

Abstract
After kidney transplantation, infection and death are important clinical complications, especially for the growing numbers of older patients with limited resilience to withstand adverse events. Evaluation of changes in gene expression in immune cells can reveal the underlying mechanisms behind vulnerability to infection.
A cohort of 60 kidney transplant recipients was evaluated. Gene expression in peripheral blood mononuclear cells 3 months after kidney transplantation was analyzed to compare differences between patients with infection and those who were infection-free in the first year post-transplant.
Pro-inflammatory genes such as IL1B, CCL4, and TNF were found to be down-regulated in post-transplant PBMC from patients who developed infection. In contrast, genes involved in metabolism, HLA genes, and transcripts involved in Type I interferon innate antiviral responses were found to be up-regulated. Promoter-based bioinformatic analyses implicated increased activity of Interferon Regulatory Factors, erythroid nuclear factor (E2), and CCAAT-enhancer binding protein (C/EBP) in patients who developed infections.
Differential patterns of gene expression were observed in patients who developed infection after kidney transplantation, with patterns distinct from changes associated with patient age, suggesting possible mechanisms behind vulnerability to infection. Assessment of gene expression in blood may offer an approach for patient risk stratification and monitoring after transplantation.
Keywords: Infections, Gene expression, Inflammation, Interferon Type I, Kidney Transplantation, Aging
Introduction
As the population ages, the numbers of patients with end stage renal diseases continues to increase, fueling increasing numbers of older kidney transplant recipients 1. However, although benefiting compared with remaining on dialysis, older kidney transplant patients experience increased rates of death, and increased rates of death due to infection, compared with younger patients.2–4 Conversely, older patients experience lower rates of acute rejection compared with younger kidney transplant recipients, suggesting that many older transplant recipients are functionally over-immunosuppressed, likely due to age-associated immune-senescence present prior to transplantation.3,5,6
The ability to evaluate level of immune function from sampling of peripheral blood would provide the opportunity to customize immunosuppression and risk stratify patients in an attempt to prevent both infection and rejection. In addition, a diagnostic approach for infection may be beneficial given the fact that infection may have an indolent and nonspecific presentation in transplant recipients, leading to delayed diagnosis and worsened clinical outcomes.7,8 Several previous studies have used peripheral blood gene expression to analyze immunological responses to infection,9–12 but none have sought to predict the incidence of infection in immunosuppressed transplant patients based on post-transplant blood samples (i.e., prior to clinically manifest infection).
Our previous work has identified changes in gene expression associated with older compared with younger kidney transplant recipients, demonstrating increased expression of pro-inflammatory transcripts and decreased expression of antiviral immune response genes.13 Age-associated immune dysfunction is known to be associated with impaired immune response important for control of infection, including decrease in dendritic cell IFN-γsecretion in response to antigen challenge, decreased numbers and impaired class switching and antibody response in B cells, and T cell immune senescence.14–17
We seek here to extend this approach by evaluating the relative differences in gene expression in patients who developed infection in this patient cohort, and to determine which transcripts are associated with infection risk independent of patient age. We utilize post-transplant blood samples, which should be easy to incorporate into post-transplant follow-up protocols if they prove effective in forecasting incident infections or providing insight into the biological pathways underlying infection risk in immunosuppressed patients.
Materials and Methods
Clinical care
Kidney transplant recipients were enrolled in an observational study approved by the UCLA Institutional Review Board. Inclusion criteria included willingness to provide informed consent, and the absence of active infection or rejection at the time of the blood draw 3 months after transplantation. As previously described, peripheral blood mononuclear cells (PBMC) were isolated and stored for batched analysis 18. This cohort was based on 23 older patients (>=60 years old) with PBMC available for analysis, which were subsequently matched to 37 younger patients ages 30–60) based on induction therapy (antithymocyte globulin (ATG) versus basiliximab) and donor type (deceased versus living). Patients received similar maintenance immunosuppression regimens with protocolized target drug levels and monitoring for infection as previously described.6 Determination of acute rejection was based on Banff definitions.19 Presence of donor specific antibody (DSA) was determined by review of single antigen HLA antibody testing over the first year post transplantation. No patients were experiencing acute rejection at the time of sample collection.
Infection
Infection was assessed by targeted chart review of enrolled patients as previously described 20. Standard definitions were used including American Thoracic Society/Infectious Diseases Society of America guidelines for pneumonia to identify cases of pneumonia, bacteremia, osteomyelitis, and viral infections 21. CMV DNAemia or end organ disease was identified according to standard guidelines 22. CMV screening was performed monthly after transplantation using the Roche COBAS® AmpliPrep/COBAS® TaqMan® CMV Test. Given the frequency of positive urine cultures and difficulty in symptom assessment in retrospective chart review, urinary tract infections were not defined based on positive urinary culture alone, and were included only with documented evidence of urinary or systemic symptoms, pyelonephritis, or bacteremia, to distinguish invasive infection from asymptomatic bacteriuria.23
Transcriptome analysis
Total RNA was isolated from PBMC in the UCLA Biological Samples Processing Core (RNeasy, Qiagen) as previously described).13 RNA was converted to fluorescent cRNA and hybridized to Illumina Human HT-12 v4 BeadArrays following the manufacturer’s standard protocol. Standard quality assurance metrics were applied to ensure validity of microarray data, which has been shown to correlate with RNA sequencing.24 Assays were performed at the UCLA Neuroscience Genomics Core as previously described.13,25
Statistical analysis
Demographic differences between older compared with younger patients were analyzed by Fisher’s exact test using JMP Pro 11 (SAS Software).
