

Abstract
Purpose:
To characterise the genotypic and phenotypic spectrum of foveal hypoplasia (FH)
Design:
Multi-centre, observational study
Subjects:
907 patients with a confirmed molecular diagnosis of albinism, PAX6, SLC38A8, FRMD7, AHR or achromatopsia (ACHM) from twelve centres in nine countries (n=523), or, extracted from publicly available datasets from previously reported literature (n=384).
Methods:
Individuals with a confirmed molecular diagnosis and availability of foveal optical coherence tomography (OCT) scans were identified from twelve centres or from the literature, between January 2011 and March 2021. A genetic diagnosis was confirmed by sequence analysis. Grading of FH was derived from OCTs.
Main outcome measures:
Grade of FH, presence or absence of photoreceptor specialisation (PRS+ vs PRS−), molecular diagnosis and visual acuity (VA).
Results:
The most common genetic etiology for typical FH in our cohort was albinism (67.5%), followed by PAX6 (21.8%), SLC38A8 (6.8%) and FRMD7 (3.5%) variants. AHR variants were rare (0.4%). Atypical FH was seen in 67.4% of ACHM cases. Atypical FH in ACHM had significantly worse VA compared to typical FH (p<0.0001). There was a significant difference in the spectrum of FH grades based on the molecular diagnosis (X2=60.4, p<0.0001). All SLC38A8 cases were PRS− (p=0.003), while all FRMD7 cases were PRS+ (p<0.0001). Analysis of albinism sub-types revealed a significant difference in the grade of FH (X2=31.4, p<0.0001) and VA (p=0.0003) between oculocutaneous albinism (OCA) compared to ocular albinism (OA) and Hermansky-Pudlak syndrome (HPS). OA and HPS demonstrated higher grades of FH and worse VA than OCA. There was a significant difference (p<0.0001) in VA between FRMD7 variants compared to other diagnoses associated with FH.
Conclusion:
We characterised the phenotypic and genotypic spectrum of FH. Atypical FH is associated with much worse prognosis compared to all other forms of FH. In typical FH, our data suggests that arrested retinal development occurs earlier in SLC38A8, OA, HPS and AHR variants and much later in FRMD7 variants. The defined time-period of foveal developmental arrest for OCA and PAX6 variants appears to demonstrate more variability. Our findings provide mechanistic insight into disorders associated with FH and also have significant prognostic and diagnostic value.
Keywords: foveal hypoplasia, optical coherence tomography, genetics, retinal development, genotype-phenotype correlation
Précis
We observe distinctive patterns of arrested foveal development based on genotype and highlight the mechanistic implications with direct clinical relevance to diagnosis and prognosis. This helps prioritise genetic testing, subsequent counselling, and support.
Introduction
The normal foveal anatomy consists of the extrusion of the plexiform layers, a formed foveal pit, cone photoreceptor outer segment (OS) lengthening and outer nuclear layer (ONL) widening relative to the parafoveal OS and ONL.1 Foveal development begins with the centrifugal displacement of the inner retinal layers, followed by the centripetal migration of the cone photoreceptors towards the incipient fovea and finally cone photoreceptor specialisation (figure 1A–E).2–4 Foveal pit formation begins mid-gestation and continues postnatally characterised by pit deepening and widening. Simultaneously, outer retinal specialisation occur at a rapid rate in the first two years of life but ONL widening continues up to the age of 13 years.4 Lengthening of the OS layer represents cone photoreceptor specialisation and reflects peak cone photoreceptor density.5 The maturation of the human fovea is a complex process and is critical for visual function.4 Many genes are involved in the development of the retina. Pathogenic variants in these genes often cause disruption to the foveal developmental process, resulting in foveal hypoplasia (FH), which describes the underdevelopment of the fovea.1
Figure 1:
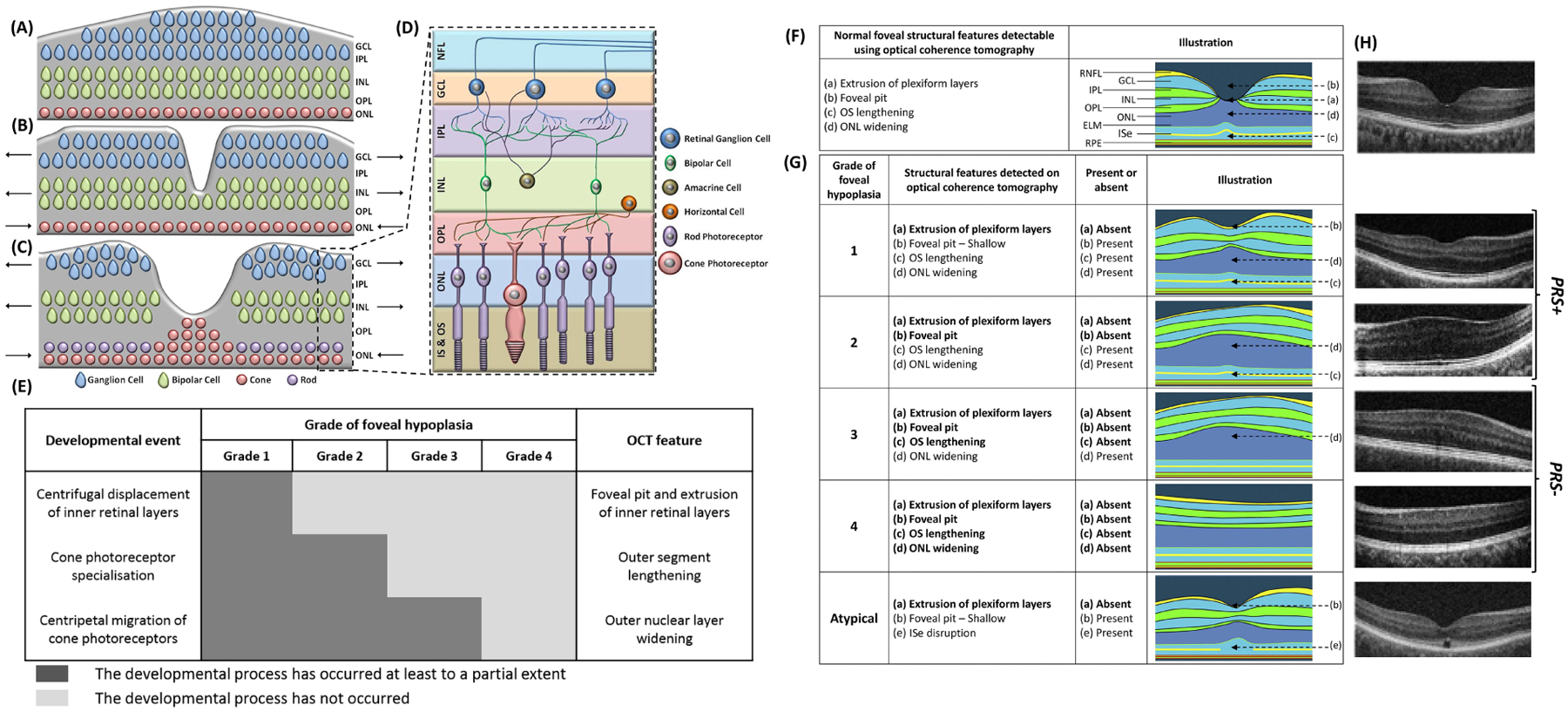
Foveal pit formation and movement of retinal cells during formation of the area of high acuity. The laminar retinal structure prior to foveal pit formation is shown in (A). The inner retinal layers were displaced centrifugally (away from the future fovea) during foveal pit formation (B). The cone photoreceptors migrate centripetally (towards the fovea) and form the pure cone area (C). Arrows point in the direction of movement of the cellular layers. The magnified laminar structure (D) with the different retinal cell types and the inner segment & outer segment (IS & OS) of the photoreceptors are shown in (D). NFL = Nerve Fibre Layer; GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer; ONL = outer nuclear layer. (A, B, C are based on developmental theory proposed by Springer and Hendrickson).39 Chart showing the 3 developmental processes involved in formation of a structural and functional fovea. In grade 1 foveal hypoplasia, all processes occur to a certain extent. However, in grade 4 foveal hypoplasia, none of these processes occur; thus, the retina resembles that of the parafovea. In grade 2 and 3 foveal hypoplasia, there is outer nuclear layer widening, but no foveal pit. The difference between grade 2 and 3 foveal hypoplasia is occurrence of cone photoreceptor specialization. Identifying these specific features on optical coherence tomography (OCT) enables