

Summary
Clubroot is one of the most important diseases for many important cruciferous vegetables and oilseed crops worldwide. Different clubroot resistance (CR) loci have been identified from only limited species in Brassica, making it difficult to compare and utilize these loci. European fodder turnip ECD04 is considered one of the most valuable resources for CR breeding. To explore the genetic and evolutionary basis of CR in ECD04, we sequenced the genome of ECD04 using de novo assembly and identified 978 candidate R genes. Subsequently, the 28 published CR loci were physically mapped to 15 loci in the ECD04 genome, including 62 candidate CR genes. Among them, two CR genes, CRA3.7.1 and CRA8.2.4, were functionally validated. Phylogenetic analysis revealed that CRA3.7.1 and CRA8.2.4 originated from a common ancestor before the whole‐genome triplication (WGT) event. In clubroot susceptible Brassica species, CR‐gene homologues were affected by transposable element (TE) insertion, resulting in the loss of CR function. It can be concluded that the current functional CR genes in Brassica rapa and non‐functional CR genes in other Brassica species were derived from a common ancestral gene before WGT. Finally, a hypothesis for CR gene evolution is proposed for further discussion.
Keywords: clubroot, turnip, CR gene, whole‐genome triplication, functional analysis
Introduction
Many Brassica species, such as oilseed rape (Brassica napus L.), kale, cabbage, cauliflower and turnip, are important crops throughout the world (Cheng et al., 2014). For example, oilseed rape is widely cultivated in China, Canada, Australia and European countries as an important source of edible oil and protein‐rich livestock feed (Wu et al., 2019). However, in recent years, production of oilseed rape in some countries such as China and Canada is seriously threatened by clubroot (Chai et al., 2014), which is a soil‐borne disease caused by the obligate protist Plasmodiophora brassicae (P. brassicae) (Nikolaev et al., 2004; Strelkov and Hwang, 2014). As one of the oldest plant diseases, clubroot is generally believed to originate from Europe (Howard et al., 2010; Watson and Baker, 1969). After infection of plant roots, P. brassicae colonization will lead to the swelling of roots and formation of galls, eventually inhibiting the uptake of nutrients and water from the soil by the roots (Li et al., 2018). Clubroot can normally cause a yield loss of 10%–15% in Brassica crops, and even over 40% loss or total crop failure in severe outbreaks (Chai et al., 2014; Dixon, 2006, 2009). For example, it was estimated that 3.2–4.0 million hectares of cruciferous crops are affected by clubroot in China each year (Chai et al., 2014; Strelkov and Hwang, 2014), and in western Canada, over 4.7 million hectares of spring canola crops are influenced (Peng et al., 2011).
Some of the resting spores of P. brassicae can remain dormant but viable in soil for at least 20 years (Dixon, 2009), making many control measures ineffective or impractical. Currently, genetic resistance is considered the most economical and effective approach for the control of clubroot. Common Brassica crops comprise three diploid species, namely Brassica rapa (A genome), Brassica nigra (B genome) and Brassica oleracea (C genome), and three allopolyploid species, including Brassica napus (AC genome), Brassica juncea (AB genome) and Brassica carinata (BC genome). All of these species have undergone whole‐genome triplication (WGT) compared with their close relative Arabidopsis thaliana (A. thaliana) (Cheng et al., 2016). In extensive screening of Brassica germplasms, clubroot‐resistance (CR) was only found from a limited number of germplasms, most of which were from A‐genome species, especially European fodder turnips. Some were found from B‐genome species while very few were from C‐genome species (Chang et al., 2019; Chen et al., 2013; Hasan and Rahman, 2018; Hatakeyama et al., 2013; Hirani et al., 2018; Huang et al., 2017; Li et al., 2016a; Matsumoto et al., 1998; Pang et al., 2018; Suwabe et al., 2003; Ueno et al., 2012; Werner et al., 2008; Yu et al., 2017). European fodder turnips have been used as the major source in CR breeding, and most of the CR loci have been mapped to chromosomes A1, A2, A3, A6 and A8 in the donor plants (Chen et al., 2013; Hatakeyama et al., 2013; Huang et al., 2017; Matsumoto et al., 1998; Pang et al., 2018; Sakamoto et al., 2008; Strelkov et al., 2018; Suwabe et al., 2003; Ueno et al., 2012; Yu et al., 2017). For example, CR turnips, including European Clubroot Differential 02 (ECD02), Siloga and ECD04, are considered to harbour more than 20 CR loci (Chen et al., 2013; Hirani et al., 2018; Neik et al., 2017; Suwabe et al., 2003) and have been successfully applied in molecular breeding (Strelkov et al., 2018). Cheng et al. (2016) analysed worldwide B. rapa accessions with diverse morphotypes and found that European turnips are the closest to the progenitor of this species. Recent reports have also indicated that the A genome of the allotetraploid species B. napus originated from European turnips (Song et al., 2020; Yang et al., 2016). However, so far, the reason for rare discovery of CR germplasms in natural B. napus remains unclear.
Plant innate immunity system comprises pathogen‐associated molecular pattern (PAMP) triggered immunity (PTI) and effector‐triggered immunity (ETI) (Wu and Zhou, 2013). For PTI, PAMPs are perceived by cell surface‐localized proteins known as pattern recognition receptors (PRRs), including receptor‐like kinases (RLKs) and receptor‐like proteins (RLPs) (Wu and Zhou, 2013). Multiple PRRs have been identified and characterized against several diseases on Brassica crops, such as blackleg (Kim et al., 2020; Larkan et al., 2015). Effectors are recognized by plant intracellular nucleotide binding site‐leucine‐rich repeat (NLR) proteins, resulting in an ETI response (Grund et al., 2019). To date, only two CR genes, Crr1a on A08 and CRa on A03, have been successfully isolated, both of which encode NLR proteins (Hatakeyama et al., 2013; Ueno et al., 2012). Crr1a is an incompletely dominant CR gene, whereas CRa is dominant (Hatakeyama et al., 2013; Ueno et al., 2012). The resistance of these CR loci usually exhibits certain specificity against different pathotypes of P. brassicae. The specific association between a resistance gene and certain pathotypes mainly depends on ETI but sometimes is also associated with PTI (Kim et al., 2016; Larkan et al., 2020; Neik et al., 2017; Yuan et al., 2021). A crop variety with a single CR locus may lose its resistance fairly quickly under selection pressure when multiple pathogen pathotypes are present in the soil. Therefore, to breed new varieties with multiple CR loci and resistance durability, it is highly necessary to physically map CR loci from different sources for isolation, functional and evolutionary analyses. However, the B. rapa genotypes with published genomes are highly susceptible to clubroot. This genome information is a good baseline reference for B. rapa (Belser et al., 2018; Zhang et al., 2018), but further information based on sequencing of CR genotypes would provide more direct references for studying and discovering new resistance sources.
In this study, we de novo assembled a reference genome for CR using ECD04 with abundant CR loci based on previous studies (Chen et al., 2013; Hirani et al., 2018; Piao et al., 2004; Werner et al., 2008). This study aims to determine (i) whether this reference map is good enough to help reconcile different names for the same CR genes in different studies, and fundamentally rectify the confusion about the number and genomic position of the physically mapped CR loci; (ii) whether this reference map can facilitate CR gene cloning and functional validation; (iii) how the WGT event affects the evolution of CR genes and the survival ability of Brassica plants against clubroot disease.
Results and discussion
De novo assembly of the ECD04 genome
The European turnip ECD04 (Brassica rapa subsp. rapifera, AA, 2n = 20) has been widely used as a germplasm for the introgression of CR genes into Brassica crops (Chen et al., 2013; Hirani et al., 2018; Piao et al., 2004; Werner et al., 2008). To explore the genomic and evolutionary basis of clubroot resistance (CR) in ECD04, we de novo assembled the ECD04 reference genome by integrating 20.84 Gb PacBio long‐read sequencing data (60‐fold coverage), 105.42 Gb Illumina paired‐end short‐read sequencing data (301‐fold coverage) and 129.15 Gb Hi‐C data (369‐fold coverage). First, a total of 1275 contigs with a total size of ~350 Mb and a N50 size of 1.50 Mb were assembled using long‐read data and then polished using short‐read data (Table 1). Subsequently 1212 polished contigs were corrected, clustered, sorted and oriented using Hi‐C data (Dudchenko et al., 2017). Finally, 346 Mb sequences accounting for 99.35% of the genome were anchored on A01–A10 pseudochromosomes (Figure 1a,b and Table 1). Compared with previously published B. rapa genomes, namely Chiifu‐401‐42 (hereafter referred to as Chiifu) and Z1, the chromosome size of the ECD04 genome was closer to that of Z1 but larger than that of Chiifu (Table S2) (Belser et al., 2018; Zhang et al., 2018). About 229.34 Mb (65.46%) sequences of the ECD04 genome could be mapped to Chiifu genome in one‐to‐one syntenic blocks (Figure S1), and 8126 (~25.96 Mb) inversions/translocations were identified in these blocks (Table S3), including 4416 (~7.59 Mb) inter‐chromosome translocations (Table S4). An inversion of 3.5 Mb on A03 chromosome (from 6.9 Mb to 10.4 Mb) was identified in the ECD04 genome, which could be validated by the Hi‐C data (Figure 1c and Figure S2). Similarly, 242.73 Mb sequences (69.28%) of ECD04 genome could be mapped to Z1 genome and 4979 structural variants (~28.08 Mb) were identified (Figure S1), including 3285 (~16.00 Mb) inter‐chromosome translocations (Table S5). Using the ECD04 genome as the reference, published sequences of 199 B. rapa accessions were mapped with an average mapping rate of over 95% (Cheng et al., 2016) (Table S7). A total of 1 818 999 SNPs and 422 526 InDels were also identified. The genome completeness was estimated to be 97.50% by the BUSCO assessment (Table 1). According to the Hi‐C heatmap of ECD04 (Figure 1b), the intrachromosomal interaction was significantly stronger than the interchromosomal interaction, and Hi‐C interaction probability showed a significant negative power correlation with genomic distance, which is similar to the results in other plants (Grob et al., 2014; Mascher et al., 2017; Figure S2a).
Table 1.
Summary of the genome assemblies for ECD04
| Genome size (Mb) | 350.34 |
| Gap ratio (%) | 0.34 |
| Contig number | 1275 |
| Total contig size (Mb) | 349.14 |
| Maximum contig size (Mb) | 14.27 |
| Average contig size (bp) | 273 834 |
| Median contig size (bp) | 61 049 |
| Contig N50 (Mb) | 1.52 |
| GC content (%) | 36.78 |
| TE proportion (%) | 44.82 |
| Annotated protein‐coding genes | 48 094 |
| Total length of Annotated protein‐coding genes (Mb) | 146.73 |
| Completeness (%, BUSCO) | 97.50 |
Figure 1.
