

Abstract
Treatment of (ArL)CoBr (ArL = 5-mesityl-1,9-(2,4,6-Ph3C6H2)dipyrrin) with a stoichiometric amount of 1-azido-4-(tert-butyl)benzene N3(C6H4-p-tBu) furnished the corresponding four-coordinate organoazide-bound complex (ArL)CoBr(N3(C6H4-p-tBu)). Spectroscopic and structural characterization of the complex indicated redox innocent ligation of the organoazide. Slow expulsion of dinitrogen (N2) was observed at room temperature to afford a ligand functionalized product via a [3+2] annulation, which can be mediated by a high-valent nitrene intermediate such as a CoIII iminyl (ArL)CoBr(·N(C6H4-p-tBu)) or CoIV imido (ArL)CoBr(N(C6H4-p-tBu)) complex. The presence of the proposed intermediate and its viability as a nitrene group transfer reagent are supported by intermolecular C–H amination and aziridination reactivities. Unlike (ArL)CoBr(N3(C6H4-p-tBu)), a series of alkyl azide-bound CoII analogues expel N2 only above 60 °C, affording paramagnetic intermediates that convert to the corresponding Co-imine complexes via α–H–atom abstraction. The corresponding N2-released structures were observed via single-crystal-to-crystal transformation, suggesting formation of a Co-nitrenoid intermediate in solid-state. Alternatively, the alkyl azide-bound congeners supported by a more sterically accessible dipyrrinato scaffold tBuL (tBuL = 5-mesityl-(1,9-di-tert-butyl)dipyrrin) facilitate intramolecular 1,3-dipolar cycloaddition as well as C–H amination to furnish 1,2,3-dihydrotriazole and substituted pyrrolidine products, respectively. For the C–H amination, we observe that the temperature required for azide activation varies depending on the presence of weak C–H bonds, suggesting that the alkyl azide adducts serve as viable species for C–H amination when the C–H bonds are (1) proximal to the azide moiety and (2) sufficiently weak to be activated.
Graphical Abstract
1. INTRODUCTION
Transition-metal-mediated C–H amination is an appealing synthetic route for direct incorporation of N-functionality into unactivated C–H bonds.1–6 For such transformations, metal nitrene complexes (e.g., imido, M(NR2−);7–9 iminyl, M(2NR−);10–19 nitrene adducts, M(3NR)20–21) have been invoked as viable N-group transfer reagents, attracting great interest for synthesizing these complexes.22 Toward this end, N2 extrusion from organoazides (N3R) represents one of the most common approaches to generate metal nitrene complexes.23 As such, while many metal nitrene compounds have been isolated, the corresponding precursors, metal-organoazide adduct complexes are relatively rare due to rapid N2 expulsion.24
Despite the transient nature of metal-organoazide adduct complexes, a few examples have been structurally characterized.25 Owing to the possible resonance structures and the dipolar character of the azide moiety, several binding modes of organoazides towards transition metals have been observed. For example, organoazides bound to an early transition metal such as tantalum,26–27 vanadium,28–29 or tungsten30 display a significantly bent N–N–N moiety along with a relatively short M–Nγ distance due to a π–back bonding interaction. This binding mode is most commonly observed when the organoazide behaves as a dianionic ligand in the form of a diazenimido (Figure 1a). In contrast, organoazides prefer to bind in a neutral (redox-innocent) fashion to late transition metals (e.g., palladium,24 iridium,31 copper,32 silver,32 ruthenium,33 and cobalt34) and exhibit a relatively linear N–N–N moiety (Figure 1b–c). Interestingly, an iron-based organoazide adduct reported by the Peters group manifests intermediate binding properties of the aforementioned two extremes by displaying a partially oxidized iron center along with a bent N–N–N moiety owing to a spin delocalization through the azide moiety (Figure 1d).35 In addition to these η1-type interactions, an η2-coordination has been observed by Hillhouse using a 1,2-bis(di-tert-butylphosphino)ethane nickel complex (Figure 1e).36 Moreover, μ2-coordination has been observed with a heterobinuclear complex composed of zirconium and iridium (Figure 1f).37
Figure 1.
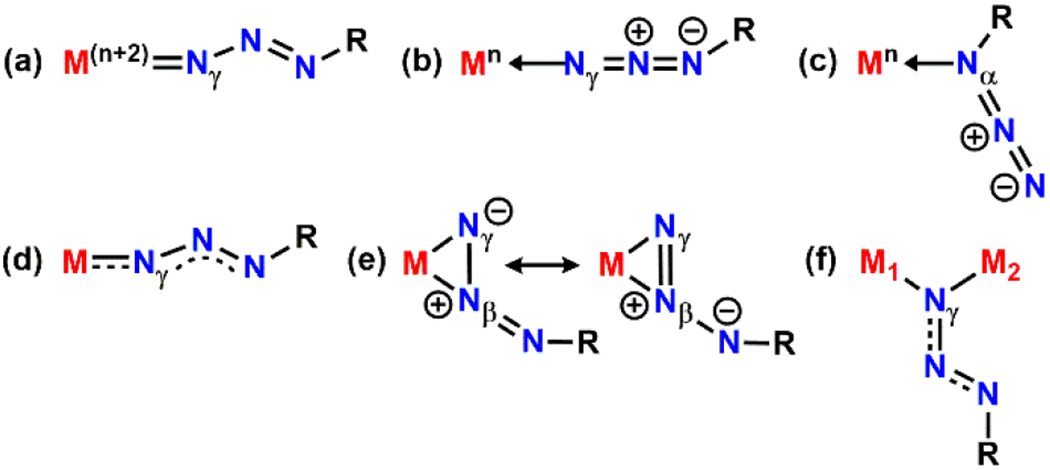
Observed binding modes of organoazides to a metal center(s).
Each binding mode can dictate a discrete decomposition pathway for conversion to the corresponding metal nitrene compounds.26–27, 29, 36–37 Moreover, in a few cases, metal-organoazide adducts, and not the corresponding metal nitrene complexes, are proposed as viable nitrene group transfer reagents as elucidated by kinetic analysis.25, 38–40 Thus, studying the nature of the binding interaction and the electronic structure of organoazide-bound metal complexes becomes critical for investigating subsequent nitrene group transfer reactions.
Our group has previously demonstrated both intra- and intermolecular C–H amination mediated by FeIII iminyl species generated from dipyrrinato FeII complexes and various organoazides.11–13, 41 In the synthesis of these ferric iminyl complexes, the corresponding organoazide-bound iron complexes have been invoked as intermediate species. However, no such intermediate was detected due to extremely fast N2 extrusion. Similarly, organoazide adducts have been often proposed as relevant precursors in other late, first-row transition metal-catalyzed C–H amination processes,15, 42–44 yet structural evidence for those species is absent.
In order to unveil the presence and structural identity of transient organoazide-bound metal complexes, we chose to investigate analogous dipyrrinato cobalt complexes as cobalt is typically harder to oxidize given its higher electronegativity relative to iron. With this approach, we report herein the synthesis and spectroscopic characterization of a series of organoazide-bound dipyrrinato CoII complexes. By utilizing a sterically encumbered dipyrrin scaffold ArL (ArL = 5-mesityl-1,9-(2,4,6-Ph3C6H2)dipyrrin), we were able to crystallographically characterize both aryl and alkyl azide-bound Co complexes. From those adduct species, we demonstrate a range of intra- and intermolecular C–H activation processes as well as N-group transfer reactivity, which is proposed to be mediated by a high-valent nitrene intermediate such as a CoIV imido or CoIII iminyl species. Indeed, using alkyl azide adducts, we were able to directly observe the proposed high-valent Co-nitrene intermediate within a crystal lattice following either thermolysis or X-ray irradiation of single crystals of the azide adducts. Alternatively, a more sterically accessible ligand tBuL (tBuL = 5-mesityl-(1,9-di-tert-butyl)dipyrrin) offers greater flexibility to analogous alkyl azide adducts enabling facile intramolecular C–H amination to afford substituted pyrrolidines. Detailed mechanistic studies on the C–H amination processes establish the competency of the azide adduct itself to undergo C–H amination without necessitating a high-valent metal nitrene intermediate.
2. RESULTS AND DISCUSSION
2.1. Synthesis of Cobalt Organoazide Adducts.
Our group has previously reported the preparation of both four- and three-coordinate dipyrrinato CoII complexes, (ArL)CoCl(py) (ArL = 5-mesityl-1,9-(2,4,6-Ph3C6H2)dipyrrin) (1)45 and (ArL)CoBr (2),46 respectively. We envisioned that treatment of such CoII complexes with an organoazide would furnish either the corresponding organoazide-bound complexes or cobalt nitrene species. Heating a solution of 1 in benzene-d6 with 1-azido-4-(tert-butyl)benzene N3(C6H4-p-tBu) at 60 °C, however, affords no reaction. In contrast, the coordinatively unsaturated complex 2 rapidly consumes the azide at room temperature to generate a new paramagnetic species as verified by 1H NMR spectroscopy.
Crystals of this product were obtained from a toluene/pentane mixture at −35 °C. X-ray diffraction analysis revealed formation of the corresponding four-coordinate aryl azide-bound complex (ArL)CoBr(N3(C6H4-p-tBu)) (3) (Scheme 1, Figure 2). The solid-state structure of 3 exhibits a distorted trigonal monopyramidal geometry (τ4 = 0.80),47 featuring an axially coordinated aryl azide with a Co–Nα distance of 2.111(5) Å, and a nearly linear Nα–Nβ–Nγ angle of 174.8(7) °. Additionally, an intramolecular π–π stacking between the aryl unit of the parent organoazide and one of the phenyl rings from the 2,4,6-triphenylphenyl substituents was observed (Figure S-1).48–49
Scheme 1.
