

Abstract
Carnitine metabolism is thought to be negatively correlated with the progression of hepatocellular carcinoma (HCC) and the specific molecular mechanism is yet to be fully elucidated. Here, we report that little characterized cysteine‐rich protein 1 (CRIP1) is upregulated in HCC and associated with poor prognosis. Moreover, CRIP1 promoted HCC cancer stem‐like properties by downregulating carnitine energy metabolism. Mechanistically, CRIP1 interacted with BBOX1 and the E3 ligase STUB1, promoting BBOX1 ubiquitination and proteasomal degradation, and leading to the downregulation of carnitine. BBOX1 ubiquitination at lysine 240 is required for CRIP1‐mediated control of carnitine metabolism and cancer stem‐like properties. Further, our data showed that acetylcarnitine downregulation in CRIP1‐overexpressing cells decreased beta‐catenin acetylation and promoted nuclear accumulation of beta‐catenin, thus facilitating cancer stem‐like properties. Clinically, patients with higher CRIP1 protein levels had lower BBOX1 levels but higher nuclear beta‐catenin levels in HCC tissues. Together, our findings identify CRIP1 as novel upstream control factor for carnitine metabolism and cancer stem‐like properties, suggesting targeting of the CRIP1/BBOX1/β‐catenin axis as a promising strategy for HCC treatment.
Keywords: cancer stemness, carnitine metabolism, cysteine‐rich intestinal protein 1, gamma‐butyrobetaine hydroxylase 1, hepatocellular carcinoma
Subject Categories: Cancer, Metabolism
Zinc finger protein CRIP1 antagonizes protective carnitine biosynthesis in liver cancer.
Introduction
Liver cancer is the sixth most commonly diagnosed cancer and ranks third as the leading cause of cancer‐related mortality worldwide. Hepatocellular carcinoma (HCC) is the most common type of primary liver malignancy, accounting for 75–85% of all cases (Li et al, 2021; Sung et al, 2021). Although significant progress has been achieved in the treatment of advanced HCC, including the use of multikinase inhibitor, sorafenib, and immune checkpoint inhibitor, nivolumab (Huang et al, 2020; Pinter et al, 2021), patients with advanced HCC still suffer from poor prognoses. The challenges and dilemmas of HCC treatment largely result from our limited understanding of the mechanisms of HCC progression.
Metabolic reprogramming is considered a hallmark of cancer. Metabolic adaptations allow cancer cells to meet demands for homeostasis and growth in response to a variety of cell‐extrinsic and cell‐intrinsic cues (Faubert et al, 2020). Altered lipid metabolism is the most prominent metabolic alteration in cancer, not only providing a structural basis for biological membranes but also regulating multiple energetic metabolisms and cell–cell communication (Snaebjornsson et al, 2020). Increasing evidence suggests that alterations in tumor lipid metabolism play an essential role in HCC progression (Satriano et al, 2019; Hall et al, 2021). The most important biological function of carnitine is transporting fatty acids into the mitochondria for β‐oxidation, a process to form acylcarnitine. The endogenous carnitine pool is composed of carnitine and various acylcarnitines (Bremer, 1983; Reuter & Evans, 2012). The level of acetylcarnitine has been reported to gradually decrease with HCC progression from stage T1 to T4 and may therefore be a diagnostic and prognostic biomarker of HCC (Lu et al, 2016). Accumulating evidence suggests that carnitine inhibits hepatocarcinogenesis via protection of mitochondria (Chang et al, 2005). In addition, carnitine supplementation has been shown to prevent diethylnitrosamine (DENA)‐induced hepatic carcinogenesis (Al‐Rejaie et al, 2009). However, the role of carnitine metabolism in regulating tumor growth remains to be clarified. Thus, in this study, we sought to determine the mechanism of carnitine metabolism in HCC progression.
Cysteine‐rich intestinal protein 1 (CRIP1) belongs to the LIM/double zinc finger protein family. LIM proteins function as DNA‐binding transcription factors or facilitate protein–protein interactions. CRIP1 was initially identified as a developmentally regulated intestinal gene and may be involved in intestinal zinc transport (Birkenmeier & Gordon, 1986; Hempe & Cousins, 1991). It has been reported to play a role in immune cell activation or differentiation (Hallquist et al, 1996; Davis et al, 1998), and was upregulated in several human malignancies, including gastric (Sun et al, 2021), cervical cancer (Zhang et al, 2018), and colorectal cancer (Zhang et al, 2019), and is involved in tumor progression. We previously reported that CRIP1 cysteine‐rich intestinal protein 1 suppresses apoptosis and chemosensitivity to 5‐fluorouracil in colorectal cancer through ubiquitin‐mediated Fas degradation (Zhang et al, 2019). However, the role of CRIP1 in HCC remains unclear. In this study, we also aimed to determine whether CRIP1 is involved in HCC and investigated the relationship between CRIP1 and carnitine metabolism.
Our findings from this present study show that CRIP1 promotes cancer stem‐like properties by downregulating carnitine metabolism. Gamma‐butyrobetaine hydroxylase 1 (BBOX1) is a critical enzyme catalyzing butyrobetaine to endogenously yield carnitine (Vaz et al, 1998). We also discovered that CRIP1 acted as a scaffold protein, interacted with BBOX1 and the E3 ligase STUB1, and accelerated ubiquitination and proteasomal degradation of the BBOX1 protein, leading to carnitine downregulation. Moreover, CRIP1 activated the Wnt/β‐catenin pathway by decreasing acetylcarnitine‐mediated β‐catenin acetylation, providing a promising therapeutic strategy for HCC.
Results
CRIP1 is highly expressed and associated with poor survival in HCC
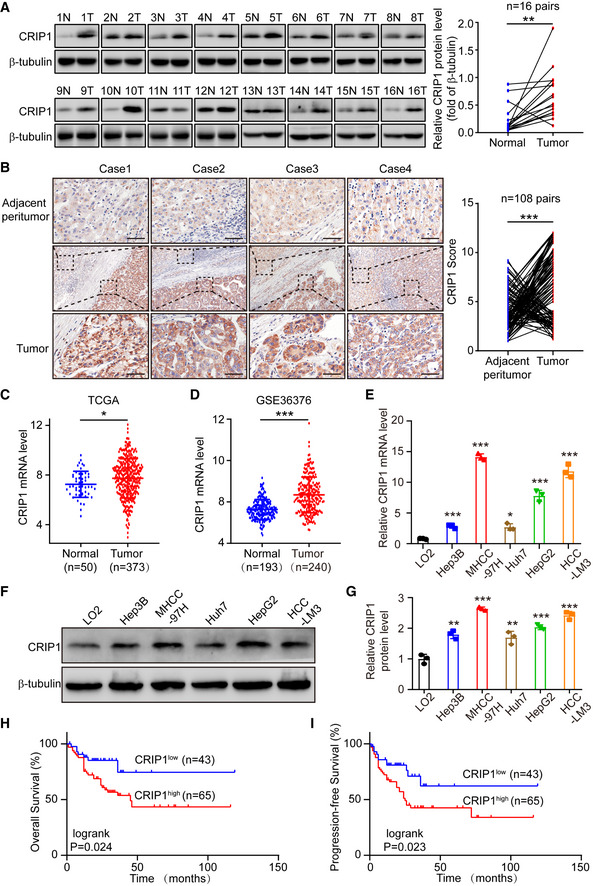
To explore the role of CRIP1 in HCC, CRIP1 expression was analyzed in human HCC samples. Western blot analysis showed that CRIP1 protein levels were elevated in HCC samples compared with corresponding nontumor tissues (Fig 1A). This result was also confirmed by immunohistochemical analysis of 108 paired HCC and adjacent nontumor tissues (Fig 1B). The Cancer Genome Atlas (TCGA)‐LIHC and Gene Expression Omnibus (GEO) databases further verified that CRIP1 mRNA levels were higher in HCC (Fig 1C and D). Next, we analyzed the mRNA and protein levels of CRIP1 in human HCC cell lines and normal LO2 hepatocytes and found that CRIP1 displayed a high expression level in most HCC cell lines compared with normal LO2 hepatocytes (Fig 1E–G). Clinicopathological analysis revealed that high CRIP1 expression was significantly associated with more aggressive tumor behaviors, including the tumor‐node‐metastasis (TNM) stage (P = 0.020), metastasis (P = 0.001), and differentiation (P = 0.007). However, no significant correlation was found between CRIP1 expression and other clinicopathological features, such as patient sex, age, tumor size, and tumor multiplicity (P > 0.05; Appendix Table S1). Further survival analysis showed that high expression of CRIP1 was associated with poor overall survival (OS) and progression‐free survival (PFS; Fig 1H and I). Together, these results clearly show that CRIP1 is upregulated in HCC tissues and HCC cells and associated with poor prognosis of HCC patients, suggesting that CRIP1 may be involved in the development and progression of HCC.
Figure 1. CRIP1 is upregulated in HCC and associated with poor survival.
-
AWestern blot analysis of CRIP1 protein expression in 16 paired HCC tumor tissues (T) and adjacent nontumor tissues (N). The graph represents CRIP1 protein expression normalized to GAPDH levels (right).
-
BRepresentative IHC images of CRIP1 protein expression in 108 paired HCC tumor tissues and adjacent nontumor tissues. Scale bars: 20 μm. Statistical analysis of immunohistochemical score of CRIP1 expression in HCC tumor tissues and paired adjacent tissues (right).
-
CCRIP1 mRNA expression in the TCGA‐LIHC dataset.
-
DmRNA levels of CRIP1 in the GSE36376 dataset from the GEO database.
-
E, FThe mRNA (E) and protein (F) levels of CRIP1 in HCC cell lines and normal LO2 hepatocytes.
-
GRelative CRIP1 protein level compared to normal LO2 hepatocytes.
-
H, IKaplan–Meier survival analyses of OS (H) and PFS (I) based on CRIP1 expression levels in 108 HCC patients.
Data information: Data represent the mean ± SD of at least three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001. Differences were tested using a two‐tailed, paired (A, B) and unpaired (C–E, G) Student's t‐test and the log‐rank test (H, I).
