

Abstract
SARS-CoV-2 is a novel betacoronavirus that has caused the global health crisis known as COVID-19. The implications of mitochondrial dysfunction with COVID-19 are discussed as well as deregulated mitochondria and inter-organelle functions as a posited comorbidity enhancing detrimental outcomes. Many environmental chemicals and endocrine-disrupting chemicals can do damage to mitochondria and cause mitochondrial dysfunction. During infection, SARS-CoV-2 via its binding target ACE2 and TMPRSS2 can disrupt mitochondrial function. Viral genomic RNA and structural proteins may also affect the normal function of mitochondria-endoplasmic reticulum-Golgi apparatus. Drugs considered for treatment of COVID-19 should consider effects on organelles including mitochondria functions. Mitochondria self-balance and clearance via mitophagy are important in SARS-CoV-2 infection, which indicate monitoring and protection of mitochondria against SARS-CoV-2 are important. Mitochondrial metabolomic analysis may provide new indicators of COVID-19 prognosis. A better understanding of the role of mitochondria during SARS-CoV-2 infection may help to improve intervention therapies and better protect mitochondrial disease patients from pathogens as well as people living with poor nutrition and elevated levels of socioeconomic stress and environmental chemicals.
Keywords: SARS-CoV-2, mitochondria, dysfunction, pollutant, socioeconomic, stress, exposome, metabolomics, immunosenescence
Graphical Abstract
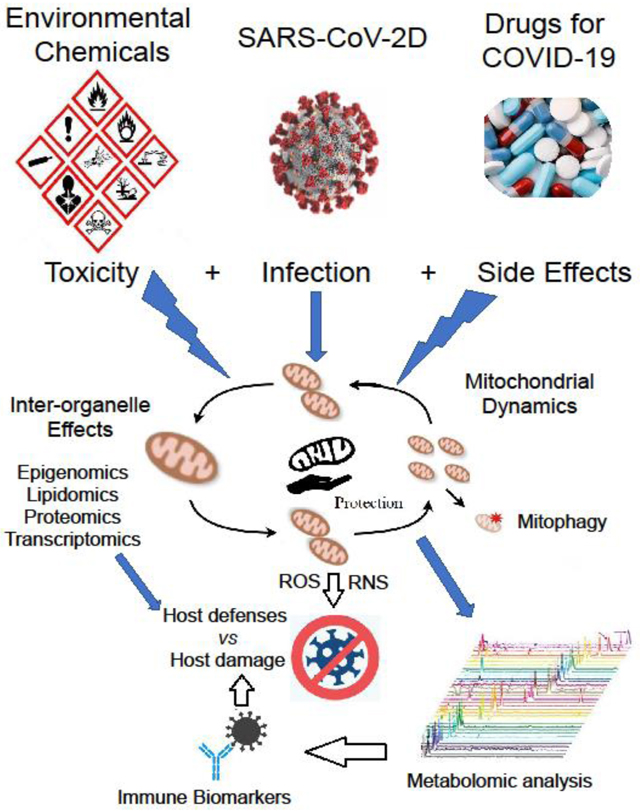
Introduction
The newly arrived novel betacoronavirus (β-CoV) named SARS-CoV-2, which causes COVID-19, is creating a more global pandemic than its β-CoV relatives that caused Severe Acute Respiratory Syndrome (SARS-CoV) in 2012 and Middle East Respiratory Syndrome (MERS-CoV) in 2002–2004.[1] Unlike SARS-CoV and MERS-CoV, SARS-CoV-2 is causing more deaths with a high incidence of acute respiratory distress syndrome (ARDS), clotting blood, and systemic dysfunctions, including organ failures, because of sepsis. Considering the dramatic increased number of infections and deaths worldwide, COVID-19 has been a global health crisis and is currently a public health challenge to humans, which requires better understanding of susceptibility and the responsible mechanisms. People living in polluted areas may be more easily infected with SARS-CoV-2 and environmental stressors, including crowded housing, socioeconomic stress and exposure to pollutants, and inadequate supply of nutrients may be conditions making COVID-19 symptoms more severe. We speculate that mitochondrial dysfunction due to the attack of environmental chemicals (ECs), such as heavy metals, endocrine-disrupting chemicals (EDCs), some drugs, and socioeconomic stress is a key factor for the higher susceptibility to SARS-CoV-2 in some populations. The hazards of environmental stresses on mitochondria and cellular coping with stress were reviewed. [2] Environmental stresses may exacerbate mitochondrial dysfunction, which is associated with inflammaging processes.[3] Environmental toxicants and psychological stress can promote chronic inflammation.[4] Older individuals, who are more susceptible to COVID-19, often have inflammaging and accumulation of senescent cells, and beside environmental stresses, senescence of the immune system (immunosenescence) causes ‘inflammaging’.[5] Sirtuins, a family of deacetylases, which require NAD+ as a cofactor, generally decline with age affecting mitochondrial biogenesis, stress resistance, and cellular senescence.[6] SIRT1 of the sirtuins is implicated in mitochondrial biogenesis and mitophagy, which likely are dependent on tissue differences, exposome, and physiological conditions.[7]
Mitochondrial disease (MtD) occurs due to mutations of mitochondrial DNA (mtDNA) and/or nuclear DNA (nDNA) that affect a heterogeneous group of disorders. MtD is generally thought to occur from inherited genetic defects. MtD manifestation may begin early or be expressed at different times throughout life. Variations in the clinical appearances of an MtD, its severity, and the different organs that may be affected have made diagnosis and treatment difficult.[8, 9] A delay in clinical symptoms has suggested involvement of toxic events from environmental pollutants such as cigarette smoke, which may evoke or enhance MtD.[10,11] Children of white and non-Hispanic ancestry with higher median income have been reported to have higher prevalence of MtD;[12] however, the phenotypes and genetic abnormalities of MtD have been increasing since earlier investigations in 1988,[13] which has raised the question of environmental influences.[14]
Mitochondria provide energy as adenosine triphosphate (ATP) to living cells and are vital for all cell activities, including immune cell trafficking, activities to kill infected cells and produce antibodies to aid capture and neutralize virus. ATP is mainly generated from the tricarboxylic acid cycle (Krebs cycle) and oxidative phosphorylation in mitochondria. When there’s a shift toward glycolysis (the “Warburg effect”) as occurs with viral infections, immune system dysregulation, sepsis and cancer,[15,16] we posit that mitochondria play a critical role either in aiding or hindering SARS-CoV-2 infection. Differential effects from mitochondria are suggested to be dependent on a person’s genetics, “exposome”,[17] and biology. During COVID-19’s pathophysiological effects, mitochondria-rich organs like brain, muscles, lung, heart, kidney, liver and the gastrointestinal system in need of more energy are vulnerable,[18] and mitochondrial dysfunction in one or more of these organs may be at high risk for severe COVID-19 outcomes from combinations of multiple environmental stressors.