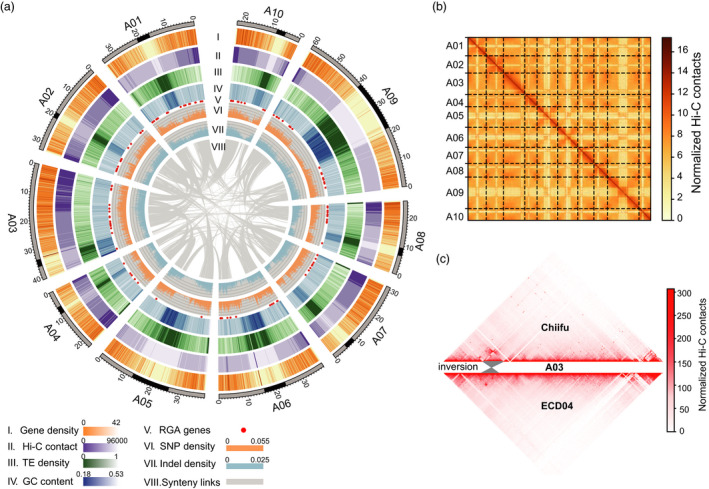
Genome features and Hi‐C interaction of ECD04. (a) Genome features of ECD04. Outer‐toinner circles indicate: gene density(I), Hi‐C contact (II), transposable‐element density (III) and GC content (IV), R genes (V), SNP density (VI), InDel density (VII) and genome synteny (VIII) of ECD04 genome. The sliding‐window size is 100 kb and the step size is 100 kb. (b) Hi‐C interaction heatmap of ECD04 genome. (c) Hi‐C validation of 3.4‐Mb inversion. The grey region is a 3.4‐Mb inversion between ECD04 (A03: 6 938 313–10 405 731) and Chiifu (A03: 6 601 636–9 974 053).
In ECD04 genome, 500 709 transposon elements (TEs) with a total length of 161.82 Mb were identified, which accounted for 46.19% of the genome assembly (Table S6). This proportion is similar to that found with Z1 genome (44.57%) and Chiifu genome (47.11%) respectively. Long terminal repeat retrotransposons (LTR‐RTs) accounted for 21.80% of the genome. A total of 19 749 intact LTR‐RT insertion events were identified and the insertion time of intact LTR‐RTs (substitution rate of 1.5 × 10−8) was calculated. Similar to those in Chiifu, LTR‐RTs in ECD04 genome underwent three waves of expansion since ECD04 diverged from B. oleracea (about 3.9 Mya), but the expansion time was not exactly the same, particularly the expansion at 0.8–1 Mya (Figure S3) (Zhang et al., 2018). In total, 48 094 protein‐coding genes were annotated on the unmasked genomic sequence, accounting for 41.88% of the genome. It was found that protein‐coding genes tended to be distributed on chromosome arms with poor SNPs, InDels, TEs, a low GC content and Hi‐C interactions, which is consistent with the findings in previous studies of other Brassica species (Song et al., 2020; Zhang et al., 2018).
Recent studies have revealed that European turnip might be the earliest domesticated Brassica crop and the progenitor of the A subgenome of B. napus (Cheng et al., 2016; Lu et al., 2019; Qi et al., 2017; Song et al., 2020; Yang et al., 2016). The ECD04 genome provides an opportunity to validate this inference. Using eight Brassica genomes, Raphanus sativus genome and A. thaliana genome, we identified 3029 single‐copy gene clusters and constructed a phylogenetic tree using A. thaliana as the outgroup. The tree structure clearly showed that ECD04 and Z1 were closer to the A subgenome of B. napus than Chiifu, but Z1 showed a closer relationship with the A subgenome of B. napus than ECD04. We deduced that the Brassica progenitor might have separated from A. thaliana ~21.25 million years (Myr) ago after undergoing polyploidizations twice (Figures S4a and S5). Similar to the case in other Brassica species, WGT has left many imprints on the ECD04 genome (Chalhoub et al., 2014; The Brassica rapa Genome Sequencing Project et al., 2011; Yang et al., 2016). Based on the homologous gene pairing between ECD04 and A. thaliana, we identified 22 ancestral crucifer karyotype blocks (A–X) in the subgenomes LF, MF1 and MF2 of ECD04 genome after WGT (Lysak et al., 2005; Figures S4–S6, Table S8).
Identification of R genes
The recognition of pathogens by plant resistance gene analogues (RGAs) in cells will result in the activation of ETI or PTI immunity (Kapos et al., 2019). RGAs contain NBS‐LRRs, RLKs and RLPs (Hammond‐Kosack and Jones, 1997). At present, two CR genes encoding NBS‐LRRs, Crr1a and CRa, have been successfully cloned (Hatakeyama et al., 2013; Ueno et al., 2012), which have been found to be localized in the clubroot‐resistant regions of Bra.A.CRa and PbBa8.1 in ECD04 genome respectively (Chen et al., 2013; Hirani et al., 2018). We predicted a total of 978 RGAs in ECD04 genome, including 213 NLRs, 658 RLKs and 107 RLPs. RGAs can be divided into different subfamilies according to differences in their C‐ and N‐terminal domains. The number and ratio of RGAs in the A or C genome/subgenome were found to vary among Brassica species (Table S9), which is similar to the findings in previous research (Figure S7; Tirnaz et al., 2020). It should be noted that the ratio of NLRs and RLPs in A genome/subgenome was the lowest compared with that in other genomes (Tables S10–S12), while the ratio of RLKs in C genome/subgenome was slightly lower than that in other genomes, especially in the HDEM genome (1.31%; Table S12).
Identification and functional analysis of CR genes
Physical mapping of CR loci
Due to the lack of reference A genome for CR, fine mapping and isolation of CR genes can be extremely challenging. The ECD04 genome is believed to contain at least nine CR loci and provides opportunities for identifying additional CR genes (Table S13). Using the assembled ECD04 genome as a reference, a total of 28 reported CR loci were compared, and their integration resulted in 15 loci distributed on A01, A02, A03, A06 and A08 chromosomes, and 12 CR loci except for CRA3.3, CRA3.5 and CRA3.6 comprised 62 candidate R genes, including 30 NLRs, 24 RLKs and 8 RLPs (Figure 2, Tables S14 and S15). Surprisingly, 11 of 15 loci were overlapped with the CR loci previously mapped in ECD04, indicating that compared with the published genomes of B. rapa highly susceptible to clubroot, the ECD04 genome with the most resistance genes would provide a direct and solid baseline reference for further CR gene mapping and cloning. The above information clearly shows that ECD04 genome may greatly help to reconcile different names used for the same genes and fundamentally rectify the confusion. Previous research has demonstrated that the enhancement of crop disease resistance might be at the cost of crop yield (Brown, 2003; Buschges et al., 1997; Denance et al., 2013; Deng et al., 2017; Gao et al., 2021; Tian et al., 2003), but no apparent negative relationship was observed between disease resistance and plant growth or development due to different balance mechanisms (Chandran et al., 2018; Cui et al., 2020; Deng et al., 2017; Li et al., 2021; Ning et al., 2017). In addition, as for ECD04 with 15 CR loci but exhibiting no obvious yield loss when acquiring the resistance trait, it remains to be clarified whether this is a new mechanism that coordinates the balance between resistance and growth in ECD04.
Figure 2.
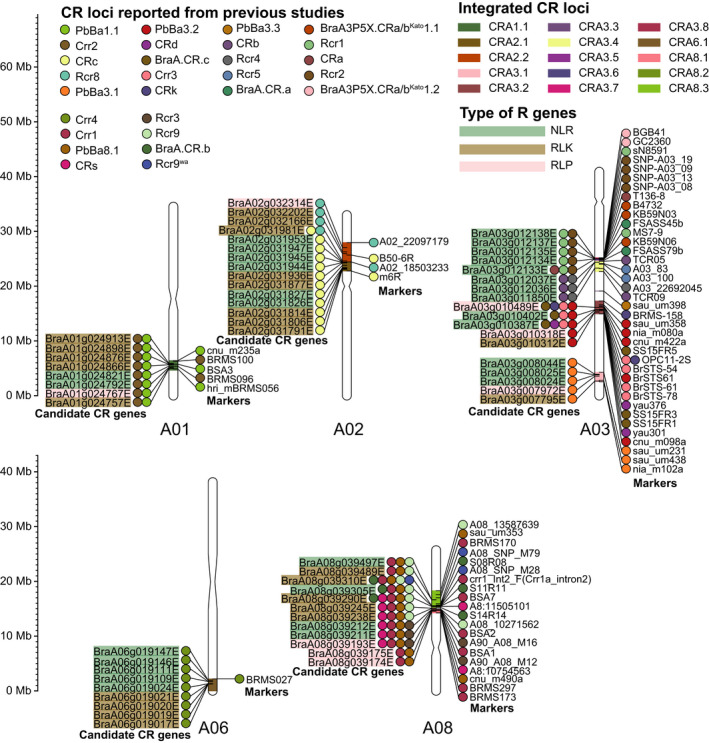
Physically mapping of CR loci. The texts on the left with three background colors represent three types of candidate CR genes including NLRs, RLPs and RLKs, respectively. Circles with 28 colors on both sides of the chromosomes represent the markers of 28 previously reported CR loci and 62 candidate genes within these CR loci, respectively. The regions with 15 colors within the chromosomes indicate 15 mapped CR loci, respectively.
Functional analysis of CRA3.7.1
BraA03g012133E, one of the candidate CR genes for CRA3.7 and therefore named as CRA3.7.1, is identical to CRa from Chinese cabbage T136‐8 and CRb‐α from European turnip ECD02 (Figure 3a; Hatakeyama et al., 2017; Ueno et al., 2012). To test the function and particularly the pathotype specificity of CRA3.7.1 in response to clubroot pathogens from different regions especially in China, the full‐length genomic sequence of CRA3.7.1 was isolated and inserted into a plant binary vector driven by the constitutive promoter of CaMV 35S for functional analysis with stable transformation of B. napus. Clubroot resistance tests against different field isolates indicated that two independent CRA3.7.1 overexpression lines (35S‐CRa‐6 and 35S‐CRa‐30) were resistant against Chengdu, Huangshan and Zhijiang clubroot pathogens (pathotype 4) (Figure 3b). We chose the resistant T1 plants to propagate and investigated plants in T2 generation for both the presence and absence of exogenous CRA3.7.1, as well as its relative expression (Figure 3c,d). Although some susceptible T2 plants (grade 1) were positive for CRA3.7.1 gene, the fully susceptible ones were negative (Figure 3c). Furthermore, the expression level of CRA3.7.1 in susceptible plants was significantly lower than those in resistant ones (Figure 3d), suggesting CRA3.7.1 as a functional CR gene. However, whether CRA3.7.1 is the only CR gene in the region remains unclear since there are other four tandem duplicated genes localized in this region and at least three of them are different from those in ECD02 (Hatakeyama et al., 2017).
Figure 3.
