

Abstract
Resin-immobilized catalysts were prepared through chirality-driven self-assembly. The method allows the resin-immobilized catalyst to be regenerated under mild conditions and in situ catalyst exchange to be carried out quantitatively. The uniqueness of the methodology was demonstrated by the preparation of a catalyst for TEMPO oxidation as well as a two-step sequential TEMPO oxidation/aldol condensation sequence enabled by facile catalyst exchange.
Catalyst recovery and recycling1 is of significance in modern synthetic chemistry as the increase in environment awareness motivates scientists to develop and employ greener approaches.2 Resin-immobilized catalysts have gained popularity due mostly to their facile recovery from the reaction media thus reducing cost and labour in preparing catalysts.3 However, their recyclability and reusability is often limited by the functional integrity of the loaded catalyst, that is, the ability to regenerate and reuse the catalyst.4,5 The exchangeability/renewability of damaged or poisoned catalysts plays a critical role in extending the life-span of supporting resins. Catalyst immobilization to a resin support is typically accomplished through either covalent6 or non-covalent interactions.7,8 The non-covalent immobilization of catalysts to the solid support reduces the number of synthetic steps, and the process is achieved through mild reaction conditions. For instance, phosphine ligands were anchored on the dendrimer support through multiple hydrogen bonds to urea adamantyl functional groups on the periphery of the dendrimer. The catalyst was successfully employed in the various Pd or Rh-catalyzed reactions.9 The bis(imidazolium)-tagged 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) catalyst was immobilized onto cross-linked polymeric imidazolium solid supports via electrostatic interactions.10 The loaded TEMPO effectively oxidized various primary and secondary alcohols, and immobilized catalysts were successfully recycled several times. Lastly, π–π stacking was employed for the immobilization of NHC–Pd and NHC–Ru catalysts onto the reduced graphene oxide (rGO) surface. It was used for the hydrogenation and nitro group reduction.11
The relatively weak interactions among non-covalent often led to immobilized catalysts prone to leaching thus limiting their applications. In contrast, covalent immobilization has reduced the risk of catalyst leaching.12 However, covalent immobilization process often involves lengthy preparation steps and demands strict compatible chemical/reaction conditions, resulting in lack of flexibility in catalyst design. Metal ligand complexation may provide an alternative to the current approaches. The comparatively higher bond strength of the metal–ligand interactions13 is expected to enhance resistance toward catalyst leaching, while its reversible nature still allows recharging or replacing catalysts under mild condition without damaging the structural integrity of the polymeric support.14 Among the metal–ligand complexation strategies employed,15 we believe chirality-directed self-assembly16 using labile covalent, chelating methylene bis(oxazoline) (box) subunits provides a highly promising but underutilized approach. Appropriately designed, the formation of heterochiral metal–ligand complex can be highly favoured over homochiral complexes. We recently reported using this high selectivity to generate structurally distinct Janus dendrimers quantitatively in one-pot process.17 Prior studies used the strategy to construct supramolecular chiral bidentate ligands in various asymmetric reactions.18 We now report applying a similar approach to loading catalysts on to a solid resin support. As illustrated in Scheme 1, generation of undesirable unbound catalyst can be effectively eliminated. (Scheme 1, A vs. C) In addition, chirality-directed self-assembly allows in situ modification of loaded catalysts through direct ligand exchange. For example, the homochiral assembly, a thermodynamically weaker complex, can be efficiently and quantitatively transformed into the heterochiral assembly by exposure to the ligand with opposite chirality. This is a challenging task with traditional metal–ligand complexes using ligand combination that lacks significant differences in stability upon complexation with the metal (Scheme 1, B vs. D).
Scheme 1.
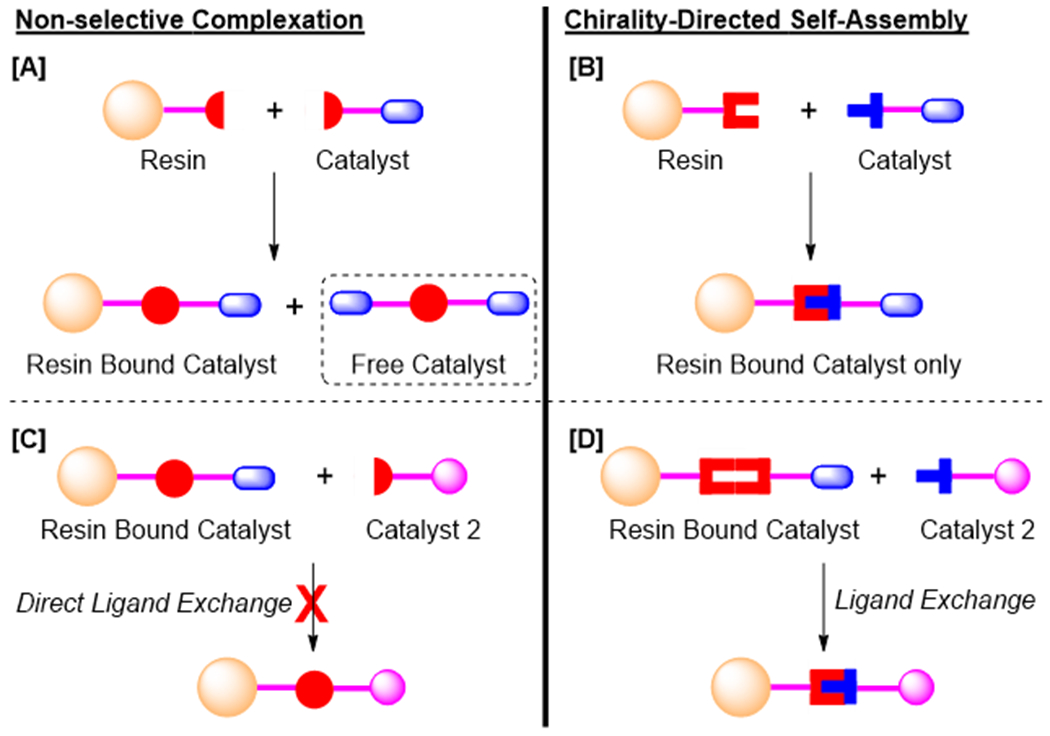
Non-selective (A and B) versus selective self-assembly (C and D).