us to understand whether the respective developmental process has occurred. F, Illustration showing the unique features of a normal fovea detectable on optical coherence tomography. G, Illustration of typical and atypical grades of foveal hypoplasia. All grades of foveal hypoplasia had incursion of inner retinal layers. Atypical foveal hypoplasia also had incursion of the inner retinal layers. Grade 1 foveal hypoplasia is associated with a shallow foveal pit, outer nuclear layer (ONL) widening, and outer segment (OS) lengthening relative to the parafoveal ONL and OS length, respectively. In Grade 2 foveal hypoplasia, all features of grade 1 are present except the presence of a foveal pit. Grade 3 foveal hypoplasia consists of all features of grade 2 foveal hypoplasia except the widening of the cone outer segment. Grade 4 foveal hypoplasia represents all the features seen in grade 3 except there is no widening of the ONL at the fovea. Finally, an atypical form of foveal hypoplasia also is described in which there is a shallower pit with disruption of the inner segment ellipsoid (ISe). (Adapted with permission from Thomas et al. 2011).1 H, original OCTs demonstrating the different grades of foveal hypoplasia. Grades 1 and 2 can be considered to show signs of photoreceptor specialisation (PRS+), however grades 3 and 4 do not show signs of photoreceptor specialisation (PRS−). ELM = external limiting membrane; GCL = ganglion cell layer; INL = inner nuclear layer; IPL = inner plexiform layer; OPL = outer plexiform layer; RNFL = retinal nerve fibre layer; RPE = retinal pigment epithelium.
FH is characterised by the continuation of inner retinal layers posterior to the foveola, and progressive loss of these foveal elements is represented by increasing grades of FH. Three key foveal development stages form the basis of The Leicester Grading System for FH, developed by Thomas et al. (2011) (figure F–H).1 The Leicester Grading System is divided into four grades of typical FH (grades 1–4) and one grade of atypical FH, which is associated with photoreceptor degeneration.
The Leicester Grading System can be applied to a diverse range of genetic disorders including albinism, achromatopsia and those caused by pathogenic variants in PAX6, SLC38A8, FRMD7 and AHR. In addition to providing insight into the degree of foveal development, identifying the grade of FH has diagnostic and prognostic implications.1,6 Recently, Rufai et al. (2020) reported that identifying the grade of FH can predict future visual acuity (VA) in preverbal children with nystagmus.6
The introduction of optical coherence tomography (OCT) has revolutionised ophthalmic diagnosis, with the ability to promptly visualise retinal morphology in a high-resolution, non-invasive manner.7,8 Furthermore, the advent of hand-held OCT has facilitated the foveal examination of paediatric patients.9 OCT is now available in most ophthalmology clinics, and subsequently has rapidly become part of routine ophthalmic assessment.7 Thus, the necessary technology to identify foveal developmental abnormalities is now accessible in most centres.
The phenotypic spectrum of albinism has previously been reported, including the variance in degree of arrested retinal development.10,11 Furthermore, high grades of FH (grades 3 and 4) have recently been consistently associated with SLC38A8 variants.12 However, to date, the full spectrum of FH linked with variants of known genes involved in retinal development, has not yet been investigated. Thus, it is unclear whether variants of certain genes known to be related to FH and nystagmus, are associated with a more underdeveloped foveal morphology. We performed a comparative study to investigate and characterise the genotypic and phenotypic spectrum of FH in albinism, achromatopsia, PAX6, SLC38A8, FRMD7 and AHR variants.
Methods
In this multi-centre study, we collated genotypic and phenotypic data for 907 patients from twelve centres in nine countries, from a cohort of patients with infantile nystagmus and/or FH over a 10-year period (2011–2021). We set up an international consortium with a special interest in foveal developmental disorders, composed of paediatric ophthalmologists, neuro-ophthalmologists, clinical geneticists, data scientists and statistical geneticists. Data was collected by our Foveal Development Investigators Group (FDIG). Inclusion criteria was defined as: (1) a molecular diagnosis and (2) report of foveal morphology on OCT. Genes known to be associated with typical FH (table 1) were selected; PAX6, SLC38A8, FRMD7, AHR and genes underlying both syndromic and non-syndromic forms of albinism (TYR, OCA2, TYRP1, SLC45A2, SLC24A5, LRMDA (C10orf11), GPR143, HPS (1–11), LYST). An overview of the phenotypical characteristics associated with each of these disorders is included in table 1. We also included the achromatopsia genes (CNGB3, CNGA3, GNAT2, PDE6C, PDE6H and ATF6) associated with atypical FH (table 1). Previously reported cases that met the inclusion criteria were additionally identified and collected from the literature between 2011–2021. We included all patients with foveal morphology documented on OCT and pathogenic variants in any one of the genes listed, irrespective of whether they had FH or normal foveal morphology. This study was approved by the local ethics committee and adhered to the tenets of Declaration of Helsinki.
