

Abstract
Sirtiun 5 (SIRT5) is a NAD+-dependent protein lysine deacylase primarily located in mitochondria. SIRT5 displays an affinity for negatively charged acyl groups and mainly catalyzes lysine deglutarylation, desuccinylation, and demalonylation while possessing weak deacetylase activity. SIRT5 substrates play crucial roles in metabolism and reactive oxygen species (ROS) detoxification, and SIRT5 activity is protective in neuronal and cardiac physiology. Moreover, SIRT5 exhibits a dichotomous role in cancer, acting as context-dependent tumor promoter or suppressor. Given its multifaceted activity, SIRT5 is a promising target in the design of activators or inhibitors that might act as therapeutics in many pathologies, including cancer, cardiovascular disorders, and neurodegeneration. To date, few cellular-active peptide-based SIRT5 inhibitors (SIRT5i) have been described, and potent and selective small-molecule SIRT5i have yet to be discovered. In this perspective, we provide an outline of SIRT5’s roles in different biological settings and describe SIRT5 modulators in terms of their mode of action, pharmacological activity, and structure–activity relationships.
1. Introduction
Following translation, proteins may undergo post-translational modifications (PTMs) on their amino acid side chains, expanding the spectrum of functions, stability, and subcellular localization. Most PTMs are dynamic, thereby enabling each protein to interchange between many functional states. In particular, the ε-N-lysine residues of proteins are subject to many PTMs, such as alkylation and acylation.1 Among these PTMs, lysine acetylation was initially identified on histone proteins in the 1960s and linked to transcriptional regulation.2 Subsequent studies identified the enzymes that catalyzed the transfer of acetyl groups to histones, called histone acetyltransferases (HATs),3 while the enzymes that catalyzed the removal of acetyl groups were named histone deacetylases (HDACs).4−6 Later investigations demonstrated that proteins other than histones7 may also undergo (de)acetylation, and recent studies have revealed other acyllysine modifications other than acetylation (e.g., lysines acylated with short-, medium-, and long-chain saturated carboxylic acids, short-chain dicarboxylic acids, and carboxylic acids with extra moieties such as 2-hydroxyisobutyric, crotonic, and lipoic acid residues).6,8 In line with this, HDACs were shown to catalyze a wider range of deacylation reactions in both protein and nonprotein substrates, such as polyamines.6
HDACs are divided into Zn2+-dependent deacylases consisting of classes I, II, and IV HDACs9 and nicotinamide adenine dinucleotide (NAD+)-dependent enzymes consisting of class III HDACs, also named sirtuins (SIRTs) due to their homology to the yeast silent information regulator 2 (Sir2).10 Some SIRT family members also possess broad-spectrum protein lysine deacylase, mono-ADP-ribosylase, and lipoamidase activities.11 Given their ability to catalyze the removal of many PTMs, sirtuins are involved in several biological processes, including DNA damage repair, aging, cell cycle regulation, gene expression, metabolism, longevity, and stress response.12−15 It is worth noting that epigenetic and metabolic pathways are tightly interconnected. Indeed, most enzymes that catalyze epigenetic modifications use crucial metabolites as cosubstrates (for example, S-adenosyl methionine, α-ketoglutarate, acetyl- and acyl-CoA, FAD/FADH2, and NAD+/NADH).16 Specifically, sirtuins require NAD+ as a cosubstrate for catalysis and are inhibited by NADH;17 as a result, they are sensitive to the intracellular NAD+/NADH ratio, thereby serving as sensors of cellular metabolic status. Under normal conditions, the NAD+/NADH ratio fluctuates modestly; nevertheless, it varies drastically under situations of nutrient deprivation, obesity, tumorigenesis, and aging. Consequently, changes in metabolism also influence gene expression and signaling pathways through the altered activity of sirtuins.
In mammals, the sirtuin family includes seven isoforms (SIRT1–7)18 that possess highly conserved NAD+-binding and catalytic domains and differing N- and C-termini, which determine their substrate preference, enzymatic activity, and subcellular localization. SIRT1, SIRT6, and SIRT7 are mostly present in the nucleus, with SIRT7 being mainly a nucleolar protein. Among them, SIRT1 may also be found in the cytosol.19−22 SIRT2 is mainly cytoplasmic, although it may shuttle in the nucleus during mitosis, and an alternatively spliced isoform is constitutively present in the nucleus.21,23,24 Finally, SIRT3, SIRT4, and SIRT5 are predominantly found in the mitochondrial matrix.19,25−28 The sirtuin-mediated deacylation reaction employs NAD+ as a cosubstrate and produces, besides the deacylated product, 2′-O-acyl-ADP-ribose and nicotinamide, which can act as a physiological sirtuin inhibitor.29 Each sirtuin isoform exhibits a preference for different ε-N-acyl-lysine PTMs (Table 1). SIRT1–3 preferentially catalyze protein lysine deacetylation reactions.19,30,31 SIRT4 exhibits lipoamidase32 and mono-ADP-ribosyltransferase27 activities and has also the ability to cleave glutaryl, 3-methylglutaryl, 3-hydroxy-3-methyl-glutaryl (HMG), and 3-methylglutaconyl groups.33,34 SIRT6 exhibits a broad spectrum of deacylase activities and a mono-ADP-ribosyltransferase action,35,36 while SIRT7 exhibits deacetylation,37−39 desuccinylation,40 and deglutarylation41 activities. SIRT5 has been shown to selectively cleave negatively charged acyl lysine modifications, such as glutarate, succinate, and malonate, both in vitro and in vivo, although it also has weak deacetylase activity (Figure 1), with a catalytic efficiency roughly 1000-fold lower than those of the deacylation reactions.18,42 While lysine acetylation is catalyzed by many enzymes belonging to the HAT family,43 the identification of acyltransferases that catalyze the transfer of nonacetyl groups has remained elusive for many years. Recently, known enzymes that catalyze a diverse subset of reactions have been revealed to also exhibit lysine acyltransferase activity.44,45 These include carnitine palmitoyl transferase 1A (CPT1A) and HATs p300/CBP and general control nondepressible 5 (GCN5), which were shown to demonstrate succinyltransferase activity.46−48 GCN5 also exhibits lysine glutaryltransferase activity.41 In the case of long-chain fatty acids, the N-terminal glycine myristoyltransferases (NMTs) 1 and 2 were recently shown to also catalyze lysine myristoylation.49 Moreover, numerous lysine acyl modifications arise through a nonenzymatic mechanism involving the direct reaction of acyl-CoA species (especially 4- and 5-carbon negatively charged dicarboxyl CoA thioesters such as succinyl-CoA, glutaryl-CoA, methylglutaryl-CoA, and HMG-CoA) with lysine ε0amino groups under physiological conditions, particularly in the mitochondrial matrix.50,51 Finally, protein lipoylation, counteracted by SIRT4,28 is catalyzed by specific enzymes that either directly transfer lipoic acid to lysine ε0amino groups (LplA) or act indirectly via a stepwise mechanism whereby octanoic acid is transferred to lysine ε-amino groups by LipB or LplA, followed by the insertion of two sulfur atoms at C6 and C8 by LipA to form a complete lipoamide.52
Table 1. Summary of the Enzymatic Activity of Each SIRT Isoform.
| SIRT isoform | enzymatic activity |
|---|---|
| SIRT1 | deacetylase |
| SIRT2 | deacetylase |
| SIRT3 | deacetylase |
| SIRT4 | delipoylase, de-HMG-ase, deglutarylase, demethylglutarylase, demethylglutaconylase, mono-ADP-ribosylase |
| SIRT5 | deglutarylase, desuccinylase, demalonylase, deacetylase |
| SIRT6 | deacylase (long fatty acyl chains), deacetylase, mono-ADP-ribosylase |
| SIRT7 | deacetylase, desuccinylase, deglutarylase |
Figure 1.

Deacylation reaction catalyzed by SIRT5. The acyl moiety is transferred to the NAD+ cosubstrate, yielding the corresponding 2′-O-acyl-ADP-ribose and nicotinamide.
Several studies have indicated that SIRT5 participates in various biochemical pathways by regulating the activity of many metabolic enzymes such as carbamoylphosphate synthetase I (CPS1), which is important in ammonia detoxification,53 and 3-hydroxy-3-methylglutaryl-CoA synthetase 2 (HMGCS2), which is involved in the formation of ketone bodies.54 As a mitochondrial sirtuin, SIRT5 plays a pivotal role in mitochondrial metabolism, regulating amino acid degradation, cellular respiration,55 reactive oxygen species (ROS) management,56 fatty acid oxidation,57,58 and glycolysis.59 As a result, SIRT5 dysregulation can lead to a variety of diseases, such as metabolic (e.g., diabetes) and neurodegenerative disorders, cardiovascular pathologies, and cancer.60−64 Due to its wide-ranging functions, the modulation of SIRT5 activity has great potential for the treatment of these diseases. Consequently, SIRT5 is a valuable target for the development of modulators that, acting as either activators or inhibitors, may have significant therapeutic potential in various contexts. Here, we present an overview of SIRT5’s characteristics, structure, functional roles in both physiological and pathological cellular processes, and pharmacological modulation with the aim of suggesting new approaches for developing new potential SIRT5 modulators that may be used to treat SIRT5-related diseases.
2. Functional and Structural Features of SIRT5
SIRT5 is widely distributed in the human body and is mostly localized in the liver, heart, kidneys, brain, muscles, and testes.19,53 At the cellular level, SIRT5 is largely present in the mitochondrial matrix, although some studies have demonstrated its presence in the cytosol, nucleus, and peroxisomes.39,57,65,66 In line with this, high levels of several succinylated,54,57,67 glutarylated,68 and malonylated59 cytosolic and nuclear proteins were reported following SIRT5 deletion in mice, while the acetylation level was not affected.11,68−71 Interestingly, in humans, there are four different isoforms encoded by the SIRT5 gene: SIRT5iso1, SIRT5iso2, and SIRT5iso3, which are localized in the mitochondria, and SIRT5iso4, which is localized in the cytosol. SIRT5iso1 is the most studied isoform, while SIRT5iso2–4 are rarely detected in human cells. Compared to SIRT5iso1, SIRTiso2 lacks 11 residues at the C-terminus, SIRT5iso3 lacks an internal sequence of 18 residues, and SIRT5iso4 lacks 108 N-terminal residues, including the mitochondrial localization tag.66,72,73
SIRT5 activity is controlled by two key metabolism regulators. The overexpression of peroxisome proliferator-activated receptor coactivator 1α (PGC-1α) leads to high levels of cellular SIRT5, whereas the activation of AMP-activated protein kinase (AMPK) causes SIRT5 downregulation.74 As previously stated, SIRT5 predominantly exhibits deglutarylase,68 desuccinylase,54,57 and demalonylase59,70 activities, but it also displays weak deacetylase activity toward different substrates (Figure 1).42,69,75 Specifically, using a CPS1-derived octapeptide appropriately modified at the lysine residues, Roessler and colleagues performed kinetic studies through a HPLC-based method to investigate the catalytic efficiencies of the various deacylation and deacetylation reactions. This analysis suggested that SIRT5 had the highest catalytic efficiency for deglutarylation (kcat/KM = 18699 M–1 s–1), followed by desuccinylation (kcat/KM = 13995 M–1 s–1) and demalonylation (kcat/KM = 3758 M–1 s–1), while the deacetylation reaction was shown to be by far the least catalytically efficient (kcat/KM = 16 M–1 s–1).76 Notably, adding a carboxylic group to the acyl chain did not produce massive changes in the apparent affinity for the SIRT5 catalytic site, since KM remained in the same order of magnitude, but did increase the catalytic rate, as exemplified by the 50–200-fold increase in kcat. Given these results, it seems that the deacetylase activity of SIRT5 is negligible compared to the deacylase activity. Moreover, multiple studies indicate that deacylation, particularly desuccinylation, is the most relevant SIRT5-catalyzed reaction at the cellular level. However, some reports point toward the SIRT5-mediated deacetylation of certain substrates. Hence, the further characterization of SIRT5’s enzymatic activity at the cellular level would be necessary to understand whether SIRT5 genuinely has substrate-specific deacetylase activity or if these findings are due to SIRT5 overexpression or cross-reactivity with antiacetyllysine antibodies. To date, many crystal structures of SIRT5 in complex with substrates or small molecules have been released,69,76−80 thereby allowing the structural and functional characterization of the enzyme and aiding the design of specific modulators. By inspecting the crystal structure of SIRT5 in complex with the H3K9succ peptide and NAD+,69 we can observe that it consists of 14 α-helices and 9 β-strands that are organized to form a Rossmann fold and a Zn2+-binding domain. Between these two domains is a cleft that forms the catalytic region, which contains the binding sites of both the protein substrate and the cosubstrate NAD+. The Rossmann fold domain is comprised of six parallel β-strands that form a central β-sheet surrounded by nine α-helices. The Zn2+-binding domain contains five small α-helices and an antiparallel β-sheet formed by the β-strands (Figure 2A). The antiparallel β-sheet is stabilized by the presence of a Zn2+ ion coordinated with four Cys residues (Cys166, Cys169, Cys207, and Cys212).
Figure 2.

(A) Structure of SIRT5 in complex with the H3K9-succinyl peptide (beige) and bound NAD+ (yellow) (PDB ID 3RIY). (B) Focus on the catalytic pocket. The key interactions of the substrate peptide and NAD+ with SIRT5 residues are indicated. Dashed orange lines indicate polar interactions.
The catalytic cleft is formed by several connecting loops between the Rossman fold and the Zn2+-binding domains. Loop S, which connects α10 of the Rossman fold domain with β6 of the Zn2+-binding domain, is crucial for substrate binding. Loop N, which connects α2 of the Rossman fold domain with α3 of the Zn2+-binding domain, is involved in NAD+ binding (Figure 2A). Many residues in this region are involved in substrate and cosubstrate binding. Among them, Phe223, Leu227, and Val254 define the hydrophobic entry gate for acyl-lysine, while Ala86, Tyr102, Arg105, and His158 directly interact with the acyl-lysine substrate. Gln140 and Asn141 interact with the ribose moiety of NAD+, whereas Asp143 binds the nicotinamide product (Figure 2B). In addition, the flexible residue Phe70 acts like a valve, facilitating NAD+ binding as well as nicotinamide release.18,69
Some of these structural features are conserved in SIRT1–3;18,81−83 for instance, the hydrophobic residues Phe223, Leu227, and Val254 are placed in the corresponding position in these orthologues. Conversely, SIRT5 possesses specific residues that characterize its substrate specificity and catalytic activity. In particular, the two nonhydrophobic residues Tyr102 and Arg105 localize deep into the substrate pocket, forming hydrogen bonds and electrostatic interactions with the negatively charged acyl-lysine substrate (Figure 2B). These residues precisely recognize glutaryl, succinyl, and malonyl groups, giving SIRT5 its specific deglutarylase, desuccinylase, and demalonylase activities, respectively.69 Another key residue for substrate recognition is Ala86, which is also specific to SIRT5 because SIRT1–3 bear a phenylalanine residue in the same position. The presence of alanine instead of phenylalanine makes the acyl-lysine binding pocket larger compared to those of other sirtuins, thereby making SIRT5 capable of binding bulkier acylated lysine substrates.69,70
3. Biological Activities and Disease Relevance of SIRT5
To date, it has been reported that SIRT5 regulates many processes involved in cellular metabolism and homeostasis. SIRT5 catalyzes NAD+-dependent deglutarylation, desuccinylation, and demalonylation of metabolic enzymes implicated in glycolysis;59 mitochondrial oxidative phosphorylation;55 fatty acid β-oxidation (FAO);57,58 ROS response;56 glutamine metabolism; and ammonia detoxification.53,84,85 In addition, SIRT5 expression is altered in a variety of cancer types, and it may behave as either a tumor promoter or a tumor suppressor. SIRT5 also plays significant roles in cardiac health maintenance and the neuronal stress response. A recent report suggested that SIRT5 is pivotal in facilitating the replication of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the etiologic agent causing the current COVID-19 pandemic.86 It is therefore apparent that SIRT5 has a rather pleiotropic nature, which is typical of other epigenetic proteins. Nonetheless, SIRT5 activity is mainly linked to the regulation of mitochondrial pathways. Hence, targeting SIRT5 would be especially useful in those settings where mitochondrial dysfunction is relevant. Moreover, the pleiotropic character of SIRT5 activities does not preclude SIRT5 from being considered a potential pharmacological target, since in certain contexts multiple SIRT50affected pathways concur to determine the same phenotype. In the next sections, we will provide detailed information on the molecular mechanisms connecting SIRT5’s activity and physiological and pathological roles and indicate the contexts where SIRT5 inhibition or activation may represent a viable therapeutic option.
3.1. Metabolism
As mentioned above, SIRT5 targets several proteins involved in glycolysis, gluconeogenesis, the tricarboxylic acid (TCA) cycle, and the electron transport chain (ETC), thus regulating many metabolic pathways. Notably, quantitative proteomic analyses showed that SIRT5 preferentially demalonylates glycolytic enzymes, including glyceraldehyde 3-phosphate dehydrogenase (GAPDH), thereby promoting glycolysis (Figure 3). In fact, Nishida et al. demonstrated that replacing Lys184 in GAPDH with glutamic acid, which mimics malonyl-lysine, leads to the inhibition of the enzymatic activity, thus suggesting that the enzyme works only after SIRT5-mediated demalonylation. Consistent with these findings, primary hepatocytes obtained from SIRT5 knockout (KO) mice displayed decreased glycolytic flux.59 These experiments indicated that SIRT5 regulates glucose metabolism. In addition, SIRT5 was also found to be involved in insulin sensitivity; indeed, high SIRT5 levels were found in adipose tissues and were linked with a high insulin response in monozygotic twins.87
Figure 3.
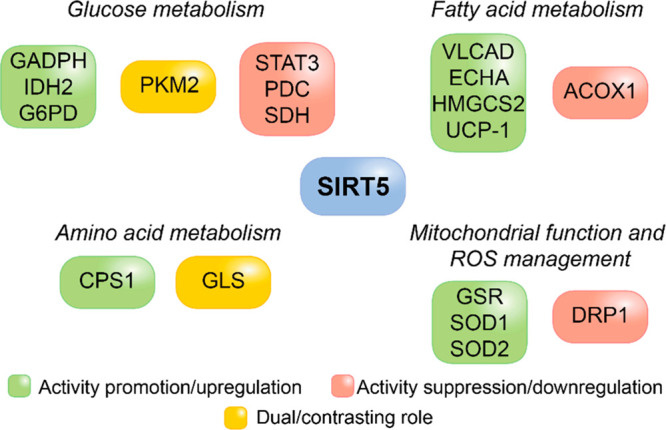
Involvement of proteins modulated by SIRT5 in the regulation of cellular metabolism, mitochondrial function, and the oxidative stress response.
Furthermore, SIRT5 has been shown to deacetylate the signal transducer and activator of transcription 3 (STAT3), suppressing its mitochondrial translocation and inhibiting its interaction with and activation of pyruvate dehydrogenase complex (PDC). This inhibits the catalytic activity of PDC, which consists of oxidizing pyruvate into acetyl-CoA, and subsequently prohibits acetyl-CoA from entering the TCA cycle.88 In the study, the authors also show that SIRT3 contributes, although to a much lesser extent, to STAT3 deacetylation. However, the biological significance of STAT3 deacetylation by SIRT3 was not further explored. In addition, the influence of SIRT5 on STAT3 deacylation (e.g., desuccinylation) was not assessed. Hence, given the weak deacetylase activity of SIRT5, we cannot exclude that it also acts as STAT3 desuccinylase. SIRT5 also inhibits PDC via direct desuccinylation (Figure 3), impairing pyruvate metabolism, causing a decrease in ATP production, and also resulting in the promotion of tumorigenesis. Consistent with this data, SIRT5 ablation resulted in increased ATP synthesis.57 However, SIRT5 loss in HEK293 cells is associated with a reduced pyruvate-dependent cellular respiration,89 thus suggesting that the role of SIRT5 in glucose metabolism is context-dependent.90
The double-faced role of SIRT5 in glycolysis has also been described in the regulation of pyruvate kinase M2 (PKM2), which transforms phosphoenolpyruvate into pyruvate. PKM2 exists in two different functional forms: as a tetramer it possesses strong pyruvate kinase activity, while as a dimer it is mainly localized in the nucleus, has weak pyruvate kinase activity, and mainly acting as a protein kinase.91−94 In a recent study, Wang and co-workers demonstrated that the SIRT5-mediated desuccinylation of PKM2 at Lys311 leads an augmented activity, thereby supporting the glycolytic flux.94 Conversely, Xiangyun and colleagues showed that SIRT5 desuccinylates PKM2 at Lys498 under oxidative stress conditions, inhibiting its activity (Figure 3), repressing glycolysis in lung cancer cells, and consequently readdressing the glucose flux into the pentose phosphate pathway.95 Another study reported that the desuccinylation of PKM2, under glucose deficiency conditions obstructs its translocation into mitochondria and facilitates the degradation of voltage-dependent anion channel 3 (VDAC3), thereby enhancing the opening of the mitochondrial permeability transition pore and finally leading to the apoptosis of colon cancer cells.96 In this case, the contrasting outcomes of these studies may depend on the different cell lines used or different types of induced stress conditions.