Our main objective for this study is to apply chirality-driven self-assembly for immobilizing catalysts onto a resin to generate renewable/exchangeable heterogeneous catalysts. Scheme 2-i illustrates the proposed application to a single-step renewable resin-immobilized catalyst and its facile regeneration and recharge. Scheme 2-ii illustrates a proposed sequential two-step catalysts sequence in which the resin undergoes facile in situ exchange to effect new catalytic activity.
Scheme 2.
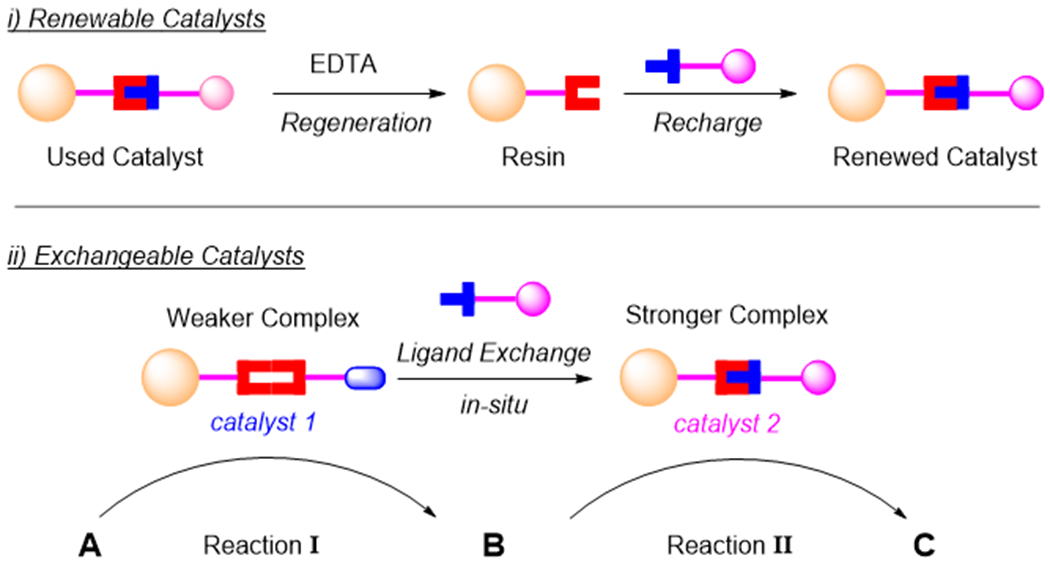
Renewability and exchangeability of the self-assembly based resin immobilized catalysts.
The immobilization of chiral methylene bis(oxazoline) ((R,R)-BOX) ligand onto Wang resin (R,R)-1 was achieved in one-step by following a known procedure from commercially available brominated Wang resin.19 As shown in Scheme 3, the functional loading capacity of chiral BOX for self-assembly was measured spectroscopically using UV/Vis through the titration of fmoc-derived (S,S)-2, and determined to be 0.28 mmol per gram of resin.20 The formation of the heterochiral Zn(II) complex (RR,SS)-3 was also monitored by the FTIR analysis; the C═N bond stretch shifts from 1655 cm−1 ((S,S)-2) to 1600 cm−1 ((RR,SS)-3).21 The shift in the C═N bond stretching band is similar to that observed with the free (i.e., unbound) heterochiral Zn(II) complexes formed in solution which results in a shift from 1654 cm−1 to 1599 cm−1.
Scheme 3.
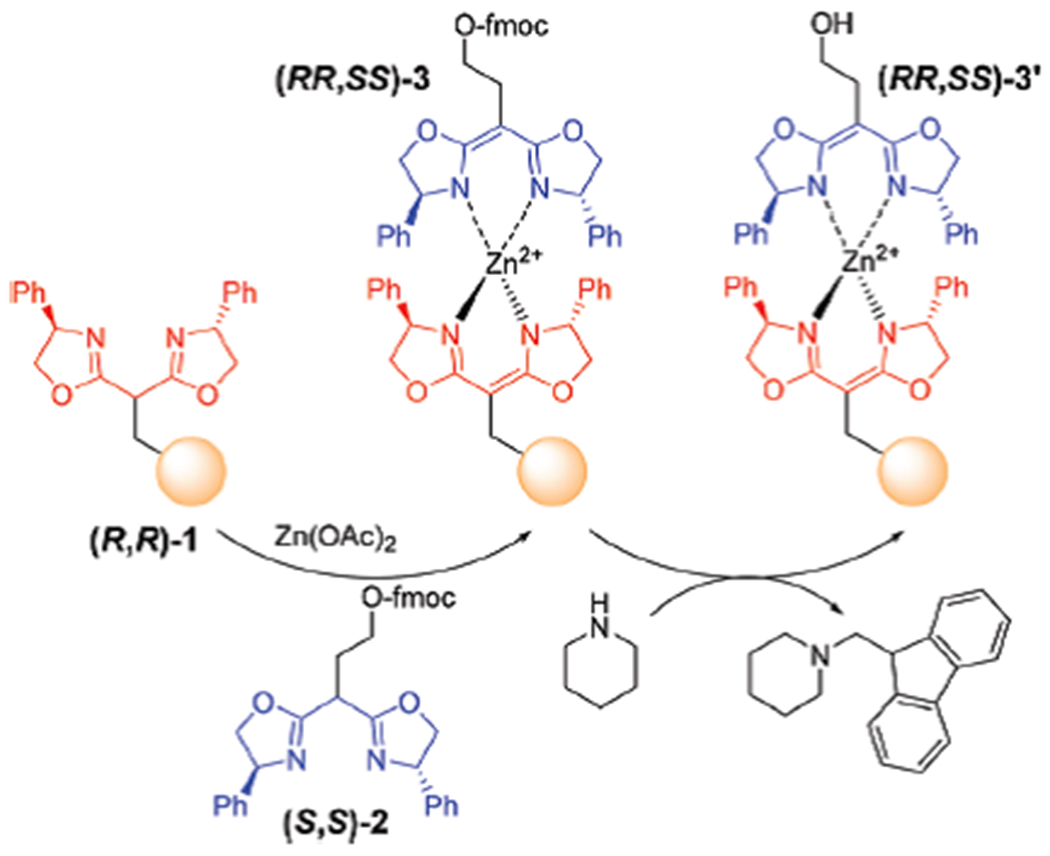
One-pot preparation of fmoc-derived resin used for quantifying the functional capacity.