Table 1:
List of genes associated with typical and atypical foveal hypoplasia with genes reported in this study highlighted in bold
| Condition | Clinical Phenotype | Gene | Location | MIM Gene ID | Phenotype title | MIM phenotype ID | Inheritance |
|---|---|---|---|---|---|---|---|
| Oculocutaneous Albinism (OCA) | A clinically and genetically heterogenous disorder. Characterised by pigmentation defects of the hair, skin, and eyes. Foveal hypoplasia, chiasmal misrouting, infantile nystagmus, iris transillumination defects, fundus hypopigmentation. | TYR | 11q14.3 | 606933 | OCA1 | 203100, 606952 | AR |
| OCA2 | 15q12-q13 | 611409 | OCA2 | 203200 | AR | ||
| TYRP1 | 9p23 | 115501 | OCA3 | 203290 | AR | ||
| SLC45A2 | 5p13.2 | 606202 | OCA4 | 606574 | AR | ||
| SLC24A5 | 15q21.1 | 609802 | OCA6 | 113750 | AR | ||
| LRMDA (C10orf11) | 10q22.2-q22.3 | 614737 | OCA7 | 615179 | AR | ||
| Ocular Albinism (OA) | Shares the same clinical characteristics as OCA, however with pigmentation defects generally limited to the eyes | GPR143 | Xp22.2 | 300808 | OA1 | 300500 | XL |
| Hermansky-Pudlak Syndrome (HPS) | A syndromic form of albinism demonstrating the same clinical characteristics as OCA, in addition to blood platelet dysfunction with prolonged bleeding | HPS1 | 10q24.2 | 604982 | HPS1 | 203300 | AR |
| AP3B1 | 5q14.1 | 603401 | HPS2 | 608233 | AR | ||
| HPS3 | 3q24 | 606118 | HPS3 | 614072 | AR | ||
| HPS4 | 22q12.1 | 606682 | HPS4 | 614073 | AR | ||
| HPS5 | 11p15.1 | 607521 | HPS5 | 614074 | AR | ||
| HPS6 | 10q24.32 | 607522 | HPS6 | 614075 | AR | ||
| DTNBP1 | 6p22.3 | 607145 | HPS7 | 614076 | AR | ||
| BLOC1S3 | 19q13.32 | 609762 | HPS8 | 614077 | AR | ||
| BLOC1S6 | 15q21.1 | 604310 | HPS9 | 614171 | AR | ||
| AP3D1 | 19p13.3 | 607246 | HPS10 | 617050 | AR | ||
| BLOC1S5 | 6p24.3 | 607289 | HPS11 | 619172 | AR | ||
| Chediak-Higashi Syndrome (CHS) | A syndromic form of albinism demonstrating the same clinical characteristics of OCA, in addition to immune deficiency and ability to bruise and bleed easily | LYST | 1q42.3 | 606897 | CHS1 | 214500 | AR |
| FHONDA | Foveal hypoplasia, chiasmal misrouting, infantile nystagmus, and anterior segment dysgenesis in some cases (minor association) | SLC38A8 | 16q23.3 | 615585 | FHONDA, FVH2 | 609218 | AR |
| Aniridia | A pan-ocular condition which can cause corneal and lens abnormalities, iris abnormalities (aniridia), raised intraocular pressure, foveal hypoplasia, infantile nystagmus and optic nerve abnormalities | PAX6 | 11p13 | 607108 | AN1, FVH1 | 106210, 136520 | AD |
| FRMD7-related infantile nystagmus | Associated with idiopathic infantile nystagmus Most commonly associated with normal foveal morphology. Rare association with foveal hypoplasia. | FRMD7 | Xq26.2 | 300628 | NYS1 | 310700 | XL |
| AHR-related foveal hypoplasia and infantile nystagmus | A recently reported condition characterised by foveal hypoplasia and infantile nystagmus (only two cases reported in the literature, from the same family) | AHR | 7p21.1 | 600253 | * | - | AR |
| Achromatopsia | Characterised by cone photoreceptor dysfunction, reduced vision and infantile nystagmus. Known to be associated with atypical foveal hypoplasia and inner segment ellipsoid disruption | CNGB3 | 8q21.3 | 605080 | ACHM3 | 262300 | AR |
| CNGA3 | 2q11.2 | 600053 | ACHM2 | 216900 | AR | ||
| GNAT2 | 1p13.3 | 139340 | ACHM4 | 613856 | AR | ||
| PDE6C | 10q23.33 | 600827 | ACHM5 | 613093 | AR | ||
| PDE6H | 12p12.3 | 601190 | ACHM6 | 610024 | AR | ||
| ATF6 | 1q23.3 | 616517 | ACHM7 | 605537 | AR |
Grading of foveal hypoplasia
FH was graded using the Leicester Grading System for FH (figure 1F,G).1 Typical FH was diagnosed as Grade 1 if OCT revealed incomplete extrusion of the inner retinal layers posterior to the foveola, the presence of a foveal pit (irrespective of depth), lengthening of the OS layer and widening of the ONL. FH was diagnosed as grade 2 if all features of grade 1 were present, except there was no longer a foveal pit. FH was diagnosed as grade 3 FH if all features of grade 2 FH were present except there was no lengthening of the OS layer. FH was diagnosed as grade 4 FH if all features of grade 3 were present except there was no ONL widening (mimicking the appearance of the peripheral retina).1 Grades 1 and 2 together can be considered to show evidence of cone photoreceptor specialisation (PRS+), whilst in grades 3 and 4 there is no evidence of cone photoreceptor specialisation (PRS−).1
Atypical FH is associated with photoreceptor degeneration and was diagnosed if OCT revealed the disruption of the inner segment ellipsoid associated with continuation of the inner retinal layers posterior to the foveola.1 Foveal scans were interpreted and graded by experienced clinicians within the FDIG.
Multi-Centre Data Collection
Patients with a confirmed molecular diagnosis and a report of foveal morphology were identified between Jan-2011 and March-2021 from twelve different sites in nine countries. Foveal scans were captured with OCT using site-specific protocols previously described (supplementary table 1).10,12,13 Patient demographic and basic ophthalmic examination findings were included, where available. Informed consent was obtained from all involved participants. Ethical approval was received from the research ethics committee (REC references: (1) 20/EM/0040, (2) 31499, (3) 10/H0406/74, (4) 12/EM/0261).
To perform genetic analysis, genomic DNA was extracted from saliva samples or peripheral blood samples, dependent on protocols at each specific site. DNA was extracted as per manufacturer guidelines. Targeted next generation sequencing (NGS) panels,10,14,15 whole exome sequencing,16 whole genome sequencing,16,17 or Sanger sequencing10,11,18,19 was then performed. Individuals with a genetic diagnosis (including those with missing heritability i.e., just one pathogenic variant) in the appropriate clinical context, were included. Details of genetic analysis protocols have previously been published.10,14,17 Briefly, our NGS analysis protocol consisted of variant calling using the GATK pipeline (https://gatk.broadinstitute.org/) and further annotation using ANNOVAR (http://www.annovar.openbioinformatics.org/).
Data Extraction from Literature Review
Data was extracted from the literature by five experienced clinicians in order to identify previous reports of cases and cohorts of individuals with a diagnosis and foveal OCT. Literature searches were carried out using the databases PubMed, Medline and Scopus. We applied a filter to search published literature between January-2011 and March-2021. FH grading was introduced in 2011 hence we did not include literature prior to this.
Search terms included “optical coherence tomography” or “foveal hypoplasia” plus the diagnosis in question (e.g., “albinism”, “PAX6”). To broaden our search strategy, we also included search terms “aniridia”, “infantile nystagmus” and the abbreviations for ocular albinism (“OA”) and oculocutaneous albinism (“OCA”). Syndromic forms of albinism; Hermansky-Pudlak syndrome (HPS), Chediak Higashi syndrome (CHS) and Griscelli syndrome (GS) were also searched for. An overview for the literature search approach can be found in supplementary figure 1. Further details of the cohort extracted from the literature is shown in supplementary table 2.
In cases where a foveal tomogram was provided but no formal comment of foveal morphology was made, an experienced clinician would interpret and grade the tomogram, using the Leicester Grading System.1 Basic demographic and clinical characteristics were also collected, where available. VA scores or fractions were converted to logarithm of minimum angle of resolution (logMAR).