As previously described, gene expression values were quantile-normalized and log2-transformed.13 34674 genes transcripts were analyzed. General linear model analyses were used on log2-transformed values relating the expression of each assayed gene transcripts to infection, while also controlling for the following variables: age, sex, transplant type (deceased versus living donor), and induction type (basiliximab versus ATG).
We used point estimates of association magnitude for each gene as input into testing for infection-related variations in the activity of pre-defined sets of genes involved in a narrow range of specifically hypothesized biological processes (e.g., inflammation, interferon-related antiviral responses, activity of specific transcription factors, etc.) using higher-order bioinformatics analyses, as previously described.13 We list specific genes with point estimates of association greater than a pre-defined cut-off.
Gene transcripts showing > 1.25-fold difference in average expression by clinical group (infection vs. no infection) served as input into higher-order bioinformatics analyses (fold-change threshold was established a priori in order to provide a suitable number of input genes for well-powered gene set analyses). The magnitude of differential gene expression for input into the higher-order bioinformatics were not quantified using p-values. Point estimates of differential expression of each gene are used because previous research has found point estimate-based screening to provide more reliable basis for analysis in such higher-order bioinformatics than does screening based on p-/q-values.26–28
We used the Transcription Element Listening System (TELiS) promoter–based bioinformatic analysis to identify potential transcription control pathways that may drive differential risk of infection.13 A prevalence matrix was generated based on predicted transcription factor binding to a given promoter’s DNA sequence, and the prevalence of those predicted binding sites then tested for over- or under-representation relative to the population mean prevalence using a single-sample z-test. Each z-value generates a two-tailed p-value in order to determine statistical significance (adapted from Cole et al) 29. Multiple hypothesis testing is not used; each transcription factor’s distribution of binding sites across all promoters is tested using a single integrated statistical test.
Transcript origin analysis was also applied to these gene lists to identify the leukocyte subtypes mediating the observed differences in gene expression.13,30 This analysis utilizes data regarding how predominantly every gene is expressed in each cell type relative to the other cell types examined, based on a separate reference study in which the different cell types were physically separated and underwent individual transcriptome profiling (Gene Expression Omnibus GSE1133), with score computation as described (essentially expressing the average expression of each gene in each cell type in terms of its SD difference from the average expression level in all other PBMC cell types). These scores are averaged for genes identified as differentially expressed to provide a cell type specificity score which applies to each cell type analyzed, and that average score is then tested for statistically significant difference from the mean cell type specificity score, as computed across all genes assayed by the microarray (the population null hypothesis value) using standard errors derived from bootstrap resampling.
Results
Demographic characteristics of patients
We studied a cohort consisting of 23 patients over age 60 (ages 60–80, median age 67) and 37 younger patients, as previously reported.13 Of these 60 total patients, 19 (32%) developed a clinically diagnosed infection in the first year after kidney transplantation, while 41 did not (Table 1 and Figure 1). Episodes of infection included viral infections such as CMV DNAemia and gastroenteritis, bacterial infections including bacteremia, pyelonephritis, and osteomyelitis, and fungal infection, namely severe oral thrush, as described previously (Table 2).18 Other than male sex, there were no demographic characteristics that were significantly associated with infection (Table 1).
Table 1.
Demographic and clinical characteristics of patients with and without infection after kidney transplantation.
| Infection (n=19) | No Infection (n=41) | p-value | |
|---|---|---|---|
| Older (>= 60) | 47.3% | 34.2% | 0.397 |
| Age,years (median, range) | 47 (36–77) | 47 (34–80) | 0.605 |
| Donor type (% Deceased donor) | 63.2% | 36.6% | 0.093 |
| Induction type (% ATG) | 36.8% | 26.8% | 0.547 |
| Sex (% Male) | 84.2% | 56.1% | 0.044 |
| Hispanic ethnicity | 79.0% | 53.7% | 0.088 |
| Race (% White) | 57.9% | 70.7% | 0.384 |
| Dialysis ever | 68.4% | 85.0% | 0.174 |
| Previous Transplant | 0.0% | 11.9% | 0.126 |
| Tacrolimus immunosuppression | 88.9% | 99.5% | 0.670 |
| MMF dose, g (median) | 2.0 | 1.5 | 0.495 |
| Prednisone use | 100% | 97.6% | 0.436 |
| DSA | 16.7% | 9.5% | 0.624 |
| Acute rejection | 5.6% | 16.7% | 0.246 |
Figure 1.