As mentioned above, the chirality-driven self-assembly enables facile construction of resin-immobilized catalyst which can be easily renewed without affecting integrity of the resin as catalytic activity deteriorated. To demonstrate simplicity in renewing immobilized catalyst, the resin-immobilized pyrrolidine catalyst22 was subject to the catalyst renewal after several use in aldol reaction.23
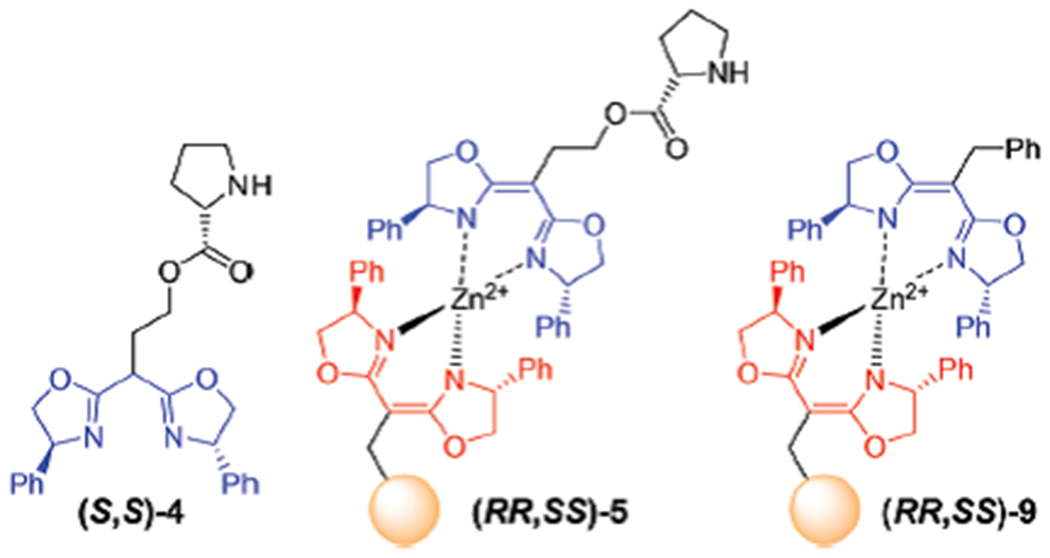
The self-assembled pyrrolidine derived resin (RR,SS)-5 was prepared by using the same strategy for (RR,SS)-3 in Scheme 3 by mixing an equimolar amount (0.28 mmol) of Zn(OAc)2, (S,S)-BOX-pyrrolidine derivative (S,S)-4 and the (R,R)-BOX functionalized resin (R,R)-1 (Scheme 4). A variety of pyrrolidine derived catalysts have been successfully used to catalyse cross aldol reactions.24 Those results serve as a useful benchmark against which the current strategy can be compared. For example, proline methyl ester catalyzes the condensation of isobutyaldehyde and 4-nitrobenzaldehyde under the conditions highlighted in Table 1 gave a good yield (92%) in modest reaction time (3 h) (entry 1). Under comparable conditions, the resin-immobilized pyrrolidine (RR,SS)-5 performs nearly identically (91% yield, 6 h) (entry 2). The outcome of enantioselectivity (54%) was also identical between free and bound chiral pyrrolidine catalysts.25 In contrast the resin lacking the crucial pyrrolidine subunit, (RR,SS)-9, gave only recovered starting materials unchanged after 3 days (entry 3). In addition, neither resin (R,R)-1 itself nor 1 : 1 Zn(II)-BOX complex of resin (R,R)-1 yielded aldol product 8 (entries 4 and 5). The pyrrolidine immobilized (RR,SS)-5 can be recycled several times; the catalytic efficiency as well as enantioselectivity deteriorated after 8 recycles (entry 6). Pyrrolidine catalysts on (RR,SS)-5 was renewed by first removing existing pyrrolidine under mild reaction conditions through a treatment with EDTA solution.18 The fresh pyrrolidine catalyst (S,S)-4 was added to this recycled (R,R)-1 to obtain renewed (RR,SS)-5. The catalytic efficiency was regained with a renewed (RR,SS)-5, maintaining the efficiency for several recycling (entries 7 and 8).
Scheme 4.
Renewability and exchangeability of the self-assembly based resin immobilized catalysts.
Table 1.