Another target of SIRT5 is the enzyme complex succinate dehydrogenase (SDH), also called respiratory complex II, which is involved in both the TCA cycle and the ETC. SDH catalyzes the oxidation of succinate to fumarate and simultaneously transforms ubiquinone to ubiquinol. SIRT5-mediated desuccinylation inhibits SDH activity (Figure 3) and consequently reduces succinate-dependent cellular respiration.57 Interestingly, Zhang and co-workers demonstrated that SIRT5 also desuccinylates various subunits of the ETC complexes and ATP-synthase after cardiolipin binding, thus promoting cellular respiration.89 Finally, SIRT5 has been shown to desuccinylate isocitrate dehydrogenase 2 (IDH2). This increases its activity for the oxidative decarboxylation of isocitrate to α-ketoglutarate in a NADP+-dependent manner, which produces NADPH and CO2 as byproducts.97,98
Concerning FAO, SIRT5 desuccinylates the very-long-chain acyl-CoA dehydrogenase (VLCAD) that catalyzes the initial step in the β-oxidation in mitochondria. Notably, SIRT5 cooperates with SIRT3, which deacetylates VLCAD at Lys299, stabilizing its localization and promoting the association of the cofactor flavin adenine dinucleotide (FAD).58 Overall, the two enzymes promote VLCAD activity (Figure 3) by facilitating the interaction with FAD and increasing its localization in the mitochondrial membrane. In line with this, reduced FAO was reported upon SIRT5 KO in mice.54 In addition, SIRT5 desuccinylates HMGCS2, thereby increasing its activity and stimulating ketone body formation under conditions of caloric restriction.54 SIRT5 supports another step of FAO by increasing the activity of enoyl-CoA hydratase (ECHA, Figure 3), which catalyzes the hydration of the double bond between C2 and C3 of enoyl-CoA.67
In mammals, there are two different types of adipose tissues: white adipose tissue (WAT) specializing in energy storage and release in the form of triglycerides and brown adipose tissue (BAT) containing multiple mitochondria devoted to the dissipation of energy through the expression of uncoupling protein 1 (UCP-1, Figure 3), which is involved in thermogenesis.99,100 Mitochondrial SIRT5 is largely expressed in BAT where it catalyzes protein demalonylation and desuccinylation, thus suggesting that it regulates BAT functions and thermogenesis.101,102 In mouse models, SIRT5 loss was found to reduce UCP-1 function, leading to protein hypersuccinylation and decreased levels of α-ketoglutarate and finally resulting in increased repressive histone methylation (H3K9me2 and H3K9me3) at the promoter region of Prdm16, a transcription factor that facilitates the expression of brown adipocyte genes.103 SIRT5 is also important in the differentiation of brown adipocytes and the conversion of white adipocytes to brown adipocytes.103 Overall, given the involvement of SIRT5 in BAT/WAT equilibrium and because BAT is a key regulator of glucose homeostasis, targeting SIRT5 may be a useful therapeutic approach against metabolic disorders such as obesity and type 2 diabetes.64
Various studies reported the key role of SIRT5 in the regulation of ammonia detoxification and amino acid catabolism through the deacylation and consequent activation of CPS1 (Figure 3).53,68,69,104 This enzyme catalyzes the conversion of ammonia into carbamoyl phosphate, the first reaction of the urea cycle.105 Under caloric restriction, SIRT5-overexpressing cells showed increased hepatic CPS1 activity due to high levels of SIRT5 mRNA in the liver.104 Conversely, SIRT5 KO mice exhibited lower CPS1 activities and enhanced ammonia levels in blood.53,69 SIRT5 also regulates ammonia production in nonliver cells, where it desuccinylates mitochondrial glutaminase (GLS); two studies have reported opposite outcomes (Figure 3). Polletta et al. demonstrated that the SIRT5-mediated desuccinylation of GLS inhibits its activity, thereby repressing the glutamine catabolism to glutamate and the generation of ammonia as a byproduct. The authors proposed Lys245 and Lys320 as possible succinylation sites that may be accessible to the SIRT5 catalytic pocket. Since it was reported that ammonia could induce autophagy and mitophagy in tumor cells, the SIRT5-mediated inhibition of GLS could overcome this protective mechanism for tumor cells, suggesting a tumor suppressor role for SIRT5 in this context.84 Conversely, another study suggested that SIRT5-mediated desuccinylation at Lys164 protects GLS from ubiquitination at Lys164 and the consequent proteasomal degradation, thereby stabilizing it and supporting glutamine catabolism.85
3.2. Mitochondrial Function and Oxidative Stress
The fact that SIRT5’s deacylating activity is reliant on NAD+, a major redox signaling molecule, supports the idea that it is a key player in the regulation of cellular redox homeostasis. Indeed, since NAD+ is a key electron acceptor in multiple enzymatic reactions, the NAD+/NADH ratio is a crucial factor for redox pathways and, therefore, the regulation of ROS levels.
Guedouari et al. reported that SIRT5 regulates many mitochondrial processes, such as elongation, fusion, and division. Indeed, SIRT5-depleted mouse embryonic fibroblasts (MEFs) displayed augmented mitochondrial fragmentation and mitophagy under starvation conditions, along with an increase of dynamin-related protein 1 (DRP1) levels (Figure 3). This indicates that SIRT5 defends mitochondria from starvation-induced autophagy and degradation.106
SIRT5 has a significant role in reducing ROS levels through modulating different enzymes. These include the previously mentioned glycolytic enzymes and glucose-6-phosphate dehydrogenase (G6PD), which converts glucose 6-phosphate to ribose 5-phosphate for the biosynthesis of nucleotides in the pentose phosphate pathway. They both produce NADPH as a byproduct, which is important for the reduction of oxidized glutathione (GSSG) to reduced glutathione (GSH). GSH in turn reduces cellular ROS levels. SIRT5 desuccinylates and deglutarylates IDH2 and G6PD, respectively, activating these enzymes (Figure 3) and promoting NADPH production.97 In line with this, SIRT5 KO or knockdown leads to significantly decreased NADPH and GSH levels, leading to an impairment of the ROS scavenging capability and increased cell vulnerability to oxidative stress.97 Furthermore, SIRT5 deficiency was shown to be correlated with lower levels of glutathione reductase (GSR),62 the enzyme that converts GSSG to GSH.107 In particular, in nonsmall cell lung cancer (NSCLC) cells, SIRT5 knockdown resulted in reduced GSR expression.62
SIRT5 attenuates oxidative stress by targeting peroxisomal acyl-CoA oxidase 1 (ACOX1), a key enzyme involved in FAO that contributes to H2O2 production.108 ACOX1 is functional as a dimer, and its dimerization is inhibited by SIRT5 desuccinylation, thereby blocking H2O2 production and mitigating oxidative stress.39 SIRT5 was also reported to regulate oxidative stress via the deacetylation of the Forkhead protein FOXO3a, thus promoting its shuttling into the nucleus and facilitating the expression of antioxidant defense-related genes.109 However, it should be noticed that FOXO3a is also deacetylated by SIRT1–3, which possess higher deacetylase activities than SIRT5. Moreover, SIRT5-mediated desuccinylation activates Cu/Zn superoxide dismutase 1 (SOD1, Figure 3), and there is a consequent increase in ROS detoxification.56 Overall, these findings suggest that SIRT5 has a pivotal role in regulating cellular mechanisms to protect cells from oxidative stress.
3.3. Neurodegeneration
Mitochondrial functions such as energy production, apoptotic signaling, redox homeostasis, and oxidative phosphorylation are crucial for neuronal health. Consequently, the dysfunction of these processes is connected with the onset of many neurodegenerative diseases, including Parkinson’s disease (PD), Alzheimer’s disease (AD), and epileptic disorders.110 In this context, SIRT5 plays neuroprotective roles, as exemplified by several studies.
Following exposure to kainate, a glutamate analogue that exters neuroexcitatory and epileptogenic effects,111 SIRT5 expression increased in the hippocampus, thereby ensuring neuroprotection against the formation of astrogliosis. Consistent with this data, the depletion of SIRT5 in kainate-exposed mice leads to hippocampal neuronal loss and a severe response to epileptic seizure, which is caused by kainate activity on glutamate receptors.40 Interestingly, the protective role of SIRT5 in this context seems unrelated to its function in ROS detoxification.
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is chemical tool widely employed to induce PD symptoms in animal models. It is a prodrug of the neurotoxin 1-methyl-4-phenylpyridinium (MPP+), which causes the degeneration of dopaminergic neurons in the substantia nigra by increasing ROS levels and inducing cell death.112,113 Notably, treatment with MPTP induced SIRT5 expression in the brain of treated mice. Conversely, a SIRT5 deficiency in mouse brain striata exacerbated the MPTP-induced loss of nigrostriatal dopaminergic neurons. This was associated with the reduced expression of the mitochondrial antioxidant enzyme manganese superoxide dismutase 2 (SOD2, Figure 3).61 These results suggest that SIRT5 activity contributes to ROS scavenging in nigrostriatal dopaminergic neurons and alleviates the effects of MPTP.
Finally, SIRT5 seems to have a protective role also in the context of AD. Indeed, AD mouse models displayed the downregulation of SIRT5 and impaired autophagy, which was reversed by SIRT5 overexpression.114 In addition, SIRT5 expression was associated with elevated SOD activity, lower ROS levels, and diminished apoptosis both in vitro and in vivo. Neuron damage and inflammation were also lower in AD brains that expressed higher SIRT5 levels, which may be a consequence of the inhibition of astrocytes and microglia activation. Overall, these results indicate that SIRT5 activity mitigates neuron damage by suppressing oxidative stress and decreasing the activity of astrocytes and microglia.
3.4. Cardiovascular Regulation
We previously mentioned that the deficiency of SIRT5 in cardiac tissue results in increased levels of succinylated lysine proteins67,115 including SDH,57 which is inhibited by SIRT5-mediated desuccinylation (Figure 3). Interestingly, SIRT5 deficiency has been associated with an increased predisposition to myocardial ischemia-reperfusion injury.116 In addition, treatment with dimethyl malonate, a precursor of the SDH inhibitor malonate, led to reduced superoxide production in SIRT5 KO hearts, confirming the key role of SIRT5 in regulating ROS levels even at the cardiac level.116 In line with this, another study with both in vitro and in vivo models confirmed that the inhibition of SDH in the heart is protective against cardiac myocardial ischemia-reperfusion damage.117
Furthermore, SIRT5 has a protective role for cardiomyocytes, since it suppresses oxidative-stress-induced apoptosis through its interaction with the antiapoptotic factor Bcl-XL.118 SIRT5 also plays a significant role in the cardiac stress response. In a model of hypertrophy induced by pressure overload as a consequence of traverse aortic constriction, SIRT5 loss was associated with a twofold increase in succinylation in more than 750 proteins, along with cardiac dysfunction and higher mortality rates.115
As mentioned above, SIRT5 activates ECHA, an enzyme crucial for myocardial fatty acid metabolism, through desuccinylation (Figure 3). Hence, SIRT5 ablation impairs cardiac FAO and reduces ATP production in conditions where energy is particularly needed, such as during physical exercise or fasting conditions. In addition, SIRT5 KO causes cardiac hypertrophy and an altered echocardiogram profile.67 Overall, these results suggest the importance of SIRT5 activity in cardiac tissue, since its deletion or downregulation may impair heart functionality.
3.5. COVID-19
Recently, SIRT5 was shown to interact with the nonstructural protein 14 (Nsp14) from SARS-CoV-2, a highly conserved enzyme required for viral replication.86 Nsp14 interacts with Nsp10, which stabilizes its N-terminal domain possessing 3′–5′-exoribonuclease activity. The Nsp14–Nsp10 complex is therefore essential for exoribonuclease activity. Nsp14 also possesses a C-terminal domain that displays RNA cap guanine N7-methyltransferase activity, which is not influenced by Nsp10 binding. SIRT5 was shown to interact with Nsp14 but not Nsp10, suggesting the formation of an alternative complex. Notably, SIRT5 catalytic activity is necessary for this interaction, as suggested by mutation experiments or pharmacological inhibition (see compound 3d in section 4.1). However, Nsp14 does not seem to be a direct substrate of SIRT5, and a clear molecular function of the Nsp14–SIRT5 complex could not be revealed. Nonetheless, at the cellular level, SIRT5 KO or pharmacological inhibition reduced SARS-CoV-2 levels. In addition, SIRT5 KO led to higher levels of immunity and a better antiviral response, indicating that SIRT5 also has a role in SARS-CoV-2 infection that goes beyond its interaction with Nsp14. Overall, this study uncovered an unusual type of interaction and points toward the key role of SIRT5 in viral replication, suggesting that SIRT5 inhibition could be a useful strategy to combat COVID-19, most likely in combination with other therapeutics.86
3.6. Double-Faced Role in Cancer
Like other human sirtuins, SIRT5 is involved in different processes, including the maintenance of genomic stability, metabolism, and tumor microenvironment regulation.119,120 Hence, it is not surprising that SIRT5 may have a tumor-promoting or tumor-suppressing role depending on the context and cancer type.
In the next sections, we report different cases in which SIRT5 exhibits either a tumor-suppressor or tumor-promoter function. Notably, in lung cancer,56,62,88,121 hepatocellular carcinoma (HCC),39,122−124 and breast cancer,84,85 SIRT5 displays a dichotomous role, further indicating that its activity is dependent strictly on the specific context and not only the type of tissue or cancer.
3.6.1. Tumor Suppressor Role of SIRT5 in Cancer
As demonstrated by in vitro and in vivo experiments, SIRT5 exerts tumor suppressor functions in glioma, where its desuccinylase activity plays pivotal roles in maintaining mitochondrial functions and arresting cell proliferation.55
Clark and colleagues reported the presence of mutant IDH1 and IDH2 in different types of cancer such as acute myeloid leukemia (AML), chondrosarcoma, and glioma.125 Instead of catalyzing the conversion of isocitrate to α-ketoglutarate, mutant IDH1 and IDH2 convert α-ketoglutarate to R-2-hydroxyglutarate.98,126 This derivative is proposed to promote cancer progression and protect tumor cells from apoptosis by inhibiting α-ketoglutarate-dependent dioxygenases127 and SDH, leading to an upsurge of succinyl-CoA levels and the consequent aberrant succinylation of mitochondrial proteins.55 Furthermore, in glioma cells presenting the R132H mutation of IDH1, protein hypersuccinylation leads to the accumulation of Bcl-2, which promotes apoptotic resistance.55 Conversely, SIRT5 overexpression in glioma cells decreased protein succinylation and reduced cell growth both in vitro and in vivo (Figure 4).55
Figure 4.

Roles of SIRT5 in cancer. The figure depicts the main proteins and pathways regulated by SIRT5, which exerts both tumor-suppressing and tumor-promoting functions. Several mechanisms are implicated, including the regulation of glycolysis, FAO, amino acid metabolism, ATP production, ROS detoxification, apoptosis, and autophagy.
In gastric cancer, SIRT5 overexpression inhibits oxoglutarate dehydrogenase (OGDH), thus decreasing ATP production, increasing ROS levels, and leading to the inhibition of cancer cell proliferation and migration.128 Furthermore, enhanced SIRT5 activity leads to cell cycle arrest at the G1/S phase in tumor cells due to the negative modulation of cyclin-dependent kinase 2 (CDK2) and the inhibition of glycolysis (Figure 4).129
A recent report by Hu et al. reported that SIRT5 acts as a tumor suppressor in pancreatic ductal adenocarcinoma (PDAC).130 PDAC cells with KRAS mutations metabolize glutamine following the GOT2/GOT1/ME1 pathway, a dispensable pathway for the other cells. It was reported that SIRT5 deacetylates aspartate aminotransferase GOT1, predominantly at Lys369, thus inhibiting its activity and decreasing the relative abundance of glutamine or glutathione metabolism intermediates. GOT1 catalyzes the conversion of α-ketoglutarate and aspartate into glutamate and oxaloacetate in the cytosol, increasing NADPH and GSH production to maintain redox homeostasis and facilitate PDAC cell growth (Figure 4). Accordingly, SIRT5 loss leads to a reduction in ROS levels and the consequent proliferation of tumor cells. Notably, it was found that SIRT5 expression is downregulated in both human PDAC tissues and murine pancreatic tumors and is associated with cancer progression and poor prognosis. Furthermore, SIRT5 KO mice expressing KRAS or KRAS/p53 oncogenic mutations exhibited an acceleration in tumor onset and significantly enhanced cancer cell proliferation in a caerulein-induced pancreatitis model in the absence of caerulein. These findings show that SIRT5 may be a tumor suppressor in this type of cancer and that its pharmacological activation (see compound 14, section 4.1) impairs GOT1 activity and reduces PDAC cell viability.130 Hence, activating SIRT5 could be a promising strategy to target PDAC.
As mentioned in the previous section, SIRT5 desuccinylates and activates SOD1, thereby exerting a key function in ROS detoxification. Lin et al. observed that SOD1 succinylation increased lung cancer cell proliferation (Figure 4). In line with this, cells expressing a SOD1 mutant resistant to succinylation showed decreased growth rates, suggesting the protective role of SIRT5 in this setting.56 In addition, in lung cancer A549 cells, SIRT5 is downregulated, resulting in the acetylation and mitochondrial translocation of STAT3. This accelerates the transformation of pyruvate to acetyl-CoA through the interaction with PDC, thus promoting ATP production that sustains cell growth.88
We previously stated that SIRT5 desuccinylates and inhibits the peroxisomal enzyme ACOX1,39 thus reducing the production of H2O2 and consequently alleviating cellular oxidative stress.108 The excessive activation of ACOX1 leads to oxidative DNA damage and alters FAO and redox homeostasis, which causes chronic hepatic disease and finally leads to the insurgence of HCC (Figure 4).39 Another study also indicated that SIRT5 expression is lower in primary liver cancer tissue compared to normal hepatic tissues.122 This causes intensified succinylation and the consequent activation of ACOX1, finally promoting HCC progression due to elevated H2O2 production and oxidative stress (Figure 4).39 Hence, these studies suggest that SIRT5 activity may prevent the development of HCC.
As previously mentioned, SIRT5 is involved in ammonia detoxification through desuccinylation and the consequent inhibition of GLS, which catalyzes the hydrolysis of glutamine to glutamate and produces ammonia as a byproduct.84 Notably, breast cancer cells MDA-MB-231 and C2C12 overexpressing SIRT5 were characterized by decreased ammonia levels, with a consequent reduction of ammonia-induced autophagy and mitophagy (Figure 4). These mechanisms play a defensive role against chemotherapy or stress mechanisms such as hypoxia or fasting.84 Importantly, in cancer cells, glutamine catabolism is necessary for ATP production and lipid biosynthesis to support cell proliferation. Indeed, glutamine is crucial for the anaplerotic replenishment of the TCA cycle through its catabolic product α-ketoglutarate.131 Hence, in these cases, SIRT5 acts as a tumor suppressor, rendering tumor cells more susceptible to chemotherapeutics and environmental stresses and causing a decrease in ATP production.