Individuals reported in publications authored by a collaborator site were omitted from the ‘literature cases’ to avoid repeated data. For the purpose of reporting results, data from study sites and the literature was combined, unless otherwise specified.
Statistical Analysis
Statistical analysis was carried out using SPSS software (IBM Corp. Released 2019. IBM SPSS Statistics for Windows, Version 26.0. Armonk, NY: IBM Corp.). An average of right and left VA measurements was calculated and used for statistical analysis. Normality testing of the VA distribution was carried out with the Shapiro-Wilk test.
The Kruskall-Wallis test was used to test for statistical differences between average VA measurements across different diagnostic groups, grades of FH, and to assess for statistical differences in average VA between the albinism subgroups; OCA, OA and HPS. To test whether cone photoreceptor specialisation was affected (PRS+ versus PRS−) we performed the Pearson chi-squared test. This allowed us to investigate whether there was a difference in the proportion of cases with and without cone photoreceptor specialisation (PRS+ versus PRS−) based on the genotype. We performed a sub-analysis (Pearson chi-squared test) within the albinism group dividing the cohort into OCA, OA and HPS. Post-hoc analysis was performed as previously described,20 and we report the adjusted residuals (adjusted z-scores) with adjusted p-values (Bonferroni correction) to control for a Type 1 error.
All analyses were considered statistically significant when a probability value of p ≤ 0.05 was identified.
Results
Overview of the cohort
This multi-centre study identified a total of 907 suitable individuals with genotypic and phenotypic data, from study sites (57.7%) and from the literature (42.3%). The mean age of the cohort was 22.7 years (SD=16.7 years), with a higher proportion of males (53.6%) compared to females (46.4%). The age and gender breakdown per diagnostic group is shown in the supplementary figure 2. In the achromatopsia cohort (n=310), atypical FH was observed in 67.4% of cases (figure 2A). Among individuals with variants in genes linked to typical FH (n=597), we observed typical FH in 81.6% (figure 2A).
Figure 2:
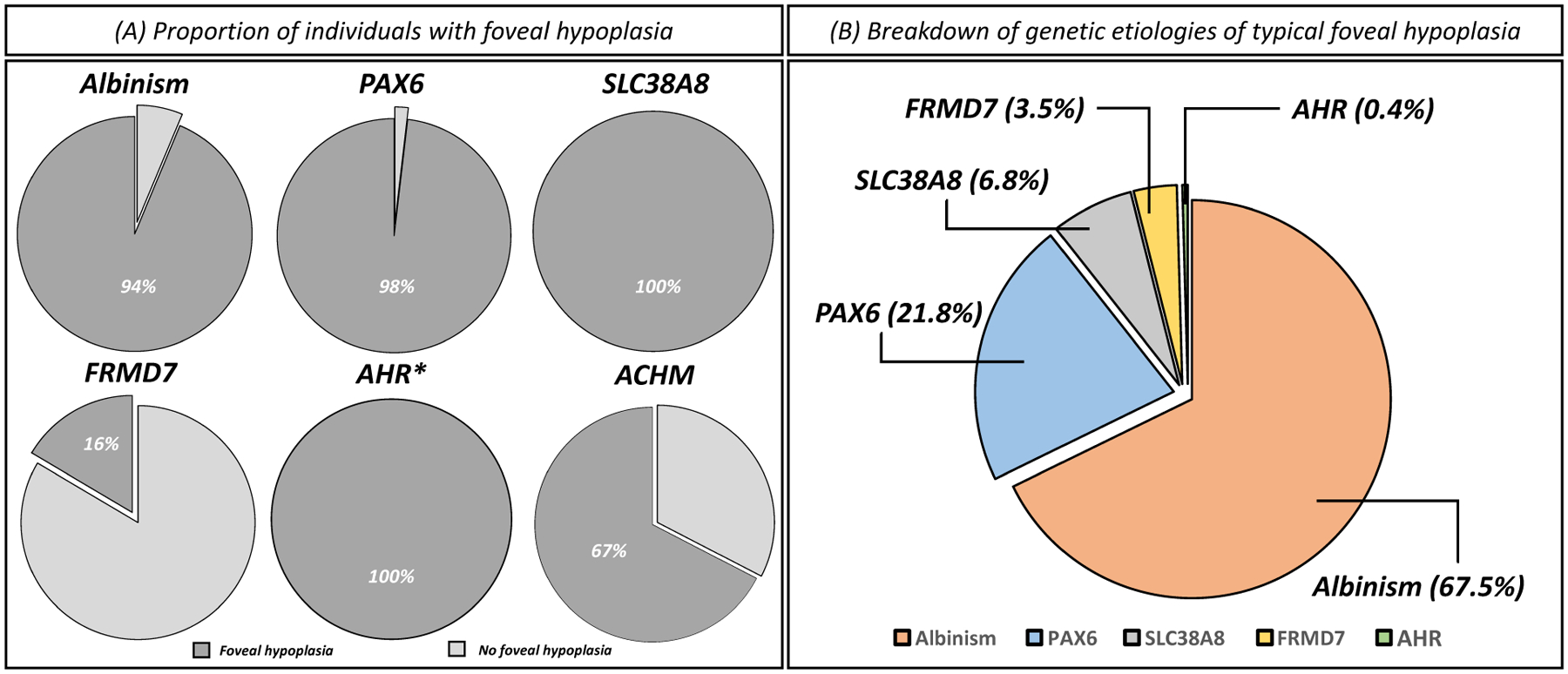
(A) Proportion of individuals with foveal hypoplasia within each genetic diagnosis. (B) Breakdown of genetic etiologies causative of typical foveal hypoplasia. *Only two cases of AHR variants with foveal hypoplasia were identified.
Comparison of VA between typical foveal hypoplasia, atypical foveal hypoplasia and normal foveal morphology
Atypical FH had a significantly worse VA (p<0.0001) compared to the typical FH group and the normal foveal morphology group (figure 3A). Further analysis with typical FH split into each grade of FH revealed significant (p<0.05) differences in VA (figure 3B) for all pairwise comparisons except between grade 2 and 3 FH.