Aldol Reactions of p-Nitrobenzaldehyde 7 and Isobutyl Aldehyde 6[a]

| |||
|---|---|---|---|
|
| |||
| entry | catalyst | Time (h)[b] | Yield (%)[c] 8 |
| 1 | proline methyl ester | 4 | 92 |
| 2 | (RR,SS)-5 | 6 | 91 |
| 3 | (R,R)-1 + (S,S)-10 | 72 | trace |
| 4 | (R,R)-1 | 72 | trace |
| 5 | (R,R)-1 + Zn(OAc)2 | 72 | trace |
| 6 | (RR,SS)-5 Repeat #8 | 8 | 84 |
| 7[d] | (RR,SS)-5 Renewed | 6 | 90 |
| 8[d] [e] | (RR,SS)-5 Renewed, repeat #3 | 6 | 92 |
p-Nitrobenzaldehyde 7 (1.1 mmol), isobutyl aldehyde 6 (1.0 mmol), acetic acid (1.0 mmol) and catalyst (20 mol%) in DMF.
Monitored by GC.
An isolated yield after column purification (Hex:EtOAc = 6:4),
Regenerated catalyst was used.
The 3rd reaction cycle using the renewed catalyst.
The heterochiral Zn(II) BOX was used as a catalyst immobilization in above studies. Interestingly, we found that an efficient resin-bound catalyst can also be obtained with the weaker homochiral Zn(II) complex. For example, the resin-immobilized TEMPO catalyst26 was prepared, and the resulting resin-bound catalyst was evaluated in the oxidation of a prototypical alcohol, p-nitrobenzyl alcohol, using DIB as TEMPO oxidant to generate N-oxoammonium cation (Scheme 5 and Table 2). The preparation of (RR,RR)-11 required three mole equivalents of (R,R)-10, followed by washing step to remove unbound TEMPO-BOX homochiral Zn(II) complex (RR,RR)-Zn(II) 10.27
Scheme 5.
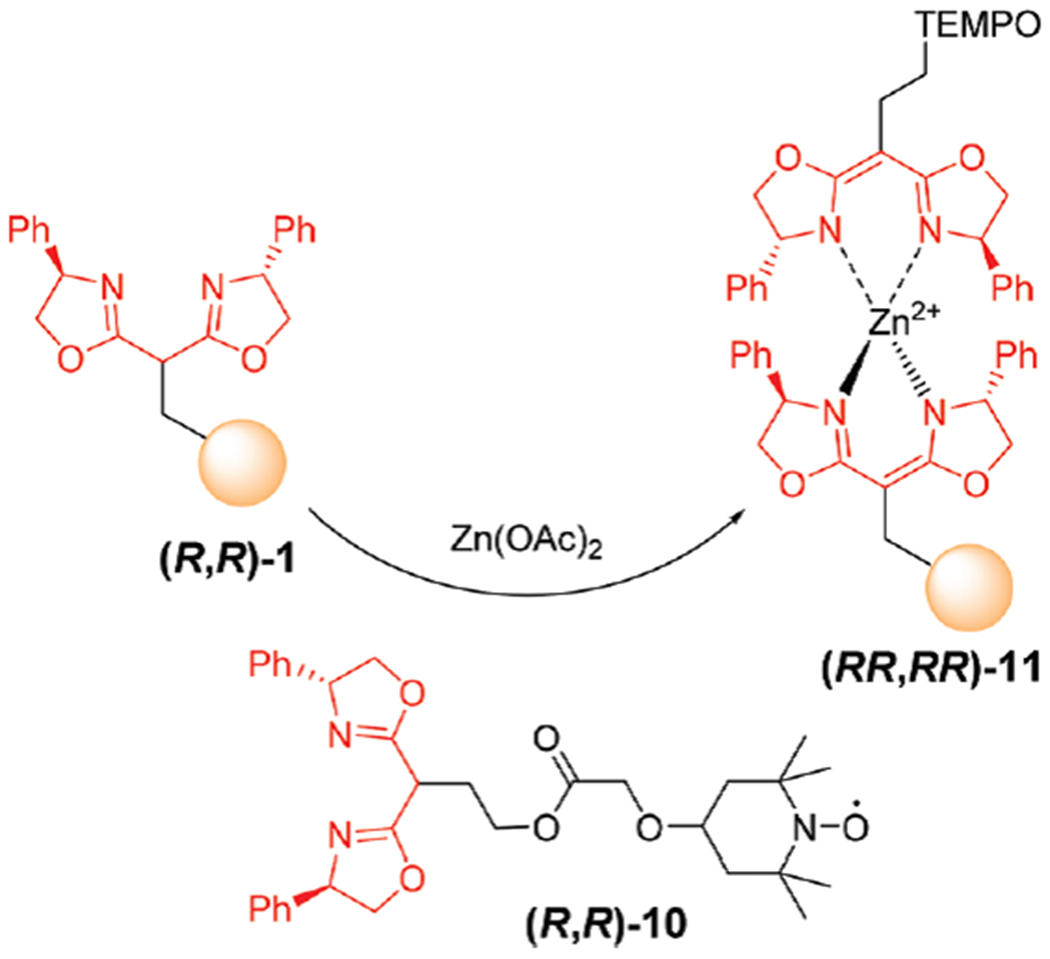
A one-pot preparation of resin immobilized TEMPO catalyst (RR,RR)-11.
Table 2.
TEMPO oxidation of p-nitrobenzalcohol 12a

| |||
|---|---|---|---|
|
| |||
| entry | catalyst | Time (h)[b] | 21 Yield (%)[c] |
| 1 | 14 | 4 | >98 |
| 2 | (RR,RR)-12 | 5.5 | >98 |
| 3 | (R,R)-1 | 24 | ND |
| 4 | (RR,RR)-1 + Zn(OAc)2 | 24 | ND |
| 5[d] | (RR,RR)-12 Repeat #8 | 8 | 87 |
| 6[d][e] | (RR,RR)-12 Renewed, Repeat #3 | 5.5 | >98 |
p-Nitrobenzaldehyde 7 (1.0 mmol), DIB (1.02 mmol) and catalyst (20 mol%) in DMF.
Monitored by GC.
An isolated yield after column purification (Hex:EtOAc = 7:3 to 5:5).
