

Abstract
The 2021 Metrics of the HUPO Human Proteome Project (HPP) show that protein expression has now been credibly detected (neXtProt PE1 level) for 18,357 (92.8%) of the 19,778 predicted proteins coded in the human genome, a gain of 483 since 2020 from reports throughout the world reanalyzed by the HPP. Conversely, the number of neXtProt PE2, PE3, and PE4 missing proteins has been reduced by 478 to 1421. This represents remarkable progress on the proteome parts list. The utilization of proteomics in a broad array of biological and clinical studies likewise continues to expand with many important findings and effective integration with other omics platforms. We present highlights from the Immunopeptidomics, Glycoproteomics, Infectious Disease, Cardiovascular, Musculo-Skeletal, Liver, and Cancers B/D-HPP teams and from the Knowledgebase, Mass Spectrometry, Antibody Profiling, and Pathology resource pillars, as well as ethical considerations important to the clinical utilization of proteomics and protein biomarkers.
Keywords: Human Proteome Project (HPP), neXtProt protein existence (PE) metrics, missing proteins (MP), non-MS PE1 proteins, uncharacterized protein existence 1 (uPE1), Chromosome-centric HPP (C-HPP), Biology and Disease-HPP (B/D-HPP), PeptideAtlas, Mass Spectrometry Interactive Virtual Environment (MassIVE), Human Protein Atlas
Graphical Abstract
PROGRESS ON THE HUMAN PROTEOME PARTS LIST
For more than a decade, the Human Proteome Project (HPP), the flagship initiative of the global Human Proteome Organization (HUPO), has pursued twin goals: credibly identifying the protein parts list, essentially, but not entirely, by mass spectrometry, and making proteomics an integral part of multi-omics studies of health and disease1, 2. The HPP consortium has stimulated international collaboration, data sharing, standardized reanalysis of datasets, guidelines for quality assurance, and progress in building and utilizing proteomics knowledge globally. The HPP is organized into 25 teams by chromosome and mitochondria, 19 teams by biological processes and diseases, and 4 resource pillars for antibody-based protein localization, mass spectrometry, knowledgebases, and pathology3.
The chromosome-centric protein parts list is curated and documented by neXtProt4. Table 1 shows the annual progress from 13,975 proteins with protein-level evidence (PE1) as of 2012-02 to 18,357 PE1 proteins in the neXtProt release of 2021-02, including an increase of 473 in the past year from 2020 to 2021. This PE1 total now represents 92.8% of the PE1,2,3,4 predicted proteins from protein-coding genes. This is quite remarkable because we have wondered for several years whether we might be approaching the limit of detection of several classes of very low abundance or membrane-embedded proteins. Moreover, we have applied progressively more stringent Guidelines for Interpretation of Mass Spectrometry Data5, 6. Conversely, Table 1 documents the reduction of PE2,3,4 “missing proteins” (classified based on expression of transcripts, expression in other species, or gene models, respectively, while lacking sufficient protein-level evidence) from 5511 in 2012-02 to 1421 in 2021-02, which is 7.2% of the PE1,2,3,4 total of 19,778.
Table 1.
neXtProt protein existence evidence levels from 2012 to 2021 showing progress in identifying PE1 proteins and PeptideAtlas canonical proteins. More stringent guidelines were imposed in 2016.
| PE Level | 2012-02 | 2013-09 | 2014-10 | 2016-01 | 2017-01 | 2018-01 | 2019-01 | 2020-01 | 2021-02 |
|---|---|---|---|---|---|---|---|---|---|
| 1: Evidence at protein level | 13,975 | 15,646 | 16,491 | 16,518 | 17,008 | 17,470 | 17,694 | 17,874 | 18,357 |
| 2: Evidence at transcript level | 5205 | 3570 | 2647 | 2290 | 1939 | 1660 | 1548 | 1596 | 1265 |
| 3: Inferred from homology | 218 | 187 | 214 | 565 | 563 | 452 | 510 | 253 | 147 |
| 4: Predicted | 88 | 87 | 87 | 94 | 77 | 74 | 71 | 50 | 9 |
| MP = PE2 + PE3 + PE4 | 5511 | 3844 | 2948 | 2949 | 2579 | 2186 | 2129 | 1899 | 1421 |
| Human PeptideAtlas canonical proteins | 12,509 | 13,377 | 14,928 | 14,569 | 15,173 | 15,798 | 16,293 | 16,655 | 16,702 |
PE1/PE1+2+3+4 = 18,357/19,778 = 92.8%
PE 2+3+4 = 1421 "missing proteins" as of 2021-02.
Figure 1 presents a schematic flow diagram of the changes in the neXtProt PE levels for predicted proteins during the past year, from 2020-02 to 2021-02. neXtProt includes a PE5 category for dubious or uncertain genes, mostly pseudogenes, which we exclude from the HPP metrics. Of the 17,874 PE1 proteins in 2020, 17 were deleted due to changes in UniProtKB/Swiss-Prot that carried into neXtProt, especially merging entries; 19 were demoted to PE2,3,4 MPs and 4 to PE5; and 17,834 were carried forward to the 2021-02 release. Meanwhile, 61 new protein entries appeared in neXtProt, of which 40 were PE1, 5 PE2, 4 PE3, 0 PE4, and also 12 PE5; there are 18 immunoglobulin chains, 13 T Cell Receptors, and 30 miscellaneous proteins. Of the 61, 21 were based on non-MS evidence; the entire list appears as Supplementary Table 1.
Figure 1.

Flow diagram of changes in numbers of PE 1,2,3,4,5 classes of predicted proteins from 2020-02 to 2021-02 neXtProt release. Blue arrows indicate increases to PE1 (blue); red arrows indicate demotions from PE1 or PE2,3,4 MPs to PE5 (red); black arrows show increases to MPs (black). In the margins are shown new entries to and removals from neXtProt (see text).
From the point of view of the Missing Proteins, there were 1899 in the 2020-02 release (Table 1, Figure 1). Of those, 481 were elevated to PE1, a remarkably large number, including 331 from PE2, 106 from PE3, and 41 from PE4. Most of this very large gain comes from the inclusion of PPI data in Swiss-Prot or neXtProt. Swiss-Prot curates, validates, and integrates PPI data from publications and from IntAct for upgrading to PE1; that decision is then incorporated by neXtProt. In 2020 an IntAct PPI dataset drove the number of PPI-based PE1 entries in Swiss-Prot from 334 to 615. The pharmaceutical industry has increased its focus on PPI targets; PPI are entering into mainstream drug discovery. neXtProt incorporated a human PPI dataset mapped to the level of interacting domains manually annotated from the literature by ENYO Pharma SA, which resulted in 150 additional PE1 upgrades. As with all other data in neXtProt, these PPI data are “FAIRified”: Findable using the neXtProt semantic SPARQL query tools; Accessible under CC-BY 4.0 license; Interoperable with all the data in neXtProt and many other resources using the same semantic web standards; and Reusable, since all publications and their experimental methods are documented using the PSI Molecular Interations (PSI-MI) controlled vocabulary.
For MS-based PE1 decisions, neXtProt incorporates peptide identifications from PeptideAtlas7, 8 (see Table 1) and MassIVE and applies HPP guidelines for protein validation (at least two uniquely-mapping, non-nested peptides of 9 or more amino acids from a single source)3, 5, 6.
UniProtKB now has 176 entries for functional immunoglobulin (Ig) genes in 3 clusters located on Chr 14 (50V, 23D, 6J, 9C = 88), Chr 2 (40V, 5J, 1C = 46), and Chr 22 (32V, 5J, and 5C = 42). The huge antibody repertoire of each human arises during B cell development though combinatorial V-(D)-J rearrangements, junctional diversity, and somatic hypermutation to yield about 1012 different immunoglobulins. UniProtKB relies on IGMT (the international ImMunoGeneTics organization at CNRS/Montpellier, France) for immunoglobulin and T cell receptor nomenclature and data. Joint with IMGT-Gene-DB, UniProtKB/Swiss-Prot presents a representative set of full-length germline Ig protein sequences, with 15 entries showing the sequences of all C regions and 122 representing all V regions. Sequences for D (5aa) and J (15-30 aa) are extremely small. An Ig is encoded by 7 genes: IGHV, IGHD, IGHJ, IGHC for the H chain and IGKV, IGKJ, or IGKC for a kappa or KGLV, IGLJ or IGLC for a lambda chain. As for other human proteins, the sequences shown match translation of the reference genome (Genome Reference Consortium GRCh38/hg38). To the best of our knowledge, there is no plan to capture immunoglobulin heterogeneity in UniProtKB/Swiss-Prot; this is the mission of the specialized, comprehensive IMGT repertoire. Reciprocal links to IMGT from UniProtKB ensure easy navigation between resources (http://www.imgt.org/IMGTrepertoire/).
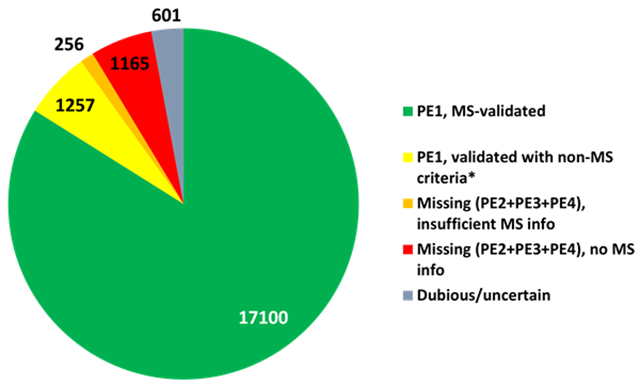
Of the 18,357 PE1 proteins, 17,100 (92.6%) are now based on mass spectrometry and 1257 (7.4%) are validated with non-MS protein evidence (Figure 2). Of the 17,100 MS-based PE1 proteins, 16,920 from the published datasets were reanalyzed and submitted to neXtProt by PeptideAtlas; 16,252 were reanalyzed by MassIVE and submitted to neXtProt; 16, 074 were in both PA and MassIVE; 17,097 in PA or MassIVE; 177 only in MassIVE and 846 only in PeptideAtlas; while 3 non-PE1 proteins meeting the HPP Guidelines 3.0 were added by neXtProt from PTM papers not reviewed by PA or MassIVE. Currently, 78% of neXtProt entries have at least one PTM associated, which represents a total of 191,837 PTM sites. Approximately half of the sites are phosphorylation sites from PeptideAtlas; the others were imported from UniProtKB or GlyConnect or manually annotated from publications (16,759 ubiquitin-like conjugation sites, 3,647 glycosylation sites, 3,354 phosphorylation sites, 2,468 acetylation sites and 1,174 methylation sites). Many more PTMs certainly are present in human samples, but their detection remains a challenge. The various resources named above, including neXtProt, make a constant effort to model and curate new PTM data in order to better capture the complexity of the human proteome.
Figure 2.

Pie chart of MS-based PE1 (blue), non-MS-based PE1 (orange), and PE2,3,4 predicted proteins lacking any or sufficient protein-level evidence, shown as PE2 as of neXtProt release 2021-02. The progress in these metrics since 2016 is depicted in detail in Figure 3.
Of the 1257 non-MS-based PE1 proteins, 62 are primarily based on Edman degradation, 30 on 3D structures in Protein Data Bank, 765 on protein-protein interactions, 42 on antibody studies, 133 on PTMs and processing, 6 on genetic mutations, and 219 from biochemical studies. Many are supported by multiple types of studies; those with MS data do not meet the HPP Guidelines.
