

Abstract
NadR is a 45-kDa bifunctional regulator protein. In vivo genetic studies indicate that NadR represses three genes involved in the biosynthesis of NAD. It also participates with an integral membrane protein (PnuC) in the import of nicotinamide mononucleotide, an NAD precursor. NadR was overexpressed and purified as a His-tagged fusion in order to study its DNA-binding properties. The protein bound to DNA fragments containing NAD box consensus sequences. NAD proved to be the relevant in vivo corepressor, but full NAD dependence of repressor activity required nucleotide triphosphates. DNA footprint analysis and gel shift assays suggest that NadR binds as a multimer to adjacent NAD boxes. The DNA-repressor complex would sequester a potential RNA polymerase binding site and thereby decrease expression of the nad regulon.
The pyridine nucleotides NAD and NADP play central roles in cellular metabolism. Because of the broad impact of NAD cellular physiology, maintenance of optimal levels of this pyridine nucleotide is critical for efficient cell growth. Genes involved in the recycling (pncB) and biosynthesis (nadB and the nadA-pnuC operon) of NAD are transcriptionally repressed in Salmonella typhimurium by the product of nadR (7), also referred to as nadI (34). Regulation by NadR presumably occurs in response to internal NAD concentrations, although this has never been shown directly (16, 32). In addition to its role as a transcriptional regulator, NadR is also important in the transport of nicotinamide mononucleotide (NMN) as an exogenous precursor of NAD (8, 19, 23, 32). The transport of this phosphorylated compound requires both NadR and PnuC, an apparent integral membrane protein. PnuC transporter activity is modulated by NadR in response to internal pyridine nucleotide levels (23, 32, 33).
The cloning and sequencing of this locus confirmed that a single gene complements both transport and transcriptional regulator functions, establishing NadR as a unique bifunctional regulator (8). Other known bifunctional transcriptional regulators, such as the biotin holoenzyme synthetase, BirA (1), and the PutA proline dehydrogenase, have enzymatic activities as their second function (21, 22). NadR, in contrast, does not appear to have an enzymatic activity but regulates the activity of the PnuC transporter. Single-strand conformational polymorphisms of PCR-amplified regions from various nadR mutants have shown that both transport and regulatory phenotypes map within the nadR open reading frame (9). Those studies also established that repressor function primarily resides within the amino-terminal half of the protein while transport function is associated with the carboxyl end. In this study, wild-type NadR and a His-tagged NadR fusion protein were purified to provide in vitro DNA binding evidence for the regulatory function of this protein.
MATERIALS AND METHODS
Bacterial strains and media.
The bacterial strains used in this work are listed in Table 1. Plasmid pGP1-2 was a gift from S. Tabor (25). The medium used was the minimal E medium, containing 0.4% glucose and Luria-Bertani (LB) medium, of Vogel and Bonner (29). Ampicillin (Ap) (60 μg/ml), chloramphenicol (Cm) (30 μg/ml), and kanamycin (Km) (50 μg/ml) were added as needed.
TABLE 1.
Strains used
| Strain | Characteristics | Source or reference |
|---|---|---|
| E. coli | ||
| EK192 | (HMS174) F−hsdR recA, Rifr (rK− mK+) | Novagen |
| EK193 | EK192(DE3) λ lysogen; lacUV5 controlled expression of T7 RNA polymerase | Novagen |
| EK194 | EK193/pLysS; T7 lysozyme under the control of the φ T7 3.8 promoter | Novagen |
| EK258 | EK192 /pFP129; His-taged NadR61 fusion | EK192 transformed with pFP129 |
| EF270 | EK194 /pFP129 | EK194 transformed with pFP129 |
| S. typhimurium | ||
| SF73 (TT6202) | pncA::Tn10 | J. R. Roth |
| SF586 (JR501) | hsdSA29 hsdSB121 hsdL6 metA22 metE551 trpC2 ilv452 rpl-102 xyl-404 galE719 fla66 nml H1-b H2-e (Fels2) | 27 |
| JF1057 | Δ(nadA) galE hutR Δ(pnuA) Δ(pnuA-nadR)4 recA1 Δ(srl) | 8 |
| JF1521 | nadB1017::MudJ | |
| JF1943 | JF1057/pFW38-46 | |
| JF1945 | JF1057/pGP1-2 (Kmr, 30°C) | |
| JF1947 | JF1057/pFW38-46 (nadR+)/pGP1-2 (T7 Pol) | 8 |
| JF3005 | nadB1017::MudJ | SF586 × P22HTint (JF1521) |
| JF3008 | pncA::Tn10, nadB1017::MudJ | JF3005 × P22HTint (SF73) |
| JF3010 | JF3005/pFP132 (nadA-O) | |
| JF3011 | JF3008/pFP204 (pncB-O) |
Plasmid constructions.