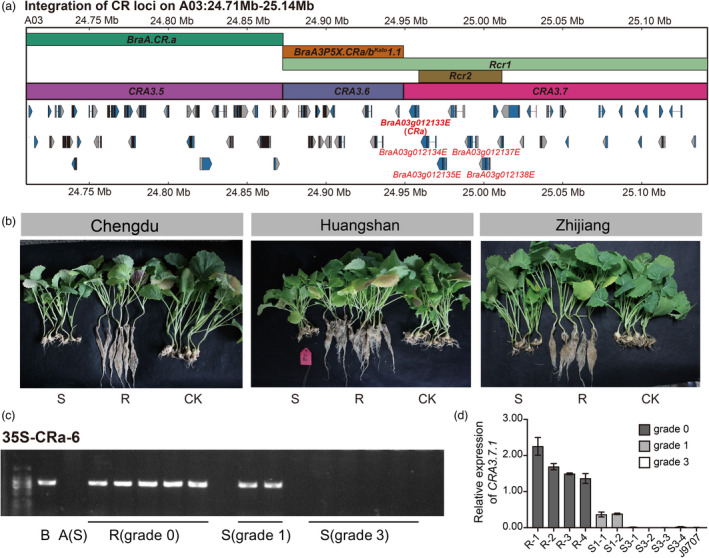
Mapping and functional verification of CRA3.7.1. (a) Physically mapping of CRA3.7. The bars with different colors indicate different CR loci. The top three bars indicate previously reported CR loci, and the bottom bar indicates the mapped CR loci. Red texts represent candidate CR genes in 24.71–25.34 Mb of chromosome A03. CRA3.7 contains five CR candidate genes (BraA03g012133E, BraA03g012134E, BraA03g012135E, BraA03g012137E and BraA03g012138E), among which BraA03g012133E (CRA3.7.1) is identical to previously cloned CRa. (b) Disease phenotype investigation of CRA3.7.1 over‐expression T1 plants after inoculation with clubroot isolates from Chengdu, Huangshan and Zhijiang. J9709 used as susceptible control (CK), R represents the resistant plants, S represents the susceptible plants. (c) Genotyping results of resistant and susceptible T2 plants. R represents the resistant lines and S represents susceptible lines. A (S), susceptible control, B, resistant control. (d) Relative expression level of CRA3.7.1 in resistant, susceptible T2 plants, J9709 as susceptible control. R‐1, R‐2, R‐3 and R‐4, the resistant plants (grade 0, dark grey); S1‐1 and S1‐2, the susceptible plants of grade 1 (light grey); S3‐1, S3‐2, S3‐3 and S3‐4 represent the susceptible plants of grade 3 (white). Grade 0‐3 indicates the disease resistance from resistant to fully susceptible.
Fine mapping and functional analysis of CRA8.2.4
We have previously located a dominant CR locus (PbBa8.1) to a physical region between 8.15 and 11.22 Mb on chromosome A08 using a BC3F2 population constructed from a cross between B. napus Huashuang5 (H5) and ECD04, with Huashuang5 as the backcross parent (Zhan et al., 2020). To further fine map PbBa8.1, which includes CRA8.1, CRA8.2 and CRA8.3 in this study (Figure 4a,b), a bulked segregant analysis sequencing (BSA‐seq) was performed (Figure 4c). Besides, 11 independent recombinants were identified from the segregating BC3F4 population comprising approximately 3000 plants using three InDel markers (4346, A08‐52 and A08‐29) with high polymorphism at different chromosomal regions (Figure 4d). The self‐pollinated seeds of these recombinants were harvested, and the progeny plants were inoculated again with Zhijiang or Huangshan isolates (pathotype 4) to check the disease resistance (Figure 4e). Three heterozygous plants identified by the marker 4346 exhibited a recombination event while the resistance to clubroot was maintained. Five homozygous plants identified by the marker A08‐52 showed susceptible phenotype (Figure 4d,e). Based on these results, the PbBa8.1 locus was narrowed down into a 1428 kb region between the markers 4346 and A08‐52, which included CRA8.1 and CRA8.2. Nine candidate CR genes were identified in these regions, including three RLKs (CRA8.1.1, CRA8.1.2 and CRA8.1.3), three RLPs (CRA8.2.1, CRA8.2.2 and CRA8.2.3) and three NLRs (CRA8.1.4, CRA8.1.5 and CRA8.2.4). CRA8.2.4 is an allele of the previously cloned CR gene Crr1a in European turnip ‘Siloga’, but with significant differences from Crr1a on DNA and protein levels (Figure 4b, Figure S8). The identity of the coding sequences of two CR genes was 82.5% and a 267‐bp deletion in LRR domain was observed in Crr1a compared with CRA8.2.4 (Figure S8). It can be deduced that these differences might contribute to the obvious differences in resistance to clubroot conferred by PbBa8.1 and Crr1a because it has been shown that PbBa8.1 and Crr1a are dominant and incompletely dominant genes respectively (Zhan et al., 2020). Therefore, functional analysis of CRA8.2.4 was further performed. The full‐length coding sequence of CRA8.2.4 was cloned into a plant binary vector driven by CaMV 35S promoter and subsequently transformed into B. napus lines with a gene transformation system in root. Phenotypic assessment at 30 days after inoculation of P. brassicae showed that out of 42 transformants with CRA8.2.4, 21 transformants were resistant, while only four out of 34 transformants with vector control were resistant (Figure 4e,f). The disease index of CRA8.2.4 transformants and the empty vector control transformants was 34.92% and 80.39% respectively (Figure 4f), suggesting a significant CR of CRA8.2.4. We randomly selected at least three independent CRA8.2.4 transformants from each disease index group and checked the transgene expression. The results revealed that transgenic plants with the highest expression of CRA8.2.4 were not infected, while those with lower or no expression of CRA8.2.4 showed a higher disease index (Figure 4g). These results further confirmed the CR of CRA8.2.4. However, at present, we cannot exclude the possibility that the other two candidate NLRs (CRA8.1.4 and CRA8.1.5), or the other three RLKs and three RLPs, or the interactions among them, might also contribute to CR.
Figure 4.
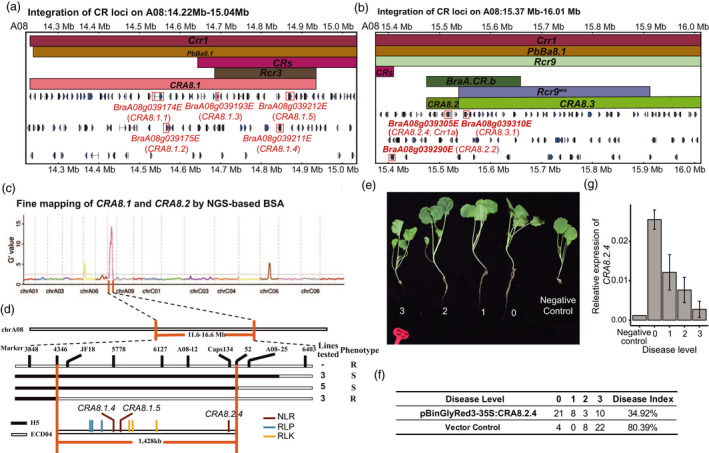
Mapping and functional verification of CRA8.2.4. (a) Physically mapping of CRA8.1. CRA8.1 consists of five CR candidate genes, including two NLRs‐BraA08g039211E (CRA8.1.4), BraA08g039212E (CRA8.1.5) and three RLPs. The bars with different colors indicate different CR loci. The top three bars indicate previously reported CR loci, and the bottom bar indicates the mapped CR loci. (b) Physically mapping of CRA8.2 and CRA8.3. The top five bars indicate previously reported CR loci, and the bottom bar indicates mapped CR loci. Red texts represent five candidate CR genes in 14.22–15.04 Mb of chromosome A08 in (a) and (b). (c) and (d) Fine mapping of CRA8.1, CRA8.2 and CRA8.3. (b) Fine mapping by BSAseq. (d) Fine mapping by linkage markers. Brown lines represent the boundaries of CR loci after fine mapping. Deep red lines represent NLR genes; blue lines represent RLP genes; yellow lines represent RLK genes. (e) Inoculation experiment of B. napus transformed with CRA8.2.4. The infection grades of different transgenic Bing409 lines, which were transformed with the candidate resistant gene CRA8.2.4 using the root transformation system after inoculation with the field isolate from the Zhijiang region. Bing409 represents the negative susceptible control to P. brassicae, with 0 to 3 representing increasing infection level. (f) Numbers of lines with different infection levels and (g) relative expression level of CRA8.2.4 in different types of plants.
To summarize, the assembled ECD04 genome is of high quality and can be used for the prediction and discovery of CR genes. Functional verification of CRa (CRA3.7.1) and CRA8.2.4 suggested that the prediction of CR genes is reliable and feasible for promoting CR breeding in Brassica species.
Evolutionary analysis of CR genes
To date, only CRA3.7.1 and CRA8.2.4 have been validated for their CR functions, while the other 60 candidate CR genes identified in this study by genome‐wide sequencing of ECD04 remain to be validated. Since only a limited number of CR loci have been found in European turnips, and most of B. rapa or B. napus germplasm/accessions appeared to be susceptible to clubroot, the factors responsible for these variabilities are still unclear. Given that P. brassicae is an ancient pathogenic organism, it is of interests to investigate the evolution of these resistance genes to understand the mechanism through which they contribute to the different clubroot‐resistant phenotypes we observed among Brassica accessions (Howard et al., 2010).
Phylogenetic analysis of CR genes
To elucidate the evolution of CR genes, an neighbour‐joining (NJ) tree was constructed including all 30 NLR genes identified in the ECD04 genome at 15 loci, mainly because both CRA3.7.1/CRa and CRA8.2.4 validated in this study are NLRs (Figure 5). Their distributions on the ancient crucifer karyotype (ACK) genomes were investigated in detail (Chen et al., 2013; Lysak et al., 2016). Interestingly, we found that 11 of the candidate CR genes derived from the U block of the ACK genome could be grouped into three clusters in the phylogenetic tree (Chen et al., 2013; Lysak et al., 2016). For example, the CR genes CRA8.2.4 and CRA3.7.1 (marked with blue stars in Figure 5), along with four adjacent tandem duplicated genes were clustered together, while the candidate CR genes CRA1.1.4 (BraA01g024821E), CRA3.4.3 (BraA03g012037E) and CRA8.1.5 were close to each other, and the candidate gene CRA3.4.2 (BraA03g012036E) was adjacent to CRA8.1.4 (Figure 5). The close clustering pattern of several CR genes indicated that candidate CR genes in each cluster might have originated from a common ancient CR gene before WGT. Since CRA8.2.4 and CRA3.7.1 have been functionally validated, based on the above results, we hypothesize that these CR genes might have existed prior to the genome triplication. To test this hypothesis, the genomic segments containing CR genes within the ECD04 genome were aligned with the homologous segments from the A. thaliana genome. The results indicated that the aligned segments of each cluster indeed exhibited collinearity and retained most of the genes in the homologous regions of A. thaliana with a similar gene distribution (Figure 6a). However, the genes homologous to CRA8.2.4 or CRA3.7.1 were not found in the first cluster of A. thaliana, while CRA1.1.4, CRA3.4.3 and CRA8.1.5 from the second cluster, and CRA8.1.4 and CRA3.4.2 from the third cluster all appeared to be synteny genes (Figure 6a).
Figure 5.
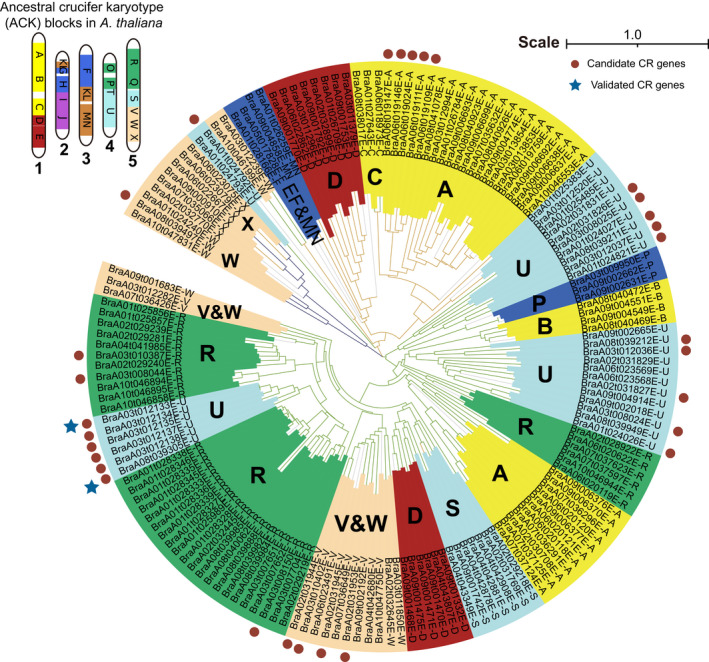
Neighbor‐joining tree of NLR genes from ECD04 genome. Clades with different colors represent different types of NLR genes including TIR‐NBS‐LRR (green), CC‐NBS‐LRR (brown) and RPW‐NBSLRR (blue). The colors of blocks indicate different ACK genome blocks (A‐X) of NLR genes. The deep red dots indicate candidate CR genes. The blue stars indicate CR genes (CRA3.7.1 and CRA8.2.4).