SIRT5 was found to be downregulated in androgen-independent prostate cancer cells (PC-3 and PC-3M), with its expression being lower in more advanced cancers. Furthermore, inhibiting SIRT5 with a peptide-based inhibitor (compound 3d, section 4.1) increased PC-3 cell migration and invasion, thereby confirming its tumor suppressive role in this context. In line with this, SIRT5 KOincreases PC-3 cell proliferation, migration, and invasion. The observed effects were ascribed to the higher activity of lactate dehydrogenase (LDH) A, which is activated upon succinylation at Lys118 and is a demonstrated substrate of SIRT5. Nonetheless, no mechanistic insight was provided regarding the role of LDHA in the onset and progression of prostate cancer.132
3.6.2. Tumor-Promoting Role of SIRT5 in Cancer
SIRT5 may also play a tumor-promoting function in lung cancer via different mechanisms. Indeed, a recent study indicated that SIRT5 is overexpressed in NSCLC cells, which is associated with poor prognosis. Consistent with this, SIRT5 downregulation suppressed tumor cell growth and differentiation121 and sensitized lung cancer cells to genotoxic drugs such as cisplatin, 5-fluorouracil, and bleomycin both in vitro and in vivo.62 Moreover, SIRT5 ablation decreased the expression of NRF2 (Figure 4), a transcription factor involved in the regulation of genes that defend cells from oxidative stress and xenobiotics, including drug resistance genes.62
SIRT5 negatively regulates the expression of SAD1/UNC84 domain protein 2 (SUN2) (Figure 4), an important component of the linker of the nucleoskeleton and cytoskeleton (LINC) complex.121 SUN2 inhibits the Warburg effect, a metabolic alteration in which ATP is produced mainly from glycolysis rather than oxidative phosphorylation, thereby generating immediate energy to support cancer cell proliferation.133 SUN2 activity facilitates the suppression of cancer cell growth, metastasis, and the increased susceptibility to apoptosis induced by cisplatin. Overall, in this context, SIRT5 seems to play an oncogenic role by impairing SUN2 activity and sustaining tumor growth via the Warburg effect.121
SIRT5 may also play a tumor-promoting role in lung cancer via inhibiting PKM2 (Figure 4). The impairment of PKM2 activity results in a diminished glycolytic flux in tumor cells but consequently promotes the pentose phosphate pathway, yielding higher NADPH levels. This thus protects cancer cells from oxidative stress and facilitates their proliferation. Moreover, PKM2 hypersuccinylation led to the repression of tumor development, consistent with the fact that SIRT5 ablation or cell treatment with the nonselective sirtuin inhibitor suramin (compound 7, section 4.1) induced PKM2 activity and thus suppressed the proliferation of A549 lung cancer cells.95 Similarly, SIRT5 KO in HCC is correlated with enhanced apoptosis and reduced cell proliferation and invasion, while its overexpression is associated with poor prognosis.124 SIRT5 was shown to negatively regulate the expression of E2F1 (Figure 4), an oncosuppressor involved in cell cycle regulation,134 thereby suggesting that HCC tumor progression could be supported by SIRT5 via the downregulation of E2F1.124
Different from what was described previously about SIRT5’s role in breast cancer, Greene et al. suggested that SIRT5 plays an oncogenic role through stabilizing GLS against ubiquitination and proteasomal degradation (Figure 4).85 This in turn supports glutamine catabolism and the consequent obtainment of α-ketoglutarate, which enters the TCA cycle that leads to ATP production. SIRT5 was shown to be upregulated during the cancerous transformation and promoted tumorigenesis and cell proliferation. In addition, increased SIRT5 expression in human breast tumors was correlated with poor prognosis for patients.85 Consistent with this, the pharmacological inhibition of SIRT5 strongly impaired the cell proliferation and anchorage-independent growth of MCF7 and MDA-MB-231 breast cancer cells (see compounds 2a and 2b in section 4.1).135
Liang et al. reported that SIRT5 was overexpressed in cultured SH-EP neuroblastoma cells, where it counteracted oxidative stress by reducing ROS levels and preventing apoptosis (Figure 4), thus exerting a tumor-promoting function.136 SIRT5 is also overexpressed in ovarian cancer,137 where it protects tumor cells from genotoxic drugs such as cisplatin by modulating the NRF2/HO-1 pathway, which in turn increases the cellular levels of the ROS scavenger GSH (Figure 4).138
Yang et al. showed that SIRT5-mediated desuccinylation at Lys280 activates the catabolizing enzyme serine hydroxymethyltransferase 2 (SHMT2), which in turn promotes tumor progression in osteosarcoma U2OS and colorectal carcinoma (CRC) HCT116 cells (Figure 4).139 Indeed, SIRT5 KO or the expression of the succinylation mimetic SHMT2 mutant (K280E) resulted in the suppression of tumor growth both in vitro and in vivo.139 SHMT2 is a crucial enzyme involved in one-carbon-unit metabolism that catalyzes the conversion of serine into glycine using tetrahydrofolate (THF) as a cosubstrate, which is converted to N5,N10-methylene-THF, a key intermediate of purine biosynthesis.140 Similarly, SIRT5 was found to activate the one-carbon-unit metabolism in melanoma. The activation of this pathway, along with the promotion of the expression of pro-survival genes such as c-MYC and MITF, was shown to sustain melanoma cell growth (Figure 4).141 In particular, SIRT5 was shown to promote the proliferation and survival of different cutaneous melanoma cell lines and a uveal melanoma cell line, a subtype that develops in the eye. In addition, SIRT5 was essential for tumor development in both melanoma mouse xenografts and the autochthonous BRAF PTEN-driven melanoma mouse model. SIRT5 was also found to regulate both the methylation and acetylation of histone, which in turn facilitate the expression of the above-mentioned c-MYC and MITF, respectively.141
In another study, high SIRT5 expression in CRC cells was associated with increased autophagy, which promotes tumor onset and progression.142 Mechanistically, SIRT5 deacetylates and activates LDHB, which promotes the conversion of lactate and NAD+ to pyruvate, NADH, and H+. The generated protons promote lysosomal acidification and consequent autophagy (Figure 4). Consistent with this, SIRT5 KO or treatment with the nonselective SIRT5 inhibitor GW5074 (see compound 11 in section 4.1) augmented LDHB acetylation at Lys329 and inhibited LDHB activity, which reduced autophagy and CRC cell growth both in vitro and in vivo. It should be noted that while the effects of SIRT5 KO are clearly related to the loss of SIRT5 activity, the consequences of GW5074 treatment cannot be unambiguously connected to SIRT5 inhibition or downregulation given the lack of selectivity of the compound. Furthermore, SIRT5-mediated deglutarylation and the consequent activation of glutamate dehydrogenase 1 (GLUD1) stimulate glutamine catabolism, supporting CRC proliferation (Figure 4).143 In line with this, SIRT5 knockdown in HCT116 and LoVo CRC cell lines led to the inhibition of cell proliferation.143 In addition, CRC cells expressing both SIRT5 and wild-type KRAS display resistance to anticancer agents like cetuximab. High SIRT5 expression in CRC patients expressing wild-type KRAS is also associated with increased tumor recurrence and poor survival.144 In this context, it was shown that drug resistance was gained by the activation of the ROS scavenger protein thioredoxin reductase 2 (TrxR2).144 Mechanistically, the SIRT5-mediated desuccinylation of succinate dehydrogenase complex subunit A (SDHA) and the inhibition of its enzymatic activity lead to higher levels of succinate, which determines TrxR2 activation (Figure 4). Through this mechanism, SIRT5 protects tumor cells from oxidative damage and promotes their proliferation.144 In line with this, SIRT5 silencing leads to the activation and hypersuccinylation of SDHA, thereby suppressing clear cell renal cell carcinoma proliferation.145
Different from what previously reported, SIRT5 was shown to possess tumor-promoter activity in prostate cancer, where it activates acetyl-CoA acetyltransferase 1 (ACAT1) and thus stimulates the mitogen-activated protein kinase (MAPK) pathway, leading to enhanced proliferation, invasion, and migration.146 SIRT5 also has a critical role in the development of AML, where its activity promotes cancer cell survival by reducing oxidative stress and sustaining oxidative phosphorylation and glutamine catabolism.147 In line with this, SIRT5 knockdown decreases colony formation and enhances apoptosis in a wide range of AML cell lines, and the pharmacological inhibition of SIRT5 impairs cell proliferation and induces apoptosis in SIRT5-dependent AML cells such as OCI-AML2, SKM-1, and MOLM-13 (see compounds 3b, 3d, and 3i, respectively, in section 4.1).147,148 Similarly, SIRT5 expression is necessary for tumor insurgence and growth in both xenograft and syngeneic AML mouse models.147 Finally, a recent study also revealed that the tumor suppressor p53 is succinylated at Lys120;149 this residue was also previously identified as an acetylation site of KAT8, Tip60, and NAT10.3,43 In this case, SIRT5 mediates p53 desuccinylation, which results in its inhibition and the consequent suppression of both the expression of p53 target genes and p53-induced apoptosis. These data suggest that SIRT5 may also act as a tumor promoter by suppressing the functions of p53.149
4. Pharmacological Modulation of SIRT5
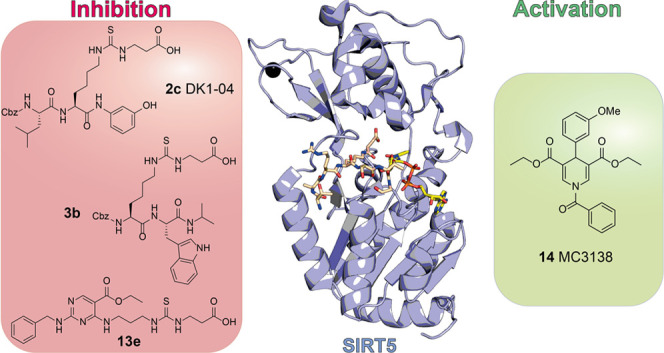
Given the involvement of SIRT5 as a regulator of different pathways, many research groups have investigated the possibility of targeting SIRT5 via inhibitors or activators. So far, research has been mainly focused on SIRT5 inhibitors used as either chemical tools to phenocopy SIRT5 knockdown or lead molecules for the development of novel potential therapeutics. On the other hand, a recent study described the first SIRT5 activator that has been used in the context of cancer, specifically PDAC, where SIRT5 plays an oncosuppressor role.130 This indicates that there is increased interest in developing both inhibitors and activators, thereby enabling a better understanding of SIRT5’s function and paving the way to personalized approaches. In the next section, we will initially examine the most relevant SIRT5 inhibitors and then discuss a recently reported activator.
4.1. SIRT5 Inhibitors
4.1.1. Peptide and Amino Acid Inhibitors
Starting from the analysis of the SIRT5 crystal structure that provided important information about its catalytic site, Roessler et al. synthesized various peptide-based analogues based on a CPS1-derived sequence (Figure 5) that served as a SIRT5 substrate in its acetylated form.76 All these compounds possess a succinyl residue at the lysine side chain, which was shown to interact with Tyr102 and Arg105 in the catalytic site. This dicarboxylic acyl portion gives the compounds the optimal chain length to allow the carboxyl group to form a salt bridge with Arg105 as well as a hydrogen bond to Tyr102. To obtain compounds that could impair NAD+ binding in the so-called C-pocket, which accommodates nicotinamide, the authors introduced bulky moieties in the C3 position of the succinyl chain. This approach initially led to compound 1a, which had a phenyl ring on C3 and possessed a KD value of 8.20 μM and Ki value of 100 μM for lysine desuccinylation. Compound 1b, bearing a n-butyl chain on succinyl C3, displayed a great increase in inhibitory potency (Ki = 17.2 μM). Both peptides were crystallized in complex with zebrafish SIRT5 (zSIRT5) and showed similar binding modes, with the substituent at C3 pointing toward the binding site. In an attempt to move the phenyl moiety further inside the C-pocket, the authors inserted a methylcarbamate linker between the phenyl and succinyl groups, yielding compounds 1c and 1d with S- and R- configurations on C3, respectively. Compound 1c displayed a KD value of 5.78 μM that was associated with a Ki value for desuccinylation of 38.1 μM, almost threefold lower compared to that of 1a. Although no thermodynamic constants were provided for compound 1d, it was shown to possess a reduced affinity for SIRT5, thereby suggesting that the R-configuration is not optimal for the interaction with the C-pocket. The cocrystal structure of zSIRT5 in complex with 1d showed that extending the linker moved the phenyl ring deeper into the C-pocket, thereby mimicking the nicotinamide binding. Hence, the augmented potency may be ascribed to both interactions with the key residues in the catalytic site and the steric hindrance that blocks the NAD+ binding. Compound 1e, consisting of a derivative of 1a bearing an additional methyl group on C3, displayed an almost 25-fold increase in inhibitory potency, with a Ki value of 4.3 μM (desuccinylation). Tested at a concentration of 50 μM against SIRT1–3, 1e displayed less than 1% inhibition for SIRT1 and SIRT3 and ∼4% inhibition for SIRT2, showing great selectivity for SIRT5 (Table 2). Another active compound, although less potent than 1e, is 1f, which has a thioacetic residue on succinyl C3 and displays a Ki value of 10.6 μM (desuccinylation).76
Figure 5.

Structures of CPS1-derived peptidic SIRT5 inhibitors.
Table 2. Most Relevant SIRT5 Inhibitors.
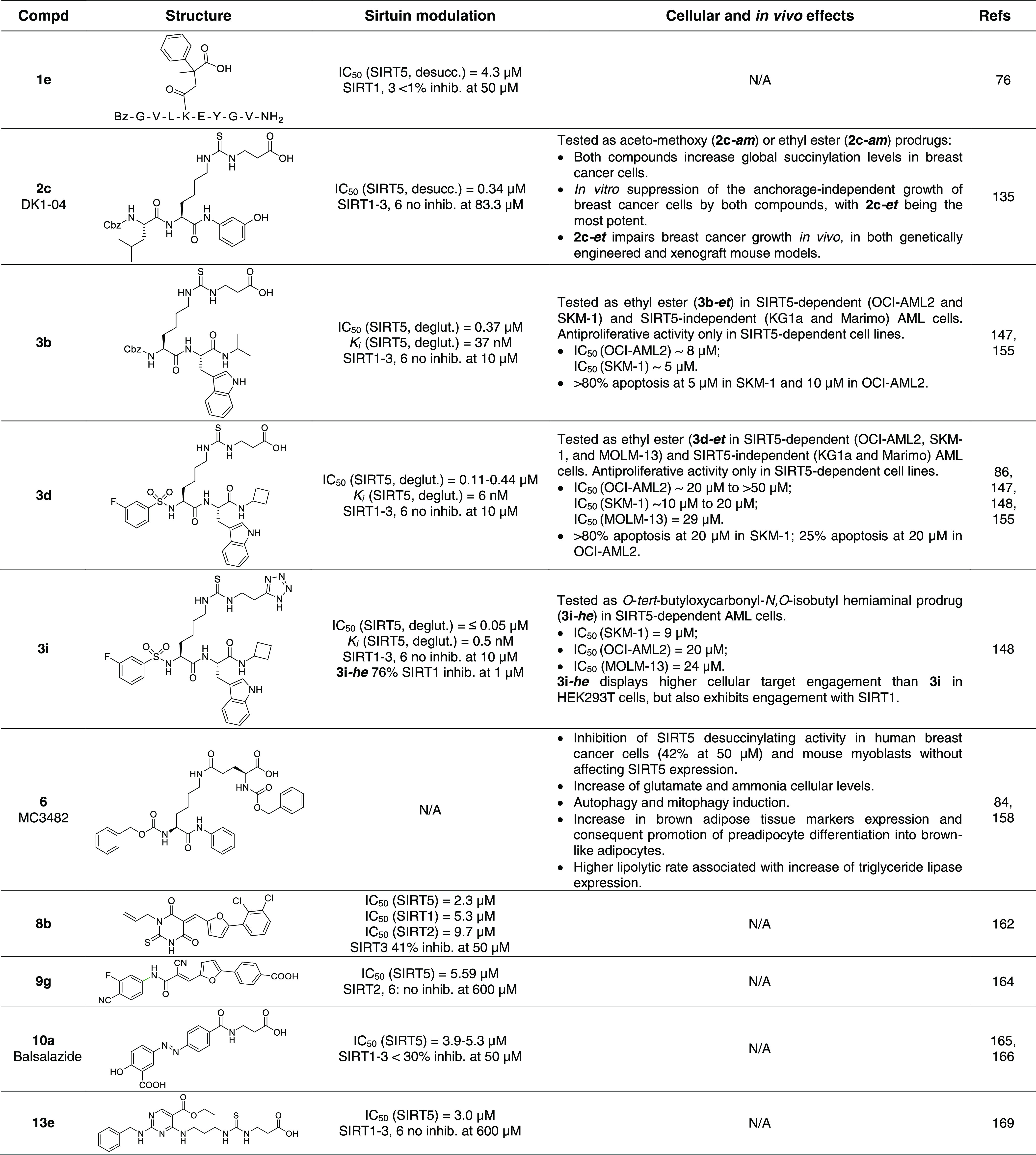
In another study, the same research team analyzed the 3-(arylthio)succinyl scaffold to improve its inhibitory efficacy toward SIRT5. Among the synthesized molecules, the (S)-3-(2-naphthylthio) succinyl derivative 1g (Figure 5) displayed strong inhibition of SIRT5 deglutarylase activity, with an IC50 value of 30.3 nM and a Ki value of 30.1 nM.150 Consistent with this data, the zSIRT5–1g cocrystal structure showed that the naphthyl moiety completelyoccupied the C-pocket. Compound 1g was also selective over other SIRT isoforms (SIRT1–3 and SIRT6) at concentrations up to 50 μM. To create a more drug-like structure, Kalbas et al. prepared compound 1h, a tripeptide with the same 3-substituted succinyl scaffold discussed above. Although it is less potent than the parent compound, it still retains promising inhibitory activity, with IC50 = 350.4 nM and a Ki = 179.8 nM (deglutarylation), thereby representing a good starting point for further development.150
With the aim of improving the cell permeability of SIRT5 inhibitors, Abril et al. modified a previously reported thiosuccinyllysine peptide (H3K9TSu, 2a, Figure 6), which was found to inhibit SIRT5 desuccinylase activity with IC50 = 5 μM while being selective over SIRT1–3 (no inhibition at 100 μM). Indeed, they gradually shortened the peptide and replaced the thioamide moiety with a thiourea function to yield compound JH-I5-2 (2b), which consisted of a lysine derivative protected by a benzyloxycarbonyl (Cbz) group at the N-terminus and a N-(3-hydroxyphenyl) carboxamide moiety at the C-terminus.135 Despite the fact that the thiourea functionality is prone to metabolic S-oxidation in vivo, which is mostly mediated by cytochrome P450 and flavin-containing monooxygenases (FMO) causing the formation of sulfoxide intermediates that may also undergo hydrolysis to the corresponding urea,151−154 compound 2b showed stronger SIRT5 inhibition, with an IC50 value of 2.1 μM for desuccinylation (Table 2). This potent inhibitory activity is probably due to the presence of the hydroxyl group on the C-terminal anilide moiety that provides an additional hydrogen bond, thereby granting tighter binding to the enzyme. DK1-04 (2c) was obtained by adding a Cbz-protected leucine residue to the N-terminus. This compound displayed the strongest inhibition of SIRT5 desuccinylase activity, with IC50 = 0.34 μM. Both 2b and 2c (Figure 6) showed selectivity for SIRT5, and no inhibition of SIRT1–3 or SIRT6 was detected at a concentration of 83.3 μM. These molecules are mechanism-based inhibitors that form a stalled covalent 1′-(S)-alkylimidate intermediate with the ADP-ribose in the active site, which blocks the catalytic mechanism. The group also synthesized two different pro-drug forms of these compounds to increase their cell permeability, which was compromised by the free carboxylic acid moiety. Hence, they developed 2b-am, 2b-et, 2c-am, and 2c-et, bearing an aceto-methoxy (am) or ethyl ester (et) group, which displayed cellular activity by increasing global lysine succinylation in MCF7 breast cancer cells at a concentration of 50 μM. 2c-Based prodrugs significantly decreased MCF7 and MDA-MB-231 breast cancer cell viability (GI50(2c-am) = 51 μM and GI50(2c-et) = 20 μM). All compounds impaired the anchorage-independent growth of the same cell lines with GI50 values between 10 and 37 μM, although 2c-based prodrugs were still more potent. In particular, the most effective prodrug was the ethyl ester derivative 2c-et, which also blocked breast cancer growth in both genetically engineered and xenograft mouse models. In the case of genetically engineered mice, 2c-et was administered at a dose of 50 mg/kg five times per week for six weeks, while in the case of xenograft mouse models it was administered at the same dose for three weeks.135
Figure 6.

Structures of H3K9Tsu-derived peptide SIRT5 inhibitors 2a–c.