Figure 3:
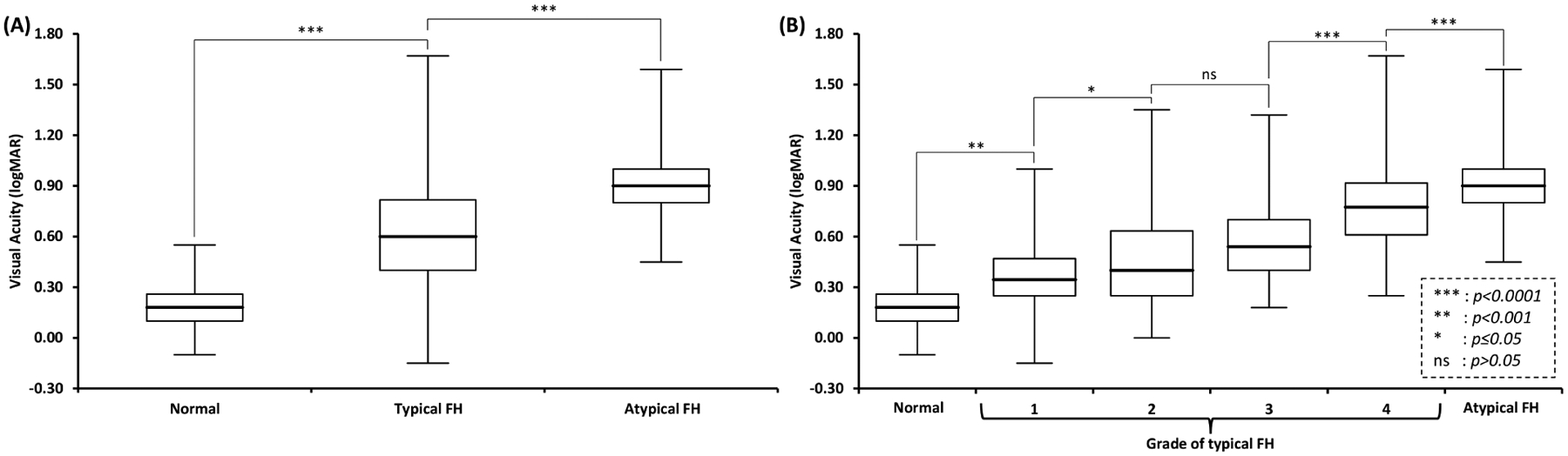
(A) Box and whisker plots of visual acuity in individuals with normal foveal morphology compared to typical foveal hypoplasia (FH) and atypical FH. Pairwise comparisons show significant differences between the three groups. (B) Pairwise comparisons with typical FH split into individual grades (1–4) shows significant differences across all groups except between grade 2 and 3 FH.
Typical FH: genetic etiologies, photoreceptor specialisation and visual acuity
Genetic etiologies
The breakdown of genetic etiologies of typical FH (n=487) included: albinism (67.5%), PAX6 variants (21.8%), SLC38A8 variants (6.8%), FRMD7 variants (3.5%) and AHR variants (0.4%) (figure 2B). Grade 4 FH was the most frequently reported grade of FH in this study (43.1%), and grade 2 was the least prevalent (14.0%) (figure 4). An overview of the spectrum of typical FH OCT tomograms associated with each diagnosis is shown in figure 4. The genotypic spectrum for the cases of typical FH is shown in supplementary table 3.
Figure 4:
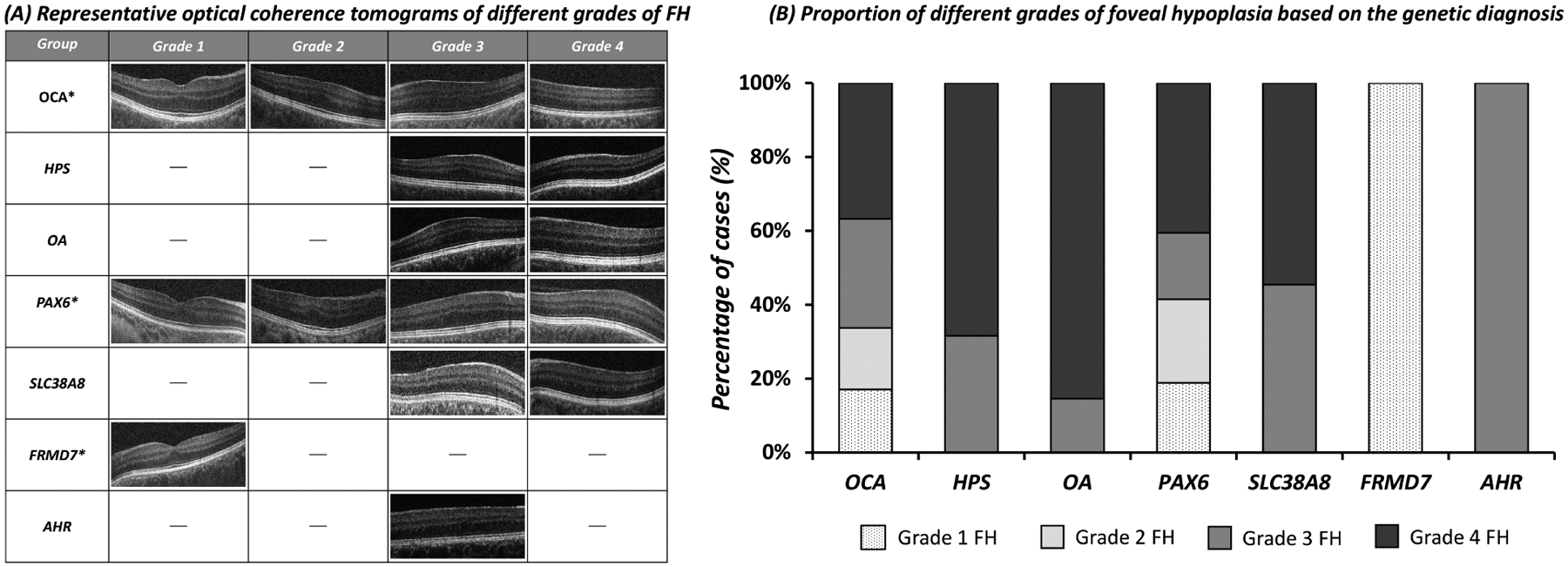
(A) Representative tomograms of typical foveal hypoplasia in different genetic etiologies. (B) The relative proportions of each grade of foveal hypoplasia within each genetic diagnosis is shown in this bar chart. Non-syndromic oculocutaneous albinism (OCA) and PAX6 variants had the full spectrum of foveal hypoplasia. However, SLC38A8 variants, GPR143 variants associated with ocular albinism (OA) and variants associated with Hermansky Pudlak Syndrome (HPS) only had grade 3 and grade 4 foveal hypoplasia. FRMD7 only had grade 1 foveal hypoplasia. AHR only had grade 3 foveal hypoplasia. *Albinism, PAX6 and FRMD7 can also have normal foveal morphology.
Photoreceptor specialisation
There was a significant difference in the grade of FH (PRS+ versus PRS−) between the genetic etiologies (X2 = 60.4, p < 0.0001). All SLC38A8 variants were PRS− cases (adjusted z-score = 4.0, p=0.003). In contrast, all FRMD7 variants were PRS+ (adjusted z-score = 6.3, p < 0.0001). Similarly, sub-analysis for albinism showed a significant difference in the grade of FH (PRS+ versus PRS−) between the albinism subtypes (X2 = 31.4, p < 0.0001). Post-hoc analysis showed that HPS (adjusted z-score = 2.7, p=0.0065) and OA (adjusted z-score = 4.5, p < 0.0001) were associated with only PRS− cases. However, OCA had a spectrum of both PRS+ and PRS− cases (adjusted z-score = 5.6, p < 0.0001).