pFW38-46 was used to produce wild-type NadR for purification. It contains an intact, wild-type nadR gene controlled by its natural promoter and a T7 promoter (8). To produce His-tagged NadR, a 1.2-kb nadR fragment was cloned into the NdeI and BamHI sites of pET15b (Novagen, Madison, Wis.). The nadR fragment was produced by PCR amplification from the chromosome of S. typhimurium by using oligonucleotide primers oligo 40 (GAGGCTCATATGTCATCGTTC; overlaps the nadR start codon) and oligo 41 (GCTGGATCCGAAGCGTATC; starts at nucleotide 1392 [8]). The primers were designed (see underlined bases) to incorporate restriction sites for NdeI (oligo 40) and BamHI (oligo 41). Cleaving the PCR product with NdeI and BamHI removes the nadR Shine-Dalgarno sequence. The resulting plasmid, pFP129, will not express nadR without specific induction by isopropyl-β-d-thiogalactopyranoside (IPTG), a feature that allowed stable maintenance of this plasmid in EF258 and EF270.
Plasmid pTF23 containing nadA on a 1.9-kb insert was described previously (26). A 1.4-kb HincII fragment from pTF23 was subcloned into pTZ19R (Pharmacia, Uppsala, Sweden), producing pFP79. A 354-bp HaeIII-HpaII fragment from pFP79 containing the predicted nadA operator was subcloned into pTZ19R, producing pFP80. A 377-bp KpnI-PstI fragment from pFP80 was used for some gel retardation experiments. A 174-bp Fnu4HI fragment from pFP80 containing both NAD boxes of the nadA operator was blunt ended with T7 DNA polymerase and ligated to HincII-digested pBluescript SK(+), producing plasmid pFP108. A 200-bp XhoI-HindIII fragment from pFP108, also containing both NAD boxes, was subcloned into pSP70 (Promega, Madison, Wis.), producing pFP132. This plasmid was used to amplify and label the nadA operator by using the SP6 and T7 primers that flank the insert. The product was a 225-bp fragment. For pncB-NadR interactions, a 300-bp SalI-NdeI fragment of pRM18.1 (28) carrying the pncB operator was gel purified and ligated into pSP70 to generate pFP204. Strains of Escherichia coli and galE mutants of Salmonella were transformed by a rapid CaCl2 method (12, 27).
Purification of wild-type NadR.
Wild-type NadR was purified from cells (JF1947) grown to late log phase in E glucose with Ap, Km, and 0.1 mM NMN by established methods (25). 35S-labelled NadR, used as a tracer during purification, was prepared from a 100-ml culture. Rifampin was added to 300 μg/ml to inhibit host RNA polymerase, and after 30 min 10 μCi of 35S-Trans label (ICN) per ml was added and the culture was then incubated for 5 min before harvesting. A small amount of labelled [35S]NadR-containing cells (0.1 g, wet weight) was added to 20 g (wet weight) of unlabelled NadR-overproducing cells in two volumes of buffer A (50 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 1 mM EDTA, and 1.0 mM dithiothreitol [DTT]) with 0.1 mM phenylmethylsulfonyl fluoride (PMSF) added just before sonication. Crude sonicate is shown in Fig. 1A, lane 2. Cleared lysate obtained after centrifugation at 20,000 × g for 20 min (4°C) is shown in Fig. 1A, lane 3. Cleared lysate was subjected to precipitation with polyethylenimine (PEI; Sigma) (10% [wt/vol], pH 8.0; 35 μl/ml of lysate) (3a). The pellet was washed in buffer A containing 0.4 M NaCl, collected by centrifugation, and homogenized in buffer A with 1.0 M NaCl to extract NadR. The NadR-containing supernatant was brought to 60% saturation with solid ammonium sulfate. Precipitated proteins, including NadR, were recovered by centrifugation and redissolved in buffer A, and the solution was clarified by centrifugation (Fig. 1A, lane 4). The clarified solution was subjected to chromatography through an Affi-Gel Blue column (1 cm wide by 15 cm long; Bio-Rad Laboratories, Hercules, Calif.). NadR eluted at 0.7 M NaCl in a 0 to 1.0 M NaCl linear gradient. NadR-containing radioactive fractions were combined and brought to 55% saturation with ammonium sulfate, and the precipitate was dissolved in 20 ml of buffer A (Fig. 1A, lane 5). This material was loaded onto a DE-52 column (Whatman) (1 cm wide by 8 cm long), and NadR eluted at 0.2 M NaCl in a 0 to 0.5 M NaCl linear gradient in buffer A. Radioactive fractions containing NadR were precipitated with ammonium sulfate (60% saturation) and resuspended in buffer A (Fig. 1A, lane 6). Partially purified NadR was stored as an ammonium sulfate suspension at 4°C.
FIG. 1.