Figure 6.
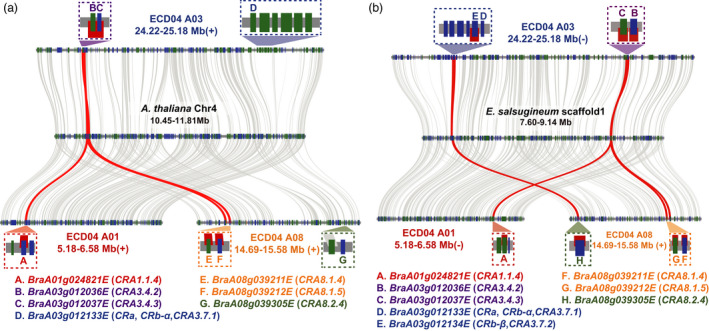
Micro‐synteny of CR loci. (a) Micro‐synteny of CR loci between ECD04 and A. thaliana. (b) Micro‐synteny of CR loci between ECD04 and E. salsugineum. Colors of texts represent different CR loci including CRA1.1 (red), CRA3.4 (purple), CRA3.7 (blue), CRA8.2 (orange) and CRA8.3 (green).
Origin and differentiation of CR genes
The above analysis indicated that the two confirmed CR genes, CRA3.7.1 and CRA8.2.4, likely originated from a common ancestral gene through WGT, considering the low probability for two resistant genes to have evolved in syntenic regions at random. To validate this hypothesis, we identified the homologous genes or footprint sequences of CRA3.7.1 and CRA8.2.4 in a cruciferous genome that has not undergone WGT. After a BLAST search of the NCBI database and construction of a phylogenetic tree (Figure S9), two tandem duplicated NLR genes, Thhalv10024211m and Thhalv10024234, were found to be homologous to CRA3.7.1 and CRA8.2.4 in the genome of Eutrema salsugineum, a cruciferous species that has not undergone WGT (Wu et al., 2012). The coding sequences of CRA3.7.1 and CRA8.2.4 have higher similarity to that of Thhalv10024234 than to that of Thhalv10024211m. The sequences encoding the TIR domain of CRA3.7.1 and CRA8.2.4 had 82% and 84.5% similarities to that of Thhalv10024234, respectively, while those sequences encoding the NBS domain showed 81.9% and 79.5% similarities (Figure S10). In addition, this result was further validated by the microsynteny analysis (Figure 6b). Taken together, these CR genes might have already existed in the Brassica species progenitor prior to WGT.
Most Brassica species are susceptible to clubroot, and CR genes have been found mostly in a few subspecies of European turnips. To better understand this phenomenon, the genomic structures of homologous genes in Brassica species susceptible to clubroot were compared with that of CRA3.7.1 and CRA8.2.4. To this end, an NJ tree was constructed using 38 genes, including the two validated CR genes, four tandem duplicated genes in CRA3.7, two orthologous genes in E. salsugineum, five tandem duplicated genes (CRb‐α, CRb‐β, CRb‐γ, CRb‐δ and CRb‐ε) from ECD02, Crr1a from Siloga and 24 orthologous genes from the sequenced genomes of susceptible Brassica spp. (Figure 7a). In the NJ tree, these genes could be divided into five clades (clades 1–5): clade 1 mainly consisted of CRA3.7.1, CRb‐α, BraA03g012134E, BraA03g012135E and other homologous genes and clade 5 comprised CRA8.2.4 and its homologous genes (Figure 7a). The genes in these two clades were from Chinese cabbage Chiifu and Z1 as well as the rapeseed varieties Darmor‐bzh and ZS11. Four materials were confirmed for their susceptibility to clubroot (Figure 7b). Gene structural comparison was performed for genes in clade 1 and clade 5, which included functionally validated CR genes in this study. For genes in clade 1, a homologous sequence (BnaA03g45000D) was found only in the Darmor‐bzh genome, yet with a large deletion (~7.4 kb) (Hatakeyama et al., 2017) (Figure S11). As for genes in clade 5, the homologous sequences of CRA8.2.4 were found in the genomes of all four susceptible materials, but with transposon insertions observed at different positions (Figures 7c and Figure S12). Therefore, all susceptible materials differ from ECD04 in the coding sequence of the validated CR genes. Based on these results, it can be speculated that most Brassica species fail to possess resistance to clubroot mainly due to the loss of function of CR genes after WGT.
Figure 7.
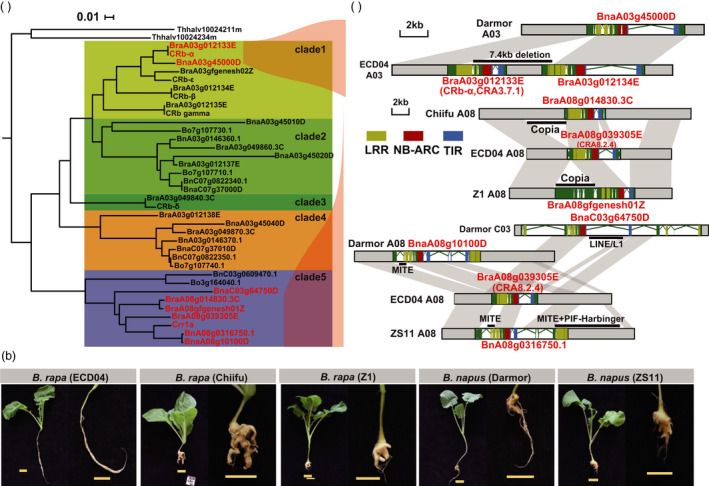
Differentiation among CR genes and homologs. (a) NJ‐tree of 38 CR genes and homologs. Five clades of NJ‐tree are labeled with different colors. The red text in clade 1 represents candidate CR genes homologous to CRA3.7.1, and the red text in clade 5 represents candidate CR genes homologous to CRA8.2.4. (b) Root inoculation results of ECD04, Chiifu, Z1, Darmor and ZS11 with P. brassicae from Zhijiang region. (c) Structural variations and transposon insertions in homologous genes of CRA3.7.1 and CRA8.2.4. The red, blue, yellow and green rectangles represent the NB‐ARC domain, the TIR domain, the LRR domain, and the exon region, respectively, and the thick black lines represents the transposon insertions.
Based on the above analysis, it remains unclear how functional CR genes have been lost in most of Brassica species after genome triplication. Due to the lack of clear evidence, it is unclear how the CR genes in the progenitors of the current Brassica evolved since 20 million years ago, and whether there are other selective pressures on CR genes except for clubroot pathogens during the evolution. Many hypotheses have been proposed to explain why CR genes only exist in some Brassica accessions such as the European fodder turnip. Here, we would also raise one hypothesis for discussion (Figure 8). Chinese cabbage, which is widely cultivated in Asia and has no functional CR genes, evolved from European turnip, which is regarded as closer to the ancestor of the A genome. Since the current CR of CR Brassica lines was derived from the CR European turnip by crossing, it can be therefore hypothesized that clubroot disease may not be a global disease either in the past or in the present world. In addition, after WGT, triplicated CR genes in Brassica species might greatly enhance their survival ability against different pathogens, including P. brassicae. As a result, the dispersal of soil‐borne P. brassicae is reduced (Figure 8, purple color). Therefore, either the lack or disappearance of clubroot pathogen in Asia and some other places or areas might lead to the functional loss of CR genes by the random insertion of TEs or other ways in some susceptible plants such as ZS11 and Chiifu.
Figure 8.
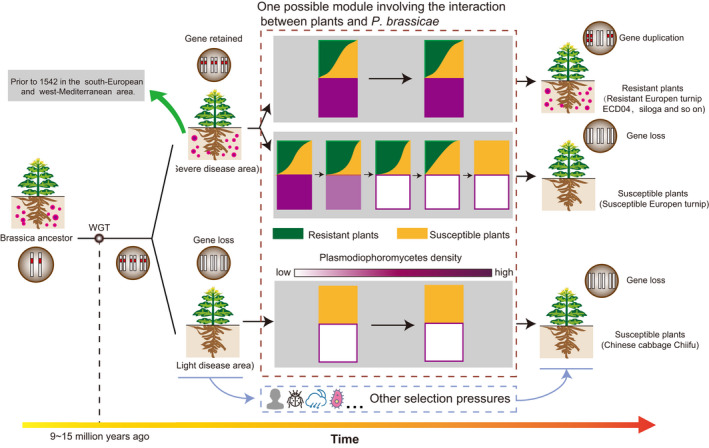
An evolutionary model of CR genes in Brasscia A‐genome plants in the long‐term struggle against clubroot. Before the Brasscia triplication event, P. brassicae (purple circles) had already existed in the soil and clubroot resistant (CR) genes (red blocks) had been present in the genome of Brassica ancestor accordingly. After triplication, the duplicated CR genes conferred Brassica plants with strong resistance to clubroot disease for better adaption to the stress environment. Later, in some areas, P. brassicae and the plants reached a state of dynamic equilibrium, resulting in the retention of CR genes, such as in ECD04; in most areas, with the gradual increase in the proportion of CR plants, P. brassicae gradually disappeared, resulting in the final functional loss of CR genes, such as susceptible European turnips. Along with human activities, Brassica plants are spread around the world. For those plants in areas without clubroot disease such as Chinese cabbage, they gradually lost CR genes.
Overall, these findings indicate that the development of durable resistance varieties may require pyramiding of different CR genes and/or increasing the copy number or expression level of CR genes through genetic engineering. Additionally, it is possible that more CR genes may be found from the germplasms with close associations to the ancestor progenitor with diverse CR functions.
Conclusion
In this study, a high‐quality genome of European turnip ECD04 (B. rapa) was developed via de novo assembly, which contains multiple CR loci. A total of 28 CR loci reported previously from Brassica A genome were mapped to 15 loci in the ECD04 genome and a uniform nomenclature was proposed to reconcile gene names and rectify confusion. In addition, 62 candidate genes were identified at these CR loci, with 12 of them being associated with at least one candidate CR gene, demonstrating the high quality of this reference genome for CR gene mapping and cloning. Besides, we validated the function of two candidate CR genes, CRA3.7.1 and CRA8.2.4, which have been widely used in elite rapeseed and Chinese cabbage varieties in China. Genomic and phylogeny comparison of these two CR genes in this study helped to explain why a lack of CR was observed in most Brassica species, a phenomenon possibly resulting from the transposable element (TE) insertion that would disrupt the gene function. Furthermore, the phylogenetic and microsyntenic analyses indicated that CRA3.7.1 and CRA8.2.4 might have originated from a common ancestral gene before WGT event, as evidenced by their homologues in one of the cruciferous species without experiencing WGT. Taken together, these results may help to explain how most present Brassica species have become susceptible to clubroot after WGT and why CR genes are retained only in a few European turnip genotypes after WGT.
