

Abstract
As the third most common vascular disease, venous thromboembolism is associated with significant mortality and morbidity. Pathogenesis underlying venous thrombosis is still not fully understood. Accumulating data suggest fibrin network structure and factor XIII-mediated crosslinking are major determinants of venous thrombus mass, composition, and stability. Understanding the cellular and molecular mechanisms mediating fibrin(ogen) and factor XIII production and function and their ability to influence venous thrombogenesis and resolution may inspire new anticoagulant strategies that target these proteins to reduce or prevent venous thrombosis in certain at-risk patients. This article summarizes fibrinogen and factor XIII biology and current knowledge of their function during venous thromboembolism.
Keywords: Fibrinogen, factor XIII, venous thrombosis, red blood cell, hemostasis
Graphical Abstract
INTRODUCTION
Venous thromboembolism (VTE), including deep vein thrombosis (DVT) and/or pulmonary embolism (PE), affects 1–2 individuals per 1000 each year globally, with relatively higher incidence in North America and Europe than in Asia.1,2 VTE is the third most common vascular disease after acute myocardial infarction and stroke, and is associated with high mortality and morbidity.3,4 Up to half of patients with a DVT develop post-thrombotic syndrome and up to 4% patients with a PE develop chronic thromboembolic pulmonary hypertension, which reduce quality of life and result in a substantial economic health-care burden.5–8
Several classes of anticoagulants are currently used to prevent VTE or reduce thrombus extension, including indirect thrombin/factor [F] Xa inhibitors, vitamin K antagonist, and direct thrombin/FXa inhibitors. Heparin binds to antithrombin and accelerates the antithrombin-dependent inactivation of several coagulation proteases (e.g., thrombin, FXa, and FIXa).9 The oral vitamin K antagonist warfarin, first approved in the 1950s, is used for treatment or secondary prevention of VTE and stroke in patients with atrial fibrillation.10 Warfarin blocks the vitamin K epoxide reductase and therefore, formation of vitamin K1 and vitamin KH2, which are essential for γ-carboxylation of vitamin K-dependent proteins including FVII, FIX, FX, and prothrombin, and anticoagulant proteins C and S. Several small molecule inhibitors arrived on the market in the early 21st century; these direct oral anticoagulants inhibit FXa or thrombin and have been increasingly used for prevention or treatment of VTE and stroke because of their superior benefit-to-risk ratio compared to heparin or warfarin.11 Clinical observations of individuals with FXII or FXI deficiency and experience with animal models of these deficiencies have sparked interest in FXII or FXI(a) inhibition as potentially safer anticoagulation strategies, and several FXI(a) inhibitors are in clinical development.12–16 By reducing the production or activity of procoagulant proteins, all of these anticoagulants can reduce thrombus extension and venous thrombosis recurrence. However, these drugs are also associated with bleeding risk, likely because they each reduce thrombin-mediated fibrin formation. This limitation has fueled a continued search for new effective drugs with improved safety profiles.
Factor XIII (FXIII) functions in the final step of the coagulation cascade, where its activated form FXIIIa catalyzes the formation of crosslinks within fibrin fibers to stabilize the clot. The use of new mouse models and development of novel technologies, including intravital microscopy to visualize blood cells, fibrin, and FXIII during thrombus formation in vivo, have revealed newly appreciated roles of fibrinogen and fibrin (collectively “fibrin[ogen]”) and FXIIIa-mediated crosslinking in venous thrombus structure, stability, composition, and mass. These findings suggest fibrin(ogen) and FXIII(a) might be effective targets for reducing venous thrombosis in certain situations.
FIBRIN(OGEN) AND FXIII IN HEMOSTASIS
Fibrin(ogen)
Fibrinogen structure and function.
Fibrinogen is one of the most abundant plasma proteins (2–4 mg/mL, 6–12 μM) and the most abundant circulating coagulation protein. Fibrinogen is expressed constitutively, but its expression can be upregulated 2–3-fold above baseline in response to inflammation.17 Fibrinogen circulates as a large (340 kDa) hexameric glycoprotein consisting of 2 each of 3 polypeptides: 2 Aα-, 2 Bβ-, and 2 γ-chains (AαBβγ)2.18 Alternative splicing within the fibrinogen γ-chain leads to a subset of molecules containing one γ’-chain (~8–15% of total circulating fibrinogen). The fibrinogen chains are synthesized and assembled in hepatocytes and the fully-formed fibrinogen hexamer is secreted into the blood (Figure 1A).19 Following vascular injury, activation of the coagulation cascade leads to production of thrombin. Thrombin proteolytically cleaves fibrinogen, which releases N-terminal fibrinopeptides from the Aα- and Bβ-chains in the central E region to generate fibrin monomers. The newly exposed “knobs” on the α- and β-chains can then insert into “pockets” in the C-terminal globular γC and βC regions of the D domain of another fibrin molecule, enabling formation of fibrin oligomers and protofibrils. Through subsequent lateral aggregation and branching events, the half-staggered, double-stranded protofibrils assemble into fiber polymers, and ultimately produce an insoluble 3-dimensional fibrin network.20 Several molecules and environmental conditions influence clot structure. In particular, thrombin has a profound effect on fibrin structure; low thrombin concentrations generate thick fibrin fibers in coarse and permeable networks that are susceptible to fibrinolysis, whereas high thrombin concentration produce thin fibrin fibers in densely-packed networks that are less permeable and resistant to fibrinolysis.21
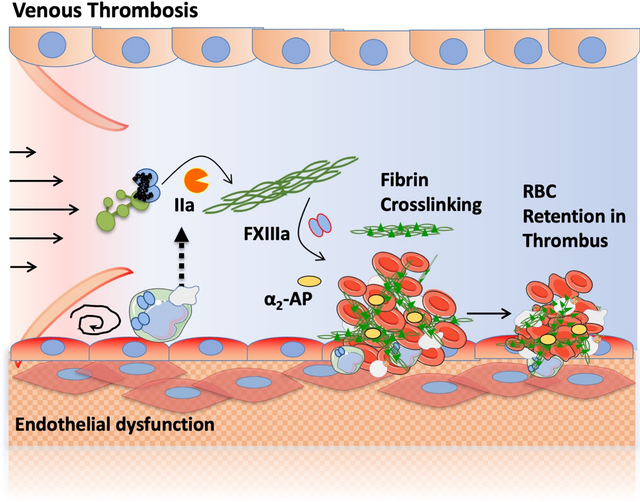
Figure 1. Fibrinogen and FXIII in hemostasis and venous thrombosis.
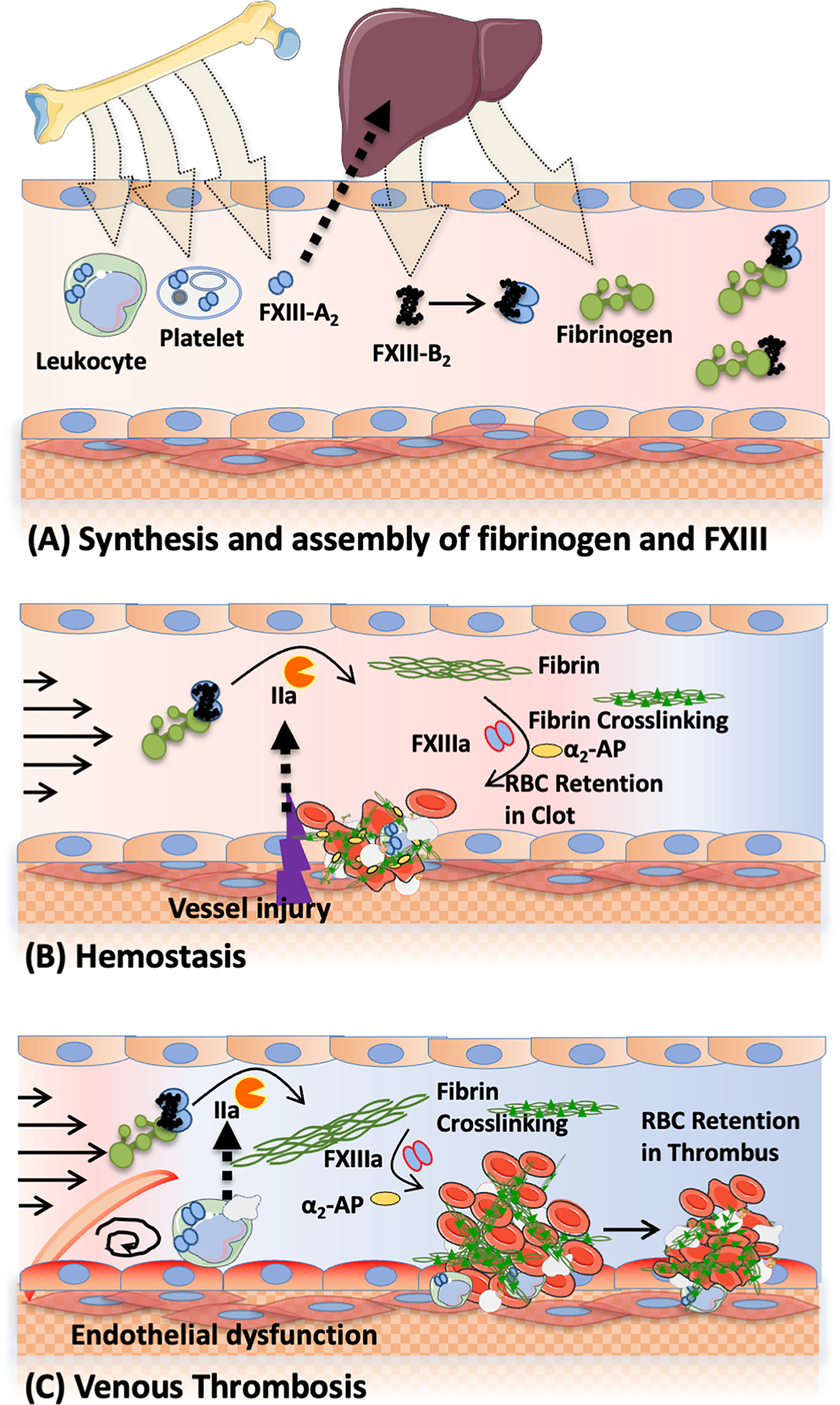
(A) FXIII-A- and B-subunits are synthesized in bone marrow and liver, respectively and assembled in plasma. Fibrinogen is also synthesized in the liver. FXIII-A2B2 circulates in plasma bound to fibrinogen. FXIII-A2 also circulates in platelets and leukocytes. (B-C) Vessel injury (hemostasis, B) or endothelial dysfunction associated with blood stasis (venous thrombosis, C) triggers the activation of coagulation and results in the production of thrombin which cleaves fibrinogen into fibrin and activates FXIII to FXIIIa. FXIIIa catalyzes crosslinks between fibrin molecules and between fibrin and antifibrinolytic proteins (i.e., α2-antiplasmin [α2-AP]). Crosslinking provides mechanical and biochemical stability to the clot and promotes retention of red blood cells within contracted clots. Controlled clot formation seals the injury site during hemostasis and facilitates wound healing, whereas formation of large intravascular thrombi occludes the vessel and leads to venous thrombosis.