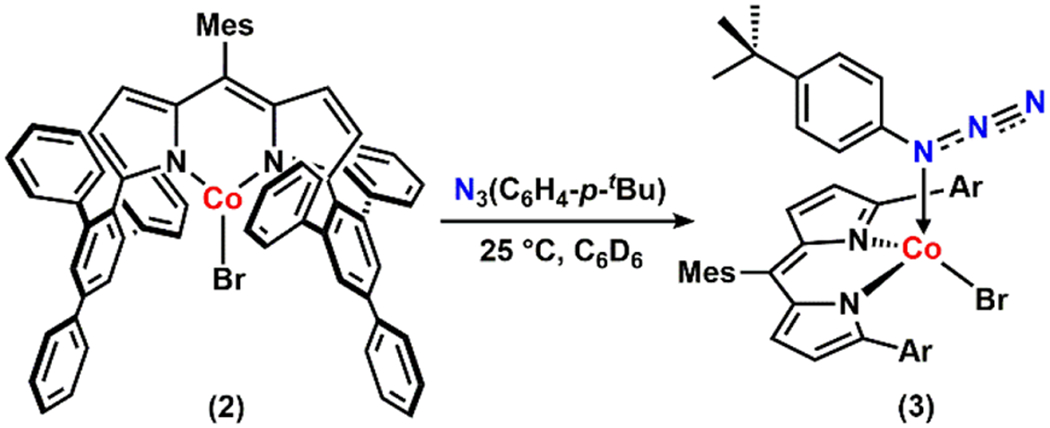
Synthesis of 3
Figure 2.
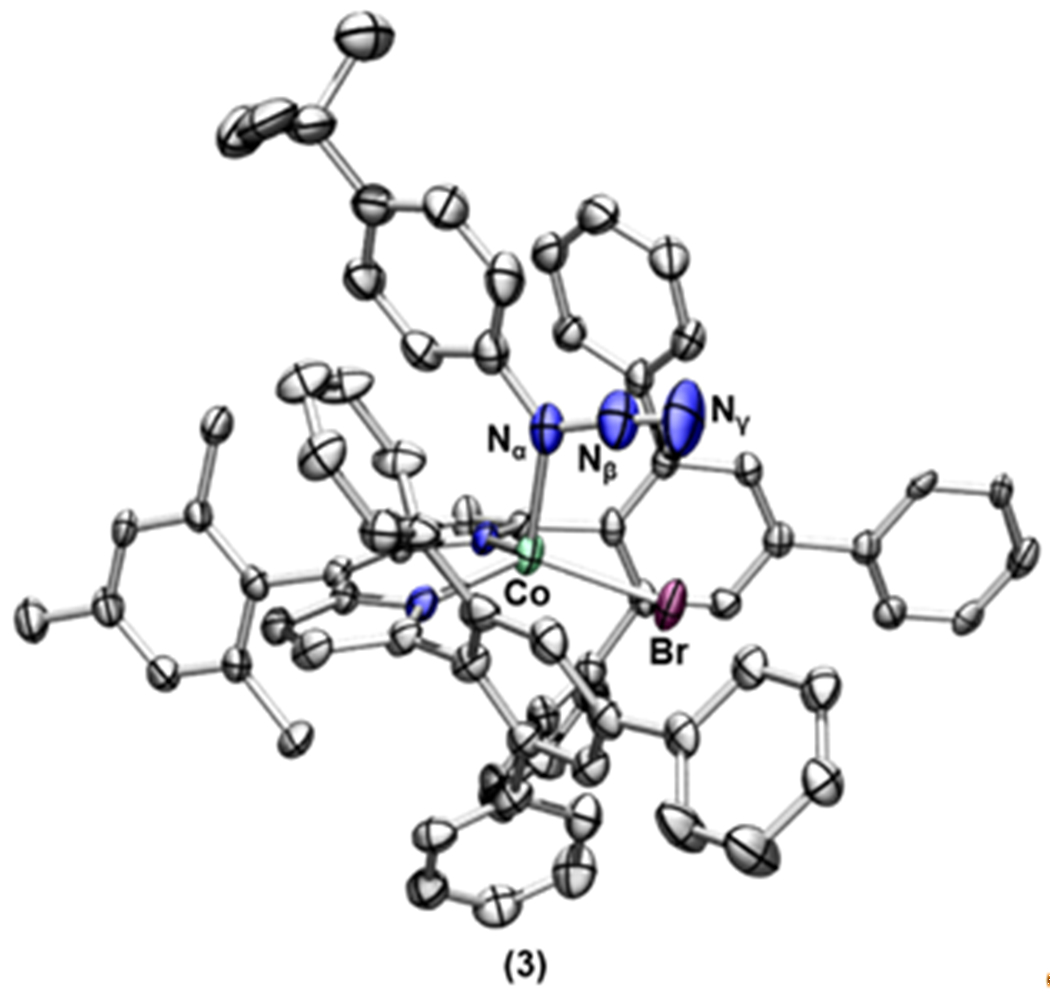
Solid-state structure of 3 at 100 K with thermal ellipsoids set to the 50% probability level (hydrogen atoms and solvent molecules are omitted for clarity; Co aquamarine, C gray, N blue, Br, maroon). Selected bond lengths (Å) and angles (deg) for 3: Co–Nα 2.111(5); Nα–Nβ 1.282(7); Nβ–Nγ 1.125(7); Nα–Nβ–Nγ 174.8(7).
We note that complex 3 displays a relatively rare binding mode of the organoazide, where the cobalt center coordinates to the Nα as opposed to terminal Nγ ligation. To date, only a few complexes feature this binding motif, most of which feature a second- or third-row transition metal such as Rh,50 Pd,24 Ag,32 Ir,31 or Au.51 Among first-row transition metals, a bis(pyrrolyl)pyridine pincer cobalt(II) complex is the only example reported to exhibit a similar interaction with 1-azidoadamantane.34 We note that neither mono nor bis ortho-substituted aryl azides exhibits observable binding to 2, highlighting the preferred Co–Nα interaction over the more sterically accessible Nγ.
2.2. Electronic Structure of 3.
As mentioned above, the binding mode of the organoazide observed for 3 is rare, especially for first-row transition metals. In addition, given that the steric profile of organoazides is not the dominating factor that dictates the binding mode, we hypothesized that 3 may favor certain electronic configurations. As such, we sought to investigate the electronic structure of 3 by characterizing the binding properties of the organoazide. First, as the bond length of Co–Nα (2.111(5) Å) is within the range of a Co–N single bond, we speculated that 3 can be best described as a Lewis acid-base pair. Moreover, we observed that aryl azides such as 2-azido-1,3,5-trifluorobenzene or 1-azido-2,3,4,5,6-pentafluorobenzene display no binding affinity towards 2, suggesting that the relatively electron deficient aryl azides are not sufficiently donating. These results are in line with the lack of reactivity between 1 and 1-azido-4-(tert-butyl)benzene as pyridine is more Lewis basic than the aryl azide, suggesting a simple dative interaction between the cobalt and the organoazide.
The solution IR spectrum of 3 in benzene revealed a distinct blue-shifted ν(N3) stretch at room temperature, indicating the absence of π-backbonding (Figure 3a). Additionally, the frozen toluene solution EPR spectrum of 3 at 4 K displays a typical hyperfine structure of a high-spin (S = 3/2) CoII center, validating the redox-innocent ligation of the azide moiety (Figure 3b). These spectroscopic data are further corroborated with single-point DFT calculations using the crystallographic coordinates of 3 to illustrate that the unpaired spin density (S = 3/2) is mostly localized on the cobalt center (Figure 3c). Thus, we believe that the bonding between the cobalt and the aryl azide can be best depicted as a redox-neutral, Lewis acid-base interaction. This electronic structure assignment is in accord with the previous report by Marynick which showed that in the absence of π-backbonding, coordination through Nα is favored as the substituted nitrogen atom (Nα) is more basic than the terminal Nγ.32
Figure 3.
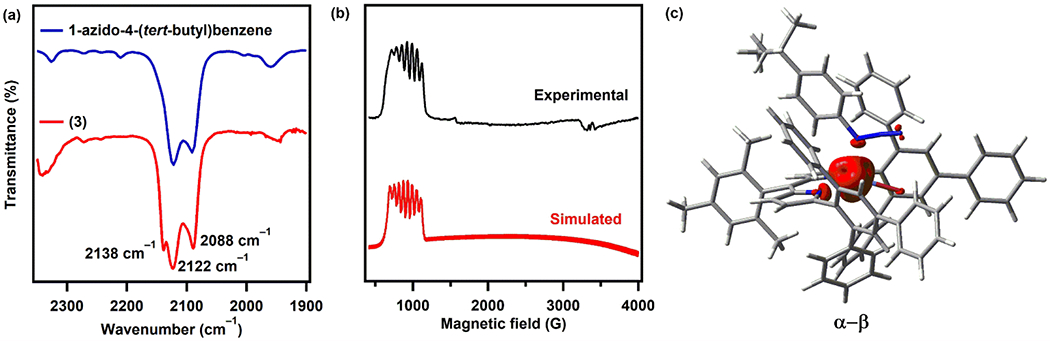
(a) Solution IR spectra of free 1-azido-4-(tert-butyl)benzene (above, blue) and 3 (below, red) in benzene. (b) EPR spectrum of 3 in frozen toluene at 4 K (above, black) and its simulation (below, red); simulation parameters: S = 3/2, g1 = 2.4, g2 = 2.4, g3 = 2.4; A1(Co) = 0 MHz, A2(Co) = 600 MHz, A3(Co) = 200 MHz. (c) Mulliken spin-density plot calculated for 3.52
2.3. N-group Transfer Reactivity of 3.
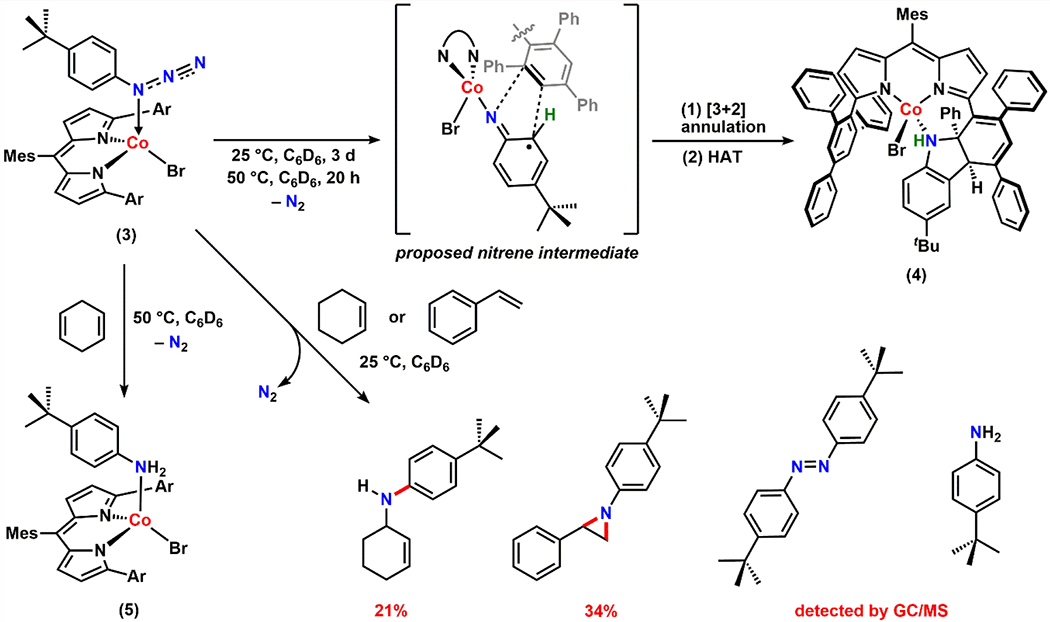
With a comprehensive understanding of the electronic properties of 3, we sought to investigate its N2 extrusion reactivity and subsequent N-group transfer chemistry. While 3 is relatively stable at low temperatures, slow decay to a new paramagnetic species was observed over 3 days at room temperature. The same transformation was completed in 20 hours at 50 °C accompanied by a color change from maroon to purple.