As the Figure 2 pie chart and Figure 3 bar chart summarize, besides the 17,100 MS-based PE1 and 1257 non-MS-based PE1 proteins, there are now just 1421 PE2,3,4 missing proteins (in gray) that remain to be confidently detected; of these 256 have some MS data but insufficient to meet the Guidelines. The 2020 Metrics paper included a discussion of the features of the most challenging remaining missing proteins2; these challenges remain cogent.
Figure 3.

Bar chart showing the growth in numbers of PE1 proteins based on MS evidence (blue), the initial diminution in numbers of PE1 proteins based on non-MS data (as many of these proteins were identified with MS) with a large increase for 2021 from PPI datasets (orange) (see discussion of Figure 1), and the striking, progressive reduction in PE2,3,4 missing proteins (grey). The numbers 84.9% and 92.8% above 2016 and 2021 represent the percentage of all PE 1,2,3,4 proteins that are PE1.
A multi-year longitudinal depiction of the pie chart from the baseline in 2016 after application of the HPP Guidelines for Interpretation of MS Data v2.1 (and v3.0 in 2019) is presented in Figure 3. The detailed figures (related to Table 1) are provided for the 2016 base year and show the increment from 2020 to 2021.
Finally, Figure 4 shows the contribution of the top 9 new datasets in PeptideAtlas that generated the increment of 47 canonical proteins (Table 1). Three of these datasets come from the NCI CPTAC3 studies of colorectal cancers, HPV-negative head & neck squamous cell cancers, and glioblastomas.
Figure 4.

Nine datasets registered in ProteomeXchange9 and incorporated into PeptideAtlas after reanalysis with the Trans-Proteomic Pipeline10-12 in the 2021-02 build that contributed the most new PeptideAtlas canonical proteins13-21. The newly MS-based PE1 proteins arose as new PE1 proteins that were previously PE2,3,4 (gray in Figure 3) or from previously non-MS-based PE1 (orange in Figure 3). Each dataset is labeled with its identifier in the respective database, the first and last authors’ names, publication year, number of new canonical proteins, and major biological features. As of 5/17/21, there are 6330 human proteomic datasets in ProteomeXchange.
A paper published too late to make the 2021 update of PeptideAtlas addressed the under-studied skin proteome and reported 10,701 proteins detected (though not using HPP Guidelines 3.0)22. This dataset will be subjected to analysis in the TransProteomicPipeline, with the HPP Guidelines, and be incorporated into the 2022 update of PeptideAtlas and neXtProt.
HIGHLIGHTS OF THE CHROMOSOME-CENTRIC HPP (C-HPP)
The C-HPP teams contributed 15 papers across a wide variety of topics and a lead editorial22 to the JPR 2020 HPP 8th Special Issue (https://pubs.acs.org/toc/jprobs/19/12) following 20 published in the JPR 2019 HPP Special Issue. In addition, 61 papers were published elsewhere during 2019/2020. Progress on the MP50 missing proteins challenge, based on the work of the entire community, is documented in Tables 1 and 2.
Table 2.
Chromosome-by-Chromosome Status of Predicted Proteins in neXtProt 2021-02-18
| Chr | PE1 | PE2 | PE3 | PE4 | PE2+3+4 | PE1/PE1-4 | % PE1/PE1-4 | Net Decrease PE2+3+4 (2016 to 2021) |
uPE1 Proteins |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1859 | 139 | 21 | 2 | 162 | 1859/2021 | 92.00% | 147 (309/162) | 148 |
| 2 | 1228 | 45 | 3 | 1 | 49 | 1228/1277 | 96.20% | 85 (134/49) | 80 |
| 3 | 1005 | 52 | 5 | 0 | 57 | 1005/1062 | 94.50% | 84 (141/57) | 60 |
| 4 | 701 | 35 | 11 | 0 | 46 | 701/747 | 93.80% | 49 (95/46) | 49 |
| 5 | 835 | 26 | 2 | 0 | 28 | 835/863 | 96.80% | 94 (122/28) | 55 |
| 6 | 940 | 54 | 4 | 1 | 59 | 940/999 | 92.80% | 77 (136/59) | 60 |
| 7 | 863 | 105 | 6 | 1 | 112 | 863/975 | 89.70% | 25 (137/112) | 52 |
| 8 | 625 | 34 | 5 | 0 | 39 | 625/664 | 94.30% | 56 (95/39) | 39 |
| 9 | 706 | 58 | 4 | 0 | 62 | 706/768 | 91.20% | 67 (129/62) | 67 |
| 10 | 681 | 47 | 0 | 1 | 48 | 681/729 | 92.80% | 67 (115/48) | 50 |
| 11 | 1069 | 169 | 53 | 0 | 222 | 1069/1291 | 82.70% | 97 (319/222) | 73 |
| 12 | 959 | 52 | 2 | 0 | 54 | 959/1013 | 94.90% | 65 (119/54) | 45 |
| 13 | 311 | 10 | 1 | 0 | 11 | 311/322 | 96.60% | 32 (43/11) | 29 |
| 14 | 632 | 75 | 12 | 2 | 89 | 632/721 | 88.80% | 4 (93/89) | 37 |
| 15 | 535 | 40 | 0 | 0 | 40 | 535/575 | 93.40% | 33 (73/40) | 34 |
| 16 | 773 | 44 | 0 | 1 | 45 | 773/818 | 94.50% | 54 (99/45) | 51 |
| 17 | 1087 | 55 | 3 | 0 | 58 | 1087/1145 | 94.70% | 90 (148/58) | 64 |
| 18 | 255 | 9 | 0 | 0 | 9 | 255/264 | 96.20% | 15 (24/9) | 11 |
| 19 | 1318 | 79 | 8 | 0 | 87 | 1318/1405 | 93.90% | 174 (261/87) | 79 |
| 20 | 504 | 31 | 0 | 0 | 0 | 504/535 | 93.70% | 51 (82/31) | 40 |
| 21 | 204 | 27 | 2 | 0 | 0 | 204/233 | 87.60% | 20 (49/29) | 11 |
| 22 | 455 | 23 | 5 | 0 | 28 | 455/483 | 95.80% | 36 (64/28) | 33 |
| X | 764 | 51 | 0 | 0 | 51 | 764/815 | 93.60% | 94 (145/51) | 105 |
| Y | 37 | 3 | 0 | 0 | 3 | 37/40 | 92.50% | 13 (16/3) | 1 |
| Mito | 15 | 0 | 0 | 0 | 0 | 15/15 | 100.00% | 0 (0/0) | |
| Unk | 2 | 2 | 0 | 0 | 2 | 4-Feb | 50.00% | 0 (2/2) | |
| ALL | 18357 | 1265 | 147 | 9 | 1421 | 18357/19778 | 92.80% | 1528 (2949/1421) | 1273 |
| Sums | 18363 | 1271 | 147 | 9 |
NOTE: There are discrepancies between the true total numbers of proteins in each PE category (ALL) and the Sums because six proteins are derived from two genes on two different chromosomes, and thus appear twice under the per-chromosome table values.
Several chromosome teams (e.g., Chr 5 and Chr 12) are active in the Cancer Moonshot and CPTAC projects and successfully analyzing these data for MPs. Chromosome 6 initiated a directed search for PE1 proteins lacking MS evidence (non-MS PE1 proteins), with several identified by MS in human bone that met the HPP Guidelines for PE1 identification by MS15. A precision medicine molecular corrector drug was developed by Chr 6 team members that restored functional levels of a mutant protein isoform of MALT123. Untreated, this mutant protein led to a rare immunodeficiency disease. The disease was phenotyped in a previous paper by proteomics and TAILS that led to this discovery and then the drug candidate.
Chromosome 10 has assembled one of the world’s largest collections of full-length Gateway plasmids representing 90% of all human protein-coding genes and are distributing the collection through their repository and distribution web portal DNASU (dnasu.org). Chr 10 with Chr 5, 15, 16, and 19 teams have been providing the IVTT-compatible plasmids for missing proteins for IVTT-assisted SRM for many years and continue to generate more plasmids for the whole community. Currently, Chr 10 has full-length plasmids for 175 missing proteins.
A total of 15 C-HPP teams have now met the MP50 Challenge (Table 2). For examples, the number of PE2,3,4 missing proteins coded on Chr 17 has been reduced from 148 in 2016 to 58 in 2021, with reduction on Chr X from 145 to 51, corresponding to 90 and 94 new PE1 proteins, respectively. The CP50 Challenge to each C-HPP team24 to characterize the function(s) of currently unannotated PE1 proteins has made modest progress, as the number of uPE1 proteins (1273 in 2021 versus 1254 in 2020) has not risen proportionately with the increase in total PE1 proteins. Table 2 shows the number of functionally unannotated PE1 proteins for each chromosome. In order to speed up work on the CP50 challenge, neXtProt has designed specific community pages to host manually verified protein function predictions. Duek et al conducted a Functionathon with students at the University of Geneva and proposed functional annotation for seven interesting uPE1 proteins. This work complements the annotations using I-TASSER/CO-FACTOR25. The HPP teams and the broad community are encouraged to discuss the available predictions, to submit new ones, and to start planning validation experiments.
HIGHLIGHTS FROM THE B/D-HPP
The Biology and Disease branch of the Human Proteome Project (B/D-HPP) aims to characterize the biological functions of human proteins and to uncover drivers of disease development and outcomes of preventive and therapeutic interventions. The B/D-HPP integrates 19 international teams of scientists that focus on distinct areas of biological and clinical relevance, including immunity, infectious diseases, cancers, glycoproteomics, cardiovascular, skeletal muscle, and liver diseases, and nutrition (https://hupo.org/B/D-HPP). These research areas are expanded and updated depending on need and the significance of emerging topics. The teams implement proteomics within multidisciplinary biological and clinical studies and disseminate appropriate proteomics methods to the broader scientific community.
Immunopeptidomics.
The comprehensive characterization of Major Histocompatibility Complex (MHC)-associated peptides has experienced dramatic progress in recent years. Immunopeptidomics is expected to impact the treatment of cancers, infectious diseases, and autoimmunity. During the COVID-19 pandemic, the SARS-CoV-2 peptidome and human leukocyte antigen (HLA) peptides associated with T cell responses were reported26-28, supporting the design of vaccines. Proteogenomics/bioinformatics analyses of HLA-bound peptides detected immunogenic cancer-specific targets from non-coding genes, transposable elements, novel ORFs, and bacterial sources, which could be targeted by cancer immunotherapy29-33. For example, 16S rRNA gene sequencing and HLA peptidomics demonstrated the presentation of bacterial peptides by tumor cells from melanoma metastases; intracellular bacteria may impact immune responses29. Integration of exome sequencing, single-cell transcriptomics, ribosome profiling, and MS analyses identified tumor-specific non-canonical HLA peptides30. Other studies are exploiting data-independent acquisition (DIA), MS/MS deep learning, and automation34-39, and large-scale resources are becoming available18, 40.
Glycoproteomics.
