

Abstract
COPD patients have increased susceptibility to airway bacterial colonisation. Haemophilus influenzae, Moraxella catarrhalis and Streptococcus pneumoniae are three of the most common respiratory bacterial species in COPD. H. influenzae colonisation, but not other bacteria, in COPD patients is associated with higher sputum neutrophil counts. Alveolar macrophages are key in clearance of bacteria as well as releasing mediators to recruit and activate other immune cells in response to infection. The aim was to characterise differences in COPD macrophage responses to H. influenzae, M. catarrhalis and S. pneumoniae, focusing on release of inflammatory and chemotactic mediators, and apoptosis regulation.
Lung macrophages and monocyte-derived macrophages from COPD patients and control subjects were exposed to H. influenzae, M. catarrhalis or S. pneumoniae. Cytokine secretion (tumour necrosis factor-α, interleukin (IL)-6, CXCL8, CCL5 and IL-1β) were measured by ELISA and quantitative reverse transcriptase PCR (RT-qPCR), and apoptosis genes MCL-1, BCL-2, BAX and BAK1 by RT-qPCR. Apoptosis and reactive oxygen species (ROS) release were also measured.
Macrophages responded differentially to the bacterial species, with increased, prolonged production of the neutrophil chemoattractant CXCL8 in response to H. influenzae and M. catarrhalis but not S. pneumoniae. S. pneumoniae initiated macrophage apoptosis and ROS release, H. influenzae and M. catarrhalis did not and increased anti-apoptosis gene expression (BCL-2 5.5-fold and MCL-1 2.4-fold, respectively).
Differential cytokine responses of macrophages to these bacterial species can explain neutrophilic airway inflammation associated with H. influenzae, but not S. pneumoniae in COPD. Furthermore, delayed macrophage apoptosis is a potential mechanism contributing to inability to clear H. influenzae.
Short abstract
Differential cytokine responses of macrophages to Haemophilus influenzae, Moraxella catarrhalis and Streptococcus pneumoniae can explain neutrophilic airway inflammation associated with H. influenzae but not S. pneumoniae in COPD https://bit.ly/3950HVZ
Introduction
Persistent cigarette smoking causes neutrophilic airway inflammation, which is further increased in patients with COPD [1]. Neutrophilic airway inflammation causes tissue remodelling, mucus hyper-secretion and alveolar destruction [2]. Indeed, a recent study demonstrated an association between the magnitude of neutrophilic infiltration in the small airway walls and the degree of loss of alveolar attachments [3]. However, COPD is a heterogeneous disease, and the degree of neutrophilic inflammation varies considerably among individuals [2].
There is increased susceptibility to bacterial infection in COPD, with common pathogens being Haemophilus influenzae, Moraxella catarrhalis and Streptococcus pneumoniae [4, 5]. Bacterial colonisation during the stable state in COPD patients is associated with higher neutrophil cell counts in sputum and bronchoalveolar lavage samples [6, 7]. Recent COPD cohort studies using quantitative polymerase chain reaction (qPCR) on sputum samples have shown an association between H. influenzae and sputum neutrophil counts, while this relationship with neutrophilia does not exist for other bacteria [8–11]. Similarly, 16S RNA-gene based microbiome analysis of sputum samples has demonstrated associations between neutrophil counts and both the Proteobacteria phylum and Haemophilus genus [12]. The clinical relevance of these findings was highlighted by a cohort study reporting a positive association between Proteobacteria abundance and mortality in COPD patients [13].
The underlying mechanisms responsible for the differential host (neutrophilic) immune response to H. influenzae, in contrast to other bacterial species, remains unclear. Alveolar macrophages may play a key role here, as these cells provide first-line host defence against bacteria through phagocytosis and the release of mediators to recruit and activate other immune cells [14]. Alveolar macrophages exposed to bacterial ligands secrete chemokines, including CXCL8, that enable neutrophil chemotaxis [15]. Alveolar macrophages from COPD patients have reduced phagocytic ability [16, 17]; this has been demonstrated for both H. influenzae and S. pneumoniae. Studies in human and murine macrophages have shown that S. pneumoniae exposure causes downregulation of expression of the anti-apoptotic protein Mcl-1, which causes increased macrophage apoptosis and bacterial killing [18, 19]. We hypothesised that the observed associations between sputum neutrophilia and H. influenzae colonisation, but not other bacterial species, are due to differential alveolar macrophage responses to bacterial species, including induction of chemokines and regulation of macrophage apoptosis.
The aim of this study was to characterise COPD macrophage responses to H. influenzae, M. catarrhalis and S. pneumoniae, focusing on the release of chemotactic mediators and the expression of proteins involved in apoptosis regulation. The purpose of these investigations was to elucidate the mechanisms responsible for the observed association in COPD cohort studies between sputum neutrophil counts and H. influenzae, but not other bacterial species [8–11].
Methods
Subjects
10 COPD patients and 10 healthy non-smoker (HNS) controls were recruited from the Medicines Evaluation Unit (Manchester University NHS Foundation Trust), and eight patients undergoing surgical resection for suspected lung cancer (Manchester University NHS Foundation Trust) were recruited (four ex-smokers (ES) without airflow obstruction and four COPD patients) (table 1). COPD patients had a smoking history of ≥10 pack-years and had evidence of airflow obstruction according to Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria [20]. All patients provided written informed consent using protocols approved by local Ethics Committees (10/H1016/25 and 20/NW/0302).