Recently, Rajabi and colleagues developed a series of ε-N-thioglutaryllysine derivatives and performed an extensive SAR study to elucidate the molecular features necessary for SIRT5 inhibition.155 Compound 3a is a submicromolar inhibitor of SIRT5 deglutarylase activity (IC50 = 0.83 μM) bearing a thioamide moiety, a Cbz-protected N-terminus, and a C-terminal L-Trp; 3a has stronger inhibitory activity than the corresponding derivative with D-Trp (90% inhibition at 100 μM).155 Compound 3b is a thiourea analogue of 3a with IC50 = 0.37 μM for SIRT5-mediated deglutarylation (Table 2). Both 3a and 3b (Figure 2) are Cbz-protected at the N-terminus (Figure 7A). The research team managed to cocrystallize these two molecules with both human and zebrafish SIRT5 and confirmed the formation of a catalytic intermediate with the ADP-ribose and key interactions with residues Tyr102 and Arg105 (Figure 7B and C). Due to the lack of specific interactions of the benzyloxycarbonyl group, they investigated other structures, which led to the development of more derivatives bearing the same scaffold as 3a and 3b with different substitutions on the N- and C-termini. This led to compounds 3c–3e (Figure 7A), which possessed a 3-fluorobenzensulfonamide at the N-terminus but differed at the C-terminus due to the substitution of the carboxamide N with a cyclopropyl, cyclobutyl, or cyclopentyl group, respectively. Among them, 3d is the most potent SIRT5 inhibitor with an IC50 value 0.11 μM for deglutarylation (another study reported an IC50 value of 0.44 μM)86 (Table 2), while compounds 3c and 3e present IC50 values of 0.26 and 0.23 μM, respectively, against SIRT5 deglutarylase activity. As mentioned above, these compounds are mechanism-based inhibitors that promote the formation of a covalent stalled intermediate with NAD+ within the active site. Hence, using only IC50 values as indication of inhibitory potency may be erroneous, as they cannot be compared to those obtained with reversible inhibitors that are based on measurements at equilibrium. Nonetheless, the authors also obtained Ki values from continuous flow experiments for the most promising molecules, which enabled a kinetic analysis and a more accurate estimation of the inhibitor potency. Specifically, 3a, 3b and 3d were shown to have a slow tight-binding mechanism of inhibition, and their Ki values are 22, 37, and 6 nM, respectively. In addition, compounds 3b–3e showed great selectivity for SIRT5 over SIRT1–3 and 6, while 3a was not tested against other isoforms.155 Among these molecules, 3b and 3d were subsequently tested in cellular assays as pro-drug esters. Indeed, to improve their cell permeability, the negatively charged carboxylic moiety was masked with an ethyl ester, yielding prodrugs 3b-et and 3d-et. 3b-et and 3d-et were tested in AML cell lines whose proliferations were either SIRT5-dependent (OCI-AML2 and SKM-1) or SIRT5-independent (KG1a and Marimo). Both molecules inhibited cell proliferation and induced the apoptosis of SIRT5-dependent cells, while they did not show any effect on SIRT5-independent AML cell lines. Among the two molecules, 3b-et was the most potent one, with IC50 values of 5–8 μM, while 3d-et showed IC50 values of 10–20 μM. Accordingly, 3b-et induced more than 80% apoptosis at 5 or 10 μM in SKM-1 or OCI-AML2, respectively, while 3d-et induced more than 80% apoptosis only at 20 μM in SKM-1 (Table 2). Notably, the effects induced by 3b-et resembled SIRT5 knockdown. In addition, mice injected with 3b-et-treated AML cells (at 12.5 or 25 μM) displayed higher survival rates compared to the controls.147
Figure 7.

(A) Structures of ε-N-thioglutaryllysine derivatives 3a–j. (B) Structure of hSIRT5 in complex with the ADP-ribose-1′-thioimidate intermediate of compound 3b (green) (PDB ID 6EQS). (C) Focus on the binding site to show how the most important residues mediate the protein–compound interaction. Dashed orange lines indicate polar interactions.
Interestingly, compound 3d was recently tested in a SARS-CoV-2 infection cellular model.86 Initial experiments performed in HEK-293 SIRT5 knockdown cells transfected with SIRT5 and Nsp14 from SARS-CoV-2 indicated that this compound was able to disrupt the SIRT5–Nsp14 interaction starting at a concentration of 25 μM. More importantly, Calu-3 cells infected with SARS-CoV-2 and treated with 3d displayed reduced viral titers and mRNA levels at 25 and 100 μM, respectively.86
Starting from compound 3d, Rajabi et al. recently developed a series of derivatives to investigate whether the bioisosteric substitution of the carboxylic acid moiety might retain the SIRT5 inhibitory potency.148 Among the derivatives, 1,2,4-oxadiazol-5(4H)-one (3f), 1,2,4-oxadiazol-5(4H)-thione (3g), 2-hydroxyisoxazole (3h), and tetrazole (3i) displayed submicromolar IC50 values for SIRT5-mediated deglutarylation (IC50(3f) ≤ 0.05 μM, IC50(3g) = 0.9 μM, IC50(3h) = 0.29 μM, and IC50(3i) ≤ 0.05 μM). The kinetics of SIRT5 inhibition by compounds 3f, 3h, and 3i was also evaluated. 3f and 3i exhibited Ki values in the low nanomolar range for (7 and 0.5 nM, respectively), while 3g exhibited a Ki value of 122 nM. All compounds were also tested against SIRT1–3 and SIRT6 and displayed negligible inhibitory activities at 10 μM, with only 3f displaying 37% SIRT1 inhibition and 3h showing 40% SIRT3 inhibition at the same concentration. Notably, compound 3j in which the alkyl spacer length was reduced to one methylene unit displayed a drop in potency (IC50 = 5.1 μM), thereby indicating the importance of both the length and the flexibility of the lysine side chain for the SIRT5 affinity of the isosteres (Figure 1), as previously shown for carboxylates.76,155 All newly developed compounds displayed poor cell permeabilities, which were comparable to that of the parent molecule 3d and one order of magnitude lower than that of its ethyl ester 3d-et. Hence, the authors prepared compound 3i-he, a prodrug of 3i bearing a masked tetrazole moiety, using an O-tert-butyloxycarbonyl-N,O-isobutyl hemiaminal functionality. Compound 3i-he was assessed for its in vitro activity toward SIRT1–3 and SIRT6 and presented 76% SIRT1 inhibition at 1 μM, showing that this masking group decreased the isoform selectivity compared to the unprotected parent molecule.
Cellular target engagement was then assessed in HEK293T cells for increasing compound concentrations (2.6 nM to 10 μM) of 3d, 3d-et, 3f, 3g, 3i, and 3i-he via an isothermal dose–response fingerprinting cellular thermal shift assay (ITDRF-CETSA) performed at a constant temperature of 52 °C. Compounds 3f and 3g showed poor target engagement, with EC50 values higher than 10 μM, while compounds 3d and 3i exhibited EC50 values of 0.9 and 1.3 μM, respectively. Notably, the prodrugs 3d-et and 3i-he bearing masked acidic groups displayed more prominent target engagement, with EC50 values of 0.25 and 0.15 μM, respectively. Full melting experiments with 3i-he (1 μM) against SIRT1, SIRT3, and SIRT5 confirmed that target engagement and suggested selectivity over the other mitochondrial isoform SIRT3, with shifts in the protein melting temperature of 5.4 °C for SIRT5 and 0.5 °C for SIRT3. However, considerable engagement was observed for SIRT1, with a shift in the protein melting temperature of 4.6 °C, thereby suggesting the incomplete hydrolysis of the masking group inside HEK293T cells. Hence, more advanced masking approaches would be necessary to improve the selectivities of the tetrazole-containing derivatives.
When tested in SIRT5-dependent SKM-1 AML cells and immortalized HEK293T cells, compounds 3d, 3f, and 3i did not exhibit any decrease in viability at concentrations up to 100 μM. Conversely, the 3d-et and 3i-he displayed IC50 values against SKM-1 cells of 21 and 9 μM, respectively. When tested in HEK293T cells, 3d-et displayed a IC50 value between 50 and 100 μM, while 3i-he showed a IC50 value higher than 100 μM with less than 35% growth inhibition at 100 μM, thereby indicating the higher cancer selectivity of 3i-he compared to 3d-et. 3d-et and 3i-he were also assessed in two further SIRT5-dependent AML cell lines, OCI-AML2 and MOLM-13. 3i-he displayed a higher efficacy in OCI-AML2 (IC50(OCI-AML2, 3d-et) > 50 μM and IC50(OCI-AML2, 3i-he) = 20 μM), while similar cell growth inhibition was observed for MOLM-13 (IC50 (MOLM-13, 3d-et) = 29 μM and IC50 (MOLM-13, 3i-he) = 24 μM).148
Given the potency and selectivity of the thiourea-type warhead, which can also circumvent the cytotoxicity issue that results from the thioamide-based derivatives, Liu and colleagues developed cyclic pentapeptides harboring a central ε-N-carboxyethylthiocarbamoyllysine residue. Compound 4a (Figure 8), the side chain-to-side chain cyclic pentapeptide depicted in Figure 3, inhibits SIRT5 desuccinylase activity with IC50 = 7.5 μM and is selective over SIRT1–3 and SIRT6 (IC50 values >1 mM).156 Compared to its linear counterpart 4b (Figure 8), compound 4a was found to be more proteolytically stable when tested in proteolytic digestion using Pronase as the protease. In addition, compound 4b was tested under the same SIRT5 inhibition assay conditions and was found to exhibit a SIRT5 inhibitory potency comparable to that of 4a with an IC50 value of 7.6 μM (desuccinylase). However, it also exhibited a notable inhibitory activity against SIRT2 (IC50 = 96.4 μM) while still being selective over SIRT1, SIRT3, and SIRT6. These data suggest that this macrocyclic bridging unit is not favorable for enhancing the SIRT5 inhibitory potency compared to its linear counterpart and does not provide a tighter binding at the enzyme active site. Nonetheless, the presence of a macrocycle confers a better selectivity profile and greatly increases the metabolic stability. Hence, the macrocycle bridging unit immediately surrounding the warhead could serve as a lead for the development of new, more potent, and selective SIRT5 inhibitors.156 In line with this, in another study the same group synthesized a series of N-terminus-to-side chain cyclic tripeptides bearing the same SIRT5 inhibitory warhead as seen in the previous work, with the idea that the various bridging units would ensure a favorable interaction in the active site and yield tighter binding to SIRT5.157 Compounds 5a–d (Figure 8) present spacers of various lengths between the N-terminal α-amino group and the side chain ε-amino group of the lysine residue and harbor an arginine residue at the N-terminus. Among them, compound 5c, which presents a succinyl bridging unit, exhibited the greatest SIRT5 inhibitory activity with IC50 = 2.2 μM, being 2–6× more potent than compounds 5a, 5b, and 5d (IC50(5a) = 13.2 μM, IC50(5b) = 6.5 μM, and IC50 (5d) = 4.0 μM; all values were measured using the succinyllysine SIRT5 substrate). Compound 5c also displayed >60-fold selectivity over SIRT1–3 and SIRT6. Furthermore, compound 5e, the linear counterpart of 5c, exhibited a more than 42-fold decrease in SIRT5 inhibition (IC50(desuccinylation) = 93.1 μM), suggesting that in this case the peptide chain macrocyclization could enhance the target binding affinity. A proteolysis assay performed using the Pronase as proteolytic enzyme again indicated the higher proteolytic stability of the cyclic peptide 5c compared to its linear counterpart 5e. In conclusion, the tripeptide 5c displayed a SIRT5 inhibitory potency more than threefold greater than the previously reported pentapeptide 4a, suggesting that this smaller peptide could be a useful starting point for further SAR investigations to obtain new, more potent, and selective SIRT5 inhibitors.157
Figure 8.
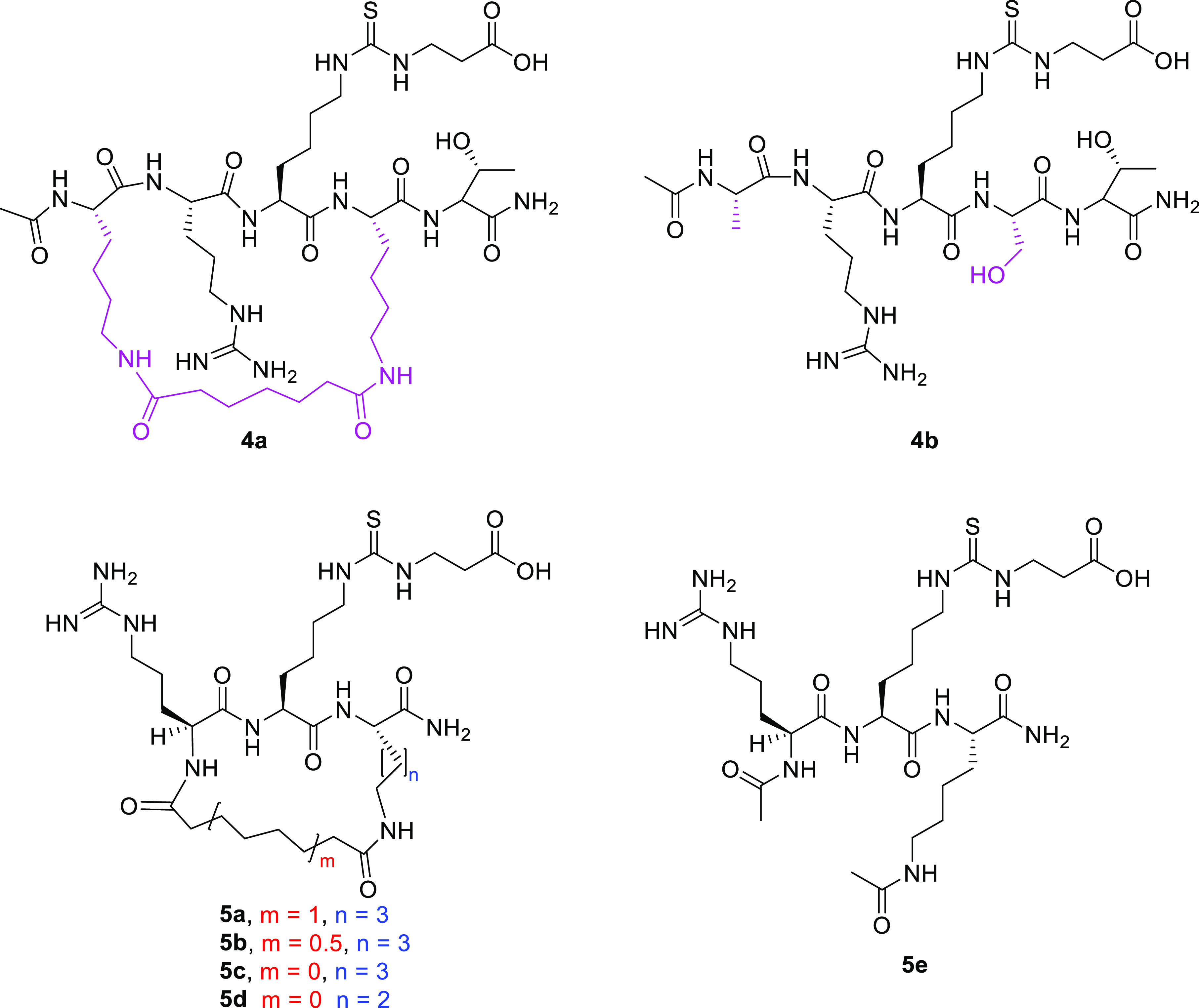
Structures of cyclic penta- and tripeptide SIRT5 inhibitors.
Polletta et al. recently developed MC3482 (6, Figure 9), an ε-N-glutaryllysine-based compound wherein the α-amine of the lysine residue is Cbz-protected while the C-terminal carboxy group forms an anilide function.84 Compound 6 was reported to be a promising inhibitor of SIRT5-mediated desuccinylation, exhibiting dose-dependent activity and reaching 42% SIRT5 inhibition at 50 μM when tested in MDA-MB-231 cells without having effect on SIRT1 and exhibiting only 8% SIRT3 inhibition at the same concentration. Moreover, both human breast cancer cells (MDA-MB-231) and mouse myoblasts (C2C12) treated with compound 6 (50 μM) displayed an increase in succinylated proteins as a result of the inhibition of SIRT5 desuccinylase activity.84 In addition, treating MDA-MB-231 and C2C12 cells with compound 6 (50 μM) led to an increase in cellular glutamate and ammonia levels via an increase GLS succinylation. These results are in line with SIRT5’s role in the regulation of ammonia production through the modulation of glutamine metabolism. In the same report, compound 6 was also shown to promote ammonia-induced autophagy and mitophagy (Table 2). In a recent study, Molinari and co-workers demonstrated that this compound was also able to stimulate the expression of brown adipose tissue markers, thus facilitating preadipocyte differentiation into brown-like adipocytes when dispensed at early stages of differentiation.158 Furthermore, treatment with compound 6 at 50 μM led to more efficient mitochondrial activity and biogenesis along with a higher lipolytic rate associated with an increase of triglyceride lipase expression, indicating that SIRT5 inhibition is a favorable strategy to treat obesity and metabolic diseases.158
Figure 9.

Structure of MC3482.
4.1.2. Small-Molecule Inhibitors
Suramin (7, Figure 10A), a well-known antiparasitic agent, was identified as one of the first sirtuin inhibitors and found to also inhibit SIRT5.159 To comprehend how this molecule binds to the enzyme and the structural and molecular mechanisms of inhibition, Schuetz et al. determined the crystal structure of SIRT5 in complex with 7. Interestingly, SIRT5 dimerizes in solution upon suramin binding and is stabilized by the suramin itself. The main interactions with the enzyme originate from the sulfonate groups of 7, which form hydrogen bonds with the side chains of Arg71, Tyr102, Arg105, and Arg141 and with the backbone amide of Phe70 (Figure 10B and C). Interestingly, Phe70 and Arg71 seem to have a role in the release of nicotinamide, thereby suggesting that 7 mimics this reaction product when interacting with SIRT5. Tyr102 and Arg105 are also involved in interactions with the acyl-lysine substrate, thus suggesting that suramin occupies the peptide’s substrate-binding site. Furthermore, the carbonyl oxygen of the amide portion that connects the naphthalene to the benzene moiety of 7 forms a hydrogen bond with His158 (Figure 10B and C), thereby mimicking the interaction between the 3′-hydroxyl group of NAD+. This was confirmed by the superimposition of the SIRT5–ADP-ribose and SIRT5–suramin complex structures that showed 7 occupied the C-pocket, thus indicating that suramin mimics the binding of the cosubstrate. In addition, the central urea portion connecting the two symmetric portions of compound 7 forms a hydrogen bond with the hydroxyl group of Tyr255 (Figure 10B and C), which is usually involved in peptide substrate binding. Collectively, these results suggest that suramin inhibits SIRT5 activity through various interactions in the active site, as it resembles the interactions of substrate, product, and cosubstrate.159
Figure 10.

(A) Structure of suramin (7). (B) Structure of hSIRT5 in complex with compound 7 (green) (PDB ID 2NYR). (C) Focus on the binding site of compound 7 showing how the key residues mediate the protein-compound interaction. Dashed orange lines indicate polar interactions.
The evidence that 7 interacts with the NAD+-binding site makes it nonselective over other isoforms possessing a similar cosubstrate binding pocket. In fact, it not only inhibits SIRT5 NAD+-dependent deacetylase activity with IC50 values of 14.2 and 22 μM,159,160 depending on the study, but also targets SIRT1 (IC50 = 0.297–2.6 μM, depending on the study)159,161 and SIRT2 (IC50 = 1.15 μM). A recent study also reported the 7-mediated inhibition of SIRT5 desuccinylation activity, with IC50 = 46.6 μM.162 Overall, to overcome this lack of selectivity, it would be necessary to preferentially target the peptide substrate-binding site to avoid binding to other NAD+-dependent enzymes. Compound 7 was also tested in A549 lung cancer cells, where it seemed to increase the activity of PKM2, an enzyme inhibited by SIRT5-mediated desuccinylation, and lead to the suppression of cancer cell proliferation.95
With the aim of improving the knowledge of the isoform selectivity of potential new SIRT5 inhibitors and discovering the key interactions that lead to greater inhibition, Maurer and co-workers screened their internal library and found thiobarbiturates were potential SIRT5 inhibitors. This series of compounds (8a–g, Table 3) displayed inhibitions for SIRT5-mediated desuccinylation in the mid to low micromolar range, although they showed similar inhibitions for SIRT1 and 2 and displayed low potencies against SIRT3.162 Compound 8a, bearing a 2-(2,3-dichlorophenyl)furanyl substitution, is a good SIRT5 inhibitor (IC50(SIRT5) = 3.6 μM); however, its selectivity profile is not ideal, since it inhibits SIRT1 with the same potency (IC50 (SIRT1) = 3.4 μM)) (Table 3). Derivative 8b, bearing an allyl substitution at the thiobarbituric nitrogen, displays improvements in terms of potency and selectivity, with an IC50 value of 2.3 μM for SIRT5 (IC50(SIRT1) = 5.3 μM, IC50 (SIRT2) = 9.7 μM, and 41% inhibition of SIRT3 at 50 μM, Table 2). Notably, compound 8b is the most potent and selective among the reported molecules. Conversely, the same allyl substitution is detrimental for compound activity in the series of compounds 8c–h bearing a benzyloxyphenyl substitution, as indicated by the high IC50 value of compound 8f (Table 3). In contrast, the alkyl substitution improves the selectivity over SIRT1 but not SIRT2 and SIRT 3, as indicated by the IC50 values of compounds 8d and 8e compared to those of 8c, 8g, and 8h, which are unsubstituted on the thiobarbituric nitrogen (Table 3).
Table 3. Structures and Inhibition Data of Thiobarbiturates 8a–ga.