Mitochondrial dysfunction results from loss of mitochondrial morphology and function and is recognized as a major pathogenic event. Mitochondria dysfunction includes inhibited mitochondrial biogenesis, which is needed for increased energy demand, loss of mitochondrial membrane potential (MMP or ∆Ψm), inhibition of oxidative phosphorylation (OXPHOS), and release of free oxygen radicals. In this review, the attacks of environmental stressors and SARS-CoV-2 on mitochondrial structures and functions during prognosis of COVID-19, impacts of drugs used to mitigate COVID-19, and meaningfulness of monitoring and protecting mitochondria against SARS-CoV-2 are presented, including involvement of inflammaging and cellular senescence. A better understanding of the role of mitochondria and inter-organelle functions during a SARS-CoV-2 infection might help to improve intervention therapies and better protect patients with MtD [19,20] from pathogens. It is important to consider use of different drugs since some might exacerbate MtD.[21] Due to the importance of mitochondria in many pathophysiological conditions, means to target compounds to mitochondria with attachment of lipophilic cations for diagnostic and therapeutic purposes are being investigated.[22]
Effects of ECs on mitochondria
ECs have access to the human body from polluted air, water, food, soil, dust, and personal care and other manufactured products. There are ECs and their metabolites in blood, urine, tissues and breast milk of many humans. As a chemical messenger system, hormones of the endocrine system target many organs throughout the body. EDCs include a wide range of natural or artificial substances that mimic and interfere with the function of the endocrine system producing adverse effects on normal physiological functions including neurological, cardiovascular and immunologic activities. Adipose tissue is considered an endocrine organ,[23] and many persistent organic pollutants (POPs) are housed in adipose tissue creating increased inflammation. White and brown adipose tissue (WAT and BAT) are affected by POPs.[24–26] Many of the ECs are included in industrial pollutions, wastewater, fertilizers, detergents, cosmetics, pesticides, and antibiotic residues that can directly or indirectly via the food chain from trace accumulation be taken into the human body. Here, we briefly mention the effects of some common ECs on mitochondria. EC induce an accumulation of mutations, which increase with age, that affect mitochondrial functions.[27,28]
Benzene
White blood cell (WBC) and platelet counts are reduced in benzene-exposed workers, especially in those with high exposure. Benzene exposure caused increased mitochondrial mass and mtDNA copy number in response to the oxidative stress induced by benzene. An increase in mtDNA, with exposed workers was inversely correlated with WBC counts.[29] Thus, these mitochondrial changes can be useful biomarkers of benzene toxicity in hematopoietic tissues and may relate to development of a leukemia.[30] Benzene metabolites through induction of oxidative stress or aryl hydrocarbon receptor (AhR) dysregulation inhibit immunosurveillance, which increases susceptibility to infection as well as cancers, especially leukemias.[31] The major mechanism of benzene metabolites on leukemogenesis is due to chromosomal alterations [32] likely related to generation of oxidative stress.[33] Mitochondrial cytochrome c oxidase subunit I variants at T6392C, G6962A and C7196A in platelet mtDNA occurred with chronic benzene poisoning.[34] In vitro analysis of mitochondria demonstrated bioactivation of benzene to toxic metabolites, which bound to DNA causing an inability of mitochondrial RNA polymerase to transcribe the genome with subsequent inhibition of translation.[35] Benzene can also inhibit mitochondrial translation most probably because transcription is inhibited, resulting in a lack of mRNA and a subsequent disaggregation of polysomes.
Formaldehyde (FAL)
FAL induces cytogenetic and immunologic effects and cytotoxicity;[36–39] it exerts G2/M arrest, and reduction in the time spent in S phase as well as induction of oxidative stress.[40] FAL can greatly reduce cellular ATP levels and MMP in a dose-dependent manner, together with the inhibition of the mitochondrial electron transport chain (ETC) involving complex I (NADH dehydrogenase) and complex IV (cytochrome c oxidase), oxidative stress-sensitive aconitase and activation of mitophagy. FAL or its products can also increase the nuclear fragmentation and the activities of the apoptosis family of caspase-9, caspase-3, caspase-7 in apoptosis and necrosis progression.[41] Increased DNA double-strand breaks (DSBs) in mitochondria associated with mitochondrial structural rearrangements were also observed after exposure to FAL. Exposure to FAL causes immune-mediated inflammation via Nuclear factor of activated T cells (NFAT), T-cell receptor calcium (TCR-calcium), Hypoxia-inducible factor 1 alpha (HIF-1), AP-1 and p38MAPK.[42]
Bisphenol A (BPA)
As one of the EDCs, BPA is widely used for production of polycarbonate plastics. It can induce oxidative stress, apoptosis and inflammation via interference with mitochondrial functions. Exposure to BPA will decrease the activities of mitochondrial ETC complexes, reduced glutathione level and the activity of superoxide dismutase; it also increases lipid peroxidation (LPO), protein oxidation, and mitochondrial superoxide generation due to its enhancement of oxidative stress.[43] An in vivo study reported dietary BPA uptake could increase the levels of oxidative stress indicators such as reactive oxygen species (ROS) and reactive nitrogen species (RNS) in mouse serum, colon and liver tissues.[44] Antioxidant indicators and total antioxidant capacity (T-AOC), as well as proinflammatory cytokines (IL-1β, IL-6, IL-8 and TNF-α) were also significantly reduced in the serum, colon, and liver tissues in the BPA group. Mitochondria-encoded genes like the Cox family and ND family, and mtDNA copies, mitochondrial respiratory chain complex activity and ATP content were significantly reduced in the colon and liver tissues of the BPA mice. In contract, gene expression and enzyme activity of caspase-3, −8, −9 and −10 were increased.[44] An in vitro study indicated that BPA triggered dysfunction and apoptosis of rat insulinoma (INS-1) cells via mitochondrial defects in β cells, with ATP depletion, cytochrome c release, loss of mitochondrial mass and membrane potential, and abnormal expression of Tfam, Nd4l, Atp6, citrate synthase, Ucp2 and Ogdh, which are involved in normal mitochondrial function and metabolism.[45]
Toxic metals
Toxic metals are widely distributed in the environment with high toxicity. Most common toxic metals threatening the public health are As, Cd, Cr, Pb, and Hg with dose-dependent and accumulation toxicity.[46] They can directly inhibit the mitochondrial functions such as metabolic processes, activity of antioxidants and free radical scavengers, increase the free radicals and Ca2+ overload, release cytochrome c (CytC) and cleaved caspase 3, damage mitochondrial structure and functions. [47] Mitochondrial electron transport chain (mtETC) is also affected in heavy-metal-induced neurotoxicity.[48] During the process of mitochondrial dysfunction, toxic metals usually promote inflammation, such as Pb, which can promote inflammatory response and secretion of proinflammatory cytokines, and increase B-cell activation/proliferation and skew T-cell help (Th) cells to Th2 cells. [49,50] Mitochondrial control of the cytosolic Ca2+ level may be upset by toxic heavy metals. [51] The thirteen genes of mitochondria affect the cellular proteasome; therefore, metal-induced epigenetic modifications may have profound effects on health,[52] in part, by disrupting the metals needed for proper mitochondrial function.