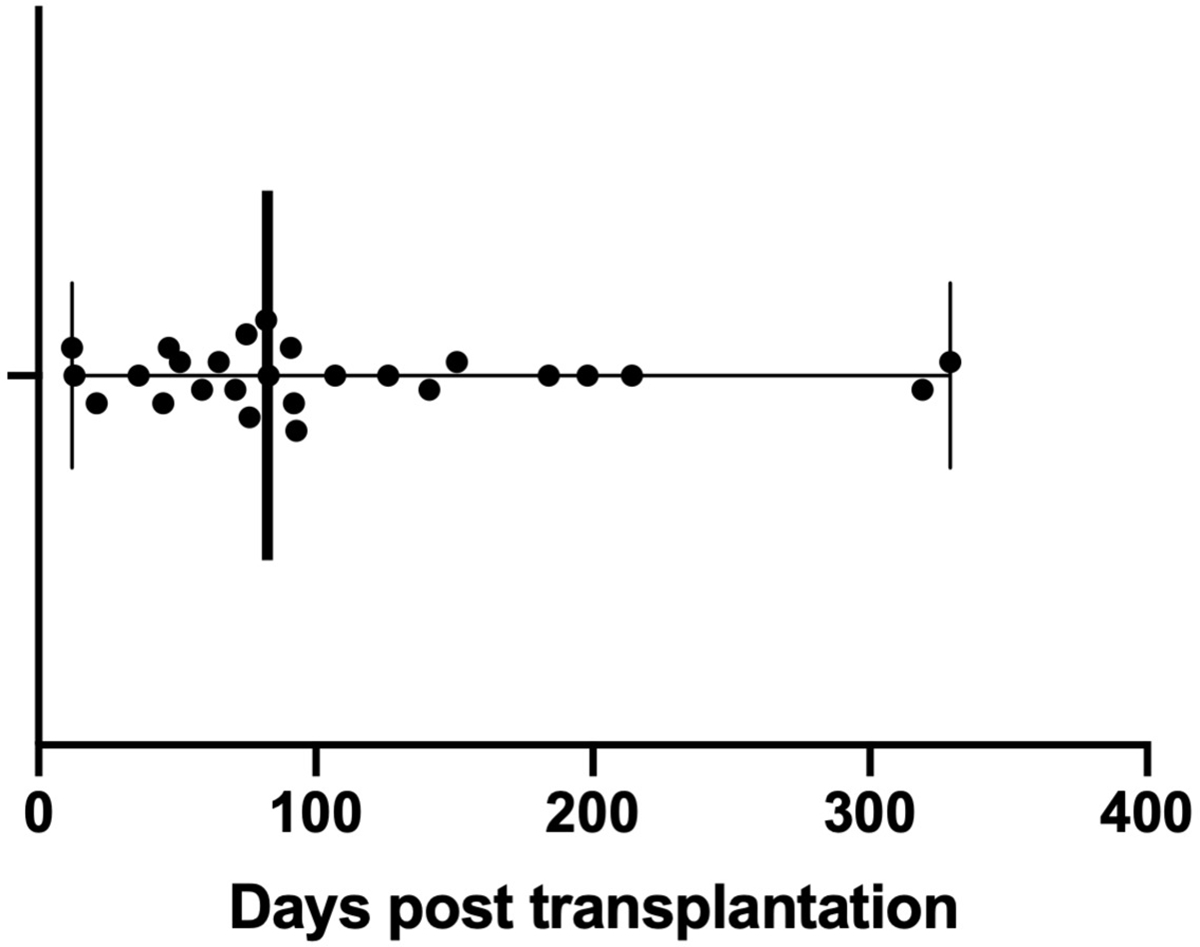
Time to infection episodes after transplantation. Bar and whiskers indicate the median and range of time to infection after kidney transplantation. Each dot represents an individual patient, with one infectious episode per patient.
Table 2.
Characterization of infectious complications identified in the patient cohort. Infection type and species indicated, if identified by culture or PCR testing
| Site | Bacteria | Viruses | Fungi | Total |
|---|---|---|---|---|
| Bone/joints | 1 (Enterococcus/Streptococcus) | 1 | ||
| Disseminated* | 2 (Unknown, Escherichia coli) | 8 (CMV) | 10 | |
| Gastrointestinal | 1 (Clostridium difficile) | 2 (Unknown, Norovirus) | 3 | |
| Pyelonephritis | 1 (Escherichia coli) | 1 | ||
| Lung | 2 (Unknown, Mycobacterium tuberculosis) | 1 (Unknown) | 3 | |
| Oral cavity | 1 (Candida sp.) | 1 | ||
| Total | 7 | 11 | 1 | 19 |
Disseminated includes bloodstream infection with or without sepsis symptoms. CMV DNAemia characterized as disseminated viral infection (PCR>137 IU/ml)
Differences in gene transcript expression in patients with infection
Genome-wide transcriptional profiling revealed multiple differences in post-transplant PBMC gene expression in patients who developed infection as compared to those who did not. Review of the top 25 genes most strongly upregulated in association with infection revealed multiple transcripts related to metabolism (particularly respiration: HBG1, HBG2, HBD, HBM, AHSP) (Table 3). We additionally noted HLA-DR and Fc fragment of IgG receptor genes important for the adaptive immune response, as well as multiple transcripts involved innate antiviral response such as interferon-induced transcripts (IFIT1L, GBP1. GBP5) and guanylate binding protein (GBP5). Additional genes up to 1.4x upregulated are shown in Supplementary Material (Appendix 1).