HLA-bound peptides identified on dendritic cells upon exposure to SARS-CoV-2 spike (S) protein displayed trimmed glycan residues27. Overall, the standardization of proteomics workflows, which is needed for reproducible, traceable, and transferable measurements, remains an important focus for the glycoproteomics team. The initiative MIRAGE (Minimum Information Required for a Glycomics Experiment) has been designing guidelines that improve the reporting and reproducibility of glycoanalytical methods, such as for liquid chromatography41. Standardization has been enhanced through a recent interlaboratory study targeting monoclonal antibodies for biopharmaceuticals development42. This collaborative strategy revealed a significant increase of paucimannosidic glycans in several types of cancers, including liver, colorectal, prostate, chronic lymphocytic leukemia, and glioblastoma cells43. Moreover, the platform Glyconnect (https://glyconnect.expasy.org/) allows gathering, monitoring, integration, and visualization of data to facilitate interpretation of collected glycoscience data44. neXtProt is integrating glycosylation sites from Glyconnect. The critical nature of protein glycosylation in human health and disease progression is illustrated by the discovery of fibronectin as a target of the glycosyltransferase fukutin-related protein (FKRP) and its link with muscular dystrophy45, and the role of hyper-truncated Asn355- and Asn391-glycans in myeloperoxidase activity and neutrophil-mediated immunity45.
Post-Translational Modifications (PTMs) in Infectious Diseases.
The characterization of PTMs as molecular toggles with critical roles in processes required for cellular homeostasis or linked to disease development has been a shared interest of B/D-HPP teams, including glycoproteomics, cancers, and cardiovascular and infectious diseases. ID investigators discovered lamin B1 site-specific acetylation at the nuclear periphery as a regulatory hub functioning in host defense during viral infection and in choice of DNA damage pathways during cell cycle progression46. An MS-based approach defined phosphoribosylated peptides alongside phosphopeptides on distinct proteoforms in lipopolysaccharide (LPS)-stimulated macrophages, pointing to possible functional cross-talk between PTMs47. Thermal proximity coaggregation (TPCA) and thermal proteome profiling have characterized overall impacts, both pro- and anti-viral, on protein-protein interactions of human cytomegalovirus48. SARS-CoV-249, and herpes simplex virus type 150. The TPCA method captured dynamic enzyme-substrate interactions, leading to discovery of a functional link between the viral DNA interferon inducible protein 16 (IFI16) and the DNA-dependent protein kinase (DNA-PK)50, enhanced computationally51. Many studies examined the biology, pathogenesis, and therapeutic intervention opportunities for SARS-CoV-2 infections, including immunohistochemistry on autopsy lung and proteomics on blood from COVID-19 subjects with acute respiratory distress syndrome (ARDS)52,55 and various bacterial infections53-56.
Cardiovascular Disorders.
Brisk progress has been made by Cardiovascular B/D-HPP researchers in both technology development and biological investigations. The Van Eyk group at Cedars-Sinai has reported a refined protocol and instructions for automated programmable plasma protein digestion in 96-well plate format, which improved the accuracy, precision, and reproducibility of targeted and discovery proteomics assays for clinical implementation57. The Kirk group at Loyola University used ubiquitination proteomics to reveal a new link between the BAG3 protein and myofilament turnover in the setting of heart failure58. The Ge group at University of Wisconsin made advances with top-down proteomics to characterize proteoform permutations and discover that different genetic variants associated with hypertrophic cardiomyopathy funnel into common patterns of altered sarcomeric proteoforms that are predictive of disease phenotypes and may represent common intervention targets59. The Gundry group at the University of Nebraska Medical Center pioneered a new reference glycan structure library for the human heart and demonstrated that specific changes in glycan structure motifs underpin cardiomyocyte differentiation60 and probably other critical cardiac functions. The Mayr group at King’s College London used targeted mass spectrometry and other techniques to show that human PCSK9 is primarily associated with HDL in the circulation, which will facilitate ongoing development of therapeutics targeting PCSK9 to lower serum LDL-cholesterol60.
Musculo-Skeletal.
This B/D-HPP team has focused on understanding the molecular mechanisms that underlie the decline of skeletal muscle strength with aging, which is a primary cause of mobility loss and frailty in the elderly. Such mechanisms have been investigated using transcriptomics61, ribosome profiling62, proteomics in muscle biopsies63, and plasma proteomics64. These multiomic studies revealed that the older human muscle is characterized by deranged energy metabolism, a pro-inflammatory environment, increased proteolysis, and changes in alternative splicing.
Liver.
A major area of interest within the Human Liver Proteome Project team has been the study of proximal biofluids as a valuable information source to assess disease setting, status, and progression. A specific procedure to analyze the human bile proteome65 aimed to discriminate biliary stenosis cases resulting from cholangiocarcinoma or from benign conditions; the combination of proteomics and metabolomics resulted in a panel of 5 proteins and 10 lipids that discriminated between patients with and without these cancers with unprecedented accuracy66. Combined analyses also elucidated pathogenic mechanisms underlying liver disorders. Methionine adenosyltransferase 1A (MAT1A) and the product of its activity, S-adenosylmethionine (SAMe), are keys to preserving a well differentiated and quiescent liver. MAT1A has been recognized as a tumor suppressor in hepatocellular carcinomas, mediated at least in part by interaction with FOXM167. Moreover, Kilanczyk et al have shown that SAMe promotes antioxidant and glutathionylation processes in cholangiocytes that prevent the autoimmune response in patients with primary biliary cholangitis68. Finally, several tools extract and visualize protein-centric functional networks, such as PINE69, and infer biological functions of uncharacterized proteins, uPE, as defined by the C-HPP, based on gene expression correlation and the PageRank algorithm UPEFinder70.
Cancers.
Cancer B/D-HPP investigators are working within large collaborative teams as part of the U.S. National Cancer Institute Clinical Proteomic Tumor Analysis Consortium (CPTAC) and the International Cancer Proteomics Consortium (ICPC). From these large-scale collaborative efforts, proteo(geno)mes are being characterized to shed light on different aspects of cancer biology, immunology, and pathology and to propose precisely targeted therapeutic interventions, as shown during this past year for glioblastomas21, head and neck cancers20, pediatric brain cancers71, breast cancers72, serous ovarian carcinomas73, and lung adenocarcinomas74, as reviewed by Nice75 from the HPP Pathology Pillar. To aid the analysis of large-scale quantitative proteomic datasets, these studies have been accompanied by continued improvement of web servers for data evaluation, visualization, and modeling (e.g., ProteomeExpert and BatchServer)76, 77.
Lastly, to facilitate the dissemination of results and newly-developed methodologies during the pandemic-imposed virtual setting this past year, the B/D-HPP has inaugurated webinars to bring together scientists from different B/D-HPP teams, open to the public. The first was dedicated to “PTMs in Human Disease” (https://www.hupo.org/Webinars-and-Virtual-Presentations).
HIGHLIGHTS FROM THE KNOWLEDGEBASE AND MASS SPECTROMETRY RESOURCE PILLARS
The Human Plasma Proteome and the Human PhosphoProteome
An updated report on the Human Plasma Proteome led by Schwenk and Deutsch (under review for this Special Issue) provides a comprehensive update on the status of the human blood plasma proteome, including several special topics—extracellular vesicles, COVID-19 studies, coagulation, proteogenomics, and ageing. The Human Plasma PeptideAtlas Build of 2021-01 now comprises 4389 canonical human proteins, and the Human Blood Extracellular Vesicle PeptideAtlas 2021-07 has 2757 canonical proteins detected from extracellular vesicles circulating in plasma, of which 2047 are in common and 703 detected only in the EV datasets. These resource pillars are also developing a Human PhosphoPeptideAtlas for phosphosites on serine, threonine, and tyrosine residues of proteins that can be identified and be related to the phosphorylating kinase, the de-phosphorylating phosphatase, cross-talk with other post-translational modifications, and activation of downstream pathways. Many of the precision oncology medications target these proteins and the dynamic phosphoproteome. Kalyuzhnyy et al78 profiled the phosphoproteome to estimate the true extent of protein phosphorylation.
Hoopmann et al79 reported the results of the Phosphopeptide Challenge organized by the HPP Mass Spectrometry Resource Pillar. A standard set of 94 phosphopeptides with phosphorylation at Ser, Thr, or Tyr residues, and their non-phosphorylated counterparts, mixed at different ratios in a neat sample and in a yeast tryptic digest background, was prepared and provided to about 100 laboratories, of which 22 reported data back. The peptides were synthesized and contributed to the HPP by SynPeptide of Shanghai, China, and IMAC beads were provided by ReSyn Biosciences of Johannesburg, South Africa. Each lab analyzed both samples with their own choice of fractionation and purification methods, instruments, and software; they reported identification and site localization of the peptides, relative abundances, and enrichment in the yeast background. The MS Pillar team then reanalyzed all of the submitted data with a single software pipeline (Trans-Proteomics Pipeline) and discussed the challenges and successes in correct identification and localization of phosphosites. True positive, false positive, and false negative identifications from each lab were analyzed across the many variables. Given the salience of phosphopeptide analyses in proteogenomic analyses, like those of CPTAC (above), these results will be quite useful. The data were uploaded to MassIVE with ProteomeXchange identifier PXD020801.
HIGHLIGHTS FROM THE PATHOLOGY RESOURCE PILLAR
The Pathology Pillar, co-led by Michael Roehrl and Edouard Nice, has been active in organizing many meetings and reaching out to other components of the HPP to stimulate future collaborations. Publications from the past year from the Roehrl Laboratory at Memorial Sloan Kettering Cancer Center include DEAD-box RNA helicase protein DDX21 as a prognosis marker for early stage colorectal cancers80, autoantigen profiles of A549 lung and Hep-2 cells81, 82, MS-based absolute quantification of amyloid proteins83, and proteome-based pathology for precision medicine84; see also Nice75.
Complementary publications from the Aebersold Laboratory and colleagues at ETH-Zurich show important insights from proteogenomic analyses. Mehnert et al85 determined the effects of specific mutations in the gene for the Dual-Specificity Tyrosine Phosphorylation-regulated Kinase Dyrk2 on various levels of the proteome, altering the composition, topology, and activity of the kinase complex and the phosphoproteomic state of the cell through altered protein-protein interactions. There is extensive plasticity of the molecular responses to gene mutations. The Tumor Profiling (TuPro) Study workflow86 examines how complex mechanisms evolve in tumors as biological responses involve the tumor microenvironment, cellular heterogeneity, and cell-cell interactions within a tumor mass. These molecular pathology studies were performed on specimens from patients with metastatic melanomas, metastatic epithelial ovarian cancers, and acute myeloid leukemias, with two million single cells per patient profiled across six technologies, together with bulk approaches. Digital pathology and imaging mass cytometry provide quantitative, single-cell, spatially resolved data particularly useful for predicting the success of therapies like immune checkpoint inhibitors that depend upon direct cell-cell interactions.
HIGHLIGHTS FROM THE ANTIBODY PILLAR/HUMAN PROTEIN ATLAS
The Antibody Resource Pillar is tightly linked to the Human Protein Atlas (HPA) project87, a major open-access knowledge resource focusing on spatial proteomics, antibody-based technologies, and integration with transcriptomics. In 2020, the HPA celebrated 20 years since the start of the project, and released a dedicated section and booklet summarizing all the major scientific milestones (www.proteinatlas.org/about/history). HPA v20 also includes a new main section, where the HPA takes the leap into single cell transcriptomics in the Single Cell Type Atlas. This new section constitutes an important complement to the Tissue Atlas, allowing integration of mRNA and protein levels in single cell types. Another recent major milestone is the release of a Cell Cycle Atlas88. By utilizing the novel data in the Single Cell Type Atlas, the cell type-specific expression patterns observed with the antibodies can be confirmed, taking antibody validation to the next level and opening up more detailed studies focusing on particular cell types89.