TABLE 1.
Demographics for COPD subjects and ex-smoking and healthy non-smoking (HNS) controls
| Monocyte-derived macrophages | Lung macrophages | |||||
| HNS | COPD | p-value | Ex-smoker | COPD | p-value | |
| Subjects n | 10 | 10 | 4 | 4 | ||
| Age years | 32 (23–56) | 67 (53–73) | <0.0001 | 74 (71–83) | 67 (59–78) | 0.49 |
| Sex: M/F | 6/4 | 5/5 | 0/4 | 2/2 | ||
| FEV1 L | 3.4±0.6 | 1.5±0.8 | <0.0001 | 2.3±1.1 | 1.7±0.4 | 0.30 |
| FEV1 % predicted | 99±11 | 53±21 | <0.0001 | 82±20 | 77±10 | 0.60 |
| FVC L | 4.5±1.1 | 3.3±1.1 | 0.03 | 3.3±1.5 | 2.6±0.6 | 0.4 |
| FEV1/FVC ratio (%) | 76±7 | 37±16 | <0.0001 | 77±6 | 65±2 | 0.1 |
| Current smokers % | NA | 20 | 0 | 50 | ||
| Pack-years | NA | 39±11 | 12±14 | 50±26 | 0.2 | |
| ICS usage % | NA | 60 | NA | 25 | ||
Data presented as median (range) or mean±sd as appropriate, unless indicated otherwise. FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; ICS: inhaled corticosteroids.
Cell culture
Experiments were initially performed using lung macrophages isolated from resected lung tissue as previously described [21] (see supplementary material). However, due to the COVID-19 pandemic lung resection samples were unobtainable and subsequent experiments were carried out using monocyte-derived macrophages (MDMs). MDMs were generated from peripheral blood mononuclear cells by culture with granulocyte–macrophage colony-stimulating factor 10 ng mL−1 for 7 days (full details in supplementary material). Macrophages were stimulated with unopsonised H. influenzae (National Collection of Type Cultures operated by Public Health England [NCTC] 12699), M. catarrhalis (NCTC 11020) or S. pneumoniae (NCTC 12977) at multiplicity of infection (MOI) stated in text or left unexposed for 4, 24 and 72 h. See supplementary material for bacterial preparation. Preliminary experiments showed 5:1 to be an optimal MOI for H. influenzae, M. catarrhalis and S. pneumoniae comparison. Also, no significant differences between MDM cytokine responses after exposure to opsonised or unopsonised bacteria were observed (supplementary figure S2).
Supernatant cytokine levels (tumour necrosis factor (TNF)-α, interleukin (IL)-6, IL-1β, C-X-C motif chemokine ligand (CXCL)-8 and chemokine (C-C motif) ligand 5 (CCL)-5) were measured by ELISA according to the manufacturer's instructions (R & D System, Abingdon, UK). The lower limits of quantification were 15.6 pg·mL−1 for TNF-α, 9.4 pg·mL−1 for IL-6, 31.3 pg·mL−1 for CXCL8 and CCL5, and 3.9 pg·mL−1 for IL-1β. Supernatant levels of reactive oxygen species (ROS) were measured using dihydrorhodamine 123 (D,123). Cells were harvested for mRNA extraction and concentrations measured (supplementary figure S3). Samples were normalised to generate equal concentrations of cDNA for qPCR analysis of TNF-α, IL-6, CXCL8, IL-1β, the anti-apoptotic genes induced myeloid leukaemia cell differentiation protein (MCL-1) and B-cell lymphoma 2 (BCL-2), the pro-apoptotic genes BCL2-associated X protein (BAX) and BCL2 homologous antagonist/killer (BAK1), and the endogenous control glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (for full methods see supplementary material).
The percentage of cells undergoing apoptosis was determined using In Situ Cell Death Detection Fluorescein kit (Roche, Welwyn, UK) (for full methods see supplementary material) with a minimum of 50 cells per condition counted across multiple fields of view to determine percentage positive staining.
Statistical analysis
Normality of data was assessed using Shapiro–Wilk normality test. For ELISA data two-way ANOVA followed by Bonferroni's multiple comparison tests were used to compare between bacterial treatments and between subject groups. One way ANOVA followed by Tukey's test was also used to compare between bacterial treatments for apoptosis and ROS release data. Gene expression data were non-parametric, and Kruskal–Wallis test with Dunn's post hoc analysis was used to compare between bacterial treatments. p<0.05 was considered statistically significant. All analyses were performed using GraphPad Prism version 5.02 (GraphPad, San Diego, CA, USA).