Table 3:
Top 25 upregulated genes in kidney transplant recipients with infection compared with those without infection
| Upregulated genes | Alias | Fold difference | Function | |
|---|---|---|---|---|
| 1 | HBG1 | Hemoglobin Subunit Gamma 1 | 2.46 | Metabolism |
| 2 | HBG2 | Hemoglobin Subunit Gamma 2 | 2.42 | Metabolism |
| 3 | CA1 | Carbonic Anhydrase 1 | 2.40 | Metabolism |
| 4 | AHSP | Alpha Hemoglobin Stabilizing Protein | 2.24 | Metabolism |
| 5 | HBD | Hemoglobin Subunit Delta | 2.13 | Metabolism |
| 6 | ALAS2 | Aminolevulinate, Delta-, Synthase 2 | 2.08 | Metabolism |
| 7 | HLA-DRB1 | Major Histocompatibility Complex, Class II, DR Beta 1 | 2.01 | Adaptive immune response |
| 8 | HLA-DRB5 | Major Histocompatibility Complex, Class II, DR Beta 5 | 1.96 | Adaptive immune response |
| 9 | FCGR1A | Fc Fragment Of IgG Receptor Ia 1 | 1.90 | Adaptive immune response |
| 10 | MMP9 | Matrix Metallopeptidase 9 | 1.84 | Innate immune response |
| 11 | ZDHHC19 | Zinc Finger DHHC-Type Containing 19 | 1.82 | Metabolism |
| 12 | SLC4A1 | Solute Carrier Family 4 Member 1 | 1.78 | Metabolism |
| 13 | HBM | Hemoglobin Subunit Mu | 1.78 | Metabolism |
| 14 | IL18RAP | Interleukin 18 Receptor Accessory Protein | 1.78 | Adaptive immune response |
| 15 | IFIT1L | Interferon Induced Protein | 1.75 | Innate antiviral response |
| 16 | FAM46C | Terminal Nucleotidyltransferase 5C | 1.74 | Metabolism |
| 17 | FCGR1C | Fc Fragment Of IgG Receptor Ic | 1.71 | Adaptive immune response |
| 18 | FCGR1B | Fc Fragment Of IgG Receptor Ib | 1.71 | Adaptive immune response |
| 19 | SELENBP1 | Selenium Binding Protein 1 | 1.68 | Metabolism |
| 20 | EPB42 | Erythrocyte Membrane Protein Band 4.2 | 1.65 | Metabolism |
| 21 | OSBP2 | Oxysterol Binding Protein 2 | 1.63 | Metabolism |
| 22 | GBP5 | Guanylate Binding Protein 5 | 1.60 | Innate antiviral response |
| 23 | SNCA | Synuclein Alpha | 1.57 | Cell signaling |
| 24 | GBP1 | Guanylate Binding Protein 1 | 1.57 | Innate antiviral response |
| 25 | ANKRD | Ankyrin Repeat Domain 22 | 1.56 | Innate immune response |
Review of the 25 genes most downregulated in association with infection revealed several transcripts important in inflammation and the adaptive immune response (Table 4). These included chemokine ligands for CCL3 (CCL3L1) and CCL4 (CCL4L1 and CCL4L2), and chemokine ligand 3 (CCL3). In addition, we noted downregulation of tumor necrosis factor (TNF); the DC activation marker CD83, which is also important in B cell activation; HLA molecules; fibroblast growth factor binding protein (FGFBP2); and CD160, important for both T cell and NK cell function. Additional genes up to 1.3x downregulated are shown in are shown in Supplementary Material (Appendix 2).