CRIP1 promotes cancer stem‐like properties in HCC
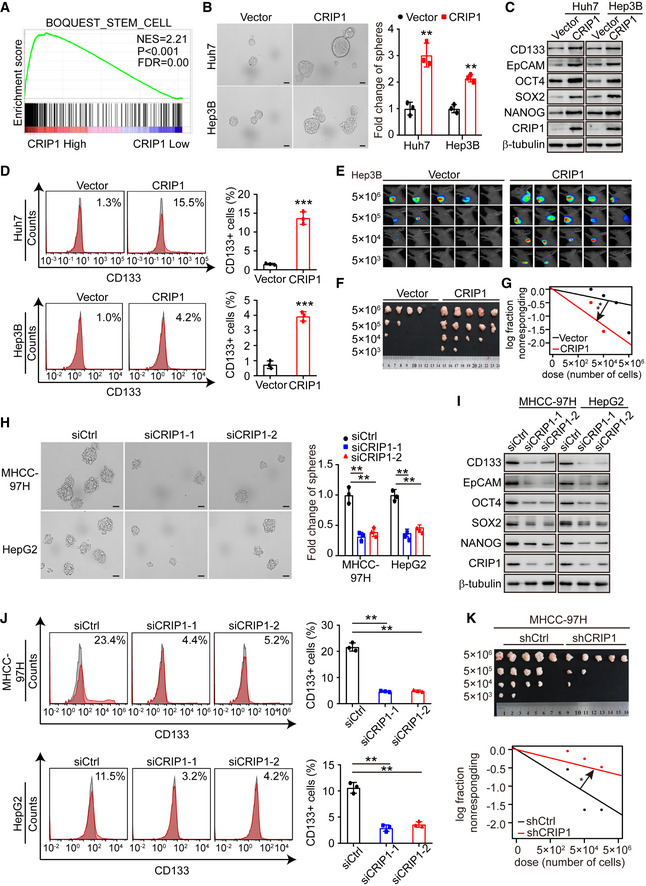
To explore the potential biological function of CRIP1 in HCC, gene set enrichment analysis (GSEA) was conducted based on CRIP1 expression from the TCGA‐LIHC database. The results revealed that high CRIP1 expression was positively associated with stem cell signaling pathways (Fig 2A). To confirm the effects of CRIP1 on the stemness features of HCC cells, we chose Huh7/Hep3B cells with relatively lower CRIP1 expression and MHCC‐97H/HepG2 cells with relatively higher CRIP1 expression (Fig 1E–G) to investigate gain‐ and loss‐of CRIP1 functional studies. HCC cell lines overexpressing CRIP1 were conducted by lentivirus infection. Simultaneously, knockdown of CRIP1 was performed by transiently transfecting CRIP1 siRNAs in HCC cells. Tumor sphere formation assay showed that CRIP1 overexpression in Huh7, Hep3B, MHCC‐97H, and HepG2 cells significantly increased the formation of spheres (Fig 2B and Appendix Fig S1A). HCC cancer stem cells have been reported to express high levels of CD133, EPCAM, and other markers including NANOG, OCT4, and SOX2 (Nio et al, 2017). Next, western blot and flow cytometric analysis were performed and the results showed that CRIP1 overexpression increased the protein levels of CD133, EpCAM, NANOG, OCT4, and SOX2, as well as the percentage of CD133+ cells (Fig 2C and D, and Appendix Fig S1B and C). In vitro and in vivo limiting dilution analysis also showed that CRIP1 overexpression enhanced the tumorigenic capacity of HCC cells (Fig 2E–G and Appendix Fig S1D). However, CRIP1 knockdown exerted the opposite effect (Fig 2H–K and Appendix Fig S1E–H). Taken together, these results suggest that CRIP1 promotes cancer stem‐like properties in HCC.
Figure 2. CRIP1 promotes HCC stemness features.
-
AGSEA from TCGA data mining.
-
BTumor sphere formation assay in CRIP1‐overexpressing Huh7 and Hep3B cells. The data are shown as the relative fold change spheres compared with the vector group (right). Scale bar: 50 μm.
-
CWestern blot analysis of HCC cancer stem cell markers in CRIP1‐overexpressing Huh7 and Hep3B cells.
-
DFlow cytometry analysis of the proportion of CD133+ cells in CRIP1‐overexpressing Huh7 and Hep3B cells.
-
E‐GIn vivo limiting dilution xenograft formation of Hep3B cells with CRIP1‐overexpressing or control cells (n = 5). In vivo bioluminescent images (E), bright‐field images (F), and the statistical analysis (G) of xenograft formation of Hep3B cells are presented.
-
HTumor sphere formation assay in CRIP1‐knockdown MHCC‐97H and HepG2 cells. The data are shown as the relative fold change spheres compared with the siCtrl group (right). Scale bar: 50 μm.
-
IWestern blot analysis of HCC cancer stem cell markers in CRIP1‐knockdown MHCC‐97H and HepG2 cells.
-
JFlow cytometry analysis of the proportion of CD133+ cells in CRIP1‐knockdown MHCC‐97H and HepG2 cells.
-
KIn vivo limiting dilution xenograft formation of CRIP1‐knockdown or control MHCC‐97H cells (n = 5). Bright‐field images (top), and the statistical analysis (bottom) of xenograft formation are presented.
Data information: Data represent the mean ± SD at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001. Differences were tested using an unpaired two‐tailed Student's t‐test (B, D, H, J). Limiting dilution assay calculations were performed using the ELDA software (G, K).
CRIP1 promotes cancer stem‐like properties via the downregulation of carnitine metabolism in HCC
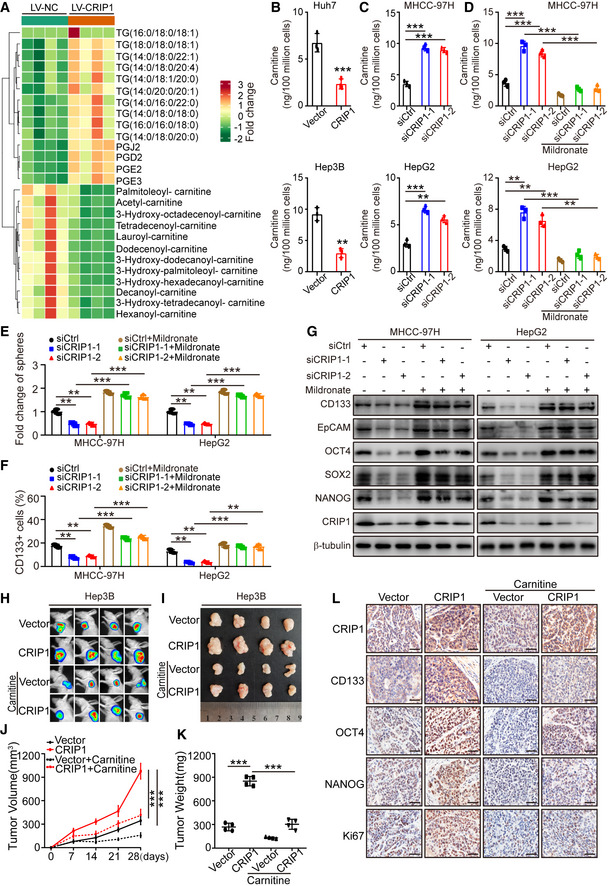
Accumulating evidence indicates that lipid metabolism alteration contributes to cancer stem‐like properties (Chen et al, 2016; Yi et al, 2018; Li et al, 2020; Capece et al, 2021). Therefore, we sought to determine whether lipid metabolism plays a role in CRIP1‐induced stem‐like properties. We performed lipidomics analysis on Huh7 cells with CRIP1 overexpression or controls. Among detected metabolites, 12 kinds of lipids were downregulated while 14 kinds of lipids were upregulated in the CRIP1‐overexpressing groups. Increased fatty acids and triglycerides were observed, but acylcarnitines decreased in the CRIP1‐overexpressing groups (Fig 3A and Appendix Fig S2A). Compared with more in‐depth literature on fatty acids and triglycerides in HCC (Li et al, 2015; Guri et al, 2017; Liu et al, 2018; Che et al, 2020), few reports described the role of acylcarnitine in HCC. Acylcarnitine generates from the binding of carnitine with carboxylic acids at its hydroxyl group, and carnitine is the main focus of our current research. Therefore, we tested whether CRIP1 regulated carnitine levels by enzyme‐linked immunosorbent assay (ELISA). We found that CRIP1 overexpression decreased carnitine levels in Huh7 and Hep3B cells whereas CRIP1 knockdown increased the level of carnitine (Fig 3B and C). These results suggest that CRIP1 downregulates carnitine metabolism.
Figure 3. CRIP1 downregulates carnitine to facilitate stemness in HCC.
-
ALipidomics analysis of CRIP1‐overexpressing Huh7 cells.
-
BCarnitine levels in CRIP1‐overexpressing Huh7 and Hep3B cells.
-
CCarnitine levels in CRIP1 knockdown MHCC‐97H and HepG2 cells.
-
DCarnitine levels in CRIP1 knockdown MHCC‐97H and HepG2 cells with or without 5 mM mildronate for 24 h.
-
ESphere formation ability in the indicated groups.
-
FFlow cytometric analysis revealed the proportions of CD133+ cells in the indicated groups.
-
GWestern blot analysis of cancer stem cell markers in the indicated groups.
-
H, IIn vivo bioluminescent images (H) and bright‐field images (I) of xenografts in mice with the indicated Hep3B cells. Carnitine was added to drinking water to a final concentration of 4.0 mg/ml.
-
J, KThe growth curve of tumors (J) and quantification of tumor weight (K) are presented (n = 4).
-
LIHC analysis of CRIP1 and HCC cancer stem cell markers. Scale bar: 20 μm.
Data information: Data represent the mean ± SD at least three independent experiments. **P < 0.01 and ***P < 0.001. Differences were tested using an unpaired two‐tailed Student's t‐test (B–F, J, K).
To determine the involvement of carnitine in the function of CRIP1 in HCC, pharmacological inhibition of γ‐butyrobetaine hydroxylase was used to inhibit carnitine biosynthesis in MHCC‐97H and HepG2 cells. Mildronate, a widely used specific inhibitor of γ‐butyrobetaine hydroxylase (Simkhovich et al, 1988), was applied to downregulate carnitine (Liao et al, 2020). The results revealed that the elevated levels of carnitine induced by CRIP1 knockdown were impaired by mildronate treatment (Fig 3D). Furthermore, mildronate restored the decreased sphere formation ability by CRIP1 knockdown (Fig 3E and Appendix Fig S2B). Moreover, mildronate treatment rescued the reduced percentages of CD133+ cells, and protein levels of CD133, EpCAM, OCT4, SOX2, and NANOG in CRIP1 knockdown cells (Fig 3F and G, and Appendix Fig S2C). To assess the role of carnitine function in vivo, we employed a xenograft tumor model using CRIP1 knockdown MHCC‐97H cells by CRIP1 shRNA. When the tumors reached 50–100 mm3, mildronate was intraperitoneally administered to the mice for 20 days. CRIP1 knockdown not only inhibited tumor growth but also suppressed cancer cell stemness in vivo. However, further administration of mildronate restored the suppressed effect of CRIP1 knockdown (Appendix Fig S2D–H).