Results
Bacterial stimulation of macrophage cytokine production
Protein secretion from COPD and control MDMs and lung macrophages
MDMs from 10 HNS and eight COPD patients and lung macrophages from four ES and four COPD patients were cultured with H. influenzae, M. catarrhalis or S. pneumoniae at increasing MOIs (0.005–5:1) for 24 h (supplementary figures S3 and S4, respectively). Full MOI curve data showed that the highest levels of cytokine induction were reached at MOI 5:1. Figure 1 shows cytokine levels at MOI 5:1; H. influenzae and M. catarrhalis caused upregulation of TNF-α, IL-6 and CXCL8 secretion compared to control (cells not exposed to bacteria), while S. pneumoniae had no effect in either MDMs or lung macrophages. In both MDMs and lung macrophages, M. catarrhalis caused significant upregulation of all cytokines. H. influenzae caused significant upregulation of cytokine secretion in HNS MDMs and lung macrophages from both ES and COPD.
FIGURE 1.

Effects of bacterial exposure on macrophage cytokine production. a–c) Monocyte-derived macrophages (MDMs) from 10 healthy nonsmokers (HNS) and eight COPD patients or d–f) lung macrophages (LM) from four ex-smokers (ES) and four COPD patients were exposed to Haemophilus influenzae (HI), Moraxella catarrhalis (MC) or Streptococcus pneumoniae (SP) at a multiplicity of infection (MOI) of 5:1 or left unexposed (Control) for 24 h. Supernatant levels of tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6) and C-X-C motif chemokine ligand (CXCL8) were measured by ELISA. Data presented as mean+sem. Levels were compared between conditions using two-way ANOVA followed by a Bonferroni's multiple comparisons test. #,##,###: significantly above unexposed control (p<0.05, 0.01, 0.001 respectively). *,**,***: significant difference between bacterial species (p<0.05, 0.01, 0.001, respectively).
Comparisons between bacterial species showed significantly higher levels of TNF-α and IL-6 secretion for H. influenzae and M. catarrhalis and CXCL8 secretion for M. catarrhalis exposed cells compared to S. pneumoniae. TNF-α levels were significantly greater with M. catarrhalis exposure compared to H. influenzae in COPD MDMs, although this was not observed in lung macrophages.
Cytokine levels were similar in HNS compared to COPD patients from MDMs (n=10 and n=8, respectively), and ES compared to COPD patients in lung macrophages (n=4 and n=4, respectively). Subanalysis of the COPD group showed no differences in cytokine levels in patients treated with inhaled corticosteroids compared to those without (supplementary figure S5).
In a subset of COPD patients (n=7) CCL5 and IL-1β levels from MDMs were also measured after 24-h exposure to all three bacteria at MOI 5:1. CCL5 and IL-1β levels were significantly increased after exposure to H. influenzae and M. catarrhalis but not S. pneumoniae (supplementary figure S6).
Additional time-profile MDM experiments (n=5 COPD) showed that CXCL8 production was significantly greater after 72 h versus 24 h exposure to H. influenzae and M. catarrhalis, and CCL5 after exposure to M. catarrhalis, while TNF-α, IL-6 and IL-1β levels were similar at these time-points (supplementary figure S7).
The experiments already described used commercially available bacterial strains. We also cultured MDMs from four HNS and four COPD patients for 24 h with H. influenzae (n=2 strains), M. catarrhalis (n=1 strain) and S. pneumoniae (n=1 strain) isolated from a single COPD patient (obtained over two clinic visits; supplementary figures S8 and S9). The induction of TNF-α, IL-6 or CXCL8 were similar between the commercial strains and clinically isolated strains for each bacterial species.
Gene expression in MDMs from COPD patients
MDMs from COPD patients (n=5) were cultured with H. influenzae, M. catarrhalis or S. pneumoniae at an MOI of 5:1 for 4, 24 and 72 h (figure 2). H. influenzae significantly upregulated expression of TNF-α, CXCL8 and IL-1β at all time-points, and IL-6 at 24 and 72 h (p<0.05 for all comparisons versus controls). Peak expression was reached at 24 h for TNF-α and IL-6, and at 72 h for CXCL8 and IL-1β. The effects of M. catarrhalis were numerically lower in comparison to H. influenzae, with significant upregulation of gene expression observed at fewer time-points. S. pneumoniae had no significant effect on expression of any cytokine.
FIGURE 2.

Effects of bacterial exposure on monocyte-derived macrophage cytokine gene expression. Monocyte-derived macrophages from five COPD patients were exposed to Haemophilus influenzae (HI), Moraxella catarrhalis (MC) or Streptococcus pneumoniae (SP) at a multiplicity of infection of 5:1 or left unexposed (control) for 4, 24 or 72 h. mRNA expression levels of a) tumour necrosis factor-α (TNF-α), b) interleukin-6 (IL-6), c) C-X-C motif chemokine ligand (CXCL8) and d) IL-1β were measured by quantitative reverse transcriptase PCR (RT-qPCR). Data presented as median±range. RT-qPCR data expression relative to endogenous control (2−ΔΔCt). *,**,***: significantly above unexposed control (p<0.05, 0.01, 0.001, respectively).
Apoptosis in MDMs from COPD patients
MDMs from COPD patients (n=6) were cultured with H. influenzae, M. catarrhalis or S. pneumoniae at an MOI of 5:1 for 24 h (figure 3). H. influenzae and M. catarrhalis caused 7% of cells to undergo apoptosis, with positive staining for TUNEL. Culture with S. pneumoniae significantly increased apoptosis compared to control (p=0.017) with 98% of cells staining positive.
FIGURE 3.

