

Abstract
HPV infections in the oral cavity that progress to cancer are on the increase in the USA. Model systems to study co-factors for progression of these infections are lacking as HPVs are species-restricted and cannot grow in preclinical animal models. We have recently developed a mouse papillomavirus (MmuPV1) oral mucosal infection model that provides opportunities to test, for the first time, the hypothesis that tobacco carcinogens are co-factors that can impact the progression of oral papillomas to squamous cell carcinoma (SCC). Four cohorts of mice per sex were included: (1) infected with MmuPV1 and treated orally with DMSO-saline; (2) infected with MmuPV1 and treated orally with the tobacco carcinogen, dibenzo[def,p]chrysene (DBP); (3) uninfected and treated orally with DMSO-saline, and (4) uninfected and treated orally with DBP. Oral swabs were collected monthly for subsequent assessment of viral load. Oral tissues were collected for in situ viral DNA/RNA detection, viral protein staining, and pathological assessment for hyperplasia, papillomas and SCC at study termination. We observed increased rates of SCC in oral tissue infected with MmuPV1 and treated with DBP when compared to mice treated with DBP or virus individually, each of which showed minimal disease. Virally-infected epithelium showed strong levels of viral DNA/RNA and viral protein E4/L1 staining. In contrast, areas of SCC showed reduced viral DNA staining indicative of lower viral copy per nucleus but strong RNA signals. Several host markers (p120 ctn, p53, S100A9) were also examined in the mouse oral tissues; of particular significance, p120 ctn discriminated normal un-infected epithelium from SCC or papilloma epithelium. In summary, we have confirmed that our infection model is an excellent platform to assess the impact of co-factors including tobacco carcinogens for oral PV cancerous progression. Our findings can assist in the design of novel prevention/treatment strategies for HPV positive vs. HPV negative disease.
Keywords: Human papillomavirus, Mouse papillomavirus, Tobacco carcinogens, Oral cavity, Squamous cell carcinoma, p120 ctn
1. Introduction
Worldwide, head and neck squamous cell carcinoma (HNSCC) is the 6th most common human cancer, and oral squamous cell carcinoma (OSCC) is the most common histological type [1–3]. Exposure to exogenous carcinogens such as tobacco smoke, smokeless tobacco, excess alcohol, and human papillomavirus (HPV) can account for 90% of OSCC [4]. HPV-associated HNSCC is on the rise in the USA and shows increased incidence for Caucasian men in the 50–70 age group [5]. Tobacco carcinogens are also associated with HNSCC but show a characteristic regional difference in oral tissues demonstrating that HPV and tobacco are distinct and separate oral carcinogens (reviewed in Ref. [6–8]). HPV-associated HNSCC is preferentially located in the oropharynx and the base of the tongue.
Co-factors for HPV-associated cervical cancer are well-characterized and include tobacco smoking, hormonal changes, co-infections (such as EBV, Chlamydia), UV exposure and immune deficiencies (reviewed in Ref. [9]). In contrast, co-factors for HPV-associated HNSCC are less well characterized (reviewed in Ref. [6–8]). Interestingly, several recent studies including a meta-analysis indicated that smoking and tobacco exposure may impact the survival and recurrence of HPV positive tumors [9,10] thereby representing a potential co-factor in oral HPV disease persistence and progression. Other co-factors that have been implicated thus far include UV-exposure [11] and HCV infections [12]. Furthermore, in vitro cell culture studies using oral keratinocytes showed co-operation between HPV and tobacco carcinogens [13–15], alcohol metabolism by streptococci [16], and EBV-co-infections [17,18]. Early diagnosis of HPV-HNSCC in patients is challenging due to patients often first presenting at the clinic with a neck mass representing an advanced stage of disease [19]. Preclinical models to study early detection of HPV precursor lesions are additionally complicated by the species restriction of HPV to humans.
We have recently developed a mouse PV (MmuPV1) model of infection of the mouse oral cavity [20–23]. Mouse oral mucosa is susceptible to infection following gentle pre-abrasion of the oral mucosa and papillomas develop at various wounded sites including the tongue, upper and lower palate. Most intriguingly, immunodeficient mice develop secondary infections in previously uninfected oral tissues that show local preference to the base of the tongue (circumvallate papillae and Von Ebners gland [21]); sites that are also targeted for HPV infections in patient oral tissues. This mouse model thus provides a novel opportunity to test the role of various co-factors in HPV-associated HNSCC. A recent study [24] examined the impact of 4-nitroquinoline-1--oxide (NQO) on oral MmuPV1 infections in immune deficient as well as immune incompetent strains of mice and clearly demonstrated that (i) chemical carcinogens can act as co-factors to accelerate oral PV-associated SCC, and (ii) that the MmuPV1 mouse preclinical model is ideal for the testing of various co-factors for HPV disease progression. To our knowledge, NQO has never been found in tobacco smoke or the environment and thus, in the current study we tested for the first time, the hypothesis that a known tobacco oral carcinogen [8] could act as one of potentially several co-factors for enhancement of oral PV disease progression.
Previous studies conducted in our laboratory demonstrated that the tobacco smoke constituent dibenzo[def,p]chrysene also known as dibenzo[a,l]pyrene (DBP) induced DNA damage and mutations in the mouse oral cavity with profiles similar to those found in p53 gene in human HNSCC [25]. In addition to tobacco smoke, DBP has been identified in various combustion systems such as diesel particulate extract and urban air, as well as soil sediments [4].
We showed that DBP up-regulates p53 and COX-2 proteins as well as several inflammatory-related genes in the mouse oral tissues [25,26]. In addition to genetic alterations, we showed that DBP altered the epigenetic landscape including genes involved in epithelial-mesenchymal transition pathway [27]. More recently, we showed that black raspberry inhibited DBP-induced oral cancer via genetic and epigenetic alterations [28]. Collectively, our previous studies clearly demonstrate that our mouse model, using a relevant tobacco carcinogen, provides a realistic platform to explore the impact of several etiological agents in the development of oral cancer. In the present study we report for the first time the effects of chronic topical application of DBP into the mouse oral cavity at a dose based on our previous study [25] and monitored the PV infections in the oral cavity for accelerated disease progression when compared to virus-alone oral infections. We also monitored several disease markers, and viral and host protein content via in situ hybridization and immunohistological staining. The outcome of the study showed that mice infected with MmuPV1 at oral sites and treated with DBP showed an increased incidence of squamous cell carcinoma (SCC) in hyperplastic epithelium and that the model is ideally suited to assess select co-factors for HPV-associated HNSCC.
2. Materials and methods
2.1. Viral infection, oral swab and oral tissue collection
All animal studies were reviewed and approved by the PSU COM IACUC, and followed NIH guidelines for care and use of animals in research. MmuPV1 infection of athymic mouse NU/J mice (Jackson) oral tissues was achieved using previously described procedures [22]. In brief, 6–10 week old mice were anesthetized on day 0 with ketamine/xylazine, and the oral cavity sites (tongue, upper and lower mucosal tissues) were gently wounded using microneedles. Animals were allowed to recover overnight. The following day, each animal was again anesthetized, and virus inoculum (20 μl viral stock solution in saline, 3 × 108 viral DNA) placed onto the pre-wounded sites with additional gentle abrasion with a plastic pipette tip. Oral swabs were collected monthly on infected mice by holding the mice with the scruff technique, then placing a small plastic brush dipped in saline into the mouth and swirled several times, then withdrawn and placed into a collection tube for DNA extraction and viral DNA quantitation using Q-PCR. Oral tissues were collected from mice at humane experimental termination endpoints and labeled as upper oral mucosa (UOM), lower oral mucosa (LOM) and tongue. The UOM and LOM also included skin lip tissues. Tissue collected also included the base of the tongue and upper pharynx.