Renewed catalyst was used.
3rd reaction cycle using the renewed catalyst. ND: not determined.
ND: not determined.
The resin-immobilized TEMPO catalyst (RR,RR)-11 oxidized alcohol 12 in 5.5 hours, which is comparable in efficiency relative to the free TEMPO catalyst (Table 2, entry 1 vs. 2). As is the case in aldol reaction, resin (R,R)-1 as well as 1 : 1 Zn(II) complex of (R,R)-1-Zn(II) did not oxidize the alcohol (entries 2 vs. 3 and 4). The (RR,RR)-11 can be re-used several times before catalytic efficiency started deteriorating (entry 5). As is the case in pyrrolidine catalyst (RR,SS)-5, the catalytic efficiency of (RR,RR)-11 can be recovered by regenerating TEMPO on the resin. The renewed catalytic activity was identical to its original (RR,RR)-11 (entry 1 vs. 6).
A two-step sequential oxidation/aldol reaction28 was next examined as a model reaction sequence that exploits catalyst exchange. The overall strategy is summarized in Scheme 6. When the oxidation and aldol reactions were conducted with free catalysts (TEMPO and pyrrolidine) sequentially in one pot, the oxidation step went smoothly, however, the aldol reaction was sluggish due to the generation of N-oxoammonium cation from TEMPO and excess (diacetoxyiodo)benzene (DIB). These by-products deactivate the pyrrolidine catalyst (Table 3, entry 2). The two catalysts are incompatible,29 and TEMPO and its reaction by-products must be removed prior to the pyrrolidine catalysis. Using the TEMPO catalyst (RR,RR)-11, oxidations of both alcohols (i.e., isobutanol and p-nitrobenzyl alcohol) underwent smoothly. Upon completion of oxidation step, pyrrolidine catalyst (RR,SS)-5 was generated through a ligand exchange between (RR,RR)-11 and (SS,SS)-4. The exchanged resin was simply washed several times to remove the TEMPO containing complex (RR,SS)-14. The two-step sequence gave the final aldol product in 80% overall isolated yield with 54% ee (Table 3, entry 1).
Scheme 6.
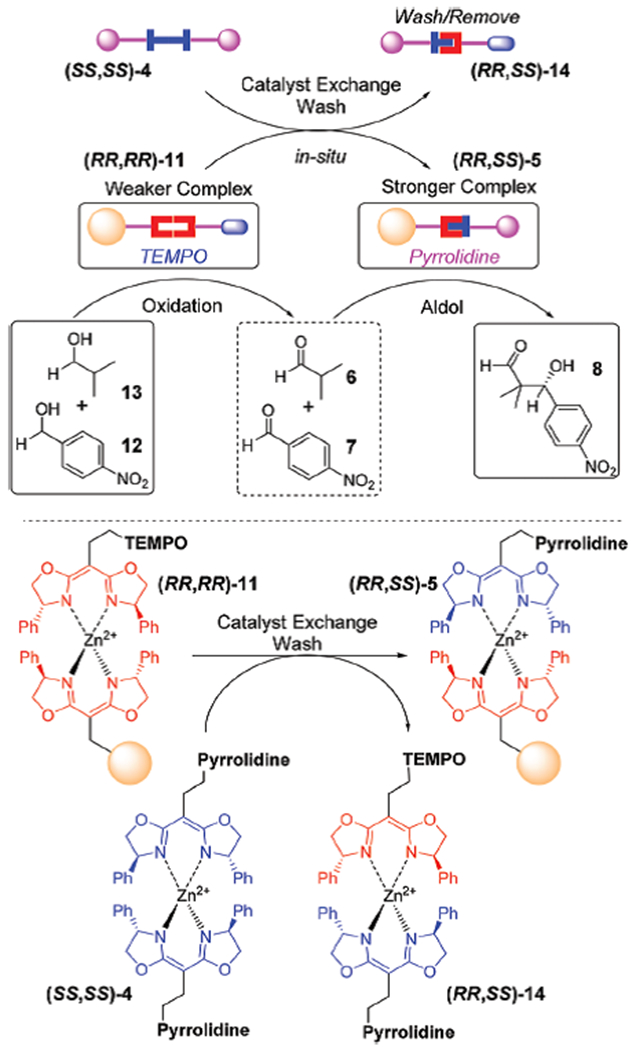
Sequential oxidation/aldol reaction using catalyst exchangeable resin.
Table 3.
Sequential Oxidation-Aldol Reaction[a]
| entry | Oxidation Catalyst | Aldol Catalyst | Time (h)[b] Ox/Aldol | Yield (%)[c] 8 |
|---|---|---|---|---|
| 1 | (RR,RR)-17 | (RR,SS)-12 | 9/12 | 80 |
| 2[d] | 14 | 9 | 9/72 | 21 |
| 3 | (RR,RR)-17 | (RR,SS)-12 | 9/72 | 25 |
| 4 | (RR,RR)-8 | (RR,SS)-12 | 72/NA | 0 |
| 5 | (RR,RR)-17 | (RR,SS)-8 | 9/72 | trace |
| 6 | (RR,SS)-17 | + (S,S)-15 | 9/72 | trace |
| 7 | (RR,RR)-17 | + (S,S)-15 | 9/12 | 81 |
Oxidation: p-nitrobenzalcohol 12 (1.0 mmol), isobutyl alcohol 14 (1.0 mmol), DIB (2.04 mmol) and catalyst (0.2 mmol) followed by aldol reaction.
Total reaction time not including catalyst isolation/exchange processes (approx. 30 min).
Isolated yield of 8.
Pyrrolidine 9 was added upon completion of oxidation by 14.