Fibrin(ogen) interaction with plasma proteins and cells.
Fibrin(ogen) provides binding sites for plasma proteins involved in clot formation (thrombin), stabilization (FXIII), and lysis (tissue-type plasminogen activator [tPA], plasmin[ogen], α2-antiplasmin, plasminogen activator inhibitor-2), and other proteins. Fibrin(ogen) also interacts with receptors on multiple cell types: αMβ2 and αXβ2 on monocytes;22–24 αIIbβ3 and glycoprotein VI (GPVI) on platelets;25,26 αvβ3, α5β1, intercellular adhesion molecule-1, and VE-cadherin on endothelial cells;27–30 and αvβ3 on fibroblasts31. These interactions mediate the diverse roles of fibrin(ogen) in hemostasis, immunity, inflammation, and infection.32 For example, fibrin(ogen) binding to monocytes enhances monocyte activation.23 Interaction of extravascular fibrin(ogen) with the toll-like receptor-4 on macrophages stimulates chemokine secretion and promotes an immune response.33 Fibrin(ogen) binding to the activated platelet integrin receptor αIIbβ3 serves as a bridge that mediates platelet aggregation, clot contraction, and thrombus consolidation. Fibrin is also a ligand for platelet collagen receptor GPVI and this interaction is associated with phosphatidylserine exposure.26 Fibrinogen can also increase the permeability of cultured endothelial cells and may contribute to microvascular leakage in cardiovascular disease.34 Assembly of fibrin(ogen) on leukocytes and endothelial cells stabilize leukocyte attachment and migration to endothelium.35 Binding and incorporation of plasma proteins and blood cells into the nascent fibrin network can alter protofibril formation and polymerization, network density, pore size/clot permeability, elasticity, and the rate of fibrinolysis, with direct consequences for the clot’s mechanical and fibrinolytic properties.20,36
Quantitative and qualitative fibrin(ogen) defects.
Fibrinogen disorders can be congenital or acquired and are classified as quantitative deficiencies marked by no or low plasma fibrinogen (afibrinogenemia or hypofibrinogenemia, respectively), or qualitative abnormalities associated with abnormal function of fibrinogen molecules present at either normal or low levels (dysfibrinogenemia or hypodysfibrinogenemia, respectively). Congenital fibrinogen disorders are typically caused by mutations within the fibrinogen structural genes FGA, FGB, and FGG. Afibrinogenemia is relatively rare and occurs in ~1 in 1 million individuals, whereas hypofibrinogenemia and dysfibrinogenemia are more common. Acquired fibrinogen disorders are usually caused by clinical situations that alter synthesis (e.g., liver disease), increase consumption (e.g., cancer, sepsis with disseminated intravascular coagulation), alter plasma concentration (e.g., hemodilution during transfusion), or promote autoantibody formation (e.g., myeloma, autoimmune disease, or drug-induced). Combinations of these mechanisms (e.g., blood loss, consumption, hemodilution, and hyperfibrinolysis) frequently occur in trauma and contribute to poor outcomes.37–39 The phenotype of individuals with fibrinogen deficiency is highly variable and can be asymptomatic or can be associated with increased bleeding and/or thrombosis. A more detailed description of fibrinogen deficiencies can be found elsewhere.40–42
FXIII
Structure and activation of plasma FXIII.
FXIII, known as “fibrin stabilizing factor,” is a member of a family of nine transglutaminase proteins. FXIII is the only member of this family that is present in plasma, as well as cells. Plasma FXIII consists of a dimer of catalytic A-subunits (FXIII-A2) and a dimer of carrier/inhibitory B-subunits (FXIII-B2) that circulate as a heterotetrameric complex (FXIII-A2B2, 320 kDa, 14–28 μg/mL). Each A-subunit is an ~83 kDa molecule with 731 amino acids arranged in four structural domains: an activation peptide (AP-FXIII [residues 1–37]), a β-sandwich domain (residues 38–184), a catalytic core domain (residues 185–515), and two β-barrel domains (residues 516–628 and 629–731). Each B-subunit is an ~80 kDa molecule consisting of 641 amino acids assembled in ten sushi domains, each held together by two internal disulfide bonds.43 The FXIII A- and B-subunits are synthesized in different tissues; the A-subunits are produced in hematopoietic cells thought to be resident tissue macrophages in the aorta,44 whereas the B-subunits are synthesized in hepatocytes (Figure 1A).45 Assembly of the FXIII-A2B2 heterotetramer occurs in the plasma (Figure 1A). The equilibrium dissociation constant (KD) for the interaction between FXIII-A2 and FXIII-B2 subunits is ~10−10 M, such that 99% of FXIII-A2 circulates in the FXIII-A2B2 complex.46 The B-subunits stabilize the A-subunits by preventing spontaneous activation, and are essential for maintaining plasma FXIII levels.47,48 Although early studies suggested FXIII-A2B2 circulates bound to the alternatively-spliced γ’ sequence in fibrinogen, more recent studies showed the FXIII-B subunits mediate binding of FXIII-A2B2 to fibrinogen residues γ390–396 in both humans and mice.49 In addition to residues γ390–396, fibrinogen residues α371–425, and particularly αGlu396 within the αC domain, have also been implicated in FXIII binding and activation.50,51 FXIII-B2 circulates in a ~2-fold molar excess relative to FXIII-A2. Essentially all FXIII-A2B2 and FXIII-B2 circulate bound to fibrinogen residues γ390–396.49 A small pool of FXIII-A2B2 is found in platelet α-granules, likely endocytosed with circulating fibrinogen.52
In concert with fibrin formation, thrombin proteolytically activates FXIII by thrombin-mediated cleavage of the Arg37-Gly38 peptide bond and dissociation of the N-terminal activation peptides. This process is accelerated by the presence of polymerized fibrin.53 Activation peptide release is followed by calcium-mediated dissociation of B-subunits from the A-subunits and exposure of the active site cysteine.
Structure, activation, and activity of cellular FXIII.
Cellular FXIII consists of only FXIII A-subunits (cFXIIIA, FXIII-A2). FXIII-A2 is present in cells of bone marrow origin and mesenchymal lineage, including osteoblasts and chondrocytes,54,55 monocytes/macrophages,56–58 and megakaryocytes and platelets59–61. Cellular FXIII-A is activated nonproteolytically by increased intracellular Ca2+, can be exposed on the membrane surface62,63, and is involved in multiple cellular functions.55 Almost half of circulating FXIII-A is present in platelets64, and FXIII-A is one of the most prevalent platelet proteins (~83,000 copies per platelet)65. Unlike most coagulation proteins in platelets that are located in the α-granules, platelet FXIII-A is localized in the cytoplasm (Figure 2).61,66 FXIII-A exposure on platelets requires stimulation by strong dual agonists (e.g., convulxin plus thrombin or thrombin receptor activation peptide), but mechanisms regulating the exposure and release of FXIII-A from platelets, as well as other cells, are unclear. Two populations of FXIII-exposing platelets are formed after strong stimulation: ballooned procoagulant (phosphatidylserine-exposing) platelets with FXIII-A on a protruding “cap,” and spread platelets with FXIII-A in a diffuse distribution (Figure 2).67,68 FXIII-A is also exposed on extracellular vesicles released from GPVI and protease-activated receptor (PAR)-activated platelets.69 Platelet FXIII-A can be activated by calpain (Ca2+-dependent cysteine proteinase) in purified systems.70 However, during platelet activation, elevated intracellular Ca2+ induces a conformational change in FXIII-A2 that produces enzymatically active FXIII without proteolytic removal of the activation peptide (FXIII-A°).63,71 After exposure and/or release, FXIII-A° can be proteolytically cleaved by thrombin to produce activated FXIII-A* (FXIIIa). Proteolytically activated FXIII-A* has higher conformational flexibility and increased affinity toward glutamine substrates, suggesting activation peptide removal might make FXIII-A* more accessible to substrates.72,73 In activated platelets, nonproteolytically activated FXIII-A° can crosslink cytoskeletal proteins including myosin, actin, filamin, and vinculin. These events occur in later stages of platelet activation and may contribute to cytoskeletal remodeling and certain phases of platelet spreading (Figure 2).74–76 Although some studies suggested platelet FXIII-A is required for platelet contraction,77,78 others have seen little or no difference in the ability of clots to contract in the absence of FXIII activity.79–82
Figure 2. Platelet FXIII-A activation and externalization.