Diesel Exhaust Particles (DEPs)
DEPs are a major contributor toward air pollution and ill health, especially for people living near highways and areas with truck traffic. Experimental aerosol exposure of mice demonstrated reduced respiration and increased hydrogen peroxide production by pulmonary macrophages. [53,54] The particle itself induces oxidative stress, e.g., silica nanoparticles without any adherent chemicals induces oxidative stress in vitro with human peripheral blood mononuclear cells, [55] and DEPs usually are accompanied by metals (Cr, Cu, Fe, Pb, Zn) and volatile organics, [56] which can further enhance oxidative stress and inflammation.
Many different types of ECs and other environmental stressors can pervert mitochondrial functions and disrupt inter-organelle activities including the nuclear membrane and nuclear functions. [57,58]
Socioeconomic stress (SES)
Early in life SES can lead to physical and psychological outcomes enhancing imbalance between the nervous and immune systems,[59] which may result in health disparities including inflammatory disorders such as respiratory distress, autoimmune diseases and cardiovascular disorders.[60,61] These adverse childhood conditions can influence health as adults including physiological processes affecting mitochondrial functions.[62, 63] Severe socioeconomic deprivation may lead to adverse childhood experiences and separately or together can adversely affect mitochondrial structure, function, mtDNA copy number and transcription. [63]
Age and mitochondrial dysfunction
Aging is one of the highest risks associated with a severe COVID-19 outcome. Increased age is often accompanied with activation of the innate immune system possessing higher inflammatory mediators and producing elevated levels of cytokines.[64] NLRP3 inflammasome and caspase-1 activation are found in age-related conditions such as metabolic disease and can cause mitochondrial damage. [65] Mitochondrial mutations are increased with age and can in turn drive mammalian aging.[66] Mitochondrial functions such as mitochondrial respiratory capacity, ATP generation and mitophagy decline occur in older people due to increased age-dependent mitochondrial dysfunction and damage. [67]
SARS-CoV-2 pathogenesis, mitochondria, and inter-organelle associations
A human promonocyte HL-CZ cell line transfected with SARS-CoV 3C-like protease (3CLpro) was suggested to develop “mitochondrial-induced apoptosis”;[68] proteomic analysis indicated that 3CLpro caused up-regulation of 73 proteins and down regulation of 21 proteins with 36% of the up-regulated proteins and 19% of down-regulated proteins being mitochondrial proteins;[68] the SARS protease was concluded to affect mitochondrial apoptosis signaling, ETC, and ATP synthesis. Another in vitro study reported downregulation of mitochondrial organization, respiration processes and mitochondrial translation in SARS-CoV-2 infected A549 adenocarcinomic human alveolar basal epithelial cells.[69] Given SARS-CoV-2 is a novel SARS-CoV, there are few other direct reports regarding the effects of SARS-CoV-2 on mitochondria. However, viral infections may affect mitochondria biogenesis and mitophagy with detriments to immune responses, neuroendocrine immune network[62,70] and immunosenescence,[71] which would create more severe COVID-19 outcomes. Mitochondrial dysfunction has been posited to affect organ failure from sepsis,[72] endothelial changes leading to cardiovascular disease,[73] and alveolar epithelial cells damage in mice with S. aureus pneumonia.[74] Absence of MKK3 kinase in mice lessened LPS-induced lung damage and presence of ROS with concomitant increase in mitochondrial biogenesis and mitophagy due to activiation of Sirt1, Pink1, and Parkin.[75] The involvement of mitochondrial dysfunction and ROS in the induction of organ damage is supported by thioredoxin-1 aiding Sepsis-induced myocardial dysfunction with attenuating mitochondrial dysfunction.[76] Patients with MtD are known to be more susceptible to many stressors,[77] which likely further alter mitochondrial functions. SARS-CoV-2 has four main structural proteins: spike (S) glycoprotein, small envelope (E) glycoprotein, membrane (M) glycoprotein, and nucleocapsid (N) protein, and several accessory proteins.[78,79] Cell entry of SARS-CoV-2 depends on targeted cells expressing angiotensin-converting enzyme 2 (ACE2) and transmembrane serine protease 2 (TMPRSS2). The S-protein of SARA-CoV-2 binds to ACE2 on cell surfaces, and in concert with TMPRSS2 is translocated into ACE2+ cells.[80] ACE2 is express on the surfaces of many cells in mitochondria-rich organs including lung, kidney, and gastrointestinal epithelia and cardiovascular endothelia. A study reported that during SARS-CoV infection ACE2 is endocytosed together with SARS-CoV, resulting in the reduction of ACE2 on the plasma membrane.[81] As a plasma membrane Zn metalloproteinase ectoenzyme, ACE2 can improve endothelial homeostasis by attenuation of NADPHox-induced production of reactive oxygen species (ROS) in mitochondria.[82] ACE2 loss by SARS-CoV will increase ROS generation due to enhanced NADPHox and lessening mitochondrial homeostasis. ACE2 also improves lipid metabolism via the IKKβ/NFκB/IRS-1 pathway in the co-regulation of endoplasmic reticulum and mitochondria.[83] ACE2 normally cleaves angiotensin 2 (AngII) to generate Ang-(1–7). AngII and its peptide hormonally act on the central nervous system (CNS) to regulate the activity of renal sympathetic nerve and renal function to maintain normal blood pressure.[84] Loss of ACE2 and the subsequent increase of serum AngII have been associated with hypertension and cardiac and renal damage.[85,86] AngII and its receptor, angiotensin receptor type 1 (AT1R) (AngII-AT1R) pathway, which is located in the signal downstream of ACE2, may lead to cytokine release syndrome (CRS) mainly via IL-6-STAT3 axis in COVID-19 severe patients.[87] AngII can also induce production of mitochondrial reactive oxygen species (mtROS), resulting in cardiovascular diseases.[88] Mitochondria help to localize and regulate transmembrane serine protease and apoptosis-related protease activation during cell bioenergetics.[89] Unbalanced mitochondrial homeostasis may result in incorrect secretion and localization of TMPRSS2, which affects the viral entry in turn. After entry into host cells, viral RNA is released and viral polyproteins are translated. Viruses are well known to hijack targeted cells’ intracellular organelles for its purposes.[90–93] Numerous viral and host cell proteins may interact to control viral protein replication as well as the host cell’s proliferative stage[94] and immune mechanisms against the virus.[95] Interestingly, the c-terminal KxHxx domain of SARS-CoV and SARS-CoV-2 (KLHYT) enhance its entrance into endoplasmic reticulum (ER) and increase glycosylation of exocytosis of virions.[96] Many of the inter-organelle (mitochondria, lysosome, peroxisome, ER and golgi) interactions may be dysregulated affecting intracellular metabolism.[95,97,98] Once more is learned about inter-organelle interactions,[99] the manner by which viruses upset the mechanisms can be further explored. For example, lysosome intracellular distribution and function is affected by bi-direction interactions with mitochondria,[100] and peroxisome and mitochondria interactions affect lipid metabolism.[101] Mitochondrial antiviral signaling protein (MAVS), located on the outer mitochondrial membrane, can promote inflammasome activation and thus enhance the inflammation.[102] Based on the infection pathway of SARS-CoV-2, mitochondrial structure and function likely are key targets causing some severe results compromising patients’ defenses leading to greater morbidity and mortality.