Single crystals of the purple product were grown from a hexane/benzene mixture (2:1) stored at − 35 °C. Interestingly, the solid-state structure of the species revealed a N2-released product (4), which is formed upon ligand functionalization. The central phenyl ring of one of the 2,4,6-triphenylphenyl substituents from ArL is dearomatized following a [3+2] annulation with the parent aryl azide fragment. The aryl moiety of the parent azide restores its aromaticity upon a subsequent H–atom transfer (HAT). Additionally, the solid-state structure highlights the syn-addition of the nitrene moiety into the phenyl group (Scheme 2, Figure 4a).
Scheme 2.
Intra- and Intermolecular Nitrene Group Transfer Reactivity of 3
Figure 4.
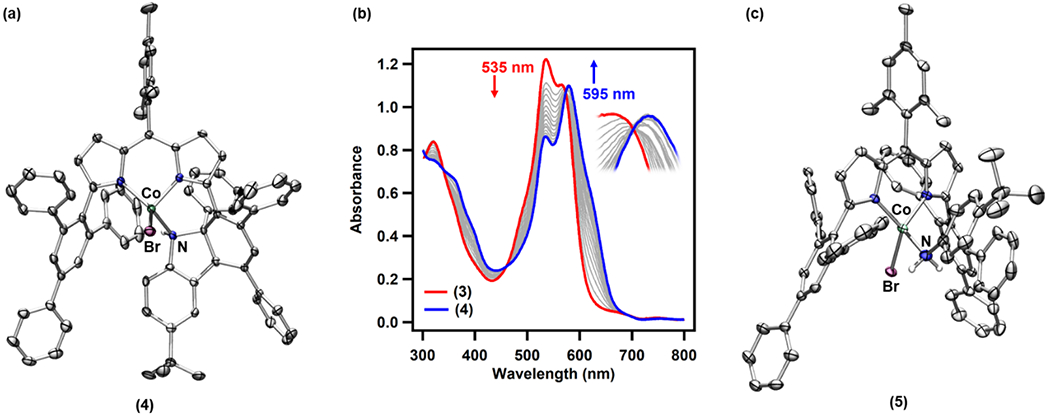
(a) Solid-state structure for 4 at 100 K with the thermal ellipsoids set at the 50% probability level (hydrogen atom except for N–H and solvent molecules are omitted for clarity; Co aquamarine, C gray, N blue, Br maroon, H white). (b) UV/vis traces for the conversion of 3 to 4 in benzene at 50 °C with spectra recorded every 30 min for 20 h. (c) Solid-state structure for 5 at 100 K with the thermal ellipsoids set at the 50% probability level (hydrogen atoms except for aniline N–H and solvent molecules are omitted for clarity; Co aquamarine, C gray, N blue, Br maroon, H white).
For this unique annulation process, either a CoIII iminyl (ArL)CoBr(·N(C6H4-p-tBu)) or a CoIV imido (ArL)CoBr(N(C6H4-p-tBu)) species could serve as a potential intermediate. Given that the aryl moiety of the parent azide is engaged in the transformation, we propose that the CoIII iminyl complex would be the more relevant intermediate, as significant radical delocalization throughout the aryl unit was observed for the iron analogue (ArL)FeCl(·N(C6H4-p-tBu)).11
The conversion from 3 to 4 exhibits no accumulation of intermediate species by 1H NMR spectroscopy and clean isosbestic points (at λ = 340, 460, and 575 nm) are observed via UV/vis spectroscopy (Figure 4b), presumably due to a fast [3+2] annulation process following loss of N2. Nevertheless, in the presence of excess 1,4-cyclohexadiene, a benzene-d6 solution of 3 exclusively furnished the corresponding aniline-bound CoII complex (ArL)CoBr(NH2(C6H4-p-tBu)) (5) at 50 °C, likely following two sequential H–atom abstraction (HAA) steps mediated by the proposed Co-nitrene intermediates (Scheme 2, Figure 4c). Furthermore, at room temperature, exposure of 3 to either 200 equiv of styrene in benzene or neat cyclohexene afforded the corresponding aziridine product (1-(4-(tert-butyl)phenyl)-2-phenylaziridine) or amination product (4-(tert-butyl)-N-(cyclohex-2-en-1-yl)aniline) in 34% and 21% yield, respectively (Scheme 2). Along with the desired products, in both cases, formation of the corresponding diazene and aniline was also detected. Similar diazene generation has been observed in several other N-group transfer reactions mediated by the corresponding metal-nitrene complexes.13, 33, 35 These results corroborate the presence of a transient Co-nitrene equivalent and demonstrate its viability as a nitrene-group delivery reagent.14–15, 53–59
Scheme 4.
Synthesis of 10, 11, and 12
2.4. Reactivity of Dipyrrinato CoII Alkyl Azide Adducts.
Encouraged by the N-group transfer reactivity of 3, we attempted to expand the C–H amination reactivity by modifying the bound organoazide. Particularly, we were inspired by our previously reported Fe-catalyzed intramolecular C–H amination which is mediated by a high-spin ferric alkyl iminyl complex.12, 60 Given that we propose 3 furnishes an analogous high-valent intermediate, we envisioned that the similar transformation affording N-heterocyclic compounds might be feasible by replacing the aryl azide of 3 with an alkyl azide.
Akin to the synthesis of 3, we were able to prepare a series of alkyl azide-bound cobalt complexes by treating 2 with a stoichiometric amount of organoazides such as benzyl azide, (4-azidobutyl)benzene, or 1-azido-4-methylpentane (Scheme 3, complexes 6, 7, and 8, respectively). α-Gem-dimethyl substituted alkyl azides, however, do not bind to 2 likely owing to the steric hindrance around the Nα atom, suggesting that the innate binding preference for Nα over Nγ is retained. Indeed, the solid-state structures of 7 and 8 revealed that each azide binds through the Nα with a Co–Nα distance 2.041(3) Å and 2.053(4) Å, respectively (Figure 5a and 5b). Similar to 3, both 7 and 8 exhibit a distorted trigonal monopyramidal geometry with τ4 = 0.84 and 0.81, respectively. Additionally, solution IR spectrum of 8 displays a distinct blue-shifted azide stretch by 46 cm−1 relative to the free azide (Figure S-2). The frozen toluene EPR spectrum of 8 features a hyperfine structure for high-spin CoII, indicating a similar dative interaction of the alkyl azide as observed for the aryl congener 3 (Figure S-6). Given that adducts of linear alkyl azides are often proposed as intermediates for the transition-metal-mediated ring-closing catalysis to generate N-heterocycles, it is noteworthy that no such adduct has been structurally characterized prior to this study.12, 42, 44, 61
Scheme 3.
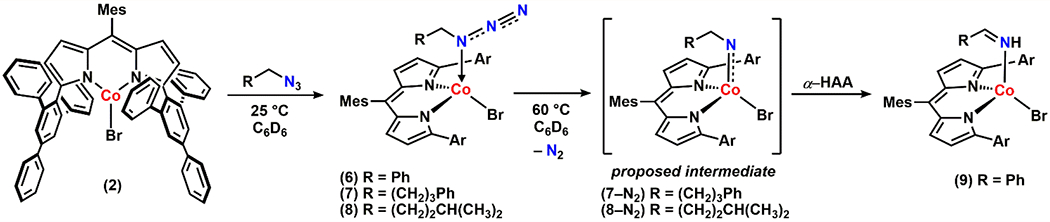
Synthesis and Reactivity of Dipyrrinato CoII Alkyl Azide Adduct Complexes
Figure 5.
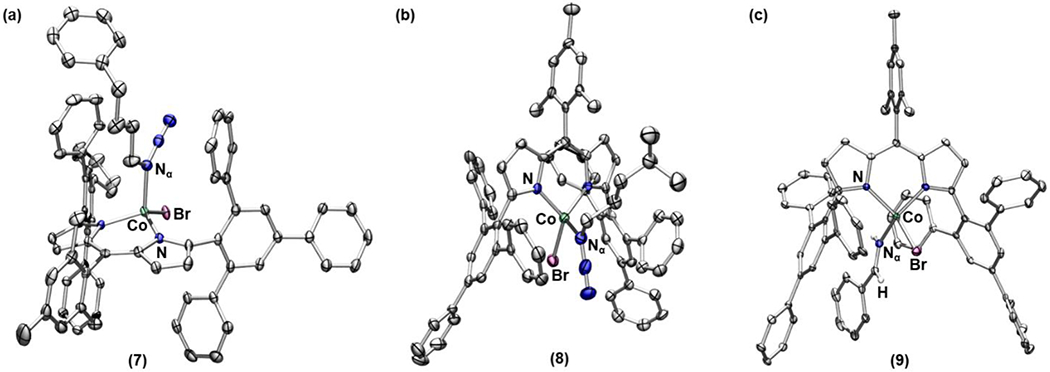
Solid-state structures for (a) 7, (b) 8 and (c) 9 at 100 K with thermal ellipsoids at the 50% probability level (hydrogen atoms except for imine moiety of 9 and solvent molecules are omitted for clarity; Co aquamarine, C gray, N blue, Br maroon, H white).