Effects of bacterial exposure on monocyte-derived macrophage (MDM) apoptosis. MDMs from six COPD patients were exposed to Haemophilus influenzae (HI), Moraxella catarrhalis (MC) or Streptococcus pneumoniae (SP) at a multiplicity of infection of 5:1, Triton-X 0.1% (positive death control) or left unexposed (control) for 24 h. Cells were stained for nuclear stain DAPI and the apoptosis marker TUNEL or left unstained (negative). The percentage of positive stained cells was determined with a minimum of 50 cells per condition counted across multiple fields of view. a) Data represent mean+sem percentage cells positive for TUNEL, and b) representative overlay images of TUNEL (green) and DAPI (blue) shown. **: significantly above unexposed control (p<0.01).
MDMs from COPD patients (n=5) were cultured with H. influenzae, M. catarrhalis or S. pneumoniae at an MOI of 5:1 for 4, 24 and 72 h (figure 4). The anti-apoptosis genes MCL-1 and BCL-2 were significantly increased at 24 h by M. catarrhalis 2.4-fold (p=0.04) and H. influenzae 5.5-fold (p<0.001), respectively. H. influenzae and M. catarrhalis also downregulated pro-apoptosis gene BAX expression at 4 h (p<0.01 both comparisons) but had no significant effect on BAK1 expression. S. pneumoniae significantly downregulated MCL-1 and BAK1 expression after 4 h (p=0.04 and 0.036, respectively) and 72 h (p<0.01 both comparisons), and BAX after 72 h (p<0.01), with no effect on BCL-2 expression.
FIGURE 4.

Effects of bacterial exposure on monocyte-derived macrophage (MDM) apoptosis gene expression. MDMs from five COPD patients were exposed to Haemophilus influenzae (HI), Moraxella catarrhalis (MC) or Streptococcus pneumoniae (SP) at a multiplicity of infection of 5:1 or left unexposed (control) for 4, 24 or 72 h. mRNA expression levels of anti-apoptosis genes a) MCL-1 and b) BCL-2 and pro-apoptosis genes c) BAX and d) BAK1 were measured by reverse transcriptase PCR (RT-qPCR). Data presented as mean+sem. Quantitative RT-qPCR data expression relative to endogenous control (2−ΔΔCt). *,**,***: significantly different compared to unexposed control (p<0.05, 0.01, 0.001, respectively).
Reactive oxygen species release in MDMs from COPD patients
MDMs from COPD patients (n=7) were cultured with H. influenzae, M. catarrhalis or S. pneumoniae at an MOI of 5:1 for 24 h and supernatant levels of ROS were determined (figure 5). H. influenzae or M. catarrhalis caused no increase in ROS release. S. pneumoniae significantly increased ROS release from MDMs (p=0.0067).
FIGURE 5.
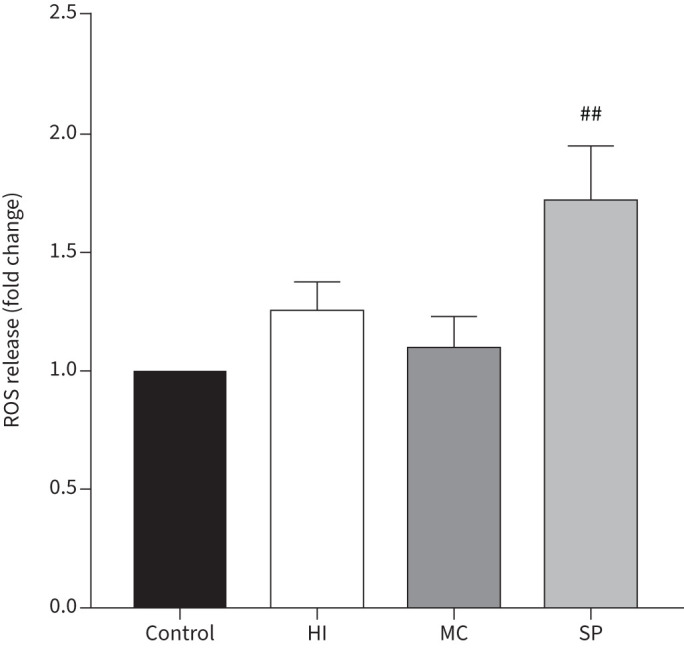
Effects of bacterial exposure on monocyte-derived macrophage (MDM) reactive oxygen species (ROS) release. MDMs from seven COPD patients were exposed to Haemophilus influenzae (HI), Moraxella catarrhalis (MC) or Streptococcus pneumoniae (SP) at a multiplicity of infection of 5:1 or left unexposed (control) for 24 h. Supernatant levels of ROS were measured. Data presented as mean+sem fold change of absorbance compared to unexposed control. ##: significantly above unexposed control (p<0.01).
Discussion
We demonstrate that human lung macrophages and MDMs show differential responses to the respiratory pathogens H. influenzae, M. catarrhalis and S. pneumoniae. H. influenzae and M. catarrhalis induce larger inflammatory cytokine responses compared to S. pneumoniae. Importantly S. pneumoniae exposure resulted in reduced expression of the anti-apoptotic marker MCL-1, associated with high levels of apoptosis (measured by TUNEL staining) and ROS release. In contrast, H. influenzae and M. catarrhalis exposure caused gene expression changes that favoured delayed apoptosis.