In addition to the large-scale effort mapping all the human proteins in various organs, tissues, cells and organelles, a major emphasis of the Antibody Resource Pillar and the HPA is developing stringent criteria for antibody validation. These criteria were recently applied in a thorough study analyzing the body-wide expression profile of the SARS-CoV-2 receptor ACE290, where both previously described and novel cell type-specific expression patterns could be confirmed with high confidence, as highlighted in the 2020 HPP Metrics paper2. Surprisingly, the expression level of ACE2 in human airway epithelia was shown to be low, which is in contrast to previous studies and highlights the importance of proper criteria for validation in antibody-based studies. Based on a workflow from the HPA project, a laboratory-based antibody assay for detection of SARS-CoV-2 specific IgG in serum and plasma has been set up, showing 99.7% sensitivity and 100% specificity91 and also useful detection in saliva92.
Meanwhile, Sivertsson et al93 explored antibody validation from a proteome-wide perspective, comparing level of confidence of the antibody-based data in the HPA with protein evidence levels. As expected, a relatively high proportion of the neXtProt missing proteins (PE2-PE4) lacked available antibodies or were only targeted by antibodies of lower reliability, suggesting that this group of proteins that remain to be detected is challenging for many reasons. Nevertheless, a subset of 56 missing proteins, as well as 171 proteins of unknown function, that were suggested, based on transcriptomics levels, to be higher expressed in specific tissues and especially male tissues/testis, were targeted by antibodies of high quality. This group of missing proteins for which reliable antibodies are available constitutes an important starting point for further assessment of evidence of existence, as well as for focused studies combining mass spectrometry with antibody-based proteomics for orthogonal validation of the specific protein identifications.
Beyond the production of antibodies for immunohistochemical and immunofluorescent characterization of expression of the human proteome, there has been substantial development of ELISA-type assays based on antibodies94 or aptamers95 as affinity binders to detect arrays of proteins in human fluids to complement or compete with targeted or non-targeted mass spectrometry-based detection and quantification. For example, Magis et al96 used 13 panels of Olink assays in a longitudinal scientific wellness study from which several cancer biomarkers were shown to identify individuals who would later be diagnosed with metastatic cancers. It is important to obtain results from analyses with each of the platforms on the same samples, both for orthogonal quality control and for better understanding of the sensitivity, specificity, advantages, and limitations of each for plasma and for selected tissue specimens. In an important step forward to align the standards and metrics of new protein assay systems, users of both Olink and Somascan participated in the 2021 meeting of the HUPO Proteomics Standards Initiative97, 98 to move toward developing guidelines for quality of detection surrounding these affinity based assays, following the model of the MS Data Interpretation Guidelines v2.15 and v3.05, 6.
Yet another development in the utilization of antibodies is the programming of kinetically controlled biomolecular probes to design antibodies directed at “undruggable” targets, starting with ion channels (specifically, transient receptor potential vanilloid 1, TRPV1) and KRAS mutations through which apoptosis can be induced99. Very limited protease action is a key step in this protocol.
ETHICAL CONSIDERATIONS FROM PROTEOME-BASED BIOMARKER DEVELOPMENTS
With the remarkable progress in proteomic technologies and throughput and aims to develop protein biomarkers for diagnosis, prognosis, and choice of therapies, authors of a pair of articles from the Matthias Mann Laboratory seek to stimulate the proteomics and multi-omics communities to think deeply about the ethical considerations that arise from research and then clinical use of protein biomarkers from mass spectrometry-based or affinity-based proteomes. Geyer et al100 utilized the data set from a 12-month study of 42 individuals in a weight loss protocol101 to show that the longitudinal pattern of individual protein concentrations and sequences of variant peptide alleles could identify each individual. Porsdam Mann et al102 illustrated applications of the four core principles of bioethics (in parentheses) as follows: informed consent to elicit data-sharing preferences (autonomy); protection of the privacy of personal health information (justice); withholding of non-actionable incidental findings (non-maleficence); and report and explanation of actionable incidental findings (beneficence). They performed a systematic review of the literature on “ethics and clinical proteomics” and identified 10 normative themes, including standards and quality control, integration of new laboratory and computer technologies, conflicting rights and duties, incidental findings, and international legal and regulatory protections and requirements. The 1975 Asilomar Conference on the development of recombinant DNA research and the current international consensus to limit uses of CRISPR-Cas9 gene editing to somatic cells in humans are notable examples of deep engagement of the biomedical community with the larger public.
Meanwhile, Nobis et al103 addressed bioethics in the context of cancer biomarker research with a focus on actual and potential benefits and harms, respect for participants, and fair, just, and moral research practices. Specifically, they emphasized the validation of biomarkers, confidentiality, identifiability of specimens, data protection, and the return of research results. The HUPO HPP is committed to share, analyze, and curate data from the various HPP projects ethically and responsibly.
OUTLOOK FOR THE NEXT SEVERAL YEARS
The extraordinary advances described above and in the papers celebrating the 10th anniversary of the HPP1-3 suggest that the HPP is asymptotically approaching its main stated goal, a high stringency blueprint of the human proteome based on high quality data and transparent, error-controlled computational methods for peptide and protein inference. It therefore seems an opportune time for the HPP to initiate community discussion of major challenges facing proteomics and to develop plans for the HPP to address them over the next years.
To this end, the HPP Scientific Advisory Board (chaired now by Ruedi Aebersold) organized a session on the occasion of the HPP Day on 22 October 2020 in the closing session of the virtual HUPO Annual Congress. The discussions centered around the following challenges that represent exciting opportunities for proteomics:
Complexity of the proteome. The HPP effort has helped the community achieve confident identification of one (sometimes several) specific translation products for most protein-coding genes, corresponding to the 92.8% classified as PE1 proteins. In reality, the number of protein species that constitute the proteome – proteoforms - is massively expanded compared to the number of protein-coding genes by mechanisms including alternative splicing, protein processing, and post-translational modifications. The proteoforms universe is largely unexplored. Furthermore, proteins assume different conformations in 3D and associate with other biomolecules to form macromolecular complexes that often constitute functional units. Importantly, these latter properties of the proteome cannot be presently predicted from their polypeptide sequence. Recently a range of methods has become available to measure them. The confident measurement of these properties of the proteome therefore will provide an exclusive opportunity for proteomics to discover new biology not revealed by DNA and RNA studies.
The Translation of Molecular Maps. The core proteome already mapped and the forthcoming maps of proteoforms and complexes need to be translated into biological function and phenotypes. It is generally well accepted that proteins either by themselves or in the form of modules are responsible for most biological functions. Knowing the proteins as chemical entities does not fully explain their biological functions. The determination of the biological function of the proteoforms and macromolecular complexes that constitute the proteome is a major opportunity for the proteomics community. In that regard and to focus the task at hand, it is useful to consider different levels of function, including the biochemical and cell biological functions of proteins. The biochemical, catalytic function of a protein, exemplified by the phosphotransferase activity of a protein kinase or the proteolytic activity of a protease, is of fundamental importance for the translation of molecular measurement into an understanding of molecular processes and mechanisms. The cell biological functions of proteins reflect their roles in the context of the living cell. The cellular function of proteins needs to explain, e.g., the set of proteins that are phosphorylated by a specific kinase—and dephosphorylated by a specific phosphatase – in a particular cellular state and then the impact of these events on the biochemical processes of the cell and then higher levels of organization. The exploration of the cellular functions of proteins and protein modules presents an outstanding opportunity for proteomics with large implications for basic and translational research.
Mainstreaming Proteomics in Life Sciences Research. Increased dissemination and uptake of proteomics techniques and knowledge resources as a mainstream component of life science research has been a goal for many years. It has been apparent for some time that proteomics uptake has been slow compared to, e.g., genomic or imaging-based approaches, even though it is now widely acknowledged that proteomic data are particularly and uniquely informative. Addressing the bottlenecks that prevent more rapid uptake and dissemination of proteomics presents a high yield opportunity for proteomics and developers of technologies and sample processing methods for a much broader community of users in an era of precision medicine.
The 2020 workshop discussions provided a rich source of ideas and information to further develop a HPP roadmap for the next few years. The three anniversary articles in Nature Communications1, Journal of Proteome Research2, and Molecular & Cellular Proteomics3 each painted a vision of strategic developments for the coming decade. Over the last few months, the HPP Executive Committee has continued these discussions and is presently planning community workshops with the goal of arriving by 2022 at a consensus HPP roadmap and actionable research plan for the next several years.
Supplementary Material
ACKNOWLEDGMENTS
We appreciate the guidance from the HPP Executive Committee and the participation of all HPP investigators. We thank the UniProt groups at SIB, EBI, and PIR for providing high-quality annotations for the human proteins in UniProtKB/Swiss-Prot. The neXtProt server is hosted by VitalIT in Switzerland, ProteomeXchange and PRIDE at the European Bioinformatics Institute in Cambridge, UK, PeptideAtlas at the Institute for Systems Biology in Seattle, and MassIVE at the University of California San Diego. G.S.O. acknowledges support from National Institutes of Health grants P30ES017885-01A1 and U24CA210967; E.W.D. from National Institutes of Health grants R01GM087221, R24GM127667, U19AG023122, and from National Science Foundation grant DBI-1933311; L.L. and neXtProt from the SIB Swiss Institute of Bioinformatics; C.M.O. by Canadian Institutes of Health Research Foundation Grant 148408 and a Canada Research Chair in Protease Proteomics and Systems Biology; M.S.B. by NHMRC Project Grant APP1010303; C.L. by the Knut and Alice Wallenberg Foundation for the Human Protein Atlas; Y.-K.P. by grants from the Korean Ministry of Health and Welfare HI13C22098 and HI16C0257; M.H.R by National Institutes of Health grants R21 CA263262, U01 CA253217, R21 CA251992, P30 CA008748 (MSKCC CCSG, Pathology Component), NIH-Leidos CPTAC contract 17X173, and Farmer Family Foundation; and I.M.C. from National Institutes of Health grant R01GM114141.
Footnotes
Supporting Information
The following supporting information is available free of charge at ACS website (http://pubs.acs.org). Supplementary Table 1, List of 61 new protein entries in neXtProt 2021-02 across protein evidence levels.