SARS-CoV-2 genomic RNA encodes nonstructural proteins (NSPs) for viral RNA synthesis and structural proteins for virion assembly. Coronavirus genomic RNA replication is regulated by RNA replicase–transcriptase complex (RdRp); CoV RdRp involves nsp12.[103] Structural proteins are translated in ribosomes then bound to the endoplasmic reticulum (ER) and presented on the surface for virion assembly. Viral genomic RNA and structural proteins are fused together from the ER through the Golgi Apparatus and transported to infected host cell surface via small vesicles, then released as exocytosis with a repeat the infection of next host cell.[104] Mitochondrial dysfunction and endoplasmic reticulum stress, Golgi Apparatus (GA) dysfunction are often found together with proinflammatory activities, indicating the mitochondria, ER, and GA tight connections both physically and physiologically. ER and GA are affected by SARS-CoV-2 during infection,[78] making the mitochondria function worse. Viruses are known modulators of mitochondria.[105] High susceptibility to SARS-CoV-2 in polluted areas makes COVID-19 symptoms worsening of are partly due to the worse mitochondrial dysfunction under the double attacks of both the SARS-CoV-2 as main reason and with assistance from ECs.
Impacts of current drugs used in COVID-19 treatment on mitochondria
There are no licensed drugs and vaccines specific for treating or preventing SARS-CoV-2 and its COVID-19 events. Current symptomatic treatment of COVID-19 is supportive. However, the U.S. Food & Drug Administration (FDA) issues some drugs as an emergency use authorization (EUA) in critical health condition. Some drugs used below have shown some benefits and might be a potential treatment. All these drugs have side effects on mitochondria or other constituents, but the extent of detrimental influences vary.
Remdesivir.
Remdesivir (RDV, formally known as GS-5734) is an anti-viral nucleotide analog and a phosphoramidate prodrug of a 1′-cyano-substitued adenosine analogue. RDV has been used for ebolavirus (EBOV) treatment and currently is showing some benefit by shortening COVID-19 patients recovery time. Currently, this is the only drug with no direct reported toxicity on mitochondria.. However, the active triphosphate form of Remdesivir (RDV-TP) has high selectivity over incorporation of its natural nucleotide counterpart ATP [106] and thus might change the cytosolic ATP/ADP ratio, which may affect the mitochondria function. RDV treatment has shown some increase in circulating liver enzymes suggestive of some liver damage; it also may cause some nausea and like all drugs and toxicants dose and dosage is critical.[107] RDV is currently showing some benefit by shortening recovery time for COVID-19 patients,[108,109] but no drug is 100% safe for everyone.
Chloroquine and Hydroxychloroquine
Chloroquine causes toxicity on hepatic and neural mitochondria; it can cause an increase of phospholipids and a decrease of cholesterol by rat hepatic mitochondria.[110] Activities of enzymes located on the mitochondrial inner membrane (MIM), respiratory control ratio and ATP generation are significantly reduced. Chloroquine can also inhibit mitochondrial function and cause mtDNA damage in neurons, together with the altered key metabolism of TCA cycle including citrate synthase and glutaminolysis.[111] Chloroquine can induce mitochondrial cristae membrane damage and cause impaired ECT complexes, which affect increased ROS in cancer cells in triple negative breast cancer treatment.[112] Chloroquine and hydroxychloroquine has been useful in treatment of systemic lupus erythematosus and rheumatoid arthritis but long-term treatment may lead to retinal toxicity especially if dosage is not well controlled. Although early reports suggested some benefit for COVID-19, more extensive evaluation has indicated they have minimal if any influence for treating COVID-19.[113] Hydroxychloroquine or azithromycin alone or combined also demonstrated no benefit.[114] Hydroxychloroquine provided no benefit difference from standard care and adverse events were greater.[115] Clinical trials for COVID-19 have been stopped in UK and USA. FDA revoked the Emergency Use Authorization (EUA) on hydroxychloroquine and chloroquine for the treatment of COVID-19 on June 15, 2020 and issued a warning on hydroxychloroquine or chloroquine taken outside of a hospital for COVID-19 due to risk of heart rhythm problems on July 1, 2020. The NIH and WHO have also stopped the studies.
Lopinavir/Ritonavir.
Lopinavir/ritonavir is a combination antiretroviral medication for the HIV prevention and treatment via inhibition of 3-chymotrypsin-like protease. Generally, they are safe with some side effects and few severe side effects including pancreatitis, metabolism dysfunction. Lopinavir/Ritonavir can induce oxidative stress and Caspase-independent apoptosis. An in vivo study on BALB/c male mice indicated Lopinavir/Ritonavir could induce ER stress via ROS-dependent JNK activation.[116] It could also induce mitochondrial damage resulting in increased ROS generation, followed by apoptosis in a neuronal cell line.[117] Lopinavir/Ritonavir can also increase serum LDL-cholesterol levels, inhibit the myocardial ubiquitin-proteasome system (UPS) and leads to elevated calcineurin and connexin 43, attenuating mitochondrial function.[118]
Ribavirin.