Several control experiments provided further information about the reactions. If the resins were not washed upon ligand exchange to remove TEMPO containing unbound (RR,SS)-14, only trace amount of aldol adduct was obtained (entry 3). Use of the catalytically inert Bn-BOX (SS,SS)-9 instead of pyrrolidine (SS,SS)-4 at ligand exchange step yielded no aldol product after 72 hours. (entry 4) When TEMPO catalyst (RR,SS)-11 was assembled through heterochiral Zn(II) assembly using opposite enantiomer of BOX ligand (S,S)-10 instead of (R,R)-10, the second aldol step failed to take place as pyrrolidine (SS,SS)-4 was unable to replace heterochiral Zn(II) complex bound TEMPO on resin (RR,SS)-11 (entry 5). The pyrrolidine catalyst (RR,SS)-5 can be transformed back to the initial non-catalytic resin (R,R)-1 through Zn(II) complex disassembly by the EDTA solution. It is noteworthy to mention that the detached BOX-pyrrolidine (S,S)-4 ligand was recovered through extraction of EDTA solution. The second-round sequential reactions with regenerated catalytic resin yielded aldol product in similar overall yield as well as enantioselectivity (entry 6).
Conclusions
In summary, we demonstrated a highly versatile approach in assembling resin-bound catalysts using chirality-directed self-assembly. The assembled catalyst is stable in the temperature range typically used in synthesis, yet the loaded catalyst can be easily detached from resin under mild conditions through a ligand exchange. Therefore, a facile regeneration of resin-immobilized catalysts is possible without negatively impacting on the functional/structural frameworks of the chiral BOX functionalized resin. In addition, by taking advantage of the stability differences between homochiral and heterochiral Zn(II) BOX complexes, resin-bound catalysts through a homochiral complex bridge can be easily swapped by simply adding a new catalyst that is bound to a homochiral BOX of the opposite enantiomer. Catalyst exchange on the resin allows the same resin to be used in sequential operations without preparing multiple sets of resin-immobilized catalysts. The simplicity in assembly enables one to build a large library of resin-immobilized catalysts in very short period of time. In addition, the application of this concept is not limited to the resin-immobilized catalyst, and it can be employed in connecting two functional groups together. Further applications generating a multi-functional resin-immobilized catalysts are under development.
Acknowledgements
We thank Professor James M. Takacs (University of Nebraska) for valuable discussion. This work is supported by the National Science Foundation under grant number 1856522. DHH is grateful to the National Institutes of Health, National Institute of General Medical Sciences (R01 GM128659) for financial support of this research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob00439a
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.(a) Miceli M, Frontera P, Macario A and Malara A, Catalysts, 2021, 11, 591–607; [Google Scholar]; (b) Susam ZD and Tanyeli C, Asian J. Org. Chem, 2021, 10, 1251–1266; [Google Scholar]; (c) Liu M, Wu J and Hou H, Chem. – Eur. J, 2019, 25, 2935–2948; [DOI] [PubMed] [Google Scholar]; (d) Ye R, Zhukhovitskiy AV, Deraedt CV, Toste FD and Somorjai GA, Acc. Chem. Res, 2017, 50, 1894–1901; [DOI] [PubMed] [Google Scholar]; (e) Moberg C, Acc. Chem. Res, 2016, 49, 2736–2745. [DOI] [PubMed] [Google Scholar]
- 2.(a) Molnar A and Papp A, Coord. Chem. Rev, 2017, 349, 1–65; [Google Scholar]; (b) Sheldon RA, Chem. Soc. Rev, 2012, 41, 1437–1451; [DOI] [PubMed] [Google Scholar]; (c) Anastas PT and Eghbali N, Chem. Soc. Rev, 2010, 39, 301–312. [DOI] [PubMed] [Google Scholar]
- 3.(a) Zhang H, Li H, Xu CC and Yang S, ACS Catal., 2019, 9, 10990–11029; [Google Scholar]; (b) Munnik P, de Jongh PE and de Jong KP, Chem. Rev, 2015, 115, 6687–6718; [DOI] [PubMed] [Google Scholar]; (c) Barak-Kulbak E, Goren K and Portnoy M, Pure Appl. Chem, 2014, 86, 1805–1818; [Google Scholar]; (d) Benaglia M, Puglisi A and Cozzi F, Chem. Rev, 2003, 103, 3401–3429. [DOI] [PubMed] [Google Scholar]
- 4.(a) Trindade AF, Gois PMP and Afonso CAM, Chem. Rev, 2009, 109, 418–514; [DOI] [PubMed] [Google Scholar]; (b) Lu J and Toy PH, Chem. Rev, 2009, 109, 815–838; [DOI] [PubMed] [Google Scholar]; (c) Heitbaum M, Glorius F and Escher I, Angew. Chem., Int. Ed, 2006, 45, 4732–4762; [DOI] [PubMed] [Google Scholar]; (d) Cozzi F, Adv. Synth. Catal, 2006, 348, 1367–1390; [Google Scholar]; (e) Benaglia M, New J. Chem, 2006, 30, 1525–1533. [Google Scholar]