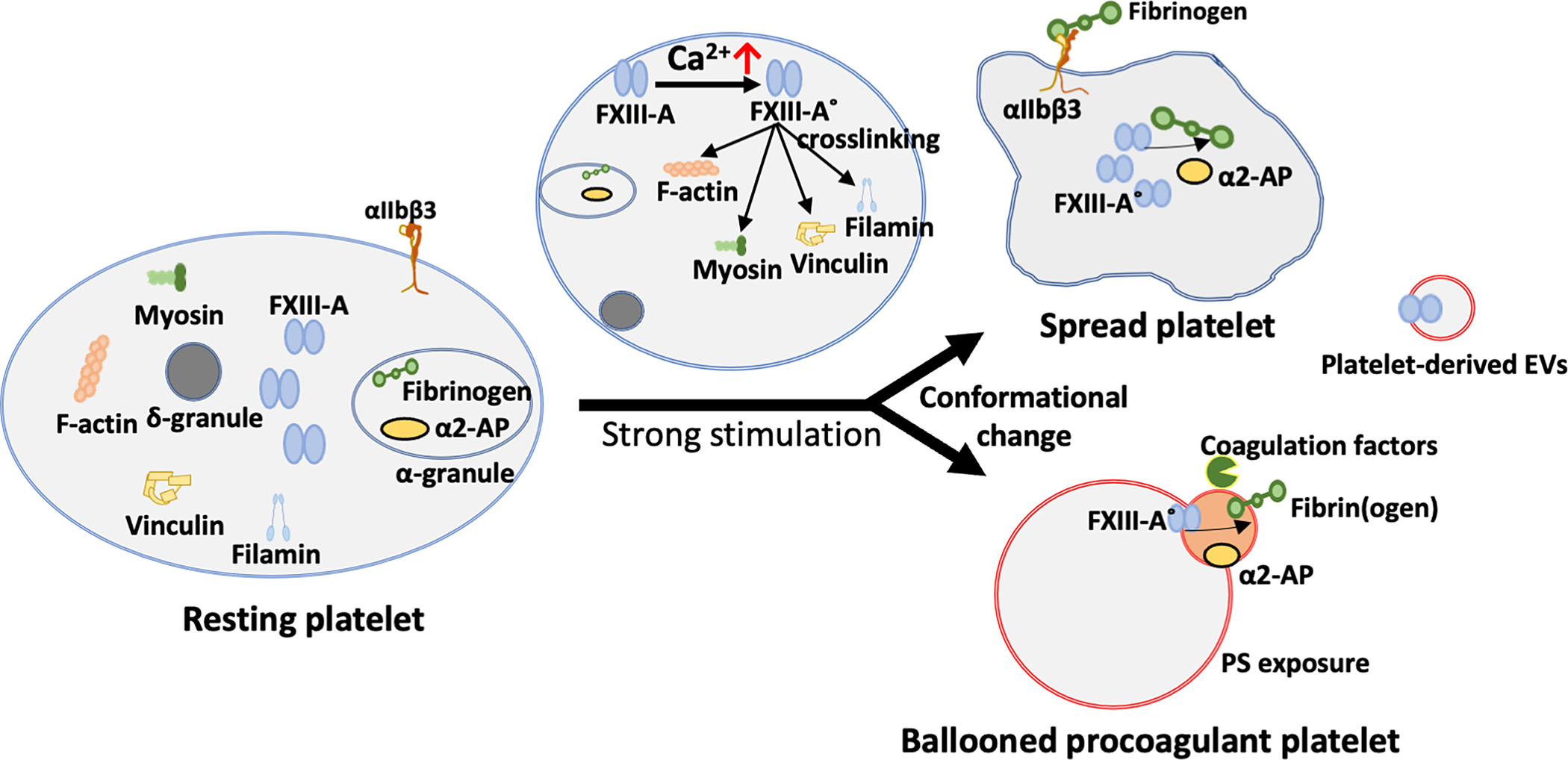
In unstimulated platelets, FXIII-A exists in the cytoplasm in a diffuse distribution. After platelet activation, increased intracellular Ca2+ nonproteolytically activates FXIII-A to FXIII-A° which crosslinks cytoskeletal proteins and contributes to cytoskeletal rearrangement and platelet conformational change. After stimulation by strong dual agonists (convulxin + thrombin), two populations of FXIII-A-exposing platelets are formed: spread platelets with FXIII-A in a diffuse distribution, and ballooned procoagulant (phosphatidylserine-exposing) platelets with FXIII-A on a protruding “cap”. FXIII-A is also exposed on extracellular vesicles (EVs).
FXIII(a) function in hemostasis.
Plasma FXIIIa catalyzes the formation of covalent ε-(γ-glutamyl)-lysine isopeptide bonds between glutamine and lysine residues. FXIIIa is a relatively promiscuous enzyme, and proteomic analysis has identified almost 150 FXIIIa substrates in plasma, including 48 that may be incorporated into the insoluble fibrin clot during coagulation.83 Plasma and cellular substrates for FXIIIa and their related crosslinking sites are reviewed elsewhere.84,85 Nonetheless, the primary physiological function of plasma FXIII(a) is well-established: 1) crosslinking fibrin γ- and α-chains into γ-chain dimers, α-chain polymers, and γ-α species to increase clot mechanical stability; and 2) crosslinking antifibrinolytic proteins (e.g., α2-antiplasmin) to fibrin(ogen) to protect clots against biochemical degradation by the fibrinolytic system (Figure 1B). Fibrin γ-chain dimer formation is a fast process that results from reciprocal intermolecular bond formation between the γ406 lysine of one γ-chain and a γ398/399 glutamine residue of another γ-chain. Crosslinking of α-chains is slower than γ-chain crosslinking and involves multiple glutamine and lysine residues.86 Crosslinking of α2-antiplasmin to fibrin (primarily at α-chain residue Lys303) lags slightly behind fibrin γ-dimer formation but precedes fibrin α-chain polymer formation.55,87,88 Platelet FXIII-A may also have antifibrinolytic activity by crosslinking α2-antiplasmin to fibrin(ogen), but this effect is only significant when plasma FXIII is below 20%.68 Similarly, FXIII(a) exposed on interleukin (IL)-4- and IL-10-stimulated monocytes may also stabilize thrombi against fibrinolytic degradation in settings where plasma FXIII concentrations are low.89
FXIII deficiency.
FXIII deficiency is a rare disorder, affecting ~1 in 2–3 million people. Congenital deficiency and FXIII activity below 3% is often identified by delayed umbilical cord bleeding and is associated with severe, life-long bleeding tendency, including intracranial bleeding in ~30% of patients.90 FXIII deficiency is also associated with abnormal wound healing and spontaneous miscarriage.90–93 Congenital FXIII deficiency can arise from mutations in genes encoding either the FXIII-A subunit (F13a1, type 2) or the FXIII-B subunit (F13b, type 1), although defects in FXIII-A account for 95% of FXIII-related bleeding disorders.91,93 Bleeding in FXIII-B deficiency is generally milder than that seen in FXIII-A-deficient patients; this has been attributed to low residual levels of plasma FXIII-A2 in FXIII-B-deficient individuals, or to antifibrinolytic activity of platelet FXIII-A which may compensate for the loss of plasma FXIII-A2B2.90,94 Acquired FXIII is more common than congenital deficiency, and can be caused by autoimmune disease, consumption (e.g., surgery, infection, inflammatory bowel disease, thrombosis), reduced synthesis (e.g., liver disease, leukemia, medication-related) and/or hemodilution. Since conventional coagulation tests available in most clinical settings are not sensitive to FXIII, FXIII deficiency is difficult to identify and may be underdiagnosed.95–97 Congenital FXIII deficiency is treated with plasma-derived FXIII-A2B2 (in patients with either A- or B-subunit deficiency) or recombinant FXIII-A2 (in patients with genetically-confirmed A-subunit deficiency). Patients with acquired FXIII deficiency associated with autoantibody development are treated with immunosuppressive agents combined with cryoprecipitate and/or FXIII concentrate.98–100
FIBRIN(OGEN) AND FXIII IN VENOUS THROMBOSIS
Pathophysiologic mechanisms in VTE.
VTE pathophysiology is usually described as the intersection of three major abnormalities (venous stasis, vascular dysfunction/injury, and blood hypercoagulability) known as Virchow’s Triad. In this conceptual model, reduced (stasis) or turbulent (nonlaminar) flow of blood around the venous valve pocket creates a hypoxic environment that activates endothelial cells and leads to abnormal expression of adhesion molecules that bind and retain leukocytes and platelets at the endothelial surface.101 Activated leukocytes, and potentially also the dysfunctional endothelial cells themselves, express tissue factor, triggering the coagulation cascade. Leukocytes, red blood cells (RBCs) and platelets that accumulate in the valve pockets promote thrombin generation and ultimately, the formation of a thrombus rich in RBCs and fibrin (Figure 1C). Venous thrombi can occlude venous flow and/or dislodge and migrate through the heart to the pulmonary vasculature.
Epidemiologic data associating changes in fibrin(ogen) and FXIII with VTE.
Previous reviews have summarized epidemiologic studies investigating relationships between fibrin(ogen) and FXIII in venous thrombosis102–104 and are only summarized here. Briefly, elevated fibrinogen is associated with increased risk of venous thrombosis. Risk persists even after adjusting for potential confounded effects of ongoing inflammation,102 but is complicated by the relative presence of the alternatively-spliced fibrinogen γ’-chain, which offers protection against VTE.104 Similar discord in the literature investigating the role of FXIII in thrombosis has arisen through complexities in interpreting FXIII activity assays, complex relationships between FXIII and fibrinogen levels, polymorphisms that alter FXIII function, and potential sex-specific effects.103 However, together the findings suggest FXIII antigen, activity, and/or genotype influence thrombosis risk in certain populations and clinical situations. For example, meta-analyses show the common FXIII Val34Leu polymorphism (rs5985) protects against VTE.105 Presence of this polymorphism leads to faster activation by thrombin, and therefore, faster fibrin crosslinking.106 Interestingly, the functional impact of the FXIII Val34Leu polymorphism is manifested through a “gene-environment interaction” in which homozygous presence of the Leu34 allele promotes the formation of clots with thicker fibers and more permeable clots when fibrinogen concentrations are high, but formation of thinner fibers and denser networks when fibrinogen concentrations are low.107 Collectively, these studies provide strong support for the premise that both fibrin(ogen) and FXIII are major contributors to VTE.
Fibrin and FXIIIa as determinants of venous thrombus formation and composition.
Conventionally, venous clots are referred as “red” RBC- and fibrin-rich thrombi, and their fibrin content has been used as an imaging target.108 Platelets also contribute to both thrombus initiation109,110 and composition. Scanning and transmission electron microscopy of thrombi retrieved from patients as well as contracted whole blood clots formed in vitro, reveal the presence of closely packed, distorted RBCs (termed “polyhedrocytes”) in clot core.111 Platelet-mediated clot contraction generates the force required to compress these resident RBCs into polyhedrocytes. RBCs in circulation or in the thrombus may contribute mechanistically to VTE by interacting with other cells, supporting thrombin generation, altering fibrin structure, and/or slowing the diffusion of lytic enzyme into the clot and enhancing clot resistance to fibrinolysis (reviewed in 112).
In experimental models of venous thrombosis, elevated fibrinogen shortens the time to vessel occlusion, increases fibrin deposition within thrombi, and increases fibrin stability.113 Clots with increased fibrin network density, including those formed in the presence of high tissue factor concentrations, retain higher numbers of RBCs.114 Notably however, FXIII(a) crosslinking of fibrin promotes RBC retention in clots independent of fibrin network density.81,114,115 Following inferior vena cava ligation, FXIII-deficient mice (F13a1−/−) produce thrombi with decreased RBC content and consequently, reduced mass than wild-type mice.81,115 Similarly, human whole blood clots retain fewer RBCs in the absence of FXIII.115 Experiments with FXIIIa inhibitors and recombinant fibrinogen variants associated this effect with the production of α-chain-rich high molecular weight crosslinks.114 Subsequent studies suggested these crosslinks are produced primarily by plasma, but not platelet, FXIII.81 The ability of clots to retain RBCs depends not only on the presence of FXIII, but also on the timing of its activation. Studies using a FXIIIa-sensitive near-infrared fluorescence imaging agent (A15 peptide) showed significant crosslinking in acute thrombi, but less incorporation in aged thrombi116, consistent with the early activation of FXIII that is synchronized with fibrin formation. Delayed FXIII activation associated with reduced FXIII (F13a1+/−) or reduced FXIII-A2B2 binding to fibrinogen (as in mice bearing mutated fibrinogen, Fibγ390−396A) decreases RBC retention in venous thrombi in mice.49,81,115 Interestingly, accelerated FXIII activation also decreases RBC retention in clots, but in a gene/environment mechanism; compared to the FXIII 34Val allele, presence of the FXIII 34Leu allele reduces the impact of elevated fibrin(ogen) on whole blood clot mass in vitro, and thus may protect from venous thrombosis in vivo.117
Fibrin and FXIIIa as determinants of PE and venous thrombus resolution.