REFERENCES
- 1.Adhikari S; Nice EC; Deutsch EW; Lane L; Omenn GS; Pennington SR; Paik YK; Overall CM; Corrales FJ; Cristea IM; Van Eyk JE; Uhlen M; Lindskog C; Chan DW; Bairoch A; Waddington JC; Justice JL; LaBaer J; Rodriguez H; He F; Kostrzewa M; Ping P; Gundry RL; Stewart P; Srivastava S; Srivastava S; Nogueira FCS; Domont GB; Vandenbrouck Y; Lam MPY; Wennersten S; Vizcaino JA; Wilkins M; Schwenk JM; Lundberg E; Bandeira N; Marko-Varga G; Weintraub ST; Pineau C; Kusebauch U; Moritz RL; Ahn SB; Palmblad M; Snyder MP; Aebersold R; Baker MS, A high-stringency blueprint of the human proteome. Nat Commun 2020, 11 (1), 5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Omenn GS; Lane L; Overall CM; Cristea IM; Corrales FJ; Lindskog C; Paik YK; Van Eyk JE; Liu S; Pennington SR; Snyder MP; Baker MS; Bandeira N; Aebersold R; Moritz RL; Deutsch EW, Research on the human proteome reaches a major milestone: >90% of predicted human proteins now credibly detected, according to the HUPO human proteome project. J Proteome Res 2020, 19 (12), 4735–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Omenn GS, Reflections on the HUPO Human Proteome Project, the flagship project of the Human Proteome Organization, at 10 years. Mol Cell Proteomics 2021, 20, 100062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zahn-Zabal M; Michel PA; Gateau A; Nikitin F; Schaeffer M; Audot E; Gaudet P; Duek PD; Teixeira D; Rech de Laval V; Samarasinghe K; Bairoch A; Lane L, The neXtProt knowledgebase in 2020: data, tools and usability improvements. Nucleic Acids Res 2020, 48 (D1), D328–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deutsch EW; Overall CM; Van Eyk JE; Baker MS; Paik YK; Weintraub ST; Lane L; Martens L; Vandenbrouck Y; Kusebauch U; Hancock WS; Hermjakob H; Aebersold R; Moritz RL; Omenn GS, Human proteome project mass spectrometry data interpretation guidelines 2.1. J Proteome Res 2016, 15 (11), 3961–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deutsch EW; Lane L; Overall CM; Bandeira N; Baker MS; Pineau C; Moritz RL; Corrales F; Orchard S; Van Eyk JE; Paik YK; Weintraub ST; Vandenbrouck Y; Omenn GS, Human proteome project mass spectrometry data interpretation guidelines 3.0. J Proteome Res 2019, 18 (12), 4108–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desiere F; Deutsch EW; King NL; Nesvizhskii AI; Mallick P; Eng J; Chen S; Eddes J; Loevenich SN; Aebersold R, The PeptideAtlas project. Nucleic Acids Res 2006, 34 (Database issue), D655–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deutsch EW; Sun Z; Campbell D; Kusebauch U; Chu CS; Mendoza L; Shteynberg D; Omenn GS; Moritz RL, State of the human proteome in 2014/2015 as viewed through PeptideAtlas: Enhancing accuracy and coverage through the AtlasProphet. J Proteome Res 2015, 14 (9), 3461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deutsch EW; Bandeira N; Sharma V; Perez-Riverol Y; Carver JJ; Kundu DJ; Garcia-Seisdedos D; Jarnuczak AF; Hewapathirana S; Pullman BS; Wertz J; Sun Z; Kawano S; Okuda S; Watanabe Y; Hermjakob H; MacLean B; MacCoss MJ; Zhu Y; Ishihama Y; Vizcaino JA, The ProteomeXchange consortium in 2020: enabling 'big data' approaches in proteomics. Nucleic Acids Res 2020, 48 (D1), D1145–D1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keller A; Eng J; Zhang N; Li XJ; Aebersold R, A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Molecular systems biology 2005, 1, 2005.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deutsch EW; Mendoza L; Shteynberg D; Farrah T; Lam H; Tasman N; Sun Z; Nilsson E; Pratt B; Prazen B; Eng JK; Martin DB; Nesvizhskii AI; Aebersold R, A guided tour of the trans-proteomic pipeline. Proteomics 2010, 10 (6), 1150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deutsch EW; Mendoza L; Shteynberg D; Slagel J; Sun Z; Moritz RL, Transproteomic pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clin. Appl 2015, 9 (7-8), 745–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nusinow DP; Szpyt J; Ghandi M; Rose CM; McDonald ER 3rd; Kalocsay M; Jane-Valbuena J; Gelfand E; Schweppe DK; Jedrychowski M; Golji J; Porter DA; Rejtar T; Wang YK; Kryukov GV; Stegmeier F; Erickson BK; Garraway LA; Sellers WR; Gygi SP, Quantitative proteomics of the cancer cell line encyclopedia. Cell 2020, 180 (2), 387–402 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vasaikar S; Huang C; Wang X; Petyuk VA; Savage SR; Wen B; Dou Y; Zhang Y; Shi Z; Arshad OA; Gritsenko MA; Zimmerman LJ; McDermott JE; Clauss TR; Moore RJ; Zhao R; Monroe ME; Wang YT; Chambers MC; Slebos RJC; Lau KS; Mo Q; Ding L; Ellis M; Thiagarajan M; Kinsinger CR; Rodriguez H; Smith RD; Rodland KD; Liebler DC; Liu T; Zhang B; Clinical Proteomic Tumor Analysis Consortium, Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 2019, 177 (4), 1035–1049 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell PA; Solis N; Kizhakkedathu JN; Matthew I; Overall CM, Proteomic and N-Terminomic TAILS analyses of human alveolar bone proteins: Improved protein extraction methodology and LysargiNase digestion strategies increase proteome coverage and missing protein identification. J Proteome Res 2019, 18 (12), 4167–4179. [DOI] [PubMed] [Google Scholar]
- 16.Bohm G; Prefot P; Jung S; Selzer S; Mitra V; Britton D; Kuhn K; Pike I; Thompson AH, Low-pH solid-phase amino labeling of complex peptide digests with TMTs improves peptide identification rates for multiplexed global phosphopeptide analysis. J Proteome Res 2015, 14 (6), 2500–10. [DOI] [PubMed] [Google Scholar]
- 17.Muller JB; Geyer PE; Colaco AR; Treit PV; Strauss MT; Oroshi M; Doll S; Virreira Winter S; Bader JM; Kohler N; Theis F; Santos A; Mann M, The proteome landscape of the kingdoms of life. Nature 2020, 582 (7813), 592–596. [DOI] [PubMed] [Google Scholar]
- 18.Sarkizova S; Klaeger S; Le PM; Li LW; Oliveira G; Keshishian H; Hartigan CR; Zhang W; Braun DA; Ligon KL; Bachireddy P; Zervantonakis IK; Rosenbluth JM; Ouspenskaia T; Law T; Justesen S; Stevens J; Lane WJ; Eisenhaure T; Lan Zhang G; Clauser KR; Hacohen N; Carr SA; Wu CJ; Keskin DB, A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat Biotechnol 2020, 38 (2), 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y; Lin Z; Tan Y; Bu F; Hao P; Zhang K; Yang H; Liu S; Ren Y, Exploration of missing proteins by a combination approach to enrich the low-abundance hydrophobic proteins from four cancer cell lines. J Proteome Res 2020, 19 (1), 401–408. [DOI] [PubMed] [Google Scholar]
- 20.Huang C; Chen L; Savage SR; Eguez RV; Dou Y; Li Y; da Veiga Leprevost F; Jaehnig EJ; Lei JT; Wen B; Schnaubelt M; Krug K; Song X; Cieslik M; Chang HY; Wyczalkowski MA; Li K; Colaprico A; Li QK; Clark DJ; Hu Y; Cao L; Pan J; Wang Y; Cho KC; Shi Z; Liao Y; Jiang W; Anurag M; Ji J; Yoo S; Zhou DC; Liang WW; Wendl M; Vats P; Carr SA; Mani DR; Zhang Z; Qian J; Chen XS; Pico AR; Wang P; Chinnaiyan AM; Ketchum KA; Kinsinger CR; Robles AI; An E; Hiltke T; Mesri M; Thiagarajan M; Weaver AM; Sikora AG; Lubinski J; Wierzbicka M; Wiznerowicz M; Satpathy S; Gillette MA; Miles G; Ellis MJ; Omenn GS; Rodriguez H; Boja ES; Dhanasekaran SM; Ding L; Nesvizhskii AI; El-Naggar AK; Chan DW; Zhang H; Zhang B; Clinical Proteomic Tumor Analysis Consortium, Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer cell 2021, 39 (3), 361–379 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang LB; Karpova A; Gritsenko MA; Kyle JE; Cao S; Li Y; Rykunov D; Colaprico A; Rothstein JH; Hong R; Stathias V; Cornwell M; Petralia F; Wu Y; Reva B; Krug K; Pugliese P; Kawaler E; Olsen LK; Liang WW; Song X; Dou Y; Wendl MC; Caravan W; Liu W; Cui Zhou D; Ji J; Tsai CF; Petyuk VA; Moon J; Ma W; Chu RK; Weitz KK; Moore RJ; Monroe ME; Zhao R; Yang X; Yoo S; Krek A; Demopoulos A; Zhu H; Wyczalkowski MA; McMichael JF; Henderson BL; Lindgren CM; Boekweg H; Lu S; Baral J; Yao L; Stratton KG; Bramer LM; Zink E; Couvillion SP; Bloodsworth KJ; Satpathy S; Sieh W; Boca SM; Schurer S; Chen F; Wiznerowicz M; Ketchum KA; Boja ES; Kinsinger CR; Robles AI; Hiltke T; Thiagarajan M; Nesvizhskii AI; Zhang B; Mani DR; Ceccarelli M; Chen XS; Cottingham SL; Li QK; Kim AH; Fenyo D; Ruggles KV; Rodriguez H; Mesri M; Payne SH; Resnick AC; Wang P; Smith RD; Iavarone A; Chheda MG; Barnholtz-Sloan JS; Rodland KD; Liu T; Ding L; Clinical Proteomic Tumor Analysis Consortium, Proteogenomic and metabolomic characterization of human glioblastoma. Cancer cell 2021, 39 (4), 509–528 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Overall CM, The HUPO high-stringency inventory of humanity's shared human proteome revealed. J Proteome Res 2020, 19 (11), 4211–4214. [DOI] [PubMed] [Google Scholar]
- 23.Quancard J; Klein T; Fung SY; Renatus M; Hughes N; Israel L; Priatel JJ; Kang S; Blank MA; Viner RI; Blank J; Schlapbach A; Erbel P; Kizhakkedathu J; Villard F; Hersperger R; Turvey SE; Eder J; Bornancin F; Overall CM, An allosteric MALT1 inhibitor is a molecular corrector rescuing function in an immunodeficient patient. Nature chemical biology 2019, 15 (3), 304–313. [DOI] [PubMed] [Google Scholar]