2.2. Exposure of infected and un-infected mice to DBP
At four weeks after virus infection male and female mice were treated by topical application into the oral cavity with DBP dissolved in DMSO (24 nmol, 3 times per week for a period of 21 weeks) as described previously [25]. Briefly, this experiment included 4 cohorts of mice per sex that were: (1) infected with MmuPV1 and treated orally with DMSO-saline; (2) infected with MmuPV1 and treated orally with the DBP; (3) uninfected and treated orally with DMSO-saline, and (4) uninfected and treated orally with DBP. At monthly intervals, oral lavages were collected for subsequent assessment of viral load via Q-PCR. Mice were monitored weekly for body weight and tumor formation around the mouth and muzzle area until tumor size reached up to 0.5 cm in diameter, a criteria used to define a humane endpoint for the study. At 25 weeks post-infection, the experiment was terminated, and mouse oral tissues were collected for histology. Tongue, UOM and LOM, including lip skin were sectioned in a microtome and 4 μm sections prepared for H&E staining and subsequent pathological assessment. Sequential slides were prepared for in situ hybridization for viral DNA, RNAscope for viral RNA, and viral and host protein immuno-histochemistry.
2.3. Viral load assessments
Viral titers in the oral swab samples were determined as previously described [22]. The swabs containing oral cells and saliva were placed into saline in collection vials and total DNA extracted using standard methods [22]. Q-PCR using primers and probe targeting the E2 region (150 bp amplimer) were amplified and quantitated for viral DNA genomes using a standard curve of amplification from purified plasmid DNA containing a single viral genome [22]. The mean and standard errors (SEM) of the number of viral genomes per sample was plotted against time for each treatment group that included MmuPV1 infections.
2.4. Histological and immunohistological analyses
Mouse oral tissues were harvested at termination of the bioassay, fixed in 10% neutral-buffered formalin and embedded in paraffin using standard procedures as described above. H&E stained tissue sections were evaluated for inflammation, epithelial hyperplasia, hyperkeratosis, cellular atypia, SCCs, papillomas and other neoplasms by a board-certified veterinary pathologist (Hannah Atkins) blinded to treatment group. Several host and viral protein markers including p120 ctn, p53, S100A9, MmuPV1-L1 and MmuPV1-E4 were tested using standard immunohistochemistry of sequential sections of selected oral tissues [21,22]. L1 was detected using an in-house mouse monoclonal antibody, MPV. B9 [21], and E4 was detected using a rabbit polyclonal antiserum to E4 (kind gift from John Doorbar [Cambridge University, UK]). Antibodies to host proteins were obtained from R&D Systems (goat polyclonal anti-p53 Cat #1355-SP: 1:200 dilution) and Abcam (goat polyclonal anti-S100A9, Cat #PAB11470: 1:20 dilution) and used as recommended by the manufacturers. Tissue sections were pre-treated using low-temperature antigen retrieval, citrate buffer (pH6) (Vector Laboratories), and incubated with corresponding primary and secondary antibodies at room temperature followed by colorimetric development as described in our previous studies [23].
2.5. Viral in situ assessment (DNA and RNA)
In situ hybridization was independently conducted to detect both viral DNA and viral RNA. Viral DNA was detected using standard methods previously described by our team [22,23]. In brief, a sub-genomic fragment of MmuPV1 (3913bp EcoRV/BamH1 fragment) was used as an in situ hybridization probe for the detection of MmuPV1 DNA in tissues. The probe was biotinylated using the random priming method. Access to target DNA was obtained with 0.2 mg/ml pepsin in 0.1 N HCl incubation at 37 °C for 8 min. The biotinylated probe was applied and heated to 95 °C for 5 min to achieve dissociation of target and probe DNA. Reannealing was allowed to occur for 2 h at 37 °C. Target-bound biotin was detected using a streptavidin AP conjugate followed by colorimetric development in BCIP/NBT [23]. Viral RNA was detected using RNAscope in which E1ΛE4 transcript was used as the probe determined from the published viral transcriptome [29]. Detection used a horseradish peroxide enzyme-labeled probe and colorimetric readout in situ.
2.6. Immunofluorescence for p120 ctn
Paraffin tissue sectioning and immunofluorescence (IF) were performed essentially as described previously by Stairs et al. [30]. Slides were incubated in the p120 ctn antibody (610,134; BD Transduction) overnight at 4°C. Secondary antibody Alexa Fluor 488-anti-mouse (A-11001; Invitrogen) incubation was for 1 h at room temperature. Slides were coverslipped/counterstained using VectaShield HardSet Antifade mounting medium with DAPI (H1500; Vector Laboratories). Quantification of IF intensity was performed using ImageJ.
2.7. Statistical analyses
Summary counts for outcomes of interest including SCC in haired skin, SCC in oral mucosa, and severe atypical hyperplasia were computed separately for the combined MmuPV1/DBP group versus DBP only to determine if there was an association between viral exposure and outcome against a common background of carcinogen exposure. Similar counts were computed separately for MmuPV1/DBP versus MmuPV1/DMSO to determine if there was an association between carcinogen exposure and outcome against a common background of viral infection. In both scenarios, odds ratios and two-sided Fisher’s exact tests were used to compare proportions of outcomes of interest in the two exposure groups.
Viral DNA levels in MmuPV1 infected mice were compared in groups defined by gender and DBP treatment status at multiple time points. In brief, the lmer function in the lme4 R package [31] was utilized to fit linear mixed effects models based on the factors gender, treatment status, and time. Because each mouse had measurements at multiple time points, a subject-specific random intercept was included in the models. Nested models were compared to assess the impact of factors of interest. Paired t-tests were applied to compare p120 ctn levels in normal tissue to either pre-malignant or malignant tissue. R 3.6.3 [32] was used to perform all statistical analyses and generate figures.
3. Results
3.1. The effects of DBP and MmuPV1 infection individually and in combination on oral pathology and tumorigenesis
The assessment of the disease status of the tissues is presented in Table 1 and Supplemental Table S1. We observed an increase in the incidence of SCC in oral tissues from mice infected with MmuPV1 and treated topically with DBP. Tissues from mice treated either with DBP alone, or virus-infected alone showed lower to no incidence of SCC, respectively (Table 1). There was also an increased incidence of SCC in the oral skin sites (lip area) versus the oral mucosa (Table 2). Mice treated with placebo (DMSO vehicle) showed no pathological features of papillomas or SCC but showed a low incidence of hyperplasia. We have observed that MmuPV1 infections alone in these athymic mice can progress to SCC [21,23] but usually take up to 8 months for this progression. The reduced time from primary infection to termination in this study (25 weeks) concurred with the lack of SCC progression in the viral-alone infected oral tissues.
Table 1.