Purification of NadR. (A) Wild-type NadR. Shown in this Coomassie blue-stained SDS–11.5% PAGE gel are the relevant fractions from the purification of NadR described in Materials and Methods. Lanes 1, low-molecular-mass markers; lane 2, crude sonicate; lane 3, cleared lysate; lane 4, 1.0 M NaCl extraction of PEI precipitate; lane 5, Affi-Gel Blue pool; lane 6, DE-52 eluate. (B) Purification of His-tagged NadR fusion protein. Shown are Coomassie blue-stained SDS–11.5% PAGE gels of EF270 uninduced crude extracts (lane 2) and crude extracts after 2 h of IPTG induction (lane 3) and Coomassie blue-stained SDS-PAGE gels of soluble protein fractions following lysis and clarification to remove insoluble material (lane 5) and after elution from the Ni2+ chelate column following ammonium sulfate precipitation and redissolving of the sample in Novagen column buffer (lane 6). Molecular mass markers are shown in lanes 1 and 4.
Purification of His-tagged NadR.
EF270 containing pFP129 was grown to an optical density at 600 nm (OD600) of 0.5 in 100 ml of LB medium containing Ap and Cm and induced with 1.0 mM IPTG for 2 h. Cells were harvested, lysed, and purified from a Ni2+-charged metal chelate column per the recommendations of the manufacturer (Novagen). Protein concentrations were determined with a Bio-Rad protein assay (2). Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) was performed by using 11.5% acrylamide (gels 18). Since the His-tagged NadR performed as well as wild-type NadR in gel retardation assays (data not shown), the 20-amino-acid tag was not removed for this study. When needed, radiolabelled His-tagged NadR was produced by growing EF270 in E medium to an OD600 of 0.5. Expression was induced with 1.0 mM IPTG for 0.5 h, after which 20 μCi of 35S-Trans label (ICN) per ml was added for 1.5 h and the culture was harvested.
Gel retardation assays.
Band shift or gel retardation assays were performed in 3% polyacrylamide gels (10, 11). The acrylamide gels were aged for 24 to 96 h or prerun (10 V/cm for 1 h). Radiolabelled DNA fragments were prepared by using pFP132, containing the nadA promoter, in a standard PCR protocol employing SP6 and T7 oligonucleotide primers, with one of the primers being end labelled with 32P by using T4 polynucleotide kinase. After PCR amplification, the labeled fragment (225 bp) was purified (Promega Wizard PCR cleanup resin; direct extraction technique). The same PCR strategy was used for the pncB operator with the substitution of pFP204 as the template, producing a 370-bp labelled fragment.
Band shift assays were performed in 30-μl reaction volumes that included approximately 5,000 cpm of labelled operator DNA, 20 mM Tris-HCl (pH 7.9), 50 mM KCl, 1.0 mM DTT, 75 μg of acetylated bovine serum albumin (BSA) per ml, 1.0 mM EDTA, and 10 mM MgCl2. Additions made to the core binding mix were NadR, NAD, and ATP at 6.4 nM, 600 μM, and 60 μM, respectively, unless indicated otherwise. When used, competitor DNA (pTZ19R) was added to 25 μg/ml. Wild-type or His-tagged NadR was added to the reaction mixture last, in a small volume (less than 5% of the total reaction volume). The mixture was incubated at 37°C for 10 min. Eight microliters of ice-cold gel loading buffer (stock solution; 33% glycerol and 0.03% bromphenol blue) was added, and the sample was mixed by gentle stirring to avoid shearing. The samples were immediately loaded onto polyacrylamide gels and electrophoresed at 4°C and 10 V/cm in Tris-borate-EDTA–3% polyacrylamide gels.
Stoichiometry and gel filtration.
To determine the molar ratio of NadR protein bound to DNA, gel retardation assays were performed by using 3H-labelled NadR and 32P-labelled nadA operator fragments (225-bp fragments; see above). The specific activities of the DNA and His-tagged NadR samples were determined to be 1.7 × 10−10 and 1.2 × 10−9 μmol/dpm, respectively. The gel was sliced into 1 cm by 1 cm squares and digested for 24 h at 42°C in 100 μl of 21% H2O2 with 16% perchlorate before addition of scintillant. Differential counts were performed in a Beckman LS6800 scintillation counter and an LKB 1219 Rackbeta liquid scintillation counter. To determine its molecular weight in solution, His-tagged NadR was loaded onto Sephacryl S-300 columns equilibrated with Novagen 0.1 eluate buffer with 1% dimethyl sulfoxide. His-tagged NadR was loaded at several concentrations (ranging from 0.05 to 2.0 mg/ml) onto a 1.5 cm (inside diameter) by 45 cm (outside diameter) column at a flow rate of 0.17 ml/min.
DNase I footprinting.