- 24.Paik YK; Lane L; Kawamura T; Chen YJ; Cho JY; LaBaer J; Yoo JS; Domont G; Corrales F; Omenn GS; Archakov A; Encarnacion-Guevara S; Lui S; Salekdeh GH; Cho JY; Kim CY; Overall CM, Launching the C-HPP neXt-CP50 pilot project for functional characterization of identified proteins with no known function. J Proteome Res 2018, 17 (12), 4042–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C; Lane L; Omenn GS; Zhang Y, Blinded testing of function annotation for uPE1 proteins by I-TASSER/COFACTOR pipeline using the 2018-2019 additions to neXtProt and the CAFA3 challenge. J Proteome Res 2019, 18 (12), 4154–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weingarten-Gabbay S; Klaeger S; Sarkizova S; Pearlman LR; Chen DY; Gallagher KME; Bauer MR; Taylor HB; Dunn WA; Tarr C; Sidney J; Rachimi S; Conway HL; Katsis K; Wang Y; Leistritz-Edwards D; Durkin MR; Tomkins-Tinch CH; Finkel Y; Nachshon A; Gentili M; Rivera KD; Carulli IP; Chea VA; Chandrashekar A; Bozkus CC; Carrington M; MGH COVID-19 Collection & Processing Team; Bhardwaj N; Barouch DH; Sette A; Maus MV; Rice CM; Clauser KR; Keskin DB; Pregibon DC; Hacohen N; Carr SA; Abelin JG; Saeed M; Sabeti PC, Profiling SARS-CoV-2 HLA-I peptidome reveals T cell epitopes from out-of-frame ORFs. Cell 2021. 184(15):3962–3980.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parker R; Partridge T; Wormald C; Kawahara R; Stalls V; Aggelakopoulou M; Parker J; Powell Doherty R; Ariosa Morejon Y; Lee E; Saunders K; Haynes BF; Acharya P; Thaysen-Andersen M; Borrow P; Ternette N, Mapping the SARS-CoV-2 spike glycoprotein-derived peptidome presented by HLA class II on dendritic cells. Cell reports 2021, 35 (8), 109179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelde A; Bilich T; Heitmann JS; Maringer Y; Salih HR; Roerden M; Lubke M; Bauer J; Rieth J; Wacker M; Peter A; Horber S; Traenkle B; Kaiser PD; Rothbauer U; Becker M; Junker D; Krause G; Strengert M; Schneiderhan-Marra N; Templin MF; Joos TO; Kowalewski DJ; Stos-Zweifel V; Fehr M; Rabsteyn A; Mirakaj V; Karbach J; Jager E; Graf M; Gruber LC; Rachfalski D; Preuss B; Hagelstein I; Marklin M; Bakchoul T; Gouttefangeas C; Kohlbacher O; Klein R; Stevanovic S; Rammensee HG; Walz JS, SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat Immunol 2021, 22 (1), 74–85. [DOI] [PubMed] [Google Scholar]
- 29.Kalaora S; Nagler A; Nejman D; Alon M; Barbolin C; Barnea E; Ketelaars SLC; Cheng K; Vervier K; Shental N; Bussi Y; Rotkopf R; Levy R; Benedek G; Trabish S; Dadosh T; Levin-Zaidman S; Geller LT; Wang K; Greenberg P; Yagel G; Peri A; Fuks G; Bhardwaj N; Reuben A; Hermida L; Johnson SB; Galloway-Pena JR; Shropshire WC; Bernatchez C; Haymaker C; Arora R; Roitman L; Eilam R; Weinberger A; Lotan-Pompan M; Lotem M; Admon A; Levin Y; Lawley TD; Adams DJ; Levesque MP; Besser MJ; Schachter J; Golani O; Segal E; Geva-Zatorsky N; Ruppin E; Kvistborg P; Peterson SN; Wargo JA; Straussman R; Samuels Y, Identification of bacteria-derived HLA-bound peptides in melanoma. Nature 2021, 592 (7852), 138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chong C; Muller M; Pak H; Harnett D; Huber F; Grun D; Leleu M; Auger A; Arnaud M; Stevenson BJ; Michaux J; Bilic I; Hirsekorn A; Calviello L; Simo-Riudalbas L; Planet E; Lubinski J; Bryskiewicz M; Wiznerowicz M; Xenarios I; Zhang L; Trono D; Harari A; Ohler U; Coukos G; Bassani-Sternberg M, Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat Commun 2020, 11 (1), 1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Q; Laverdure JP; Lanoix J; Durette C; Cote C; Bonneil E; Laumont CM; Gendron P; Vincent K; Courcelles M; Lemieux S; Millar DG; Ohashi PS; Thibault P; Perreault C, Proteogenomics Uncovers a Vast Repertoire of Shared Tumor-Specific Antigens in Ovarian Cancer. Cancer Immunol Res 2020, 8 (4), 544–555. [DOI] [PubMed] [Google Scholar]
- 32.Ehx G; Larouche JD; Durette C; Laverdure JP; Hesnard L; Vincent K; Hardy MP; Theriault C; Rulleau C; Lanoix J; Bonneil E; Feghaly A; Apavaloaei A; Noronha N; Laumont CM; Delisle JS; Vago L; Hebert J; Sauvageau G; Lemieux S; Thibault P; Perreault C, Atypical acute myeloid leukemia-specific transcripts generate shared and immunogenic MHC class-I-associated epitopes. Immunity 2021, 54 (4), 737–752 e10. [DOI] [PubMed] [Google Scholar]
- 33.Demmers LC; Kretzschmar K; Van Hoeck A; Bar-Epraim YE; van den Toorn HWP; Koomen M; van Son G; van Gorp J; Pronk A; Smakman N; Cuppen E; Clevers H; Heck AJR; Wu W, Single-cell derived tumor organoids display diversity in HLA class I peptide presentation. Nat Commun 2020, 11 (1), 5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pak H; Michaux J; Huber F; Chong C; Stevenson BJ; Muller M; Coukos G; Bassani-Sternberg M, Sensitive immunopeptidomics by leveraging available large-scale multi-HLA spectral libraries, data-independent acquisition, and MS/MS prediction. Mol Cell Proteomics 2021, 20, 100080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang L; McAlpine PL; Heberling ML; Elias JE, Automated ligand purification platform accelerates immunopeptidome analysis by mass spectrometry. J Proteome Res 2021, 20 (1), 393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jappe EC; Garde C; Ramarathinam SH; Passantino E; Illing PT; Mifsud NA; Trolle T; Kringelum JV; Croft NP; Purcell AW, Thermostability profiling of MHC-bound peptides: a new dimension in immunopeptidomics and aid for immunotherapy design. Nat Commun 2020, 11 (1), 6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stopfer LE; Mesfin JM; Joughin BA; Lauffenburger DA; White FM, Multiplexed relative and absolute quantitative immunopeptidomics reveals MHC I repertoire alterations induced by CDK4/6 inhibition. Nat Commun 2020, 11 (1), 2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfammatter S; Bonneil E; Lanoix J; Vincent K; Hardy MP; Courcelles M; Perreault C; Thibault P, Extending the comprehensiveness of immunopeptidome analyses using isobaric peptide labeling. Analytical chemistry 2020, 92 (13), 9194–9204. [DOI] [PubMed] [Google Scholar]
- 39.Wilhelm M; Zolg DP; Graber M; Gessulat S; Schmidt T; Schnatbaum K; Schwencke-Westphal C; Seifert P; de Andrade Kratzig N; Zerweck J; Knaute T; Braunlein E; Samaras P; Lautenbacher L; Klaeger S; Wenschuh H; Rad R; Delanghe B; Huhmer A; Carr SA; Clauser KR; Krackhardt AM; Reimer U; Kuster B, Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat Commun 2021, 12 (1), 3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcu A; Bichmann L; Kuchenbecker L; Kowalewski DJ; Freudenmann LK; Backert L; Muhlenbruch L; Szolek A; Lubke M; Wagner P; Engler T; Matovina S; Wang J; Hauri-Hohl M; Martin R; Kapolou K; Walz JS; Velz J; Moch H; Regli L; Silginer M; Weller M; Loffler MW; Erhard F; Schlosser A; Kohlbacher O; Stevanovic S; Rammensee HG; Neidert MC, HLA Ligand Atlas: A benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J Immunother Cancer 2021, 9 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell MP; Abrahams JL; Rapp E; Struwe WB; Costello CE; Novotny M; Ranzinger R; York WS; Kolarich D; Rudd PM; Kettner C, The minimum information required for a glycomics experiment (MIRAGE) project: LC guidelines. Glycobiology 2019, 29 (5), 349–354. [DOI] [PubMed] [Google Scholar]
- 42.De Leoz MLA; Duewer DL; Fung A; Liu L; Yau HK; Potter O; Staples GO; Furuki K; Frenkel R; Hu Y; Sosic Z; Zhang P; Altmann F; Grunwald-Grube C; Shao C; Zaia J; Evers W; Pengelley S; Suckau D; Wiechmann A; Resemann A; Jabs W; Beck A; Froehlich JW; Huang C; Li Y; Liu Y; Sun S; Wang Y; Seo Y; An HJ; Reichardt NC; Ruiz JE; Archer-Hartmann S; Azadi P; Bell L; Lakos Z; An Y; Cipollo JF; Pucic-Bakovic M; Stambuk J; Lauc G; Li X; Wang PG; Bock A; Hennig R; Rapp E; Creskey M; Cyr TD; Nakano M; Sugiyama T; Leung PA; Link-Lenczowski P; Jaworek J; Yang S; Zhang H; Kelly T; Klapoetke S; Cao R; Kim JY; Lee HK; Lee JY; Yoo JS; Kim SR; Suh SK; de Haan N; Falck D; Lageveen-Kammeijer GSM; Wuhrer M; Emery RJ; Kozak RP; Liew LP; Royle L; Urbanowicz PA; Packer NH; Song X; Everest-Dass A; Lattova E; Cajic S; Alagesan K; Kolarich D; Kasali T; Lindo V; Chen Y; Goswami K; Gau B; Amunugama R; Jones R; Stroop CJM; Kato K; Yagi H; Kondo S; Yuen CT; Harazono A; Shi X; Magnelli PE; Kasper BT; Mahal L; Harvey DJ; O'Flaherty R; Rudd PM; Saldova R; Hecht ES; Muddiman DC; Kang J; Bhoskar P; Menard D; Saati A; Merle C; Mast S; Tep S; Truong J; Nishikaze T; Sekiya S; Shafer A; Funaoka S; Toyoda M; de Vreugd P; Caron C; Pradhan P; Tan NC; Mechref Y; Patil S; Rohrer JS; Chakrabarti R; Dadke D; Lahori M; Zou C; Cairo C; Reiz B; Whittal RM; Lebrilla CB; Wu L; Guttman A; Szigeti M; Kremkow BG; Lee KH; Sihlbom C; Adamczyk B; Jin C; Karlsson NG; Ornros J; Larson G; Nilsson J; Meyer B; Wiegandt A; Komatsu E; Perreault H; Bodnar ED; Said N; Francois YN; Leize-Wagner E; Maier S; Zeck A; Heck AJR; Yang Y; Haselberg R; Yu YQ; Alley W; Leone JW; Yuan H; Stein SE, NIST interlaboratory study on glycosylation analysis of monoclonal antibodies: Comparison of results from diverse analytical methods. Mol Cell Proteomics 2020, 19 (1), 11–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chatterjee S; Lee LY; Kawahara R; Abrahams JL; Adamczyk B; Anugraham M; Ashwood C; Sumer-Bayraktar Z; Briggs MT; Chik JHL; Everest-Dass A; Forster S; Hinneburg H; Leite KRM; Loke I; Moginger U; Moh ESX; Nakano M; Recuero S; Sethi MK; Srougi M; Stavenhagen K; Venkatakrishnan V; Wongtrakul-Kish K; Diestel S; Hoffmann P; Karlsson NG; Kolarich D; Molloy MP; Muders MH; Oehler MK; Packer NH; Palmisano G; Thaysen-Andersen M, Protein paucimannosylation is an enriched N-glycosylation signature of human cancers. Proteomics 2019, 19 (21-22), e1900010. [DOI] [PubMed] [Google Scholar]
- 44.Alocci D; Mariethoz J; Gastaldello A; Gasteiger E; Karlsson NG; Kolarich D; Packer NH; Lisacek F, GlyConnect: Glycoproteomics goes visual, interactive, and analytical. J Proteome Res 2019, 18 (2), 664–677. [DOI] [PubMed] [Google Scholar]