Visual acuity
There was a significant difference in VA between genetic etiologies associated with typical FH (H(3)=21.3, p < 0.0001) (figure 5). Multiple comparisons revealed that this was due to significant differences in VA between FRMD7 and albinism (median difference = 0.30 logMAR, p = 0.003), PAX6 (median difference = 0.40 logMAR, p = 0.0004) and SLC38A8 (median difference = 0.31 logMAR, p = 0.004) (figure 5A). The FRMD7 group demonstrated the best VA whilst the poorest median VA was associated with PAX6 variants. Similarly, there was a significant difference in VA between the albinism subtypes (H(2)=21.8, p < 0.0001). Multiple comparisons revealed that this was due to significantly better VA in OCA compared to OA (median difference = 0.14 logMAR, p = 0.008) and HPS (median difference = 0.28 logMAR, p = 0.006) (figure 5B). Sub-analysis of data only from study sites (i.e., excluding cases from the literature) showed similar results for both cone photoreceptor specialisation and VA with genetic etiology (supplementary table 4).
Figure 5:
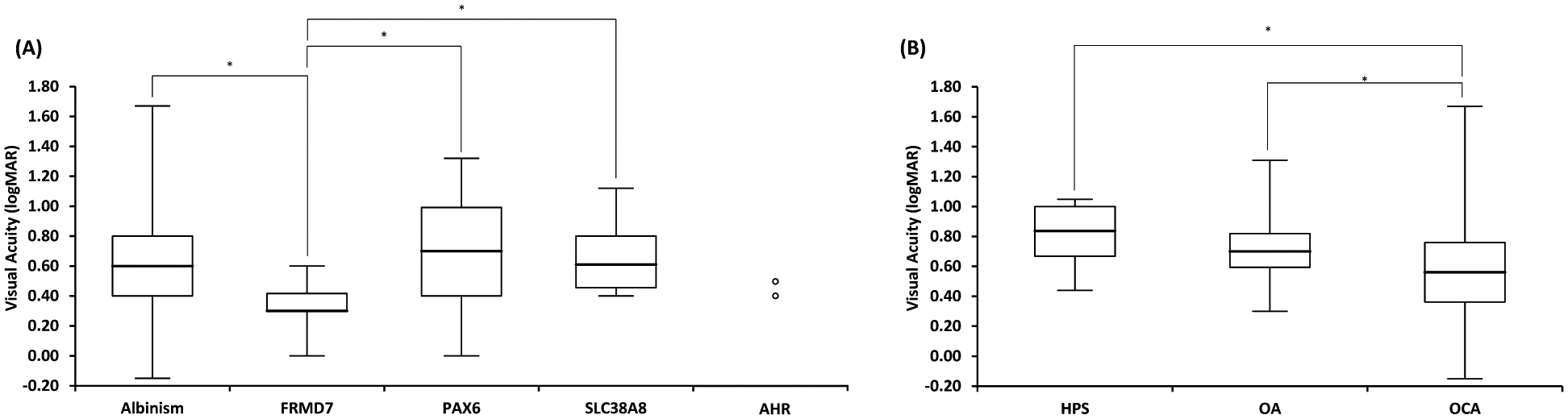
(A) Box and whisker plots of visual acuity in each genetic etiology associated with foveal hypoplasia. Only individuals with foveal hypoplasia from each diagnostic group were included. (B) Box and whisker plots of visual acuity for albinism subtypes: Non-syndromic Oculocutaneous Albinism (OCA), Hermansky Pudlak Syndrome (HPS) and Ocular Albinism (OA) due to GPR143 variants. Box represents the interquartile range, line through the box represents the median and extent of whiskers represents the range. Significant comparisons (p≤0.05) are shown with *.
Discussion
This multi-centre observational study represents the largest cohort of patients with FH providing significant insight into retinal development and visual prognosis based on a molecular diagnosis. We identified that albinism and PAX6 variants are associated with a wide spectrum of arrested retinal development (grade 1–4 FH). Interestingly within the albinism group, OA and HPS had higher grades of FH (grade 3 and 4 FH), whilst a spectrum of FH (grade 1–4 FH) was observed in OCA. A narrow spectrum of FH was identified in SLC38A8 variants (grade 3–4 FH) and FRMD7 variants (grade 1 FH). Only two patients with AHR variants were reported in the literature, both with grade 3 FH, thus more data on AHR variants must be analysed to determine any association with degree of arrested retinal development. Consistent with the grade of FH, the FRMD7 cohort had the best VA. Taken together, this highlights the central role of determining foveal morphology in patients due to its strong correlation to genetics and visual prognosis. Moreover, in scenarios where phenotypic data is lacking (for example an uncooperative patient), but genotype is available, our data can guide clinicians in providing visual prognosis based on the correlations we describe in this study.14,16 There is a paucity of studies systematically comparing the different genetic etiologies associated with FH.1,4 Previously this has been limited to comparatively smaller cohorts or without a molecular diagnosis.1,4 Consistent with previous studies characterising foveal development in albinism or PAX6 variants,1,10,22 we highlight a spectrum of FH seen in both of these genetic conditions. This spectrum of FH is indicative of a variable timeframe of foveal developmental arrest in albinism and PAX6 variants. On the contrary, SLC38A8 and AHR variants were consistently associated with high grades of FH (grade 3 and 4),12,21 thus we hypothesize that arrested retinal development occurs at a more defined time period, earlier in development.12 FRMD7 variants were associated with grade 1 FH or normal foveal morphology,23 therefore likely indicative of retinal developmental arrest at a much later, defined time-point.
High grades of FH (grade 3 and 4) were consistently associated with OA and HPS. In contrast, OCA demonstrated a spectrum of FH (grade 1–4 FH). Genes involved in non-syndromic OCA are generally enzymes or ion channels/exchangers.24 Previous in vitro studies have shown that hypomorphic variants in TYR can exhibit reduced enzymatic activity of tyrosine hydroxylase and DOPA oxidase.25 Thus, the FH spectrum seen in OCA could be attributed to the variable enzymatic function. In the presence of residual enzyme activity partial pigmentation maybe present as previously described in OCA1B patients.26 Conversely, HPS genes encode proteins that regulate intracellular vesicle trafficking, whilst OA is caused by variants in GPR143, which is an intracellular G-protein coupled receptor.24 Both GPR143 and HPS genes are considered to be crucial for melanosome biogenesis. Previous in vitro studies show that deletions and nonsense GPR143 variants produce either no protein or rapidly degraded truncated proteins. Similarly, the majority of GPR143 missense variants cause significant protein misfolding with inability to exit the endoplasmic reticulum, thus these variants are thought to have a similar pathogenesis to the large deletion and splice pathogenic variants.27,28 We hypothesize that this cellular phenotype and impact on melanosome biogenesis translates to a more severe clinical phenotype with earlier arrested retinal development and worse visual prognosis.
The proportion of atypical FH in the achromatopsia group is similar to previous studies, with Thomas et al (2011) previously identifying 69% of achromatopsia individuals with atypical FH.29 Interestingly earlier work has shown that the foveal avascular zone is intact in achromatopsia30 This implies that normal cone photoreceptor development and function is an important process for structural development of the fovea. The outer retinal changes in achromatopsia can be progressive based on longitudinal studies.31 However, further studies are needed to understand the relationship between cone photoreceptor dysfunction and foveal development.