Primer pair T7 and SP6, with one primer end labelled with 32P, was used in standard PCRs to generate NAD box-containing target fragments from pFP132 (nadA) or pFP204 (pncB). DNA-binding reactions for footprinting were carried out in 130-μl reaction volumes containing 20 mM Tris-HCl (pH 7.9), 50 mM KCl, 1.0 mM DTT, 75 μg of acetylated BSA per ml, 1.0 mM EDTA, 10 mM MgCl2, 25 μg of pTZ19R competitor DNA per ml, 32P-labelled operator DNA fragment at 175 cpm/μl, and ATP at 60 μM. His-tagged NadR and NAD were added as indicated in a given experiment. The mixture was incubated at room temperature for 30 min, and 30 μl was removed for a gel retardation assay. The remaining 100 μl was treated with 0.1 U of DNase I (BRL) and incubated at room temperature for 4 min. A 46-μl aliquot of DNase stop solution (55 mM EDTA, 6.8 M NH4CH3COOH, and 440 μg of tRNA per ml) was added, and the DNA was ethanol precipitated. The DNA pellet was dissolved in formamide gel loading buffer (Promega), heated for 5 min at 75°C, and loaded onto a 6% Long Ranger sequencing gel (FMC, Rockland, Maine).
Determination of dissociation constants.
The apparent affinities between NadR and the nadA and pncB operators were determined from DNase I protection studies. Binding isotherms were generated from points calculated at different NadR concentrations from the following equation:
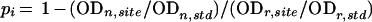 |
In this equation, pi represents the fractional protection of the DNA backbone against nuclease cleavage at a specific NadR concentration (DNase I protection). Two types of bands were chosen for comparison within each experimental (n) and reference (no DNase) (r) lane. One band or set of bands represented NadR- protected regions (site). The second set of bands (control bands) was those located in unprotected regions (std). Each site and std band was quantified by densitometric analysis.
RESULTS
Purification of wild-type and His-tagged NadR.
Wild-type NadR was found to be overexpressed in S. typhimurium when examined using a two-plasmid system in which one plasmid (pFW38-46) carried the nadR gene under the control of a T7 phage promoter and a second plasmid (pGP1-2) encoded an inducible T7 RNA polymerase gene (25). Wild-type NadR produced in this strain (JF1947) was purified to approximately 70% (Fig. 1A). Although this preparation performed well in gel retardation experiments (see below), attempts to further purify wild-type NadR failed. Consequently, a His-tag fusion to nadR was constructed in the pET15b vector (Novagen). His-tagged NadR was easily purified to approximately 95% (Fig. 1B) and functioned similarly to wild-type NadR preparations.
NadR specifically binds to nad regulon operator regions.
Previous DNA sequencing studies identified an inverted repeat in the predicted operator of each NadR-regulated gene (6, 8, 28, 31). Subsequent comparisons between these putative operator regions revealed a consensus sequence, referred to as the NAD box, consisting of TGTTTA and its inverted repeat separated by 5 to 6 base pairs, or approximately one-half of a helical turn. The nadA and nadB operators contain two NAD boxes overlapping potential RNA polymerase binding sites (box 1) and ribosome binding sites (RBS) (box 2), whereas the pncB loci contain only one complete NAD box which overlaps an RBS. Because of this conservation, the NAD box sequence was predicted to define the NadR binding site. Gel retardation assays performed using NadR protein and nadA operator-containing fragments from pFP79 revealed that NadR specifically bound to a 354-bp HaeIII-HpaII region that included the putative NAD box region (Fig. 2). His-tagged NadR preparations bound to the same fragment (data not shown).
FIG. 2.

DNA band shifts of specific nad operator-containing fragments. Plasmids pTF23 (A) and pFP79 (B) contain multiple bacterial promoters, including that of nadA (hatched box). Each plasmid was digested with the restriction enzymes shown (bottom). The wild-type NadR preparation exhibited specific binding to DNA fragments containing the nadA promoter/operator. Gel retardation assays were performed by incubating 100 nM DNA with 200 nM wild-type NadR with or without 666 μM NAD+ (as indicated in upper panels) in the gel retardation mix (20 mM Tris-HCl [pH 7.5], 50 mM KCl, 1.0 mM DTT, 1.0 mM EDTA, 75 μg of acetylated BSA [Promega] per ml, 10 mM MgCl2). Arrowheads indicate free DNA and NadR-DNA complexes for nadA operator-containing fragments. Gels were stained with ethidium bromide.
NTPs diminish NAD-independent binding of NadR to operator DNA.
Although the preliminary gel retardation results suggested that NadR must interact with NAD to effectively bind the operator region (Fig. 2), we observed that wild-type NadR exhibited NAD-independent binding to operator DNA after extended storage (for more than 1 month) of the enzyme at 4°C (data not shown). The same phenomenon was observed with freshly purified His-tagged NadR (Fig. 3A, lane 1). This result suggested that an effector molecule associated with NadR may have been inactivated during storage (wild-type NadR) or lost during purification (His-tagged NadR). DNA sequence analysis of nadR revealed the presence of a consensus sequence, starting at amino acid position 237, that is indicative of a mononucleotide binding site (8, 20). Because of this observation, various nucleotides were tested for their ability to maintain NadR as an NAD-dependent DNA-binding protein. ATP prevented the NAD-independent binding of NadR to operator DNA (Fig. 3A; compare lanes 1 and 2) but did not diminish the effectiveness of NAD as a corepressor (Fig. 3A; compare lane 2 with lane 7 and lane 6 with lane 7). This result was the same whether wild-type or His-tagged NadR preparations were tested. We also examined whether other nucleotide triphosphates (NTPs) could inhibit NAD-independent binding. GTP, CTP, and UTP all reversed NAD-independent DNA binding (Fig. 3A, lanes 3, 4, and 5) while not interfering with NAD-dependent binding (Fig. 3A, lanes 8, 9, and 10). Mononucleotides had no effect, while dinucleotides did have a slight effect (data not shown). As a result of this finding, all subsequent gel retardation assays contained ATP unless indicated otherwise.