- 45.Wood AJ; Lin CH; Li M; Nishtala K; Alaei S; Rossello F; Sonntag C; Hersey L; Miles LB; Krisp C; Dudczig S; Fulcher AJ; Gibertini S; Conroy PJ; Siegel A; Mora M; Jusuf P; Packer NH; Currie PD, FKRP-dependent glycosylation of fibronectin regulates muscle pathology in muscular dystrophy. Nat Commun 2021, 12 (1), 2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murray-Nerger LA; Justice JL; Rekapalli P; Hutton JE; Cristea IM, Lamin B1 acetylation slows the G1 to S cell cycle transition through inhibition of DNA repair. Nucleic Acids Res 2021, 49 (4), 2044–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daniels CM; Kaplan PR; Bishof I; Bradfield C; Tucholski T; Nuccio AG; Manes NP; Katz S; Fraser IDC; Nita-Lazar A, Dynamic ADP-Ribosylome, phosphoproteome, and interactome in LPS-activated macrophages. J Proteome Res 2020, 19 (9), 3716–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hashimoto Y; Sheng X; Murray-Nerger LA; Cristea IM, Temporal dynamics of protein complex formation and dissociation during human cytomegalovirus infection. Nat Commun 2020, 11 (1), 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Selkrig J; Stanifer M; Mateus A; Mitosch K; Barrio-Hernandez I; Rettel M; Kim H; Voogdt CGP; Walch P; Kee C; Kurzawa N; Stein F; Potel C; Jarzab A; Kuster B; Bartenschlager R; Boulant S; Beltrao P; Typas A; Savitski MM, SARS-CoV-2 infection remodels the host protein thermal stability landscape. Molecular systems biology 2021, 17 (2), e10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Justice JL; Kennedy MA; Hutton JE; Liu D; Song B; Phelan B; Cristea IM, Systematic profiling of protein complex dynamics reveals DNA-PK phosphorylation of IFI16 en route to herpesvirus immunity. Sci Adv 2021, 7 (25). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Federspiel JD; Cook KC; Kennedy MA; Venkatesh SS; Otter CJ; Hofstadter WA; Jean Beltran PM; Cristea IM, Mitochondria and peroxisome remodeling across cytomegalovirus infection time viewed through the lens of Inter-ViSTA. Cell reports 2020, 32 (4), 107943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Price DR; Benedetti E; Hoffman KL; Gomez-Escobar L; Alvarez-Mulett S; Capili A; Sarwath H; Parkhurst CN; Lafond E; Weidman K; Ravishankar A; Cheong JG; Batra R; Büyüközkan M; Chetnik K; Easthausen I; Schenck EJ; Racanelli AC; Reed HO; Laurence JC; Josefowicz SZ; Lief L; Choi ME; Rafii S; Schmidt F; Borczuk AC; Krumsiek J; Choi AMK, The maladaptive vascular response in COVID-19 acute respiratory distress syndrome and recovery. medRxiv 2021, 2021.05.20.21257542. [Google Scholar]
- 53.Song L; Song M; Rabkin CS; Williams S; Chung Y; Van Duine J; Liao LM; Karthikeyan K; Gao W; Park JG; Tang Y; Lissowska J; Qiu J; LaBaer J; Camargo MC, Helicobacter pylori immunoproteomic profiles in gastric cancer. J Proteome Res 2021, 20 (1), 409–419. [DOI] [PubMed] [Google Scholar]
- 54.Barderas R; Srivastava S; LaBaer J, Protein Microarray-Based Proteomics for Disease Analysis. Methods in molecular biology 2021, 2344, 3–6. [DOI] [PubMed] [Google Scholar]
- 55.Meyer TC; Michalik S; Holtfreter S; Weiss S; Friedrich N; Volzke H; Kocher T; Kohler C; Schmidt F; Broker BM; Volker U, A Comprehensive view on the human antibody repertoire against staphylococcus aureus antigens in the general population. Front Immunol 2021, 12, 651619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lachen-Montes M; Corrales FJ; Fernandez-Irigoyen J; Santamaria E, Proteomics insights into the molecular basis of SARS-CoV-2 infection: What we can learn from the human olfactory axis. Front Microbiol 2020, 11, 2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fu Q; Johnson CW; Wijayawardena BK; Kowalski MP; Kheradmand M; Van Eyk JE, A plasma sample preparation for mass spectrometry using an automated workstation. J Vis Exp 2020, (158). [DOI] [PubMed] [Google Scholar]
- 58.Martin TG; Myers VD; Dubey P; Dubey S; Perez E; Moravec CS; Willis MS; Feldman AM; Kirk JA, Cardiomyocyte contractile impairment in heart failure results from reduced BAG3-mediated sarcomeric protein turnover. Nat Commun 2021, 12 (1), 2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tucholski T; Cai W; Gregorich ZR; Bayne EF; Mitchell SD; McIlwain SJ; de Lange WJ; Wrobbel M; Karp H; Hite Z; Vikhorev PG; Marston SB; Lal S; Li A; Dos Remedios C; Kohmoto T; Hermsen J; Ralphe JC; Kamp TJ; Moss RL; Ge Y, Distinct hypertrophic cardiomyopathy genotypes result in convergent sarcomeric proteoform profiles revealed by top-down proteomics. Proceedings of the National Academy of Sciences of the United States of America 2020, 117 (40), 24691–24700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ashwood C; Waas M; Weerasekera R; Gundry RL, Reference glycan structure libraries of primary human cardiomyocytes and pluripotent stem cell-derived cardiomyocytes reveal cell-type and culture stage-specific glycan phenotypes. J Mol Cell Cardiol 2020, 139, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tumasian RA 3rd; Harish A; Kundu G; Yang JH; Ubaida-Mohien C; Gonzalez-Freire M; Kaileh M; Zukley LM; Chia CW; Lyashkov A; Wood WH 3rd; Piao Y; Coletta C; Ding J; Gorospe M; Sen R; De S; Ferrucci L, Skeletal muscle transcriptome in healthy aging. Nat Commun 2021, 12 (1), 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tharakan R; Ubaida-Mohien C; Piao Y; Gorospe M; Ferrucci L, Ribosome profiling analysis of human skeletal muscle identifies reduced translation of mitochondrial proteins with age. RNA Biol 2021, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adelnia F; Ubaida-Mohien C; Moaddel R; Shardell M; Lyashkov A; Fishbein KW; Aon MA; Spencer RG; Ferrucci L, Proteomic signatures of in vivo muscle oxidative capacity in healthy adults. Aging Cell 2020, 19 (4), e13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zampino M; Tanaka T; Ubaida-Mohien C; Fantoni G; Candia J; Semba RD; Ferrucci L, A plasma proteomic signature of skeletal muscle mitochondrial function. Int J Mol Sci 2020, 21 (24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ciordia S; Alvarez-Sola G; Rullan M; Urman JM; Avila MA; Corrales FJ, Digging deeper into bile proteome. J Proteomics 2021, 230, 103984. [DOI] [PubMed] [Google Scholar]
- 66.Urman JM; Herranz JM; Uriarte I; Rullan M; Oyon D; Gonzalez B; Fernandez-Urien I; Carrascosa J; Bolado F; Zabalza L; Arechederra M; Alvarez-Sola G; Colyn L; Latasa MU; Puchades-Carrasco L; Pineda-Lucena A; Iraburu MJ; Iruarrizaga-Lejarreta M; Alonso C; Sangro B; Purroy A; Gil I; Carmona L; Cubero FJ; Martinez-Chantar ML; Banales JM; Romero MR; Macias RIR; Monte MJ; Marin JJG; Vila JJ; Corrales FJ; Berasain C; Fernandez-Barrena MG; Avila MA, Pilot multi-omic analysis of human bile from benign and malignant biliary strictures: A machine-learning approach. Cancers (Basel) 2020, 12 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J; Li Y; Liu Q; Huang Y; Li R; Wu T; Zhang Z; Zhou J; Huang H; Tang Q; Huang C; Zhao Y; Zhang G; Jiang W; Mo L; Zhang J; Xie W; He J, Sirt6 alleviated liver fibrosis by deacetylating conserved Lysine 54 on Smad2 in hepatic stellate cells. Hepatology 2021, 73 (3), 1140–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kilanczyk E; Banales JM; Wunsch E; Barbier O; Avila MA; Mato JM; Milkiewicz M; Milkiewicz P, S-adenosyl-L-methionine (SAMe) halts the autoimmune response in patients with primary biliary cholangitis (PBC) via antioxidant and S-glutathionylation processes in cholangiocytes. Biochim Biophys Acta Mol Basis Dis 2020, 1866 (11), 165895. [DOI] [PubMed] [Google Scholar]
- 69.Sundararaman N; Go J; Robinson AE; Mato JM; Lu SC; Van Eyk JE; Venkatraman V, PINE: An automation tool to extract and visualize protein-centric functional networks. J Am Soc Mass Spectrom 2020, 31 (7), 1410–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gonzalez-Gomariz J; Serrano G; Tilve-Alvarez CM; Corrales FJ; Guruceaga E; Segura V, UPEFinder: A bioinformatic tool for the study of uncharacterized proteins based on gene expression correlation and the PageRank algorithm. J Proteome Res 2020, 19 (12), 4795–4807. [DOI] [PubMed] [Google Scholar]
- 71.Petralia F; Tignor N; Reva B; Koptyra M; Chowdhury S; Rykunov D; Krek A; Ma W; Zhu Y; Ji J; Calinawan A; Whiteaker JR; Colaprico A; Stathias V; Omelchenko T; Song X; Raman P; Guo Y; Brown MA; Ivey RG; Szpyt J; Guha Thakurta S; Gritsenko MA; Weitz KK; Lopez G; Kalayci S; Gumus ZH; Yoo S; da Veiga Leprevost F; Chang HY; Krug K; Katsnelson L; Wang Y; Kennedy JJ; Voytovich UJ; Zhao L; Gaonkar KS; Ennis BM; Zhang B; Baubet V; Tauhid L; Lilly JV; Mason JL; Farrow B; Young N; Leary S; Moon J; Petyuk VA; Nazarian J; Adappa ND; Palmer JN; Lober RM; Rivero-Hinojosa S; Wang LB; Wang JM; Broberg M; Chu RK; Moore RJ; Monroe ME; Zhao R; Smith RD; Zhu J; Robles AI; Mesri M; Boja E; Hiltke T; Rodriguez H; Zhang B; Schadt EE; Mani DR; Ding L; Iavarone A; Wiznerowicz M; Schurer S; Chen XS; Heath AP; Rokita JL; Nesvizhskii AI; Fenyo D; Rodland KD; Liu T; Gygi SP; Paulovich AG; Resnick AC; Storm PB; Rood BR; Wang P; Children's Brain Tumor Network; Clinical Proteomic Tumor Analysis Consortium, Integrated proteogenomic characterization across major histological types of pediatric brain cancer. Cell 2020, 183 (7), 1962–1985 e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krug K; Jaehnig EJ; Satpathy S; Blumenberg L; Karpova A; Anurag M; Miles G; Mertins P; Geffen Y; Tang LC; Heiman DI; Cao S; Maruvka YE; Lei JT; Huang C; Kothadia RB; Colaprico A; Birger C; Wang J; Dou Y; Wen B; Shi Z; Liao Y; Wiznerowicz M; Wyczalkowski MA; Chen XS; Kennedy JJ; Paulovich AG; Thiagarajan M; Kinsinger CR; Hiltke T; Boja ES; Mesri M; Robles AI; Rodriguez H; Westbrook TF; Ding L; Getz G; Clauser KR; Fenyo D; Ruggles KV; Zhang B; Mani DR; Carr SA; Ellis MJ; Gillette MA; Clinical Proteomic Tumor Analysis Consortium, Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. Cell 2020, 183 (5), 1436–1456 e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu Y; Pan J; Shah P; Ao M; Thomas SN; Liu Y; Chen L; Schnaubelt M; Clark DJ; Rodriguez H; Boja ES; Hiltke T; Kinsinger CR; Rodland KD; Li QK; Qian J; Zhang Z; Chan DW; Zhang H; Clinical Proteomic Tumor Analysis Consortium, Integrated proteomic and glycoproteomic characterization of human high-grade serous ovarian carcinoma. Cell reports 2020, 33 (3), 108276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gillette MA; Satpathy S; Cao S; Dhanasekaran SM; Vasaikar SV; Krug K; Petralia F; Li Y; Liang WW; Reva B; Krek A; Ji J; Song X; Liu W; Hong R; Yao L; Blumenberg L; Savage SR; Wendl MC; Wen B; Li K; Tang LC; MacMullan MA; Avanessian SC; Kane MH; Newton CJ; Cornwell M; Kothadia RB; Ma W; Yoo S; Mannan R; Vats P; Kumar-Sinha C; Kawaler EA; Omelchenko T; Colaprico A; Geffen Y; Maruvka YE; da Veiga Leprevost F; Wiznerowicz M; Gumus ZH; Veluswamy RR; Hostetter G; Heiman DI; Wyczalkowski MA; Hiltke T; Mesri M; Kinsinger CR; Boja ES; Omenn GS; Chinnaiyan AM; Rodriguez H; Li QK; Jewell SD; Thiagarajan M; Getz G; Zhang B; Fenyo D; Ruggles KV; Cieslik MP; Robles AI; Clauser KR; Govindan R; Wang P; Nesvizhskii AI; Ding L; Mani DR; Carr SA; Clinical Proteomic Tumor Analysis Consortium, Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 2020, 182 (1), 200–225 e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nice EC, The status of proteomics as we enter the 2020s: Towards personalised/precision medicine. Anal Biochem 2020, 113840. [DOI] [PubMed] [Google Scholar]