To further validate the role of carnitine in CRIP1‐induced cancer stem‐like properties, we examined whether supplementation with carnitine could reverse the effect of CRIP1 overexpression in Huh7 and Hep3B cells. Sphere formation assay identified that carnitine supplement effectively restrained the elevated sphere formation ability in CRIP1‐overexpressing Huh7 and Hep3B cells (Appendix Fig S2I). Additionally, the increased percentages of CD133+ cells and protein levels of cancer stem cell markers by CRIP1 overexpression were remarkably decreased after treatment of carnitine (Appendix Fig S2J and K). To investigate the role of carnitine supplement in HCC in vivo, CRIP1‐overexpressing Hep3B cells were implanted subcutaneously into nude mice. When the tumors reached 50–100 mm3, mice were orally administrated with carnitine for 20 days. Compared with the vector group, CRIP1 overexpression significantly increased the growth and the weight of HCC xenograft tumors. However, this kind of effect was reversed by carnitine supplementation (Fig 3H–K). In addition, we performed immunohistochemistry (IHC) analysis to characterize HCC cancer stem cell markers in tumor xenograft samples. The staining of HCC cancer stem cell markers significantly enhanced in the CRIP1 overexpression groups, but this kind of phenomenon was also reversed by treatment with carnitine (Fig 3L). In summary, these results suggest that carnitine metabolism is essential for CRIP1‐mediated cancer stem‐like properties in HCC.
CRIP1 promotes interactions between BBOX1 and the E3 ligase STUB1 to accelerate BBOX1 degradation via the ubiquitin‐proteasome pathway
Next, we explored how CRIP1 regulated carnitine metabolism. Carnitine can be absorbed from the diet or endogenously synthesized through a five‐step enzymatic reaction. Gamma‐butyrobetaine hydroxylase 1 (BBOX1) is a critical enzyme in the last step of hydroxylation on butyrobetaine to yield carnitine (Bremer, 1983; Vaz et al, 1998). Therefore, we tested whether CRIP1 downregulated carnitine by regulating BBOX1. The qRT–PCR assay revealed that the mRNA level of BBOX1 was not changed in CRIP1‐overexpressing Huh7 and Hep3B cells or CRIP1 knockdown MHCC‐97H and HepG2 cells (Appendix Fig S3A and B). However, CRIP1 overexpression evidently downregulated BBOX1 protein levels (Fig 4A). In contrast, CRIP1 knockdown markedly elevated BBOX1 protein expression (Fig 4B), indicating that CRIP1 regulates the expression of BBOX1 post‐transcriptionally. To determine whether BBOX1 is essential for CRIP1‐induced carnitine metabolism, BBOX1 was overexpressed in CRIP1‐overexpressing Huh7 and Hep3B cells but silenced in CRIP1‐knockdown MHCC‐97H and HepG2 cells. We found that BBOX1 overexpression could restore the reduced BBOX1 protein expression and carnitine levels in CRIP1‐overexpressing Huh7 and Hep3B cells (Fig 4C and Appendix Fig S3C), while BBOX1 knockdown strongly repressed the elevated carnitine levels by CRIP1 knockdown in MHCC‐97H and HepG2 cells (Appendix Fig S3D–F). Next, we sought to validate whether BBOX1 contributed to CRIP1‐induced stemness. Sphere formation assay indicated that BBOX1 depletion restored the suppressed sphere formation ability in CRIP1 knockdown MHCC‐97H and HepG2 cells (Appendix Fig S3G). Consistently, flow cytometric and western blot analyses were performed and the results verified that BBOX1 depletion rescued the decreased percentage of CD133+ cells and protein levels of CD133, EpCAM, NANOG, OCT4, and SOX2 by CRIP1 knockdown (Appendix Fig S3H and I). These data suggest that BBOX1 is essential for CRIP1‐mediated carnitine metabolism and stemness.
Figure 4. CRIP1 promotes STUB1‐mediated degradation of BBOX1.
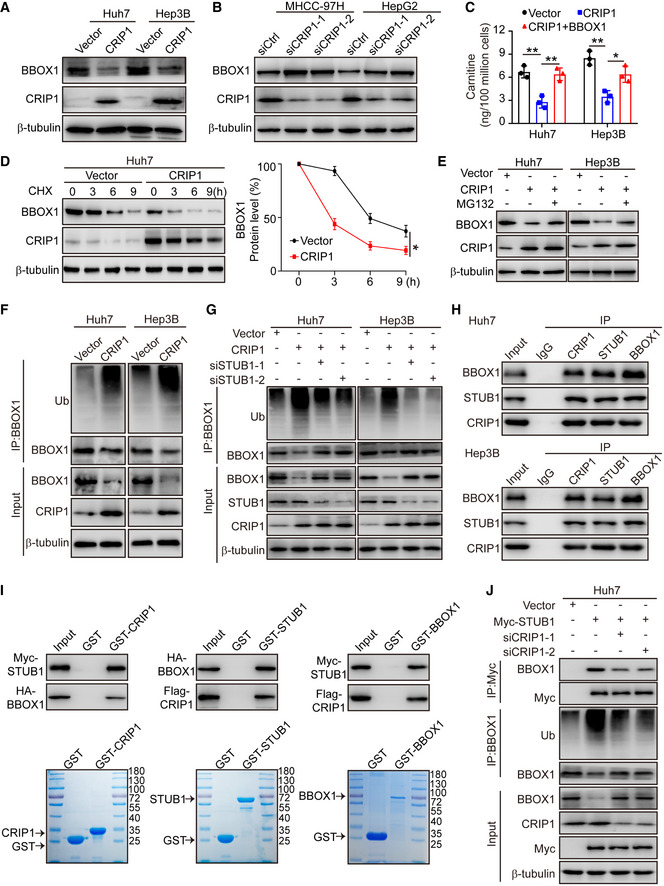
-
AWestern blot analysis of BBOX1 protein levels in CRIP1‐overexpressing Huh7 and Hep3B cells.
-
BWestern blot analysis of BBOX1 protein levels in CRIP1 knockdown MHCC‐97H and HepG2 cells.
-
CELISA showing carnitine levels in CRIP1‐overexpressing Huh7 and Hep3B cells transfected with BBOX1.
-
DWestern blot analysis of BBOX1 in CRIP1‐overexpressing Huh7 cells treated with CHX (100 μg/ml) for the indicated durations (left). The graph shows quantification of relative BBOX1 levels (right).
-
EWestern blot analysis of BBOX1 protein levels in CRIP1‐overexpressing Huh7 and Hep3B cells treated with MG132 (10 μM) for 10 h.
-
FCo‐immunoprecipitation and immunoblot analyses of BBOX1 ubiquitination in CRIP1‐overexpressing Huh7 and Hep3B cells.
-
GCo‐immunoprecipitation and immunoblot analyses of BBOX1 ubiquitination in CRIP1‐overexpressing Huh7 and Hep3B cells transfected with STUB1 siRNAs.
-
HCo‐immunoprecipitation and immunoblot analyses of the interaction among endogenous CRIP1, STUB1, and BBOX1 in Huh7 and Hep3B cells.
-
IGST pulldown assay analysis of the association of CRIP1 with BBOX1 and STUB1.
-
JCo‐immunoprecipitation and immunoblot analyses of the interaction between STUB1 and BBOX1 and BBOX1 ubiquitination in Huh7 cells transfected with STUB1‐Myc and CRIP1 siRNAs.
Data information: Data represent the mean ± SD of at least three independent experiments. *P < 0.05 and **P < 0.01. Differences were tested using an unpaired two‐tailed Student's t‐test (C, D).
To investigate how BBOX1 is post‐transcriptionally regulated by CRIP1, we performed protein half‐life experiments using cycloheximide (CHX) to inhibit new protein synthesis and found that higher CRIP1 levels led to a shorter half‐life of BBOX1 (Fig 4D). In contrast, the knockdown of CRIP1 increased BBOX1 stability (Appendix Fig S4A), indicating that CRIP1 downregulates BBOX1 through protein degradation. The 26S proteasome is responsible for most protein degradation in both the cytosol and the nucleus. Our findings showed that MG132, an inhibitor of the 26S protostome, could reverse the downregulation of the BBOX1 protein in CRIP1‐overexpressing Huh7 and Hep3B cells (Fig 4E), indicating that BBOX1 is regulated by CRIP1 through the proteasome pathway. Consistent with this result, CRIP1 overexpression increased the ubiquitination of BBOX1 in Huh7 and Hep3B cells (Fig 4F). In addition, ubiquitinated BBOX1 accumulated in the presence of MG132 while CRIP1 knockdown abolished the MG132‐induced accumulation of ubiquitinated BBOX1 in MHCC‐97H cells (Appendix Fig S4B). These results suggest that the CRIP1‐facilitated degradation of BBOX1 is dependent on the proteasome pathway.
Since CRIP1 lacks E3 ubiquitin ligase activity, we speculated that an E3 ligase may be involved in CRIP1‐mediated proteasomal degradation of BBOX1. STUB1 was estimated to have the highest confidence as an E3 ligase for BBOX1 in the UbiBrowser database (Appendix Fig S4C). We further confirmed that STUB1 promoted BBOX1 proteasomal degradation (Appendix Fig S4D). To explore whether CRIP1 facilitated degradation of BBOX1 through STUB1, we knocked down STUB1 in CRIP1‐overexpressing Huh7 and Hep3B cells and found that knockdown of STUB1 reversed the CRIP1‐enhanced ubiquitination in CRIP1‐overexpressing cells (Fig 4G), suggesting that CRIP1 promotes BBOX1 degradation through the specific E3 ligase STUB1.
LIM domain proteins functioning as scaffolding proteins can mediate protein–protein interactions (Dawid et al, 1998), and the STUB1 protein level was not affected by CRIP1 overexpression (Fig 4G). We speculated that CRIP1 may function as a scaffolding protein facilitating interactions between BBOX1 and the E3 ligase STUB1. Co‐immunoprecipitation confirmed the interaction among endogenous CRIP1, STUB1, and BBOX1 in Huh7 and Hep3B cells (Fig 4H). GST pull‐down assays identified that CRIP1 directly bound to STUB1 and BBOX1 (Fig 4I). We also observed that CRIP1 knockdown substantially attenuated the interaction between STUB1 and BBOX1 and the ubiquitination of BBOX1 in STUB1‐overexpressing Huh7 cells. However, the STUB1‐repressed BBOX1 protein levels were markedly enhanced after CRIP1 knockdown (Fig 4J). In summary, these data suggest that CRIP1 functions as a scaffold to mediate the STUB1/BBOX1 interaction and STUB1‐mediated degradation of BBOX1.