These differential cytokine responses, particularly the neutrophil chemoattractant CXCL8, provide a mechanistic explanation for the clinical observations that H. influenzae is associated with increased neutrophil numbers in COPD patients. In contrast, S. pneumoniae caused greater macrophage apoptosis which is likely to be the cause of lower chemokine secretion. The effects of H. influenzae and M. catarrhalis with regard to delayed apoptosis provides a mechanism by which these bacteria may undergo intracellular survival within the macrophage [22] by evading the microbial killing mechanisms associated with cellular apoptosis [18], and thereby persist as colonising bacteria in the airways.
The time-profile of gene and protein expression showed that CXCL8 and IL-1β had a much later peak (at least 72 h) compared to TNF-α and IL-6, which peaked much earlier. We have previously reported similar patterns of prolonged secretion of CXCL8, in contrast to other inflammatory mediators, from alveolar macrophages [23–25]. CXCL8 plays a major role as a neutrophil chemoattractant in neutrophilic lung inflammation in COPD [15, 26], and the results here demonstrate a mechanism by which H. influenzae and M. catarrhalis, but not S. pneumoniae, can cause excessive neutrophil recruitment into the lungs via macrophage activation.
COPD cohort studies have demonstrated a stronger association for neutrophilic inflammation with H. influenzae compared to M. catarrhalis during the stable state [4, 9, 27]. M. catarrhalis has been linked to increased interferon signalling, while H. influenzae presence is associated with neutrophilia [4] and increased levels of TNF-α, CXCL8, IL-1β and myeloperoxidase in sputum [9, 27]. However, we did not observe clear differences between H. influenzae and M. catarrhalis for TNF-α, IL-6, CXCL8 and IL-1β protein secretion, although there was evidence of greater TNF-α, CXCL8 and IL-1β gene expression for H. influenzae. These cytokine protein results do not provide an explanation for the greater neutrophilic inflammation profile observed in COPD cohort studies with H. influenzae compared to M. catarrhalis when sampling was performed in the stable state. A possible explanation is that M. catarrhalis is present in less COPD patients in the stable state but is a cause of exacerbations when it may cause excessive inflammation [4]. M. catarrhalis has shorter periods of colonisation [28], while some H. influenzae strains give rise to long-term persistence [29], and the potential to upregulate further the neutrophilic inflammation that exists in COPD.
Although levels of TNF-α, IL-6 and CXCL8 are increased in the airways of COPD patients [30, 31], we observed no differences between COPD and control macrophages. This is consistent with previously published data utilising lung macrophages from COPD patients with no differences in baseline or lipopolysaccharide-stimulated levels of TNF-α, IL-6 or CXCL8 [21] and H. influenzae antigen-stimulated TNF-α and CXCL8 in MDMs [32]. However, the absolute numbers of alveolar macrophages are increased in the airways of COPD patients compared to controls [33], which ex vivo cell cultures do not take into account. Therefore, the increased macrophage numbers may account for the overall raised cytokine levels in the lungs of COPD patients. Comparisons between COPD and control macrophages were limited to cytokine protein levels. It would also be of interest to assess potential differences between groups in cytokine gene expression, apoptosis or ROS release.
We observed similar effects with H. influenzae and M. catarrhalis on apoptosis and anti-apoptosis gene expression profiles, suggesting that this is not the mechanistic reason for differences between these bacterial species in their ability to persist in the airways by evading the host immune response. Other potential explanations are that H. influenzae reduces expression of the bacterial recognition markers CD36, CD206 and CD163 in alveolar macrophages [23], which may allow evasion of host defence. Additionally, H. influenzae strains identified as persistent colonisers in COPD show phase variation in simple sequence repeats in genes encoding virulence functions, including adhesins (HMW1A/HMW2A), modifications of lipooligosaccharide and iron uptake (haemoglobin–haptoglobin binding protein) [34].
A previous study investigated alveolar macrophage responses to respiratory bacteria [35]. Although no direct comparisons or statistical analysis were performed, visualisation of the figures suggests lower levels of both CXCL8 and TNF-α were induced in response to S. pneumoniae compared to H. influenzae or M. catarrhalis [35]. Here, we confirm statistical significance for these differences in COPD macrophages.
The lack of stimulatory effect of S. pneumoniae on cytokine release could potentially be due to macrophage death and a reduction in cell numbers. Indeed, we observed a reduction in the quantity of mRNA collected from S. pneumoniae-exposed cells at 24 and 72 h suggesting fewer cells. However, the mRNA collected was normalised to generate equal concentrations of cDNA for the PCR assays, and the expression data presented are relative to housekeeping genes which would also be reduced. Therefore, while it is likely that there were less cells present at the later time-points for S. pneumoniae exposure, those which were present were expressing lower levels of cytokines. Also the mRNA concentrations at 4 h were similar between the control and all experiments with bacteria, highlighting that the differences seen for cytokine mRNA expression at 4 h is not due to a reduced number of cells.