FIG. 3.

Effects of ribonucleotides (A) and pyridine nucleotides (B) on NadR DNA binding. Panels present band shift results obtained by using His-tagged NadR and radiolabelled nadA operator DNA fragments (225-bp PCR product from pFP132). Basic assay conditions were as described in Materials and Methods, with 6.5 nM NadR being used in each reaction mixture. Panel A shows the effect of ribonucleotides on NAD-independent DNA binding by NadR. NAD and various ribonucleotides were added as indicated. Panel B shows the identity of the NadR corepressor. Basic assay conditions were the same as for panel A except that 60 μM ATP was added to all binding reaction mixtures. The indicated pyridine nucleotide cycle intermediates (nucleotide) were all added at 660 μM. QA, quinolinic acid; NA, nicotinic acid; NAMN, nicotinic acid mononucleotide; NAm, nicotinamide; NAAD, nicotinic adenine dinucleotide.
NAD is the corepressor for NadR.
There are several compounds within the pyridine nucleotide cycle that could fill the role of corepressor for this system. Indirect genetic evidence implicating NAD as the corepressor of the NAD regulon has been reported (32). However, no direct evidence has been reported. Consequently, in vitro DNA band shift assays were used to prove that NAD, and no other pyridine nucleotide cycle intermediate, is the corepressor for NadR. The results indicated that only NAD+ serves as a corepressor at relevant in vivo levels (Fig. 3B, lane 8). All other pyridine nucleotide intermediates were invalidated as potential candidates. While quinolinic acid, nicotinic acid, nicotinic acid mononucleotide, nicotinamide, and nicotinic adenine dinucleotide showed no detectible corepressor activity, NMN, NADH, and NADP were slightly active at 660 μM, a concentration 10- to 100-fold higher than their measured in vivo levels. At physiological levels they had no effect (data not shown). Thus, the data suggest that NAD+ is the in vivo corepressor for this system.
Stoichiometry.
Column chromatography was used to determine the oligomeric state of NadR in solution. The majority (80%) of NadR (45 kDa) migrated with an Mr of 90 kDa under native conditions on a Sephacryl (Pharmacia) S-300 column, suggesting that, in solution, NadR exists as a dimer (data not shown). 3H- or 35S-labelled NadR and 32P-labelled operator DNA were utilized to determine the number of NadR molecules present in a protein-DNA complex. Gel slices from one dual-label gel retardation experiment produced 575 dpm from [32P]DNA and 281 dpm from [3H]NadR. This represented 9.6 × 10−8 μmol of DNA and 3.4 × 10−7 μmol of protein, for a 1:3.5 molar ratio. A second experiment with 35S-labelled NadR displayed 429 dpm from 32P (7.3 × 10−8 μmol of DNA) and 486 dpm from 35S (3.2 × 10−7 μmol of protein). The average from these experiments is four NadR molecules per nadA operator. This agrees with a model in which a dimer of NadR binds to each NAD box within the operator region.
DNase I footprinting.
The results of DNase I footprinting experiments are shown in Fig. 4 (nadA) and Fig. 5 (pncB). Each figure shows the sequence protected by NadR in the nontemplate (i.e., coding) and template strands. Two regions were protected in nadA, while only one was protected within pncB. Each protected sequence was centered around the NAD boxes. The presence of DNase I-hypersensitive sites (Fig. 4) between the two protected NAD box regions in nadA-O is indicative of torsional strain placed on the DNA by bending (15). This finding provides additional support for the idea that NadR forms a DNA loop within the nad operator. In contrast, the pncB operator, which only has only one complete NAD box, did not display any hypersensitive sites.
FIG. 4.

DNA footprint of NadR on nadA operator. Basic conditions for the assay performed by using His-tagged NadR are the same as described in the legend to Fig. 3B. Lanes marked G, A, T, and C are a DNA sequencing ladder of the coding strand (A) and template strand (B) of the 225-bp nadA operator-containing PCR fragment (pFP132). Lanes 1 to 4 show the results of DNase I protection assays. For panel A, lanes 1 and 2 contain 22 nM His-tagged NadR, and lanes 3 and 4 contain 11 nM His-tagged NadR. Lanes 1 and 3 contain no NAD. Lanes 2 and 4 contain 660 μM NAD. For panel B, lane 1 contains no NadR while lanes 2 and 3 contain 11 nM His-tagged NadR. Only lane 3 has 660 μM NAD. The DNA sequence of the nadA operator is shown to the left in each panel. Consensus −10 and −35 RNA polymerase recognition sequences are indicated. Bases protected against and hypersensitive to DNase I treatment are illustrated adjacent to the printed sequence by boxes and circles, respectively. A rectangle next to the footprint indicates the location of the protected region. Brackets around printed sequence indicate bases consistent with consensus NAD box sequences (TGTTTA and inverted repeat).