We explored whether there was a difference in VA based on a molecular diagnosis. Albinism, SLC38A8 and PAX6 variants had similar median VA’s (0.60–0.70 logMAR), whereas FRMD7 had significantly better median VA of 0.30 logMAR. FRMD7 variants were consistently associated with grade 1 FH or normal foveal morphology, thus demonstrating OS lengthening. OS lengthening is a surrogate marker for peak foveal cone density.5 Thus, the OS lengthening in FRMD7 indicates a more tightly packed cone photoreceptor mosaic with foveal specialisation compared to other genetic etiologies, thus resulting in good visual prognosis. This is consistent with previous literature describing better median VA in FRMD7 compared to albinism.32 Albinism, PAX6 and SLC38A8 had worse VA’s which could be due to the predominantly higher grades of FH (grade 3 and 4 FH) observed, this represents a lack of OS lengthening (or PRS−).12 Although the median VA for albinism and PAX6 group were 0.60 logMAR and 0.70 logMAR respectively, a large distribution of VA was observed, thus reflecting the spectrum of FH grade (grade 1–4 FH) associated with both conditions. Furthermore, PAX6 variants are associated with pan-ocular phenotypes and significant phenotypic heterogeneity, thus likely providing a further explanation for the variance in VA.4,33 In the achromatopsia group, individuals with atypical FH demonstrated worse VA compared to those with typical FH or normal foveal morphology. This is consistent with our previous reports and is likely due to the cone photoreceptor dysfunction.1 The grade of FH has been recognised to facilitate the prediction of future VA in preverbal children.1,6 Identifying the grade of FH therefore provides significant prognostic value.6
A limitation of our study was the exclusion of data from individuals without a molecular diagnosis, but with a report of foveal morphology on OCT. Individuals with deep intronic variants or novel variants therefore may have been missed from our analysis. Similarly, data extracted from the literature represents a subset of patients which met the inclusion criteria therefore may not be representative of the entire cohorts reported within each study. Whilst every effort was made to record BCVA, it is possible that in PAX6 the presence of panocular phenotypes (e.g., cataracts and anterior segment dysgenesis) could also be contributory factors to the reduced VA, in addition to the presence of FH. Similarly, nystagmus can be variable between cases, and this could also be contributary to reduced VA. Furthermore, it is possible that our cohort has some selection bias as we included patients that had FH as determined by an OCT, thus potentially excluding patients with media opacities, such as cataracts and associated keratopathy. The inclusion of data obtained prior to the introduction of the Leicester Grading System in 2011 posed a challenge, due to the inconsistency with reported degree of arrested retinal development, attributed to the lack of consensus on a suitable grading system for foveal underdevelopment.1,34–37. Moreover, most reports prior to 2011 utilised time-domain OCT with limited spatial resolution for sufficient FH grading. We therefore only included data from 2011 onwards, to ensure the standardised grading of FH, by using the same classification system across all disorders.1 Wilk et al. (2014) proposed the subdivision of grade 1 FH into grade 1a (extrusion of plexiform layers but with a nearly normal pit) and 1b (extrusion of plexiform layers with a shallow pit),38 which has since been included as part of the Leicester Grading System.6 However, due to its recent introduction, many centres and subsequent publications are yet to adopt the new subclassification, therefore we categorised grade of FH only as grade 1–4 to maintain consistency with grading. Often in clinical environments, patients may be identified for analysis due to an obvious phenotype, for example, those demonstrating nystagmus. This may give rise to selection bias, where more severe grades of FH have been overrepresented in our cohort. Our study design included only qualitative reports of foveal morphology and genotype. Further studies to perform quantitative analysis across the retinal developmental disorders are required, to investigate the potentially more subtle relationships with foveal morphology, function and genotype. Furthermore, our study looked at genotype on a gene level only, therefore future work investigating the genotype-phenotype relationships on a variant level is required.
In summary, this multi-centre collaborative study has utilised the largest dataset so far, to reveal genotypic correlations with foveal development and its consequence to vision. This provides mechanistic insight into how genes involved in foveal development interact at different temporal points resulting in varying degrees of arrested retinal development. Variants of SLC38A8, GPR143, AHR and genes involved in HPS are associated with high grades of FH and poor cone photoreceptor specialisation which translates to earlier foveal developmental arrest and worse visual prognosis. This has significant diagnostic and prognostic value and can help with prioritisation of genetic testing, subsequent counselling, and support.
Supplementary Material
Supplementary figure 1: An overview of the literature search strategy to collect previously published data of individuals with a confirmed molecular diagnosis and report on foveal morphology.
Supplementary figure 2: (A) Diamond plots showing the average age and 95% confidence intervals for each diagnostic group. (B) Relative proportions of males and females within each diagnostic group.
Acknowledgements:
This research used the ALICE High Performance Computing Facility at the University of Leicester. We thank Dr Guohong Zhao and Dr Sohaib Rufai for their support with this study.
Financial Support:
This study was supported by the Medical Research Council (MRC), London, UK (grant number: MR/J004189/1, MRC/N004566/1 and MC_PC_17171), Fight for Sight (grant ref: 5009/5010 and 24NN181), Ulverscroft Foundation, Korea Centers for Disease Control and Prevention (2018-ER6902-02), the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1C1C1007965), The Rebecca D. Considine Research Institute, Akron Children’s Hospital. FKC is supported by the NH&MRC Fellowship (MRF1142962) and the NH&MRC Project Grant (GNT1188694, GNT1116360). ECE is an HHMI Investigator. RCHJ is supported by the Miocevich Retina Fellowship. BPB is supported by the Intramural Program at the National Eye Institute, NIH. BD is supported by the NIHR. HJK is supported by a Wellcome Trust Fellowship. MGT is supported by the NIHR (CL-2017-11-003).