- 76.Zhu T; Chen H; Yan X; Wu Z; Zhou X; Xiao Q; Ge W; Zhang Q; Xu C; Xu L; Ruan G; Xue Z; Yuan C; Chen GB; Guo T, ProteomeExpert: a docker image based web-server for exploring, modeling, visualizing, and mining quantitative proteomic data sets. Bioinformatics 2021, 37(2):273–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu T; Sun R; Zhang F; Chen GB; Yi X; Ruan G; Yuan C; Zhou S; Guo T, BatchServer: A web server for batch effect evaluation, visualization, and correction. J Proteome Res 2021, 20 (1), 1079–1086. [DOI] [PubMed] [Google Scholar]
- 78.Kalyuzhnyy A; Eyers PA; Eyers CE; Sun Z; Deutsch EW; Jones AR, Profiling the human phosphoproteome to estimate the true extent of protein phosphorylation. bioRxiv 2021.04.14.439901, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoopmann MR; Kusebauch U; Palmblad M; Bandeira N; Shteynberg DD; He L; Xia B; Stoychev SH; Omenn GS; Weintraub ST; Moritz RL, Insights from the first phosphopeptide challenge of the MS resource pillar of the HUPO Human Proteome Project. J Proteome Res 2020, 19 (12), 4754–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tanaka A; Wang JY; Shia J; Zhou Y; Ogawa M; Hendrickson RC; Klimstra DS; Roehrl MH, DEAD-box RNA helicase protein DDX21 as a prognosis marker for early stage colorectal cancer with microsatellite instability. Scientific reports 2020, 10 (1), 22085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang JY; Zhang W; Roehrl MW; Roehrl VB; Roehrl MH, An autoantigen profile of human A549 lung cells reveals viral and host etiologic molecular attributes of autoimmunity in COVID-19. J Autoimmun 2021, 120, 102644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang JY; Zhang W; Rho JH; Roehrl MW; Roehrl MH, A proteomic repertoire of autoantigens identified from the classic autoantibody clinical test substrate HEp-2 cells. Clin Proteomics 2020, 17, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ogawa M; Shintani-Domoto Y; Nagashima Y; Ode KL; Sato A; Shimizu Y; Ohashi K; Roehrl MHA; Ushiku T; Ueda HR; Fukayama M, Mass spectrometry-based absolute quantification of amyloid proteins in pathology tissue specimens: Merits and limitations. PloS one 2020, 15 (7), e0235143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roehrl MH; Roehrl VB; Wang JY, Proteome-based pathology: the next frontier in precision medicine. Expert Rev Precis Med Drug Dev 2021, 6 (1), 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mehnert M; Ciuffa R; Frommelt F; Uliana F; van Drogen A; Ruminski K; Gstaiger M; Aebersold R, Multi-layered proteomic analyses decode compositional and functional effects of cancer mutations on kinase complexes. Nat Commun 2020, 11 (1), 3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Irmisch A; Bonilla X; Chevrier S; Lehmann KV; Singer F; Toussaint NC; Esposito C; Mena J; Milani ES; Casanova R; Stekhoven DJ; Wegmann R; Jacob F; Sobottka B; Goetze S; Kuipers J; Sarabia Del Castillo J; Prummer M; Tuncel MA; Menzel U; Jacobs A; Engler S; Sivapatham S; Frei AL; Gut G; Ficek J; Miglino N; Tumor Profiler C; Aebersold R; Bacac M; Beerenwinkel N; Beisel C; Bodenmiller B; Dummer R; Heinzelmann-Schwarz V; Koelzer VH; Manz MG; Moch H; Pelkmans L; Snijder B; Theocharides APA; Tolnay M; Wicki A; Wollscheid B; Ratsch G; Levesque MP, The Tumor Profiler Study: integrated, multi-omic, functional tumor profiling for clinical decision support. Cancer cell 2021, 39 (3), 288–293. [DOI] [PubMed] [Google Scholar]
- 87.Uhlen M; Fagerberg L; Hallstrom BM; Lindskog C; Oksvold P; Mardinoglu A; Sivertsson A; Kampf C; Sjostedt E; Asplund A; Olsson I; Edlund K; Lundberg E; Navani S; Szigyarto CA; Odeberg J; Djureinovic D; Takanen JO; Hober S; Alm T; Edqvist PH; Berling H; Tegel H; Mulder J; Rockberg J; Nilsson P; Schwenk JM; Hamsten M; von Feilitzen K; Forsberg M; Persson L; Johansson F; Zwahlen M; von Heijne G; Nielsen J; Ponten F, Proteomics. Tissue-based map of the human proteome. Science 2015, 347 (6220), 1260419. [DOI] [PubMed] [Google Scholar]
- 88.Mahdessian D; Cesnik AJ; Gnann C; Danielsson F; Stenstrom L; Arif M; Zhang C; Le T; Johansson F; Shutten R; Backstrom A; Axelsson U; Thul P; Cho NH; Carja O; Uhlen M; Mardinoglu A; Stadler C; Lindskog C; Ayoglu B; Leonetti MD; Ponten F; Sullivan DP; Lundberg E, Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature 2021, 590 (7847), 649–654. [DOI] [PubMed] [Google Scholar]
- 89.Karlsson M; Zhang C; Méar L; Zhong W; Digre A; Katona B; Sjöstedt E; Butler L; Odeberg J; Dusart P; Edfors F; Oksvold P; von Feilitzen K; Zwahlen M; Arif M; Altay O; Li X; Ozcan M; Mardonoglu A; Fagerberg L; Mulder J; Luo Y; Ponten F; Uhlén M; Lindskog C, A single–cell type transcriptomics map of human tissues. Science Advances 2021, 7 (31), eabh2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hikmet F; Mear L; Edvinsson A; Micke P; Uhlen M; Lindskog C, The protein expression profile of ACE2 in human tissues. Molecular systems biology 2020, 16 (7), e9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rudberg AS; Havervall S; Manberg A; Jernbom Falk A; Aguilera K; Ng H; Gabrielsson L; Salomonsson AC; Hanke L; Murrell B; McInerney G; Olofsson J; Andersson E; Hellstrom C; Bayati S; Bergstrom S; Pin E; Sjoberg R; Tegel H; Hedhammar M; Phillipson M; Nilsson P; Hober S; Thalin C, SARS-CoV-2 exposure, symptoms and seroprevalence in healthcare workers in Sweden. Nat Commun 2020, 11 (1), 5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alkharaan H; Bayati S; Hellstrom C; Aleman S; Olsson A; Lindahl K; Bogdanovic G; Healy K; Tsilingaridis G; De Palma P; Hober S; Manberg A; Nilsson P; Pin E; Sallberg Chen M, Persisting salivary IgG against SARS-CoV-2 at 9 months after mild COVID-19: A complementary approach to population surveys. J Infect Dis 2021, 224(3):407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sivertsson A; Lindstrom E; Oksvold P; Katona B; Hikmet F; Vuu J; Gustavsson J; Sjostedt E; von Feilitzen K; Kampf C; Schwenk JM; Uhlen M; Lindskog C, Enhanced validation of antibodies enables the discovery of missing proteins. J Proteome Res 2020, 19 (12), 4766–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Assarsson E; Lundberg M; Holmquist G; Bjorkesten J; Thorsen SB; Ekman D; Eriksson A; Rennel Dickens E; Ohlsson S; Edfeldt G; Andersson AC; Lindstedt P; Stenvang J; Gullberg M; Fredriksson S, Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PloS one 2014, 9 (4), e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rohloff JC; Gelinas AD; Jarvis TC; Ochsner UA; Schneider DJ; Gold L; Janjic N, Nucleic acid ligands with protein-like side chains: Modified aptamers and their use as diagnostic and therapeutic agents. Mol Ther Nucleic Acids 2014, 3, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Magis AT; Rappaport N; Conomos MP; Omenn GS; Lovejoy JC; Hood L; Price ND, Untargeted longitudinal analysis of a wellness cohort identifies markers of metastatic cancer years prior to diagnosis. Scientific reports 2020, 10 (1), 16275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Orchard S; Hermjakob H; Apweiler R, The proteomics standards initiative. Proteomics 2003, 3 (7), 1374–6. [DOI] [PubMed] [Google Scholar]
- 98.Deutsch EW; Orchard S; Binz PA; Bittremieux W; Eisenacher M; Hermjakob H; Kawano S; Lam H; Mayer G; Menschaert G; Perez-Riverol Y; Salek RM; Tabb DL; Tenzer S; Vizcaino JA; Walzer M; Jones AR, Proteomics standards initiative: Fifteen years of progress and future work. J Proteome Res 2017, 16 (12), 4288–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trkulja CL; Jungholm O; Davidson M; Jardemark K; Marcus MM; Hagglund J; Karlsson A; Karlsson R; Bruton J; Ivarsson N; Srinivasa SP; Cavallin A; Svensson P; Jeffries GDM; Christakopoulou MN; Reymer A; Ashok A; Willman G; Papadia D; Johnsson E; Orwar O, Rational antibody design for undruggable targets using kinetically controlled biomolecular probes. Sci Adv 2021, 7 (16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Geyer PE; Mann SP; Treit PV; Mann M, Plasma proteomes can be re-identifiable and potentially contain personally sensitive and incidental findings. Mol Cell Proteomics 2021, 100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Geyer PE; Wewer Albrechtsen NJ; Tyanova S; Grassl N; Iepsen EW; Lundgren J; Madsbad S; Holst JJ; Torekov SS; Mann M, Proteomics reveals the effects of sustained weight loss on the human plasma proteome. Molecular systems biology 2016, 12 (12), 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mann SP; Treit PV; Geyer PE; Omenn GS; Mann M, Ethical principles, constraints and opportunities in clinical proteomics. Mol Cell Proteomics 2021, 100046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nobis N; Grizzle WE; Sodeke S, Bioethics and Cancer Biomarker Research. In Biomarkers in Cancer Screening and Early Detection, First ed.;Srivastava S, Ed. John Wiley & Sons, Inc.: 2017; pp 277–282. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.