]
- 60.Kalish JM, Boodhansingh KE, Bhatti TR, Ganguly A, Conlin LK, Becker SA, Givler S, Mighion L, Palladino AA, Adzick NS, De Leon DD, Stanley CA & Deardorff MA. Congenital hyperinsulinism in children with paternal 11p uniparental isodisomy and Beckwith-Wiedemann syndrome. J Med Genet 2016. 53 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Some or all datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request.