Materials and methods
Plant materials
An elite line of clubroot resistant European turnip ECD04 was derived from the European Clubroot Differential set, which consisted of five genotypes each of B. rapa, B. napus and B. oleracea, and have been widely used to identify strains of P. brassicae (Buczacki et al., 1975; Pang et al., 2020; Toxopeus et al., 1986). Seeds of ECD04 were obtained from Shenyang Agricultural University. The seeds of CR variety Huashuang 5R including PbBa8.1 derived from ECD04 were generated in Huazhong Agricultural University (Zhan et al., 2020). The seeds of the rest of the plants in this study were collected and kept at National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan.
Illumina and PacBio library construction and sequencing
High‐molecular weight DNA was extracted from 3‐week‐old seedlings. Illumina library construction and sequencing were performed at Novogene, China. Illumina paired‐end sequencing libraries were constructed following the manufacturer’s standard protocol (Illumina) and sequenced on the Illumina HiSeq platform. Paired‐end reads of 150‐bp were generated with an insert size of 350 bp. Construction and sequencing of PacBio library were performed at BGI, China. DNA fragments larger than 20 kb were selected by BluePippin electrophoresis (Sage Sciences). SMRTbell libraries were constructed as previously described and sequenced on the PacBio Sequel platform (Pacific Biosciences; Pendleton et al., 2015).
Hi‐C library construction and sequencing
About 1.5 g of 3‐week‐old plant seedlings was extracted for Hi‐C experiment. The Hi‐C experiment procedure was similar to that in the previous study with some steps being improved for efficiency (Xie et al., 2015). To digest extra protein and make the nuclei more permeable, the nuclei were resuspended in 150 μL of 0.5% SDS buffer and incubated at 62 °C for 5 min. Chromatin was digested for 12 h with 20 units of DpnII restriction enzyme (NEB) at 37 °C and the resuspended mixture was incubated at 62 °C for 20 min to inactivate the restriction digestion. The DNA pieces between 300 and 500 bp were excised and purified using Ampure XP beads (Beckman Coulter). The library was constructed by an Illumina TruSeq DNA Sample Prep Kit and sequenced by Illumina Hiseq Xten with 2 × 150‐bp reads.
Genome assembly of ECD04
The de novo assembly of the ECD04 genome was performed using Canu (v1.5) based on PacBio reads with default parameters (Koren et al., 2017). The assembled contigs were self‐polished with PacBio reads using the arrow program of GenomicConsensus package (https://github.com/PacificBiosciences/GenomicConsensus). The contigs were then polished using Pilon (v1.22) with default parameters (Walker et al., 2014). The final contigs were used for Hi‐C scaffolding. For Hi‐C scaffolding, the clean Hi‐C reads were first mapped to the contigs using Juicer (Durand et al., 2016). The contigs were then corrected, clustered, ordered and oriented using the 3D‐DNA pipeline (Dudchenko et al., 2017). Centromeric repeat sequences, including CentBr, CRB, TR238 and PCRBr, were mapped to the ECD04 genome using NUCmer to identify the locations of centromeres (Table S1; Mason et al., 2016).
Genome annotation of ECD04
The repeat sequences of the ECD04 genome were identified using a combination of database‐based and de novo approaches. A repeat library of ECD04 was constructed using RepeatModeler (v1.0.11) (http://www.repeatmasker.org/RepeatModeler/). In addition, LTR retrotransposons were identified using LTR_FINDER (v1.0.5) with default parameters and added to the library (Xu and Wang, 2007). For the determination of DNA level, RepeatMasker (v4.0.9) (http://www.repeatmasker.org/RepeatMasker/) was used to search against RepBase as the repeat library. For protein‐level determination, RepeatProteinMask was used to perform an RMBlast search by mapping to the TE protein database. Tandem repeats were identified using the TRF software (v2.5) (Benson, 1999).
RNA was collected from the roots, stems and leaves of ECD04 plants and subjected to transcriptome sequencing. The gene structures of the ECD04 genome were annotated by a combination of three approaches (ab initio prediction, homologue proteins and transcriptome data) using MAKER (v2.31.10) (Cantarel et al., 2008). Ab initio gene prediction was performed using AUGUSTUS (v3.3.2) (Stanke et al., 2004) and GlimmerHMM (v3.0.4) (Majoros et al., 2004). Protein sequences from B. rapa, B. oleracea, B. napus and A. thaliana were used for the gene annotation based on homologous proteins of the ECD04 genome using Exonerate (v2.2.0). RNA‐seq data were mapped to the ECD04 genome and assembled into transcripts using HISAT2 (v2.1.0) (Kim et al., 2015) and StringTie (v1.3.5) (Pertea et al., 2015), respectively. Gene functions were annotated by mapping to the GO, KEGG_ALL, KEGG_KO, Swiss‐Prot, TrEMBL, NR and COG databases using InterProScan and BLASTP (−evalue = 1e‐5) (Camacho et al., 2009; Mulder and Apweiler, 2007).
Phylogenetic tree construction and divergence time estimation
All genes from 10 genomes including B. rapa (Chiifu‐401‐41, v3.0), B. rapa (Z1), B. nigra, B. oleracea (HDEM), B. oleracea (To1000, v2.0), B. napus (Darmor, v4.1), B. napus (ZS11), R. sativus (XYB‐2), E. salsugineum and A. thaliana (TAIR10) were clustered using OrthoFinder (v2.2.6) (Emms and Kelly, 2019) with the parameter ‘‐p blast’. Subsequently, 3043 single‐copy gene groups aligned using MUSCLE (v3.8.1551). Then, the best‐fit model of protein evolution was estimated by ProtTest (v3.4.2) (Darriba et al., 2011), and the JTT+G+I+F module was estimated to be the best model. For determination of likelihood, we used RAxML (v8.2.12) with the JTT+G+I+F model and 1000 replicate searches (Stamatakis, 2014). The divergence time for the species was then estimated based on alignment among all single‐copy orthologous genes using the MCMCTree program in the PAML package (v4.9h) (Yang, 2007).
Comparative genome analysis
Comparative analysis of ECD04, Chiifu and Z1 genomes was performed using the NUCmer program of the MUMmer package (v4.0.0beta2) (Marcais et al., 2018) with the parameter ‘‐‐mum ‐c 700’, and then the blocks with one‐to‐one alignment were filtered using the delta‐filter program with the parameter ‘‐1’. All one‐to‐one alignment blocks were extracted using show‐coords for manual checking. Those blocks with different directions were identified as inversions. In addition, the alignment blocks in different positions were extracted to assess their flanking blocks. If the alignment blocks were noncollinear flanking sequences, they were retained as putative translocations.
Gene synteny analysis
The raw orthologues were identified using blastp with the parameter ‘‐evalue 1e‐10 ‐num_alignments 1’. Syntenic blocks of A. thaliana and ECD04 were constructed using MCScan (https://github.com/tanghaibao/jcvi/wiki/MCscan‐(Python‐version)) based on the raw orthologues with the parameter ‘‐‐cscore=.99’. Multiple ECD04 chromosomal segments that matched the A. thaliana chromosomal segment were then partitioned into three subgenomes: LF, MF1 and MF2.
Identification and phylogenetic analysis of RGA genes
Resistance gene analogue genes of three types including RLKs, RLPs and NLRs were identified using the RGAugury pipeline as previously described (Li et al., 2016b; Tirnaz et al., 2020). NLRs were divided into eight subgroups according to their domain architecture, named as NBS(N), CNL, TNL, TN, CN, NL, TX (TIR with unclassified domains) and Other by RGAugury pipeline. Then, the RPW domain (PF05659) was identified using hmmsearch in HMMER(v3.1b2) (Mistry et al., 2013) with default parameters (Bateman et al., 2000). Finally, NLRs were divided into 10 subgroups including RNL, CNL, TNL, NL, RN, CN, TN, N, TX and Other. The phylogenetic tree was constructed for the NLRs. NLRs were first aligned using ClustalW (v2.1) (Thompson et al., 2002), and a NJ tree was constructed using TreeBeST (v1.9.2) with 1000 bootstrap replicates and visualized using iTOL (Letunic and Bork, 2019). As for RLKs and RLPs, the Lys domain (PF00062) was identified using hmmsearch program, and then RLKs and RLPs were divided into three subclasses: LRR‐RLK/RLP, LysM‐RLK/RLP and other‐RLK/RLP as previously described (Tirnaz et al., 2020).
Physically mapping of CR loci and identification of candidate genes
A total of 28 CR loci were published in 16 previous studies (Chen et al., 2013; Fredua‐Agyeman et al., 2020; Hatakeyama et al., 2013; Hirai et al., 2004; Hirani et al., 2018; Huang et al., 2017, 2019; Laila et al., 2019; Matsumoto et al., 1998; Pang et al., 2018; Piao et al., 2004; Sakamoto et al., 2008; Suwabe et al., 2003, 2006; Ueno et al., 2012; Yu et al., 2017). The close primer pairs of 28 CR loci were mapped to the ECD04 genome. To physically map these loci from different studies, the following standard was performed in general: if the two close loci on the same chromosome share no candidate CR genes, they would be defined as independent loci. After physically mapping, the locus was named as CRAxy according to the following rules: (i) CRA represents the CR from A‐genome Brassica species; (ii) x represents the 10 different chromosome numbers of the A genome; and (iii) y represents the different and sequent loci on the same chromosome from one end to the other.
Primer sequences used to locate these loci from different studies were mapped to the ECD04 genome by e‐PCR to locate these loci in the ECD04 genome (Schuler, 1997). Then, these loci were integrated according to whether the two loci contained the same R genes. In total, 28 CR loci were physically mapped to 15 loci.
DNA and RNA extraction
DNA extraction was performed from young leaves using a CTAB protocol as previously described (Bowers et al., 2003). The quality and quantity of DNA were determined by spectrometry using the 260/280 nm absorbance ratio, and the DNA concentration was determined at 260 nm with a DS‐11 spectrophotometer (DeNovix Inc, Wilmington, DE 19810USA). Roots of ECD04 plants after inoculation with P. brassicae pathotype 4 were collected for RNA extraction. The extracted RNA was mixed in equal amounts and then converted into cDNA, which was used as a template for gene cloning.
Vector construction
To construct pCAMBIA1305‐35S:CRa, the full‐length coding sequence (4609 bp) of CRA3.7.1 (gCRa) was amplified with primer pairs CRaF1 and CRaR1 using genomic DNA of ECD04 as templates, and cloned into the pBINRED3 vector digested by EcoRI/XhoI. Then, the integrated fragment containing the 35S promoter, gCRa and T‐NOS was amplified using CRaF2 and CRaR2 from the pBINRED3‐CRa plasmid and cloned into pCAMBIA1305 binary vector digested by BamHI/PstI. All vectors were constructed using One Step Cloning Kit (Vazyme, China) and all primer sequences were listed in Table S16. Primers designed according to both ends of CRA8.2.4 were used to amplify the full‐length CDS of CRA8.2.4 using cDNA as templates. The cloned fragments were then ligated into the multiple cloning site after the 35S promoter of the pBinGlyRed3 vector via homologous recombination, resulting in pBinGlyRed3‐35S:CRA8.2.4.