Table 4:
Top 25 downregulated genes in kidney transplant recipients with infection compared with those without infection
| Downregulated genes | Alias | Fold difference | Function | |
|---|---|---|---|---|
| 1 | CCL4L1 | C-C Motif Chemokine Ligand 4 Like 1 | 1.72 | Pro-inflammatory |
| 2 | IL1B | Interleukin 1 Beta | 1.70 | Pro-inflammatory |
| 3 | CCL4L2 | C-C Motif Chemokine Ligand 4 Like 2 | 1.69 | Pro-inflammatory |
| 4 | CCL3L1 | C-C Motif Chemokine Ligand 3 Like 1 | 1.67 | Pro-inflammatory |
| 5 | GSTM2 | Glutathione S-Transferase Mu 2 | 1.64 | Metabolism |
| 6 | CCL3L3 | C-C Motif Chemokine Ligand 3 Like 3 | 1.63 | Pro-inflammatory |
| 7 | GSTM1 | Glutathione S-Transferase Mu 1 | 1.62 | Metabolism |
| 8 | CCL3 | C-C Motif Chemokine Ligand 3 | 1.60 | Pro-inflammatory |
| 9 | TNF | Tumor Necrosis Factor | 1.54 | Pro-inflammatory |
| 10 | SNORD3D | Small Nucleolar RNA, C/D Box 3D | 1.52 | Metabolism |
| 11 | CD83 | CD83 Molecule | 1.50 | Adaptive immune response |
| 12 | SNORD13 | Small Nucleolar RNA, C/D Box | 1.48 | Metabolism |
| 13 | HLA-A29.1 | Major Histocompatibility Complex, Class I, A | 1.48 | Adaptive immune response |
| 14 | RNU11 | RNA, U11 Small Nuclear | 1.47 | Metabolism |
| 15 | HLA-DQB1 | Major Histocompatibility Complex, Class II, DQ Beta 1 | 1.45 | Adaptive immune response |
| 16 | SNORD3A | Small Nucleolar RNA, C/D Box 3A | 1.44 | Metabolism |
| 17 | MYOM2 | Myomesin 2 | 1.43 | Structure |
| 18 | FGFBP2 | Fibroblast Growth Factor Binding Protein 2 | 1.42 | Adaptive immune response |
| 19 | SGK1 | Serum/Glucocorticoid Regulated Kinase 1 | 1.40 | Metabolism |
| 20 | SGK | Serum/Glucocorticoid Regulated Kinase | 1.39 | Metabolism |
| 21 | OLIG1 | Oligodendrocyte Transcription Factor 1 | 1.39 | Transcriptional regulation |
| 22 | CD160 | CD160 Molecule | 1.38 | Adaptive immune response |
| 23 | FCRLA | Fc Receptor Like A | 1.38 | Adaptive immune response |
| 24 | RPPH1 | Ribonuclease P RNA Component H1 | 1.38 | Transcriptional regulation |
| 25 | GPR56 | Adhesion G Protein-Coupled Receptor G1 | 1.38 | Cell signaling |
Expression-based monitoring of transcription factor activity for infection
We used the “Transcription Element Listening System” (TELiS) bioinformatics analysis to evaluate upregulated and downregulated transcripts with respect to the distribution of transcription factor-binding motifs (TFBMs) present in their core promoter sequences in order to identify alterations in transcription control pathways that might drive the observed empirical transcriptome differences, we used. Transcription factors implicated as upregulated in association with infection included interferon regulatory factor 2 (IRF2; TFBM ratio in up- vs. down-regulated genes = 2.77 ± 1.35 fold, p=0.001) and interferon regulatory factor 1 (IRF1; 1.90 ± 1.32, p=0.022), consistent with the observed increase in innate antiviral response related gene expression. In addition, two other transcriptional factors related to immune activation were implicated, erythroid nuclear factor (E2) (2.24 ± 1.28, p=0.001) and CCAAT-enhancer binding protein (C/EBP) (1.65 ± 1.24, p=0.023). Transcription factor indications downregulated in association with subsequent infection included Nuclear transcription factor Y (0.43 ± 1.4, p=0.013), Ikaros 3 (0.46 ± 1.31, p=0.005), Myb-related protein B (0.55 ± 1.25, p=0.008), and NFkappaB (0.56 ± 1.3, p=0.028), which are important in lymphoid development and regulation.
Evaluation of cellular origin of transcripts
Transcript origin analysis identified monocytes as the most prominent source of gene transcripts upregulated in association with infection, although this trend did not reach statistical significance (Figure 2). Analysis of downregulated genes identified B cells and dendritic cells as primary cellular origins (p=0.030 and 0.041, respectively) (Figure 2).
Figure 2.