Ribavirin, a guanine analogue, is an antiviral medication to treat RSV infection, hepatitis C and some viral hemorrhagic fevers by inhibiting viral RNA-dependent RNA polymerase. A few severe side effects include hematological and metabolism dysfunctions and allergic reactions. Ribavirin can inhibit mitochondrial DNA replication and cause mtDNA damage. It can also lead to increased intracellular and mitochondrial concentrations of dideoxynucleotide ATP (ddATP).[119]
Monitor and protection on mitochondria against SARS-CoV-2
Environmental stressors, co-morbidities, COVID-19, and drugs may mutually contribute toward making mitochondrial dysfunction worse. This means we need to pay much more attention to mitochondrial functions and biomarkers of exposome influences during COVID-19 pandemic, especially in high pollution area. There are many methods to monitor mitochondrial integrity and function, such as mitochondria activity assay, mitochondria mediated metabolism and related enzymes assays, ATP generation assays, cytochrome C releasing assays, MMP assays, ADP/ATP ratio assays, mitochondrial apoptosis/autophagy pathway assays, oxygen consumption assays and glycolysis assays. Many vitamins and minerals are required to support and protect healthy mitochondrial functions. These include but are not limit to: Vitamins as Biotin, Vitamin B family (B1, B2, B3, B5, B6), Vitamin C, Vitamin E; Minerals as Fe, Mg, Mn, Se, S, Zn, Cu; Amino Acids as L-Carnitine; CoQ10 and α-Lipoic acid,[77,120] and melatonin.[121] Vitamin C can reduce intracellular ROS level while Vitamin E can mitigate the toxicity arising from lipid peroxides. Melatonin drives mitochondrial conversion of pyruvate to acetyl-coenzyme A (acetyl-CoA) via mitochondrial melatonergic pathway and inhibits viral replication.[121] These nutrients are available in dietary foods for a healthy person but moderate increases may be required as additional supplements to COVID-19 patients. Other healthy lifestyles like enough sleep, appropriate exercise, body mass index (BMI) control and stress relax are also encouraged. Health is dependent on an individual’s exposome, biology and behavior.[122]
Mitochondria dynamics and the regulated metabolomics as new indicator of COVID-19 prognosis
COVID-19 severe patients often have metabolic diseases. Metabolic abnormalities worsen patients’health. Infection of SARS-CoV-2 can modify the metabolism of many immune cells. Monocytes and macrophages, two enriched immune cells in the lungs, become highly glycolytic after SARS-CoV-2 infection, which triggers mitochondrial ROS production and hypoxia-inducible factor-1α (HIF-1α) activation, which promotes glycolysis. This process can directly inhibit T cell responses and reduce epithelial cell survival. [123] Patients who have suffered from the severe or critical conditions of COVID-19 usually had higher proinflammatory cytokine levels of interleukin-6 (IL-6), which is suggested to be a key promoter of COVID-19 mediated CRS [124] along with IL-1β, IL-2, IL-8, IL-17, G-CSF, GM-CSF, IP10, MCP1, MIP1ɑ (CCL3) and TNFα. Besides cytokines, mitochondria dynamics in host cells are also important. Mitochondria dynamics refers to mitochondria self- balance and clearance regulation. Mitochondrial fission and fusion play critical roles in maintaining functional mitochondria and both are needed to keep balance. During infection, different viruses will shift host cell mitochondrial dynamics towards either fission or fusion by interacting with related factors such as DRP1, MFN, and OPA1. SARS-CoV ORF-9b protein can promote mitochondrial fusion by fission inhibition via interacting with DRP1 and facilitating its proteasomal degradation.[125] However, fission is required for new mitochondria generation. Disable mitochondrial fission will make the total number of mitochondria decreased, making mitochondrial functions even worse in mitochondrial disease patients. As a selective autophagy of mitochondria to eliminate damaged mitochondria, mitophagy is critical for mitochondrial quality control. Generally, mitochondrial fission/fragmentation will promote mitophagy, especially in early stage of infection.[126] Mitochondria are response for generation of metabolites via TCA cycle under physiological and pathological conditions.[127] SARS-CoV-2 will usurp some of these metabolites. Metabolomics would be useful to monitor and measure metabolites in immune cells before and after activation in the presence and absence of SARA-Cov-2 and as COVID-19 progresses to a more severe status to determine metabolite differences. Recently, serum iron was suggested as an ideal indicator for severity of hypoxemic respiratory failure in COVID-19 patients.[128] Iron is utilized in mitochondria as iron-sulfur clusters or heme prosthetic groups required for heme synthesis. Mitochondria play a key role in iron homeostasis including iron import, utilization, storage, and export. [129] A cell’s gene expression and metabolic processes are diverted by both environmental stressors, which includes pathogens. The term “exposome” was introduced to put environmental exposures on par with a person’s genome affecting a person’s health.[130] It is now widely accepted that genetics and environment affect many human diseases. The difficulty of fully appreciating effects of environment on health may come from not completely connecting the exposome to the biology of human health. A more current understanding of the exposome describes it as where chemistry meets biology.[131] The term ”idiosome” may be a more appropriate than exposome, because it represents how each person’s genetics, microbiota, lifetime of environmental exposures and behaviors, affect a person’s epigenome, biology, and health. A person’s biomarkers in response to environmental exposures are becoming a better individualistic means to assess health outcome (personalized medicine). Biomarkers defining a person are the molecules analyzed by researching genomics, lipidomics, metabonomics, proteomics, and transcriptomics. Further, it’s the overall pattern of biomarkers that best define an idiosome and a person’s health status; usually, no one biomarker is adequate.
Summary
In conclusion, mitochondria are potentially fragile organelles during any infection [132] and may be especially compromised by SARS-CoV-2; mitochondria may need special attention and care. Although mitochondria have high importance to many functions and are emphasized herein all aspects of a cell’s biochemistry, ecology and structure impacts physiology, which is modified by environmental stress from chemicals, biologicals, physical and psychological stressors. Based on the suggested influences of environmental stressors of many different forms affecting susceptibility to infections, it should be no surprise that people living with more stresses are less able to deal with COVID-19. Age, sex, and co-morbidities such as hypertension, diabetes and obesity are risk factors, and these parameters connect with inflammaging,[133] oxidative stress, [134] and mitochondria.[135] Health disparities due to life with socioeconomic stress, an inadequate supply of appropriate nutrients, and exposure to ECs are suggested to be more susceptible to COVID-19 due to dysfunctional mitochondria, which may be creating the greater susceptibility and less improvement from interventions. However, the greater severity of COVID-19 amongst men and Black, Asian and Minority Ethnic (BAME) individuals remains questionable.[136] Additional evidence for cause to effect relies on molecular and biological investigation of genetic and exposome influences and mitochondrial dysfunction as suggested herein. These concerns have been linked with the reported incidence and morbidity from COVID-19 amongst African Americans. [137,138] Essential care and medication to protect mitochondria is necessary for a healthier life. The recent differences in how we defend ourselves against SARS-CoV-2 and cope with COVID-19 has re-emphasized the “omic” differences amongst us, which need to be further explored and understood.
Acknowledgement
This review was aided by support from Wadsworth Center and a NIH grant (ES025584) to DAL.
Footnotes
Conflict of interest
No declared conflict of interest.