The contributions of different Toll-like receptor (TLR) stimulations varies between different bacterial wall components and bacterial species [35]. S. pneumoniae is a Gram-positive bacteria with a different wall structure to the Gram-negative H. influenzae and M. catarrhalis. TLR2 has been shown to play a major part in Gram-positive bacterial recognition [36], while TLR2- and TLR4-mediated cytokine responses are diminished in COPD alveolar macrophages leading to impaired H. influenzae clearance [35]. It is likely that differential TLR signalling plays a role in the mechanisms involved in our findings, and it would be valuable to investigate specific TLR signalling mechanisms involved in the differential bacterial responses we observed.
It is interesting that Streptococcus is one of the most abundant genera in healthy individuals [37], while S. pneumoniae levels measured by PCR in the sputum of healthy controls is higher compared to both H. influenzae and M. catarrhalis [11]. It is possible that the lower induction of pro-inflammatory mediators allows the presence of some S. pneumoniae strains to be tolerated at relatively high levels in the airways.
S. pneumoniae induces apoptosis in alveolar macrophages [38, 39], which is required for effective bacterial clearance [18]. Mcl-1 is a key anti-apoptotic protein involved in the switch from cell viability to apoptosis in alveolar macrophages [18]. Mcl-1 forms heterodimers with pro-apoptotic Bcl-2 family members such as Bax and Bak to inhibit mitochondrial membrane permeabilisation [40]. The decrease of Mcl-1 expression in macrophages after 14–16 h of exposure to S. pneumoniae causes apoptosis and therefore killing and clearance [18]. Overexpression of MCL-1 in a murine model decreases the caspase-dependent mitochondrial ROS production associated with this late-phase intracellular killing of S. pneumoniae, and thereby increases neutrophil recruitment [41]. While we observed a decrease in the pro-apoptotic gene BAK-1 upon S. pneumoniae exposure, it is possible this is not a true reflection of apoptotic state, and the ratio of BAX/Bcl-2 protein may have been a better measurement. However, consistent with these previous reports, we showed a decrease in the expression of MCL-1, and importantly an increase in macrophage apoptosis and ROS production following exposure to S. pneumoniae.
COPD macrophages are defective in apoptosis-associated killing and clearance of S. pneumoniae infection regulated by Mcl-1 [19]. MCL-1 expression in alveolar macrophages from COPD patients is higher both at baseline and following exposure to S. pneumoniae leading to decreased apoptosis compared to controls. Higher levels of Mcl-1 in COPD also reduced mROS production after bacterial challenge [19]. We did not study regulation of Mcl-1 in COPD versus controls, as this has been well documented as above. However, we demonstrate differential regulation of apoptosis and anti-apoptosis genes by S. pneumoniae, in comparison to H. influenzae and M. catarrhalis, that can contribute to different outcomes with regard to the host immune response.
A limitation of this analysis is that owing to the COVID-19 pandemic the availability of surgical samples and as such alveolar macrophages was limited leading to the use of MDMs for many of the experiments. While MDMs are not truly identical to alveolar macrophages, MDMs from COPD patients show dysfunctional behaviour compared to controls [42, 43] and therefore represent a relevant model. Also, although we showed differential bacterial effects on the release of ROS by the macrophages, the assay utilised here was not specific, and it would be of interest to further investigate the effects on caspase-1-induced mitochondrial ROS, which is key in the mechanism of bacterial clearance and dysfunctional in COPD macrophages [19, 43].
We show differential macrophage cytokine secretion in response to the common bacterial species found in COPD, which can explain the excessive neutrophilic airway inflammation observed with H. influenzae, but not S. pneumoniae. Furthermore, the regulation of macrophage apoptosisprovides a potentially important mechanism contributing to persistent H. influenzae colonisation in COPD.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00044-2022.SUPPLEMENT (691.5KB, pdf)
Acknowledgements
The authors would like to acknowledge the Manchester Allergy, Respiratory and Thoracic Surgery Biobank and the North West Lung Centre Charity for supporting this project. In addition, we would like to thank the study participants for their contribution.
Footnotes
Provenance: Submitted article, peer reviewed.
Data availability: The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author contributions: S. Lea, J. Baker, R. Gaskell, D. Pindolia and A.B. Dikwa performed experimentation. R. Shah was the lead surgeon for the study. S. Lea, A. Beech and D. Singh designed the study. All authors were involved in analysis and interpretation of the data, and preparation of the manuscript, with major contributions from S. Lea and D. Singh. All authors read and approved the final manuscript.
Conflict of interest: S. Lea has nothing to disclose.
Conflict of interest: A. Beech has nothing to disclose.
Conflict of interest: J. Baker has nothing to disclose.
Conflict of interest: R. Gaskell has nothing to disclose.
Conflict of interest: D. Pindolia has nothing to disclose.
Conflict of interest: A.B. Dikwa has nothing to disclose.
Conflict of interest: R. Shah has nothing to disclose.
Conflict of interest: D. Singh has received sponsorship to attend and speak at international meetings, and honoraria for lecturing or attending advisory boards from Aerogen, AstraZeneca, Boehringer Ingelheim, Chiesi, Cipla, CSL Behring, Epiendo, Genentech, GlaxoSmithKline, Glenmark, Gossamerbio, Kinaset, Menarini, Novartis, Pulmatrix, Sanofi, Teva, Theravance and Verona.