Ubiquitination of the K240 site in BBOX1 is crucial for CRIP1‐induced cancer stem‐like properties
To determine the specific sites of BBOX1 ubiquitination by the STUB1 E3 ligase, we performed mass spectrometric ubiquitination analysis of purified BBOX1 protein. We identified that the ubiquitination site at lysine 240 (K240) in BBOX1 was ubiquitinated (Fig 5A), which was also estimated to be a potential ubiquitination site in the BDM‐PUB and PhosphoSitePlus databases (Appendix Fig S5A and B). To determine whether K240 is a potential ubiquitination site for BBOX1 ubiquitinated by STUB1, we generated a ubiquitination‐defective mutant of BBOX1 (BBOX1 K240R) in which the lysine was changed to arginine at position 240. Compared with wild‐type (WT) BBOX1, the proteasomal degradation of BBOX1 K240R mutant by STUB1 was dramatically reduced in Huh7 and Hep3B cells (Fig 5B). Consistently, the protein level of WT BBOX1 was decreased by STUB1 in a dose‐dependent manner. In contrast, BBOX1 K240R mutant protein levels remained largely unchanged in increasing STUB1‐overexpressing Huh7 and Hep3B cells (Fig 5C). These results suggest that BBOX1 is ubiquitinated at K240 by STUB1.
Figure 5. BBOX1 ubiquitination at K240 mediated by STUB1 promotes cancer stem‐like properties induced by CRIP1.

-
ABBOX1 ubiquitination modification at Lys240 by MS/MS.
-
BCo‐immunoprecipitation and immunoblot analyses of BBOX1 ubiquitination in STUB1‐overexpressing Huh7 and Hep3B cells transfected with BBOX1‐WT or BBOX1 K240R mutant.
-
CWestern blot analysis of BBOX1‐WT or K240R mutant protein expression in Huh7 and Hep3B cells transfected with increasing amounts of STUB1.
-
D‐GCarnitine levels (D), tumor sphere formation ability and quantification of spheres (E), flow cytometric analysis of the proportion of CD133+ cells (F), and protein levels of HCC cancer stem cell markers (G) in STUB1‐overexpressing Huh7 and Hep3B cells transfected with BBOX1‐WT or BBOX1 K240R mutant. Scale bar: 50 μm.
Data information: Data represent the mean ± SD of at least three independent experiments. **P < 0.01 and ***P < 0.001. Differences were tested using an unpaired two‐tailed Student's t‐test (D–F).
We confirmed that CRIP1 ubiquitinated BBOX1 through the STUB1 E3 ligase (Fig 4G) and next examined whether the K240 site of BBOX1 was required for oncogenic functions of CRIP1 in HCC. The WT or K240R mutant of BBOX1 was transfected into CRIP1‐overexpressing Huh7 and Hep3B cells. Compared with WT BBOX1, K240R mutant resulted in higher levels of carnitine in CRIP1‐overexpressing Huh7 and Hep3B cells (Fig 5D), indicating that the K240 site is crucial for CRIP1‐mediated carnitine metabolism. Moreover, sphere formation assay showed that K240R mutant BBOX1 exhibited a stronger inhibition of sphere formation ability in CRIP1‐overexpressing Huh7 and Hep3B cells compared with WT BBOX1 (Fig 5E). Consistently, K240R mutant BBOX1 dramatically reduced the increased percentage of CD133+ cells and protein levels of CD133, EpCAM, NANOG, OCT4, and SOX2 (Fig 5F and G). Altogether, these findings suggest that the K240 site ubiquitination in BBOX1 mediated by STUB1 promotes CRIP1‐induced cancer stem‐like properties via downregulation of carnitine metabolism.
CRIP1 promotes cancer stem‐like properties by activating the Wnt/β‐catenin pathway through carnitine‐induced decrease of β‐catenin acetylation
We have described the detailed mechanism by which CRIP1 regulates carnitine metabolism by targeting BBOX1. Next, we explored how CRIP1 facilitates cancer stem‐like properties through carnitine metabolism in HCC. The primary function of carnitine is transporting esterified fatty acids into the mitochondrial matrix to facilitate β‐oxidation (Bremer, 1983). To determine whether β‐oxidation is involved in CRIP1‐mediated cancer stem‐like properties, sphere formation assay was performed and revealed that inhibition of CPT1A, which is a key enzyme of fatty acid β‐oxidation, did not rescue the inhibitory sphere formation ability induced by CRIP1 knockdown (Appendix Fig S6A and B), suggesting that CRIP1‐induced cancer stem‐like properties are independent of fatty acid β‐oxidation. Acetylcarnitine, a most abundant type of acylcarnitine, has been reported to be a candidate diagnostic and prognostic biomarker of hepatocellular carcinoma (Lu et al, 2016). We observed that mildronate inhibited the increased acetylcarnitine by CRIP1 knockdown (Fig 6A), showing that CRIP1 reduces acetylcarnitine through the downregulation of carnitine. Increasing evidence indicates that acetylcarnitine may act as a donor of acetyl groups to proteins (Madiraju et al, 2009; Nasca et al, 2013), and deacetylation of β‐catenin has been reported to help maintain the self‐renewal of liver cancer stem cells (Chen et al, 2019). To determine the involvement of the Wnt/β‐catenin pathway in the function of CRIP1 in HCC, GSEA was performed using TCGA‐LIHC dataset and the result revealed that high CRIP1 expression was positively associated with the Wnt pathway (Fig 6B). Furthermore, we treated CRIP1‐overexpressing cells with the Wnt/β‐catenin inhibitor iCRT3. Sphere formation assay showed that iCRT3 markedly reduced the enhanced sphere formation ability in CRIP1‐overexpressing Huh7 cells (Fig 6C). Similarly, the elevated percentage of CD133+ cells, and protein levels of CD133, EpCAM, NANOG, OCT4, and SOX2 by CRIP1 overexpression were attenuated by iCRT3 (Fig 6D and E), indicating that the Wnt/β‐catenin pathway is involved in CRIP1‐induced cancer stem‐like properties in HCC.
Figure 6. CRIP1 activates the Wnt/β‐catenin pathway by decreasing β‐catenin acetylation.
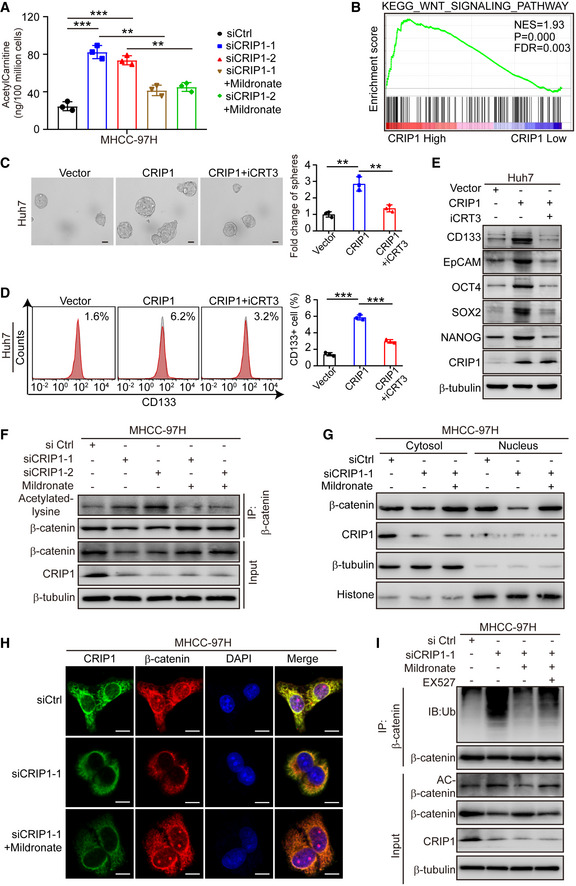
-
AELISA analysis of acetylcarnitine levels in CRIP1‐knockdown cells treated with mildronate (5 mM) for 24 h.
-
BGSEA based on CRIP1 expression in the TCGA‐LIHC database.
-
C, DRepresentative images of tumor sphere formation and quantification of spheres (C) and flow cytometric analysis of the proportion of CD133+ cells (D) in CRIP1‐overexpressing Huh7 cells with or without iCRT3 (50 μM) for 24 h. Scale bar: 50 μm.
-
EWestern blot analysis of HCC cancer stem cell markers in CRIP1‐overexpressing Huh7 cells with or without iCRT3 (50 μM) for 24 h.
-
FCo‐immunoprecipitation and immunoblot analyses of β‐catenin acetylation in CRIP1 knockdown MHCC‐97H cells treated with mildronate (5 mM) for 24 h.
-
GWestern blot analysis of cytoplasmic and nuclear fractions of β‐catenin in CRIP1 knockdown MHCC‐97H cells treated with mildronate (5 mM) for 24 h.
-
HConfocal microscopy images of β‐catenin in CRIP1 knockdown MHCC‐97H cells treated with mildronate (5 mM) for 24 h. Scale bar: 10 μm.
-
ICo‐immunoprecipitation and immunoblot analyses of β‐catenin ubiquitination in CRIP1 knockdown MHCC‐97H cells treated with mildronate (5 mM) and EX527 (20 μM) for 24 h.
Data information: Data represent the mean ± SD of at least three independent experiments. **P < 0.01 and ***P < 0.001. Differences were tested using an unpaired two‐tailed Student's t‐test (A, C and D).
To investigate whether CRIP1 could decrease β‐catenin acetylation via downregulation of acetylcarnitine, western blot was performed and revealed that β‐catenin acetylation was decreased in CRIP1‐overexpressing Huh7 cells (Appendix Fig S6C). Conversely, mildronate markedly inhibited the increase of β‐catenin acetylation in CRIP1‐knockdown MHCC‐97H cells (Fig 6F). As demonstrated by our data (Appendix Fig S6B), β‐oxidation is not involved in CRIP1‐mediated cancer stem‐like properties. However, to completely exclude the effect of β‐oxidation on Wnt/β‐catenin pathway, we further examined the levels of acetyl‐CoA, which is the production of β‐oxidation, in CRIP1 overexpressing and knockdown cells. The results determined that acetyl‐CoA was not influenced by CRIP1 (Appendix Fig S6D). Western blot analysis further indicated that inhibition of CPT1A did not rescue the elevated β‐catenin acetylation by CRIP1 knockdown (Appendix Fig S6E), suggesting that CRIP1 decreases β‐catenin acetylation by downregulation of carnitine metabolism rather than β‐oxidation.
Nuclear accumulation of β‐catenin is critical for downstream gene expression. Deacetylation of β‐catenin has been reported to facilitate β‐catenin nuclear translocation (Chen et al, 2019). We next explored whether CRIP1‐mediated β‐catenin acetylation affected nuclear accumulation of β‐catenin. The results showed that CRIP1 overexpression enhanced the ratio of nuclear β‐catenin and β‐catenin protein levels in both the cytoplasm and nucleus by CRIP1 overexpression (Appendix Fig S6F). However, decrease in β‐catenin nuclear accumulation, induced by CRIP1 knockdown, was restored by mildronate (Fig 6G). These findings were further confirmed by immunofluorescence (Fig 6H and Appendix Fig S6G), indicating that CRIP1 promotes β‐catenin nuclear accumulation via decreasing β‐catenin acetylation. The deacetylase SIRT1 has been reported to be involved in the deacetylation of β‐catenin and further prevents its proteasomal degradation (Chen et al, 2019). To determine whether decrease of β‐catenin acetylation by CRIP1‐induced carnitine metabolism also prevents its proteasomal degradation, we knocked down CRIP1 in MHCC‐97H cells treated with mildronate and EX527, an inhibitor of SIRT1. The results revealed that EX527 reversed the decrease of β‐catenin acetylation and ubiquitination in CRIP1‐knockdown cells treated with mildronate (Fig 6I), while the levels of carnitine and acetylcarnitine were not affected (Appendix Fig S6H), indicating that the decrease of β‐catenin acetylation via CRIP1‐mediated acetylcarnitine downregulation prevented its ubiquitination and proteasomal degradation. Taken together, these results suggest that CRIP1 decreases β‐catenin acetylation by downregulating acetylcarnitine to promote cancer stem‐like properties in HCC.