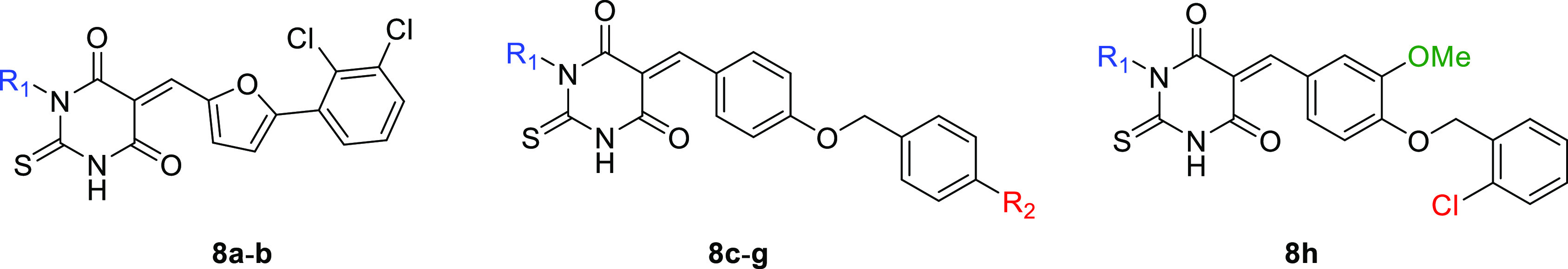
| IC50 (μM) |
||||||
|---|---|---|---|---|---|---|
| compd | R1 | R2 | SIRT1 | SIRT2 | SIRT3 | SIRT5 |
| 8a | –H | 3.4 | 10.5 | 30% inhib. @ 50 μM | 3.6 | |
| 8b | —CH2CH=CH2 | 5.3 | 9.7 | 41% inhib. @ 50 μM | 2.3 | |
| 8c | –H | –H | 10.5 | 9.8 | 29.3 | 12.6 |
| 8d | –CH3 | –H | 56.5 | 10.0 | 22% inhib. @ 50 μM | 17.8 |
| 8e | -–H2CH3 | –H | 53.2 | 14.4 | 25% inhib. @ 50 μM | 12.9 |
| 8f | —CH2CH=CH2 | –H | 26.8 | no inhib. @ 50 μM | 13% inhib. @ 50 μM | 67.3 |
| 8g | –H | –Br | 9.9 | 3.4 | 30.3 | 6.2 |
| 8h | –H | 6.7 | 7.5 | 46.4 | 12.4 | |
SIRT5’s IC50 values were measured against its desuccinylase activity.
Overall, these results indicate that the 2-(2,3-dichlorophenyl)furanyl substitution is more favorable than the benzyloxyphenyl one. The research team also performed docking studies to characterize the interactions of the compounds within the active site of SIRT5. They found that the thiobarbiturate ring fits into the substrate-binding site and forms hydrogen bonds with Tyr102, Arg105, and Gln140, thus mimicking the substrate succinyl group. In particular, such interaction is stabilized by strong electrostatic contacts between the basic guanidinium group of Arg105 and the acidic thiobarbiturate.162 Two related thiobarbiturates were recently identified as non-nucleoside inhibitors of the H3K79 histone methyltransferase DOT1L using ligand-based and structure-based combined approaches, thus suggesting the promiscuous nature of these compounds.163
Starting from a virtual screening aimed at finding novel SIRT5-selective inhibitors, Liu and colleagues initially identified compounds 9a and 9b (Table 4), which inhibited SIRT5-mediated desuccinylation with IC50 values of 18.30 and 9.26 μM, respectively.164 According to docking studies, the carboxylate group of the two compounds forms hydrogen bonds and electrostatic interactions with Tyr102 and Arg105. To improve the inhibitory potency and identify the structural features key to the activity, various 9b analogues were prepared, all of which had the same central (E)-2-cyano-N-phenyl-3-(5-phenylfuran-2-yl)acrylamide core with different substitutions at the amide (moiety A) and at C5 furan ring position (moiety B). According to the IC50 measurements, for moiety B, the para-benzoic acid substitution is preferred to the meta-benzoic acid substitution, and a further alkyl substitution on the phenyl ring of the para-benzoic acid is detrimental (see compounds 9c–e, Table 4). Hence, the presence of a carboxylic acid at the para-position most likely provides the right orientation for the compound to interact with residues Tyr102 and Arg105 in the active site of SIRT5. Regarding moiety A, the presence of electron-withdrawing groups (EWGs) at the meta- and para-phenyl ring positions seemed to increase the inhibitory potency, as suggested by the lower IC50 values of 9f–i compared to those of 9c and 9j (Table 4). The most potent compound, 9g (IC50 = 5.59 μM), was also assessed against SIRT2 and SIRT6, where it displayed no inhibition up to 600 μM (Table 2). Furthermore, its inhibitory activity was not affected by the NAD+ concentration, suggesting that 9g is not competitive toward NAD+ but acts via competitive inhibition with the succinyl-lysine substrate. The docking analysis indicated that it likely forms hydrogen bonds with the side chains of Tyr102 and Arg105 and backbone amides of Leu227 and Try255, with the fluorine atom forming a halogen bond with Asn226.164
Table 4. Structures and IC50 Values (μM) for the Desuccinylase Activity of SIRT5 toward (E)-2-Cyano-N-phenyl-3-(5-phenylfuran-2-yl)acrylamide Derivatives.


In the search for selective inhibitors, Guetschow et al. carried out a high-throughput screening of a library of 1280 compounds using microchip electrophoresis and found 8 molecules able to inhibit SIRT5’s desuccinylating activity.165 Among them, balsalazide (10a, Figure 11) was reported to have an IC50 value of 3.9 μM. Compound 10a is an approved nonsteroidal anti-inflammatory drug currently employed for the treatment of inflammatory bowel disease. This molecule presents a salicylic moiety that is connected by a central azo group, with a benzamide substituted with a β-alanine side chain. To gain more insights into the binding mode of 10a and explore the possibility of further optimization, Glas and colleagues set out to perform a SAR study using 10a as the lead compound.166 They initially performed docking calculations for 10a bound to a previously reported SIRT5–succinyl-lysine-based peptide cocrystal structure (PDB ID 3RIY)69 in the presence of NAD+. They found that 10a not only forms hydrogen bonds with Tyr102 and Arg105 residues through its carboxylate group but also forms hydrogen bonds with a hydroxyl group of the cosubstrate NAD+ and with the backbone residues Val221 and Glu225 via its amide moiety. These results suggest that the side chain of 10a, derived from β-alanine, is likely the moiety that contributes to the affinity and thus the inhibitory effect of 10a, while the role of the salicylic group remains to be assessed. With the aim of investigating which functional groups of 10a were essential for its inhibitory activity, the research group synthesized a series of 13 analogues.166 The initial evaluation of 10a toward SIRT5’s desuccinylation activity yielded an IC50 value of 5.3 μM and 83% inhibition at 50 μM (Table 2). Removing functional groups from the salicylic portion of balsalazide yielded the phenol derivative 10b, the benzoic acid derivative 10c, and the phenyl analogue 10d (Figure 11), which displayed 73%, 63%, and 62% inhibition of SIRT5’s desuccinylation activity at 50 μM, respectively. Conversely, removing the carboxamide moiety led to compounds with reduced inhibitory activities (30% or lower at 50 μM). These results confirm the hypothesis that the carboxylate group of the β-alanine side chain is crucial for the interactions in the active site, while modifications in the salicylic acid moiety are partially tolerated. In addition, these compounds were tested against the other SIRT isoforms at 50 μM and showed very low inhibitory activities, thereby showing they were SIRT5-selective. Furthermore, the authors indicated that 10a and 10b do not compete with the cosubstrate NAD+ or the synthetic substrate ZKsA. Unfortunately, 10a hardly solubilizes in water and it is likely to be hydrolyzed through enzymatic degradation, hence it can not be used as a possible drug to target SIRT5.166
Figure 11.

Structures of compounds 10a–d, 11, and 12.
Suenkel et al. tested a series of previously reported SIRT inhibitors for their effects on SIRT5 deacetylation and desuccinylation.160 Among them, GW5074 (11, Figure 11), an indole derivative previously reported as Raf-1 kinase (IC50(Raf-1) = 9 nM)167 and a SIRT2 inhibitor (IC50(SIRT2) = 12.5 μM),168 inhibited SIRT5 desuccinylation activity with an IC50 value of 19.5 μM using a succinylated peptide derived from peroxiredoxin 1 (succPrx1). Interestingly, when tested at the same concentration range for its inhibitory effects on SIRT5 deacetylation activity using acPrx1, 11 showed an IC50 value of about 200–400 μM. These data indicate that acPrx1 is a weak SIRT5 substrate and that only 11 inhibits SIRT5 desuccinylation significantly. When a different acetylated peptide (acCPS1) was used, 11 inhibited SIRT5 deacetylation activity with an IC50 of 97.8 μM, which is still fourfold higher than the IC50 for the inhibition of SIRT5’s desuccinylation activity. Collectively, these results suggest that both the substrate sequence and the acyl modification influence the compound inhibitory potency.160 Compound 11 was also tested in the CRC cell line HCT116, where it reduced SIRT5 levels, consequently decreased the activity of its substrate LDHB, and finally decreased autophagy and cell proliferation.142 Notably, 11 showed similar results in terms of its influence on SIRT5 expression, autophagy, and tumor growth in mouse xenograft models. Overall, although promising, this study did not demonstrate that the effects of 11 are a consequence of SIRT5 inhibition but rather indicated that it was able to modulate its expression.
Yang et al. reported 16 fluorogenic peptide SIRT substrates that were tested against SIRT isoforms to determine their sensitivity and efficiency through fluorescence-based assays used to identify SIRT inhibitors.79 Since three succinyl-modified substrates showed high sensitivities and selectivities for SIRT5, three of them were cocrystallized with the enzyme. Crystallographic analyses revealed that these peptides placed the succinyl-lysine moiety in the substrate-binding site of SIRT5, forming hydrogen bonds with residues Tyr102, Arg105, Val221, Gly224, and Glu225 and π–π stacking interactions with the residues Leu227, Met259, Asn226, and Tyr255. These promising substrates led the authors to perform an in-house library screening, which identified TW-37 (12, Figure 11), an inhibitor of Bcl-2 family members, as a SIRT5 inhibitor. Docking studies indicated that 12 binds into SIRT5 substrate-binding pocket as well as the C-pocket. The IC50 values against SIRT5 were determined using three of the peptide substrates previously developed and were 21.9, 6.6, and 6.1 μM. In addition, compound 12 displayed no inhibition toward SIRT1–3.79 Hence, this molecule represents a new starting point for the development of dual SIRT5 and Bcl-2 inhibitors that may be relevant in cancer types where both proteins play a critical role.
Another study identified SIRT5 inhibitors by joining a heteroaromatic ring to a 3-thioureidopropanoic acid warhead through an aminoethyl linker to mimic the interactions of ε-N-glutaryllysine within the SIRT5 active site.169 Among the synthesized molecules, compounds 13a and 13b bearing a pyridine scaffold and a 2-benzylamino substitution, respectively, were about threefold less potent than compound 13c (IC50(desuccinylation) = 9.6 μM), where pyridine was replaced by pyrimidine (Table 5). Starting from 13c, various modifications were performed to gain SAR information. Increasing the length of the linker, as in the case of 13d bearing a 2-phenetylamino substitution, led to a decrease in the potency. Conversely, replacing the nitro group at the C5 (R3) position of the pyrimidinyl ring with an ethoxycarbonyl group (13e) enhanced the inhibitory activity (IC50 = 3.0 μM), while the absence of a substitution (13f) or replacement with fluorine (13g) led to a drop in the inhibitory potency (Table 4), probably due to the lack of interactions with the active site. A similar trend was observed when the 2-benzylamino on the pyrimidinyl ring was replaced with a (2-(1H-indol-3-yl)ethyl)amino portion. Indeed, the C5-nitro derivative 13h was about 3-4× less potent than 13i and 13j, which harbored ethoxycarbonyl and carboxylate substitutions at C5, respectively (Table 4). Interestingly, moving the carboxylate group from C5 to C4 (13k) led to a fourfold drop in inhibitory activity. These findings indicate that the presence of a carboxylate or ethoxycarbonyl moiety as R3 is favorable for compound activity. Furthermore, the most potent compounds (13e, 13i, and 13j) showed no inhibitory activities against SIRT1–3 or SIRT6 up to concentrations of 600 μM. The docking analysis of 13j binding to hSIRT5 indicated that the carboxylate group forms hydrogen bonds and electrostatic interactions with Tyr102 and Arg105 as well as hydrogen bonds with Val221, Glu225, and Tyr255, thus suggesting that 13j acts by mimicking the acyl-lysine substrate via a competitive mechanism of inhibition. Moreover, 13j may react with the cosubstrate NAD+ to form a more stable ADP-ribose-1′-thioimidate intermediate.169
Table 5. Structures and IC50 Values (μM) for the Desuccinylase Activity of SIRT5 toward 3-Thioureidopropanoic Acid Derivatives.


4.2. SIRT5 Activator
Although enhancing SIRT5 activity may benefit its role in human homeostasis, research in this field has been slower compared to SIRT5 inhibition, and only one small molecule has been described so far. Hu and colleagues recently reported the first small-molecule SIRT5 activator, termed MC3138 (14, Figure 12).130 This 1,4-dihydropyridine compound is structurally related to previously reported SIRT1 activators170−172 but displays selective SIRT5 activation, since it does not show any activity toward SIRT1 and SIRT3. Compound 14 increased SIRT5 deacetylase activity ∼1.5-fold at 10 μM, ∼3-fold at 50 μM, and ∼4-fold at 200 μM. Treating different PDAC cell lines with 14 led to a deacetylation profile like that caused by SIRT5 overexpression, resulting in the inhibition of GOT1 enzymatic activity. Compound 14 also decreased PDAC cell viability, with IC50 values ranging between 25.4 and 236.9 μM, and reduced metabolite levels involved in the glutamine, glutathione, and pyrimidine metabolic pathways. Selective SIRT5 activation at the cellular level was confirmed by experiments in the mouse PDAC cell lines KPC and KPCS. While KPC cells were sensitive to 14, KPCS cells, which do not express SIRT5, were indeed resistant to compound 14 treatment, thereby indicating a causal correlation between SIRT5 activation and the anticancer properties of 14. Furthermore, association of 14 and gemcitabine, an approved chemotherapeutic drug used for PDAC, resulted in synergistic effects at different concentrations in different human PDAC cell lines; this association also reduced tumor size and tumor weight in vivo and was well-tolerated in mice.
Figure 12.

Structure of the SIRT5 activator MC3138 (14).
5. Conclusions
A growing body of research has shown that SIRT5 primarily performs protein deglutarylation, desuccinylation, and demalonylation at the cellular level and, given its main subcellular localization, preferentially targets mitochondrial proteins (see Figure 3 for an overview of proteins modulated by SIRT5). This is in line with the critical roles that SIRT5 has been shown to play in maintaining cellular homeostasis. These include regulating glycolysis, the TCA cycle, oxidative phosphorylation, FAO, ketone body formation, amino acid catabolism, and ROS management. SIRT5 enzymatic activities are particularly important for brain and heart health, particularly in the context of aging and in response to environmental and oxidative stress.
In the context of cancer, SIRT5 plays a dichotomous role, since it either suppresses or promotes cancer initiation or progression depending on various factors such as tissue or cell type and transformation stage. This is a consequence, at least in part, of the SIRT5-mediated regulation of ATP production and oxidative stress. Indeed, ROS detoxification may be beneficial in healthy cells to avoid DNA damage and prevent tumorigenesis. On the other hand, the same mechanism protects tumor cells from apoptosis, supports cell proliferation, and may reduce the susceptibility to genotoxic chemotherapeutics.
Given the manifold functions of SIRT5, potent and selective chemical tools that may act as either inhibitors or activators are urgently needed. Indeed, the selective modulation of SIRT5 may help researchers to further understand its roles in both physiological and pathological settings. In addition, potent and selective modulators may have the potential to be brought to the clinic to treat specific pathologies in which SIRT5 has a central position. Nonetheless, the road to the discovery of such modulators is at its infancy, in line with the fact that SIRT5’s activity and biological roles were validated quite recently compared to those of other sirtuins such as SIRT1.
Among SIRT5 inhibitors, a great deal of work has been carried out starting from peptide substrate analogues, which has led to promising peptide-based inhibitors. These consist of thiourea-based molecules such as 2c,1353b, 3d,155 and 3i,148 which display submicromolar SIRT5 inhibition (Table 2). These molecules were all administered as prodrugs to mask the negative charge of the carboxylic group and increase cellular permeability. Compounds 2c and 3b, administered as ethyl esters, displayed very promising activities in breast cancer and AML cell lines, respectively. Moreover, 2c-et was also effective in both genetically engineered and xenograft mouse models of AML,135 while 3b-et was effective when administered to ex vivo AML cells subsequently injected in mice.147 Notably, compounds 3b-et, 3d-et, and 3i-he showed cellular effects only in SIRT5-dependent AML cell lines, thereby indicating a connection between SIRT5 inhibition and the observed anticancer effects. Nonetheless, further masking groups need to be explored to further increase cell permeability and avoid off-target effects such as in case of 3i-he, which displays SIRT1 inhibition in vitro along with cellular target engagement. As for 2c-et, although it determined cellular hypersuccinylation, further target engagement or genetic experiments would be necessary to confirm a causal correlation between SIRT5 inhibition and its phenotypic effects.
Regarding small-molecule SIRT5 inhibitors, there is still a great amount of research to be done. Indeed, only a few compounds have shown IC50 values in the low micromolar range. The 3-thioureidopropanoic acid derivative 13e is the most potent and selective, with an IC50 value of 3.0 μM and no activity toward SIRT1–3 or SIRT6 at 600 μM.169 This molecule has been obtained in the context of a medicinal chemistry campaign aimed at finding glutaryl-lysine mimicking molecules. Unfortunately, no biological data have been provided, hence it is unclear whether this class of molecules may have any cellular effect and in which context they may be useful. Other inhibitors that displayed activities in the low micromolar range and isoform selectivity are 9g(164) and balsalazide (10a)166 (Table 2). Nonetheless, 9g has only been tested toward SIRT2 and SIRT6, while 10a is an approved anti-inflammatory drug that acts as a prodrug and therefore possesses alternative modes of action beyond SIRT5 inhibition.173 Initial attempts to optimize 10a in terms of its potency and pharmacokinetic properties failed, hence further development is still needed. Among small-molecule inhibitors, only the unselective compound 11 was assayed in cellular models and displayed some effects related to SIRT5 inhibition; however, these effects seemed mostly associated to modulation of SIRT5 levels, and a direct inhibitory effect was not proven.142
Among SIRT5 inhibitors, compounds 3b-et, 3d, 6, 7, 9g, 10a, 11, and 12 are commercially available, although it should be noted that compounds 3b-et, 3d, and 6, are the only inhibitors that can be regarded as SIRT5-selective.
Nonetheless, more work would be necessary to deliver nanomolar inhibitors of SIRT5. To this end, cocrystal structures of SIRT5 in complex with currently known inhibitors provide valuable information for further drug development. In particular, the structures of hSIRT5 in complex with compound 3b and 7 indicated the residues that should be targeted to develop an effective inhibitor. These include the key residues Tyr102 and Arg105, which are involved in substrate recognition, but also surrounding residues present in the catalytic cleft such as His150 and Tyr255, which interact with both 3b and 7.
The road to the release of potent SIRT5 activators is also still at its early stages, since only one molecule (14) has been described so far.130 Notably, this compound displays promising anticancer activity in PDAC cell lines as a consequence of SIRT5 activation and represents an optimal lead molecule for the development of a 1,4-dihydropyridine-based series of SIRT5 activators.
To date, the best lead structures for SIRT5 modulation are represented by the peptide-based inhibitors 2c-et, 3b-et, and 3i-he and the 1,4-dihydropyridine activator 14. Further efforts will be necessary to improve the potency, selectivity, and drug-likeness of currently available modulators. To this end, available cocrystal structures, along with high-throughput screening approaches, will likely aid the quick and reliable development of new chemotypes. For instance, the SIRT5–3b/ADP-ribose-1′-thioimidate intermediate structure could be used to develop new peptidomimetics in which the peptide groups are removed via isosteric substitution to decrease the peptide character of the molecule and nonessential side chains are modified to improve the pharmacokinetic properties. Moreover, to further increase the compound selectivity and potency, scaffold hopping174 strategies could be applied to yield new compounds bearing different cores while retaining the pivotal groups for SIRT5 binding.
In summary, although the field of SIRT5 modulation is still at its infancy, the availability of many SIRT5 crystal structures suggests that structure-based drug design approaches are possible. This strategy, in combination with modern computational and biophysical methods, bears great promise for the development of new molecules that could be used as valuable chemical probes for studying the biology or potential therapeutics of SIRT5.
More generally, the potential applicability of SIRT5 modulators in specific pathologies requires concerted efforts to gain a better idea of the roles of SIRT5 in different contexts and to clarify the relevance of its catalytic activity in both physiological and pathological states. This is particularly relevant in cancer, where SIRT5 may act as either tumor promoter or tumor suppressor even in the same cancer type.