Interestingly, these alkyl azide-bound CoII complexes display distinct kinetic profiles from 3. First, unlike 3, none of these complexes (6-8) expels N2 at room temperature. Instead, upon heating to 60 °C in benzene-d6, quantitative formation of the corresponding imine adduct was observed (Scheme 3). In the case of 6, the corresponding benzyl imine bound complex (9) was crystallographically characterized with a Co–Nα distance of 2.029(2) Å (Figure 5c). A similar transformation has been observed in our previous studies on C–H amination catalysis mediated by either a ferric iminyl10, 12 or a cobalt imido46 species via rapid intramolecular α–H–atom abstraction.57 Thus, the reactivity of 6-8 is in line with our proposed generation of a CoIII iminyl or CoIV imido intermediate (Scheme 3). Moreover, unlike the conversion process of 3 to 4 showing no accumulation of intermediates, a paramagnetic intermediate was observed by 1H NMR spectroscopy while heating a benzene-d6 solution of 8 at 60 °C to afford the corresponding imine adduct (Figure S-7). We speculated that the observed transient intermediate could be the proposed cobalt nitrene complex. However, our attempts to isolate the intermediate were unsuccessful due to the rapid subsequent α–HAA.
2.5.1. In Crystallo Thermal Extrusion of N2 from 8.
We envisioned that observation of the N2-released intermediate (e.g., 7–N2 and 8–N2 in Scheme 3) may be accomplished in a crystal lattice upon thermolysis.62–63 Given that 8 reacts above 60 °C in solution, a single crystal of 8 was heated to 345 K on an X-ray diffractometer for 5–10 min followed by cooling back to 100 K for full data collection. The thermolysis was repeated four times on the same single crystal of 8 until the crystal decomposed and each data set was compared to the initial 100 K X-ray structure to monitor relative changes in occupancy of the nitrogen atoms (Nα, Nβ, and Nγ) and the bond distance between Co and the bound nitrogen atom (Nα).
Indeed, noticeable changes in occupancy of the Nβ and Nγ were observed while the space group of the crystal (monoclinic P21/c) did not change. Specifically, the occupancies of Nβ and Nγ decreased by 16% (relative conversion) upon the first heating and by an additional 4% (relative conversion) over the subsequent three heating iterations to afford an overall 20% conversion. Based on the relatively subtle changes in the occupancies of nitrogen atoms over the last three heating iterations, we propose that the solid-state reaction reached maximum conversion within a few minutes.64 Nevertheless, because the quality of the single crystal sample decays upon N2 escape from the crystal lattice, we cannot fully exclude the possibility that the lower quality of the crystal inhibits observation of higher conversions.65–66 The results suggest that loss of N2 occurred upon cleavage of the Nα –Nβ bond as indicated in a reaction-difference map (Table S-2 and Figure S-26). The reaction difference map indicates the difference between the two data sets obtained before and after the four iterations of heating of the same single crystal of 8. While almost no change has been detected for the ancillary ligand (ArL), a significantly shorter Co–Nα distance 1.82(3) Å as compared to the initial distance of 2.053(4) Å was observed from the final model of 8–N2 which is refined based on the changes in the reaction-difference map (Figures 6a). We do not refine electron density for the extruded N2, which is likely lost from the crystal at the elevated temperatures used to promoted N–N cleavage.67
Figure 6.
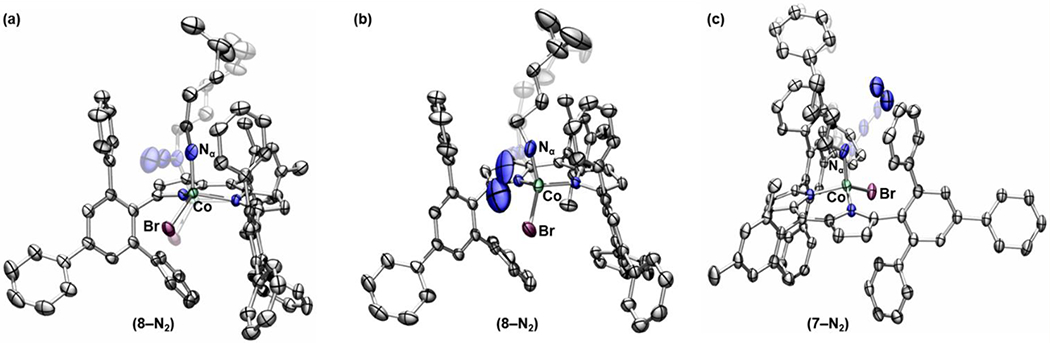
Solid-state structures for (a) 8–N2 collected at 100 K following heating to 345 K, (b) 8–N2, and (c) 7–N2 collected at 100 K upon X-ray irradiation with thermal ellipsoids at the 50% probability level (hydrogen atoms and solvent molecules are omitted for clarity; Co aquamarine, C gray, N blue, Br maroon).
Given that one of our previously reported four-coordinate CoIII complexes, a metallacycloindoline (ArL)Co(κ2-NHC6H2-2,4-Me2-6-CH2) exhibits a Co–N bond length of 1.845(3) Å,45 the observed Co–Nα bond length (1.82(3) Å) suggests that 8–N2 is likely a CoIII iminyl rather than a CoIV imido complex. Furthermore, computational analysis on 8–N2 reveals significant spin density localized on the Nα, suggesting that the nitrene (NR) unit is more likely an open-shell iminyl (2NR1−) as opposed to a closed-shell imido (NR2−), proposing that 8–N2 can be best described as a Co(III) iminyl rather than a Co(IV) imido (Table S-4 and S-5).
2.5.2. In Crystallo X-ray Induced N2–Extrusion from 7 and 8.
In order to gain further evidence for the proposed intermediate and minimize the heat-induced crystal decay, we have performed the similar experiments at a significantly lower temperature (100 K) upon irradiation with a high-flux X-ray source.68–69 Indeed, in crystallo N2 loss was also observed when crystallographic data was collected using a synchrotron source (0.41328 Å, 30 keV) at the Advanced Photon Source (APS), Argonne National Laboratory (ANL). Prolonged irradiation of each of 7 and 8 with a high-flux synchrotron source led to the X-ray induced conversion to 7–N2 (69% conversion) and 8–N2 (65% conversion), respectively. Conversion was calculated based on occupancy changes of the Nβ and Nγ (Figures S-28, S-29). Both 7–N2 and 8–N2 exhibit significantly contracted Co–Nα distances of 1.840(7) Å and 1.787(8) Å, respectively, which is in agreement with the preliminary result observed upon thermolysis (Figure 6b–c). In contrast to the result from thermolysis and presumably due to the lower temperature at which it was generated, the extruded N2 moiety was observed along with the proposed iminyl complex. Based on the observation that evolved N2 is not observed in 8-heat(1–4) (presumably due to diffusion out of the crystalline sample), we allowed the N2 occupancy to refine independent of the occupancy of the iminyl fragment. X-ray induced elimination of N2 from 7 or 8 was not observed using rotating anode sources, and thus these observations indicate that N3 activation is induced by sufficiently high-flux X-ray irradiation. Similar flux-induced chemical reactions have been observed in disulfide cleavage reactions during prolonged X-ray irradiation of protein samples and has been suggested to arise as a mechanism to dissipate absorbed X-ray energy.70,71–72
These results closely parallel recent observations of photochemical and X-ray induced loss of N2 from Ru2 azide 63 and Rh2 adamantyl azide 50, 73 complexes, which resulted in nitride and nitrene intermediates, respectively. Combined with the present example of N2 loss from a cobalt complex featuring organic azide ligands, this suite of reports highlights the potential from in crystallo chemical transformations to provide access to crystalline samples of reactive intermediates that are insufficiently stable to be handled by more classical methods. Further, while in the Ru2 and Rh2 examples, significant conversion was obtained in the solid state, the current case highlights the potential to derive chemically meaningful structural information even from incompletely converted crystalline samples.
The significantly contracted Co–Nα distances lead us to formulate 7–N2 and 8–N2 as the corresponding Co-iminyl complexes. Given high errors in our X-ray studies as a result of rapid crystal decomposition during data collection and the fact that detecting the presence of H–atoms is often limited by single crystal X-ray diffraction techniques, we cannot completely exclude the possibility that the observed N2-released structures (7–N2 and 8–N2) could be either the corresponding CoIII amide (NHR) or CoII imine (NH=CHR’) complexes. Our assignment is supported by the observation that both 7 and 8 are stable at room temperature (as indicated by 1H NMR spectroscopy (Figure S-16)) and their chemical composition has been corroborated by CHN elemental analysis, which excludes co-crystallization of 7 or 8 along with their corresponding CoIII amide or CoII imine complex. Moreover, neither 7 nor 8 exhibits intermolecular HAA reactivity as mentioned in section 2.4, suggesting that formation of a CoIII amide complex is highly unlikely, especially in the solid-state. Lastly, as mentioned above, 7–N2 and 8–N2 display significantly different Co–Nα distances with respect to the one from the authentic imine adduct 9 (2.029(2) Å). However, we emphasize that further studies and spectroscopic characterization will be necessary to better assign these compounds’ identity and understand their electronic structure.
2.6. Ligand Modification for a Sterically Accessible CoII Synthon.
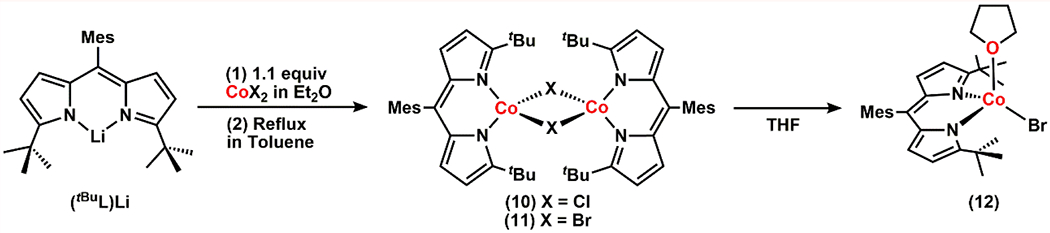
With a better understanding of the structures and reactivity of Co-organoazide adducts and the resulting Co-nitrene complexes, we hypothesized that the steric hindrance of ArL limits the flexibility of the alkyl chain of 7 and 8, thus precluding the desired cyclization. As such, we attempted to synthesize a sterically more accessible dipyrrinato CoII synthon. Metalation of (tBuL)Li (tBuL = 5-mesityl-(1,9-di-tert-butyl)dipyrrin) with anhydrous CoCl2 or CoBr2 in diethyl ether followed by refluxing in toluene afforded the dimeric species [(tBuL)CoCl]2 (10) and [(tBuL)CoBr]2 (11), respectively (Scheme 4, Figure 7a). Complexes 10 and 11 are suitable synthons to explore further reactivity with organoazides as they do not feature exogenous solvent molecules which can potentially hamper the desired binding of organoazide substrates. Moreover, such dimers can readily dissociate into the corresponding solvent-bound monomeric species, when exposed to Lewis basic molecules, as exemplified by the thf-adduct 12 (Scheme 4, Figure 7b). This result suggests a similar dissociation may be possible in the presence of organoazides to generate the corresponding monomeric organoazide-bound complex.