References
- 1.Song Z, Xu Y, Bao L, Zhang L, Yu P, Qu Y, Zhu H, Zhao W, Han Y, Qin C, Viruses. 2019, 11, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, Van Houten B, Mostoslavsky R, Bultman SJ, Baccarelli AA, Begley TJ, Sobol RW, Hirschey MD, Ideker T, Santos JH, Copeland WC, Tice RR, Balshaw DM, Tyson FL. Environ Health Perspect, 2014, 122, 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Landi F, Bernabei R, Marzetti E, Int. J. Mol. Sci. 2017, 18, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, Ferrucci L, Gilroy DW, Fasano A, Miller GW, Miller AH, Mantovani A, Weyand CM, Barzilai N, Goronzy JJ, Rando TA, Effros RB, Lucia A, Kleinstreuer N, Slavich GM, Nat Med. 2019, 25, 1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G, Ann N Y Acad Sci. 2000, 908, 244. [DOI] [PubMed] [Google Scholar]
- 6.Hwang JW, Yao H, Caito S, Sundar IK, Rahman I, Free Radic Biol Med, 2013, 61, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang BL, Mol Cells, 2016, 39, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor RW, Turnbull DM, Nat Rev Genet. 2005, 6, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW, J Pathol. 2017, 241, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyer JN, Leung MCK, Rooney JP, Sendoel A, Hengartner MO, Kisby GE, Bess AS, Toxicol Sci. 2013, 134, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Hattab AW, Zarante AM, Almannai M, Scaglia F, Mol Genet Metab. 2017, 122, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCormack SE, Xiao R, Kilbaugh TJ, Karlsson M, Ganetzky RD, Cunningham ZZ, Goldstein A, Falk MJ, Damrauer SM, Mol Genet Metab. 2017, 121, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chinnery PF, EMBO Mol Med. 2015, 7, 1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zolkipli-Cunningham Z, Falk MJ, Toxicology. 2017, 391, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bar-Or D, Carrick M, Tanner A 2nd, Lieser MJ, Rael LT, Brody E, J Crit Care. 2018, 43, 197. [DOI] [PubMed] [Google Scholar]
- 16.Liberti MV, Locasale JW, Trends Biochem Sci. 2016, 41, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dennis KK, Jones DP, Am Biol Teach. 2016, 78, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li MY, Li L, Zhang Y, Wang XS, Infect Dis Poverty. 2020, 9, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCormick EM, Zolkipli-Cunningham Z, Falk MJ, Curr Opin Pediatr. 2018, 30, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW, J Pathol. 2017, 241, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finsterer J, Segall L, Drug Chem Toxicol. 2010, 33, 138. [DOI] [PubMed] [Google Scholar]
- 22.Zielonka J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, Cheng G, Lopez M, Kalyanaraman B, Chem Rev. 2017, 117, 10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galic S, Oakhill JS, Steinberg GR, Mol Cell Endocrinol. 2010, 316, 129. [DOI] [PubMed] [Google Scholar]
- 24.Jackson E, Shoemaker R, Larian N, Cassis L, Compr Physiol. 2017, 7, 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gregorio D, Busiello RA, Aceves M. A. Burgos, Lepretti M, Paolella G, Lionetti L, Front Physiol, 2018, 9: 1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heindel JJ, Blumberg B, Cave M, Machtinger R, Mantovani A, Mendez MA, Nadal A, Palanza P, Panzica G, Sargis R, Vandenberg LN, Vom Saal F, Reprod Toxicol, 2017, 68, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chistiakov DA, Sobenin IA, Bobryshev YV, Orekhov AN, World J Cardiol, 2012, 4, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sobenin IA, Chistiakov DA, Bobryshev YV, Postnov AY, Orekhov AN, Curr Pharm Des, 2013, 19, 5942. [DOI] [PubMed] [Google Scholar]
- 29.Shen M, Zhang L, Bonner MR, Liu CS, Li G, Vermeulen R, Dosemeci M, Yin S, Lan Q, Environ Mol Mutagen. 2008, 49, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khalade A, Jaakkola MS, Pukkala E, Jaakkola JJK, Environ Health. 2010, 9, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McHale CM, Zhang L, Smith MT, Carcinogenesis, 2012, 33, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Synder R, Witz G, Goldstein BD, Environ Health Perspect. 1993, 100, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L, He X, Bi Y, Ma Q, Chem Res Toxicol, 2012, 25, 1303. [DOI] [PubMed] [Google Scholar]
- 34.Wang D, Yang X, Zhang Y, Lin D, Li P, Zhang Z, Huang X, Gu D, Loo JFC, J Thoracic Dis. 2018, 10, 6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalf GF, Rushmore T, Snyder R, Chem Biol Interact. 1982, 42, 353. [DOI] [PubMed] [Google Scholar]
- 36.He JL, Jin LF, Jin HY, Biomed Environ Sci. 1998, 11, 87. [PubMed] [Google Scholar]
- 37.Shaham J, Gurvich R, Kaufman Z, Mutat Res. 2002, 514, 115. [DOI] [PubMed] [Google Scholar]
- 38.Ye X, Yan W, Xie H, Zhao M, Ying C, Mutat Res. 2005, 588, 22. [DOI] [PubMed] [Google Scholar]
- 39.Costa S, García-Lestón J, Coelho M, Coelho P, Costa C, Silva S, Porto B, Laffon B, Teixeira JP, J Toxicol Environ Health A. 2013, 76, 217. [DOI] [PubMed] [Google Scholar]
- 40.Nadalutti CA, Stefanick DF,, Zhao M, Horton JK, Prasad R, Brooks AM, Griffith JD, Wilson SH, Sci Rep. 2020, 10, 5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zerin T, Kim JS, Gil HW, Song HY, Hong SY, Cell Biol Toxicol. 2015, 31, 261. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Liu X, McHale C, Li R, Zhang L, Wu Y, Ye X, Yang X, Ding S, PLos One. 2013, 8, e74974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan S, Beigh S, Chaudhari BP, Sharma S, Abdi S. Aliul Hasan, Ahmad S, Ahmad F, Parvez S, Raisuddin S, Environ Toxicol. 2016, 31, 1922. [DOI] [PubMed] [Google Scholar]
- 44.Wang K, Zhao Z, Ji W, Biomed Pharmacother. 2019, 117, 109182. [DOI] [PubMed] [Google Scholar]
- 45.Lin Y, Sun X, Qiu L, Wei J, Huang Q, Fang C, Ye T, Kang M, Shen H, Dong S, Cell Death & Disease. 2013, 4, e460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tchounwou PB, Yedjou CG, Patlolla AK, Sutton DJ, EXS. 2012, 101, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sabolić I, Nephron Physiology. 2006, 104, 107. [DOI] [PubMed] [Google Scholar]
- 48.Belyaeva EA, Sokolova TV, Emelyanova LV, Zakharova IO, ScientificWorldJournal. 2012, 2012, 136063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heo Y, Parsons PJ, Lawrence DA, Toxicol Appl Pharmacol. 1996, 138, 149. [DOI] [PubMed] [Google Scholar]
- 50.Kasten-Jolly J, Heo Y, Lawrence DA, Toxicol Appl Pharmacol. 2010, 247, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kasten-Jolly J, Lawrence DA, J Toxicol Environ Health, Part B, 2018, 21, 400. [DOI] [PubMed] [Google Scholar]