PE is the most serious complication of DVT and happens when part or all the thrombus detaches from the venous vessel wall, travels to a pulmonary artery, and prevents oxygen exchange. Failure of timely dissolution of PE can result in chronic thromboembolic pulmonary hypertension, right heart failure, and cardiogenic shock.118 Clinical observations119–123 and experimental studies with mice124–126 support roles for both fibrin(ogen) and FXIII(a) in maintaining venous thrombus stability and preventing embolization.
Plasmas from patients with a history of VTE produce clots with abnormal characteristics, including reduced permeability and delayed clot lysis.119 Even in patients with DVT who have discontinued anticoagulation, the formation of plasma clots with reduced permeability and prolonged clot lysis in vitro are associated with increased risk of recurrent DVT.127 These same characteristics are also associated with increased risk of developing chronic thromboembolic pulmonary hypertension, post-thrombotic syndrome, and persistent venous obstruction.120,121,128–131 The fibrinogen β chain (FGB [rs1800790]) and FXIII Val34Leu polymorphisms have been implicated as determinants of altered clot properties in acute PE.123
Using intravital video microscopy and lung histology of a FeCl3-induced model of acute thrombosis, Gross and co-workers found reduced thrombus stability and increased emboli in mice treated with the direct thrombin inhibitor dabigatran and in FXIII-deficient mice, leading to the hypothesis that by reducing thrombin production, anticoagulant treatment reduces activation of FXIII and the thrombin-activatable fibrinolysis inhibitor and decreases thrombus stability.132 They subsequently showed that supplementation with FXIII stabilizes venous thrombi and decreases embolization without altering thrombus size.124 We confirmed the role of FXIII in venous thrombus stabilization using a new mouse model of VTE. In this model, DVT are first induced by ligating the inferior vena cava (generating blood stasis) to slowly produce large RBC-enriched thrombi similar to human DVT, and then these thrombi are allowed to embolize by releasing the ligature. Using this model, we observed that complete FXIII deficiency increases PE incidence, but partial deficiency (i.e., F13a1+/−) does not,125 likely due to preservation of fibrin crosslinking in mice with reduced FXIII.81 Mice with mutations in the fibrin γ-chain crosslinking sites (FGG3X) also show increased PE following FeCl3-induced femoral vein thrombosis.126 Collectively, these studies suggest the thrombus-stabilizing effect of FXIII is manifested at least in part through mechanical stability provided by fibrin γ-γ crosslink formation. FXIIIa may also protect against PE through its ability to crosslink α2-antiplasmin to the clot and increase thrombus biochemical stability.
Interestingly, the ability of FXIII to stabilize thrombi may also potentiate PE sequelae by hindering resolution of thrombi that embolize. Compared to wild-type mice, mice with α2-antiplasmin deficiency (SERPINF2 −/−) have fewer thrombi in the lungs and decreased mortality after photochemical-induced PE in the jugular vein.133 In a model in which preformed thrombi are deployed in the jugular vein, α2-antiplasmin inhibition facilitates thrombus dissolution similar to effects of recombinant tPA, without increasing bleeding.134
FIBRIN(OGEN) AND FXIII AS POTENTIAL THERAPEUTIC TARGETS
All existing anticoagulants reduce thrombin generation or activity, and therefore inhibit thrombin-mediated platelet activation and fibrin formation. Consequently, each of these anticoagulants are associated with bleeding.101 Given the prominent roles of fibrin(ogen) and FXIII in thrombus formation, composition, size, permeability, and stability, it is interesting to consider the potential of fibrin(ogen) and FXIII as therapeutic targets. These approaches, which target coagulation at a step downstream of current therapeutic targets, may allow for normal thrombin generation and therefore, preservation of hemostasis in high-risk settings. For example, in patients with recurrent VTE in spite of apparently therapeutic levels of anticoagulants, fibrin(ogen) and/or FXIII(a)-targeting strategies may be an effective adjunct therapy for preventing recurrent VTE. Moreover, by reducing thrombus stability and facilitating clot dissolution, strategies that target fibrin(ogen) and/or FXIII may reduce long-term sequelae of VTE, including post-thrombotic syndrome and chronic thromboembolic pulmonary hypertension.
Fibrin(ogen) reduction as a therapeutic strategy.
In addition to thrombosis, fibrin(ogen) also contributes to inflammatory and immune diseases and malignancy. Accordingly, a large body of work with genetically engineered mice expressing reduced or functionally altered fibrin(ogen) has shown benefit of fibrinogen reduction in diverse clinical settings. However, means to reduce fibrin(ogen) therapeutically are not established for widespread clinical use. Ancrod, a defibrinogenating agent purified from venom of the Malaysian pit viper, has been tested as a therapy for treating acute ischemic stroke.135 However, clinical trials showed mixed results, including an increased risk of bleeding136–138, and this approach invokes concerns about potentially biologically active fibrin(ogen) degradation products inducing pathological effects in the patients. Single-stranded antisense oligonucleotides and small interfering RNA (siRNA) targeting specific fibrinogen chains that reduce circulating fibrinogen without generating fibrin(ogen) degradation products have shown benefit in mouse models of cancer, diet-induced obesity, endotoxemia, peritonitis, and tumor metastasis, without compromising hemostasis.139,140 Thus, these strategies may also be useful for reducing venous thrombosis in settings of heightened venous thrombosis risk, including constitutively elevated fibrinogen or hyperfibrinogenemia secondary to an inflammatory process.
FXIII inhibition as a therapeutic strategy.
FXIII-directed antagonists may have a relatively wide therapeutic range. FXIII reduction decreases thrombus mass in a dose-dependent manner125, whereas patients with at least 30 U/dL FXIIIa activity are usually asymptomatic and spontaneous bleeding only occurs when FXIIIa activity is below 15 IU/dL.141,142 The finding that plasma FXIII, but not platelet FXIIIA, promotes RBC retention in venous thrombi81 suggests a FXIII(a) inhibitor would only need to reach the plasma compartment to effectively reduce thrombus mass. Indeed, the relative protection of platelet FXIII from plasma inhibitors may provide sufficient transglutaminase activity to crosslink hemostatic clots and reduce bleeding risk with this strategy.
Several potential inhibitors of FXIII(a) or its crosslinked substrates (e.g., α2-antiplasmin)143 have been tested in vitro and in animal models. These include antibodies that inhibit FXIII activation, competitive substrates that reduce crosslinking of fibrin and other plasma proteins, and direct inhibitors that bind to the active site of FXIIIa and inhibit its activity.134,144 Peptide-based inhibitors of FXIII(a), including leech-derived tridegin and synthetic peptidic transglutaminase-inhibiting Michael acceptors, that decrease FXIIIa activity have gained particular interest.145–148 The drug-like FXIIIa inhibitor ZED3197, which has selectivity over other transglutaminases, decreases thrombus weight and facilitates flow restoration in a rabbit venous stasis model without increasing bleeding.146 Given the nature of venous thrombus formation, FXIII-targeting strategy for VTE prevention will require specific inhibitors with longer half-life than current molecules. Leveraging the dependence of circulating plasma FXIII-A on FXIII-B47,48, Strilchuk et al recently achieved sustained depletion of plasma FXIII-A using siRNA against hepatic FXIII-B.48 This approach led to enhanced reperfusion in a mouse model of carotid artery thrombosis, suggesting this method may also reduce thrombus mass in models of venous thrombosis.
CONCLUSIONS
The studies summarized here have identified mechanisms by which fibrin(ogen) and FXIII contribute to venous thrombus size, composition, and stability. Mechanisms include function-driving gene polymorphisms, interactions between fibrin(ogen) and FXIII that modify fibrin network structure and mechanical and/or biochemical stability, and the ability of clots to retain RBCs during platelet-mediated contraction. Abnormalities in fibrin structure and/or crosslinking may promote PE and/or facilitate the resolution of venous thrombi or PE. Although still quite speculative, development of molecules that target and modify fibrin(ogen) and FXIII(a) may alter the course of venous thrombogenesis and/or resolution. Future research is necessary to understand the effect of fibrin(ogen) and FXIII interaction with blood cells and the extracellular matrix on initiation, development, and resolution of VTE.
HIGHLIGHTS.
Fibrin(ogen) interactions with plasma proteins and blood cells mediates the diverse roles of fibrin(ogen) in hemostasis, immunity, inflammation, and infection.
Factor XIII catalyzes the formation of covalent bonds between glutamine and lysine residues in fibrin and other proteins, which protects clots against biochemical degradation and mechanical disruption, and promotes retention of red blood cells in contracted clots.
Both fibrin(ogen) and factor XIII contribute to venous thromboembolism.
Targeting fibrin(ogen) or factor XIII may decrease the incidence and size of venous thrombi and reduce the pathologic consequences of venous thrombosis.
ACKNOWLEDGEMENTS
The authors thank Drs. Kadri Kangro, Stephanie E. Reitsma, Lori A. Holle, and Matthew J. Flick for reading the manuscript.
FUNDING
This work was supported by funding from the National Institutes of Health (R01HL126974 to ASW).
ABBREVIATIONS
- VTE
venous thromboembolism
- DVT
deep vein thrombosis
- PE
pulmonary embolism
- F
factor
- FXIII
factor XIII
- GPVI
glycoprotein VI
- FXIIIa
activated factor XIII
- RBC
red blood cell
- siRNA
small interfering RNA
Footnotes
CONFLICT OF INTEREST DISCLOSURE
Neither of the authors have relevant potential conflict of interest.