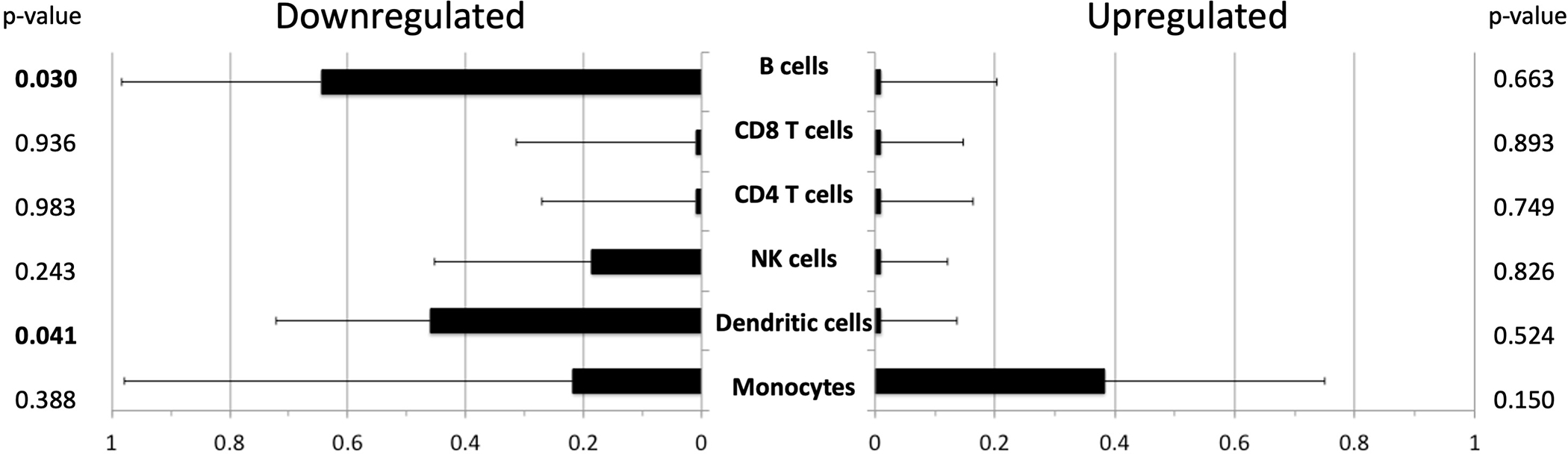
Transcript origin analysis for genes differentially expressed in patients with infection compared with patients without infection. See Methods section for details regarding the cell type identification. Bar graph indicates mean diagnosticity score for each cell type plus standard error. p-values shown on right hand of Figure. p values <0.05 are bolded for emphasis. Left hand panel demonstrates transcript origin analysis for genes upregulated, and right hand panel demonstrates downregulated genes.
We additionally used Transcriptome Representation Analysis to determine whether differences in leukocyte subset abundance might be responsible for the gene transcript abundance changes observed. This analysis revealed that for patients with and without infection, there were no statistically significant differences in cell type abundance for CD8+ T cells, CD4+ T cells, NK cells, dendritic cells, or monocytes, indicating that differences in cell type numbers was not responsible for the differences in gene transcript up- or down-regulation reported above.
Discussion
To identify the molecular basis for differential risk of infection in immunosuppressed kidney transplant patients, we conducted genome-wide transcriptional profiling of PBMC samples collected post-transplant. Infection is an important clinical outcome limiting patient success after transplantation, and we sought to identify biological pathways that might contribute to differential outcomes. Strikingly, expression of many genes important in inflammation and the adaptive immune response were downregulated in patients prior to manifest infection, primarily driven by decreased expression in B cells and dendritic cells, suggesting impaired antigen presentation may be an important mechanism behind vulnerability to infection in patients on immunosuppression (Table 4 and Figure 1). These observations expand on our previous work relating to differential gene expression in older compared with younger kidney transplant recipients, where we noted that older patients expressed increased proinflammatory but decreased antiviral immune response associated genes.13
In addition to down-regulation of adaptive immune system biology, subsequent development of infection was associated with up-regulation of innate antiviral response genes and genes involved in metabolism. The upregulation of genes involved in Type I interferon responses suggests that subclinical viral infections may potentially already be reactivated even in advance of clinical symptoms. The upregulation of hemoglobin genes (Table 3) in patients with infection suggests the possibility that subclinical immunologic activation may trigger iron sequestration via hepcidin, leading to increased hematopoiesis.31 The fact that monocytes are the cell type primarily implicated in upregulation of transcription fits with the model of innate cell activation in infection. In contrast, down regulation of pro-inflammatory cytokines, chemokines, and chemokine ligands was observed (Table 4). These results suggest two possibilities; one involving the potential for prolonged stimulation from a clinically occult infection, leading to a compensatory decrease in inflammation. Another possibility is that impaired inflammatory response is associated with vulnerability to infection, as acute inflammation is associated with resolution of infection, while chronic inflammation is counter-productive to resolution of infection.32 It is notable that the cell types responsible for this downregulation are identified as B cells and dendritic cells, both antigen presenting cells, suggesting the possibility that ineffective antigen presentation drives vulnerability to infection after transplantation, a syndrome that has been described in sepsis and pneumonia, and associated with immune aging.14,15,17,33
Direct viral infection of dendritic cells, as may be seen in CMV infection, may be another driver of defective antigen presentation via down regulation of surface molecules, impairing dendritic cell function.34–36 Interestingly, interferon regulatory factor (IRF), one of the transcription factors we identified to be upregulated in transplant patients with infection has also been implicated in CMV infection of myeloid cells and immune evasion of antiviral immune response.37 This is significant given that many of the infection experienced in this cohort were related to CMV.