- 52.Sharma N, Pasala MS, Prakash A, Environ Mol Mutagen, 2019, 60, 668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gibbs JL, Dallon BW, Lewis J, Walton CM, Arroyo JA, Reynolds PR, Bikman BT, Int. J. Molec. Sci 2019, 20, 5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim HB, Ichinose T, Miyabara Y, Takano H, Kumagai Y, Shimojyo N, Devalia JL, Sagai M. Involvement of superoxide and nitric oxide on airway inflammation and hyperresponsiveness induced by diesel exhaust particles in mice. Free Radic Biol Med 1998; 25: 635–644. [DOI] [PubMed] [Google Scholar]
- 55.Mendoza A, Torres-Hernandez JA, Ault JG, Pedersen-Lane JH, Gao D, Lawrence DA. Cell Stress Chaperones, 2014, 19, 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, Wang Y, Bai Y, Wang P, Zhao Y, J. Energy Institute, 2019, 92, 1864. [Google Scholar]
- 57.Muir R, Diot A, Poulton, Bioessays, 2016, 38, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soto-Heredero G, Baixauli F, Mittelbrunn M, Front. Cell Dev. Biol 2017, 5, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bick J, Naumova O, Hunter S, Barbot B, Lee M, Luthar SS, Raefski A, Grigorenko EL, Dev Psychopathol, 2012, 24, 1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shonkoff JP, Boyce W. Thomas, McEwen BS, JAMA, 2009, 301, 2252. [DOI] [PubMed] [Google Scholar]
- 61.Tawakol A, Osborne MT, Wang Y, Hammed B, Tung B, Patrich T, Oberfeld B, Ishai A, Shin LM, Nahrendorf M, Warner ET, Wasfy J, Fayad ZA, Koenen K, Ridker PM, Pitman RK, Armstrong KA, J Am Coll Cardiol, 2019, 73, 3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Picard M, McEwen BS, Psychosom Med, 2018, 80, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morris G, Berk M, Maes M, Carvalho AF, Puri BK, Mol Neurobiol, 2019, 56, 5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S, Mech Ageing Dev, 2007, 128, 92. [DOI] [PubMed] [Google Scholar]
- 65.De Nardo D, Latz E, Trends Immunol, 2011, 32, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hiona A, Leeuwenburgh C, Exp Gerontol, 2008, 43, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun N, Youle RJ, Finkel T, Mol Cell, 2016, 61, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lai CC, Jou MJ, Huang SY, Li SW, Wan L, Tsai FJ, Lin CW, Proteomics, 2007, 7, 1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singh K, Chen YC, Judy JT, Seifuddin F, Tunc I, Pirooznia M, BioRxiv. Preprint 2020, May 14, 10.1101/2020.05.13.092536 [DOI] [Google Scholar]
- 70.Lee F, Lawrence DA, J Toxicol Environ Health B Crit Rev. 2018, 21, 24. [DOI] [PubMed] [Google Scholar]
- 71.Müller-Werdan U, Prondzinsky R, Werdan K, Curr Opin Crit Care 2016, 22, 453. [DOI] [PubMed] [Google Scholar]
- 72.Singer M, Virulence 2014, 5, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Y-F, Dugas TR, Toxicol Lett. 2019, 317, 13. [DOI] [PubMed] [Google Scholar]
- 74.Suliman HB, Kraft B, Bartz R, Chen L, Welty-Wolf KE, Piantadosi CA, Am J Physiol Lung Cell Mol Physiol. 2017. 313, L699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mannam P, Shinn AS, Srivastava A, Neamu RF, Walker WE, Bohanon M, Merkel J, Kang M-J, Dela Cruz CS, Ahasic AM, Pisani MA, Trentalange M, West AP, Shadel GS, Elias JA, Lee PJ, Am J Physiol Lung Cell Mol Physiol. 2014, 306, L604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sánchez-Villamil JP, D’Annunzio V, Finocchietto P, Holod S, Rebagliati I, Pérez H, Peralta JG, Gelpi RJ, Poderoso JJ, Carreras MC, Int J Biochem Cell Biol. 2016, 81, 323. [DOI] [PubMed] [Google Scholar]
- 77.MitoFIRST Handbook. An Introductory Guide United Mitochondrial Disease Foundation. 2016, https://www.umdf.org/wp-content/uploads/2016/10/mito_first.pdf
- 78.Astuti I, Ysrafil, Diabetes Metab Syndr. 2020, 14, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P, Meng J, Zhu Z, Zhang Z, Wang J, Sheng J, Quan L, Xia Z, Tan W, Cheng G, Jiang T, Cell Host Microbe. 2020, 27, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA, Drosten C, Pöhlmann S, Cell. 2020, 181, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM, Nat Med. 2005, 11, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lovren F, Pan Y, Quan A, Teoh H, Wang G, Shukla PC, Levitt KS, Oudit GY, Al-Omran M, Stewart DJ, Slutsky AS, Peterson MD, Backx PH, Penninger JM, Verma S, Am J Physiol Heart Circ Physiol. 2008, 295, H1377. [DOI] [PubMed] [Google Scholar]
- 83.Cao X, Lu XM, Tuo X, Liu JY, Zhang YC, Song LN, Cheng ZQ, Yang JK, Xin Z, Lipids Health Dis. 2019, 18, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sata Y, Head GA, Denton K, May CN, Schlaich MP, Front Med (Lausanne). 2018, 5, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Varagic J, Ahmad S, Nagata S, Ferrario CM, Curr Hypertens Rep. 2014, 16, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mizuiri S, Ohashi Y, World J Nephrol. 2015, 4, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hirano T, Murakami M, Immunity. 2020, 52, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dikalov SI, Nazarewicz RR, Antioxid Redox Signal. 2013, 19, 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Susin SA, Zamzami N, Castedo M, Daugas E, Wang HG, Geley S, Fassy F, Reed JC, Kroemer G, J Exp Med. 1997, 186, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh KK, Chaubey G, Chen JY, Suravajhala P, Am J Physiol Cell Physiol. 2020, 319, C258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Glingston RS, Deb R, Kumar S, Nagotu S, Microbes Infect. 2019, 21, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Romero-Brey I, Bartenschlager R, Viruses. 2016, 8, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Knoops K, Kikkert M, van den Worm SH, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ, PLoS Biol. 2008, 6, e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.De Wilde AH, Snijder EJ, Kikkert M, van Hemert MJ, Roles of Host Gene and Non-coding RNA Expression in Virus Infection. 2018, 419, 1. [Google Scholar]
- 95.Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH, Kehrl JH, J Immunol. 2014,193, 3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McBride CE, Li J, Machamer CE, J Virol. 2007, 81, 2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang L, Wei L, Jiang D, Wang J, Cong X, Fei R, Artif Cells Blood Substit Immobil Biotechnol. 2007, 35, 237. [DOI] [PubMed] [Google Scholar]