REFERENCES
- 1.Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, Hylek EM, Kakkar A, Konstantinides SV, McCumber M, et al. Thrombosis: a major contributor to the global disease burden. J Thromb Haemost. 2014;12:1580–1590. doi: 10.1111/jth.12698 [DOI] [PubMed] [Google Scholar]
- 2.Wendelboe AM, Raskob GE. Global burden of thrombosis. CircRes. 2016;118:1340–1347. doi: 10.1161/CIRCRESAHA.115.306841 [DOI] [PubMed] [Google Scholar]
- 3.Di Nisio M, van Es N, Büller HR. Deep vein thrombosis and pulmonary embolism. Lancet. 2016;388:3060–3073. doi: 10.1016/S0140-6736(16)30514-1 [DOI] [PubMed] [Google Scholar]
- 4.Khan F, Tritschler T, Kahn SR, Rodger MA. Venous thromboembolism. Lancet. 2021;398:64–77. doi: 10.1016/s0140-6736(20)32658-1 [DOI] [PubMed] [Google Scholar]
- 5.Galanaud JP, Monreal M, Kahn SR. Epidemiology of the post-thrombotic syndrome. Thromb Res. 2018;164:100–109. doi: 10.1016/j.thromres.2017.07.026 [DOI] [PubMed] [Google Scholar]
- 6.Hoeper MM, Madani MM, Nakanishi N, Meyer B, Cebotari S, Rubin LJ. Chronic thromboembolic pulmonary hypertension. The Lancet Respiratory medicine. 2014;2:573–582. doi: 10.1016/s2213-2600(14)70089-x [DOI] [PubMed] [Google Scholar]
- 7.Grosse SD, Nelson RE, Nyarko KA, Richardson LC, Raskob GE. The economic burden of incident venous thromboembolism in the United States: A review of estimated attributable healthcare costs. Thromb Res. 2016;137:3–10. doi: 10.1016/j.thromres.2015.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts LN, Patel RK, Donaldson N, Bonner L, Arya R. Post-thrombotic syndrome is an independent determinant of health-related quality of life following both first proximal and distal deep vein thrombosis. Haematologica. 2014;99:e41–e43. doi: 10.3324/haematol.2013.089870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wardrop D, Keeling D. The story of the discovery of heparin and warfarin. Br J Haematol. 2008;141:757–763. doi: 10.1111/j.1365-2141.2008.07119.x [DOI] [PubMed] [Google Scholar]
- 10.Lim GB. Warfarin: from rat poison to clinical use. Nat Rev Cardiol. 2017. doi: 10.1038/nrcardio.2017.172 [DOI] [PubMed] [Google Scholar]
- 11.Chen A, Stecker E, Warden BA. Direct oral anticoagulant use: A practical guide to common clinical challenges. J Am Heart Assoc. 2020;9:e017559. doi: doi: 10.1161/JAHA.120.017559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davoine C, Bouckaert C, Fillet M, Pochet L. Factor XII/XIIa inhibitors: Their discovery, development, and potential indications. Eur J Med Chem. 2020;208:112753. doi: 10.1016/j.ejmech.2020.112753 [DOI] [PubMed] [Google Scholar]
- 13.Weitz JI, Strony J, Ageno W, Gailani D, Hylek EM, Lassen MR, Mahaffey KW, Notani RS, Roberts R, Segers A, et al. Milvexian for the prevention of venous thromboembolism. N Engl J Med. 2021;385:2161–2172. doi: 10.1056/NEJMoa2113194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Büller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, Segers A, Verhamme P, Weitz JI. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015;372:232–240. doi: 10.1056/NEJMoa1405760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lorentz CU, Tucker EI, Verbout NG, Shatzel JJ, Olson SR, Markway BD, Wallisch M, Ralle M, Hinds MT, McCarty OJT, et al. The contact activation inhibitor AB023 in heparin-free hemodialysis: results of a randomized phase 2 clinical trial. Blood. 2021;138:2173–2184. doi: 10.1182/blood.2021011725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeLoughery EP, Olson SR, Puy C, McCarty OJT, Shatzel JJ. The safety and efficacy of novel agents targeting factors XI and XII in early phase human trials. Semin Thromb Hemost. 2019;45:502–508. doi: 10.1055/s-0039-1692439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roshal M Chapter 123 - Thrombin time and fibrinogen determination. In: Shaz BH, Hillyer CD, Roshal M, Abrams CS, eds. Transfusion Medicine and Hemostasis (Second Edition). San Diego: Elsevier; 2013:793–798. [Google Scholar]
- 18.Pieters M, Wolberg AS. Fibrinogen and fibrin: An illustrated review. Res Pract Thromb Haemost. 2019;3:161–172. doi: 10.1002/rth2.12191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tennent GA, Brennan SO, Stangou AJ, O’Grady J, Hawkins PN, Pepys MB. Human plasma fibrinogen is synthesized in the liver. Blood. 2007;109:1971–1974. doi: 10.1182/blood-2006-08-040956 [DOI] [PubMed] [Google Scholar]
- 20.Weisel JW, Litvinov RI. Mechanisms of fibrin polymerization and clinical implications. Blood. 2013;121:1712–1719. doi: 10.1182/blood-2012-09-306639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev. 2007;21:131–142. doi: 10.1016/j.blre.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 22.Altieri DC, Agbanyo FR, Plescia J, Ginsberg MH, Edgington TS, Plow EF. A unique recognition site mediates the interaction of fibrinogen with the leukocyte integrin Mac-1 (CD11b/CD18). J Biol Chem. 1990;265:12119–12122. [PubMed] [Google Scholar]
- 23.Kaneider NC, Mosheimer B, Günther A, Feistritzer C, Wiedermann CJ. Enhancement of fibrinogen-triggered pro-coagulant activation of monocytes in vitro by matrix metalloproteinase-9. Thromb J. 2010;8:2. doi: 10.1186/1477-9560-8-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ugarova TP, Yakubenko VP. Recognition of fibrinogen by leukocyte integrins. Ann N Y Acad Sci. 2001;936:368–385. doi: 10.1111/j.1749-6632.2001.tb03523.x [DOI] [PubMed] [Google Scholar]
- 25.Bennett JS. Platelet-fibrinogen interactions. Ann N Y Acad Sci. 2001;936:340–354. doi: 10.1111/j.1749-6632.2001.tb03521.x [DOI] [PubMed] [Google Scholar]
- 26.Alshehri OM, Hughes CE, Montague S, Watson SK, Frampton J, Bender M, Watson SP. Fibrin activates GPVI in human and mouse platelets. Blood. 2015;126:1601–1608. doi: 10.1182/blood-2015-04-641654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suehiro K, Gailit J, Plow EF. Fibrinogen is a ligand for integrin α5β1 on endothelial cells*. J Biol Chem. 1997;272:5360–5366. doi: 10.1074/jbc.272.8.5360 [DOI] [PubMed] [Google Scholar]
- 28.Languino LR, Duperray A, Joganic KJ, Fornaro M, Thornton GB, Altieri DC. Regulation of leukocyte-endothelium interaction and leukocyte transendothelial migration by intercellular adhesion molecule 1-fibrinogen recognition. Proc Natl Acad Sci U S A. 1995;92:1505–1509. doi: 10.1073/pnas.92.5.1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viela F, Speziale P, Pietrocola G, Dufrêne YF. Mechanostability of the fibrinogen bridge between Staphylococcal surface protein ClfA and endothelial cell integrin αVβ3. Nano Lett. 2019;19:7400–7410. doi: 10.1021/acs.nanolett.9b03080 [DOI] [PubMed] [Google Scholar]
- 30.Yakovlev S, Gao Y, Cao C, Chen L, Strickland DK, Zhang L, Medved L. Interaction of fibrin with VE-cadherin and anti-inflammatory effect of fibrin-derived fragments. J Thromb Haemost. 2011;9:1847–1855. doi: 10.1111/j.1538-7836.2011.04438.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gailit J, Clarke C, Newman D, Tonnesen MG, Mosesson MW, Clark RAF. Human fibroblasts bind directly to fibrinogen at RGD sites through integrin αvβ3. Exp Cell Res. 1997;232:118–126. doi: 10.1006/excr.1997.3512 [DOI] [PubMed] [Google Scholar]
- 32.Luyendyk JP, Schoenecker JG, Flick MJ. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood. 2019;133:511–520. doi: 10.1182/blood-2018-07-818211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-Like receptor 4. J Immunol. 2001;167:2887–2894. doi: 10.4049/jimmunol.167.5.2887 [DOI] [PubMed] [Google Scholar]
- 34.Tyagi N, Roberts AM, Dean WL, Tyagi SC, Lominadze D. Fibrinogen induces endothelial cell permeability. Mol Cell Biochem. 2008;307:13–22. doi: 10.1007/s11010-007-9579-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sriramarao P, Languino LR, Altieri DC. Fibrinogen mediates leukocyte-endothelium bridging in vivo at low shear forces. Blood. 1996;88:3416–3423. [PubMed] [Google Scholar]
- 36.Lord ST. Molecular mechanisms affecting fibrin structure and stability. Arterioscler Thromb Vasc Biol. 2011;31:494–499. doi: 10.1161/ATVBAHA.110.213389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schlimp CJ, Schöchl H. The role of fibrinogen in trauma-induced coagulopathy. Hamostaseologie. 2014;34:29–39. doi: 10.5482/hamo-13-07-0038 [DOI] [PubMed] [Google Scholar]
- 38.Hayakawa M Dynamics of fibrinogen in acute phases of trauma. J Intensive Care. 2017;5:3. doi: 10.1186/s40560-016-0199-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rourke C, Curry N, Khan S, Taylor R, Raza I, Davenport R, Stanworth S, Brohi K. Fibrinogen levels during trauma hemorrhage, response to replacement therapy, and association with patient outcomes. J Thromb Haemost. 2012;10:1342–1351. doi: 10.1111/j.1538-7836.2012.04752.x [DOI] [PubMed] [Google Scholar]
- 40.May JE, Wolberg AS, Lim MY. Disorders of fibrinogen and fibrinolysis. Hematol Oncol Clin North Am. 2021;35:1197–1217. doi: 10.1016/j.hoc.2021.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casini A, Neerman-Arbez M, de Moerloose P. Heterogeneity of congenital afibrinogenemia, from epidemiology to clinical consequences and management. Blood Rev. 2021;48:100793. doi: 10.1016/j.blre.2020.100793 [DOI] [PubMed] [Google Scholar]