The sponsor or funding organization had no role in the design or conduct of this research
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: MGT, HJK, FAP, VS and ZT are all consultants for Leica Microsystems
References
- 1.Thomas MG et al. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology 118, 1653–1660, doi: 10.1016/j.ophtha.2011.01.028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hendrickson A, Possin D, Vajzovic L & Toth CA Histologic development of the human fovea from midgestation to maturity. Am J Ophthalmol 154, 767–778 e762, doi: 10.1016/j.ajo.2012.05.007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hendrickson AE & Yuodelis C The morphological development of the human fovea. Ophthalmology 91, 603–612, doi: 10.1016/s0161-6420(84)34247-6 (1984). [DOI] [PubMed] [Google Scholar]
- 4.Thomas MG, Papageorgiou E, Kuht HJ & Gottlob I Normal and abnormal foveal development. The British journal of ophthalmology, doi: 10.1136/bjophthalmol-2020-316348 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Wilk MA, Wilk BM, Langlo CS, Cooper RF & Carroll J Evaluating outer segment length as a surrogate measure of peak foveal cone density. Vision Res 130, 57–66, doi: 10.1016/j.visres.2016.10.012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rufai SR et al. Can Structural Grading of Foveal Hypoplasia Predict Future Vision in Infantile Nystagmus?: A Longitudinal Study. Ophthalmology 127, 492–500, doi: 10.1016/j.ophtha.2019.10.037 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujimoto J & Swanson E The Development, Commercialization, and Impact of Optical Coherence Tomography. Investigative ophthalmology & visual science 57, OCT1–OCT13, doi: 10.1167/iovs.16-19963 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang D et al. Optical coherence tomography. Science 254, 1178–1181, doi: 10.1126/science.1957169 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee H et al. In Vivo Foveal Development Using Optical Coherence Tomography. Investigative ophthalmology & visual science 56, 4537–4545, doi: 10.1167/iovs.15-16542 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Kruijt CC et al. The Phenotypic Spectrum of Albinism. Ophthalmology 125, 1953–1960, doi: 10.1016/j.ophtha.2018.08.003 (2018). [DOI] [PubMed] [Google Scholar]
- 11.McCafferty BK et al. Clinical Insights Into Foveal Morphology in Albinism. J Pediatr Ophthalmol Strabismus 52, 167–172, doi: 10.3928/01913913-20150427-06 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuht HJ et al. SLC38A8 mutations result in arrested retinal development with loss of cone photoreceptor specialization. Hum Mol Genet 29, 2989–3002, doi: 10.1093/hmg/ddaa166 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hvid K et al. Prevalence and causes of infantile nystagmus in a large population-based Danish cohort. Acta Ophthalmol, doi: 10.1111/aos.14354 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Thomas MG, Maconachie G, Sheth V, McLean RJ & Gottlob I Development and clinical utility of a novel diagnostic nystagmus gene panel using targeted next-generation sequencing. Eur J Hum Genet 25, 725–734, doi: 10.1038/ejhg.2017.44 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei A et al. NGS-based 100-gene panel of hypopigmentation identifies mutations in Chinese Hermansky-Pudlak syndrome patients. Pigment Cell Melanoma Res 29, 702–706, doi: 10.1111/pcmr.12534 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Rim JH et al. Accuracy of Next-Generation Sequencing for Molecular Diagnosis in Patients With Infantile Nystagmus Syndrome. JAMA Ophthalmol 135, 1376–1385, doi: 10.1001/jamaophthalmol.2017.4859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gronskov K et al. A pathogenic haplotype, common in Europeans, causes autosomal recessive albinism and uncovers missing heritability in OCA1. Sci Rep 9, 645, doi: 10.1038/s41598-018-37272-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas MG et al. The clinical and molecular genetic features of idiopathic infantile periodic alternating nystagmus. Brain 134, 892–902, doi: 10.1093/brain/awq373 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas S et al. Autosomal-dominant nystagmus, foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation. Eur J Hum Genet 22, 344–349, doi: 10.1038/ejhg.2013.162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schumacker TM B. RE Multiple Regression Approach to Analyzing Contingency Tables: Post Hoc and Planned Comparison Procedures. The Journal of Experimental Education 64, 79–93 (1995). [Google Scholar]
- 21.Mayer AK et al. Homozygous stop mutation in AHR causes autosomal recessive foveal hypoplasia and infantile nystagmus. Brain 142, 1528–1534, doi: 10.1093/brain/awz098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sannan NS et al. Correlation of novel PAX6 gene abnormalities in aniridia and clinical presentation. Can J Ophthalmol 52, 570–577, doi: 10.1016/j.jcjo.2017.04.006 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Thomas MG et al. Abnormal retinal development associated with FRMD7 mutations. Hum Mol Genet 23, 4086–4093, doi: 10.1093/hmg/ddu122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montoliu L & Marks MS A new type of syndromic albinism associated with mutations in AP3D1. Pigment Cell Melanoma Res 30, 5–7, doi: 10.1111/pcmr.12543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaki M et al. Molecular and functional studies of tyrosinase variants among Indian oculocutaneous albinism type 1 patients. The Journal of investigative dermatology 131, 260–262, doi: 10.1038/jid.2010.274 (2011). [DOI] [PubMed] [Google Scholar]
- 26.King RA & Witkop CJ Detection of heterozygotes for tyrosinase-negative oculocutaneous albinism by hairbulb tyrosinase assay. American journal of human genetics 29, 164–168 (1977). [PMC free article] [PubMed] [Google Scholar]
- 27.d’Addio M et al. Defective intracellular transport and processing of OA1 is a major cause of ocular albinism type 1. Hum Mol Genet 9, 3011–3018, doi: 10.1093/hmg/9.20.3011 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Shen B, Rosenberg B & Orlow SJ Intracellular distribution and late endosomal effects of the ocular albinism type 1 gene product: consequences of disease-causing mutations and implications for melanosome biogenesis. Traffic 2, 202–211, doi: 10.1034/j.1600-0854.2001.020306.x (2001). [DOI] [PubMed] [Google Scholar]
- 29.Thomas MG, Kumar A, Kohl S, Proudlock FA & Gottlob I High-resolution in vivo imaging in achromatopsia. Ophthalmology 118, 882–887, doi: 10.1016/j.ophtha.2010.08.053 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Linderman RE et al. Preservation of the Foveal Avascular Zone in Achromatopsia Despite the Absence of a Fully Formed Pit. Investigative ophthalmology & visual science 61, 52, doi: 10.1167/iovs.61.10.52 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas MG, McLean RJ, Kohl S, Sheth V & Gottlob I Early signs of longitudinal progressive cone photoreceptor degeneration in achromatopsia. The British journal of ophthalmology 96, 1232–1236, doi: 10.1136/bjophthalmol-2012-301737 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Kumar A et al. Clinical and oculomotor characteristics of albinism compared to FRMD7 associated infantile nystagmus. Investigative ophthalmology & visual science 52, 2306–2313, doi: 10.1167/iovs.10-5685 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Hingorani M, Williamson KA, Moore AT & van Heyningen V Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Investigative ophthalmology & visual science 50, 2581–2590, doi: 10.1167/iovs.08-2827 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Chong GT et al. Abnormal foveal morphology in ocular albinism imaged with spectral-domain optical coherence tomography. Arch Ophthalmol 127, 37–44, doi: 10.1001/archophthalmol.2008.550 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Marmor MF, Choi SS, Zawadzki RJ & Werner JS Visual insignificance of the foveal pit: reassessment of foveal hypoplasia as fovea plana. Arch Ophthalmol 126, 907–913, doi: 10.1001/archopht.126.7.907 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mietz H, Green WR, Wolff SM & Abundo GP Foveal hypoplasia in complete oculocutaneous albinism. A histopathologic study. Retina 12, 254–260, doi: 10.1097/00006982-199212030-00011 (1992). [DOI] [PubMed] [Google Scholar]
- 37.Seo JH et al. Correlation of visual acuity with foveal hypoplasia grading by optical coherence tomography in albinism. Ophthalmology 114, 1547–1551, doi: 10.1016/j.ophtha.2006.10.054 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Wilk MA et al. Relationship between foveal cone specialization and pit morphology in albinism. Investigative ophthalmology & visual science 55, 4186–4198, doi: 10.1167/iovs.13-13217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Springer AD & Hendrickson AE Development of the primate area of high acuity. 1. Use of finite element analysis models to identify mechanical variables affecting pit formation. Vis Neurosci 21, 53–62, doi: 10.1017/s0952523804041057 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1: An overview of the literature search strategy to collect previously published data of individuals with a confirmed molecular diagnosis and report on foveal morphology.
Supplementary figure 2: (A) Diamond plots showing the average age and 95% confidence intervals for each diagnostic group. (B) Relative proportions of males and females within each diagnostic group.