Acknowledgments
This work was supported by Italian Ministry of University FISR2019_00374 MeDyCa (A.M.), Progetto di Ateneo “Sapienza” 2017 no. RM11715C7CA6CE53, and Regione Lazio PROGETTI DI GRUPPI DI RICERCA 2020-A0375-2020-36597 (D.R.).
Glossary
Abbreviations Used
- ACAT1
acetyl-CoA acetyltransferase 1
- ACOX1
acyl-CoA oxidase 1
- AMPK
AMP-activated protein kinase
- BAT
brown adipose tissue
- Bcl-XL
B-cell lymphoma-extra large
- BRAF
serine/threonine protein kinase B-Raf
- CoA
coenzyme A
- COVID-19
coronavirus disease 2019
- CPS1
carbamoylphosphate synthetase I
- CPT1A
carnitine palmitoyl transferase 1A
- CRC
colorectal cancer
- DRP1
dynamin-related protein 1
- E2F1
E2 transcription factor 1
- ECHA
enoyl-CoA hydratase
- ETC
electron transport chain
- EDG
electron-donating group
- EWG
electron-withdrawing group
- FAO
fatty acid β-oxidation
- FMO
flavin-containing monooxygenases
- FOXO3a
Forkhead box O3a
- G6PD
glucose-6-phosphate dehydrogenase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GCN5
general control nondepressible 5
- GLS
glutaminase
- GLUD1
glutamate dehydrogenase 1
- GOT
glutamic-oxaloacetic transaminase
- GSH
reduced glutathione
- GSR
glutathione reductase
- GSSG
oxidized glutathione
- HAT
histone acetyltransferase
- HCC
hepatocellular carcinoma
- HMGCS2
3-hydroxy-3-methylglutaryl-CoA synthetase 2
- HO-1
heme oxygenase 1
- IDH
isocitrate dehydrogenase
- KAT8
lysine acetyltransferase 8
- kcat
catalytic constant
- KD
dissociation constant
- KO
knockout
- KRAS
Kirsten rat sarcoma virus
- LDH
lactate dehydrogenase
- LINC
linker of nucleoskeleton and cytoskeleton
- ME1
NADP-dependent malic enzyme
- MEFs
mouse embryonic fibroblasts
- MITF
microphthalmia-associated transcription factor
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NAT10
N-acetyltransferase 10
- NMT
N-terminal glycine myristoyltransferase
- NRF2
nuclear factor erythroid 2-related factor
- Nsp
nonstructural protein
- OGDH
oxoglutarate dehydrogenase
- PDAC
pancreatic ductal adenocarcinoma
- PDC
pyruvate dehydrogenase complex
- PGC-1α
peroxisome proliferator-activated receptor coactivator-1α
- PKM2
pyruvate kinase M2
- PTEN
phosphatase and tensin homologue
- PTM
post-translational modification
- ROS
reactive oxygen species
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SDH
succinate dehydrogenase
- SDHA
succinate dehydrogenase complex subunit A
- SHMT2
serine hydroxymethyltransferase 2
- Sir2
silent information regulator 2
- SIRT
sirtuin
- SOD
superoxide dismutase
- STAT3
signal transducer and activator of transcription 3
- SUN2
SAD1/UNC84 domain protein 2
- TCA
tricarboxylic acid
- TrxR2
thioredoxin reductase 2
- THF
tetrahydrofolate
- UCP-1
uncoupling protein 1
- VDAC3
voltage-dependent anion channel 3
- VLCAD
very long-chain acyl-CoA dehydrogenase
- WAT
white adipose tissue
Biographies
Francesco Fiorentino graduated from Sapienza University of Rome (Italy) with a degree in Medicinal Chemistry in 2016. He received his Ph.D. in Biophysical Chemistry at the University of Oxford (UK) in 2020 under the supervision of Prof. Dame Carol Robinson, where he worked on elucidating the structure and regulation of membrane proteins using mass spectrometry. Following a one-year postdoc in the same lab, he joined the Mai group at Sapienza University of Rome as a Postdoctoral Researcher. His research activity is focused on investigating the molecular mechanisms that underpin protein function and modulation. To this end, he applies native mass spectrometry and other biophysical techniques to investigate the protein complexes involved in the epigenetic regulation of cellular homeostasis and bacterial membrane biogenesis.
Carola Castiello graduated from Sapienza University of Rome (Italy) with a degree in Medicinal Chemistry in 2020. Her thesis under the supervision of Prof. Antonello Mai focused on the synthesis and biological evaluation of dual inhibitors that target epigenetic enzymes. In 2021, she worked the same group as research assistant. She is now a Ph.D. student in Pharmaceutical Sciences under the supervision of Prof. Mai and works on the design and the synthesis of multitarget small-molecule compounds for epigenetic targets.
Antonello Mai graduated from Sapienza University of Rome (Italy) with a degree in Pharmacy in 1984. He received his Ph.D. in Pharmaceutical Sciences in 1992 under the supervision of Prof. M. Artico. In 1998, he was appointed Associate Professor of Medicinal Chemistry at the same University. In 2011, he was appointed Full Professor of Medicinal Chemistry at the Faculty of Pharmacy and Medicine at Sapienza University of Rome. He published more than 300 papers in peer-reviewed high-impact-factor journals. His research interests include the synthesis and biological evaluation of new bioactive small-molecule compounds, in particular modulators of epigenetic targets, for use as chemotherapeutic agents against cancer, metabolic disorders, neurodegenerative diseases, and parasitic infections. In addition, he works the fields of antibacterial or antimycobacterial, antiviral, and CNS agents.
Dante Rotili graduated from Sapienza University of Rome (Italy) with a degree in Medicinal Chemistry in 2003. He received his Ph.D. in Pharmaceutical Sciences at the same University in 2007. In 2009 and 2010, he was research associate at the Department of Chemistry of the University of Oxford, where he worked in collaboration with Prof. C. Schofield to develop chemoproteomic probes for the functional annotation of 2-oxoglutarate-dependent enzymes. In 2020, he was appointed as Associate Professor of Medicinal Chemistry at Sapienza University of Rome. In 2017, he received the Italian National Habilitation to Full Professor of Medicinal Chemistry. His research activity has mainly been focused on the development of modulators of epigenetic enzymes with potential applications in cancer and neurodegenerative, metabolic, and infectious diseases.
Author Contributions
○ These authors contributed equally to this work.
The authors declare no competing financial interest.
References
- Azevedo C.; Saiardi A. Why always lysine? The ongoing tale of one of the most modified amino acids. Adv. Biol. Regul. 2016, 60, 144–150. 10.1016/j.jbior.2015.09.008. [DOI] [PubMed] [Google Scholar]
- Phillips D. M. The presence of acetyl groups of histones. Biochem. J. 1963, 87 (2), 258–263. 10.1042/bj0870258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino F.; Mai A.; Rotili D. Lysine acetyltransferase inhibitors: structure–activity relationships and potential therapeutic implications. Future Med. Chem. 2018, 10 (9), 1067–1091. 10.4155/fmc-2017-0244. [DOI] [PubMed] [Google Scholar]
- Park S.-Y.; Kim J.-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52 (2), 204–212. 10.1038/s12276-020-0382-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mautone N.; Zwergel C.; Mai A.; Rotili D. Sirtuin modulators: where are we now? A review of patents from 2015 to 2019. Expert Opin. Ther. Pat. 2020, 30 (6), 389–407. 10.1080/13543776.2020.1749264. [DOI] [PubMed] [Google Scholar]
- Ho T. C. S.; Chan A. H. Y.; Ganesan A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63 (21), 12460–12484. 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834. 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Barnes C. E.; English D. M.; Cowley S. M. Acetylation & Co: An expanding repertoire of histone acylations regulates chromatin and transcription. Essays in Biochem. 2019, 63 (1), 97–107. 10.1042/EBC20180061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H.; Lin H. Sirtuins in epigenetic regulation. Chem. Rev. 2015, 115 (6), 2350–2375. 10.1021/cr500457h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S. I.; Armstrong C. M.; Kaeberlein M.; Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403 (6771), 795–800. 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Lombard D. B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit. Rev. Biochem. Mol. Biol. 2018, 53 (3), 311–334. 10.1080/10409238.2018.1458071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafa V.; Rotili D.; Forgione M.; Cuomo F.; Serretiello E.; Hailu G. S.; Jarho E.; Lahtela-Kakkonen M.; Mai A.; Altucci L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenetics 2016, 8, 61. 10.1186/s13148-016-0224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosciuk T.; Wang M.; Hong J. Y.; Lin H. Updates on the epigenetic roles of sirtuins. Curr. Opin. Chem. Biol. 2019, 51, 18–29. 10.1016/j.cbpa.2019.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; He J.; Liao M.; Hu M.; Li W.; Ouyang H.; Wang X.; Ye T.; Zhang Y.; Ouyang L. An overview of Sirtuins as potential therapeutic target: Structure, function and modulators. Eur. J. Med. Chem. 2019, 161, 48–77. 10.1016/j.ejmech.2018.10.028. [DOI] [PubMed] [Google Scholar]
- Fiorentino F.; Mautone N.; Menna M.; D’Acunzo F.; Mai A.; Rotili D. Sirtuin modulators: past, present, and future perspectives. Future Med. Chem. 2022, 14 (12), 915–939. 10.4155/fmc-2022-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J.; Johnson J.; Hanigan C.; Locasale J. Interactions between epigenetics and metabolism in cancers. Front. Oncol. 2012, 2, 163. 10.3389/fonc.2012.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine D. C.; Kuo H.-Y.; Hong H.-K.; Cedernaes J.; Hepler C.; Wright A. G.; Sommars M. A.; Kobayashi Y.; Marcheva B.; Gao P.; Ilkayeva O. R.; Omura C.; Ramsey K. M.; Newgard C. B.; Barish G. D.; Peek C. B.; Chandel N. S.; Mrksich M.; Bass J. NADH inhibition of SIRT1 links energy state to transcription during time-restricted feeding. Nat. Metab. 2021, 3 (12), 1621–1632. 10.1038/s42255-021-00498-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Ma X.; He Y.; Yuan C.; Chen Q.; Li G.; Chen X. Sirtuin 5: a review of structure, known inhibitors and clues for developing new inhibitors. Sci. China Life Sci. 2017, 60 (3), 249–256. 10.1007/s11427-016-0060-7. [DOI] [PubMed] [Google Scholar]
- Michishita E.; Park J. Y.; Burneskis J. M.; Barrett J. C.; Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16 (10), 4623–4635. 10.1091/mbc.e05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford E.; Voit R.; Liszt G.; Magin C.; Grummt I.; Guarente L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20 (9), 1075–1080. 10.1101/gad.1399706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero A.; Scher M. B.; Lee D. H.; Sutton A.; Cheng H. L.; Alt F. W.; Serrano L.; Sternglanz R.; Reinberg D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20 (10), 1256–1261. 10.1101/gad.1412706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R.; Chua K. F.; Lombard D. B.; Pang W. W.; Fischer M. R.; Gellon L.; Liu P.; Mostoslavsky G.; Franco S.; Murphy M. M.; Mills K. D.; Patel P.; Hsu J. T.; Hong A. L.; Ford E.; Cheng H. L.; Kennedy C.; Nunez N.; Bronson R.; Frendewey D.; Auerbach W.; Valenzuela D.; Karow M.; Hottiger M. O.; Hursting S.; Barrett J. C.; Guarente L.; Mulligan R.; Demple B.; Yancopoulos G. D.; Alt F. W. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124 (2), 315–329. 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Rack J. G. M.; VanLinden M. R.; Lutter T.; Aasland R.; Ziegler M. Constitutive Nuclear Localization of an Alternatively Spliced Sirtuin-2 Isoform. J. Mol. Biol. 2014, 426 (8), 1677–1691. 10.1016/j.jmb.2013.10.027. [DOI] [PubMed] [Google Scholar]
- North B. J.; Verdin E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PloS One 2007, 2 (8), e784. 10.1371/journal.pone.0000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B.; North B. J.; Frye R. A.; Ott M.; Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 2002, 158 (4), 647–657. 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyango P.; Celic I.; McCaffery J. M.; Boeke J. D.; Feinberg A. P. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2002, 99 (21), 13653–13658. 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis M. C.; Mostoslavsky R.; Haigis K. M.; Fahie K.; Christodoulou D. C.; Murphy A.; Valenzuela D. M.; Yancopoulos G. D.; Karow M.; Blander G.; Wolberger C.; Prolla T. A.; Weindruch R.; Alt F. W.; Guarente L. SIRT4 Inhibits Glutamate Dehydrogenase and Opposes the Effects of Calorie Restriction in Pancreatic β Cells. Cell 2006, 126 (5), 941–954. 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- Tomaselli D.; Steegborn C.; Mai A.; Rotili D. Sirt4: A Multifaceted Enzyme at the Crossroads of Mitochondrial Metabolism and Cancer. Front. Oncol. 2020, 10, 474. 10.3389/fonc.2020.00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S. I.; Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: Implications for metabolic diseases. Trends Pharmacol. Sci. 2010, 31 (5), 212–220. 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Lombard D. B. For Certain, SIRT4 Activities!. Trends Biochem. Sci. 2017, 42 (7), 499–501. 10.1016/j.tibs.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard D. B.; Alt F. W.; Cheng H. L.; Bunkenborg J.; Streeper R. S.; Mostoslavsky R.; Kim J.; Yancopoulos G.; Valenzuela D.; Murphy A.; Yang Y.; Chen Y.; Hirschey M. D.; Bronson R. T.; Haigis M.; Guarente L. P.; Farese R. V. Jr; Weissman S.; Verdin E.; Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27 (24), 8807–8814. 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias R. A.; Greco T. M.; Oberstein A.; Budayeva H. G.; Chakrabarti R.; Rowland E. A.; Kang Y.; Shenk T.; Cristea I. M. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 2014, 159 (7), 1615–1625. 10.1016/j.cell.2014.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannek M.; Simic Z.; Fuszard M.; Meleshin M.; Rotili D.; Mai A.; Schutkowski M.; Steegborn C. Crystal structures of the mitochondrial deacylase Sirtuin 4 reveal isoform-specific acyl recognition and regulation features. Nat. Commun. 2017, 8, 1513. 10.1038/s41467-017-01701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. A.; Huynh F. K.; Fisher-Wellman K.; Stuart J. D.; Peterson B. S.; Douros J. D.; Wagner G. R.; Thompson J. W.; Madsen A. S.; Green M. F.; Sivley R. M.; Ilkayeva O. R.; Stevens R. D.; Backos D. S.; Capra J. A.; Olsen C. A.; Campbell J. E.; Muoio D. M.; Grimsrud P. A.; Hirschey M. D. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metabolism 2017, 25 (4), 838–855. 10.1016/j.cmet.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino F.; Mai A.; Rotili D. Emerging Therapeutic Potential of SIRT6 Modulators. J. Med. Chem. 2021, 64 (14), 9732–9758. 10.1021/acs.jmedchem.1c00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino F.; Carafa V.; Favale G.; Altucci L.; Mai A.; Rotili D. The Two-Faced Role of SIRT6 in Cancer. Cancers (Basel) 2021, 13 (5), 1156. 10.3390/cancers13051156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber M. F.; Michishita-Kioi E.; Xi Y.; Tasselli L.; Kioi M.; Moqtaderi Z.; Tennen R. I.; Paredes S.; Young N. L.; Chen K.; Struhl K.; Garcia B. A.; Gozani O.; Li W.; Chua K. F. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487 (7405), 114–118. 10.1038/nature11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Seiler J.; Santiago-Reichelt M.; Felbel K.; Grummt I.; Voit R. Repression of RNA Polymerase I upon Stress Is Caused by Inhibition of RNA-Dependent Deacetylation of PAF53 by SIRT7. Mol. Cell 2013, 52 (3), 303–313. 10.1016/j.molcel.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Chen X. F.; Tian M. X.; Sun R. Q.; Zhang M. L.; Zhou L. S.; Jin L.; Chen L. L.; Zhou W. J.; Duan K. L.; Chen Y. J.; Gao C.; Cheng Z. L.; Wang F.; Zhang J. Y.; Sun Y. P.; Yu H. X.; Zhao Y. Z.; Yang Y.; Liu W. R.; Shi Y. H.; Xiong Y.; Guan K. L.; Ye D. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19 (5), e45124. 10.15252/embr.201745124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Liu L. SIRT5 deficiency enhances susceptibility to kainate-induced seizures and exacerbates hippocampal neurodegeneration not through mitochondrial antioxidant enzyme SOD2. Front. Cell. Neurosci. 2016, 10, 171. 10.3389/fncel.2016.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X.; Liu Z.; Zhang W.; Gladysz K.; Fung Y. M. E.; Tian G.; Xiong Y.; Wong J. W. H.; Yuen K. W. Y.; Li X. D. Glutarylation of Histone H4 Lysine 91 Regulates Chromatin Dynamics. Mol. Cell 2019, 76 (4), 660–675. 10.1016/j.molcel.2019.08.018. [DOI] [PubMed] [Google Scholar]
- Roessler C.; Tüting C.; Meleshin M.; Steegborn C.; Schutkowski M. A Novel Continuous Assay for the Deacylase Sirtuin 5 and Other Deacetylases. J. Med. Chem. 2015, 58 (18), 7217–7223. 10.1021/acs.jmedchem.5b00293. [DOI] [PubMed] [Google Scholar]
- Fiorentino F.; Mai A.; Rotili D. Lysine Acetyltransferase Inhibitors From Natural Sources. Front. Pharmacol. 2020, 11, 1243. 10.3389/fphar.2020.01243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y.; Du S.; Liu Z.; Liu M.; Xu Z.; Wang X.; Kee J. X.; Yi F.; Sun H.; Yao S. Q. Chemical Biology Tools for Protein Lysine Acylation. Angew. Chem. Int. Ed. 2022, 61 (21), e202200303. 10.1002/anie.202200303. [DOI] [PubMed] [Google Scholar]
- Blasl A. T.; Schulze S.; Qin C.; Graf L. G.; Vogt R.; Lammers M. Post-translational lysine ac(et)ylation in health, ageing and disease. Biol. Chem. 2022, 403 (2), 151–194. 10.1515/hsz-2021-0139. [DOI] [PubMed] [Google Scholar]