Clinical significance of the CRIP1‐BBOX1‐β‐catenin axis in HCC
Finally, we examined the protein levels of CRIP1 and BBOX1 using a tissue microarray (TAM) containing 68 paired human HCC tumors and adjacent normal tissues. IHC staining showed that CRIP1 was highly expressed in HCC tissue. In contrast, BBOX1 expression was significantly lower in HCC tissue than that in adjacent normal tissue (Appendix Fig S7A). This result was further confirmed by the mRNA expression of BBOX1 in the TCGA and GEO databases (Appendix Fig S7B and C). Moreover, patients with high BBOX1 expression had significantly better survival according to the TCGA database (Appendix Fig S7D), suggesting that BBOX1 is negatively associated with HCC progression.
We further examined the correlations between CRIP1 and BBOX1, β‐catenin, CD133, OCT4, and NANOG using above TAM. The results showed that high CRIP1 expression tended to be associated with low BBOX1 expression and high expression levels of β‐catenin, CD133, OCT4, and NANOG (Fig 7A), whereas low CRIP1 expression had the opposite effect in HCC tissues.
Figure 7. Clinical significance of the CRIP1‐BBOX1‐β‐catenin axis in HCC.

-
AIHC analysis of CRIP1, BBOX1, β‐catenin, CD133, OCT4, and NANOG using TAM (left). Scale bars: 20 μm. Correlation analysis of CRIP1 expression with BBOX1, β‐catenin, CD133, OCT4, and NANOG expression in HCC tissues (right).
-
BIn vivo bioluminescence images of in mice with CRIP1‐overexpressing Hep3B cells. EX527 (30 mg/kg/day) was intraperitoneally administered daily for 20 days.
-
CThe growth curves of tumors in the indicated groups (n = 4).
-
DIHC analysis of CRIP1, BBOX1, β‐catenin, CD133, OCT4, and NANOG in tumor xenograft samples. Scale bar: 20 μm.
-
EIHC analyses of Ki‐67 in the indicated groups.
-
FSchematic depicting the mechanism by which CRIP1 downregulates carnitine and promotes cancer cell stem‐like properties via STUB1‐induced BBOX1 proteasomal degradation, leading to decrease of β‐catenin acetylation and nuclear accumulation of β‐catenin.
Data information: Data represent the mean ± SD of at least three independent experiments. **P < 0.01 and ***P < 0.001. Chi‐square test was performed to determine the correlation (A). Differences were tested using an unpaired two‐tailed Student's t‐test (C, E).
We next explored the potential therapeutic effect of the CRIP1‐BBOX1‐β‐catenin axis in HCC. Lysine acetyltransferases (KATs) and lysine deacetylases (KDACs) are responsible for the dynamic balance of protein acetylation. KDAC inhibitors have emerged as attractive therapeutic candidates for treating several types of cancers (Menzies et al, 2016). Our findings showed that CRIP1 inhibited β‐catenin acetylation via the downregulation of BBOX1‐mediated carnitine. In addition, EX527, a KDAC inhibitor, rescued the decrease of β‐catenin acetylation in CRIP1 knockdown cells treated with mildronate (Fig 6I). To determine whether EX527 could reverse the role of the CRIP1‐BBOX1‐β‐catenin axis in HCC, we treated CRIP1‐overexpressing Huh7 and Hep3B cells with EX527. Sphere formation assay showed that EX527 inhibited the increased sphere‐forming ability by CRIP1 overexpression (Appendix Fig S8A). Furthermore, the upregulated percentage of CD133+ cells and protein levels of cancer stem cell markers were attenuated in CRIP1‐overexpressing Huh7 and Hep3B cells after EX527 treatment (Appendix Fig S8B and C). Moreover, EX527 decreased the increased growth and weight of HCC xenograft tumors induced by CRIP1 overexpression in vivo (Fig 7B and C, and Appendix Fig S8D and E). In addition, we performed immunohistochemistry analysis to characterize the effect of the CRIP1‐BBOX1‐β‐catenin axis on HCC cancer stem‐like properties in tumor xenograft samples. The results further confirmed that EX527 rescued the elevated effect of the CRIP1‐BBOX1‐β‐catenin axis on HCC stemness and proliferation ability (Fig 7D and E). In summary, these results indicated that the CRIP1‐BBOX1‐β‐catenin axis is activated in HCC and that inhibition of β‐catenin deacetylation may be a potential therapeutic target for HCC.
Discussion
Lipid metabolic reprogramming is crucial for cancer stemness maintenance. Wnt protein can be palmitoylated by lipid modification and activating the Wnt/β‐catenin pathway (Willert et al, 2003; Proffitt et al, 2013). In addition, YAP/TAZ can be stabilized by stearoyl‐CoA‐desaturase1, thus, activating the Hippo pathway (Noto et al, 2017), revealing that lipid metabolism is closely related to cancer stem‐like properties. In this study, we also confirmed that lipid metabolism was involved in cancer stem properties and revealed, for the first time, that CRIP1 promoted cancer stem‐like properties via downregulating the carnitine metabolism. Carnitine supplementation has been shown to be beneficial for several diseases (Malaguarnera et al, 2010; Enoki et al, 2017; Bruls et al, 2019). Recent studies also highlighted the antitumor effect of carnitine or acylcarnitine on cancers, including prostate cancer (Baci et al, 2019) and colon cancer (Roscilli et al, 2013). In addition, carnitine has been reported to inhibit hepatic carcinogenesis (Chang et al, 2005; Al‐Rejaie et al, 2009) and acetylcarnitine is a promising biomarker reflecting HCC diagnosis and progression (Lu et al, 2016). In contrast, acylcarnitine accumulation was reported to promote nonalcoholic fatty liver disease (NAFLD)‐related carcinogenesis (Fujiwara et al, 2018). The detailed mechanism underlying the effect of carnitine in different settings remains unclear and the related controversy also needs to be resolved. Our data showed that carnitine supplementation could inhibit the enhanced cancer stem‐like properties both in vitro and in vivo. Specifically, carnitine metabolism was downregulated by CRIP1, activating the Wnt/β‐catenin pathway and then promoting cancer stem‐like properties. The possible reason for the different role of carnitine in HCC could be that cancer cells can adapt to different environments to support their growth by metabolic reprogramming and different etiology‐related liver cancers may have heterogeneous metabolic patterns.
CRIP1, a member of the LIM/double‐zinc finger protein family, may function as a tumor driver or suppressor in different tumor types. Emerging evidence shows that CRIP1 is highly expressed in gastric cancer tissues, and patients with high CRIP1 expression have a poor prognosis (Sun et al, 2021). We previously discovered that CRIP1 suppresses apoptosis in colorectal cancer by promoting Fas ubiquitination and degradation (Zhang et al, 2019). Furthermore, CRIP1 was found to promote invasion and metastasis through excessive zinc‐induced epithelial‐mesenchymal transition in colorectal cancer (He et al, 2019). CRIP1 promoted cell migration and epithelial‐mesenchymal transition of cervical cancer by activating the Wnt/β‐catenin signaling pathway (Zhang et al, 2018). In contrast, CRIP1 may act as a tumor suppressor in the proliferation and invasion of breast cancer (Ludyga et al, 2013). However, the role of CRIP1 in HCC has not been reported. In this study, our data showed that CRIP1 acted as an oncogenic driver in HCC and that high CRIP1 expression was associated with poor prognosis. Furthermore, CRIP1 promoted cancer stem‐like properties by inhibiting carnitine metabolism. The biological role of CRIP1 in HCC was explored for the first time in this present study, and the relationship between CRIP1 and carnitine metabolism was also investigated in detail. BBOX1 is a critical enzyme catalyzing the precursor of carnitine butyrobetaine to yield carnitine (Vaz et al, 1998). In this study, we found that CRIP1 decreased BBOX1 protein, but not mRNA levels, and inhibited the stability of BBOX1. CRIP1 has been reported to facilitate the interaction between BRCA2 and RAD51, implying that CRIP1 may act as a scaffolding protein (Sun et al, 2021). Our novel findings revealed that CRIP1 interacted with BBOX1 and the E3 ligase STUB1, promoting the degradation of BBOX1 and downregulating carnitine. The data also provided evidence that CRIP1 is a scaffolding protein that regulates the stability of BBOX1. We further found that BBOX1 was ubiquitinated at K240 by STUB1 and that CRIP1 promoted cancer stem‐like properties depending on the ubiquitination of the K240 site in BBOX1. However, lysine 240 may be the main but not the only ubiquitination site in BBOX1 ubiquitinated by STUB1. Thus, the data presented provide new perspectives for understanding the mechanism of CRIP1‐induced carnitine metabolism.
The primary function of carnitine and acylcarnitine is to transport fatty acids into the mitochondrial matrix for β‐oxidation (Bremer, 1983). Fatty acid β‐oxidation may promote or inhibit HCC progression depending on different conditions. HIF‐1‐mediated suppression of fatty acid oxidation is critical for cancer progression (Huang et al, 2014). In this study, we found that CRIP1 promoted cancer stem‐like properties independent of β‐oxidation. Further, CRIP1 decreased acetylcarnitine by downregulating carnitine. Acetylcarnitine may act as a tumor suppressor by inhibiting crucial proinflammatory and proangiogenic factors (Baci et al, 2018; Baci et al, 2019). However, we found that acetylcarnitine downregulation by CRIP1 overexpression reduces β‐catenin acetylation, leading to nuclear accumulation of β‐catenin, and thus, facilitating cancer stem‐like properties. Acetylcarnitine has been suggested to provide acetyl groups to p65/NF‐κB and histone proteins for acetylation (Madiraju et al, 2009; Nasca et al, 2013). Our data are the first to indicate that acetylcarnitine may be an acetyl donor for β‐catenin and demonstrate a novel approach for activating the Wnt/β‐catenin pathway through carnitine metabolism.