Support statement: This report is independent research supported by the North West Lung Centre Charity and National Institute for Health Research (NIHR) Clinical Research Facility at Manchester University NHS Foundation Trust. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the North West Lung Centre Charity, the NIHR or the Department of Health. A. Beech, J. Baker, A.B. Dikwah and D. Singh are supported by the NIHR Manchester Biomedical Research Centre. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Stockley JA, Walton GM, Lord JM, et al. Aberrant neutrophil functions in stable chronic obstructive pulmonary disease: the neutrophil as an immunotherapeutic target. Int Immunopharmacol 2013; 17: 1211–1217. doi: 10.1016/j.intimp.2013.05.035 [DOI] [PubMed] [Google Scholar]
- 2.Quint JK, Wedzicha JA. The neutrophil in chronic obstructive pulmonary disease. J Allergy Clin Immunol 2007; 119: 1065–1071. doi: 10.1016/j.jaci.2006.12.640 [DOI] [PubMed] [Google Scholar]
- 3.Polosukhin VV, Gutor SS, Du RH, et al. Small airway determinants of airflow limitation in chronic obstructive pulmonary disease. Thorax 2021; 76: 1079–1088. doi: 10.1136/thoraxjnl-2020-216037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z, Maschera B, Lea S, et al. Airway host-microbiome interactions in chronic obstructive pulmonary disease. Respir Res 2019; 20: 113. doi: 10.1186/s12931-019-1085-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkinson TMA, Aris E, Bourne S, et al. A prospective, observational cohort study of the seasonal dynamics of airway pathogens in the aetiology of exacerbations in COPD. Thorax 2017; 72: 919–927. doi: 10.1136/thoraxjnl-2016-209023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolsum U, Donaldson GC, Singh R, et al. Blood and sputum eosinophils in COPD; relationship with bacterial load. Respir Res 2017; 18: 88. doi: 10.1186/s12931-017-0570-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sethi S, Maloney J, Grove L, et al. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006; 173: 991–998. doi: 10.1164/rccm.200509-1525OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beech AS, Lea S, Kolsum U, et al. Bacteria and sputum inflammatory cell counts; a COPD cohort analysis. Respir Res 2020; 21: 289. doi: 10.1186/s12931-020-01552-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Locantore N, Haldar K, et al. Inflammatory endotype-associated airway microbiome in chronic obstructive pulmonary disease clinical stability and exacerbations: a multicohort longitudinal analysis. Am J Respir Crit Care Med 2021; 203: 1488–1502. doi: 10.1164/rccm.202009-3448OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bafadhel M, Haldar K, Barker B, et al. Airway bacteria measured by quantitative polymerase chain reaction and culture in patients with stable COPD: relationship with neutrophilic airway inflammation, exacerbation frequency, and lung function. Int J Chron Obstruct Pulmon Dis 2015; 10: 1075–1083. doi: 10.2147/COPD.S80091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beech A, Lea S, Li J, et al. Airway bacteria quantification using polymerase chain reaction combined with neutrophil and eosinophil counts identifies distinct COPD endotypes. Biomedicines 2021; 9: 1337. doi: 10.3390/biomedicines9101337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayhew D, Devos N, Lambert C, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax 2018; 73: 422–430. doi: 10.1136/thoraxjnl-2017-210408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dicker AJ, Huang JTJ, Lonergan M, et al. The sputum microbiome, airway inflammation, and mortality in chronic obstructive pulmonary disease. J Allergy Clin Immunol 2021; 147: 158–167. doi: 10.1016/j.jaci.2020.02.040 [DOI] [PubMed] [Google Scholar]
- 14.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011; 11: 723–737. doi: 10.1038/nri3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaur M, Singh D. Neutrophil chemotaxis caused by chronic obstructive pulmonary disease alveolar macrophages: the role of CXCL8 and the receptors CXCR1/CXCR2. J Pharmacol Exp Ther 2013; 347: 173–180. doi: 10.1124/jpet.112.201855 [DOI] [PubMed] [Google Scholar]
- 16.Berenson CS, Kruzel RL, Eberhardt E, et al. Phagocytic dysfunction of human alveolar macrophages and severity of chronic obstructive pulmonary disease. J Infect Dis 2013; 208: 2036–2045. doi: 10.1093/infdis/jit400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor AE, Finney-Hayward TK, Quint JK, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J 2010; 35: 1039–1047. doi: 10.1183/09031936.00036709 [DOI] [PubMed] [Google Scholar]
- 18.Marriott HM, Bingle CD, Read RC, et al. Dynamic changes in Mcl-1 expression regulate macrophage viability or commitment to apoptosis during bacterial clearance. J Clin Invest 2005; 115: 359–368. doi: 10.1172/JCI200521766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bewley MA, Preston JA, Mohasin M, et al. Impaired mitochondrial microbicidal responses in chronic obstructive pulmonary disease macrophages. Am J Respir Crit Care Med 2017; 196: 845–855. doi: 10.1164/rccm.201608-1714OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report: GOLD executive summary. Eur Respir J 2017; 49: 1700214. 10.1183/13993003.00214-2017. [DOI] [PubMed] [Google Scholar]