Pathological assessment of long-term treatment of oral MmuPV1 infected mice with DBP.
| Group, gender (n) | Treatment | Number of tissues with the following pathologies |
|||||
|---|---|---|---|---|---|---|---|
| Tissues | Hyperkeratosis | Hyperplasia | Atypical hyperplasia | Papilloma (H&E and ISH) | SCC | ||
| 1. Male (9) | MmuPV1 + DBP | Tongue | 2 | 3 | 3 | 3 | 0 |
| UOM | 7 | 3 | 8 | 8 | 7 | ||
| LOM | 7 | 5 | 6 | 7 | 1 | ||
| 2. Male (9) | DBP | Tongue | 0 | 0 | 0 | N/A | 0 |
| UOM | 1 | 1 | 2 | 1 | |||
| LOM | 0 | 1 | 1 | 0 | |||
| 3. Male (3) | MmuPV1 + DMSO | Tongue | 0 | 1 | 1 | 3 | 0 |
| UOM | 2 | 3 | 2 | 3 | 0 | ||
| LOM | 3 | 1 | 3 | 3 | 0 | ||
| 4. Male (2) | DMSO | Tongue | 0 | 0 | 0 | ND | 0 |
| UOM | 1 | 1 | 0 | 0 | |||
| LOM | 0 | 0 | 0 | 0 | |||
| 5. Female (7) | MmuPV1 + DBP | Tongue | 2 | 1 | 5 | 2 | 0 |
| UOM | 6 | 1 | 7 | 6 | 2 | ||
| LOM | 3 | 0 | 4 | 6 | 5 | ||
| 6. Female (10) | DBP | Tongue | 2 | 3 | 1 | N/A | 0 |
| UOM | 5 | 4 | 4 | 1 | |||
| LOM | 5 | 5 | 2 | 0 | |||
| 7. Female (3) | MmuPV1 + DMSO | Tongue | 1 | 0 | 2 | 3 | 0 |
| UOM | 3 | 1 | 2 | 3 | 0 | ||
| LOM | 2 | 1 | 3 | 2 | 0 | ||
| 8. Female (3) | DMSO | Tongue | 1 | 1 | 1 | ND | 0 |
| UOM | 1 | 1 | 0 | 0 | |||
| LOM | 1 | 1 | 1 | 0 | |||
DMSO (dimethylsulfoxide); UOM (Upper Oral Mucosa); LOM (Lower Oral Mucosa); SCC (Squamous Cell Carcinoma); DBP (dibenzo[a,l]pyrene); ND, none detected.
Table 2.
Location (sites) and incidence with SCC and CISa.
| Group (n) | Treatment | Number of mice with SCC | Location (sites) and incidence with SCC and CIS |
||
|---|---|---|---|---|---|
| SCC in HS | SCC in OM | CIS (HS or OM) | |||
| 1. Male (9) | MmuPV1+DBP | 8 | 5/9 | 3/9 | 1/9 |
| 2. Male (9) | DBP | 1 | 1/9 | 0/9 | 0/9 |
| 5. Female (7) | MmuPV1+DBP | 7 | 6/7 | 1/7 | 0/7 |
| 6. Female (10) | DBP | 1 | 0/10 | 1/10 | 0/10 |
SCC, Squamous Cell Carcinoma; CIS, Carcinoma in situ; OM, oral mucosa; HS, haired skin (lip area).
For histological outcomes at haired lip skin sites for all mice there was a statistically significant difference between the MmuPV1-infected mice treated with DBP compared to these sites in mice treated with DBP alone when assessing for the presence of SCC (p = 0.000162, Table S1). Similarly, for all mice, SCC was more prevalent in MmuPV1-infected mice treated with DBP versus mice infected with MmuPV1 alone (p = 0.0124). Interestingly, the SCC incidence at the haired lip sites were significantly increased for female mice (p = 0.000566 and p = 0.033 respectively) but not male mice at this site (p = 0.123 and p = 0.205 respectively). For SCC incidence at oral mucosal sites, there was no statistical difference between the above two treatment groups (males, females and both genders combined, Table S1). Hyperplasia in the oral tissues was increased in DBP-treated mice infected with MmuPV1 when compared with DBP-treated uninfected mice (p = 0.00919). This latter difference was reflected with significance in male but not female mice (p = 0.00905 versus p = 1).
3.2. Immunohistological and in situ hybridization detection of viral markers
Viral DNA, RNA and viral proteins (L1 and E4) were determined using probes established for MmuPV1 nucleic acids and antibodies to L1 and E4, respectively as described in the methods section. Sequential slides from selected tissues were stained for viral DNA and RNA to establish some information that could suggest a change in the viral life cycle. For example, integrated viral DNA copy number is significantly reduced in cancerous cells such that these signals are often below the threshold of detection using standard in situ DNA hybridization methods. In contrast, viral RNA could be amplified in such cells and thus readily detected using RNAscope. Using in situ hybridization analysis for both viral DNA and RNA on these tissues, we noted that there were some epithelial cells demonstrating strong signals for both DNA and RNA (indicative of benign, productive papillomatous tissues). However, there were also regions of epithelial cells that showed very low signals for viral DNA, but strong, widespread staining for viral RNA (Fig. 1) indicative of PV precancerous tissues.
Fig. 1. RNA and DNA in situ hybridization of oral papilloma/tumor tissues (Mouse 1–3 UOM) showing papillomavirus origin of tumor tissue.
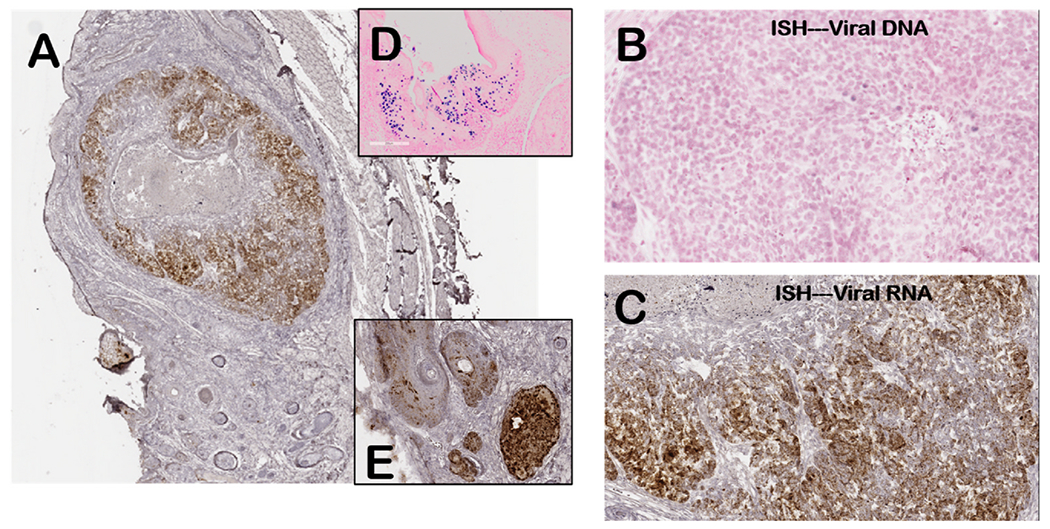
(A) lower magnification (40X) of tumor with RNA in situ for viral E4 transcripts (brown stain). Insert images show viral DNA signal in benign papilloma (D) and viral RNA in papilloma tissue (E). (B) DNA in situ hybridization (200X) showing tumor area with rare weakly-staining nuclei (blue stain) indicating reduced viral DNA copy numbers compared to benign papilloma tissue (D). (C) RNA in situ hybridization (200X) of the tumor showing areas of viral RNA staining (brown) as well as areas of reduced staining compared to benign papilloma (E). Representative tumor tissue from group 1 mouse.
Immunohistology for viral proteins E4 (Fig. 2) and L1 (Fig. 3) also demonstrated that tumor tissues showed reduced staining for both proteins when compared to benign papilloma tissues. Although areas of tumor tissue showed no L1 staining and reduced E4 staining, viral DNA and RNA signatures were found in these tissues (Fig. 1), confirming that these tumor tissues were MmuPV1 in origin.
Fig. 2. Immunohistochemical staining of representative oral tumor tissues stained for MmuPV1 E4 protein (red-brown signal).