Plant transformation, transgenic plant identification and pathogen inoculation
Brassica napus plant J9709 was used as the transgenic receptor material for pCAMBIA‐35S:CRa by Agrobacterium tumefaciens‐mediated genetic transformation of explanted hypocotyl tissue culturing as previously described (Moloney et al., 1989), and the transformants were selected on the medium containing hygromycin (50 mg/mL). The T2 offspring of two different positive T0 plants (35S‐CRa‐6 and 35S‐CRa‐30) were inoculated with P. brassica (pathotype 4 determined by William’s system) collected from Huangshan, Zhijiang and Chengdu. The resistance of the plants to P. brassicae was assessed as previously described (Pang et al., 2020). The T2 plants were used for transgene validation with the primers of CRa‐2F and CRa‐2R (Table S16). The PCR reaction conditions were 95 °C for 3 min; 95 °C for 30 min, 55 °C for 30 s, 72 °C for 1 min, 35 cycles; 72 °C for 10 min.
The expression levels of transgenes in different transformants were detected with qPCR. Total RNA was extracted from per sample using Eastep Super Total RNA Extraction Kit (Promega Biotech Co, Ltd. Beijing, China). The RevertAid First Strand cDNA Synthesis Kit (Vazyme) was used for cDNA synthesis, following the manufacturer’s protocol. Sequences of qRT primers are provided in Table S16. CFX384 Touch Real‐Time PCR Detection System (BioRad Laboratories, Shanghai, China) was used for qPCR reaction with ChamQ Universal SYBR qPCR Master Mix (Vazyme). PCR parameters were set as follows: initial denaturation at 95 °C for 3 min; 40 cycles of denaturation step at 95 °C for 10 s, annealing step at 52 °C for 20 s, extension step at 72 °C for 30 s, followed by melting curve measurement with 0.5 °C increment from 65 to 95 °C. The amplification results were analysed by LinRegPCR (Academic Medical Centre, Amsterdam, The Netherlands). The ΔΔC t method was used for quantification of gene relative expression (Livak and Schmittgen, 2001), using Actin‐7 (LOC106418315) as reference gene. Mean value and standard deviation (STDEV) were calculated from three biological replicates.
To transform CRA8.2.4 into B. napus plants, clubroot susceptible B. napus 409S was sown on MS solid medium. Seven days later, the lower hypocotyl was excised, and the upper cotyledon part was infected with Agrobacterium rhizogenes bacterial solution containing the empty pBinGlyRed3 vector and the pBinGlyRed3‐35S:CRA8.2.4 overexpression vector. After co‐cultivation on MS medium in dark for two days, the explants were rinsed several times with MS liquid medium containing cefotaxime (100 mg/L), blotted dry on sterile filter paper and then cultivated on MS solid medium containing 100 mg/L cefotaxime at 25 °C under light. Once the length of new roots reached 2 mm, the plants were transferred to soil containing P. brassicae for inoculation. After 30 days of cultivation in the greenhouse, the resistance of the plants to P. brassicae was assessed as previously described (Pang et al., 2020). The expression levels of transgenes in different transformants were detected with qPCR by using the primers of CRA8.2.4F and CRA8.2.4R (Table S16) with an anneal temperature of 52 °C.
Conservation analysis of the resistance genes
To compare the orthologous genes of CRA3.7.1 (BraA03g012133E) and CRA8.2.4 (BraA08g039305E), mVISTA was used to align these sequences and identify the conserved regions (Poliakov et al., 2014). In particular, the CRA3.7.1 and CRA8.2.4 orthologous gene in the Z1 genome were too short, but high conservation was detected in these regions. Hence, Fgenesh (http://linux1.softberry.com/berry.phtml) was used for the prediction of genes on the orthologous fragment in the Z1 genome (Solovyev et al., 2006). Orthologous genes of CRA3.7.1 and CRA8.2.4 were named as BraA03gfgenesh01Z and BraA08gfgenesh01Z respectively.
Conflict of interest
The authors declare no competing financial interests.
Author contributions
CZ, QY and ZP designed and supervised this project. YJ, NS, CCN, YZ, LZ, BW and JX planted and sampled the plant materials. JG and ZZ created sequencing libraries. QL and LY carried out inoculation experiments. YJ, ZZ, QL, JG, QZ and ZH performed gene mapping. YJ, BD, ZS, LZ, YL, L Y, JS, XZ and HW performed fine mapping and functional analysis of CR genes. ZY performed main bioinformatic analyses. DL, FY, JW, JX, SL and XZ participated in data analysis. CZ, QY, ZY YJ and PC wrote the manuscript. All authors approved the manuscript.
Supporting information
Figure S1 Genome alignment between the Chiifu‐ECD04 and Z1‐ ECD04 genomes.
Figure S2 Hi‐C data analysis of ECD04.
Figure S3 The insert time of intact LTR retrotransposons in the ECD04 genome.
Figure S4 Phylogenetic tree of ECD04 and retention of ancestral genes in ECD04 genome.
Figure S5 The evolution process of the B. rapa (ECD04) genome.
Figure S6 ACK blocks in the B. rapa (ECD04) and B. oleracea (To1000) genomes.
Figure S7 Comparison of R gene ratios and genome sizes.
Figure S8 Sequence alignment of BraA08g039305E (CRA8.2.4) and Crr1a.
Figure S9 Neighbour‐joining tree of homologous genes of CR genes.
Figure S10 Sequence alignment of homologous genes of CRA3.7.1.
Figure S11 Sequence alignment of genomic regions in CRA3.7.1 and its homologues using the mVISTA program with “ECD04” as a reference.
Figure S12 Sequence alignment of genomic regions in CRA8.2.4 and its homologues.
Table S1 The distribution of centromeres in ECD04 genome.
Table S2 The distribution of centromeres in ECD04 genome.
Table S3 Structural variation summary of Chiifu and Z1 compared with ECD04 genome.
Table S4 Inter‐chromosomal translocations between ECD04 genome and Chiifu genome.
Table S5 Inter‐chromosomal translocations between ECD04 genome and Z1 genome.
Table S6 Summary of the transposable elements in ECD04 genome.
Table S7 Mapping rate of 199 B. rapa accessions and ECD04.
Table S8 ACK blocks in ECD04 genome.
Table S9 List of 978 RGA genes in ECD04 genome.
Table S10 Statistics of predicted RGA genes in 11 genome/subgenomes.
Table S11 Ratios of predicted NLR genes in 11 genome/subgenomes and previous study.
Table S12 Ratios of predicted RLK and RLP genes in 11 genome/subgenomes and previous study.
Table S13 List of 15 integrated CR loci from 28 CR loci have been identified and reported.
Table S14 Physical position in ECD04 genome of 15 integrated CR loci.
Table S15 List of candidate CR genes in 15 integrated CR loci.
Table S16 Primers used in this study.
Acknowledgements
This project was supported by grants from the National Natural Science Foundation of China (grants no. U20A2034 and 31871659) and the China Agriculture Research System (CARS‐12) to CZ. This project was supported by grants from Fundamental Research Funds for the Central Universities (grant no. 2662018PY068) to QY. We thank Dr. Gary Peng from Agriculture and Agri‐Food Canada, Saskatoon Research Centre, Canada, for critical reading the manuscript. We thank Dr. Shilin Zhu and Miss. Lu Wang for help on bioinformatics analysis.
Yang, Z. , Jiang, Y. , Gong, J. , Li, Q. , Dun, B. , Liu, D. , Yin, F. , Yuan, L. , Zhou, X. , Wang, H. , Wang, J. , Zhan, Z. , Shah, N. , Nwafor, C. C. , Zhou, Y. , Chen, P. , Zhu, L. , Li, S. , Wang, B. , Xiang, J. , Zhou, Y. , Li, Z. , Piao, Z. , Yang, Q. and Zhang, C. (2022) R gene triplication confers European fodder turnip with improved clubroot resistance. Plant Biotechnol. J., 10.1111/pbi.13827
Contributor Information
Zhongyun Piao, Email: zypiao@syau.edu.cn.
Qingyong Yang, Email: yqy@mail.hzau.edu.cn.
Chunyu Zhang, Email: zhchy@mail.hzau.edu.cn.
Data availability statement
All the raw sequencing data sets generated during the present study are available in the NCBI BioProject under the accession number PRJNA672906. The genome assemblies and annotation files are available at the website http://bna.hzau.edu.cn/download_genomic_seq.