- 42.Neerman-Arbez M, de Moerloose P, Casini A. Laboratory and genetic investigation of mutations accounting for congenital fibrinogen disorders. Semin Thromb Hemost. 2016;42:356–365. doi: 10.1055/s-0036-1571340 [DOI] [PubMed] [Google Scholar]
- 43.Komáromi I, Bagoly Z, Muszbek L. Factor XIII: novel structural and functional aspects. J Thromb Haemost. 2011;9:9–20. doi: 10.1111/j.1538-7836.2010.04070.x [DOI] [PubMed] [Google Scholar]
- 44.Beckers CML, Simpson KR, Griffin KJ, Brown JM, Cheah LT, Smith KA, Vacher J, Cordell PA, Kearney MT, Grant PJ, et al. Cre/lox studies identify resident macrophages as the major source of circulating coagulation factor XIII-A. Arterioscler Thromb Vasc Biol. 2017;37:1494–1502. doi: 10.1161/ATVBAHA.117.309271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wölpl A, Lattke H, Board PG, Arnold R, Schmeiser T, Kubanek B, Robin-Winn M, Pichelmayr R, Goldmann SF. Coagulation factor XIII A and B subunits in bone marrow and liver transplantation. Transplantation. 1987;43:151–153. doi: 10.1097/00007890-198701000-00032 [DOI] [PubMed] [Google Scholar]
- 46.Katona E, Penzes K, Csapo A, Fazakas F, Udvardy ML, Bagoly Z, Orosz ZZ, Muszbek L. Interaction of factor XIII subunits. Blood. 2014;123:1757–1763. doi: 10.1182/blood-2013-10-533596 [DOI] [PubMed] [Google Scholar]
- 47.Mary A, Achyuthan KE, Greenberg CS. b-chains prevent the proteolytic inactivation of the a-chains of plasma factor XIII. Biochim Biophys Acta. 1988;966:328–335. doi: 10.1016/0304-4165(88)90082-7 [DOI] [PubMed] [Google Scholar]
- 48.Strilchuk AW, Meixner SC, Leung J, Safikhan NS, Kulkarni JA, Russell HM, van der Meel R, Sutherland MR, Owens AP, Palumbo JS, et al. Sustained depletion of FXIII-A by inducing acquired FXIII-B deficiency. Blood. 2020;136:2946–2954. doi: 10.1182/blood.2020004976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Byrnes JR, Wilson C, Boutelle AM, Brandner CB, Flick MJ, Philippou H, Wolberg AS. The interaction between fibrinogen and zymogen FXIII-A(2)B(2) is mediated by fibrinogen residues gamma 390–396 and the FXIII-B subunits. Blood. 2016;128:1969–1978. doi: 10.1182/blood-2016-04-712323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Credo RB, Curtis CG, Lorand L. Alpha-chain domain of fibrinogen controls generation of fibrinoligase (coagulation factor XIIIa). Calcium ion regulatory aspects. Biochemistry. 1981;20:3770–3778. doi: 10.1021/bi00516a016 [DOI] [PubMed] [Google Scholar]
- 51.Smith KA, Adamson PJ, Pease RJ, Brown JM, Balmforth AJ, Cordell PA, Ariëns RAS, Philippou H, Grant PJ. Interactions between factor XIII and the αC region of fibrinogen. Blood. 2011;117:3460–3468. doi: 10.1182/blood-2010-10-313601 [DOI] [PubMed] [Google Scholar]
- 52.Marx G, Korner G, Mou X, Gorodetsky R. Packaging zinc, fibrinogen, and factor XIII in platelet α-granules. J Cell Physiol. 1993;156:437–442. doi: 10.1002/jcp.1041560302 [DOI] [PubMed] [Google Scholar]
- 53.Greenberg CS, Achyuthan KE, Rajagopalan S, Pizzo SV. Characterization of the fibrin polymer structure that accelerates thrombin cleavage of plasma factor XIII. Arch Biochem Biophys. 1988;262:142–148. doi: 10.1016/0003-9861(88)90176-2 [DOI] [PubMed] [Google Scholar]
- 54.Nurminskaya M, Kaartinen MT. Transglutaminases in mineralized tissues. Front Biosci. 2006;11:1591–1606. doi: 10.2741/1907 [DOI] [PubMed] [Google Scholar]
- 55.Muszbek L, Bereczky Z, Bagoly Z, Komaromi I, Katona E. Factor XIII: A coagulation factor with multiple plasmatic and cellular functions. Physiol Rev. 2011;91:931–972. doi: 10.1152/physrev.00016.2010 [DOI] [PubMed] [Google Scholar]
- 56.Muszbek L, Adány R, Szegedi G, Polgár J, Kávai M. Factor XIII of blood coagulation in human monocytes. Thromb Res. 1985;37:401–410. doi: 10.1016/0049-3848(85)90069-6 [DOI] [PubMed] [Google Scholar]
- 57.Henriksson P, Becker S, Lynch G, McDonagh J. Identification of intracellular factor XIII in human monocytes and macrophages. J Clin Invest. 1985;76:528–534. doi: 10.1172/jci112002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adány R, Glukhova MA, Kabakov AY, Muszbek L. Characterisation of connective tissue cells containing factor XIII subunit a. J Clin Pathol. 1988;41:49–56. doi: 10.1136/jcp.41.1.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwartz ML, Pizzo SV, Hill RL, McKee PA. The subunit structures of human plasma and platelet factor XIII (fibrin-stabilizing factor). J Biol Chem. 1971;246:5851–5854. [PubMed] [Google Scholar]
- 60.Bohn H The human fibrin-stabilizing factors. Mol Cell Biochem. 1978;20:67–75. doi: 10.1007/BF00241384 [DOI] [PubMed] [Google Scholar]
- 61.Sixma JJ, van den Berg A, Schiphorst M, Geuze HJ, McDonagh J. Immunocytochemical localization of albumin and factor XIII in thin cryo sections of human blood platelets. Thromb Haemost. 1984;51:388–391. [PubMed] [Google Scholar]
- 62.Polgár J, Hidasi V, Muszbek L. Non-proteolytic activation of cellular protransglutaminase (placenta macrophage factor XIII). Biochem J. 1990;267:557–560. doi: 10.1042/bj2670557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muszbek L, Polgár J, Boda Z. Platelet factor XIII becomes active without the release of activation peptide during platelet activation. Thromb Haemost. 1993;69:282–285. [PubMed] [Google Scholar]
- 64.Katona EE, Ajzner E, Tóth K, Kárpáti L, Muszbek L. Enzyme-linked immunosorbent assay for the determination of blood coagulation factor XIII A-subunit in plasma and in cell lysates. J Immunol Methods. 2001;258:127–135. doi: 10.1016/s0022-1759(01)00479-3 [DOI] [PubMed] [Google Scholar]
- 65.Burkhart JM, Vaudel M, Gambaryan S, Radau S, Walter U, Martens L, Geiger J, Sickmann A, Zahedi RP. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120:e73–82. doi: 10.1182/blood-2012-04-416594 [DOI] [PubMed] [Google Scholar]
- 66.Lopaciuk S, Lovette KM, McDonagh J, Chuang HYK, McDonagh RP. Subcellular distribution of fibrinogen and factor XIII in human blood platelets. Thromb Res. 1976;8:453–465. doi: 10.1016/0049-3848(76)90223-1 [DOI] [PubMed] [Google Scholar]
- 67.Mitchell JL, Mutch NJ. Novel aspects of platelet factor XIII function. Thromb Res. 2016;141:S17–S21. doi: 10.1016/S0049-3848(16)30356-5 [DOI] [PubMed] [Google Scholar]
- 68.Mitchell JL, Lionikiene AS, Fraser SR, Whyte CS, Booth NA, Mutch NJ. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood. 2014;124:3982–3990. doi: 10.1182/blood-2014-06-583070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Somodi L, Beke Debreceni I, Kis G, Cozzolino M, Kappelmayer J, Antal M, Panyi G, Bárdos H, Mutch NJ, Muszbek L. Activation mechanism dependent surface exposure of cellular factor XIII on activated platelets and platelet microparticles. J Thromb Haemost. 2022. doi: 10.1111/jth.15668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ando Y, Imamura S, Yamagata Y, Kitahara A, Saji H, Murachi T, Kannagi R. Platelet factor XIII is activated by calpain. Biochem Biophys Res Commun. 1987;144:484–490. doi: 10.1016/S0006-291X(87)80535-1 [DOI] [PubMed] [Google Scholar]
- 71.Muszbek L, Haramura G, Polgar J. Transformation of cellular factor-XIII into an active zymogen transglutaminase in thrombin-stimulated platelets. Thromb Haemost. 1995;73:702–705. [PubMed] [Google Scholar]
- 72.Anokhin BA, Dean WL, Smith KA, Flick MJ, Ariëns RAS, Philippou H, Maurer MC. Proteolytic and nonproteolytic activation mechanisms result in conformationally and functionally different forms of coagulation factor XIII A. FEBS J. 2020;287:452–464. doi: 10.1111/febs.15040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anokhin BA, Stribinskis V, Dean WL, Maurer MC. Activation of factor XIII is accompanied by a change in oligomerization state. FEBS J. 2017;284:3849–3861. doi: 10.1111/febs.14272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serrano K, Devine DV. Intracellular factor XIII crosslinks platelet cytoskeletal elements upon platelet activation. Thromb Haemost. 2002;88:315–320. [PubMed] [Google Scholar]
- 75.Asijee GM, Muszbek L, Kappelmayer J, Polgár J, Horváth A, Sturk A. Platelet vinculin: a substrate of activated factor XIII. Biochim Biophys Acta. 1988;954:303–308. doi: 10.1016/0167-4838(88)90085-4 [DOI] [PubMed] [Google Scholar]
- 76.Adány R, Bárdos H. Factor XIII subunit A as an intracellular transglutaminase. Cell Mol Life Sci. 2003;60:1049–1060. doi: 10.1007/s00018-003-2178-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kasahara K, Souri M, Kaneda M, Miki T, Yamamoto N, Ichinose A. Impaired clot retraction in factor XIII A subunit-deficient mice. Blood. 2010;115:1277–1279. doi: 10.1182/blood-2009-06-227645 [DOI] [PubMed] [Google Scholar]
- 78.Kasahara K, Kaneda M, Miki T, Iida K, Sekino-Suzuki N, Kawashima I, Suzuki H, Shimonaka M, Arai M, Ohno-Iwashita Y, et al. Clot retraction is mediated by factor XIII-dependent fibrin-αIIbβ3-myosin axis in platelet sphingomyelin-rich membrane rafts. Blood. 2013;122:3340–3348. doi: 10.1182/blood-2013-04-491290 [DOI] [PubMed] [Google Scholar]