Figure 7.
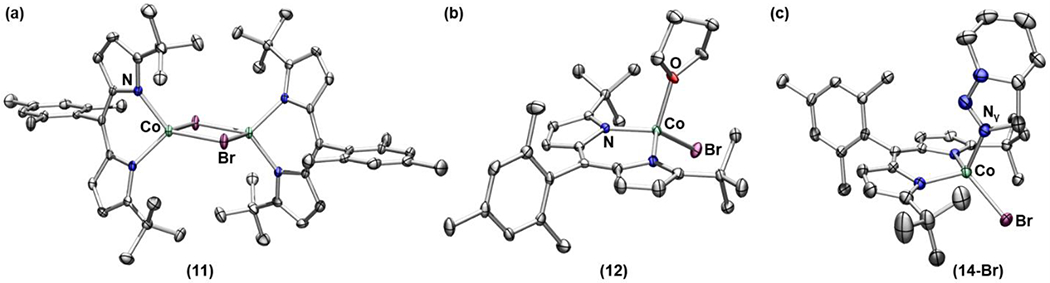
(a) Solid-state structures for (a) 11, (b) 12, and (c) 14-Br at 100 K with thermal ellipsoids at the 50% probability level (hydrogen atoms and solvent molecules are omitted for clarity; Co aquamarine, C gray, N blue, O red, Br maroon).
Indeed, upon treatment of either 10 or 11 with a stoichiometric amount of 6-azidohex-1-ene, the immediate formation of the corresponding azide adduct (13) was confirmed by 1H NMR spectroscopy (Scheme 5). Interestingly, a benzene-d6 solution of 13 readily converts into a new paramagnetic species at room temperature, identified as the corresponding 1,2,3-dihydrotriazole-bound cobalt complex (14) via X-ray diffraction studies (Scheme 5, Figure 7c). The solid-state structure of 14-Br displays the product bound through the Nγ instead of Nα, presumably due to the steric encumbrance around the tri-substituted Nα. It is noteworthy that the reaction occurs at room temperature, since in the absence of metal complexes, a similar transformation occurs only upon refluxing with an activated olefin moiety.74 Furthermore, as 1,3-dipolar cycloadditions are generally facilitated by Lewis-acids,75–78 the observation is in line with our assignment that the cobalt center is Lewis acidic. The observed cycloaddition transformation supports our initial hypothesis that smaller substituents on the dipyrrin platform enhance the flexibility of the alkyl chain.
Scheme 5.
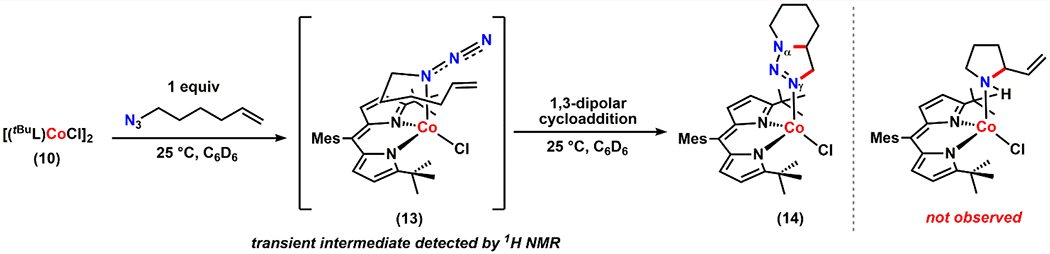
1,3-Dipolar Cycloaddition Reactivity
Encouraged by this result, we next subjected a series of alkyl azides lacking internal olefins such as (4-azidobutyl)benzene, 1-azido-4-methylpentane, or 1-azidobutane to a benzene-d6 solution of 10. In all cases, the corresponding alkyl azide-bound complexes were generated immediately at room temperature as judged by 1H NMR spectroscopy (Scheme 6, complexes 15, 17, and 19). Akin to 2, neither 10 nor 11 binds α-gem-dimethyl substituted alkyl azides suggesting the binding propensity towards the Nα–atom is maintained. Furthermore, solution IR spectra of these adducts in benzene exhibit a blue-shifted azide stretch validating that the azide coordination is redox innocent (Figures S-3, S-4, and S-5).
Scheme 6.
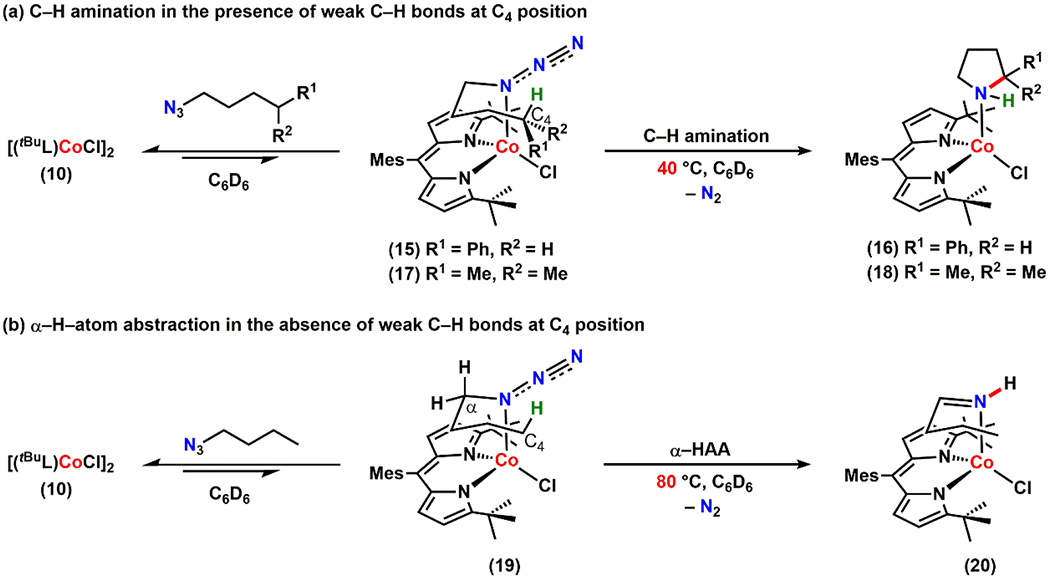
Intramolecular C–H Amination or Activation
In contrast to 7 or 8, however, our attempts to isolate any of these azide-bound species were unsuccessful due to a greater driving force to regenerate the initial dimeric species (10 or 11) during crystallization, emphasizing the reversible binding of the organoazide. Nonetheless, the maximum value in the Job plot between 11 and 1-azido-4-methylpentane appeared at 0.5 mole fraction of a total concentration of CoII center ([CoII]total = 2[11]) suggesting a 1:1 complexation of a single CoII center and each of the organoazides as a monomeric species (e.g., 17) (Figure S-9). Moreover, the changes in 1H NMR chemical shifts of 11 as a function of concentration of organoazides to the corresponding monomeric organoazide adducts are almost identical regardless of the identity of the aliphatic group of the azide substrates, suggesting the nearly same binding affinities of the azides to a CoII center (Figure S-10).
Gratifyingly, upon heating a benzene-d6 solution of 15 and 17 at 40 °C, we observed clean formation of the corresponding pyrrolidine-bound complexes (16) and (18), respectively (Scheme 6a, Figure 8a–b). It is notable that loss of N2 from 15 and 17 occurs at a lower temperature compared to the reactivity observed from 7 and 8 and proceeds without any imine formation. Surprisingly, at the same temperature (40 °C), no reaction was observed for 19, which possesses a stronger, primary C–H bond at the C4 position of the alkyl chain. Instead, 19 expels N2 upon heating to 80 °C to afford the corresponding imine adduct (20) via α–H–atom abstraction (Scheme 6b, Figure 8c). Given that N2 elimination is irreversible, these observations suggest the following: (1) the temperature required for N2 expulsion and subsequent cyclization depends on the C–H bond strength on the fourth carbon (C4) of the linear alkyl azide and (2) there is no formation of an N2-released intermediate (such as a CoIV imido or CoIII iminyl species) at relatively low temperatures as the azide moiety of 19 remains intact at 40 °C.
Figure 8.
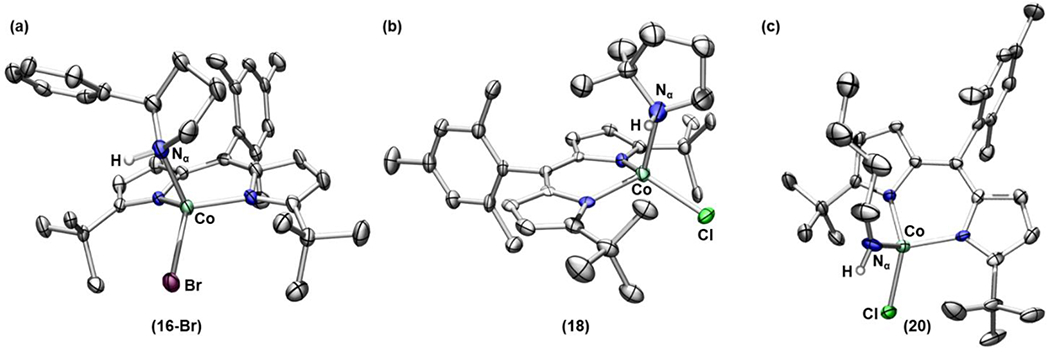
Solid-state structures for (a) 16-Br, (b) 18, and (c) 20 at 100 K with thermal ellipsoids at the 50 % probability level (hydrogen atoms except for N–H or imine C–H and solvent molecules are omitted for clarity; Co aquamarine, C gray, N blue, Br maroon, Cl green, H white.)