FIG. 5.

DNA footprint of NadR on the pncB operator. Basic assay conditions are the same as described in the legend for Fig. 3B. Lanes marked G, A, T, and C are a DNA sequencing ladder of the coding strand (panel A) and template strand (panel B) of the 370-bp pncB operator-containing PCR fragment (produced from pFP204). Lanes 1 to 5 show the results of DNase I protection assays. For each panel, lanes 2 and 3 contain 92 nM His-tagged NadR and lanes 4 and 5 contain 46 nM His-tagged NadR. Lanes 1, 3, and 5 contain no NAD. Lanes 2 and 4 contain 660 μM NAD. The DNA sequence of the pncB operator is shown to the left in each panel. Consensus −10 and −35 RNA polymerase recognition sequences are indicated. Bases protected from DNase I treatment are illustrated adjacent to the printed sequence by boxes. There are no NadR-induced hypersensitive bases. A rectangle next to the footprint indicates the location of the protected region. Brackets around printed sequence indicate bases consistent with consensus NAD box sequences (TGTTTA and inverted repeat).
Dissociation constants.
In addition to locating the regions within nad operators that bind NadR, DNase I protection also provided a means for determining NadR-DNA binding constants in solution (3). Results presented in Fig. 6 indicate that the NadR/nadA-O apparent dissociation constant is 3 nM in solution (in the presence of 660 μM NAD). In the absence of NAD, no protection of the nadA operator was observed, even at 4,000 nM NadR. The apparent dissociation constant for NadR/pncB-O was 18 nM, reflecting a sixfold-lower affinity of this operator for NadR as predicted from earlier results (16, 17). No evidence of cooperativity was observed. Both NAD boxes were equally affected at the nadA operator upon dilution of the NadR preparation.
FIG. 6.

Equilibrium curves of NadR binding to nadA and pncB operators. The data are percent saturation of NadR bound to DNA based on the percent protection from DNase I cleavage (midpoint of each curve approximates Kd(app)). Data points were generated by titrating His-tagged NadR on the nadA and pncB operators in solution and determining relative band intensities in protected and unprotected regions by using phosphor screen technology and autoradiograms. DNA binding conditions were the same as described in the legend for Fig. 4 with the addition of NAD. Closed squares, nontemplate (coding) strand of nadA; diamonds, template strand of nadA; open squares, nontemplate (coding) strand of pncB; stars, template strand of pncB.
DISCUSSION
The simplest working model for NadR function as supported by the available data is that NadR undergoes a reversible conformational shift between repressor and transport facilitator in response to NAD levels. The in vitro studies prove that, in the presence of NTPs, NAD is required for NadR to effectively bind appropriate operator DNA sequences. Once in its repressor conformation, NadR will bind to an NAD box consisting of TGTTTA and its inverted repeat. The nadA and nadB operators contain two NAD boxes overlapping potential −10 RNA polymerase binding sites (box 1) and RBS (box 2). Protein-protein interactions between the two bound NadR dimers would create an NadR tetramer and a DNA loop structure that sequesters the −10 RNA polymerase binding site.
The data also offer a possible explanation for the variable degrees of repression observed when the nadA and nadB genes (15- to 30-fold [16]) are compared to the pncB gene (2- to 4-fold [17]). DNA sequence analyses of these genes reveal that nadA and nadB each contain two complete NAD boxes, separated by 42 and 20 bp, respectively. However, the pncB operator contains only one complete and one incomplete NAD box, separated by 24 bp. The results shown in Fig. 5 suggest that in pncB, NadR binds only to the complete NAD box. Thus, NadR would only weakly block RNA polymerase movement on pncB. This is supported by the higher dissociation constant of NadR for pncB compared to nadA (Fig. 6).
NadR appears to be compartmentalized into several functional domains. The repressor and transport activities are located within the amino- and carboxyl-terminal ends, respectively (9). The central region appears important for signaling the transition between the two proposed forms of this protein. A consensus sequence diagnostic for mononucleotide binding sites (GGESSGKSTL) occurs within the central region of NadR (position 237 [8]) and may form part of the binding site for NTPs and/or NAD. The classic dinucleotide binding site, Gly-X-Gly-XX-Gly, does not occur within NadR, nor is there significant sequence similarity to the NAD binding sites of ADP-ribosylating toxins (5). However, there is a family of NAD-dependent aldehyde dehydrogenases where NAD utilizes a consensus sequence, (GLIVMFA)E(SILMTAC)(GS)G(KNLM)(SADN)(TAPFV), as part of its binding site (GCG version 9.1; Motifs Software, Madison, Wis.) (4, 13, 14, 24, 30). NadR may utilize a similar strategy since the sequence within NadR starting at amino acid 238 matches this consensus (GESSGKST) and, in fact, overlaps the mononucleotide site noted above. The coincident location of potential ATP and NAD binding sites may be a key factor in the modulation of NadR by these nucleotides.