- Kurmi K.; Hitosugi S.; Wiese E. K.; Boakye-Agyeman F.; Gonsalves W. I.; Lou Z.; Karnitz L. M.; Goetz M. P.; Hitosugi T. Carnitine Palmitoyltransferase 1A Has a Lysine Succinyltransferase Activity. Cell Rep. 2018, 22 (6), 1365–1373. 10.1016/j.celrep.2018.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Guo Y. R.; Liu K.; Yin Z.; Liu R.; Xia Y.; Tan L.; Yang P.; Lee J. H.; Li X. J.; Hawke D.; Zheng Y.; Qian X.; Lyu J.; He J.; Xing D.; Tao Y. J.; Lu Z. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 2017, 552 (7684), 273–277. 10.1038/nature25003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorro Shahidian L.; Haas M.; Le Gras S.; Nitsch S.; Mourão A.; Geerlof A.; Margueron R.; Michaelis J.; Daujat S.; Schneider R. Succinylation of H3K122 destabilizes nucleosomes and enhances transcription. EMBO Rep. 2021, 22 (3), e51009. 10.15252/embr.202051009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosciuk T.; Price I. R.; Zhang X.; Zhu C.; Johnson K. N.; Zhang S.; Halaby S. L.; Komaniecki G. P.; Yang M.; DeHart C. J.; Thomas P. M.; Kelleher N. L.; Fromme J. C.; Lin H. NMT1 and NMT2 are lysine myristoyltransferases regulating the ARF6 GTPase cycle. Nat. Commun. 2020, 11, 1067. 10.1038/s41467-020-14893-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner G.; Hirschey M. D. Nonenzymatic Protein Acylation as a Carbon Stress Regulated by Sirtuin Deacylases. Mol. Cell 2014, 54 (1), 5–16. 10.1016/j.molcel.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner G. R.; Bhatt D. P.; O’Connell T. M.; Thompson J. W.; Dubois L. G.; Backos D. S.; Yang H.; Mitchell G. A.; Ilkayeva O. R.; Stevens R. D.; Grimsrud P. A.; Hirschey M. D. A Class of Reactive Acyl-CoA Species Reveals the Non-Enzymatic Origins of Protein Acylation. Cell Metab. 2017, 25 (4), 823–837.e8. 10.1016/j.cmet.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland E. A.; Snowden C. K.; Cristea I. M. Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr. Opin. Chem. Biol. 2018, 42, 76–85. 10.1016/j.cbpa.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T.; Lomb D. J.; Haigis M. C.; Guarente L. SIRT5 Deacetylates Carbamoyl Phosphate Synthetase 1 and Regulates the Urea Cycle. Cell 2009, 137 (3), 560–570. 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin M. J.; He W.; Nishida Y.; Newman J. C.; Carrico C.; Danielson S. R.; Guo A.; Gut P.; Sahu A. K.; Li B.; Uppala R.; Fitch M.; Riiff T.; Zhu L.; Zhou J.; Mulhern D.; Stevens R. D.; Ilkayeva O. R.; Newgard C. B.; Jacobson M. P.; Hellerstein M.; Goetzman E. S.; Gibson B. W.; Verdin E. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18 (6), 920–933. 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; He X.; Ye D.; Lin Y.; Yu H.; Yao C.; Huang L.; Zhang J.; Wang F.; Xu S.; Wu X.; Liu L.; Yang C.; Shi J.; He X.; Liu J.; Qu Y.; Guo F.; Zhao J.; Xu W.; Zhao S. NADP+-IDH Mutations Promote Hypersuccinylation that Impairs Mitochondria Respiration and Induces Apoptosis Resistance. Mol. Cell 2015, 60 (4), 661–675. 10.1016/j.molcel.2015.10.017. [DOI] [PubMed] [Google Scholar]
- Lin Z. F.; Xu H. B.; Wang J. Y.; Lin Q.; Ruan Z.; Liu F. B.; Jin W.; Huang H. H.; Chen X. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem. Biophys. Res. Commun. 2013, 441 (1), 191–195. 10.1016/j.bbrc.2013.10.033. [DOI] [PubMed] [Google Scholar]
- Park J.; Chen Y.; Tishkoff D. X.; Peng C.; Tan M.; Dai L.; Xie Z.; Zhang Y.; Zwaans B. M. M.; Skinner M. E.; Lombard D. B.; Zhao Y. SIRT5-Mediated Lysine Desuccinylation Impacts Diverse Metabolic Pathways. Mol. Cell 2013, 50 (6), 919–930. 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Bharathi S. S.; Rardin M. J.; Uppala R.; Verdin E.; Gibson B. W.; Goetzman E. S. SIRT3 and SIRT5 regulate the enzyme activity and cardiolipin binding of very long-chain Acyl-CoA dehydrogenase. PLoS One 2015, 10 (3), e0122297. 10.1371/journal.pone.0122297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y.; Rardin M. J.; Carrico C.; He W.; Sahu A. K.; Gut P.; Najjar R.; Fitch M.; Hellerstein M.; Gibson B. W.; Verdin E. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 2015, 59 (2), 321–332. 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C. C.; Lin P. M.; Lin S. F.; Hsu C. H.; Lin H. C.; Hu M. L.; Hsu C. M.; Yang M. Y. Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumor Biol. 2013, 34 (3), 1847–1854. 10.1007/s13277-013-0726-y. [DOI] [PubMed] [Google Scholar]
- Liu L.; Peritore C.; Ginsberg J.; Shih J.; Arun S.; Donmez G. Protective role of SIRT5 against motor deficit and dopaminergic degeneration in MPTP-induced mice model of Parkinson’s disease. Behav. Brain Res. 2015, 281, 215–221. 10.1016/j.bbr.2014.12.035. [DOI] [PubMed] [Google Scholar]
- Lu W.; Zuo Y.; Feng Y.; Zhang M. SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumor Biol. 2014, 35 (11), 10699–10705. 10.1007/s13277-014-2372-4. [DOI] [PubMed] [Google Scholar]
- Parihar P.; Solanki I.; Mansuri M. L.; Parihar M. S. Mitochondrial sirtuins: emerging roles in metabolic regulations, energy homeostasis and diseases. Exp. Gerontol. 2015, 61, 130–141. 10.1016/j.exger.2014.12.004. [DOI] [PubMed] [Google Scholar]
- Wang C. H.; Wei Y. H. Roles of Mitochondrial Sirtuins in Mitochondrial Function, Redox Homeostasis, Insulin Resistance and Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21 (15), 5266. 10.3390/ijms21155266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y. Q.; Li T. T.; Liu X. Y.; Li Z. H.; Fu Y. C. SIRT1 and SIRT5 activity expression and behavioral responses to calorie restriction. J. Cell Biochem. 2011, 112 (12), 3755–3761. 10.1002/jcb.23315. [DOI] [PubMed] [Google Scholar]
- Matsushita N.; Yonashiro R.; Ogata Y.; Sugiura A.; Nagashima S.; Fukuda T.; Inatome R.; Yanagi S. Distinct regulation of mitochondrial localization and stability of two human Sirt5 isoforms. Genes Cells 2011, 16 (2), 190–202. 10.1111/j.1365-2443.2010.01475.x. [DOI] [PubMed] [Google Scholar]
- Sadhukhan S.; Liu X.; Ryu D.; Nelson O. D.; Stupinski J. A.; Li Z.; Chen W.; Zhang S.; Weiss R. S.; Locasale J. W.; Auwerx J.; Lin H. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. U.S.A. 2016, 113 (16), 4320–4325. 10.1073/pnas.1519858113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M.; Peng C.; Anderson K. A.; Chhoy P.; Xie Z.; Dai L.; Park J.; Chen Y.; Huang H.; Zhang Y.; Ro J.; Wagner G. R.; Green M. F.; Madsen A. S.; Schmiesing J.; Peterson B. S.; Xu G.; Ilkayeva O. R.; Muehlbauer M. J.; Braulke T.; Mühlhausen C.; Backos D. S.; Olsen C. A.; McGuire P. J.; Pletcher S. D.; Lombard D. B.; Hirschey M. D.; Zhao Y. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19 (4), 605–617. 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Zhou Y.; Su X.; Yu J. J.; Khan S.; Jiang H.; Kim J.; Woo J.; Kim J. H.; Choi B. H.; He B.; Chen W.; Zhang S.; Cerione R. A.; Auwerx J.; Hao Q.; Lin H. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334 (6057), 806–809. 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C.; Lu Z.; Xie Z.; Cheng Z.; Chen Y.; Tan M.; Luo H.; Zhang Y.; He W.; Yang K.; Zwaans B. M. M.; Tishkoff D.; Ho L.; Lombard D.; He T. C.; Dai J.; Verdin E.; Ye Y.; Zhao Y. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteom. 2011, 10 (12), M111.012658. 10.1074/mcp.M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger K. A.; Martin A. S.; Hirschey M. D. Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol. 2017, 13 (4), 213–225. 10.1038/nrneph.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlknecht U.; Ho A. D.; Letzel S.; Voelter-Mahlknecht S. Assignment of the NAD-dependent deacetylase sirtuin 5 gene (SIRT5) to human chromosome band 6p23 by in situ hybridization. Cytogenet. Genome Res. 2006, 112 (3–4), 208–212. 10.1159/000089872. [DOI] [PubMed] [Google Scholar]
- Du Y.; Hu H.; Hua C.; Du K.; Wei T. Tissue distribution, subcellular localization, and enzymatic activity analysis of human SIRT5 isoforms. Biochem. Biophys. Res. Commun. 2018, 503 (2), 763–769. 10.1016/j.bbrc.2018.06.073. [DOI] [PubMed] [Google Scholar]
- Buler M.; Aatsinki S. M.; Izzi V.; Uusimaa J.; Hakkola J. SIRT5 is under the control of PGC-1o: And AMPK and is involved in regulation of mitochondrial energy metabolism. FASEB J. 2014, 28 (7), 3225–3237. 10.1096/fj.13-245241. [DOI] [PubMed] [Google Scholar]
- North B. J.; Verdin E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004, 5 (5), 224. 10.1186/gb-2004-5-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler C.; Nowak T.; Pannek M.; Gertz M.; Nguyen G. T.; Scharfe M.; Born I.; Sippl W.; Steegborn C.; Schutkowski M. Chemical probing of the human sirtuin 5 active site reveals its substrate acyl specificity and peptide-based inhibitors. Angew. Chem., Int. Ed. Engl. 2014, 53 (40), 10728–10732. 10.1002/anie.201402679. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz B. G.; Dai H.; Koppetsch K. J.; Qian D.; Jiang F.; Mao C.; Perni R. B. Synthesis of Carba-NAD and the Structures of Its Ternary Complexes with SIRT3 and SIRT5. J. Org. Chem. 2012, 77 (17), 7319–7329. 10.1021/jo301067e. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Zhang H.; He B.; Du J.; Lin H.; Cerione R. A.; Hao Q. The bicyclic intermediate structure provides insights into the desuccinylation mechanism of human sirtuin 5 (SIRT5). J. Biol. Chem. 2012, 287 (34), 28307–28314. 10.1074/jbc.M112.384511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. L.; Wang H. L.; Yan Y. H.; Liu S.; Yu Z. J.; Huang M. Y.; Luo Y.; Zheng X.; Yu Y.; Li G. B. Sensitive fluorogenic substrates for sirtuin deacylase inhibitor discovery. Eur. J. Med. Chem. 2020, 192, 112201. 10.1016/j.ejmech.2020.112201. [DOI] [PubMed] [Google Scholar]
- Gertz M.; Nguyen G. T. T.; Fischer F.; Suenkel B.; Schlicker C.; Fränzel B.; Tomaschewski J.; Aladini F.; Becker C.; Wolters D.; Steegborn C. A Molecular Mechanism for Direct Sirtuin Activation by Resveratrol. PLoS One 2012, 7 (11), e49761. 10.1371/journal.pone.0049761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport A. M.; Huber F. M.; Hoelz A. Structural and Functional Analysis of Human SIRT1. J. Mol. Biol. 2014, 426 (3), 526–541. 10.1016/j.jmb.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniot S.; Weyand M.; Steegborn C. Structures, substrates, and regulators of mammalian Sirtuins - opportunities and challenges for drug development. Front. Pharm. 2012, 3, 16. 10.3389/fphar.2012.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L.; Wei W.; Jiang Y.; Peng H.; Cai J.; Mao C.; Dai H.; Choy W.; Bemis J. E.; Jirousek M. R.; Milne J. C.; Westphal C. H.; Perni R. B. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 2009, 284 (36), 24394–24405. 10.1074/jbc.M109.014928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polletta L.; Vernucci E.; Carnevale I.; Arcangeli T.; Rotili D.; Palmerio S.; Steegborn C.; Nowak T.; Schutkowski M.; Pellegrini L.; Sansone L.; Villanova L.; Runci A.; Pucci B.; Morgante E.; Fini M.; Mai A.; Russo M. A.; Tafani M. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 2015, 11 (2), 253–270. 10.1080/15548627.2015.1009778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene K. S.; Lukey M. J.; Wang X.; Blank B.; Druso J. E.; Lin M. J.; Stalnecker C. A.; Zhang C.; Abril Y. N.; Erickson J. W.; Wilson K. F.; Lin H.; Weiss R. S.; Cerione R. A. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 2019, 116 (52), 26625–26632. 10.1073/pnas.1911954116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M.; Chen I. P.; Vallejo-Gracia A.; Kim I. J.; Bielska O.; Lam V. L.; Hayashi J. M.; Cruz A.; Shah S.; Gross J. D.; Krogan N. J.; Schilling B.; Ott M.; Verdin E., SIRT5 is a proviral factor that interacts with SARS-CoV-2 Nsp14 protein. bioRxiv (Microbiology) January 5, 2022, 474979. 10.1101/2022.01.04.474979 [DOI] [PMC free article] [PubMed]
- Jukarainen S.; Heinonen S.; Rämö J. T.; Rinnankoski-Tuikka R.; Rappou E.; Tummers M.; Muniandy M.; Hakkarainen A.; Lundbom J.; Lundbom N.; Kaprio J.; Rissanen A.; Pirinen E.; Pietiläinen K. H. Obesity Is Associated With Low NAD(+)/SIRT Pathway Expression in Adipose Tissue of BMI-Discordant Monozygotic Twins. J. Clin. Endocrinol. Metab. 2016, 101 (1), 275–283. 10.1210/jc.2015-3095. [DOI] [PubMed] [Google Scholar]
- Xu Y. S.; Liang J. J.; Wang Y.; Zhao X. Z. J.; Xu L.; Xu Y. Y.; Zou Q. C.; Zhang J. M.; Tu C. E.; Cui Y. G.; Sun W. H.; Huang C.; Yang J. H.; Chin Y. E. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci. Rep. 2016, 6, 39517. 10.1038/srep39517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Bharathi S. S.; Rardin M. J.; Lu J.; Maringer K. V.; Sims-Lucas S.; Prochownik E. V.; Gibson B. W.; Goetzman E. S. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J. Biol. Chem. 2017, 292 (24), 10239–10249. 10.1074/jbc.M117.785022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z.; Liu G.-H.; Qu J. Mitochondrial sirtuins, metabolism, and aging. J. Genet. Genomics 2022, 49 (4), 287–298. 10.1016/j.jgg.2021.11.005. [DOI] [PubMed] [Google Scholar]
- Gao X.; Wang H.; Yang J. J.; Liu X.; Liu Z. R. Pyruvate Kinase M2 Regulates Gene Transcription by Acting as a Protein Kinase. Mol. Cell 2012, 45 (5), 598–609. 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Xia Y.; Hawke D.; Li X.; Liang J.; Xing D.; Aldape K.; Hunter T.; Alfred Yung W. K.; Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150 (4), 685–696. 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.; Li X.; Yang W.; Hawke D. H.; Zheng Y.; Xia Y.; Aldape K.; Wei C.; Guo F.; Chen Y.; Lu Z. PKM2 Regulates Chromosome Segregation and Mitosis Progression of Tumor Cells. Mol. Cell 2014, 53 (1), 75–87. 10.1016/j.molcel.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Wang K.; Xu W.; Zhao S.; Ye D.; Wang Y.; Xu Y.; Zhou L.; Chu Y.; Zhang C.; Qin X.; Yang P.; Yu H. SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1β Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep. 2017, 19 (11), 2331–2344. 10.1016/j.celrep.2017.05.065. [DOI] [PubMed] [Google Scholar]
- Xiangyun Y.; Xiaomin N.; Linping G.; Yunhua X.; Ziming L.; Yongfeng Y.; Zhiwei C.; Shun L. Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget 2017, 8 (4), 6984–6993. 10.18632/oncotarget.14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H.; Ning X.; Yu C.; Ji X.; Jin Y.; McNutt M. A.; Yin Y. Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress. Cell Death Dis. 2019, 10 (3), 170. 10.1038/s41419-018-1271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L.; Wang F.; Sun R.; Chen X.; Zhang M.; Xu Q.; Wang Y.; Wang S.; Xiong Y.; Guan K. L.; Yang P.; Yu H.; Ye D. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 2016, 17 (6), 811–822. 10.15252/embr.201541643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T.; Khawaja M. R.; DiNardo C. D.; Atkins J. T.; Janku F. Targeting Isocitrate Dehydrogenase (IDH) in cancer. Discov. Med. 2016, 21 (117), 373–380. [PubMed] [Google Scholar]
- Frontini A.; Cinti S. Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab. 2010, 11 (4), 253–256. 10.1016/j.cmet.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Townsend K. L.; Tseng Y. H. Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab. 2014, 25 (4), 168–177. 10.1016/j.tem.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Meyer J. G.; Cai W.; Softic S.; Li M. E.; Verdin E.; Newgard C.; Schilling B.; Kahn C. R. Regulation of UCP1 and Mitochondrial Metabolism in Brown Adipose Tissue by Reversible Succinylation. Mol. Cell 2019, 74 (4), 844–857. 10.1016/j.molcel.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T.; Wang F.; Stieren E.; Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280 (14), 13560–7. 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- Shuai L.; Zhang L. N.; Li B. H.; Tang C. L.; Wu L. Y.; Li J.; Li J. Y. SIRT5 Regulates Brown Adipocyte Differentiation and Browning of Subcutaneous White Adipose Tissue. Diabetes 2019, 68 (7), 1449–1461. 10.2337/db18-1103. [DOI] [PubMed] [Google Scholar]
- Ogura M.; Nakamura Y.; Tanaka D.; Zhuang X.; Fujita Y.; Obara A.; Hamasaki A.; Hosokawa M.; Inagaki N. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1. Biochem. Biophys. Res. Commun. 2010, 393 (1), 73–78. 10.1016/j.bbrc.2010.01.081. [DOI] [PubMed] [Google Scholar]
- Haussinger D. Nitrogen metabolism in liver: structural and functional organization and physiological relevance. Biochem. J. 1990, 267 (2), 281–290. 10.1042/bj2670281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedouari H.; Daigle T.; Scorrano L.; Hebert-Chatelain E. Sirtuin 5 protects mitochondria from fragmentation and degradation during starvation. Biochim. Biophys. Acta - Mol. Cell Res. 2017, 1864 (1), 169–176. 10.1016/j.bbamcr.2016.10.015. [DOI] [PubMed] [Google Scholar]
- Balendiran G. K.; Dabur R.; Fraser D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22 (6), 343–352. 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- Zeng J.; Li D. Expression and purification of his-tagged rat peroxisomal acyl-CoA oxidase I wild-type and E421 mutant proteins. Protein Expr. Purif. 2004, 38 (1), 153–160. 10.1016/j.pep.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhu Y.; Xing S.; Ma P.; Lin D. SIRT5 prevents cigarette smoke extract-induced apoptosis in lung epithelial cells via deacetylation of FOXO3. Cell Stress Chaperones 2015, 20 (5), 805–810. 10.1007/s12192-015-0599-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Wang L.; Liu J.; Xie F.; Su B.; Wang X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants (Basel) 2017, 6 (2), 25. 10.3390/antiox6020025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Yu S.; Simonyi A.; Sun G. Y.; Sun A. Y. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol. Neurobiol. 2005, 31 (1–3), 003–016. 10.1385/MN:31:1-3:003. [DOI] [PubMed] [Google Scholar]
- Przedborski S.; Jackson-Lewis V.; Naini A. B.; Jakowec M.; Petzinger G.; Miller R.; Akram M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): A technical review of its utility and safety. J. Neurochem. 2001, 76 (5), 1265–1274. 10.1046/j.1471-4159.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- Williams A. C.; Ramsden D. B. Autotoxicity, methylation and a road to the prevention of Parkinson’s disease. J. Clin. Neurosci. 2005, 12 (1), 6–11. 10.1016/j.jocn.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Wu S.; Wei Y.; Li J.; Bai Y.; Yin P.; Wang S. SIRT5 Represses Neurotrophic Pathways and Abeta Production in Alzheimer’s Disease by Targeting Autophagy. ACS Chem. Neurosci. 2021, 12 (23), 4428–4437. 10.1021/acschemneuro.1c00468. [DOI] [PubMed] [Google Scholar]