Thus, we hypothesize that activation of the azide moiety can be induced by the interaction with C–H bonds when those bonds are weak enough to be activated, suggesting that the azide adducts themselves are viable species for C–H amination. To further support this hypothesis, we heated a benzene-d6 solution of 19 in the presence of excess 1,4-cyclohexadiene to investigate whether an external weak C–H bond can promote loss of N2 at a lower temperature. Indeed, under these conditions, slow conversion to the corresponding amine adduct (21) was observed at 40 °C (Scheme 7, Figure S-11). Recalling that 19 expels N2 only above 80 °C in the absence of 1,4-cyclohexadiene, this result supports the idea that the interaction of the azide moiety with a weak C–H bond is a key step towards N2 release and subsequent C–H amination.
Scheme 7.
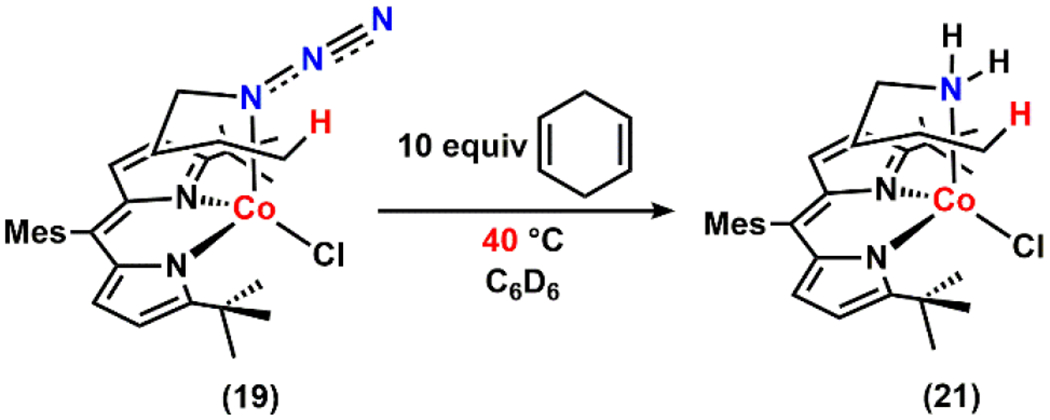
Intermolecular H–Atom Abstraction from 19
Taken together, we propose two distinct pathways for activation of the azide moiety followed by intramolecular C–H amination or C–H activation. First, in the presence of relatively weak C–H bonds (e.g., benzylic or tertiary C–H bonds), we believe that formation of pyrrolidines is directly mediated by the azide adducts, rather than a high-valent intermediate such as a CoIV imido or CoIII iminyl species (Scheme 8, pathway I). Such direct nitrene group transfer reactivity has been previously reported from a few proposed metal-organoazide adduct complexes. For example, benzylic C–H bond amination and olefin aziridination catalyzed by Co38 and Ru40 porphyrin complexes, respectively, are proposed to be mediated by azide adduct species as opposed to the corresponding metal nitrene complexes. Likewise, C–H activation or group transfer reactions promoted by a simple oxidant adduct is not only limited to organoazide-bound complexes but also observed for iodoimine (ArI=NTs),79 iodosylarene (ArIO) or iodoxyarene (ArIO2) adducts,80–81 suggesting that metal-ligand multiply bonded species are not always the active group transfer reagents. While we cannot fully exclude the presence of a high-energy intermediate complex such as a reorganized form of the azide adducts for this specific route (pathway I), we propose that these cobalt-organoazide adducts serve as active group transfer reagents. Furthermore, we believe that a key feature enabling pathway I is the sterically accessible dipyrrin scaffold tBuL facilitates the positioning of the weak C–H bonds at C4 proximal to the azide moiety for concomitant C–H activation and C–N bond formation; whereas proximal orientation of the substrate C–H bonds is not possible for the more sterically encumbered ArL. Secondly, in the absence of proximal weak C–H bonds, C–H activation (i.e., α–H–atom abstraction) may be mediated by the corresponding CoIV imido or CoIII iminyl intermediates, which can be generated at relatively high temperatures via thermal N2 extrusion (Scheme 8, pathway II). We propose that such intermediates generated at high temperatures would structurally resemble the Co-nitrene complexes described in Sections 2.5.1. and 2.5.2.
Scheme 8.
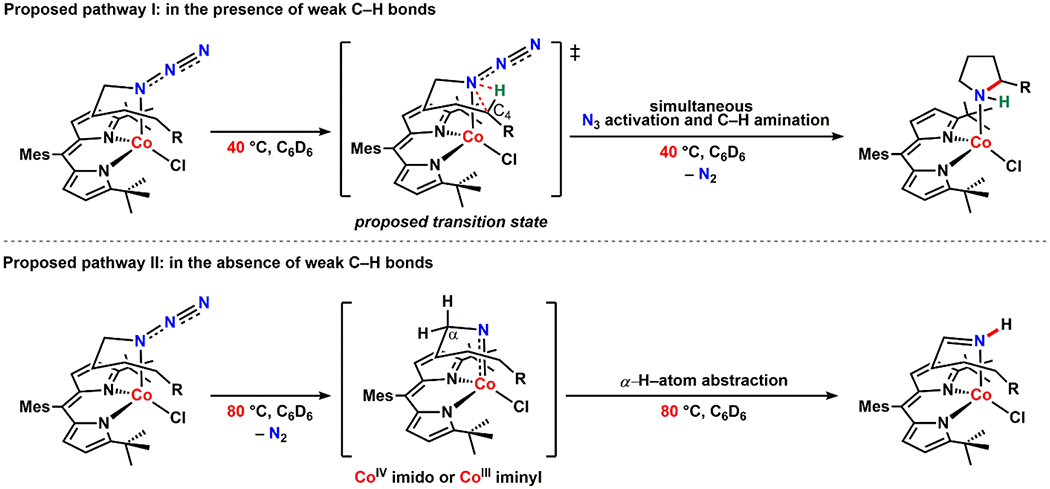
Proposed Pathways for Intramolecular C–H Amination or Activation
3. CONCLUSIONS
In this report, we demonstrated the synthesis of a series of organoazide-bound dipyrrinato CoII complexes and highlighted that their nitrene group transfer reactivity can be tuned by the (1) identity of the bound organoazide (aryl azide versus alkyl azide) and (2) steric properties of the substituents on the dipyrrin scaffold (2,4,6-triphenylphenyl versus tert-butyl). First, by utilizing a sterically encumbered ligand ArL, the binding mode of the organoazides was characterized by single-crystal X-ray diffraction, solution IR spectroscopy, and EPR spectroscopy (at 4 K) to identify a redox neutral coordination through the Nα preferentially over Nγ. Although such binding mode is consistent among varying the R substituents (aryl versus alkyl) of the organoazides, we observed notably different kinetics for N2 extrusion as well as scope of N-group transfer reactivity. For instance, the aryl azide-bound complex 3 eliminates N2 at room temperature to undergo either intra- or intermolecular C–H amination. We propose that such transformations are mediated by a CoIV imido or CoIII iminyl species, yet no accumulation of intermediates was spectroscopically detected. In contrast, the analogous complex 8 featuring an aliphatic azide expels N2 only above 60 °C, and an intermediate species was observed during the conversion into the corresponding imine compound. Although isolation of the intermediate was not feasible in solution due to the facile subsequent α–H–atom abstraction, we were able to cleave the N–N bond within a crystal lattice to observe the corresponding N2-released structure, which we have assigned as the corresponding Co-nitrenoid (i.e., CoIII(2NR), CoIV(NR)) complex. Future work will focus on studying the electronic structure of the Co-nitrene complexes via in-depth spectroscopic characterizations.
Lastly, by employing a more sterically accessible CoII synthon 10 or 11, intramolecular 1,3-dipolar cycloaddition as well as C–H amination to generate the 1,2,3-dihyrotriazole and substituted pyrrolidine products, respectively, were accomplished. Most importantly, our observations on reactivity patterns of alkyl azide adducts supported by the smaller dipyrrin scaffold (15, 17, and 19) suggest that organoazide-bound complexes themselves are competent for the desired C–H amination when weak C–H bonds are positioned proximal to the azide moiety. This demonstrates that organoazide adducts can also be viable species for C–H amination along with the typically invoked N-group transfer reagents such as metal nitrene complexes. We believe this study provides great insight towards understanding the nature and reactivity of commonly not isolable organoazide-bound metal complexes for C–H amination processes.
Supplementary Material
ACKNOWLEDGMENT
T.A.B. acknowledges this work was supported by a grant from the NIH (GM-115815), the Dreyfus Foundation (Teacher-Scholar Award), and Harvard University. Y.B. gratefully acknowledges a Kwanjeong Graduate Student Fellowship. D.C.P. thanks the Welch Foundation (A-1907) and the U.S. Department of Energy (DE-SC0018977) for financial support. The authors thank Yu-Sheng Chen for helpful discussion. Crystal structures of 7–N2 and 8–N2 in section 2.5.2 were determined at ChemMatCARS Sector 15, which is housed at the APS, which is principally supported by the NSF (Grant NSF/ CHE-1346572). Use of the PILATUS3 X CdTe 1M detector is supported by the NSF (Grant NSF/DMR-1531283). Use of the APS was supported by the U.S. DOE under Contract No. DE-AC02–06CH11357.
Footnotes
The authors declare no competing financial interests.
This material is available free of charge via the Internet at http://pubs.acs.org.