The findings from our study in patients with infection contrast with the differences in gene expression observed in older compared with younger patients.13 In the comparison of older versus younger patients, older patients demonstrated increased abundance of pro-inflammatory transcripts including cytokines and chemokines related to increased expression from monocytes. Older patients also demonstrated downregulation of genes important in the antiviral immune response. Enriched transcription factor binding sites were similarly different in our analysis of older versus younger patients in comparison with this study on infection.
This work demonstrates unique insights into mechanism of vulnerability to infection in patients on immunosuppression, providing a potential approach for risk stratification and adjustment of immunosuppression to avoid the adverse clinical outcome of infection. The achievement of this goal would have large clinical impact given that in addition to direct morbidity and mortality caused by infection, infection often triggers rejection, via a pro-inflammatory or heterologous immune effect, worsened by the often concurrent reduction of immunosuppression.38 Future studies will examine longitudinal sample collection to better characterize how gene expression changes before, during, and after infectious episodes. In addition, gene expression changes associated with infection can be analyzed in parallel with those associated with rejection in order to define a specific clinical profile.
Limitations to this study include its cross sectional nature, and the heterogeneity of infection types. However, one advantage of single center study where immunosuppression protocols, antibiotic prophylaxis, and patterns of diagnosis and response to infection are standardized. In addition, given the time period of study, we were not able to study patients who developed novel coronavirus infection (COVID-19), although we would predict that IFN signalizing is likely to be important in protection against a viral infection like COVID-19.
Future multicenter centers could correct for these limitations, and could also focus on a specific cell type such as B or T cells to better understand how transcriptional changes in specific cells are associated with infection. Additional future studies will examine whether signaling differs by infection type, e.g. bacterial versus viral infection. This work would be complemented by analysis in parallel with immune phenotyping and epigenetic analysis to provide a complete model of immunologic changes associated with vulnerability to infection. This type of translational work will allow for improved clinical outcomes and avoidance of infection in the growing numbers of older kidney transplant recipients.
Supplementary Material
Funding sources:
This work was supported by the National Institutes of Health [R03AG050946] (J.S.), [R21AG055879] (J.S.), [U01AI124319] (E.F.R.), [U19AI128913] (E.F.R.), and Mendez Transplant Institute (J.S.).
Footnotes
The authors declare no conflicts of interest.
References
- 1.Hart A, Smith JM, Skeans MA, et al. OPTN/SRTR 2018 Annual Data Report: Kidney. Am J Transplant. 2020;20 Suppl s1:20–130. [DOI] [PubMed] [Google Scholar]
- 2.Meier-Kriesche HU, Ojo AO, Hanson JA, Kaplan B. Exponentially increased risk of infectious death in older renal transplant recipients. Kidney International. 2001;59(4):1539–1543. [DOI] [PubMed] [Google Scholar]
- 3.Heinbokel T, Elkhal A, Liu G, Edtinger K, Tullius SG. Immunosenescence and organ transplantation. Transplant Rev (Orlando). 2013;27(3):65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knoll GA. Kidney Transplantation in the Older Adult. YAJKD. 2013;61(5):790–797. [DOI] [PubMed] [Google Scholar]
- 5.Tullius SG, Tran H, Guleria I, Malek SK, Tilney NL, Milford E. The Combination of Donor and Recipient Age is Critical in Determining Host Immunoresponsiveness and Renal Transplant Outcome. Transactions of the Meeting of the American Surgical Association. 2010;128:275–289. [DOI] [PubMed] [Google Scholar]
- 6.Schaenman JM, Rossetti M, Sidwell T, et al. Increased T cell immunosenescence and accelerated maturation phenotypes in older kidney transplant recipients. Human Immunology. 2018;79(9):659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pagalilauan GL, Limaye AP. Infections in transplant patients. Med Clin North Am. 2013;97(4):581–600- x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fishman JA. Infection in Organ Transplantation. American Journal of Transplantation. 2017;17(4):856–879. [DOI] [PubMed] [Google Scholar]
- 9.Mahajan P, Kuppermann N, Mejias A, et al. Association of RNA Biosignatures With Bacterial Infections in Febrile Infants Aged 60 Days or Younger. JAMA. 2016;316(8):846–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herberg JA, Kaforou M, Wright VJ, et al. Diagnostic Test Accuracy of a 2-Transcript Host RNA Signature for Discriminating Bacterial vs Viral Infection in Febrile Children. JAMA. 2016;316(8):835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharya S, Rosenberg AF, Peterson DR, et al. Transcriptomic Biomarkers to Discriminate Bacterial from Nonbacterial Infection in Adults Hospitalized with Respiratory Illness. Sci Rep. 2017;7(1):6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lydon EC, Henao R, Burke TW, et al. Validation of a host response test to distinguish bacterial and viral respiratory infection. EBioMedicine. 2019;48:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaenman JM, Rossetti M, Lum E, et al. Differences in Gene Expression in Older Compared With Younger Kidney Transplant Recipients. Transplant Direct. 2019;5(4):e436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frasca D, Blomberg BB. Aging Affects Human B Cell Responses. J Clin Immunol. 2011;31(3):430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 2013;13(12):875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pera A, Campos C, López N, et al. Immunosenescence: Implications for response to infection and vaccination in older people. Maturitas. 2015;82(1):50–55. [DOI] [PubMed] [Google Scholar]