- 5.(a) Lebreton L, Egger M and Slat B, Sci. Rep, 2019, 9, 1–10; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sheldon RA and Woodley JM, Chem. Rev, 2018, 118, 801–838; [DOI] [PubMed] [Google Scholar]; (c) Jambeck JR, Geyer R, Wilcox C, Siegler TR, Perryman M, Andrady A, Narayan R and Law KL, Science, 2015, 347, 768–771. [DOI] [PubMed] [Google Scholar]
- 6.(a) Moccia F, Rigamonti L, Messori A, Zanotti V and Mazzoni R, Molecules, 2021, 26, 2728–2754; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu J and Toy PH, Chem. Rev, 2009, 109, 815–838; [DOI] [PubMed] [Google Scholar]; (c) Zhao XS, Bao XY, Guo W and Lee FY, Mater. Today, 2006, 9, 32–39; [Google Scholar]; (d) McMorn P and Hutchings GJ, Chem. Soc. Rev, 2004, 33, 108; [DOI] [PubMed] [Google Scholar]; (e) Leadbeater NE and Marco M, Chem. Rev, 2002, 102, 3217–3274; [DOI] [PubMed] [Google Scholar]; (f) McNamara CA, Dixon MJ and Bradley M, Chem. Rev, 2002, 102, 3275–3300; [DOI] [PubMed] [Google Scholar]; (g) De Vos DE, Vankelecom IFJ and Jacobs PA, Chiral Catalysts Immobilization and Recycling, Wiley-VCH, Weinheim, 2000. [Google Scholar]
- 7.(a) Lombardo M and Trombini C, Green Chem., 2009, 3, 1–79; [Google Scholar]; (b) Barbaro P and Liguori F, Chem. Rev, 2009, 109, 515–529; [DOI] [PubMed] [Google Scholar]; (c) Heitbaum M, Glorius F and Escher I, Angew. Chem., Int. Ed, 2006, 45, 4732–4762; [DOI] [PubMed] [Google Scholar]; (d) Horn J, Michalek F, Tzschucke CC and Bannwarth W, Top. Curr. Chem, 2004, 242, 43–75. [DOI] [PubMed] [Google Scholar]
- 8.(a) Zhang B and Reek JN, Chem. – Asian J, 2021, 16, 3851–3863; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Parvulescu VI and Garcia H, Catal. Sci. Technol, 2018, 8, 4834–4857. [Google Scholar]
- 9.(a)Chen R, Bronger RP, Kamer PC, van Leeuwen PW and Reek JN, J. Am. Chem. Soc, 2004, 126, 14557–14566; [DOI] [PubMed] [Google Scholar]; (b) de Groot D, de Waal BF, Reek JN, Schenning AP, Kamer PC, Meijer EW and van Leeuwen PW, J. Am. Chem. Soc, 2001, 123, 8453–8458. [DOI] [PubMed] [Google Scholar]
- 10.Beejapur HA, Giacalone F, Noto R, Franchi P, Lucarini M and Gruttadauria M, ChemCatChem, 2013, 5, 2991–2999. [Google Scholar]
- 11.(a) Sabater S, Mata JA and Peris E, Organometallics, 2015, 34, 1186–1190; [Google Scholar]; (b) Ruiz-Botella S and Peris E, Chem. – Eur. J, 2015, 21, 15263–15271; [DOI] [PubMed] [Google Scholar]; (c) Sabater S, Mata JA and Peris E, ACS Catal., 2014, 4, 2038–2047. [Google Scholar]
- 12.(a) Lu J and Toy PH, Chem. Rev, 2009, 109, 815–838; [DOI] [PubMed] [Google Scholar]; (b) Corma A and Garcia H, Adv. Synth. Catal, 2006, 348, 1391–1412; [Google Scholar]; (c) Astruc D, Lu F and Aranzaes JR, Angew. Chem., Int. Ed, 2005, 44, 7852–7872; [DOI] [PubMed] [Google Scholar]; (d) Chen YC, Wu TF, Jiang L, Deng JG, Liu H, Zhu J and Jiang YZ, J. Org. Chem, 2005, 70, 1006–1010. [DOI] [PubMed] [Google Scholar]
- 13.Gruttadauria M, Giacalone F and Noto R, Green Chem., 2013, 15, 2608–2618. [Google Scholar]
- 14.(a) Cho SH, Ma BQ, Nguyen ST, Hupp JT and Albrecht-Schmitt TE, Chem. Commun, 2006, 2563–2565; [DOI] [PubMed] [Google Scholar]; (b) Wu CD, Hu A, Zhang L and Lin WB, J. Am. Chem. Soc, 2005, 127, 8940–8941; [DOI] [PubMed] [Google Scholar]; (c) Yang JW, Han HY, Roh EJ, Lee SG and Song CE, Org. Lett, 2002, 4, 4685–4688; [DOI] [PubMed] [Google Scholar]; (d) Nagashima T and Davies HML, Org. Lett, 2002, 4, 1989–1992; [DOI] [PubMed] [Google Scholar]; (e) Gerstberger G, Palm C and Anwander R, Chem. – Eur. J, 1999, 5, 997–1005; [Google Scholar]; (f) Fraile JM, García JI, Lázaro B and Mayoral JA, Chem. Commun, 1998, 1807–1808. [Google Scholar]
- 15.(a) Kluwer AM, Simons C, Knijnenburg Q, van der Vlugt JI, de Bruin B and Reek JN, Dalton Trans., 2013, 42, 3609–3616; [DOI] [PubMed] [Google Scholar]; (b) Marras F, van Leeuwen PW and Reek JN, Chem. – Eur. J, 2011, 17, 7460–7471; [DOI] [PubMed] [Google Scholar]; (c) Gruijters BWT, Broeren MAC, van Delft FL, Sijbesma RP, Hermkens PHH and Rutjes FPJT, Org. Lett, 2006, 8, 3163–3166. [DOI] [PubMed] [Google Scholar]