References
- Bateman, A. , Birney, E. , Durbin, R. , Eddy, S.R. , Howe, K.L. and Sonnhammer, E.L. (2000) The Pfam protein families database. Nucleic Acids Res. 28, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belser, C. , Istace, B. , Denis, E. , Dubarry, M. , Baurens, F.C. , Falentin, C. , Genete, M. et al. (2018) Chromosome‐scale assemblies of plant genomes using nanopore long reads and optical maps. Nat. Plants, 4, 879–887. [DOI] [PubMed] [Google Scholar]
- Benson, G. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers, J.E. , Chapman, B.A. , Rong, J. and Paterson, A.H. (2003) Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature, 422, 433–438. [DOI] [PubMed] [Google Scholar]
- Brown, J.K. (2003) A cost of disease resistance: paradigm or peculiarity? Trends Genet. 19, 667–671. [DOI] [PubMed] [Google Scholar]
- Buczacki, S.T. , Toxopeus, H. , Mattusch, P. , Johnston, T.D. , Dixon, G.R. and Hobolth, L.A. (1975) Study of physiologic specialization in Plasmodiophora brassicae: proposals for attempted rationalization through an international approach. Trans. Brit. Mycol. Soc. 65, 295–303. [Google Scholar]
- Büschges, R. , Hollricher, K. , Panstruga, R. , Simons, G. , Wolter, M. , Frijters, A. , van Daelen, R. et al. (1997) The barley Mlo gene: a novel control element of plant pathogen resistance. Cell, 88, 695–705. [DOI] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. and Madden, T.L. (2009) BLAST+: architecture and applications. BMC Bioinformatics, 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel, B.L. , Korf, I. , Robb, S.M. , Parra, G. , Ross, E. , Moore, B. , Holt, C. et al. (2008) MAKER: an easy‐to‐use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai, A.L. , Xie, X.W. , Shi, Y.X. and Li, B.J. (2014) Special issue: research status of clubroot (Plasmodiophora brassicae) on cruciferous crops in China. Can. J. Plant Pathol. 36, 142–153. [Google Scholar]
- Chalhoub, B. , Denoeud, F. , Liu, S. , Parkin, I.A. , Tang, H. , Wang, X. , Chiquet, J. et al. (2014) Plant genetics. Early allopolyploid evolution in the post‐Neolithic Brassica napus oilseed genome. Science, 345, 950–953. [DOI] [PubMed] [Google Scholar]
- Chandran, V. , Wang, H. , Gao, F. , Cao, X.L. , Chen, Y.P. , Li, G.B. , Zhu, Y. et al. (2018) miR396‐OsGRFs module balances growth and rice blast disease‐resistance. Front. Plant Sci. 9, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, A. , Lamara, M. , Wei, Y. , Hu, H. , Parkin, I.A.P. , Gossen, B.D. , Peng, G. et al. (2019) Clubroot resistance gene Rcr6 in Brassica nigra resides in a genomic region homologous to chromosome A08 in B. rapa . BMC Plant Biol. 19, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Jing, J. , Zhan, Z. , Zhang, T. , Zhang, C. and Piao, Z. (2013) Identification of novel QTLs for isolate‐specific partial resistance to Plasmodiophora brassicae in Brassica rapa . PLoS One, 8, e85307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F. , Sun, R. , Hou, X. , Zheng, H. , Zhang, F. , Zhang, Y. , Liu, B. et al. (2016) Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication in Brassica rapa and Brassica oleracea . Nat. Genet. 48, 1218–1224. [DOI] [PubMed] [Google Scholar]
- Cheng, F. , Wu, J. and Wang, X. (2014) Genome triplication drove the diversification of Brassica plants. Hortic. Res. 1, 14024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, C. , Wang, J.J. , Zhao, J.H. , Fang, Y.Y. , He, X.F. , Guo, H.S. and Duan, C.G. (2020) A Brassica miRNA regulates plant growth and immunity through distinct modes of action. Mol. Plant, 13, 231–245. [DOI] [PubMed] [Google Scholar]
- Darriba, D. , Taboada, G.L. , Doallo, R. and Posada, D. (2011) ProtTest 3: fast selection of best‐fit models of protein evolution. Bioinformatics, 27, 1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denance, N. , Sanchez‐Vallet, A. , Goffner, D. and Molina, A. (2013) Disease resistance or growth: the role of plant hormones in balancing immune responses and fitness costs. Front. Plant Sci. 4, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, Y. , Zhai, K. , Xie, Z. , Yang, D. , Zhu, X. , Liu, J. , Wang, X. et al. (2017) Epigenetic regulation of antagonistic receptors confers rice blast resistance with yield balance. Science, 355, 962–965. [DOI] [PubMed] [Google Scholar]
- Dixon, G.R. (2006) The biology of Plasmodiophora brassicae Wor. – a review of recent advances. Acta Hortic. 706, 271–282. [Google Scholar]
- Dixon, G.R. (2009) The occurrence and economic impact of Plasmodiophora brassicae and clubroot disease. J. Plant Growth Regul. 28, 194–202. [Google Scholar]
- Dudchenko, O. , Batra, S.S. , Omer, A.D. , Nyquist, S.K. , Hoeger, M. , Durand, N.C. , Shamim, M.S. et al. (2017) De novo assembly of the Aedes aegypti genome using Hi‐C yields chromosome‐length scaffolds. Science, 356, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, N.C. , Shamim, M.S. , Machol, I. , Rao, S.S. , Huntley, M.H. , Lander, E.S. and Aiden, E.L. (2016) Juicer provides a one‐click system for analyzing loop‐resolution Hi‐C experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D.M. and Kelly, S. (2019) OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredua‐Agyeman, R. , Yu, Z. , Hwang, S.F. and Strelkov, S.E. (2020) Genome‐wide mapping of loci associated with resistance to clubroot in Brassica napus ssp. napobrassica (rutabaga) accessions from nordic countries. Front. Plant Sci. 11, 742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, M. , He, Y. , Yin, X. , Zhong, X. , Yan, B. , Wu, Y. , Chen, J. et al. (2021) Ca(2+) sensor‐mediated ROS scavenging suppresses rice immunity and is exploited by a fungal effector. Cell, 184, 5391–5404 e5317. [DOI] [PubMed] [Google Scholar]
- Grob, S. , Schmid, M.W. and Grossniklaus, U. (2014) Hi‐C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila . Mol. Cell, 55, 678–693. [DOI] [PubMed] [Google Scholar]
- Grund, E. , Tremousaygue, D. and Deslandes, L. (2019) Plant NLRs with integrated domains: unity makes strength. Plant Physiol. 179, 1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond‐Kosack, K.E. and Jones, J.D. (1997) Plant disease resistance genes. Annu. Rev. Plant Physiol. Plant Mol. Biol. 48, 575–607. [DOI] [PubMed] [Google Scholar]
- Hasan, M.J. and Rahman, H. (2018) Resynthesis of Brassica juncea for resistance to Plasmodiophora brassicae pathotype 3. Breed. Sci. 68, 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama, K. , Niwa, T. , Kato, T. , Ohara, T. , Kakizaki, T. and Matsumoto, S. (2017) The tandem repeated organization of NB‐LRR genes in the clubroot‐resistant CRb locus in Brassica rapa L. Mol. Genet. Genomics, 292, 397–405. [DOI] [PubMed] [Google Scholar]
- Hatakeyama, K. , Suwabe, K. , Tomita, R.N. , Kato, T. , Nunome, T. , Fukuoka, H. and Matsumoto, S. (2013) Identification and characterization of Crr1a, a gene for resistance to clubroot disease (Plasmodiophora brassicae Woronin) in Brassica rapa L. PLoS One, 8, e54745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai, M. , Harada, T. , Kubo, N. , Tsukada, M. , Suwabe, K. and Matsumoto, S. (2004) A novel locus for clubroot resistance in Brassica rapa and its linkage markers. Theor. Appl. Genet. 108, 639–643. [DOI] [PubMed] [Google Scholar]
- Hirani, A.H. , Gao, F. , Liu, J. , Fu, G. , Wu, C. , McVetty, P.B.E. , Duncan, R.W. et al. (2018) Combinations of independent dominant loci conferring clubroot resistance in all four turnip accessions (Brassica rapa) from the European clubroot differential set. Front. Plant Sci. 9, 1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard, R.J. , Strelkov, S.E. and Harding, M.W. (2010) Clubroot of cruciferous crops – new perspectives on an old disease†. Can. J. Plant Pathol. 32, 43–57. [Google Scholar]
- Huang, Z. , Peng, G. , Liu, X. , Deora, A. , Falk, K.C. , Gossen, B.D. , McDonald, M.R. et al. (2017) Fine mapping of a clubroot resistance gene in Chinese cabbage using SNP markers identified from bulked segregant RNA sequencing. Front. Plant Sci. 8, 1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Z. , Peng, G. , Gossen, B.D. and Yu, F. (2019) Fine mapping of a clubroot resistance gene from turnip using SNP markers identified from bulked segregant RNA‐Seq. Mol. Breed. 39, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapos, P. , Devendrakumar, K.T. and Li, X. (2019) Plant NLRs: from discovery to application. Plant Sci. 279, 3–18. [DOI] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. and Salzberg, S.L. (2015) HISAT: a fast spliced aligner with low memory requirements. Nat. Methods, 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.H. , Qi, D. , Ashfield, T. , Helm, M. and Innes, R.W. (2016) Using decoys to expand the recognition specificity of a plant disease resistance protein. Science, 351, 684–687. [DOI] [PubMed] [Google Scholar]
- Kim, W. , Prokchorchik, M. , Tian, Y. , Kim, S. , Jeon, H. and Segonzac, C. (2020) Perception of unrelated microbe‐associated molecular patterns triggers conserved yet variable physiological and transcriptional changes in Brassica rapa ssp. pekinensis . Hortic. Res. 7, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren, S. , Walenz, B.P. , Berlin, K. , Miller, J.R. , Bergman, N.H. and Phillippy, A.M. (2017) Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Res. 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laila, R. , Park, J.I. , Robin, A.H.K. , Natarajan, S. , Vijayakumar, H. , Shirasawa, K. , Isobe, S. et al. (2019) Mapping of a novel clubroot resistance QTL using ddRAD‐seq in Chinese cabbage (Brassica rapa L.). BMC Plant Biol. 19, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkan, N.J. , Ma, L. and Borhan, M.H. (2015) The Brassica napus receptor‐like protein RLM2 is encoded by a second allele of the LepR3/Rlm2 blackleg resistance locus. Plant Biotechnol. J. 13, 983–992. [DOI] [PubMed] [Google Scholar]
- Larkan, N.J. , Ma, L. , Haddadi, P. , Buchwaldt, M. , Parkin, I.A.P. , Djavaheri, M. and Borhan, M.H. (2020) The Brassica napus wall‐associated kinase‐like (WAKL) gene Rlm9 provides race‐specific blackleg resistance. Plant J. 104, 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. and Bork, P. (2019) Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Li, X. , Xuan, Y. , Jiang, J. , Wei, Y. and Piao, Z. (2018) Genome wide identification and expression profiling of SWEET genes family reveals its role during Plasmodiophora brassicae‐induced formation of clubroot in Brassica rapa . Front. Plant Sci. 9, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Luo, Y. , Chen, B. , Xu, K. , Zhang, F. , Li, H. , Huang, Q. et al. (2016a) A genome‐wide association study reveals new loci for resistance to clubroot disease in Brassica napus . Front. Plant Sci. 7, 1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P. , Quan, X. , Jia, G. , Xiao, J. , Cloutier, S. and You, F.M. (2016b) RGAugury: a pipeline for genome‐wide prediction of resistance gene analogs (RGAs) in plants. BMC Genom. 17, 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Zheng, Y.P. , Zhou, X.H. , Yang, X.M. , He, X.R. , Feng, Q. , Zhu, Y. et al. (2021) Rice miR1432 fine‐tunes the balance of yield and blast disease resistance via different modules. Rice (N Y), 14, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lu, K. , Wei, L. , Li, X. , Wang, Y. , Wu, J. , Liu, M. , Zhang, C. et al. (2019) Whole‐genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 10, 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysak, M.A. , Koch, M.A. , Pecinka, A. and Schubert, I. (2005) Chromosome triplication found across the tribe Brassiceae . Genome Res. 15, 516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysak, M.A. , Mandakova, T. and Schranz, M.E. (2016) Comparative paleogenomics of crucifers: ancestral genomic blocks revisited. Curr. Opin. Plant Biol. 30, 108–115. [DOI] [PubMed] [Google Scholar]
- Majoros, W.H. , Pertea, M. and Salzberg, S.L. (2004) TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene‐finders. Bioinformatics, 20, 2878–2879. [DOI] [PubMed] [Google Scholar]
- Marcais, G. , Delcher, A.L. , Phillippy, A.M. , Coston, R. , Salzberg, S.L. and Zimin, A. (2018) MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher, M. , Gundlach, H. , Himmelbach, A. , Beier, S. , Twardziok, S.O. , Wicker, T. , Radchuk, V. et al. (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature, 544, 427–433. [DOI] [PubMed] [Google Scholar]