- 17.Molony RD, Malawista A, Montgomery RR. Reduced dynamic range of antiviral innate immune responses in aging. Exp Gerontol. 2018;107:130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaenman JM, Rossetti M, Sidwell T, et al. Increased T Cell Immunosenescence and Accelerated Maturation Phenotypes in Older Kidney Transplant Recipients. Human Immunology. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haas M, Loupy A, Lefaucheur C, et al. The Banff 2017 Kidney Meeting Report: Revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Transplant. 2018;18(2):293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang EC, Rossetti M, Sidwell T, et al. Differences in Proinflammatory Cytokines and Monocyte Subtypes in Older as Compared With Younger Kidney Transplant Recipients. Transplantation Direct. 2018;4(3):e348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.American Thoracic S, Infectious Diseases Society of A. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. American Journal of Respiratory and Critical Care Medicine. Vol 1712005:388–416. [DOI] [PubMed] [Google Scholar]
- 22.Ljungman P, Boeckh M, Hirsch HH, et al. Definitions of Cytomegalovirus Infection and Disease in Transplant Patients for Use in Clinical Trials. CLIN INFECT DIS. 2017;64(1):87–91. [DOI] [PubMed] [Google Scholar]
- 23.Nicolle LE, Gupta K, Bradley SF, et al. Clinical Practice Guideline for the Management of Asymptomatic Bacteriuria: 2019 Update by the Infectious Diseases Society of America. Clin Infect Dis. 2019;68(10):e83–e110. [DOI] [PubMed] [Google Scholar]
- 24.Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cole SW, Capitanio JP, Chun K, Arevalo JMG, Ma J, Cacioppo JT. Myeloid differentiation architecture of leukocyte transcriptome dynamics in perceived social isolation. Proceedings of the National Academy of Sciences. 2015;112(49):15142–15147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole SW, Galic Z, Zack JA. Controlling false-negative errors in microarray differential expression analysis: a PRIM approach. Bioinformatics. 2003;19(14):1808–1816. [DOI] [PubMed] [Google Scholar]
- 27.Norris AW, Kahn CR. Analysis of gene expression in pathophysiological states: balancing false discovery and false negative rates. Proc Natl Acad Sci U S A. 2006;103(3):649–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi L, Jones WD, Jensen RV, et al. The balance of reproducibility, sensitivity, and specificity of lists of differentially expressed genes in microarray studies. BMC Bioinformatics. 2008;9 Suppl 9:S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cole SW, Yan W, Galic Z, Arevalo J, Zack JA. Expression-based monitoring of transcription factor activity: the TELiS database. Bioinformatics. 2005;21(6):803–810. [DOI] [PubMed] [Google Scholar]
- 30.Cole SW, Hawkley L, Aravalo J, Cacciopo J. Transcript origin analysis identifies antigen-presenting cells as primary targets of sociallyregulated gene expression in leukocytes. Proceedings of the National Academy of Sciences. 2011;108(7):3080–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganz T Macrophages and Iron Metabolism. Microbiology Spectrum. 2016;4:1–10. [DOI] [PubMed] [Google Scholar]
- 32.Franceschi C, Campisi J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2014;69(Suppl 1):S4–S9. [DOI] [PubMed] [Google Scholar]
- 33.Roquilly A, McWilliam HEG, Jacqueline C, et al. Local Modulation of Antigen-Presenting Cell Development after Resolution of Pneumonia Induces Long-Term Susceptibility to Secondary Infections. Immunity. 2017;47(1):135–147 e135. [DOI] [PubMed] [Google Scholar]
- 34.Gredmark-Russ S, Soderberg-Naucler C. Dendritic cell biology in human cytomegalovirus infection and the clinical consequences for host immunity and pathology. virulence. 2012;3(7):621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinclair J, Reeves M. The intimate relationship between human cytomegalovirus and the dendritic cell lineage. Front Microbiol. 2014;5:389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silvin A, Yu CI, Lahaye X, et al. Constitutive resistance to viral infection in human CD141(+) dendritic cells. Sci Immunol. 2017;2(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brinkmann MM, Dag F, Hengel H, Messerle M, Kalinke U, Cicin-Sain L. Cytomegalovirus immune evasion of myeloid lineage cells. Med Microbiol Immunol. 2015;204(3):367–382. [DOI] [PubMed] [Google Scholar]
- 38.Ali JM, Bolton EM, Bradley JA, Pettigrew GJ. Allorecognition Pathways in Transplant Rejection and Tolerance. Transplantation Journal. 2013;96(8):681–688. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.