- 16.(a) Takacs JM, Moteki SA and Reddy DS, Supramolecular Catalysis, ed. van Leeuwen PWNM, Wiley-VCH, Weinheim, 2008, pp. 235–253; [Google Scholar]; (b) Atkins JM, Moteki SA, DiMagno SG and Takacs JM, Org. Lett, 2006, 8, 2759–2762; [DOI] [PubMed] [Google Scholar]; (c) Takacs JM, Hrvatin PM, Atkins JM, Reddy DS and Clark JL, New J. Chem, 2005, 29, 263–265. [Google Scholar]
- 17.Zhou J, Cole AM, Menuey EM, Kilway KV and Moteki SA, Chem. Commun, 2021, 57, 6404–6407. [DOI] [PubMed] [Google Scholar]
- 18.(a) Thacker NC, Moteki SA and Takacs JM, ACS Catal., 2012, 2, 2743–2752; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moteki SA, Toyama K, Liu Z, Ma J, Holmes AE and Takacs JM, Chem. Commun, 2012, 48, 263–265; [DOI] [PubMed] [Google Scholar]; (c) Moteki SA and Takacs JM, Angew. Chem., Int. Ed, 2008, 47, 894–897; [DOI] [PubMed] [Google Scholar]; (d) Takacs JM, Reddy DS, Moteki SA, Wu D and Palencia H, J. Am. Chem. Soc, 2004, 126, 4494–4495. [DOI] [PubMed] [Google Scholar]
- 19.Carreiro EP, Moura NMM and Burke AJ, Eur. J. Org. Chem, 2012, 518–528. [Google Scholar]
- 20.Buszek KR and Brown N, J. Org. Chem, 2007, 72, 3125–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Carneiro L, Silva AR, Shuttleworth PS, Budarin V and Clark JH, Molecules, 2014, 19, 11988–11998; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Silva AR, Guimaraes V, Carvalho AP and Pires J, Catal. Sci. Technol, 2013, 3, 659–672. [Google Scholar]
- 22.(a) Toma S, Green Chem., 2012, 15, 18–57; [Google Scholar]; (b) Carpenter RD, Fettinger JC, Lam KS and Kurth MJ, Angew. Chem., Int. Ed, 2008, 47, 6407–6410. [DOI] [PubMed] [Google Scholar]
- 23.(a) Martinez A, Zumbansen K, Dohring A, Gemmeren M and List B, Synlett, 2014, 932–934; [Google Scholar]; (b) Kano T, Sugimoto H and Maruoka K, J. Am. Chem. Soc, 2011, 133, 18130–18133; [DOI] [PubMed] [Google Scholar]; (c) List B, Hoang L and Martin HJ, Proc. Natl. Acad. Sci. U. S. A, 2004, 101, 5839–5842; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mase N, Tanaka F and Barbas CF, Org. Lett, 2003, 5, 4369–4372; [DOI] [PubMed] [Google Scholar]; (e) List B, Lerner RA and Barbas CF III, J. Am. Chem. Soc, 2000, 122, 2395–2396. [Google Scholar]
- 24.(a) Yang H, Liu D, Yu Q, Xia S, Yu D, Zhang M, Sun B and Zhang F, Eur. J. Org. Chem, 2019, 852–856; [Google Scholar]; (b) Limnios D and Kokotos C, RSC Adv., 2013, 3, 4496–4499; [Google Scholar]; (c) Marigo M and Melchiorre P, ChemCatChem, 2010, 2, 621–623; [Google Scholar]; (d) Zheng C, Wu Y, Wang X and Zhao G, Adv. Synth. Catal, 2008, 350, 2690–2694; [Google Scholar]; (e) Chimni S and Mahajan D, Tetrahedron, 2005, 61, 5019–5025; [Google Scholar]; (f) Cordova A, Notz W and Barbas CF, Chem. Commun, 2002, 3024–3025. [DOI] [PubMed] [Google Scholar]
- 25.The % enantiomeric excess using l-proline under the same condition was 80%.
- 26.(a) Sun T, Liang H, Liu S, Tang E and Fu C, J. Nanopart. Res, 2020, 22, 163; [Google Scholar]; (b) Zheng Z, Wang J, Zhang M, Xu L and Ji J, ChemCatChem, 2013, 5, 307–312; [Google Scholar]; (c) Saito K, Hirose K, Okayasu T, Nishide H and Hearn MTW, RSC Adv., 2013, 3, 9752–9756; [Google Scholar]; (d) Suzuki Y, Iinuma M, Moriyama K and Togo H, Synlett, 2012, 1250–1256; [Google Scholar]; (e) Fey T, Fischer H, Bachmann S, Albert K and Bolm C, J. Org. Chem, 2001, 66, 8154–8159. [DOI] [PubMed] [Google Scholar]
- 27.(a) Okada T, Asawa T, Sugiyama Y, Iwai T, Kirihara M and Kimura Y, Tetrahedron, 2016, 72, 2818–2827; [Google Scholar]; (b) Beejapur HA, Campisciano V, Giacalone F and Gruttadauria M, Adv. Synth. Catal, 2015, 357, 51–58; [Google Scholar]; (c) Beejapur HA, Giacalone F, Noto R, Franchi P, Lucarini M and Gruttadauria M, ChemCatChem, 2013, 5, 2991–2999; [Google Scholar]; (d) Zhu C and Wei Y, Adv. Synth. Catal, 2012, 354, 313–320; [Google Scholar]; (e) Lu N and Lin YC, Tetrahedron Lett., 2007, 48, 8823–8828; [Google Scholar]; (f) Karimipour GR, Shadegan HA and Ahmadpour R, J. Chem. Res, 2007, 4, 252–256. [Google Scholar]
- 28.(a) Wang Y, Wang C, Cheng Q, Su Y, Li H, Xiao R, Tan C and Liu G, Green Chem., 2021, 23, 7773–7779; [Google Scholar]; (b) Fan H, Yang Y, Song J, Ding G, Wu C, Yang G and Han B, Green Chem., 2014, 16, 600–604; [Google Scholar]; (c) Akagawa K, Takigawa S, Mano E and Kudo K, Tetrahedron Lett., 2011, 52, 770–773. [Google Scholar]
- 29.(a) Ueda Y, Ito H, Fujita D and Fujita M, J. Am. Chem. Soc, 2017, 139, 6090–6093; [DOI] [PubMed] [Google Scholar]; (b) Sasano Y, Nagasawa S, Yamazaki M, Shibuya M, Park J and Iwabuchi Y, Angew. Chem., Int. Ed, 2014, 53, 3236–3240. [DOI] [PubMed] [Google Scholar]