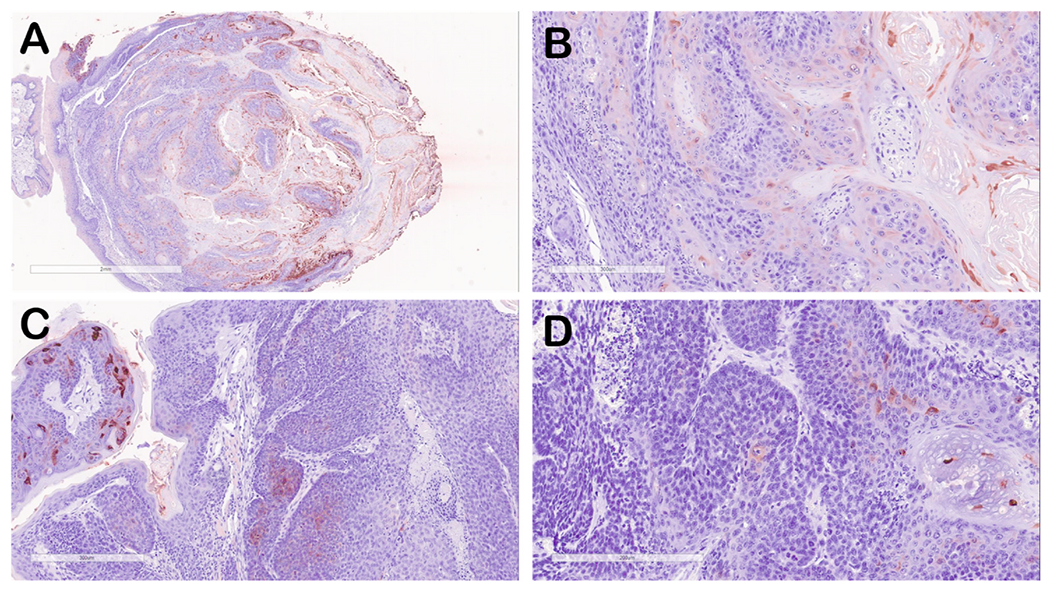
(A) lower magnification (4X) of tumor from mouse 8-2 UOM, Group 5). (B) E4 staining of tumor from mouse 8–2 (20X) showing areas of E4 staining representing areas of benign papilloma as well as tissue that is negative for E4 staining. (C) E4 staining of tumor tissue from mouse 1–3 (Group 1) showing a region of strongly stained benign papilloma (left) and tumor tissue showing minimal E4 protein. (D) E4 staining of tumor tissue from mouse 10–1 (UOM, Group 5) showing minimal E4 protein staining.
Fig. 3.
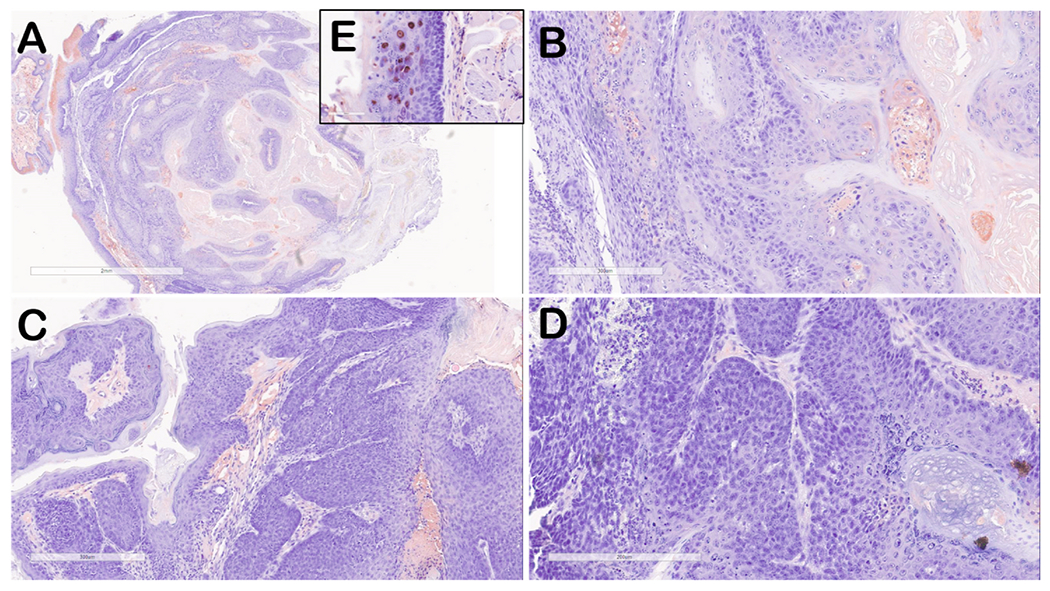
Immunohistochemical staining of representative oral tumor tissues stained for MmuPV1 L1 protein (red-brown signal) adjacent sections of tissue showing the same regions in Fig. 2.
(A) lower magnification (4X) of tumor from mouse 8-2 UOM, Group 5). (B) L1 staining of tumor from mouse 8-2 (20X) showing minimal L1 staining representing areas of benign papilloma as well as tissue that is negative for L1 staining. (C) L1 staining of tumor tissue from mouse 1-3 (Group 1) showing a region of minimally stained benign papilloma (left) and tumor tissue negative for L1 protein. (D) L1 staining of tumor tissue from mouse 10-1 (UOM, Group 5) showing tumor tissue negative for L1 protein. Insert (E) shows strong nuclear L1 staining typical for benign papilloma tissues.
3.3. Viral genomes in oral swabs
Oral swabs were collected at various times points after infection and viral genome content was assessed using Q-PCR. Data show that the viral genomes increased in number over time in all virus-infected mice and that the mean viral genome content was similar in both virus-infected vs virus-infected + DBP-treated mice (Fig. 4A and B). No viral genomes were detected in any of the mice in the other treatment groups that did not receive MmuPV1 infections.
Fig. 4. QPRC analysis of MmuPV1 genomes in oral swabs.
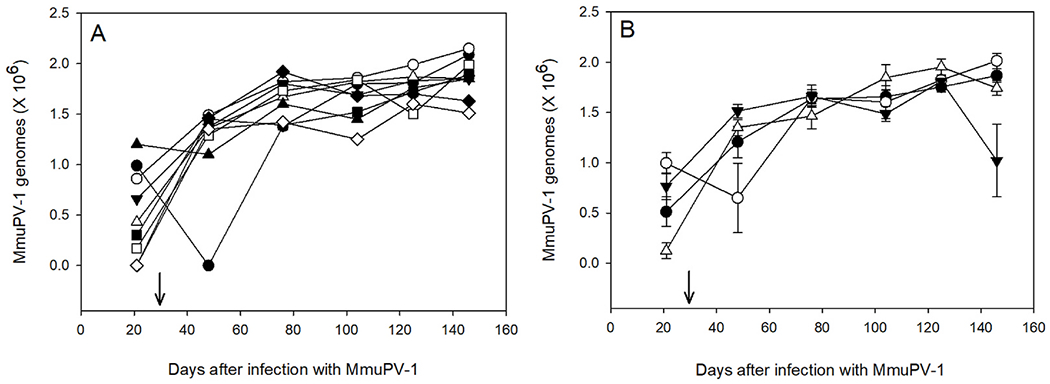
A. Selected MmuPV1-infected mice treated with DBP (Group 1). Each symbol represents a single mouse followed over time for the duration of the study. B. Mean ± SEM of QPCR for mice in group 1 (●), group 3 (○), group 5 (▼) and group 7 (▽). Arrow indicates the beginning of oral treatments with DBP. The infections were also confirmed histologically (Table 1) and in situ hybridization for viral DNA (Fig. 1).