- 79.Tutwiler V, Litvinov RI, Lozhkin AP, Peshkova AD, Lebedeva T, Ataullakhanov FI, Spiller KL, Cines DB, Weisel JW. Kinetics and mechanics of clot contraction are governed by the molecular and cellular composition of the blood. Blood. 2016;127:149–159. doi: 10.1182/blood-2015-05-647560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tang Z, Kattula S, Holle LA, Cooley BC, Lin F-C, Wolberg AS. Factor XIII deficiency does not prevent FeCl3-induced carotid artery thrombus formation in mice. Res Pract Thromb Haemost. 2020;4:111–116. doi: 10.1002/rth2.12278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kattula S, Byrnes JR, Martin SM, Holle LA, Cooley BC, Flick MJ, Wolberg AS. Factor XIII in plasma, but not in platelets, mediates red blood cell retention in clots and venous thrombus size in mice. Blood Adv. 2018;2:25–35. doi: 10.1182/bloodadvances.2017011890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rijken DC, Abdul S, Malfliet J, Leebeek FWG, de Willige SU. Compaction of fibrin clots reveals the antifibrinolytic effect of factor XIII. J Thromb Haemost. 2016;14:1453–1461. doi: 10.1111/jth.13354 [DOI] [PubMed] [Google Scholar]
- 83.Nikolajsen CL, Dyrlund TF, Poulsen ET, Enghild JJ, Scavenius C. Coagulation factor XIIIa substrates in human plasma: identification and incorporation into the clot. J Biol Chem. 2014;289:6526–6534. doi: 10.1074/jbc.M113.517904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Richardson VR, Cordell P, Standeven KF, Carter AM. Substrates of factor XIII-A: roles in thrombosis and wound healing. Clinical science (London, England : 1979). 2013;124:123–137. doi: 10.1042/cs20120233 [DOI] [PubMed] [Google Scholar]
- 85.Schmitt LR, Henderson R, Barrett A, Darula Z, Issaian A, D’Alessandro A, Clendenen N, Hansen KC. Mass spectrometry-based molecular mapping of native FXIIIa cross-links in insoluble fibrin clots. J Biol Chem. 2019;294:8773–8778. doi: 10.1074/jbc.AC119.007981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mouapi KN, Bell JD, Smith KA, Ariëns RA, Philippou H, Maurer MC. Ranking reactive glutamines in the fibrinogen αC region that are targeted by blood coagulant factor XIII. Blood. 2016;127:2241–2248. doi: 10.1182/blood-2015-09-672303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tamaki T, Aoki N. Cross-linking of alpha 2-plasmin inhibitor and fibronectin to fibrin by fibrin-stabilizing factor. Biochim Biophys Acta. 1981;661:280–286. doi: 10.1016/0005-2744(81)90016-4 [DOI] [PubMed] [Google Scholar]
- 88.Mosesson MW, Siebenlist KR, Hernandez I, Lee KN, Christiansen VJ, McKee PA. Evidence that α2-antiplasmin becomes covalently ligated to plasma fibrinogen in the circulation: a new role for plasma factor XIII in fibrinolysis regulation. J Thromb Haemost. 2008;6:1565–1570. doi: 10.1111/j.1538-7836.2008.03056.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alshehri FSM, Whyte CS, Tuncay A, Williams ML, Wilson HM, Mutch NJ. Monocytes expose factor XIII-A and stabilize thrombi against fibrinolytic degradation. Int J Mol Sci. 2021;22. doi: 10.3390/ijms22126591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Karimi M, Bereczky Z, Cohan N, Muszbek L. Factor XIII deficiency. Semin Thromb Hemost. 2009;35:426–438. doi: 10.1055/s-0029-1225765 [DOI] [PubMed] [Google Scholar]
- 91.Muszbek L, Bagoly Z, Cairo A, Peyvandi F. Novel aspects of factor XIII deficiency. Curr Opin Hematol. 2011;18:366–372. doi: 10.1097/MOH.0b013e3283497e3e [DOI] [PubMed] [Google Scholar]
- 92.Inbal A, Lubetsky A, Krapp T, Castel D, Shaish A, Dickneitte G, Modis L, Muszbek L, Inbal A. Impaired wound healing in factor XIII deficient mice. Thromb Haemost. 2005;94:432–437. doi: 10.1160/th05-04-0291 [DOI] [PubMed] [Google Scholar]
- 93.Hsieh L, Nugent D. Factor XIII deficiency. Haemophilia. 2008;14:1190–1200. doi: 10.1111/j.1365-2516.2008.01857.x [DOI] [PubMed] [Google Scholar]
- 94.Hashiguchi T, Saito M, Morishita E, Matsuda T, Ichinose A. Two genetic defects in a patient with complete deficiency of the b-subunit for coagulation factor XIII. Blood. 1993;82:145–150. doi: 10.1182/blood.V82.1.145.bloodjournal821145 [DOI] [PubMed] [Google Scholar]
- 95.Dorgalaleh A, Tabibian S, Hosseini MS, Farshi Y, Roshanzamir F, Naderi M, Kazemi A, Zaker F, Aghideh AN, Shamsizadeh M. Diagnosis of factor XIII deficiency. Hematology. 2016;21:430–439. doi: 10.1080/10245332.2015.1101975 [DOI] [PubMed] [Google Scholar]
- 96.Durda MA, Wolberg AS, Kerlin BA. State of the art in factor XIII laboratory assessment. Transfus Apher Sci. 2018;57:700–704. doi: 10.1016/j.transci.2018.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lawrie AS, Green L, Mackie IJ, Liesner R, Machin SJ, Peyvandi F. Factor XIII--an under diagnosed deficiency--are we using the right assays? J Thromb Haemost. 2010;8:2478–2482. doi: 10.1111/j.1538-7836.2010.04028.x [DOI] [PubMed] [Google Scholar]
- 98.Kessel R, Hu C, Shore-Lesserson L, Rand J, Manwani D. A child with acquired factor XIII deficiency: case report and literature review. Haemophilia. 2013;19:814–826. doi: 10.1111/hae.12145 [DOI] [PubMed] [Google Scholar]
- 99.Hayashi T, Kadohira Y, Morishita E, Asakura H, Souri M, Ichinose A. A case of acquired FXIII deficiency with severe bleeding symptoms. Haemophilia. 2012;18:618–620. doi: 10.1111/j.1365-2516.2012.02763.x [DOI] [PubMed] [Google Scholar]
- 100.Beckman JD, Kasthuri RS, Wolberg AS, Ma AD. Challenges in diagnosis and management of acquired factor XIII (FXIII) inhibitors. Haemophilia. 2018;24:e417–e420. doi: 10.1111/hae.13603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wolberg AS, Rosendaal FR, Weitz JI, Jaffer IH, Agnelli G, Baglin T, Mackman N. Venous thrombosis. Nat Rev Dis Primers. 2015;1:17. doi: 10.1038/nrdp.2015.6 [DOI] [PubMed] [Google Scholar]
- 102.Kamphuisen PW, Eikenboom JC, Vos HL, Pablo R, Sturk A, Bertina RM, Rosendaal FR. Increased levels of factor VIII and fibrinogen in patients with venous thrombosis are not caused by acute phase reactions. Thromb Haemost. 1999;81:680–683. [PubMed] [Google Scholar]
- 103.Byrnes JR, Wolberg AS. Newly-recognized roles of factor XIII in thrombosis. Semin Thromb Hemost. 2016;42:445–454. doi: 10.1055/s-0036-1571343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maners J, Gill D, Pankratz N, Laffan MA, Wolberg AS, de Maat MPM, Ligthart S, Tang W, Ward-Caviness CK, Fornage M, et al. A Mendelian randomization of γ’ and total fibrinogen levels in relation to venous thromboembolism and ischemic stroke. Blood. 2020;136:3062–3069. doi: 10.1182/blood.2019004781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wells PS, Anderson JL, Scarvelis DK, Doucette SP, Gagnon F. Factor XIII Val34Leu Variant Is Protective against Venous Thromboembolism: A HuGE Review and Meta-Analysis. Am J Epidemiol. 2006;164:101–109. doi: 10.1093/aje/kwj179 [DOI] [PubMed] [Google Scholar]
- 106.Ariëns RA, Philippou H, Nagaswami C, Weisel JW, Lane DA, Grant PJ. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood. 2000;96:988–995. [PubMed] [Google Scholar]
- 107.Lim BC, Ariëns RA, Carter AM, Weisel JW, Grant PJ. Genetic regulation of fibrin structure and function: complex gene-environment interactions may modulate vascular risk. Lancet. 2003;361:1424–1431. doi: 10.1016/s0140-6736(03)13135-2 [DOI] [PubMed] [Google Scholar]
- 108.Hara T, Bhayana B, Thompson B, Kessinger CW, Khatri A, McCarthy JR, Weissleder R, Lin CP, Tearney GJ, Jaffer FA. Molecular imaging of fibrin deposition in deep vein thrombosis using fibrin-targeted near-infrared fluorescence. JACC Cardiovascular imaging. 2012;5:607–615. doi: 10.1016/j.jcmg.2012.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.von Brühl M-L, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. Exp Med. 2012;209:819–835. doi: 10.1084/jem.20112322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mwiza JMN, Lee RH, Paul DS, Holle LA, Cooley BC, Nieswandt B, Schug WJ, Kawano T, Mackman N, Wolberg AS, et al. Both G protein-coupled and immunoreceptor tyrosine-based activation motif receptors mediate venous thrombosis in mice. Blood. 2022. doi: 10.1182/blood.2022015787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cines DB, Lebedeva T, Nagaswami C, Hayes V, Massefski W, Litvinov RI, Rauova L, Lowery TJ, Weisel JW. Clot contraction: compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood. 2014;123:1596–1603. doi: 10.1182/blood-2013-08-523860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Walton BL, Byrnes JR, Wolberg AS. Fibrinogen, red blood cells, and factor XIII in venous thrombosis. J Thromb Haemost. 2015;13 Suppl 1:S208–S215. doi: 10.1111/jth.12918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Machlus KR, Cardenas JC, Church FC, Wolberg AS. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood. 2011;117:4953–4963. doi: 10.1182/blood-2010-11-316885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Byrnes JR, Duval C, Wang YM, Hansen CE, Ahn B, Mooberry MJ, Clark MA, Johnsen JM, Lord ST, Lam WA, et al. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin alpha-chain crosslinking. Blood. 2015;126:1940–1948. doi: 10.1182/blood-2015-06-652263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aleman MM, Byrnes JR, Wang J-G, Tran R, Lam WA, Di Paola J, Mackman N, Degen JL, Flick MJ, Wolberg AS. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest. 2014;124:3590–3600. doi: 10.1172/JCI75386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jaffer FA, Tung CH, Wykrzykowska JJ, Ho NH, Houng AK, Reed GL, Weissleder R. Molecular imaging of factor XIIIa activity in thrombosis using a novel, near-infrared fluorescent contrast agent that covalently links to thrombi. Circulation. 2004;110:170–176. doi: 10.1161/01.cir.0000134484.11052.44 [DOI] [PubMed] [Google Scholar]