General experimental considerations and procedures, multinuclear NMR data, IR spectra, EPR spectra, computational details (PDF)
Crystallographic data (CIF)
REFERENCES
- 1.Davies HML; Long MS, Recent advances in catalytic intramolecular C–H aminations. Angew. Chem. Int. Ed 2005, 44 (23), 3518–3520. [DOI] [PubMed] [Google Scholar]
- 2.Jason AH, Recent advances in metal-mediated carbon-nitrogen bond formation reactions: aziridination and amidation. Curr. Org. Chem 2005, 9 (7), 657–669. [Google Scholar]
- 3.Davies HML; Manning JR, Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collet F; Dodd RH; Dauban P, Catalytic C–H amination: recent progress and future directions. Chem. Commun 2009, 0 (34), 5061–5074. [DOI] [PubMed] [Google Scholar]
- 5.Zalatan DN; Bois JD, Metal-catalyzed oxidations of C–H to C–N bonds. In: Yu J-Q; Shi Z, (eds) C–H activation. Topics in Current Chemistry, vol 292. Springer, Berlin, Heidelberg, 2009; 347–378. [DOI] [PubMed] [Google Scholar]
- 6.Müller P; Fruit C, Enantioselective catalytic aziridinations and asymmetric nitrene insertions into C–H Bonds. Chem. Rev 2003, 103 (8), 2905–2920. [DOI] [PubMed] [Google Scholar]
- 7.Saouma CT; Peters JC, M≡E and M=E complexes of iron and cobalt that emphasize three-fold symmetry (E=O, N, NR). Coord. Chem. Rev 2011, 255 (7), 920–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehn MP; Peters JC, Mid- to high-valent imido and nitrido complexes of iron. J. Inorg. Biochem 2006, 100 (4), 634–643. [DOI] [PubMed] [Google Scholar]
- 9.Zhang L; Deng L, C–H bond amination by iron-imido/nitrene species. Chin. Sci. Bull 2012, 57 (19), 2352–2360. [Google Scholar]
- 10.Hennessy ET; Liu RY; Iovan DA; Duncan RA; Betley TA, Iron-mediated intermolecular N-group transfer chemistry with olefinic substrates. Chem. Sci 2014, 5 (4), 1526–1532. [Google Scholar]
- 11.Wilding MJT; Iovan DA; Wrobel AT; Lukens JT; MacMillan SN; Lancaster KM; Betley TA, Direct comparison of C–H bond amination efficacy through manipulation of nitrogen-valence centered redox: imido versus iminyl. J. Am. Chem. Soc 2017, 139 (41), 14757–14766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hennessy ET; Betley TA, Complex N-heterocycle synthesis via iron-catalyzed, direct C–H bond amination. Science 2013, 340 (6132), 591–595. [DOI] [PubMed] [Google Scholar]
- 13.Iovan DA; Betley TA, Characterization of iron-imido species relevant for N-group transfer chemistry. J. Am. Chem. Soc 2016, 138 (6), 1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goswami M; Lyaskovskyy V; Domingos SR; Buma WJ; Woutersen S; Troeppner O; Ivanović-Burmazović I; Lu H; Cui X; Zhang XP; Reijerse EJ; DeBeer S; van Schooneveld MM; Pfaff FF; Ray K; de Bruin B, Characterization of porphyrin-Co(III)-‘nitrene radical’ species relevant in catalytic nitrene transfer reactions. J. Am. Chem. Soc 2015, 137 (16), 5468–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lyaskovskyy V; Suarez AIO; Lu H; Jiang H; Zhang XP; de Bruin B, Mechanism of cobalt(II) porphyrin-catalyzed C–H amination with organic azides: radical nature and H–atom abstraction ability of the key cobalt(III)–nitrene intermediates. J. Am. Chem. Soc 2011, 133 (31), 12264–12273. [DOI] [PubMed] [Google Scholar]
- 16.Kuijpers PF; van der Vlugt JI; Schneider S; de Bruin B, Nitrene radical intermediates in catalytic synthesis. Chem. Eur. J 2017, 23 (56), 13819–13829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li C; Lang K; Lu H; Hu Y; Cui X; Wojtas L; Zhang XP, Catalytic radical process for enantioselective amination of C(sp3)–H bonds. Angew. Chem. Int. Ed 2018, 57 (51), 16837–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu H; Li C; Jiang H; Lizardi CL; Zhang XP, Chemoselective amination of propargylic C(sp3)–H bonds by cobalt(II)-based metalloradical catalysis. Angew. Chem. Int. Ed 2014, 53 (27), 7028–7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang H; Lang K; Lu H; Wojtas L; Zhang XP, Asymmetric radical bicyclization of allyl azidoformates via cobalt(II)-based metalloradical catalysis. J. Am. Chem. Soc 2017, 139 (27), 9164–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conradie J; Ghosh A, Electronic structure of an iron-porphyrin–nitrene complex. Inorg. Chem 2010, 49 (1), 243–248. [DOI] [PubMed] [Google Scholar]
- 21.Corona T; Ribas L; Rovira M; Farquhar ER; Ribas X; Ray K; Company A, Characterization and reactivity studies of a terminal copper–nitrene species. Angew. Chem. Int. Ed 2016, 55 (45), 14005–14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berry JF, Terminal nitrido and imido complexes of the late transition metals. Comments Inorg. Chem 2009, 30 (1-2), 28–66. [Google Scholar]
- 23.Bräse S; Gil C; Knepper K; Zimmermann V, Organic azides: an exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed 2005, 44 (33), 5188–5240. [DOI] [PubMed] [Google Scholar]
- 24.Barz M; Herdtweck E; Thiel WR, Transition metal complexes with organoazide ligands: synthesis, structural chemistry, and reactivity. Angew. Chem. Int. Ed 1998, 37 (16), 2262–2265. [DOI] [PubMed] [Google Scholar]
- 25.Cenini S; Gallo E; Caselli A; Ragaini F; Fantauzzi S; Piangiolino C, Coordination chemistry of organic azides and amination reactions catalyzed by transition metal complexes. Coord. Chem. Rev 2006, 250 (11), 1234–1253. [Google Scholar]
- 26.Proulx G; Bergman RG, Synthesis and structure of a terminal metal azide complex: an isolated intermediate in the formation of imidometal complexes from organic azides. J. Am. Chem. Soc 1995, 117 (23), 6382–6383. [Google Scholar]
- 27.Proulx G; Bergman RG, Synthesis, structures, and kinetics and mechanism of decomposition of terminal metal azide complexes: isolated intermediates in the formation of imidometal complexes from organic azides. Organometallics 1996, 15 (2), 684–692. [Google Scholar]
- 28.Fickes MG; Davis WM; Cummins CC, Isolation and structural characterization of the terminal mesityl azide complex V(N3Mes)(I)(NRArF)2 and its conversion to a vanadium(V) imido complex. J. Am. Chem. Soc 1995, 117 (23), 6384–6385. [Google Scholar]
- 29.Harman WH; Lichterman MF; Piro NA; Chang CJ, Well-defined vanadium organoazide complexes and their conversion to terminal vanadium imides: structural snapshots and evidence for a nitrene capture mechanism. Inorg. Chem 2012, 51 (18), 10037–10042. [DOI] [PubMed] [Google Scholar]
- 30.Guillemot G; Solari E; Floriani C; Rizzoli C, Nitrogen-to-metal multiple bond functionalities: the reaction of calix[4]arene–W(IV) with azides and diazoalkanes. Organometallics 2001, 20 (4), 607–615. [Google Scholar]
- 31.Albertin G; Antoniutti S; Baldan D; Castro J; García-Fontán S, Preparation of benzyl azide complexes of iridium(III). Inorg. Chem 2008, 47 (2), 742–748. [DOI] [PubMed] [Google Scholar]
- 32.Dias HVR; Polach SA; Goh S-K; Archibong EF; Marynick DS, Copper and silver complexes containing organic azide ligands: syntheses, structures, and theoretical investigation of [HB(3,5-(CF3)2Pz)3]CuNNN(1-Ad) and [HB(3,5-(CF3)2Pz)3]AgN(1-Ad)NN (where Pz = Pyrazolyl and 1-Ad = 1-Adamantyl). Inorg. Chem 2000, 39 (17), 3894–3901. [DOI] [PubMed] [Google Scholar]
- 33.Takaoka A; Moret M-E; Peters JC, A Ru(I) metalloradical that catalyzes nitrene coupling to azoarenes from arylazides. J. Am. Chem. Soc 2012, 134 (15), 6695–6706. [DOI] [PubMed] [Google Scholar]
- 34.Grant LN; Carroll ME; Carroll PJ; Mindiola DJ, An unusual cobalt azide adduct that produces a nitrene species for carbon–hydrogen insertion chemistry. Inorg. Chem 2016, 55 (16), 7997–8002. [DOI] [PubMed] [Google Scholar]
- 35.Mankad NP; Müller P; Peters JC, Catalytic N–N coupling of aryl azides to yield azoarenes via trigonal bipyramid iron–nitrene intermediates. J. Am. Chem. Soc 2010, 132 (12), 4083–4085. [DOI] [PubMed] [Google Scholar]
- 36.Waterman R; Hillhouse GL, η2-Organoazide complexes of nickel and their conversion to terminal imido complexes via dinitrogen extrusion. J. Am. Chem. Soc 2008, 130 (38), 12628–12629. [DOI] [PubMed] [Google Scholar]
- 37.Hanna TA; Baranger AM; Bergman RG, Addition of organic 1,3-dipolar compounds across a heterobinuclear bond between early and late transition metals: mechanism of nitrogen loss from an organoazido complex to form a bridging imido complex. Angew. Chem. Int. Ed. Engl 1996, 35 (6), 653–655. [Google Scholar]
- 38.Ragaini F; Penoni A; Gallo E; Tollari S; Li Gotti C; Lapadula M; Mangioni E; Cenini S, Amination of benzylic C–H bonds by arylazides catalyzed by CoII–porphyrin complexes: a synthetic and mechanistic study. Chem. Eur. J 2003, 9 (1), 249–259. [DOI] [PubMed] [Google Scholar]
- 39.Fantauzzi S; Gallo E; Caselli A; Ragaini F; Macchi P; Casati N; Cenini S, Origin of the deactivation in styrene aziridination by aryl azides, catalyzed by ruthenium porphyrin complexes. Structural characterization of a Δ2-1,2,3-triazoline RuII(TPP)CO complex. Organometallics 2005, 24 (20), 4710–4713. [Google Scholar]
- 40.Fantauzzi S; Gallo E; Caselli A; Piangiolino C; Ragaini F; Cenini S, The (porphyrin)ruthenium-catalyzed aziridination of olefins using aryl azides as nitrogen sources. Eur. J. Org. Chem 2007, 2007 (36), 6053–6059. [Google Scholar]
- 41.King ER; Hennessy ET; Betley TA, Catalytic C–H bond amination from high-spin iron imido complexes. J. Am. Chem. Soc 2011, 133 (13), 4917–4923. [DOI] [PubMed] [Google Scholar]
- 42.Shing K-P; Liu Y; Cao B; Chang X-Y; You T; Che C-M, N-Heterocyclic carbene iron(III) porphyrin-catalyzed intramolecular C(sp3)–H amination of alkyl azides. Angew. Chem. Int. Ed 2018, 57 (37), 11947–11951. [DOI] [PubMed] [Google Scholar]