- 98.Anderson G, Reiter RJ, Rev Med Virol. 2020, 30, e2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gottschling DE, Nyström T, Cell. 2017, 169, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wong YC, Ysselstein D, Krainc D, Nature. 2018, 554, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shai N, Yifrach E, van Roermund CWT, Cohen N, Bibi C, IJlst L, Cavellini L, Meurisse J, Schuster R, Zada L, Mari MC, Reggiori FM, Hughes AL, Escobar-Henriques M, Cohen MM, Waterham HR, Wanders RJA, Schuldiner M, Zalckvar E, Nat Commun. 2018, 9, 1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. Cell, 2013, 153, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Snijder EJ, Decroly E, Ziebuhr J, Adv Virus Res. 2016, 96, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Robinson M, Schor S, Barouch-Bentov R, Einav S, Cell Mol Life Sci. 2018, 75, 3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anand SK, Tikoo SK, Adv Virol. 2013, 2013, 738794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gordon CJ, Tchesnokov EP, Woolner E, Perry JK, Feng JY, Porter DP, Gotte M, J Biol Chem. 2020, 295, 6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eastman RT, Roth JS, Brimacombe KR, Simeonov A, Shen M, Patnaik S, Hall MD, ACS Cent Sci. 2020, 6, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, Hohmann E, Chu HY, Luetkemeyer A, Kline S, de Castilla DL, Finberg RW, Dierberg K, Tapson V, Hsieh L, Patterson TF, Paredes R, Sweeney DA, Short WR, Touloumi G, Lye DC, Ohmagari N, Oh MD, Ruiz-Palacios GM, Benfield T, Fätkenheuer G, Kortepeter MG, Atmar RL, Creech CB, Lundgren J, Babiker AG, Pett S, Neaton JD, Burgess TH, Bonnett T, Green M, Makowski M, Osinusi A, Nayak S, Lane HC, N Engl J Med. 2020, May 22;NEJMoa2007764. doi: 10.1056/NEJMoa2007764. Online ahead of print. [DOI] [Google Scholar]
- 109.Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, Fu S, Gao L, Cheng Z, Lu Q, Hu Y, Luo G, Wang K, Lu Y, Li H, Wang S, Ruan S, Yang C, Mei C, Wang Y, Ding D, Wu F, Tang X, Ye X, Ye Y, Liu B, Yang J, Yin W, Wang A, Fan G, Zhou F, Liu Z, Gu X, Xu J, Shang L, Zhang Y, Cao L, Guo T, Wan Y, Qin H, Jiang Y, Jaki T, Hayden FG, Horby PW, Cao B, Wang C, Lancet, 2020, 395, 1568. [Google Scholar]
- 110.Deepalakshmi PD, Parasakthy K, Shanthi S, Devaraj NS, Indian J Exp Biol. 1994, 32, 797. [PubMed] [Google Scholar]
- 111.Redmann M, Benavides GA, Berryhill TF, Wani WY, Ouyang X, Johnson MS, Ravi S, Barnes S, Darley-Usmar VM, Zhang J, Redox Biol. 2017, 11, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liang DH, Choi DS, Ensor JE, Kaipparettu BA, Bass BL, Chang JC, Cancer Lett. 2016, 376, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ferner RE, Aronson JK, BMJ. 2020, 369, m1432. [DOI] [PubMed] [Google Scholar]
- 114.Rosenberg ES, Dufort EM, Udo T, Wilberschied LA, Kumar J, Tesoriero J, Weinberg P, Kirkwood J, Muse A, DeHovitz J, Blog DS, Hutton B, Holtgrave DR, Zucker HA, JAMA. 2020, 323, 2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tang W, Cao Z, Han M, Wang Z, Chen J, Sun W, Wu Y, Xiao W, Liu S, Chen E, Chen W, Wang X, Yang J, Lin J, Zhao Q, Yan Y, Xie Z, Li D, Yang Y, Liu L, Qu J, Ning G, Shi G, Xie Q, BMJ. 2020, 369, m1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Taura M, Kariya R, Kudo E, Goto H, Iwawaki T, Amano M, Suico MA, Kai H, Mitsuya H, Okada S, Free Radic Biol Med, 2013, 65, 778. [DOI] [PubMed] [Google Scholar]
- 117.Gratton R, Tricarico PM, Guimaraes RL, Celsi F, Crovella S, Curr HIV Res, 2018, 16, 106. [DOI] [PubMed] [Google Scholar]
- 118.Tricarico PM, de Oliveira Franca RF, Pacor S, Ceglia V, Crovella S, Celsi F, Cell Physiol Biochem. 2016, 39, 1463. [DOI] [PubMed] [Google Scholar]
- 119.Salmon-Céron D, Chauvelot-Moachon L, Abad S, Silbermann B, Sogni P, Lancet. 2001, 357, 1803. [DOI] [PubMed] [Google Scholar]
- 120.Du J, Zhu M, Bao H, Li B, Dong Y, Xiao C, Zhang GY, Henter I, Rudorfer M, Vitiello B, Crit Rev Food Sci Nutr. 2016, 56, 2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Anderson G, Reiter RJ, Rev Med Virol. 2020, 30, e2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Miller GW, Jones DP, Toxicol Sci. 2014, 137, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, Prodonoff JS, Carregari VC, de Biagi Junior CAO, Crunfli F, Restrepo J.L. Jimenez, Vendramini PH, Reis-de-Oliveira G, Bispo Dos Santos K, Toledo-Teixeira DA, Parise PL, Martini MC, Marques RE, Carmo HR, Borin A, Coimbra LD, Boldrini VO, Brunetti NS, Vieira AS, Mansour E, Ulaf RG, Bernardes AF, Nunes TA, Ribeiro LC, Palma AC, Agrela MV, Moretti ML, Sposito AC, Pereira FB, Velloso LA, Vinolo MAR, Damasio A, Proença-Módena JL, Carvalho RF, Mori MA, Martins-de-Souza D, Nakaya HI, Farias AS, Moraes-Vieira PM, Cell Metab, 2020. Jul 17;S1550–4131(20)30365-X. doi: 10.1016/j.cmet.2020.07.007. Online ahead of print. [DOI] [Google Scholar]
- 124.Moore JB, June CH, Science. 2020, 368, 473. [DOI] [PubMed] [Google Scholar]
- 125.Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH, Kehrl JH, J Immunol. 2014, 193, 3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang L, Qin Y, Chen M, Autophagy. 2018, 14, 1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Martínez-Reyes I, Chandel NS, Nat Commun. 2020, 11, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shah A, Frost JN, Aaron L, Donovan K, Drakesmith H & Collaborators, Critical Care. 2020, 24, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Paul BT, Manz DH, Torti FM, Torti SV, Expert Rev Hematol. 2017, 10, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wilder CP Cancer Epidemiol Biomarkers Prev, 2005, 14, 1847. [DOI] [PubMed] [Google Scholar]
- 131.Vermeulen R, Schymanski EL, Barabási AL, Miller GW, Science, 2020, 367, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Anand SK, Tikoo SK, Adv Virol. 2013, 2013, 738794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zuo L, Prather ER, Stetskiv M, Garrison DE, Meade JR, Peace TI, Zhou T, Int J Mol Sci. 2019, 20, 4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Oguntibeju OO, Int J Physiol Pathophysiol Pharmacol, 2019, 11, 45. [PMC free article] [PubMed] [Google Scholar]
- 135.Bhatti JS, Bhatti GK, Reddy PH, Biochem Biophys Acta, 2017, 1863, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Raisi-Estabragh Z, McCracken C, Bethell MS, Cooper J, Cooper C, Caulfield MJ, Munroe PB, Harvey NC, Petersen SE, J Public Health (Oxf). 2020, Jun 19;fdaa095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yancy CW, JAMA, 2020, 323, 1891. [DOI] [PubMed] [Google Scholar]
- 138.Ferdinand KW, Nasser SA, J Am Coll Cardiol. 2020, 75, 2746. [DOI] [PMC free article] [PubMed] [Google Scholar]