- 21.Higham A, Booth G, Lea S, et al. The effects of corticosteroids on COPD lung macrophages: a pooled analysis. Respir Res 2015; 16: 98. doi: 10.1186/s12931-015-0260-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Craig JE, Cliffe A, Garnett K, et al. Survival of nontypeable Haemophilus influenzae in macrophages. FEMS Microbiol Lett 2001; 203: 55–61. doi: 10.1111/j.1574-6968.2001.tb10820.x [DOI] [PubMed] [Google Scholar]
- 23.Khalaf RM, Lea SR, Metcalfe HJ, et al. Mechanisms of corticosteroid insensitivity in COPD alveolar macrophages exposed to NTHi. Respir Res 2017; 18: 61. doi: 10.1186/s12931-017-0539-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Metcalfe HJ, Lea S, Hughes D, et al. Effects of cigarette smoke on Toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages. Clin Exp Immunol 2014; 176: 461–472. doi: 10.1111/cei.12289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plumb J, Robinson L, Lea S, et al. Evaluation of glucocorticoid receptor function in COPD lung macrophages using beclomethasone-17-monopropionate. PLoS One 2013; 8: e64257. doi: 10.1371/journal.pone.0064257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukaida N. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol 2003; 284: L566–L577. doi: 10.1152/ajplung.00233.2002 [DOI] [PubMed] [Google Scholar]
- 27.Singh R, Mackay AJ, Patel AR, et al. Inflammatory thresholds and the species-specific effects of colonising bacteria in stable chronic obstructive pulmonary disease. Respir Res 2014; 15: 114. doi: 10.1186/s12931-014-0114-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy TF, Brauer AL, Aebi C, et al. Antigenic specificity of the mucosal antibody response to Moraxella catarrhalis in chronic obstructive pulmonary disease. Infect Immun 2005; 73: 8161–8166. doi: 10.1128/IAI.73.12.8161-8166.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy TF, Brauer AL, Schiffmacher AT, et al. Persistent colonization by Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004; 170: 266–272. doi: 10.1164/rccm.200403-354OC [DOI] [PubMed] [Google Scholar]
- 30.Keatings VM, Collins PD, Scott DM, et al. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153: 530–534. doi: 10.1164/ajrccm.153.2.8564092 [DOI] [PubMed] [Google Scholar]
- 31.Soler N, Ewig S, Torres A, et al. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur Respir J 1999; 14: 1015–1022. doi: 10.1183/09031936.99.14510159 [DOI] [PubMed] [Google Scholar]
- 32.Berenson CS, Wrona CT, Grove LJ, et al. Impaired alveolar macrophage response to Haemophilus antigens in chronic obstructive lung disease. Am J Respir Crit Care Med 2006; 174: 31–40. doi: 10.1164/rccm.200509-1461OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 2645–2653. doi: 10.1056/NEJMoa032158 [DOI] [PubMed] [Google Scholar]
- 34.Pettigrew MM, Ahearn CP, Gent JF, et al. Haemophilus influenzae genome evolution during persistence in the human airways in chronic obstructive pulmonary disease. Proc Natl Acad Sci USA 2018; 115: E3256–E3265. doi: 10.1073/pnas.1719654115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berenson CS, Kruzel RL, Eberhardt E, et al. Impaired innate immune alveolar macrophage response and the predilection for COPD exacerbations. Thorax 2014; 69: 811–818. doi: 10.1136/thoraxjnl-2013-203669 [DOI] [PubMed] [Google Scholar]
- 36.Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and gram-positive bacterial cell wall components. Immunity 1999; 11: 443–451. doi: 10.1016/S1074-7613(00)80119-3 [DOI] [PubMed] [Google Scholar]
- 37.Morris A, Beck JM, Schloss PD, et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med 2013; 187: 1067–1075. doi: 10.1164/rccm.201210-1913OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dockrell DH, Lee M, Lynch DH, et al. Immune-mediated phagocytosis and killing of Streptococcus pneumoniae are associated with direct and bystander macrophage apoptosis. J Infect Dis 2001; 184: 713–722. doi: 10.1086/323084 [DOI] [PubMed] [Google Scholar]
- 39.Dockrell DH, Marriott HM, Prince LR, et al. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J Immunol 2003; 171: 5380–5388. doi: 10.4049/jimmunol.171.10.5380 [DOI] [PubMed] [Google Scholar]
- 40.Cuconati A, Mukherjee C, Perez D, et al. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev 2003; 17: 2922–2932. doi: 10.1101/gad.1156903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Preston JA, Bewley MA, Marriott HM, et al. Alveolar macrophage apoptosis-associated bacterial killing helps prevent murine pneumonia. Am J Respir Crit Care Med 2019; 200: 84–97. doi: 10.1164/rccm.201804-0646OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bewley MA, Budd RC, Ryan E, et al. Opsonic phagocytosis in chronic obstructive pulmonary disease is enhanced by Nrf2 agonists. Am J Respir Crit Care Med 2018; 198: 739–750. doi: 10.1164/rccm.201705-0903OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belchamber KBR, Singh R, Batista CM, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J 2019; 54: 1802244. doi: 10.1183/13993003.02244-2018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00044-2022.SUPPLEMENT (691.5KB, pdf)