A number of different statistical models were examined to determine which variables impacted viral DNA levels including: (1) no variables; (2) time alone; (3) time + gender + an interaction; (4) time + treatment + an interaction, and (5) time + gender + treatment + all possible interactions. In summary, model 2 is preferred over model 1, which suggests the viral levels change over time. Model 3 is preferred over model 2, but model 4 is not preferred over model 2, suggesting that there is a gender effect after controlling for time (i.e., DNA levels are different by gender when controlling for time), but not a treatment effect after controlling for time (i.e., DNA levels are not different by treatment when controlling for time). Model 5 is preferred over both models 3 and 4 suggesting that DNA levels are different in groups defined by gender/treatment when controlling for time.
3.4. Immunohistology of host cell markers
Several host markers were selected to assess the disease status of the oral tissues. Some markers were also selected based upon RNA sequencing (RNAseq) analyses of mouse papillomavirus infected tissues [20]. We tested p53 and S100A9 based on papillomavirus biology and noted that p53 staining was essentially low (Fig. 5) to background levels suggestive that MmuPV1 tumors like other PV-associated cancers do not show upregulation of p53. S100A9 is also a marker for both stressed epithelium and some cancers. Similarly, monocytes/neutrophils showed low staining in tumor and adjacent normal epithelium but strong staining of individual cells in areas of tumor and papilloma epithelium (Fig. 6) indicating an infiltrate of neutrophils and/or macrophages. We have previously observed this infiltrate in MmuPV1 infected tissues [17].
Fig. 5. Immunohistostaining of mouse oral tumor tissue for p53 (brown signal).
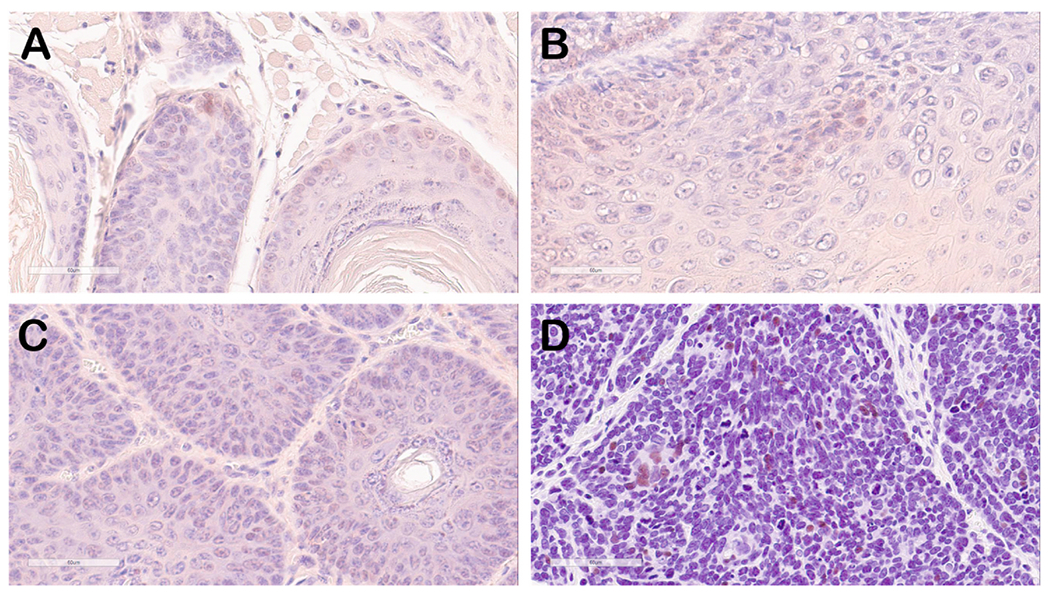
Selected mouse oral tumor tissues (A, B and C) were stained with anti-p53 antisera that also recognized mouse p53 protein. Weak to background signals in some nuclei in areas of benign papilloma can be seen. The positive control tissue (D) representing human breast cancer shows strong, intermittent nuclear staining.
Fig. 6.
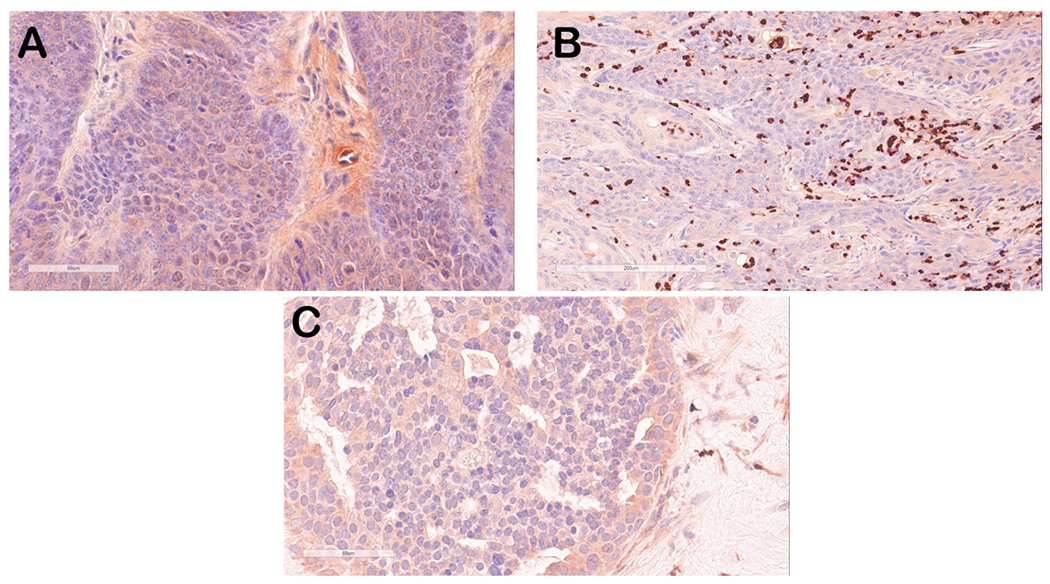
Immunostaining of mouse oral tumor tissue for S100A9 (red-brown signal). Selected mouse oral tumor tissues (A, B) were stained with anti-S100A9 antisera and showed weak to background cytoplasmic staining in tumor epithelial tissue (A) but strong staining of infiltrating neutrophils/monocytes in some tumor tissues (B), a finding that we have observed previously in some of the MmuPV1-infected mouse tissues [20]. Human breast cancer tissues also showed weak cytoplasmic staining of epithelium (C).
3.5. p120 quantitation and histology endpoints
We also introduced p120 ctn as a new marker to assess the impact of tobacco exposure and viral infection on levels of this protein which is known to be down-regulated at the protein level in oral and oropharyngeal cancers [33–36]. The outcome of p120 ctn staining shows strong staining in normal oral epithelium, and significant down-regulation in benign papilloma as well as in malignant tumor tissues indicating that the absence of p120 ctn is an excellent diagnostic indicator for proliferating epithelium associated with oral papillomavirus disease and oral SCC (Fig. 7 and Table 3).
Fig. 7. Immunofluorescence staining of mouse oral tumor tissues for p120ctn using a mouse monoclonal antibody and Alexa Fluor-488 anti-mouse secondary antibody.
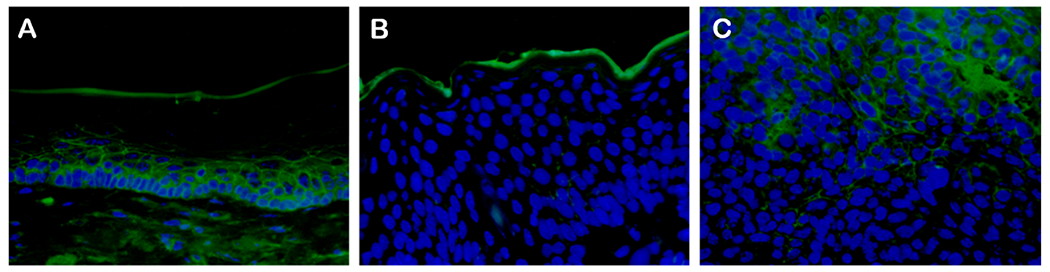
One of four similar mouse oral tissues (mouse UOM 13–3) is shown. Areas of uninfected epithelium (A) showed strong staining whereas benign papilloma tissue (B) and tumor tissues (C) showed reduced staining that is (quantitated in Table 3).