- Hershberger K. A.; Abraham D. M.; Martin A. S.; Mao L.; Liu J.; Gu H.; Locasale J. W.; Hirschey M. D. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J. Biol. Chem. 2017, 292 (48), 19767–19781. 10.1074/jbc.M117.809897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylston J. A.; Sun J.; Chen Y.; Gucek M.; Sack M. N.; Murphy E. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 88, 73–81. 10.1016/j.yjmcc.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani E. T.; Pell V. R.; Gaude E.; Aksentijević D.; Sundier S. Y.; Robb E. L.; Logan A.; Nadtochiy S. M.; Ord E. N. J.; Smith A. C.; Eyassu F.; Shirley R.; Hu C. H.; Dare A. J.; James A. M.; Rogatti S.; Hartley R. C.; Eaton S.; Costa A. S. H.; Brookes P. S.; Davidson S. M.; Duchen M. R.; Saeb-Parsy K.; Shattock M. J.; Robinson A. J.; Work L. M.; Frezza C.; Krieg T.; Murphy M. P. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515 (7527), 431–435. 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.; Che W.; Zheng C.; Liu W.; Wen J.; Fu H.; Tang K.; Zhang J.; Xu Y. SIRT5: A safeguard against oxidative stress-induced apoptosis in cardiomyocytes. Cell. Physiol. Biochem. 2013, 32 (4), 1050–1059. 10.1159/000354505. [DOI] [PubMed] [Google Scholar]
- Chalkiadaki A.; Guarente L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15 (10), 608–624. 10.1038/nrc3985. [DOI] [PubMed] [Google Scholar]
- Bringman-Rodenbarger L. R.; Guo A. H.; Lyssiotis C. A.; Lombard D. B. Emerging Roles for SIRT5 in Metabolism and Cancer. Antioxid. Redox Signal. 2018, 28 (8), 677–690. 10.1089/ars.2017.7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv X. B.; Liu L.; Cheng C.; Yu B.; Xiong L.; Hu K.; Tang J.; Zeng L.; Sang Y. SUN2 exerts tumor suppressor functions by suppressing the Warburg effect in lung cancer. Sci. Rep. 2016, 5, 17940. 10.1038/srep17940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.-X.; Yi Y.; Li Y.-W.; Cai X.-Y.; He H.-W.; Ni X.-C.; Zhou J.; Cheng Y.-F.; Jin J.-J.; Fan J.; Qiu S.-J. Down-regulation of sirtuin 3 is associated with poor prognosis in hepatocellular carcinoma after resection. BMC Cancer 2014, 14, 297. 10.1186/1471-2407-14-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal A.; Xudong Z.; Zhenyu J.; Saretzki G. Mitochondrial sirtuins in stem cells and cancer. FEBS J. 2022, 289 (12), 3393–3415. 10.1111/febs.15879. [DOI] [PubMed] [Google Scholar]
- Chang L.; Xi L.; Liu Y.; Liu R.; Wu Z.; Jian Z. SIRT5 promotes cell proliferation and invasion in hepatocellular carcinoma by targeting E2F1. Mol. Med. Rep. 2018, 17 (1), 342–349. 10.1016/j.jgg.2021.11.005. [DOI] [PubMed] [Google Scholar]
- Clark O.; Yen K.; Mellinghoff I. K. Molecular pathways: Isocitrate dehydrogenase mutations in cancer. Clin. Cancer Res. 2016, 22 (8), 1837–1842. 10.1158/1078-0432.CCR-13-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L.; White D. W.; Gross S.; Bennett B. D.; Bittinger M. A.; Driggers E. M.; Fantin V. R.; Jang H. G.; Jin S.; Keenan M. C.; Marks K. M.; Prins R. M.; Ward P. S.; Yen K. E.; Liau L. M.; Rabinowitz J. D.; Cantley L. C.; Thompson C. B.; Vander Heiden M. G.; Su S. M. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462 (7274), 739–744. 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Yang H.; Liu Y.; Yang Y.; Wang P.; Kim S. H.; Ito S.; Yang C.; Wang P.; Xiao M. T.; Liu L. X.; Jiang W. Q.; Liu J.; Zhang J. Y.; Wang B.; Frye S.; Zhang Y.; Xu Y. H.; Lei Q. Y.; Guan K. L.; Zhao S. M.; Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19 (1), 17–30. 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Yang P.; Zhao X.; Jiang M.; Hu S.; Ouyang Y.; Zeng L.; Wu J. OGDH mediates the inhibition of SIRT5 on cell proliferation and migration of gastric cancer. Exp. Cell Res. 2019, 382 (2), 111483. 10.1016/j.yexcr.2019.06.028. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Li L.; Tang Y.; Xie D.; Wu K.; Wei W.; Xiao Q. CDK2 positively regulates aerobic glycolysis by suppressing SIRT5 in gastric cancer. Cancer Sci. 2018, 109 (8), 2590–2598. 10.1111/cas.13691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T.; Shukla S. K.; Vernucci E.; He C.; Wang D.; King R. J.; Jha K.; Siddhanta K.; Mullen N. J.; Attri K. S.; Murthy D.; Chaika N. V.; Thakur R.; Mulder S. E.; Pacheco C. G.; Fu X.; High R. R.; Yu F.; Lazenby A.; Steegborn C.; Lan P.; Mehla K.; Rotili D.; Chaudhary S.; Valente S.; Tafani M.; Mai A.; Auwerx J.; Verdin E.; Tuveson D.; Singh P. K. Metabolic Rewiring by Loss of Sirt5 Promotes Kras-Induced Pancreatic Cancer Progression. Gastroenterology 2021, 161 (5), 1584–1600. 10.1053/j.gastro.2021.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H. C.; Yu Y. C.; Sung Y.; Han J. M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52 (9), 1496–1516. 10.1038/s12276-020-00504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon O. K.; Bang I. H.; Choi S. Y.; Jeon J. M.; Na A.-Y.; Gao Y.; Cho S. S.; Ki S. H.; Choe Y.; Lee J. N.; Ha Y.-S.; Bae E. J.; Kwon T. G.; Park B.-H.; Lee S. SIRT5 Is the desuccinylase of LDHA as novel cancer metastatic stimulator in aggressive prostate cancer. Genomics Proteomics Bioinformatics 2022, 10.1016/j.gpb.2022.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science 1956, 123 (3191), 309–314. 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Zhan L.; Huang C.; Meng X. M.; Song Y.; Wu X. Q.; Miu C. G.; Zhan X. S.; Li J. Promising roles of mammalian E2Fs in hepatocellular carcinoma. Cell. Signal. 2014, 26 (5), 1075–1081. 10.1016/j.cellsig.2014.01.008. [DOI] [PubMed] [Google Scholar]
- Abril Y. L. N.; Fernandez I. R.; Hong J. Y.; Chiang Y. L.; Kutateladze D. A.; Zhao Q.; Yang M.; Hu J.; Sadhukhan S.; Li B.; He B.; Remick B.; Bai J. J.; Mullmann J.; Wang F.; Maymi V.; Dhawan R.; Auwerx J.; Southard T.; Cerione R. A.; Lin H.; Weiss R. S. Pharmacological and genetic perturbation establish SIRT5 as a promising target in breast cancer. Oncogene 2021, 40 (9), 1644–1658. 10.1038/s41388-020-01637-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F.; Wang X.; Ow S. H.; Chen W.; Ong W. C. Sirtuin 5 is Anti-apoptotic and Anti-oxidative in Cultured SH-EP Neuroblastoma Cells. Neurotox. Res. 2017, 31 (1), 63–76. 10.1007/s12640-016-9664-y. [DOI] [PubMed] [Google Scholar]
- He Q.; Chen K.; Ye R.; Dai N.; Guo P.; Wang L. Associations of sirtuins with clinicopathological variables and prognosis in human ovarian cancer. Oncol. Lett. 2020, 19 (4), 3278–3288. 10.3892/ol.2020.11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Wang S.; Gai J.; Guan J.; Li J.; Li Y.; Zhao J.; Zhao C.; Fu L.; Li Q. SIRT5 Promotes Cisplatin Resistance in Ovarian Cancer by Suppressing DNA Damage in a ROS-Dependent Manner via Regulation of the Nrf2/HO-1 Pathway. Front. Oncol. 2019, 9, 754. 10.3389/fonc.2019.00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Wang Z.; Li X.; Liu B.; Liu M.; Liu L.; Chen S.; Ren M.; Wang Y.; Yu M.; Wang B.; Zou J.; Zhu W. G.; Yin Y.; Gu W.; Luo J. Shmt2 desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Res. 2018, 78 (2), 372–386. 10.1158/0008-5472.CAN-17-1912. [DOI] [PubMed] [Google Scholar]
- Ducker G. S.; Rabinowitz J. D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25 (1), 27–42. 10.1016/j.cmet.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giblin W.; Bringman-Rodenbarger L.; Guo A. H.; Kumar S.; Monovich A. C.; Mostafa A. M.; Skinner M. E.; Azar M.; Mady A. S.; Chung C. H.; Kadambi N.; Melong K. A.; Lee H. J.; Zhang L.; Sajjakulnukit P.; Trefely S.; Varner E. L.; Iyer S.; Wang M.; Wilmott J. S.; Soyer H. P.; Sturm R. A.; Pritchard A. L.; Andea A. A.; Scolyer R. A.; Stark M. S.; Scott D. A.; Fullen D. R.; Bosenberg M. W.; Chandrasekaran S.; Nikolovska-Coleska Z.; Verhaegen M. E.; Snyder N. W.; Rivera M. N.; Osterman A. L.; Lyssiotis C. A.; Lombard D. B. The deacylase SIRT5 supports melanoma viability by influencing chromatin dynamics. J. Clin. Invest. 2021, 131 (12), e138926. 10.1172/JCI138926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L.; Yan H.; An S.; Shen M.; Jia W.; Zhang R.; Zhao L.; Huang G.; Liu J. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 2019, 13 (2), 358–375. 10.1002/1878-0261.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Q.; Wang H. L.; Xu J.; Tan J.; Fu L. N.; Wang J. L.; Zou T. H.; Sun D. F.; Gao Q. Y.; Chen Y. X.; Fang J. Y. Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nat. Commun. 2018, 9, 545. 10.1038/s41467-018-02951-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z.; Liu X.; Chen T.; Gao W.; Wu Z.; Hu Z.; Wei D.; Gao C.; Li Q. Targeting a Sirt5-Positive Subpopulation Overcomes Multidrug Resistance in Wild-Type Kras Colorectal Carcinomas. Cell Rep. 2018, 22 (10), 2677–2689. 10.1016/j.celrep.2018.02.037. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Qi Y.; Wang L.; Zheng Z.; Zhang Y.; Zheng J. SIRT5-mediated SDHA desuccinylation promotes clear cell renal cell carcinoma tumorigenesis. Free Radic. Biol. Med. 2019, 134, 458–467. 10.1016/j.freeradbiomed.2019.01.030. [DOI] [PubMed] [Google Scholar]
- Guan J.; Jiang X.; Gai J.; Sun X.; Zhao J.; Li J.; Li Y.; Cheng M.; Du T.; Fu L.; Li Q. Sirtuin 5 regulates the proliferation, invasion and migration of prostate cancer cells through acetyl-CoA acetyltransferase 1. J. Cell Mol. Med. 2020, 24 (23), 14039–14049. 10.1111/jcmm.16016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D.; Franzini A.; Pomicter A. D.; Halverson B. J.; Antelope O.; Mason C. C.; Ahmann J. M.; Senina A. V.; Vellore N. A.; Jones C. L.; Zabriskie M. S.; Than H.; Xiao M. J.; van Scoyk A.; Patel A. B.; Clair P. M.; Heaton W. L.; Owen S. C.; Andersen J. L.; Egbert C. M.; Reisz J. A.; D’Alessandro A.; Cox J. E.; Gantz K. C.; Redwine H. M.; Iyer S. M.; Khorashad J. S.; Rajabi N.; Olsen C. A.; O’Hare T.; Deininger M. W. SIRT5 Is a Druggable Metabolic Vulnerability in Acute Myeloid Leukemia. Blood Cancer Discovery 2021, 2 (3), 266–287. 10.1158/2643-3230.BCD-20-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabi N.; Hansen T. N.; Nielsen A. L.; Nguyen H. T.; Baek M.; Bolding J. E.; Bahlke O.; Petersen S. E. G.; Bartling C. R. O.; Strømgaard K.; Olsen C. A. Investigation of Carboxylic Acid Isosteres and Prodrugs for Inhibition of the Human SIRT5 Lysine Deacylase Enzyme. Angew. Chem., Int. Ed. 2022, 61, e202115805. 10.1002/anie.202115805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Rong F.; Tang J.; Zhu C.; Chen X.; Jia S.; Wang Z.; Sun X.; Deng H.; Zha H.; Ouyang G.; Xiao W. Repression of p53 function by SIRT5-mediated desuccinylation at Lysine 120 in response to DNA damage. Cell Death Differ. 2022, 29 (4), 722–736. 10.1038/s41418-021-00886-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbas D.; Liebscher S.; Nowak T.; Meleshin M.; Pannek M.; Popp C.; Alhalabi Z.; Bordusa F.; Sippl W.; Steegborn C.; Schutkowski M. Potent and Selective Inhibitors of Human Sirtuin 5. J. Med. Chem. 2018, 61 (6), 2460–2471. 10.1021/acs.jmedchem.7b01648. [DOI] [PubMed] [Google Scholar]
- Stevens G. J.; Hitchcock K.; Wang Y. K.; Coppola G. M.; Versace R. W.; Chin J. A.; Shapiro M.; Suwanrumpha S.; Mangold B. L. K. In Vitro Metabolism of N-(5-Chloro-2-methylphenyl)-N′-(2-methylpropyl)thiourea: Species Comparison and Identification of a Novel Thiocarbamide–Glutathione Adduct. Chem. Res. Toxicol. 1997, 10 (7), 733–741. 10.1021/tx9700230. [DOI] [PubMed] [Google Scholar]
- Neal R. A.; Halpert J. Toxicology of thiono-sulfur compounds. Annu. Rev. Pharmacol. Toxicol. 1982, 22, 321–39. 10.1146/annurev.pa.22.040182.001541. [DOI] [PubMed] [Google Scholar]
- Poulsen L. L.; Hyslop R. M.; Ziegler D. M. S-oxygenation of N-substituted thioureas catalyzed by the pig liver microsomal FAD-containing monooxygenase. Arch. Biochem. Biophys. 1979, 198 (1), 78–88. 10.1016/0003-9861(79)90397-7. [DOI] [PubMed] [Google Scholar]
- Ziegler D. M. Flavin-containing monooxygenases: Catalytic mechanism and substrate specificities. Drug Metab. Rev. 1988, 19 (1), 1–32. 10.3109/03602538809049617. [DOI] [PubMed] [Google Scholar]
- Rajabi N.; Auth M.; Troelsen K. R.; Pannek M.; Bhatt D. P.; Fontenas M.; Hirschey M. D.; Steegborn C.; Madsen A. S.; Olsen C. A. Mechanism-Based Inhibitors of the Human Sirtuin 5 Deacylase: Structure-Activity Relationship, Biostructural, and Kinetic. Insight. Angew. Chem. Int. Ed. Engl. 2017, 56 (47), 14836–14841. 10.1002/anie.201709050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Huang Y.; Zheng W. A Selective Cyclic Peptidic Human SIRT5 Inhibitor. Molecules (Basel) 2016, 21 (9), 1217. 10.3390/molecules21091217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.; Zheng W. Cyclic Tripeptide-based Potent and Selective Human SIRT5 Inhibitors. Med. Chem. 2020, 16 (3), 358–367. 10.2174/1573406415666190603101937. [DOI] [PubMed] [Google Scholar]
- Molinari F.; Feraco A.; Mirabilii S.; Saladini S.; Sansone L.; Vernucci E.; Tomaselli G.; Marzolla V.; Rotili D.; Russo M. A.; Ricciardi M. R.; Tafuri A.; Mai A.; Caprio M.; Tafani M.; Armani A. SIRT5 Inhibition Induces Brown Fat-Like Phenotype in 3T3-L1 Preadipocytes. Cells 2021, 10 (5), 1126. 10.3390/cells10051126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz A.; Min J.; Antoshenko T.; Wang C. L.; Allali-Hassani A.; Dong A.; Loppnau P.; Vedadi M.; Bochkarev A.; Sternglanz R.; Plotnikov A. N. Structural Basis of Inhibition of the Human NAD+-Dependent Deacetylase SIRT5 by Suramin. Structure 2007, 15 (3), 377–389. 10.1016/j.str.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Suenkel B.; Fischer F.; Steegborn C. Inhibition of the human deacylase Sirtuin 5 by the indole GW5074. Bioorg. Med. Chem. Lett. 2013, 23 (1), 143–146. 10.1016/j.bmcl.2012.10.136. [DOI] [PubMed] [Google Scholar]
- Trapp J.; Meier R.; Hongwiset D.; Kassack M. U.; Sippl W.; Jung M. Structure-activity studies on suramin analogues as inhibitors of NAD+-dependent histone deacetylases (sirtuins). ChemMedChem. 2007, 2 (10), 1419–1431. 10.1002/cmdc.200700003. [DOI] [PubMed] [Google Scholar]
- Maurer B.; Rumpf T.; Scharfe M.; Stolfa D. A.; Schmitt M. L.; He W.; Verdin E.; Sippl W.; Jung M. Inhibitors of the NAD(+)-dependent protein desuccinylase and demalonylase Sirt5. ACS Med. Chem. Lett. 2012, 3, 1050. 10.1021/ml3002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino M.; Rotili D.; Patsilinakos A.; Forgione M.; Tomaselli D.; Alby F.; Arimondo P. B.; Mai A.; Ragno R. Disruptor of telomeric silencing 1-like (DOT1L): disclosing a new class of non-nucleoside inhibitors by means of ligand-based and structure-based approaches. J. Comput. Aided Mol. Des. 2018, 32 (3), 435–458. 10.1007/s10822-018-0096-z. [DOI] [PubMed] [Google Scholar]
- Liu S.; Ji S.; Yu Z. J.; Wang H. L.; Cheng X.; Li W. J.; Jing L.; Yu Y.; Chen Q.; Yang L. L.; Li G. B.; Wu Y. Structure-based discovery of new selective small-molecule sirtuin 5 inhibitors. Chem. Biol. Drug Des. 2018, 91 (1), 257–268. 10.1111/cbdd.13077. [DOI] [PubMed] [Google Scholar]
- Guetschow E. D.; Kumar S.; Lombard D. B.; Kennedy R. T. Identification of sirtuin 5 inhibitors by ultrafast microchip electrophoresis using nanoliter volume samples. Anal. Bioanal. Chem. 2016, 408 (3), 721–731. 10.1007/s00216-015-9206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glas C.; Dietschreit J. C. B.; Wossner N.; Urban L.; Ghazy E.; Sippl W.; Jung M.; Ochsenfeld C.; Bracher F. Identification of the subtype-selective Sirt5 inhibitor balsalazide through systematic SAR analysis and rationalization via theoretical investigations. Eur. J. Med. Chem. 2020, 206, 112676. 10.1016/j.ejmech.2020.112676. [DOI] [PubMed] [Google Scholar]
- Lackey K.; Cory M.; Davis R.; Frye S. V.; Harris P. A.; Hunter R. N.; Jung D. K.; McDonald O. B.; McNutt R. W.; Peel M. R.; Rutkowske R. D.; Veal J. M.; Wood E. R. The discovery of potent cRaf1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2000, 10 (3), 223–226. 10.1016/S0960-894X(99)00668-X. [DOI] [PubMed] [Google Scholar]
- Trapp J.; Jochum A.; Meier R.; Saunders L.; Marshall B.; Kunick C.; Verdin E.; Goekjian P.; Sippl W.; Jung M. Adenosine mimetics as inhibitors of NAD+-dependent histone deacetylases, from kinase to sirtuin inhibition. J. Med. Chem. 2006, 49 (25), 7307–7316. 10.1021/jm060118b. [DOI] [PubMed] [Google Scholar]
- Yang F.; Su H.; Deng J.; Mou L.; Wang H.; Li R.; Dai Q. Q.; Yan Y. H.; Qian S.; Wang Z.; Li G. B.; Yang L. Discovery of new human Sirtuin 5 inhibitors by mimicking glutaryl-lysine substrates. Eur. J. Med. Chem. 2021, 225, 113803. 10.1016/j.ejmech.2021.113803. [DOI] [PubMed] [Google Scholar]
- Valente S.; Mellini P.; Spallotta F.; Carafa V.; Nebbioso A.; Polletta L.; Carnevale I.; Saladini S.; Trisciuoglio D.; Gabellini C.; Tardugno M.; Zwergel C.; Cencioni C.; Atlante S.; Moniot S.; Steegborn C.; Budriesi R.; Tafani M.; Del Bufalo D.; Altucci L.; Gaetano C.; Mai A. 1,4-Dihydropyridines Active on the SIRT1/AMPK Pathway Ameliorate Skin Repair and Mitochondrial Function and Exhibit Inhibition of Proliferation in Cancer Cells. J. Med. Chem. 2016, 59 (4), 1471–91. 10.1021/acs.jmedchem.5b01117. [DOI] [PubMed] [Google Scholar]
- Mai A.; Valente S.; Meade S.; Carafa V.; Tardugno M.; Nebbioso A.; Galmozzi A.; Mitro N.; De Fabiani E.; Altucci L.; Kazantsev A. Study of 1,4-Dihydropyridine Structural Scaffold: Discovery of Novel Sirtuin Activators and Inhibitors. J. Med. Chem. 2009, 52 (17), 5496–5504. 10.1021/jm9008289. [DOI] [PubMed] [Google Scholar]
- Spallotta F.; Cencioni C.; Straino S.; Nanni S.; Rosati J.; Artuso S.; Manni I.; Colussi C.; Piaggio G.; Martelli F.; Valente S.; Mai A.; Capogrossi M. C.; Farsetti A.; Gaetano C. A nitric oxide-dependent cross-talk between class I and III histone deacetylases accelerates skin repair. J. Biol. Chem. 2013, 288 (16), 11004–11012. 10.1074/jbc.M112.441816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S. A.; Moss A. C. Balsalazide disodium for the treatment of ulcerative colitis. Expert Rev. Gastroenterol. Hepatol. 2008, 2 (2), 177–184. 10.1586/17474124.2.2.177. [DOI] [PubMed] [Google Scholar]
- Hoffmann T.; Gastreich M. The next level in chemical space navigation: going far beyond enumerable compound libraries. Drug Discovery Today 2019, 24 (5), 1148–1156. 10.1016/j.drudis.2019.02.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Walter M.; Chen I. P.; Vallejo-Gracia A.; Kim I. J.; Bielska O.; Lam V. L.; Hayashi J. M.; Cruz A.; Shah S.; Gross J. D.; Krogan N. J.; Schilling B.; Ott M.; Verdin E., SIRT5 is a proviral factor that interacts with SARS-CoV-2 Nsp14 protein. bioRxiv (Microbiology) January 5, 2022, 474979. 10.1101/2022.01.04.474979 [DOI] [PMC free article] [PubMed]