- 117.Kattula S, Bagoly Z, Tóth NK, Muszbek L, Wolberg AS. The factor XIII-A Val34Leu polymorphism decreases whole blood clot mass at high fibrinogen concentrations. J Thromb Haemost. 2020;18:885–894. doi: 10.1111/jth.14744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Goyal. VVA. Acute Pulmonary Embolism. In: StatPearls Publishing; 2021. [PubMed] [Google Scholar]
- 119.Undas A, Zawilska K, Ciesla-Dul M, Lehmann-Kopydlowska A, Skubiszak A, Ciepluch K, Tracz W. Altered fibrin clot structure/function in patients with idiopathic venous thromboembolism and in their relatives. Blood. 2009;114:4272–4278. doi: 10.1182/blood-2009-05-222380 [DOI] [PubMed] [Google Scholar]
- 120.Marsh JJ, Chiles PG, Liang NC, Morris TA. Chronic thromboembolic pulmonary hypertension-associated dysfibrinogenemias exhibit disorganized fibrin structure. Thromb Res. 2013;132:729–734. doi: 10.1016/j.thromres.2013.09.024 [DOI] [PubMed] [Google Scholar]
- 121.Planquette B, Sanchez O, Marsh JJ, Chiles PG, Emmerich J, Le Gal G, Meyer G, Wolfson T, Gamst AC, Moore RE, et al. Fibrinogen and the prediction of residual obstruction manifested after pulmonary embolism treatment. Eur Respir J. 2018;52. doi: 10.1183/13993003.01467-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ząbczyk M, Natorska J, Undas A. Factor XIII and fibrin clot properties in acute venous thromboembolism. Int J Mol Sci. 2021;22. doi: 10.3390/ijms22041607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Klajmon A, Chmiel J, Ząbczyk M, Pociask E, Wypasek E, Malinowski KP, Undas A, Natorska J. Fibrinogen β chain and FXIII polymorphisms affect fibrin clot properties in acute pulmonary embolism. Eur J Clin Invest. 2022;52:e13718. doi: 10.1111/eci.13718 [DOI] [PubMed] [Google Scholar]
- 124.Shaya SA, Gani DM, Weitz JI, Kim PY, Gross PL. Factor XIII prevents pulmonary emboli in mice by stabilizing deep vein thrombi. Thromb Haemost. 2019;119:992–999. doi: 10.1055/s-0039-1685141 [DOI] [PubMed] [Google Scholar]
- 125.Kattula S, Sang Y, de Ridder G, Silver AC, Bouck EG. Novel venous thromboembolism mouse model to evaluate the role of complete and partial factor XIII deficiency in pulmonary embolism risk. J Thromb Haemost. 2021. doi: 10.1111/jth.15510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Duval C, Baranauskas A, Feller T, Ali M, Cheah LT, Yuldasheva NY, Baker SR, McPherson HR, Raslan Z, Bailey MA, et al. Elimination of fibrin γ-chain cross-linking by FXIIIa increases pulmonary embolism arising from murine inferior vena cava thrombi. Proc Natl Acad Sci U S A. 2021;118:e2103226118. doi: 10.1073/pnas.2103226118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cieslik J, Mrozinska S, Broniatowska E, Undas A. Altered plasma clot properties increase the risk of recurrent deep vein thrombosis: a cohort study. Blood. 2018;131:797–807. doi: 10.1182/blood-2017-07-798306 [DOI] [PubMed] [Google Scholar]
- 128.Undas A, Ciesla-Dul M, Drazkiewicz T, Sadowski J. Altered fibrin clot properties are associated with residual vein obstruction: Effects of lipoprotein(a) and apolipoprotein(a) isoform. Thromb Res. 2012;130:E184–E187. doi: 10.1016/j.thromres.2012.06.005 [DOI] [PubMed] [Google Scholar]
- 129.Mazur P, Gawęda B, Natorska J, Ząbczyk M, Undas A, Sadowski J, Kopeć G, Waligóra M, Podolec P, Kapelak B. Fibrin structure in organized thrombotic material removed during pulmonary artery endarterectormy: the effect of vessel calibre. J Thromb Thrombolysis. 2016;42:212–217. doi: 10.1007/s11239-016-1382-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Siudut J, Grela M, Wypasek E, Plens K, Undas A. Reduced plasma fibrin clot permeability and susceptibility to lysis are associated with increased risk of postthrombotic syndrome. J Thromb Haemost. 2016;14:784–793. doi: 10.1111/jth.13264 [DOI] [PubMed] [Google Scholar]
- 131.Morris TA, Marsh JJ, Chiles PG, Magaña MM, Liang NC, Soler X, Desantis DJ, Ngo D, Woods VL Jr. High prevalence of dysfibrinogenemia among patients with chronic thromboembolic pulmonary hypertension. Blood. 2009;114:1929–1936. doi: 10.1182/blood-2009-03-208264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shaya SA, Saldanha LJ, Vaezzadeh N, Zhou J, Ni R, Gross PL. Comparison of the effect of dabigatran and dalteparin on thrombus stability in a murine model of venous thromboembolism. J Thromb Haemost. 2016;14:143–152. doi: 10.1111/jth.13182 [DOI] [PubMed] [Google Scholar]
- 133.Matsuno H, Okada K, Ueshima S, Matsuo O, Kozawa O. Alpha2-antiplasmin plays a significant role in acute pulmonary embolism. J Thromb Haemost. 2003;1:1734–1739. doi: 10.1046/j.1538-7836.2003.00252.x [DOI] [PubMed] [Google Scholar]
- 134.Singh S, Houng A, Reed GL. Releasing the brakes on the fibrinolytic system in pulmonary emboli: Unique effects of plasminogen activation and α2-antiplasmin inactivation. Circulation. 2017;135:1011–1020. doi: 10.1161/circulationaha.116.024421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Brown W, Lyden PD. 51 - Intravenous Thrombolysis. In: Grotta JC, Albers GW, Broderick JP, Kasner SE, Lo EH, Mendelow AD, Sacco RL, Wong LKS, eds. Stroke (Sixth Edition). London: Elsevier; 2016:826–848. [Google Scholar]
- 136.Sherman DG, Atkinson RP, Chippendale T, Levin KA, Ng K, Futrell N, Hsu CY, Levy DE, Participants ftS. Intravenous ancrod for treatment of acute ischemic stroke the STAT study: a randomized controlled trial. JAMA. 2000;283:2395–2403. doi: 10.1001/jama.283.18.2395 [DOI] [PubMed] [Google Scholar]
- 137.Hennerici MG, Kay R, Bogousslavsky J, Lenzi GL, Verstraete M, Orgogozo JM. Intravenous ancrod for acute ischaemic stroke in the European stroke treatment with ancrod trial: a randomised controlled trial. Lancet. 2006;368:1871–1878. doi: 10.1016/s0140-6736(06)69776-6 [DOI] [PubMed] [Google Scholar]
- 138.Levy DE, del Zoppo GJ, Demaerschalk BM, Demchuk AM, Diener HC, Howard G, Kaste M, Pancioli AM, Ringelstein EB, Spatareanu C, et al. Ancrod in acute ischemic stroke: results of 500 subjects beginning treatment within 6 hours of stroke onset in the ancrod stroke program. Stroke. 2009;40:3796–3803. doi: 10.1161/strokeaha.109.565119 [DOI] [PubMed] [Google Scholar]
- 139.Juang LJ, Hur WS, Silva LM, Strilchuk AW, Francisco B, Leung J, Robertson MK, Groeneveld DJ, La Prairie B, Chun EM, et al. Suppression of fibrin(ogen)-driven pathologies in disease models through controlled knockdown by lipid nanoparticle delivery of siRNA. Blood. 2022;139:1302–1311. doi: 10.1182/blood.2021014559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hur WS, Paul DS, Bouck EG, Negrón OA, Mwiza JM, Poole LG, Cline-Fedewa HM, Clark EG, Juang LJ, Leung J, et al. Hypofibrinogenemia with preserved hemostasis and protection from thrombosis in mice with an Fga truncation mutation. Blood. 2022;139:1374–1388. doi: 10.1182/blood.2021012537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Peyvandi F, Palla R, Menegatti M, Siboni SM, Halimeh S, Faeser B, Pergantou H, Platokouki H, Giangrande P, Peerlinck K, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost. 2012;10:615–621. doi: 10.1111/j.1538-7836.2012.04653.x [DOI] [PubMed] [Google Scholar]
- 142.Menegatti M, Palla R, Boscarino M, Bucciarelli P, Muszbek L, Katona E, Makris M, Peyvandi F, Grp P-RS. Minimal factor XIII activity level to prevent major spontaneous bleeds. J Thromb Haemost. 2017;15:1728–1736. doi: 10.1111/jth.13772 [DOI] [PubMed] [Google Scholar]
- 143.Reed GL, Houng AK, Singh S, Wang D. α2-Antiplasmin: New Insights and Opportunities for Ischemic Stroke. Semin Thromb Hemost. 2017;43:191–199. doi: 10.1055/s-0036-1585077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schmitz T, Bäuml CA, Imhof D. Inhibitors of blood coagulation factor XIII. Anal Biochem. 2020;605:113708. doi: 10.1016/j.ab.2020.113708 [DOI] [PubMed] [Google Scholar]
- 145.Bäuml CA, Schmitz T, Paul George AA, Sudarsanam M, Hardes K, Steinmetzer T, Holle LA, Wolberg AS, Pötzsch B, Oldenburg J, et al. Coagulation factor XIIIa inhibitor tridegin: on the role of disulfide bonds for folding, stability, and function. J Med Chem. 2019;62:3513–3523. doi: 10.1021/acs.jmedchem.8b01982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pasternack R, Büchold C, Jähnig R, Pelzer C, Sommer M, Heil A, Florian P, Nowak G, Gerlach U, Hils M. Novel inhibitor ZED3197 as potential drug candidate in anticoagulation targeting coagulation FXIIIa (F13a). J Thromb Haemost. 2020;18:191–200. doi: 10.1111/jth.14646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Heil A, Weber J, Büchold C, Pasternack R, Hils M. Differences in the inhibition of coagulation factor XIII-A from animal species revealed by Michael Acceptor- and thioimidazol based blockers. Thromb Res. 2013;131:e214–222. doi: 10.1016/j.thromres.2013.02.008 [DOI] [PubMed] [Google Scholar]
- 148.Finney S, Seale L, Sawyer RT, Wallis RB. Tridegin, a new peptidic inhibitor of factor XIIIa, from the blood-sucking leech Haementeria ghilianii. Biochem J. 1997;324 ( Pt 3):797–805. doi: 10.1042/bj3240797 [DOI] [PMC free article] [PubMed] [Google Scholar]