Table 3.
Quantitation of immunofluorescence signal in selected mouse oral tumor tissues stained with anti-p120 ctn.
| Mouse ID | Normal | Premalignant | Malignant |
|---|---|---|---|
| NM 119 LOM_Tongue 8–2 21 | 7.765 | 0.004 | 0.03 |
| NM 119_1–2_13 | 6.321 | 5.833 | 2.892 |
| NM 119 LOM_Tongue 10–1 21 | 8.789 | 0.003 | 0.089 |
| NM 119 UOM 11–1 21 | 10.465 | 0.708 | 0.714 |
| Average | 8.203 + 0.688 | 4.5374 + 3.098 | 3.7804 + 2.8963 |
See statistical analyses in Table S1.
Select tissues from mice that were identified as SCC were stained for p120 and statistical assessments of the quantitation of p120 signals demonstrated a trend in signal reduction in papillomatous epithelium versus normal epithelium when an outlier (mouse 1–3) is removed (Table 3). The p values for all mice were not significant (p = 0.347) for normal versus pre-malignant and significant (p = 0.05) with the one outlier removed.
3.6. Impact of sex on tumorigenesis
Both sexes of athymic mice were orally infected with MmuPV1 and treated topically with DBP to determine if there were any sex-biased outcomes as seen in patient populations with HPV oral infections. There were no clear differences in SCC in oral sites between the sexes (Table 1). Although a limited number of mice per group were used in this study, there were indications of increased hyperplasia in male mice infected with MmuPV1 and treated with DBP compared with male mice treated with DBP alone (section 3.4. above). Some statistical support for a gender difference in viral copy number in oral swabs over time was indicated (section 3.3. above), but our analysis approach does not allow us to make conclusions about directionality, i.e., whether viral copy numbers are consistently higher in one gender vs. the other over time. Indeed, it may be the case that our result is driven by variability of the gender-specific viral copy number measurements.
4. Discussion
Oral HPV-associated cancers are rising in incidence, biased towards males in the 50–70 age group, and often present clinically at late stages of disease. Unlike HPV-associated cervical cancers, the co-factors that promote oral HPV disease progression are not well characterized. One of the major challenges to assessing oral PV disease and co-factors is the absence of a preclinical model that supports HPV infections directly. Our recently developed mouse oral PV infection model [20,21] now allows a more systematic assessment of disease progression, including the role of co-factors, immune response, hormonal influence and co-infections to be conducted [8,20,37–40]. In a recent study, Wei et al. [24], presented data on the impact of NQO as a chemical carcinogen co-factor for oral PV disease progression and showed increased incidence of SCC in both immune deficient and immune competent mice. These data confirm that the MmuPV1 mouse model is a powerful preclinical model to assess the impact of various co-factors in controlled oral PV infections and demonstrated that tongue PV infections can become SCC following topical applications of an oral carcinogen. Additional biomarkers in these tongue lesions including pS6, pERK and MCM7 were upregulated similarly to the upregulation in HPV-associated cancers thus further validating the mouse MmuPV1 system as a model for HPV HNSCC [24]. This recent study provided highly useful mechanistic insights on the combined effect of viral infection and the chemical carcinogen NQO; however, to our knowledge, NQO is not found in tobacco smoke.
In the current study we were interested to employ the carcinogen, DBP, that has been identified in tobacco smoke and the environment; DBP is also known to induce oral cancer in mice [8]. Thus, we focused on determining its potential role as a possible co-factor for oral PV disease progression and some mechanistic analyses of changes in the infected tissues that may impact MmuPV1 oral infections. The data present evidence that the oral tobacco carcinogen DBP, may have a dual role in HNSCC that includes functioning as a co-factor for HPV-associated oral cancer. These data further confirm that the mouse MmuPV1 infection model provides an excellent preclinical system to systematically assess various co-factors of oral mucosal PV disease and supports the role of UV light [37,41], hormones [42], immune suppression [39,40], oral tobacco carcinogen DBP (this study) and the synthetic oral carcinogen NQO [4] for oral PV disease persistence and progression.
Viral markers including viral DNA, RNA, E4 and L1 protein showed that benign papillomavirus infections strongly stained for all 4 markers (Figs. 1–3). In contrast, the pre-malignant and malignant tumor tissue showed reduced viral RNA, E4 and L1 protein suggesting a reduction in viral copy (Figs. 1–3). These data support that the oral tumor tissues retain a viral origin and suggest that follow-up experiments are warranted to further examine the status of the viral genomes (such as possible integration). Our preliminary RNAseq analyses of MmuPV1 tissues have indicated that some viral RNAs show evidence of fusion with host genes indicating the potential for MmuPV1 to integrate (unpublished observations).
Selected host markers also indirectly confirmed the papillomavirus origin of the oral tumors. For example, p53 staining of the tumor tissues was uniformly low indicating that these tumors were not triggered exclusively by the tobacco carcinogen as these latter tumor types showed upregulation of p53 in our previous studies in mice in the absence of MmuPV1 infections [25]. We also observed some novel findings using a tumor marker, p120 ctn, which shows downregulation in various tumors including esophageal cancers [33,35]. Information on the expression levels of p120 ctn in oral tumors of papillomavirus origin are lacking. In our studies presented here, there is strong evidence that p120 ctn can provide new information on the status of PV oral disease and suggest follow-up studies on human oral cancers to assess whether the down-regulation of this marker can be used to distinguish HPV-associated versus HPV-negative oral cancer.
Detection of markers in biological fluids can provide additional mechanistic insights on the role of virus and tobacco individually and in combination. For example, Calprotein (S100A8/A9, a heterodimeric complex), is constitutively expressed in myeloid cells and the stratified mucosal epithelial lining of the oropharyngeal and genitourinary mucosae; it can also be detected in biological fluids [43,44]. The downregulation of the S100A8/A9 dimer was correlated with disease progressions in HNSCC and proposed as a prognosis marker for OPSCC patients [43,44]. Thus, our future studies will be focused on determining whether these markers (S100A8/A9) are altered in plasma; such information will be highly useful for clinical applications. Other epithelial markers including K14 and MCM7 that have been reported to be upregulated in OSCC will also be examined in future studies [24].
We were interested also in whether oral MmuPV1 infections show sex differences in infection rates, viral load and disease progression in oral mucosa. There are noted sex differences in oral HPV infections that show increased rates in men [5] and anal HPV disease that show increased rates in women [5]. We did not observe a gender bias in frequency of SCC in oral lesions chronically treated with DBP in the present study although the incidence of hyperplasia and hyperkeratosis in DBP treated mice appeared to be increased in female mice. However, some previous studies have reported strain- and sex-specific effects of the tobacco constituent, 1,3-butadiene leading to hypermethylation in the liver and lung in female CAST/EiJ mice [45]. Other studies have concluded that the formation of bulky aromatic DNA adducts was not dependent on several factors such as patient age, sex, pulmonary tuberculosis, cancer progression (TNM), environmental pollution or mechanical irritation [46]. Further studies on possible gender-specific differences on the impact of oral HPV-infection on tobacco carcinogen-induced DNA damage in the oral cavity are thus warranted. One additional observation for follow up experimentation is the possibility that the SCC tissues observed here may be driven either by papillomavirus oncogenes or perhaps represent a mixed phenotype with DBP playing a major role for some cancerous cells that retain viral sequences.