KDAC inhibitors have been used for malignant tumor treatment, including vorinostat (Rodriguez et al, 2020), romidepsin (O'Connor et al, 2019), chidamide (Jiang et al, 2019), entinostat (Pili et al, 2017), and more. SIRT1 acts as the deacetylase for the deacetylation of β‐catenin and promotes HCC cancer stem‐like properties (Chen et al, 2019). In this study, we observed that EX527, a specific inhibitor of SIRT1, disrupted the role of the CRIP1‐BBOX1‐β‐catenin axis in HCC. However, EX527 decreased tumor growth and Ki‐67‐positive‐cells proportionally to a similar extent in CRIP1‐overexpressing and vector control cells, indicating that EX527 may be efficacious but not selective for CRIP1‐overexpressing HCC tumors. EX527 has been suggested to inhibit cancer progression in a mouse model when combined with other drugs (Chen et al, 2017). Notably, EX527 has been applied in a clinical trial for endometriosis treatment (NCT04184323). Our results showed that the downregulation of carnitine activated the Wnt/β‐catenin pathway via decreasing β‐catenin acetylation, thereby providing a theoretical foundation for the clinical application of KDAC inhibitors.
To our knowledge, the present study represents the first report investigating the role and mechanism of CRIP1 in HCC. The regulation of carnitine metabolism in HCC was also described in detail. Meanwhile, our data provide new perspectives on the CRIP1/BBOX1/β‐catenin axis in cancer stem‐like properties. However, some limitations should be considered when interpreting our findings. First, human HCC tissue samples with long‐term prognostic data were only collected and analyzed, and the sera of these patients could not be acquired. The relationship between carnitine and prognosis could not be further analyzed without serum samples. Second, CRIP1 interacted with BBOX1 and the E3 ligase STUB1, but the specific binding domain remains unclear and still needs to be addressed in further investigations.
In conclusion, we showed a previously unrecognized role of CRIP1 in the downregulation of carnitine by promoting BBOX1 degradation via the E3 ubiquitin ligase STUB1 which decreased β‐catenin acetylation and induced cancer stem‐like properties in HCC (Fig 7F). Furthermore, this study provides a molecular basis and rationale for targeting the CRIP1‐BBOX1‐β‐catenin axis as a novel therapeutic strategy to treat HCC.
Materials and Methods
Cell culture and reagents
HEK293T cell, human normal hepatocyte cell line LO2, and the human liver cancer cell lines Huh7, MHCC‐97H, Hep3B, and HepG2 were purchased from Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). HCCLM3 cells were purchased from the Liver Cancer Institute of Fudan University (Shanghai, China). Short tandem repeat (STR) cell line authentication measures were applied to ensure the integrity of HCC cell lines. All cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco, USA) with 10% fetal bovine serum at 37°C in a humidified atmosphere containing 5% CO2. Mildronate, MG‐132, iCRT3, carnitine, and EX527 were purchased from Selleckchem Inc. Cycloheximide was purchased from MedChemExpress (MCE). The antibodies used are listed in Appendix Table S2.
Plasmids, siRNAs, shRNAs, and transfection
Flag‐CRIP1, WT and Mut HA‐BBOX1, His‐BBOX1, Myc‐STUB1, plasmids were purchased from Vigene Biosciences Inc. (Jinan, China). Plasmids were amplified and purified with the EndoFree Plasmid Mini Kit (TIANGEN, Beijing, China). siRNAs targeting CRIP1 and STUB1 were synthesized by Gene Pharma Company (Shanghai, China). shRNAs targeting BBOX1 and CPT1A were synthesized by Tsingke Technology Company (Shanghai, China). All plasmids, siRNAs and shRNAs were transfected into cells with Lipofectamine 3000 (Invitrogen, USA). All siRNA and shRNAs sequences are listed in Appendix Table S3.
Human HCC samples
Primary HCC paraffin‐embedded biopsy tissues (n = 108) were obtained from the Affiliated Tumor Hospital of Guangzhou Medical University (Guangzhou, China). The data of patients with detailed clinical characteristics and long‐term follow‐up data from January 2010 to December 2019 were retrieved. Prior to the surgery, no patients received any local or systemic anticancer treatments. HCC tumor specimens (n = 68) for tissue microarrays (TMAs) were collected at the Nangfang Hospital, Southern Medical University (Guangzhou, China). This study was approved by the Southern Medical University for Biomedical Research Ethics Committee, and all of the patients provided informed consent.
IHC
Paraffin‐embedded tissue slides were baked at 60°C for 2 h, deparaffinized in xylene and rehydrated in alcohol. Endogenous peroxidase was blocked with 3% hydrogen peroxide for 10 min. Antigen retrieval was performed in citrate buffer, pH 6.0, in a steamer for 5 min. The slides were then blocked with 5% BSA for 1 h and incubated at 4°C overnight with the indicated primary antibodies. The following day, the tissue sections were incubated with HRP‐conjugated secondary antibody for 1 h and detected using DAB substrates for 1 min followed by hematoxylin staining.
The staining was evaluated by two pathologists blinded to the study results. The staining scoring criteria were as follows: staining intensity (negative = 0, weak = 1, moderate = 2 and strong = 3) and the extent of stained cells (0% = 0, 1–24% = 1, 25–49% = 2, 50–74% = 3 and 75–100% = 4). The multiple of the intensity and extent score was used as the final score (0–12).
Lentiviral infection
LV‐CRIP1 constructed with Ubi‐MCS‐SV40‐firefly‐luciferase‐IRES‐puromycin vector and LV‐CRIP1 shRNA constructed with hU6‐MCS‐Ubiquitin‐firefly‐luciferase‐IRES‐puromycin vector were obtained from GeneChem (Shanghai, China). HCC cells were infected with viral supernatants containing 5 μg/ml polybrene according to the manufacturer's instructions and selected with 2 μg/ml puromycin treatment. The expression levels of CRIP1 were confirmed by RT–qPCR and western blot.
Lipidomics analysis
Huh7 cells expressing LV‐NC and LV‐CRIP1 were collected and extracted with chloroform:methanol (2:1, 1 ml). The aqueous and organic layers were separated by centrifugation (12,000 g, 10 min). The organic lipid‐containing layer was analyzed by untargeted liquid chromatography‐mass spectrometry (LC–MS), performed on a UOLC equipped Sciex 5500 QTRAP (AB Sciex, Boston, USA). Differential lipids were identified in the LV‐CRIP1 group.
Enzyme‐linked immune‐sorbent assay (ELISA)
For carnitine and acetylcarnitine measurement, cells were washed with cold PBS twice and diluted with PBS to a concentration of 100 million/ml. The samples were placed at −20°C overnight. Destruction of the cell membrane was carried out after repeated freeze–thaw cycles. Then, the cells were centrifuged at 4°C and 5,000 × g for 5 min. The supernatant was analyzed using an ELISA Kit to detect carnitine or acetylcarnitine levels (Cloud‐Clone, Wuhan, China) and acetyl‐CoA levels (Elabscience, Wuhan, China), according to the manufacturer's instructions.
Immunoprecipitation
Cells were washed with ice‐cold PBS and lysed in ice‐cold IP buffer (20 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, and 1% Triton X‐100) containing protease inhibitor cocktail and PMSF. The cell lysates were centrifuged at 12,000 g for 30 min at 4°C, then incubated with protein A/G magnetic beads for pre‐clearing at 4°C for 2 h. Next, the samples were incubated with fresh protein A/G magnetic beads and 1–4 μg antibody followed by incubation for 8–12 h at 4°C. Immunoprecipitates were extensively washed with IP wash buffer (10 mM Tris at pH 7.5, 150 mM NaCl, 1 mM EDTA, and 0.2% Triton X‐100) and subjected to immunoblotting with the indicated antibodies.
Ubiquitination assays
For the polyubiquitinated BBOX1 and β‐catenin assay, cells were harvested and lysed with RIPA buffer containing a proteinase inhibitor. The diluted lysates were then subjected to IP of endogenous BBOX1 and β‐catenin protein. The levels of BBOX1 and β‐catenin ubiquitination were detected by immunoblotting with an anti‐Ub antibody.
GST pull‐down assay
The sequences encoding CRIP1, BBOX1, and STUB1 were cloned into a pGEX‐4T‐1 vector containing the open reading frame (ORF) of the GST tag. The GST‐CRIP1, GST‐BBOX1, and GST‐STUB1 recombinant plasmids were transformed into Escherichia coli BL21 (DE3). The expression of GST or GST‐fusion proteins was induced by IPTG when the cells reached an optical density of approximately 0.6. Then, the cells were harvested and an appropriate amount of PBS supplemented with protease inhibitor cocktail was added to perform ultrasonic fracture in an ice‐water bath. Next, the mixture was centrifuged at 4°C and 12,000 × g for 30 min. The clear lysate was incubated with glutathione sepharose in a rotating incubator at 4°C overnight. The proteins were purified after washing three times with PBS. Then, purified GST‐fusion protein or the GST control was combined with the cell lysates of HEK293T cells transfected with Flag‐CRIP1, HA‐BBOX1, and Myc‐STUB1 at 4°C overnight, respectively. The bead‐bound protein complexes were washed four times and were detected by western blot.
Mass spectrometry analysis of the BBOX1 ubiquitylation site
For ubiquitination site mapping of BBOX1, purified BBOX1‐His fusion protein from HEK293T cell line by Ni‐NTA column was separated by SDS–PAGE and subjected to in‐gel digestion with trypsin (Promega). Collected MS/MS raw files were converted to MGF files using the Proteome Discoverer (version 1.4) and analyzed using the Mascot 2.3 search engine (Matrix Science, Boston, USA). Ubiquitin modification on the lysine and missed tryptic cleavage at the modified site were used, in which the data were queried against the Uniprot human database and the amino acid sequence of BBOX1.
Xenograph mouse model
BALB/c‐nu/nu mice (male, bodyweight: 18–22 g, 4 weeks old) were purchased from the Guangdong Medical Laboratory Animal Center. The mice were maintained in a specific pathogen‐free environment. All animal experiments were performed according to the Guide for the Institutional Animal Ethical Committee, Experimental Animal Center of Southern Medical University. Approximately 1 × 107 CRIP1‐overexpressin Hep3B cells or CRIP1‐knockdown MHCC‐97H cells were subcutaneously injected into the flanks of the nude mice and were then monitored for the development of tumors by measurements of their tumor weight, tumor length (L), and width (W). Tumor size (V) was evaluated by calipers every 7 days with the modified ellipsoidal formula: V = L × W2/2. For EX527 or carnitine treatment. When the tumors reached a volume of approximately 50–100 mm3, the control/CRIP1 mice were randomly divided into groups and treated with EX527 (30 mg/kg) dissolved in 5% DMSO/PEG300 via intraperitoneal injection or carnitine was added to their drinking water to obtain a final concentration of 4.0 mg/ml for 20 days. For mildronate treatment, mildronate (400 mg/kg) was given through intraperitoneal injection for 20 days. On day 28 after HCC cells injection, the mice were intraperitoneally injected with luciferin (150 mg/kg per mouse) to detect their tumor size using the In Vivo Imaging System (FX PRO, Bruker, Billerica, MA, USA). At the end of the experiment, the tumor tissues were formalin‐fixed and paraffin‐embedded for histological analysis.