ACKNOWLEDGMENTS
We thank P. Couling and N. Nixon for typing the manuscript and M. Spector, B. Stitt, and H. Winkler for insightful discussions. Special thanks to C. Grubmeyer for help in purifying the wild-type NadR protein.
This work was supported by Public Health Service grant GM39018 from the National Institutes of Health.
REFERENCES
- 1.Barker D F, Campbell A M. Genetic and biochemical characterization of the birA gene and its product: evidence for a direct role of biotin holoenzyme synthetase in repression of the biotin operon in Escherichia coli. J Mol Biol. 1981;146:469–492. doi: 10.1016/0022-2836(81)90043-7. [DOI] [PubMed] [Google Scholar]
- 2.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 3.Brenowitz M, Senear D, Jamison E, Dalma-Weiszhausz D. Footprinting of nucleic acid-protein complexes. New York, N.Y: Academic Press, Inc.; 1993. [Google Scholar]
- 3a.Burgess R R. Use of polyethyleneimine in purification of DNA binding proteins. Methods Enzymol. 1991;208:3–10. doi: 10.1016/0076-6879(91)08003-z. [DOI] [PubMed] [Google Scholar]
- 4.Cook R J, Lloyd R S, Wagner C. Isolation and characterization of cDNA clones for rat liver 10-formyltetrahydrofolate dehydrogenase. J Biol Chem. 1991;266:4965–4973. [PubMed] [Google Scholar]
- 5.Domenighini M, Montecucco C, Ripka W C, Rappuoli R. Computer modelling of the NAD binding sites of ADP-ribosylating toxins: active-site structure and mechanism of NAD binding. Mol Microbiol. 1991;5:23–31. doi: 10.1111/j.1365-2958.1991.tb01822.x. [DOI] [PubMed] [Google Scholar]
- 6.Flachmann R, Kunz N, Seifert J, Gütlich M, Wientjes F-J, Läufer A, Gassen H G. Molecular biology of pyridine nucleotide biosynthesis in Escherichia coli: cloning and characterization of quinolinate synthesis genes nadA and nadB. Eur J Clin Chem Clin Biochem. 1988;175:221–228. doi: 10.1111/j.1432-1033.1988.tb14187.x. [DOI] [PubMed] [Google Scholar]
- 7.Foster J W, Holley-Guthrie E A, Warren F. Regulation of NAD metabolism in Salmonella typhimurium: genetic analysis and cloning of the nadR repressor locus. Mol Gen Genet. 1987;208:279–287. doi: 10.1007/BF00330454. [DOI] [PubMed] [Google Scholar]
- 8.Foster J W, Park Y K, Penfound T, Fenger T, Spector M P. Regulation of NAD metabolism in Salmonella typhimurium: molecular sequence analysis of the bifunctional nadR regulator and the nadA-pnuC operon. J Bacteriol. 1990;172:4187–4196. doi: 10.1128/jb.172.8.4187-4196.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foster J W, Penfound T A. The bifunctional NadR regulator of Salmonella typhimurium: location of regions involved with DNA binding, nucleotide transport and intramolecular communication. FEMS Microbiol Lett. 1993;112:179–184. doi: 10.1111/j.1574-6968.1993.tb06445.x. [DOI] [PubMed] [Google Scholar]
- 10.Fried M G, Crothers D M. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 1981;9:6505–6525. doi: 10.1093/nar/9.23.6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garner M, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the E. coli lactose operon regulatory system. Nucleic Acids Res. 1981;9:3047–3060. doi: 10.1093/nar/9.13.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hackett P B, Fuchs J A, Messing J W. An introduction to recombinant DNA techniques: basic experiments in gene manipulations. Menlo Park, Calif: Benjamin/Cummings, Inc.; 1984. [Google Scholar]
- 13.Hempel J, Harper K, Lindahl R. Inducible (class 3) aldehyde dehydrogenase from rat hepatocellular carcinoma and 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated liver: distant relationship to the class 1 and 2 enzymes from mammalian liver cytosol/mitochondria. Biochemistry. 1988;28:1160–1167. doi: 10.1021/bi00429a034. [DOI] [PubMed] [Google Scholar]
- 14.Hidalgo E, Chen Y-M, Lin E C C, Aguilar J. Molecular cloning and DNA sequencing of the Escherichia coli K-12 ald gene encoding aldehyde dehydrogenase. J Bacteriol. 1991;173:6118–6123. doi: 10.1128/jb.173.19.6118-6123.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hochschild A. Detecting cooperative protein-DNA interactions and DNA loop formation by footprinting. Methods Enzymol. 1991;208:343–361. doi: 10.1016/0076-6879(91)08019-e. [DOI] [PubMed] [Google Scholar]