5. Conclusions
We showed for the first time, an increased rate of SCC in oral tissues infected with MmuPV1 and treated with a relevant tobacco carcinogen (DBP) when compared to mice treated with DBP alone. In addition to the assessment of several biomarkers which are associated with viral infection and tobacco smoke exposure, we showed for the first time, that p120 ctn can provide new information on the status of PV oral disease. However, future studies are urgently needed to assess whether p120 ctn can be used as a marker to distinguish HPV positive vs HPV negative disease. Better mechanistic insights on the interaction between infection and smoking in the development of oral cancer can assist in the discovery of biomarkers that can be targeted in cancer prevention strategies.
Supplementary Material
Acknowledgement
This work was supported by the following: Penn State Cancer Institute Program Project Development Award Sponsored by Highmark Community Health Reinvestment Fund; The Jake Gittlen Memorial Golf Tournament; National Institute of Dental and Craniofacial Research grant no. DE028650 (JH); National Cancer Institute grant no. CA173465 (KE); and a Research Scholar Grant, RSG-16-219-01-TBG, from the American Cancer Society (DBS). Dibenzo[def,p]chrysene was synthesized in the Penn State Cancer Institute Organic Synthesis Core.
Abbreviations:
- HPV
human papillomavirus
- MmuPV1
mouse papillomavirus
- SCC
squamous cell carcinoma
- DBP
dibenzo[def,p]chrysene (also known as dibenzo[a,l]pyrene)
- NQO
4-nitroquinoline-1-oxide
- UOM
upper oral mucosa
- LOM
lower oral mucosa
- HS
haired skin
Footnotes
CRediT authorship contribution statement
Neil D. Christensen: Conceptualization, Formal analysis, Writing - original draft, designed, conceptualized and supervised the study. analyzed and interpreted the data. wrote the manuscript. Kun-Ming Chen: performed the experiments conducted in the laboratory as well as those involved in animal treatments. Jiafen Hu: Conceptualization, Formal analysis, Writing - original draft, designed, conceptualized and supervised the study. analyzed and interpreted the data. wrote the manuscript. Douglas B. Stairs: Formal analysis, Writing - original draft, analyzed and interpreted the data. wrote the manuscript. Yuan-Wan Sun: performed the experiments conducted in the laboratory as well as those involved in animal treatments. Cesar Aliaga: performed the experiments conducted in the laboratory as well as those involved in animal treatments. Karla K. Balogh: performed the experiments conducted in the laboratory as well as those involved in animal treatments. Hannah Atkins: examined and provided the histopathological data. Debra Shearer: performed the experiments conducted in the laboratory as well as those involved in animal treatments. Jingwei Li: performed the experiments conducted in the laboratory as well as those involved in animal treatments. Sarah A. Brendle: performed the experiments conducted in the laboratory as well as those involved in animal treatments. Krishne Gowda: synthesized and ample amount of the carcinogen used for the animal bioassay. Shantu Amin: synthesized and ample amount of the carcinogen used for the animal bioassay. Vonn Walter: statistically evaluated the results of this study. Raphael Viscidi: Writing - review & editing, critically reviewed and edited the final version of the manuscript, All authors discussed the results and commented on the manuscript. Karam El-Bayoumy: Conceptualization, Formal analysis, Writing - original draft, designed, conceptualized and supervised the study. analyzed and interpreted the data. wrote the manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cbi.2020.109321.
References
- [1].Tanaka T, Ishigamori R, Understanding carcinogenesis for fighting oral cancer, J Oncol 2011 (2011) 603740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A, Global cancer statistics, 2012, Ca - Cancer J. Clin 65 (2015) 87–108. [DOI] [PubMed] [Google Scholar]
- [3].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2017, Ca - Cancer J. Clin 67 (2017) 7–30. [DOI] [PubMed] [Google Scholar]
- [4].El-Bayoumy K, Chen KM, Zhang SM, Sun YW, Amin S, Stoner G, Guttenplan JB, Carcinogenesis of the oral cavity: environmental causes and potential prevention by black raspberry, Chem. Res. Toxicol 30 (2017) 126–144. [DOI] [PubMed] [Google Scholar]
- [5].Van Dyne EA, Henley SJ, Saraiya M, Thomas CC, Markowitz LE, Benard VB, Trends in human papillomavirus-associated cancers - United States, 1999-2015, MMWR Morb. Mortal. Wkly. Rep 67 (2018) 918–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hsieh JC, Wang HM, Wu MH, Chang KP, Chang PH, Liao CT, Liau CT, Review of emerging biomarkers in head and neck squamous cell carcinoma in the era of immunotherapy and targeted therapy, Head Neck 41 (Suppl 1) (2019) 19–45. [DOI] [PubMed] [Google Scholar]
- [7].Guerrero-Preston R, Baez A, Blanco A, Berdasco M, Fraga M, Esteller M, Global DNA methylation: a common early event in oral cancer cases with exposure to environmental carcinogens or viral agents, Puert. Rico Health Sci. J 28 (2009) 24–29. [PubMed] [Google Scholar]
- [8].El-Bayoumy K, Christensen ND, Hu J, Viscidi R, Stairs DB, Walter V, Chen KM, Sun YW, Muscat JE, Richie JP, An integrated research approach for preventing oral cavity and oropharyngeal cancers: two etiologies with distinct and shared mechanisms of carcinogenesis, Canc. Prev. Res 13 (2020) 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ottria L, Candotto V, Cura F, Baggi L, Arcuri C, Nardone M, Gaudio RM, Gatto R, Spadari F, Carinci F, HPV acting on E-cadherin, p53 and p16: literature review, J. Biol. Regul. Homeost. Agents 32 (2018) 73–79. [PubMed] [Google Scholar]
- [10].Smith EM, Rubenstein LM, Haugen TH, Pawlita M, Turek LP, Complex etiology underlies risk and survival in head and neck cancer human papillomavirus, tobacco, and alcohol: a case for multifactor disease, J Oncol 2012 (2012) 571862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Godar DE, Tang R, Merrill SJ, Pharyngeal and cervical cancer incidences significantly correlate with personal UV doses among whites in the United States, Anticancer Res. 34 (2014) 4993–4999. [PubMed] [Google Scholar]
- [12].Rangel JB, Thuler LCS, Pinto J, Prevalence of hepatitis C virus infection and its impact on the prognosis of head and neck cancer patients, Oral Oncol. 87 (2018) 138–143. [DOI] [PubMed] [Google Scholar]
- [13].Kim MS, Shin KH, Baek JH, Cherrick HM, Park NH, HPV-16, tobacco-specific N-nitrosamine, and N-methyl-N’-nitro-N-nitrosoguanidine in oral carcinogenesis, Canc. Res 53 (1993) 4811–4816. [PubMed] [Google Scholar]
- [14].Trushin N, Alam S, El-Bayoumy K, Krzeminski J, Amin SG, Gullett J, Meyers C, Prokopczyk B, Comparative metabolism of benzo[a]pyrene by human keratinocytes infected with high-risk human papillomavirus types 16 and 18 as episomal or integrated genomes, J. Carcinog 11 (2012) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Merne M, Rautava J, Ruutu M, Syrjanen S, Smokeless tobacco increases aneuploidy in oral HPV16 E6/E7-transformed keratinocytes in vitro, J. Oral Pathol. Med 43 (2014) 685–690. [DOI] [PubMed] [Google Scholar]
- [16].Tao L, Pavlova SI, Gasparovich SR, Jin L, Schwartz J, Alcohol metabolism by oral streptococci and interaction with human papillomavirus leads to malignant transformation of oral keratinocytes, Adv. Exp. Med. Biol 815 (2015) 239–264. [DOI] [PubMed] [Google Scholar]