For in vivo limiting dilution assays, Hep3B cells were transduced with control (CV146‐Vector) or LV‐CRIP1 (CV146‐CRIP1) lentivirus. Then, the cells were injected subcutaneously into 4‐week‐old BALB/c nude mice in a limited dilution series (5 × 106, 5 × 105, 5 × 104, 5 × 103 cells/mouse). On day 42 after cells injection, the tumor volume was detected by bioluminescent imaging. Then, the mice were sacrificed, the tumors were isolated, and the tumor formation incidence was calculated using extreme limiting dilution analysis (ELDA).
Real‐time quantitative PCR
Total cellular RNA was extracted using TRIzol reagent (Takara). A total of 1 μg purified RNA in each sample was used for cDNA synthesis using PrimeScript™ RT Master Mix (Takara). Reverse transcription products were amplified by a LightCycler 480 Fast Real‐Time PCR detection System (Roche) using SYBR Green PCR Master Mix (Takara). The PCR conditions were as follows: 95°C for 30 s, followed by 40 cycles of denaturation at 95°C for 5 s, and annealing at 60°C for 30 s. Data were calculated using method. The gene expression levels were normalized to that of GAPDH. The primer pairs used in this study are listed in Appendix Table S4.
Western blot analysis
Cells were washed with ice‐cold PBS and lysed in RIPA lysis buffer supplemented with protease inhibitor cocktail and PMSF. For nuclear protein extraction, the assays were performed according to the manufacturer's instructions for the Nuclear and Cytoplasmic Extraction Kit (Invitrogen). The lysates were centrifuged at 12,000 g for 30 min. The protein concentration was measured by BCA assay (Invitrogen), and proteins were denatured immediately before SDS–PAGE and then transferred to PVDF membranes. After blocking in TBST buffer containing 5% nonfat milk, membranes were detected with the indicated primary antibodies overnight at 4°C. The next day, the membranes were incubated with secondary HRP‐conjugated antibodies for 1 h at room temperature after washing. The staining was visualized using ECL substrate with a 4200SF detector (Tanon) according to the manufacturer's directions.
Protein half‐life assay
To evaluate whether CRIP1 destabilized the BBOX1 protein, Huh7 cells expressing CRIP1 and MHCC‐97H cells transfected with CRIP1 siRNAs were treated with 50 μg/ml CHX (Sigma–Aldrich, USA) for 0 to 9 h before collection. Endogenous BBOX1 levels were detected by western blotting.
Sphere formation assay
For the sphere formation assay, 2 × 103 cells were seeded into ultralow attachment 6‐well plates. The cells were grown in serum‐free DMEM/F‐12 medium (Invitrogen) supplemented with 4 μg/ml insulin (Sigma‐Aldrich), 20 ng/ml EGF (PeproTech), and 20 ng/ml basic FGF (Invitrogen). After incubation for 10 days, the sphere number was counted by microscopy. For in vitro limiting dilution assays, cells were plated in 96‐well plates at 10, 25, 50, 100, or 200 cells per well, with eight replicates for each cell number. Ten days later, wells without spheres were counted. Limiting dilution analysis was performed using extreme limiting dilution analysis (http://bioinf.wehi.edu.au/software/elda/).
Flow cytometry analysis
Hepatocellular carcinoma cells were harvested and washed twice with cold PBS. The cells were incubated with PE anti‐human CD133 antibody for 30 min in the dark at room temperature. Then, the cells were washed with PBS three times by centrifugation at 500 × g for 5 min and resuspended in 0.5 ml of cell staining buffer. The samples were then analyzed by a FACSCalibur flow cytometer (BD Biosciences, CA, USA) and FlowJo software.
Immunofluorescence (IF) staining
The cells plated in glass‐bottom dishes were washed with PBS and fixed in 4% paraformaldehyde for 15 min. After fixation, the cells were permeabilized with 0.2% Triton X‐100 for 10 min and blocked with 2% BSA for 1 h. The cells were incubated with specific primary antibodies at 4°C overnight. After washing three times with PBS, the cells were stained with secondary antibodies. Nuclei were counterstained with DAPI. Images were captured using a laser confocal microscope (FV1000. Olympus).
Data mining
RNA array datasets from the NCBI/GEO database (https://www.ncbi.nlm.nih.gov/gds/) and TCGA‐LIHC database (https://xenabrowser.net/datapages/?dataset=TCGA.LIHC) were used to analyze the expression of CRIP1 and BBOX1 in normal and lung tumor tissues. Gene set enrichment analysis (GSEA) of CRIP1 was performed using RNA‐seq sets from the TCGA‐LIHC database. According to the expression and median value of the CRIP1 gene, HCC patients were divided into two groups: a high expression group and a low expression group. The survival curve of BBOX1 was drawn from the Kaplan–Meier Plotter database (http://kmplot.com/analysis/).
Statistical analysis
All experiments, except for those involving mice, were performed in at least three independent biological replicates, with technical replicates for each experiment. Data are expressed as the mean ± SD. For data with a normal distribution, unpaired or paired two‐tailed Student's t‐tests were used to compare the significance of differences between two groups of independent samples. For the in vivo experiments related to animals, the number of biological replicates is 4 or 5. Animals were allocated to control experimental groups using a blinding and randomization method. Association of CRIP1 expression with clinicopathological data from HCC patients was assessed using chi‐square test. The survival rate was determined using the Kaplan–Meier method. Statistics were performed by GraphPad Prism 8.0. A P‐value less than 0.05 was considered statistically significant. All P‐values are indicated in the figures (#P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001).
Ethics approval and consent to participate
All experiments involving patients are endorsed by the Ethics Committee of Southern Medical University and complied with the Declaration of Helsinki. No informed consent was required because data were going to be analyzed anonymously. All animal experiments involved ethical and humane treatment under a license from the Guangdong Provincial Bureau of Science.
Author contributions
Jing Wang: Investigation; methodology. Yan Zhou: Investigation; methodology. Donghui Zhang: Resources; visualization. Weiyi Zhao: Resources; visualization. Yishi Lu: Methodology. Chaoqun Liu: Data curation. Wandie Lin: Data curation. Yujie Zhang: Formal analysis. Kunling Chen: Formal analysis. Hui Wang: Conceptualization; funding acquisition; project administration; writing – review and editing. Liang Zhao: Conceptualization; supervision; funding acquisition; project administration; writing – review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
JW and YZ performed the experiments, analyzed data, and drafted the manuscript. WZ and DZ collected HCC tissues and performed IHC. CL, WL helped perform animal experiments. YZ and KC contributed to data analysis. YL conducted the lentiviral infection. HW and LZ designed experiments and revised the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Acknowledgements
We would like to thank AJE [aje.com] for English language editing. This work was supported by the National Natural Science Foundation of China (No. 81972813), Guangdong Basic and Applied Basic Research Foundation (2019A1515010974, 2020A1515011389, 2021A1515111190), and Beijing Xisike Clinical Oncology Research Foundation (Y‐Roche2019/2‐0025).
The EMBO Journal (2022) 41: e110218
Data availability
The lipidomics raw data was lost due to machine transitions from Thermo C30 Column and Sciex QTRAP to Thermo Vanquish and Thermo Orbitrap Exploris 120. The processed data has been included as Dataset EV1.
References
- Al‐Rejaie SS, Aleisa AM, Al‐Yahya AA, Bakheet SA, Alsheikh A, Fatani AG, Al‐Shabanah OA, Sayed‐Ahmed MM (2009) Progression of diethylnitrosamine‐induced hepatic carcinogenesis in carnitine‐depleted rats. World J Gastroenterol 15: 1373–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baci D, Bruno A, Bassani B, Tramacere M, Mortara L, Albini A, Noonan DM (2018) Acetyl‐l‐carnitine is an anti‐angiogenic agent targeting the VEGFR2 and CXCR4 pathways. Cancer Lett 429: 100–116 [DOI] [PubMed] [Google Scholar]
- Baci D, Bruno A, Cascini C, Gallazzi M, Mortara L, Sessa F, Pelosi G, Albini A, Noonan DM (2019) Acetyl‐L‐carnitine downregulates invasion (CXCR4/CXCL12, MMP‐9) and angiogenesis (VEGF, CXCL8) pathways in prostate cancer cells: Rationale for prevention and interception strategies. J Exp Clin Cancer Res 38: 464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenmeier EH, Gordon JI (1986) Developmental regulation of a gene that encodes a cysteine‐rich intestinal protein and maps near the murine immunoglobulin heavy chain locus. Proc Natl Acad Sci USA 83: 2516–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer J (1983) Carnitine—metabolism and functions. Physiol Rev 63: 1420–1480 [DOI] [PubMed] [Google Scholar]
- Bruls YM, de Ligt M, Lindeboom L, Phielix E, Havekes B, Schaart G, Kornips E, Wildberger JE, Hesselink MK, Muoio D et al (2019) Carnitine supplementation improves metabolic flexibility and skeletal muscle acetylcarnitine formation in volunteers with impaired glucose tolerance: a randomised controlled trial. EBioMedicine 49: 318–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capece D, D'Andrea D, Begalli F, Goracci L, Tornatore L, Alexander JL, Di Veroli A, Leow SC, Vaiyapuri TS, Ellis JK et al (2021) Enhanced triacylglycerol catabolism by carboxylesterase 1 promotes aggressive colorectal carcinoma. J Clin Invest 131: e137845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Nishikawa M, Nishiguchi S, Inoue M (2005) L‐carnitine inhibits hepatocarcinogenesis via protection of mitochondria. Int J Cancer 113: 719–729 [DOI] [PubMed] [Google Scholar]
- Che L, Chi W, Qiao Y, Zhang J, Song X, Liu Y, Li L, Jia J, Pilo MG, Wang J et al (2020) Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 69: 177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, Hess S, Machida K (2016) NANOG metabolically reprograms tumor‐initiating stem‐like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab 23: 206–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Zhang B, Xu H, Sun Y, Shi Y, Luo Y, Jia H, Wang F (2017) Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK‐1775‐induced DNA damage and apoptosis. Oncogene 36: 6863–6872 [DOI] [PubMed] [Google Scholar]