- 16.Holley E A, Spector M P, Foster J W. Regulation of NAD biosynthesis in Salmonella typhimurium: expression of nad-lac gene fusions and identification of a nad regulatory locus. Microbiology. 1985;131:2759–2770. doi: 10.1099/00221287-131-10-2759. [DOI] [PubMed] [Google Scholar]
- 17.Kinney D M, Foster J W. Identification of cis-acting regulatory region in the pncB locus of Salmonella typhimurium. Mol Gen Genet. 1985;199:512–517. doi: 10.1007/BF00330767. [DOI] [PubMed] [Google Scholar]
- 18.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Liu G, Foster J, Manlapaz-Ramos P, Olivera B M. Nucleoside salvage pathway for NAD biosynthesis in Salmonella typhimurium. J Bacteriol. 1982;152:1111–1116. doi: 10.1128/jb.152.3.1111-1116.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Möller W, Amons R. Phosphate-binding sequences in nucleotide binding proteins. FEBS Lett. 1985;186:1–7. doi: 10.1016/0014-5793(85)81326-0. [DOI] [PubMed] [Google Scholar]
- 21.Muro-Pastor A M, Maloy S. Proline dehydrogenase activity of the transcriptional repressor PutA is required for induction of the put operon by proline. J Biol Chem. 1995;270:9819–9827. doi: 10.1074/jbc.270.17.9819. [DOI] [PubMed] [Google Scholar]
- 22.Ostrovsky P C, Maloy S. Protein phosphorylation on serine, threonine, and tyrosine residues modulates membrane-protein interactions and transcriptional regulation in Salmonella typhimurium. Genes Dev. 1995;9:2034–2041. doi: 10.1101/gad.9.16.2034. [DOI] [PubMed] [Google Scholar]
- 23.Spector M P, Hill J M, Holley E A, Foster J W. Genetic characterization of pyridine nucleotide uptake mutants of Salmonella typhimurium. Microbiology. 1985;131:1313–1322. doi: 10.1099/00221287-131-6-1313. [DOI] [PubMed] [Google Scholar]
- 24.Steele M I, Lorenz D, Hatter K, Park A, Sokatch J R. Characterization of the mmsAB operon of Pseudomonas aeruginosa PAO encoding methylmalonate-semialdehyde dehydrogenase and 3-hydroxyisobutyrate dehydrogenase. J Biol Chem. 1992;267:13585–13592. [PubMed] [Google Scholar]
- 25.Tabor S, Richardson C C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc Natl Acad Sci USA. 1985;82:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tirgari S, Spector M P, Foster J W. Genetics of NAD metabolism in Salmonella typhimurium and cloning of the nadA and pnuC loci. J Bacteriol. 1986;167:1086–1088. doi: 10.1128/jb.167.3.1086-1088.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai S P, Hartin R J, Ryu J-I. Transformation in restriction-deficient Salmonella typhimurium LT2. J Gen Microbiol. 1989;135:2561–2567. doi: 10.1099/00221287-135-9-2561. [DOI] [PubMed] [Google Scholar]
- 28.Vinitsky A, Teng H, Grubmeyer C T. Cloning and nucleic acid sequence of the Salmonella typhimurium pncB gene and structure of nicotinate phosphoribosyltransferase. J Bacteriol. 1991;173:536–540. doi: 10.1128/jb.173.2.536-540.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogel H J, Bonner D M. Acetylornithase of Escherichia coli: partial purification and some properties. J Biol Chem. 1956;218:97–106. [PubMed] [Google Scholar]
- 30.Weretilnyk E A, Hanson A D. Molecular cloning of a plant betaine-aldehyde dehydrogenase, an enzyme implicated in adaptation to salinity and drought. Proc Natl Acad Sci USA. 1990;87:2745–2749. doi: 10.1073/pnas.87.7.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wubbolts M G, Terpstra P, Van Beilen J B, Kingma J, Meesters H A R, Witholt B. Variation of cofactor levels in Escherichia coli: sequence analysis and expression of the pncB gene encoding nicotinic acid phosphoribosyltransferase. J Biol Chem. 1990;265:17665–17672. [PubMed] [Google Scholar]
- 32.Zhu N, Olivera B M, Roth J R. Activity of the nicotinamide mononucleotide transport system is regulated in Salmonella typhimurium. J Bacteriol. 1991;173:1311–1320. doi: 10.1128/jb.173.3.1311-1320.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu N, Olivera B M, Roth J R. Genetic characterization of the pnuC gene, which encodes a component of the nicotinamide mononucleotide transport system in Salmonella typhimurium. J Bacteriol. 1989;171:4402–4409. doi: 10.1128/jb.171.8.4402-4409.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu N, Olivera B M, Roth J R. Identification of a repressor gene involved in the regulation of NAD de novo biosynthesis in Salmonella typhimurium. J Bacteriol. 1988;170:117–125. doi: 10.1128/jb.170.1.117-125.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]