- Mason, A.S. , Rousseau‐Gueutin, M. , Morice, J. , Bayer, P.E. , Besharat, N. , Cousin, A. , Pradhan, A. et al. (2016) Centromere locations in Brassica A and C genomes revealed through half‐tetrad analysis. Genetics, 202, 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto, E. , Yasui, C. , Ohi, M. and Tsukada, M. (1998) Linkage analysis of RFLP markers for clubroot resistance and pigmentation in Chinese cabbage (Brassica rapa ssp. pekinensis). Euphytica, 104, 79–86. [Google Scholar]
- Mistry, J. , Finn, R.D. , Eddy, S.R. , Bateman, A. and Punta, M. (2013) Challenges in homology search: HMMER3 and convergent evolution of coiled‐coil regions. Nucleic Acids Res. 41, e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney, M.M. , Walker, J.M. and Sharma, K.K. (1989) High efficiency transformation of Brassica napus using Agrobacterium vectors. Plant Cell Rep. 8, 238–242. [DOI] [PubMed] [Google Scholar]
- Mulder, N. and Apweiler, R. (2007) InterPro and InterProScan. In Comparative Genomics ( Bergman, N.H. , ed.), pp. 59–70. Totowa, NJ: Humana Press. [Google Scholar]
- Neik, T.X. , Barbetti, M.J. and Batley, J. (2017) Current status and challenges in identifying disease resistance genes in Brassica napus . Front. Plant Sci. 8, 1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev, S.I. , Berney, C. , Fahrni, J.F. , Bolivar, I. , Polet, S. , Mylnikov, A.P. , Aleshin, V.V. et al. (2004) The twilight of Heliozoa and rise of Rhizaria, an emerging supergroup of amoeboid eukaryotes. Proc. Natl Acad. Sci. USA, 101, 8066–8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning, Y. , Liu, W. and Wang, G.L. (2017) Balancing immunity and yield in crop plants. Trends Plant Sci. 22, 1069–1079. [DOI] [PubMed] [Google Scholar]
- Pang, W. , Fu, P. , Li, X. , Zhan, Z. , Yu, S. and Piao, Z. (2018) Identification and mapping of the clubroot resistance gene CRd in Chinese cabbage (Brassica rapa ssp. pekinensis). Front. Plant Sci. 9, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, W. , Liang, Y. , Zhan, Z. , Li, X. and Piao, Z. (2020) Development of a sinitic clubroot differential set for the pathotype classification of Plasmodiophora brassicae . Front. Plant Sci. 11, 568771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton, M. , Sebra, R. , Pang, A.W. , Ummat, A. , Franzen, O. , Rausch, T. , Stutz, A.M. et al. (2015) Assembly and diploid architecture of an individual human genome via single‐molecule technologies. Nat. Methods, 12, 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, G. , McGregor, L. , Lahlali, R. , Gossen, B.D. , Hwang, S.F. , Adhikari, K.K. , Strelkov, S.E. et al. (2011) Potential biological control of clubroot on canola and crucifer vegetable crops. Plant Pathol. 60, 566–574. [Google Scholar]
- Pertea, M. , Pertea, G.M. , Antonescu, C.M. , Chang, T.C. , Mendell, J.T. and Salzberg, S.L. (2015) StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat. Biotechnol. 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao, Z.Y. , Deng, Y.Q. , Choi, S.R. , Park, Y.J. and Lim, Y.P. (2004) SCAR and CAPS mapping of CRb, a gene conferring resistance to Plasmodiophora brassicae in Chinese cabbage (Brassica rapa ssp. pekinensis). Theor. Appl. Genet. 108, 1458–1465. [DOI] [PubMed] [Google Scholar]
- Poliakov, A. , Foong, J. , Brudno, M. and Dubchak, I. (2014) GenomeVISTA—an integrated software package for whole‐genome alignment and visualization. Bioinformatics, 30, 2654–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, X. , An, H. , Ragsdale, A.P. , Hall, T.E. , Gutenkunst, R.N. , Chris Pires, J. and Barker, M.S. (2017) Genomic inferences of domestication events are corroborated by written records in Brassica rapa . Mol. Ecol. 26, 3373–3388. [DOI] [PubMed] [Google Scholar]
- Sakamoto, K. , Saito, A. , Hayashida, N. , Taguchi, G. and Matsumoto, E. (2008) Mapping of isolate‐specific QTLs for clubroot resistance in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Theor. Appl. Genet. 117, 759–767. [DOI] [PubMed] [Google Scholar]
- Schuler, G.D. (1997) Sequence mapping by electronic PCR. Genome Res. 7, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev, V. , Kosarev, P. , Seledsov, I. and Vorobyev, D. (2006) Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 7(Suppl 1), S10.11–S10.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J.M. , Guan, Z. , Hu, J. , Guo, C. , Yang, Z. , Wang, S. , Liu, D. et al. (2020) Eight high‐quality genomes reveal pan‐genome architecture and ecotype differentiation of Brassica napus . Nat. Plants, 6, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Steinkamp, R. , Waack, S. and Morgenstern, B. (2004) AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelkov, S.E. and Hwang, S.F. (2014) Clubroot in the Canadian canola crop: 10 years into the outbreak. Can. J. Plant Pathol. 36, 27–36. [Google Scholar]
- Strelkov, S.E. , Hwang, S.F. , Manolii, V.P. , Cao, T.S. , Fredua‐Agyeman, R. , Harding, M.W. , Peng, G. et al. (2018) Virulence and pathotype classification of Plasmodiophora brassicae populations collected from clubroot resistant canola (Brassica napus) in Canada. Can. J. Plant Pathol. 40, 284–298. [Google Scholar]
- Suwabe, K. , Tsukazaki, H. , Iketani, H. , Hatakeyama, K. , Fujimura, M. , Nunome, T. , Fukuoka, H. et al. (2003) Identification of two loci for resistance to clubroot (Plasmodiophora brassicae Woronin) in Brassica rapa L. Theor. Appl. Genet. 107, 997–1002. [DOI] [PubMed] [Google Scholar]
- Suwabe, K. , Tsukazaki, H. , Iketani, H. , Hatakeyama, K. , Kondo, M. , Fujimura, M. , Nunome, T. et al. (2006) Simple sequence repeat‐based comparative genomics between Brassica rapa and Arabidopsis thaliana: the genetic origin of clubroot resistance. Genetics, 173, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Brassica rapa Genome Sequencing Project, C. , Wang, X. , Wang, H. , Wang, J. , Sun, R. , Wu, J. , Liu, S. et al. (2011) The genome of the mesopolyploid crop species Brassica rapa . Nat. Genet. 43, 1035. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D. , Gibson, T.J. and Higgins, D.G. (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics, Chapter 2, Unit 2.3. [DOI] [PubMed] [Google Scholar]
- Tian, D. , Traw, M.B. , Chen, J.Q. , Kreitman, M. and Bergelson, J. (2003) Fitness costs of R‐gene‐mediated resistance in Arabidopsis thaliana . Nature, 423, 74–77. [DOI] [PubMed] [Google Scholar]
- Tirnaz, S. , Bayer, P.E. , Inturrisi, F. , Zhang, F. , Yang, H. , Dolatabadian, A. , Neik, T.X. et al. (2020) Resistance gene analogs in the Brassicaceae: identification, characterization, distribution, and evolution. Plant Physiol. 184, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toxopeus, H. , Dixon, G.R. and Mattusch, P. (1986) Physiological specialization in Plasmodiophora brassicae: an analysis by international experimentation. Trans. Br. Mycol. Soc. 87, 279–287. [Google Scholar]
- Ueno, H. , Matsumoto, E. , Aruga, D. , Kitagawa, S. , Matsumura, H. and Hayashida, N. (2012) Molecular characterization of the CRa gene conferring clubroot resistance in Brassica rapa . Plant Mol. Biol. 80, 621–629. [DOI] [PubMed] [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. , Cuomo, C.A. et al. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, A.G. and Baker, K.F. (1969) Possible gene centers for resistance in the genus Brassica to Plasmodiophora brassicae . Econ. Bot. 23, 245–252. [Google Scholar]
- Werner, S. , Diederichsen, E. , Frauen, M. , Schondelmaier, J. and Jung, C. (2008) Genetic mapping of clubroot resistance genes in oilseed rape. Theor. Appl. Genet. 116, 363–372. [DOI] [PubMed] [Google Scholar]
- Wu, D. , Liang, Z. , Yan, T. , Xu, Y. , Xuan, L. , Tang, J. , Zhou, G. et al. (2019) Whole‐genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence. Mol. Plant, 12, 30–43. [DOI] [PubMed] [Google Scholar]
- Wu, H.J. , Zhang, Z. , Wang, J.Y. , Oh, D.H. , Dassanayake, M. , Liu, B. , Huang, Q. et al. (2012) Insights into salt tolerance from the genome of Thellungiella salsuginea . Proc. Natl Acad. Sci. USA, 109, 12219–12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. and Zhou, J.M. (2013) Receptor‐like kinases in plant innate immunity. J. Integr. Plant Biol. 55, 1271–1286. [DOI] [PubMed] [Google Scholar]
- Xie, T. , Zheng, J.F. , Liu, S. , Peng, C. , Zhou, Y.M. , Yang, Q.Y. and Zhang, H.Y. (2015) De novo plant genome assembly based on chromatin interactions: a case study of Arabidopsis thaliana . Mol. Plant, 8, 489–492. [DOI] [PubMed] [Google Scholar]
- Xu, Z. and Wang, H. (2007) LTR_FINDER: an efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Res. 35, W265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Liu, D. , Wang, X. , Ji, C. , Cheng, F. , Liu, B. , Hu, Z. et al. (2016) The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 48, 1225–1232. [DOI] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yu, F. , Zhang, X. , Peng, G. , Falk, K.C. , Strelkov, S.E. and Gossen, B.D. (2017) Genotyping‐by‐sequencing reveals three QTL for clubroot resistance to six pathotypes of Plasmodiophora brassicae in Brassica rapa . Sci. Rep. 7, 4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, M. , Jiang, Z. , Bi, G. , Nomura, K. , Liu, M. , Wang, Y. , Cai, B. et al. (2021) Pattern‐recognition receptors are required for NLR‐mediated plant immunity. Nature, 592, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, Z. , Jiang, Y. , Shah, N. , Hou, Z. , Zhou, Y. , Dun, B. , Li, S. et al. (2020) Association of clubroot resistance locus PbBa8.1 with a linkage drag of high erucic acid content in the seed of the European turnip. Front. Plant Sci. 11, 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Cai, X. , Wu, J. , Liu, M. , Grob, S. , Cheng, F. , Liang, J. et al. (2018) Improved Brassica rapa reference genome by single‐molecule sequencing and chromosome conformation capture technologies. Hortic. Res. 5, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Genome alignment between the Chiifu‐ECD04 and Z1‐ ECD04 genomes.
Figure S2 Hi‐C data analysis of ECD04.
Figure S3 The insert time of intact LTR retrotransposons in the ECD04 genome.
Figure S4 Phylogenetic tree of ECD04 and retention of ancestral genes in ECD04 genome.
Figure S5 The evolution process of the B. rapa (ECD04) genome.
Figure S6 ACK blocks in the B. rapa (ECD04) and B. oleracea (To1000) genomes.
Figure S7 Comparison of R gene ratios and genome sizes.
Figure S8 Sequence alignment of BraA08g039305E (CRA8.2.4) and Crr1a.
Figure S9 Neighbour‐joining tree of homologous genes of CR genes.
Figure S10 Sequence alignment of homologous genes of CRA3.7.1.
Figure S11 Sequence alignment of genomic regions in CRA3.7.1 and its homologues using the mVISTA program with “ECD04” as a reference.
Figure S12 Sequence alignment of genomic regions in CRA8.2.4 and its homologues.
Table S1 The distribution of centromeres in ECD04 genome.
Table S2 The distribution of centromeres in ECD04 genome.
Table S3 Structural variation summary of Chiifu and Z1 compared with ECD04 genome.
Table S4 Inter‐chromosomal translocations between ECD04 genome and Chiifu genome.
Table S5 Inter‐chromosomal translocations between ECD04 genome and Z1 genome.
Table S6 Summary of the transposable elements in ECD04 genome.
Table S7 Mapping rate of 199 B. rapa accessions and ECD04.
Table S8 ACK blocks in ECD04 genome.
Table S9 List of 978 RGA genes in ECD04 genome.
Table S10 Statistics of predicted RGA genes in 11 genome/subgenomes.
Table S11 Ratios of predicted NLR genes in 11 genome/subgenomes and previous study.
Table S12 Ratios of predicted RLK and RLP genes in 11 genome/subgenomes and previous study.
Table S13 List of 15 integrated CR loci from 28 CR loci have been identified and reported.
Table S14 Physical position in ECD04 genome of 15 integrated CR loci.
Table S15 List of candidate CR genes in 15 integrated CR loci.
Table S16 Primers used in this study.
Data Availability Statement
All the raw sequencing data sets generated during the present study are available in the NCBI BioProject under the accession number PRJNA672906. The genome assemblies and annotation files are available at the website http://bna.hzau.edu.cn/download_genomic_seq.