- [17].Guidry JT, Myers JE, Bienkowska-Haba M, Songock WK, Ma X, Shi M, Nathan CO, Bodily JM, Sapp MJ, Scott RS, Inhibition of epstein-barr virus replication in human papillomavirus-immortalized keratinocytes, J. Virol 93 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Makielski KR, Lee D, Lorenz LD, Nawandar DM, Chiu YF, Kenney SC, Lambert PF, Human papillomavirus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes, Virology 495 (2016) 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Berman TA, Schiller JT, Human papillomavirus in cervical cancer and oropharyngeal cancer: one cause, two diseases, Cancer 123 (2017) 2219–2229. [DOI] [PubMed] [Google Scholar]
- [20].Hu J, Cladel NM, Budgeon LR, Balogh KK, Christensen ND, The mouse papillomavirus infection model, Viruses (2017) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Cladel NM, Budgeon LR, Balogh KK, Cooper TK, Hu J, Christensen ND, Mouse papillomavirus MmuPV1 infects oral mucosa and preferentially targets the base of the tongue, Virology 488 (2016) 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hu J, Budgeon LR, Cladel NM, Balogh K, Myers R, Cooper TK, Christensen ND, Tracking vaginal, anal and oral infection in a mouse papillomavirus infection model, J. Gen. Virol 96 (2015) 3554–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cladel NM, Budgeon LR, Cooper TK, Balogh KK, Hu J, Christensen ND, Secondary infections, expanded tissue tropism, and evidence for malignant potential in immunocompromised mice infected with Mus musculus papillomavirus 1 DNA and virus, J. Virol 87 (2013) 9391–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wei T, Buehler D, Ward-Shaw E, Lambert PF, An infection-based murine model for papillomavirus-associated head and neck cancer, mBio (2020) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guttenplan JB, Kosinska W, Zhao ZL, Chen KM, Aliaga C, DelTondo J, Cooper T, Sun YW, Zhang SM, Jiang K, Bruggeman R, Sharma AK, Amin S, Ahn K, El-Bayoumy K, Mutagenesis and carcinogenesis induced by dibenzo[a,l] pyrene in the mouse oral cavity: a potential new model for oral cancer, Int. J. Canc 130 (2012) 2783–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen KM, Schell TD, Richie JP Jr., Sun YW, Zhang SM, Calcagnotto A, Aliaga C, Gowda K, Amin S, El-Bayoumy K, Effects of chronic alcohol consumption on DNA damage and immune regulation induced by the environmental pollutant dibenzo[a,l]pyrene in oral tissues of mice, J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev 35 (2017) 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sun YW, Chen KM, Imamura Kawasawa Y, Salzberg AC, Cooper TK, Caruso C, Aliaga C, Zhu J, Gowda K, Amin S, El-Bayoumy K, Hypomethylated Fgf3 is a potential biomarker for early detection of oral cancer in mice treated with the tobacco carcinogen dibenzo[def,p]chrysene, PloS One 12 (2017), e0186873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen KM, Sun YW, Kawasawa YI, Salzberg AC, Zhu J, Gowda K, Aliaga C, Amin S, Atkins H, El-Bayoumy K, Black raspberry inhibits oral tumors in mice treated with the tobacco smoke constituent dibenzo(def, p)chrysene via genetic and epigenetic alterations, Canc. Prev. Res. 13 (2020) 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xue XY, Majerciak V, Uberoi A, Kim BH, Gotte D, Chen X, Cam M, Lambert PF, Zheng ZM, The full transcription map of mouse papillomavirus type 1 (MmuPV1) in mouse wart tissues, PLoS Pathog. 13 (2017), e1006715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stairs DB, Nakagawa H, Klein-Szanto A, Mitchell SD, Silberg DG, Tobias JW, Lynch JP, Rustgi AK, Cdx1 and c-Myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward Barrett’s esophagus, PloS One 3 (2008), e3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bates D, Mächler M, Bolker B, Walker S, Fitting linear mixed-effects models using lme4, J. Stat. Software 1 (1) (2015), 2015. [Google Scholar]
- [32].R. Core Team, R: A Language and Environment for Statistical Computing, R. Foundation for Statistical Computing, Vienna, Austria, 2020. https://www.R-project.org. [Google Scholar]
- [33].Stairs DB, Bayne LJ, Rhoades B, Vega ME, Waldron TJ, Kalabis J, Klein-Szanto A, Lee JS, Katz JP, Diehl JA, Reynolds AB, Vonderheide RH, Rustgi AK, Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene, Canc. Cell 19 (2011) 470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ma LW, Zhou ZT, He QB, Jiang WW, Phosphorylated p120-catenin expression has predictive value for oral cancer progression, J. Clin. Pathol 65 (2012) 315–319. [DOI] [PubMed] [Google Scholar]
- [35].Chung Y, Lam AK, Luk JM, Law S, Chan KW, Lee PY, Wong J, Altered E-cadherin expression and p120 catenin localization in esophageal squamous cell carcinoma, Ann. Surg Oncol 14 (2007) 3260–3267. [DOI] [PubMed] [Google Scholar]
- [36].Lo Muzio L, Pannone G, Santarelli A, Bambini F, Mascitti M, Rubini C, Testa NF, Dioguardi M, Leuci S, Bascones A, Reynolds AB, Mariggio MA, Is expression of p120ctn in oral squamous cell carcinomas a prognostic factor? Oral Surg Oral Med Oral Pathol Oral Radiol 115 (2013) 789–798. [DOI] [PubMed] [Google Scholar]
- [37].Uberoi A, Yoshida S, Frazer IH, Pitot HC, Lambert PF, Role of ultraviolet radiation in papillomavirus induced disease, PLoS Pathog. 12 (2016), e1005664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Spurgeon ME, Uberoi A, McGregor SM, Wei T, Ward-Shaw E, Lambert PF, A novel in vivo infection model to study papillomavirus-mediated disease of the female reproductive tract, mBio 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sundberg JP, Stearns TM, Joh J, Proctor M, Ingle A, Silva KA, Dadras SS, Jenson AB, Ghim SJ, Immune status, strain background, and anatomic site of inoculation affect mouse papillomavirus (MmuPV1) induction of exophytic papillomas or endophytic trichoblastomas, PloS One 9 (2014), e113582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Handisurya A, Day PM, Thompson CD, Bonelli M, Lowy DR, Schiller JT, Strain-specific properties and T cells regulate the susceptibility to papilloma induction by Mus musculus papillomavirus 1, PLoS Pathog. 10 (2014), e1004314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Uberoi A, Yoshida S, Lambert PF, Development of an in vivo infection model to study Mouse papillomavirus-1 (MmuPV1), J. Virol Methods 253 (2018) 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Spurgeon ME, Lambert PF, Mus musculus papillomavirus 1: a new frontier in animal models of papillomavirus pathogenesis, J. Virol 94 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J, S100A8/A9 in inflammation 9 (2018) 1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Argyris PP, Slama ZM, Ross KF, Khammanivong A, Herzberg MC, Calprotectin and the initiation and progression of head and neck cancer, J. Dent. Res 97 (2018) 674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lewis L, Chappell GA, Kobets T, O’Brian BE, Sangaraju D, Kosyk O, Bodnar W, Tretyakova NY, Pogribny IP, Rusyn I, Sex-specific differences in genotoxic and epigenetic effects of 1,3-butadiene among mouse tissues, Arch. Toxicol 93 (2019) 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pabiszczak M, Szmeja Z, Szyfter K, Szyfter W, Analysis of aromatic DNA adducts in oral cavity and pharyngeal cancer, Otolaryngol. Pol 54 (2000) 151–156. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.