- 43.Goswami M; Geuijen P; Reek JNH; de Bruin B, Application of [Co(corrole)]− complexes in ring-closing C–H amination of aliphatic azides via nitrene radical intermediates. Eur. J. Inorg. Chem 2017, 2018 (5), 617–626. [Google Scholar]
- 44.Bagh B; Broere DLJ; Sinha V; Kuijpers PF; van Leest NP; de Bruin B; Demeshko S; Siegler MA; van der Vlugt JI, Catalytic synthesis of N-heterocycles via direct C(sp3)–H amination using an air-stable iron(III) species with a redox-active ligand. J. Am. Chem. Soc 2017, 139 (14), 5117–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.King ER; Sazama GT; Betley TA, Co(III) imidos exhibiting spin crossover and C–H bond activation. J. Am. Chem. Soc 2012, 134 (43), 17858–17861. [DOI] [PubMed] [Google Scholar]
- 46.Baek Y; Betley TA, Catalytic C–H amination mediated by dipyrrin cobalt imidos. J. Am. Chem. Soc 2019, 141 (19), 7797–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang L; Powell DR; Houser RP, Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans 2007, (9), 955–964. [DOI] [PubMed] [Google Scholar]
- 48.Johnson ER; Keinan S; Mori-Sánchez P; Contreras-García J; Cohen AJ; Yang W, Revealing noncovalent interactions. J. Am. Chem. Soc 2010, 132 (18), 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Contreras-García J; Johnson ER; Keinan S; Chaudret R; Piquemal J-P; Beratan DN; Yang W, NCIPLOT: A program for plotting noncovalent interaction regions. J. Chem. Theory Comput 2011, 7 (3), 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neese F ORCA - An ab initio, density functional and semiempirical electronic structure package, version 2.9-00; Universitat Bonn: Bonn, Germany, 2009. [Google Scholar]
- 51.Das A; Chen Y-S; Reibenspies JH; Powers DC, Characterization of a Reactive Rh2 Nitrenoid by Crystalline Matrix Isolation. J. Am. Chem. Soc 2019, 141, 16232–16236. [DOI] [PubMed] [Google Scholar]
- 52.Dash C; Yousufuddin M; Cundari TR; Dias HVR, Gold-Mediated Expulsion of Dinitrogen from Organic Azides. J. Am. Chem. Soc 2013, 135, 15479–15488. [DOI] [PubMed] [Google Scholar]
- 53.Jin L-M; Lu H; Cui Y; Lizardi CL; Arzua TN; Wojtas L; Cui X; Zhang XP, Selective radical amination of aldehydic C(sp2)–H bonds with fluoroaryl azides via Co(II)-based metalloradical catalysis: synthesis of N-fluoroaryl amides from aldehydes under neutral and nonoxidative conditions. Chem. Sci 2014, 5 (6), 2422–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olivos Suarez AI; Jiang H; Zhang XP; de Bruin B, The radical mechanism of cobalt(II) porphyrin-catalyzed olefin aziridination and the importance of cooperative H-bonding. Dalton Trans. 2011, 40 (21), 5697–5705. [DOI] [PubMed] [Google Scholar]
- 55.Subbarayan V; Jin L-M; Cui X; Zhang XP, Room temperature activation of aryloxysulfonyl azides by [Co(II)(TPP)] for selective radical aziridination of alkenes via metalloradical catalysis. Tetrahedron Lett. 2015, 56 (23), 3431–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tao J; Jin L-M; Zhang XP, Synthesis of chiral N-phosphoryl aziridines through enantioselective aziridination of alkenes with phosphoryl azide via Co(II)-based metalloradical catalysis. Beilstein J. Org. Chem 2014, 10, 1282–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao G-Y; Jones JE; Vyas R; Harden JD; Zhang XP, Cobalt-catalyzed aziridination with diphenylphosphoryl azide (DPPA): direct synthesis of N-phosphorus-substituted aziridines from Alkenes. J. Org. Chem 2006, 71 (17), 6655–6658. [DOI] [PubMed] [Google Scholar]
- 58.Harden JD; Ruppel JV; Gao G-Y; Zhang XP, Cobalt-catalyzed intermolecular C–H amination with bromamine-T as nitrene source. Chem. Commun 2007, (44), 4644–4646. [DOI] [PubMed] [Google Scholar]
- 59.Aguila MJB; Badiei YM; Warren TH, Mechanistic insights into C–H amination via dicopper nitrenes. J. Am. Chem. Soc 2013, 135 (25), 9399–9406. [DOI] [PubMed] [Google Scholar]
- 60.Iovan DA; Wilding MJT; Baek Y; Hennessy ET; Betley TA, Diastereoselective C–H bond amination for disubstituted pyrrolidines. Angew. Chem. Int. Ed 2017, 56 (49), 15599–15602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Broere DLJ; de Bruin B; Reek JNH; Lutz M; Dechert S; van der Vlugt JI, Intramolecular redox-active ligand-to-substrate single-electron transfer: radical reactivity with a palladium(II) complex. J. Am. Chem. Soc 2014, 136 (33), 11574–11577. [DOI] [PubMed] [Google Scholar]
- 62.Powers DC; Anderson BL; Hwang SJ; Powers TM; Pérez LM; Hall MB; Zheng S-L; Chen Y-S; Nocera DG, Photocrystallographic observation of halide-bridged intermediates in halogen photoeliminations. J. Am. Chem. Soc 2014, 136 (43), 15346–15355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Das A; Reibenspies JH; Chen Y-S; Powers DC, Direct characterization of a reactive lattice-confined Ru2 nitride by photocrystallography. J. Am. Chem. Soc 2017, 139 (8), 2912–2915. [DOI] [PubMed] [Google Scholar]
- 64.Zheng S-L; Pham O; Vande Velde CML; Gembicky M; Coppens P, Competitive isomerization and dimerization in co-crystals of 1,1,6,6-tetraphenyl-2,4-hexadiyne-1,6-diol and sorbic acid: a new look at stereochemical requirements for [2+2] dimerization. Chem. Commun 2008, (22), 2538. [DOI] [PubMed] [Google Scholar]
- 65.Zheng S-L; Wang Y; Yu Z; Lin Q; Coppens P, Direct Observation of a Photoinduced Nonstabilized Nitrile Imine Structure in the Solid State. J. Am. Chem. Soc 2009, 131 (50), 18036–18037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abdelmoty I; Buchholz V; Di L; Guo C; Kowitz K; Enkelmann V; Wegner G; Foxman BM, Polymorphism of Cinnamic and α-Truxillic Acids: New Additions to an Old Story. Crystal Growth & Design 2005, 5 (6), 2210–2217. [Google Scholar]
- 67.More specifically, for the reactive fragment, all non-H atoms of the product were located in the reaction-difference maps, calculated with coefficients Fo(heat)-Fo(initial), and then refined with restraints on the reacted fragment’s bond lengths and constraints of the atomic displacement parameters to the corresponding values of the unreacted fragment. If necessary, the restraints of the atomic displacement parameters have been applied for such disorder refinement. For the non-reactive fragment, the restraints on bond lengths, as well as the restraints of the atomic displacement parameters, have been applied for the disorder refinement as necessary. In order to get a reasonable bond length for the severely disordered hexane and avoid a non-positive definite of atomic displacement parameters, the restraints of DFIX and ISOR were applied.
- 68.Das A; Van Trieste GP; Powers DC, Crystallography of Reactive Intermediates. Comment. Inorg. Chem 2020, 1–43. [Google Scholar]
- 69.Ohashi Y; Sasada Y, X-ray analysis of Co-C bond cleavage in the crystalline state. Nature 1977, 267 (5607), 142–144. [DOI] [PubMed] [Google Scholar]
- 70.Garman E, Radiation damage in macromolecular crystallography: what is it and why should we care? Acta Cryst. D 2010, 66 (4), 339–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.In the refinement of 7–N2 and 8–N2, we utilized restraints on both the unconverted starting material components of the crystals and on the N2 that is eliminated. This strategy is required because conversion of azide to iminyl radical and N2 generates crystalline samples in which the starting materials and products are effectively compositionally disordered (see Figures S-30 and S-31 for raw electron density maps). In order to derive chemically meaningful data from these electron density maps, the employed restraints are critical.
- 72.Christensen J; Horton PN; Bury CS; Dickerson JL; Taberman H; Garman EF; Coles SJ Radiation damage in small-molecule crystallography: fact not fiction. IUCRJ. 2019, 6, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Das A; Maher AG; Telser J; Powers DC, Observation of a Photogenerated Rh2 Nitrenoid Intermediate in C–H Amination. J. Am. Chem. Soc 2018, 140, 10412–10415. [DOI] [PubMed] [Google Scholar]
- 74.Molander GA; Bibeau CT, Thermal rearrangements of α-(ω-azidoalkyl) enones. Tetrahedron Lett. 2002, 43 (31), 5385–5388. [Google Scholar]
- 75.Suga H; Ebiura Y; Fukushima K; Kakehi A; Baba T, Efficient catalytic effects of lewis acids in the 1,3-dipolar cycloaddition reactions of carbonyl ylides with imines. J. Org. Chem 2005, 70 (26), 10782–10791. [DOI] [PubMed] [Google Scholar]
- 76.Huisgen R, Kinetics and mechanism of 1,3-dipolar cycloadditions. Angew. Chem. Int. Ed. Engl 1963, 2 (11), 633–645. [Google Scholar]
- 77.Firestone RA, Mechanism of 1,3-dipolar cycloadditions. J. Org. Chem 1968, 33 (6), 2285–2290. [Google Scholar]
- 78.Merino P; Tejero T; Laguna M; Cerrada E; Moreno A; Lopez JA, An investigation of the Lewis acid mediated 1,3-dipolar cycloaddition between N-benzyl-C-(2-pyridyl)nitrone and allylic alcohol. Direct entry to isoxazolidinyl C-nucleosides. Org. Biomol. Chem 2003, 1 (13), 2336–2342. [DOI] [PubMed] [Google Scholar]
- 79.Zdilla MJ; Abu-Omar MM, Mechanism of catalytic aziridination with manganese corrole: the often postulated high-valent Mn(V) imido is not the group transfer reagent. J. Am. Chem. Soc 2006, 128 (51), 16971–16979. [DOI] [PubMed] [Google Scholar]
- 80.Hill EA; Kelty ML; Filatov AS; Anderson JS, Isolable iodosylarene and iodoxyarene adducts of Co and their O-atom transfer and C–H activation reactivity. Chem. Sci 2018, 9 (19), 4493–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jeong D; Ohta T; Cho J, Structure and reactivity of a mononuclear nonheme manganese(III)–iodosylarene complex. J. Am. Chem. Soc 2018, 140 (47), 16037–16041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.