- Chen X, Huan H, Liu C, Luo Y, Shen J, Zhuo Y, Zhang Z, Qian C (2019) Deacetylation of beta‐catenin by SIRT1 regulates self‐renewal and oncogenesis of liver cancer stem cells. Cancer Lett 463: 1–10 [DOI] [PubMed] [Google Scholar]
- Davis BA, Blanchard RK, Lanningham‐Foster L, Cousins RJ (1998) Structural characterization of the rat cysteine‐rich intestinal protein gene and overexpression of this LIM‐only protein in transgenic mice. DNA Cell Biol 17: 1057–1064 [DOI] [PubMed] [Google Scholar]
- Dawid IB, Breen JJ, Toyama R (1998) LIM domains: multiple roles as adapters and functional modifiers in protein interactions. Trends Genet 14: 156–162 [DOI] [PubMed] [Google Scholar]
- Enoki Y, Watanabe H, Arake R, Fujimura R, Ishiodori K, Imafuku T, Nishida K, Sugimoto R, Nagao S, Miyamura S et al (2017) Potential therapeutic interventions for chronic kidney disease‐associated sarcopenia via indoxyl sulfate‐induced mitochondrial dysfunction. J Cachexia Sarcopenia Muscle 8: 735–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Solmonson A, DeBerardinis RJ (2020) Metabolic reprogramming and cancer progression. Science 368: eaaw5473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara N, Nakagawa H, Enooku K, Kudo Y, Hayata Y, Nakatsuka T, Tanaka Y, Tateishi R, Hikiba Y, Misumi K et al (2018) CPT2 downregulation adapts HCC to lipid‐rich environment and promotes carcinogenesis via acylcarnitine accumulation in obesity. Gut 67: 1493–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, Jenoe P, Heim MH, Riezman I, Riezman H et al (2017) mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 32: e812 [DOI] [PubMed] [Google Scholar]
- Hall Z, Chiarugi D, Charidemou E, Leslie J, Scott E, Pellegrinet L, Allison M, Mocciaro G, Anstee QM, Evan GI et al (2021) Lipid remodeling in hepatocyte proliferation and hepatocellular carcinoma. Hepatology 73: 1028–1044 [DOI] [PubMed] [Google Scholar]
- Hallquist NA, Khoo C, Cousins RJ (1996) Lipopolysaccharide regulates cysteine‐rich intestinal protein, a zinc‐finger protein, in immune cells and plasma. J Leukoc Biol 59: 172–177 [DOI] [PubMed] [Google Scholar]
- He G, Zhu H, Yao Y, Chai H, Wang Y, Zhao W, Fu S, Wang Y (2019) Cysteine‐rich intestinal protein 1 silencing alleviates the migration and invasive capability enhancement induced by excessive zinc supplementation in colorectal cancer cells. Am J Transl Res 11: 3578–3588 [PMC free article] [PubMed] [Google Scholar]
- Hempe JM, Cousins RJ (1991) Cysteine‐rich intestinal protein binds zinc during transmucosal zinc transport. Proc Natl Acad Sci USA 88: 9671–9674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LT, Li X, Zhang L, Sun L, He X, Zhong X, Jia D, Song L, Semenza GL et al (2014) HIF‐1‐mediated suppression of acyl‐CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep 8: 1930–1942 [DOI] [PubMed] [Google Scholar]
- Huang A, Yang XR, Chung WY, Dennison AR, Zhou J (2020) Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther 5: 146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Li W, Hu X, Zhang Q, Sun T, Cui S, Wang S, Ouyang Q, Yin Y, Geng C et al (2019) Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor‐positive breast cancer (ACE): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol 20: 806–815 [DOI] [PubMed] [Google Scholar]
- Li H, Feng Z, He ML (2020) Lipid metabolism alteration contributes to and maintains the properties of cancer stem cells. Theranostics 10: 7053–7069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Huang Q, Long X, Zhang J, Huang X, Aa J, Yang H, Chen Z, Xing J (2015) CD147 reprograms fatty acid metabolism in hepatocellular carcinoma cells through Akt/mTOR/SREBP1c and P38/PPARalpha pathways. J Hepatol 63: 1378–1389 [DOI] [PubMed] [Google Scholar]
- Li N, Wu P, Shen Y, Yang C, Zhang L, Chen Y, Wang Z, Jiang J (2021) Predictions of mortality related to four major cancers in China, 2020 to 2030. Cancer Commun 41: 404–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C, Zhang Y, Fan C, Herring LE, Liu J, Locasale JW, Takada M, Zhou J, Zurlo G, Hu L et al (2020) Identification of BBOX1 as a therapeutic target in triple‐negative breast cancer. Cancer Discov 10: 1706–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Liang Y, Song R, Yang G, Han J, Lan Y, Pan S, Zhu M, Liu Y, Wang Y et al (2018) Long non‐coding RNA NEAT1‐modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol Cancer 17: 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Li N, Gao L, Xu YJ, Huang C, Yu K, Ling Q, Cheng Q, Chen S, Zhu M et al (2016) Acetylcarnitine is a candidate diagnostic and prognostic biomarker of hepatocellular carcinoma. Cancer Res 76: 2912–2920 [DOI] [PubMed] [Google Scholar]
- Ludyga N, Englert S, Pflieger K, Rauser S, Braselmann H, Walch A, Auer G, Hofler H, Aubele M (2013) The impact of cysteine‐rich intestinal protein 1 (CRIP1) in human breast cancer. Mol Cancer 12: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju P, Pande SV, Prentki M, Madiraju SR (2009) Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics 4: 399–403 [DOI] [PubMed] [Google Scholar]
- Malaguarnera M, Gargante MP, Russo C, Antic T, Vacante M, Malaguarnera M, Avitabile T, Li Volti G, Galvano F (2010) L‐carnitine supplementation to diet: a new tool in treatment of nonalcoholic steatohepatitis—a randomized and controlled clinical trial. Am J Gastroenterol 105: 1338–1345 [DOI] [PubMed] [Google Scholar]
- Menzies KJ, Zhang H, Katsyuba E, Auwerx J (2016) Protein acetylation in metabolism – metabolites and cofactors. Nat Rev Endocrinol 12: 43–60 [DOI] [PubMed] [Google Scholar]
- Nasca C, Xenos D, Barone Y, Caruso A, Scaccianoce S, Matrisciano F, Battaglia G, Mathe AA, Pittaluga A, Lionetto L et al (2013) L‐acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc Natl Acad Sci USA 110: 4804–4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nio K, Yamashita T, Kaneko S (2017) The evolving concept of liver cancer stem cells. Mol Cancer 16: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noto A, De Vitis C, Pisanu ME, Roscilli G, Ricci G, Catizone A, Sorrentino G, Chianese G, Taglialatela‐Scafati O, Trisciuoglio D et al (2017) Stearoyl‐CoA‐desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 36: 4573–4584 [DOI] [PubMed] [Google Scholar]
- O'Connor OA, Ozcan M, Jacobsen ED, Roncero JM, Trotman J, Demeter J, Masszi T, Pereira J, Ramchandren R, Beaven A et al (2019) Randomized phase III study of alisertib or Investigator's choice (selected single agent) in patients with relapsed or refractory peripheral T‐cell lymphoma. J Clin Oncol 37: 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pili R, Quinn DI, Hammers HJ, Monk P, George S, Dorff TB, Olencki T, Shen L, Orillion A, Lamonica D et al (2017) Immunomodulation by Entinostat in renal cell carcinoma patients receiving high‐dose interleukin 2: A multicenter, single‐arm, phase I/II trial (NCI‐CTEP#7870). Clin Cancer Res 23: 7199–7208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinter M, Scheiner B, Peck‐Radosavljevic M (2021) Immunotherapy for advanced hepatocellular carcinoma: a focus on special subgroups. Gut 70: 204–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proffitt KD, Madan B, Ke Z, Pendharkar V, Ding L, Lee MA, Hannoush RN, Virshup DM (2013) Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT‐driven mammary cancer. Cancer Res 73: 502–507 [DOI] [PubMed] [Google Scholar]
- Reuter SE, Evans AM (2012) Carnitine and acylcarnitines: pharmacokinetic, pharmacological and clinical aspects. Clin Pharmacokinet 51: 553–572 [DOI] [PubMed] [Google Scholar]
- Rodriguez CP, Wu QV, Voutsinas J, Fromm JR, Jiang X, Pillarisetty VG, Lee SM, Santana‐Davila R, Goulart B, Baik CS et al (2020) A phase II trial of pembrolizumab and Vorinostat in recurrent metastatic head and neck squamous cell carcinomas and salivary gland cancer. Clin Cancer Res 26: 837–845 [DOI] [PubMed] [Google Scholar]
- Roscilli G, Marra E, Mori F, Di Napoli A, Mancini R, Serlupi‐Crescenzi O, Virmani A, Aurisicchio L, Ciliberto G (2013) Carnitines slow down tumor development of colon cancer in the DMH‐chemical carcinogenesis mouse model. J Cell Biochem 114: 1665–1673 [DOI] [PubMed] [Google Scholar]
- Satriano L, Lewinska M, Rodrigues PM, Banales JM, Andersen JB (2019) Metabolic rearrangements in primary liver cancers: cause and consequences. Nat Rev Gastroenterol Hepatol 16: 748–766 [DOI] [PubMed] [Google Scholar]
- Simkhovich BZ, Shutenko ZV, Meirena DV, Khagi KB, Mezapuke RJ, Molodchina TN, Kalvins IJ, Lukevics E (1988) 3‐(2,2,2‐Trimethylhydrazinium)propionate (THP)‐‐a novel gamma‐butyrobetaine hydroxylase inhibitor with cardioprotective properties. Biochem Pharmacol 37: 195–202 [DOI] [PubMed] [Google Scholar]
- Snaebjornsson MT, Janaki‐Raman S, Schulze A (2020) Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab 31: 62–76 [DOI] [PubMed] [Google Scholar]
- Sun H, Zhou R, Zheng Y, Wen Z, Zhang D, Zeng D, Wu J, Huang Z, Rong X, Huang N et al (2021) CRIP1 cooperates with BRCA2 to drive the nuclear enrichment of RAD51 and to facilitate homologous repair upon DNA damage induced by chemotherapy. Oncogene 40: 5342–5355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71: 209–249 [DOI] [PubMed] [Google Scholar]
- Vaz FM, van Gool S, Ofman R, Ijlst L, Wanders RJ (1998) Carnitine biosynthesis: identification of the cDNA encoding human gamma‐butyrobetaine hydroxylase. Biochem Biophys Res Commun 250: 506–510 [DOI] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR 3rd, Nusse R (2003) Wnt proteins are lipid‐modified and can act as stem cell growth factors. Nature 423: 448–452 [DOI] [PubMed] [Google Scholar]
- Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, Zhou Y, Zeng Z, Peng S, Li X et al (2018) Emerging role of lipid metabolism alterations in cancer stem cells. J Exp Clin Cancer Res 37: 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhou R, Zhang W, Yao X, Li W, Xu L, Sun X, Zhao L (2019) Cysteine‐rich intestinal protein 1 suppresses apoptosis and chemosensitivity to 5‐fluorouracil in colorectal cancer through ubiquitin‐mediated Fas degradation. J Exp Clin Cancer Res 38: 120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LZ, Huang LY, Huang AL, Liu JX, Yang F (2018) CRIP1 promotes cell migration, invasion and epithelial‐mesenchymal transition of cervical cancer by activating the Wnt/betacatenin signaling pathway. Life Sci 207: 420–427 [DOI] [PubMed] [Google Scholar]