

Abstract
Objectives
Crohn's disease (CD) initiation and pathogenesis are believed to involve an environmental trigger in a genetically susceptible person that results in an immune response against commensal gut bacteria, leading to a compromised intestinal epithelial barrier and a cycle of inflammation. However, it has been difficult to study the contribution of all factors together in a physiologically relevant model and in a heterogenous patient population.
Methods
We developed an autologous colonic monolayer model that incorporated the immune response from the same donor and a commensal bacteria, Faecalibacterium prausnitzii. Two‐dimensional monolayers were grown from three‐dimensional organoids generated from intestinal biopsies, and the epithelial integrity of the epithelium was measured using transepithelial electrical resistance. We determined the effect of immune cells alone, bacteria alone and the co‐culture of immune cells and bacteria on integrity.
Results
Monolayers derived from CD donors had impaired epithelial integrity compared to those from non‐inflammatory bowel disease (IBD) donors. This integrity was further impaired by culture with bacteria, but not immune cells, despite a higher frequency of inflammatory phenotype peripheral T cells in CD donors. Variability in epithelial integrity was higher in CD donors than in non‐IBD donors.
Conclusion
We have developed a new autologous model to study the complexity of CD, which allows for the comparison of the barrier properties of the colonic epithelium and the ability to study how autologous immune cells directly affect the colonic barrier and whether this is modified by luminal bacteria. This new model allows for the study of individual patients and could inform treatment decisions.
Keywords: bacteria, Crohn's disease, cytokines, epithelium, organoid, T cells
We developed an autologous model to study epithelial integrity in human Crohn's disease (CD). We cultured immune cells and the commensal bacterium, Faecalibacterium prausnitzii, with patient‐matched epithelial monolayers generated from three‐dimensional organoids derived from intestinal biopsies. We showed inherent differences in integrity in monolayers from CD patients and a further reduction upon culture with bacteria but not immune cells.

Introduction
Inflammatory bowel diseases (IBD) are chronic inflammatory diseases, comprising ulcerative colitis, Crohn's disease (CD) and IBD‐unspecified that mainly affect the gut. 1 Patients with IBD experience a range of symptoms, including diarrhoea, abdominal pain, fatigue and weight loss 2 and, therefore, experience a severe decrease in quality of life. The aetiology of CD is unknown, but it is thought that the combination of four factors is responsible: an environmental trigger in a genetically susceptible person results in an immune response against commensal gut bacteria. 1 This combination of factors results in an inflammatory response and increased intestinal permeability. Which of these is the primary cause is unknown, but it is thought that the net result is increased translocation of gut microbial antigens into the lamina propria and a cycle of chronic/recurring inflammation. Several treatment choices are available with the most modern using recombinant antibodies directed against cytokines and other immune proteins. 3 However, the heterogeneity of CD means that standardised treatments for all patients do not exist (up to 2/3 of patients become unresponsive to treatment 4 ); rather, trial and error approaches are used as clinicians work through the armamentarium of drugs available. 5 The mechanisms and initial cause of disease pathology, course of disease and efficacy of therapies all vary between individuals.
In order to study the complexity of this multifactorial disease in a heterogeneous patient population, we devised an autologous colonoid monolayer co‐culture model. This model is specific for each individual, comprising differentiated epithelial cells derived from patient biopsies and matched autologous immune cells from peripheral blood. We use primary cells rather than cell lines to identify potential mechanisms of disease. 6 , 7 This model provides a tool to study the appropriate exposure of the intestinal epithelium to bacteria and immune cells, an advantage over three‐dimensional organoids. 8 Finally, the model allows for the study of individual patients and highlights the variability between individuals with a complex and multifactorial disease.
We developed this model to test the feasibility of studying immune and bacterial factors driving disease pathology in individuals. We performed preliminary experiments to study (1) inherent epithelial integrity and immune activation in CD versus healthy donors, (2) the effect of bacteria on epithelial integrity and immune function and (3) the ability to study individual patients undergoing different treatments.
Results
The integrity of intestinal epithelial monolayers from CD patients is compromised compared with those from non‐IBD donors
We first compared the integrity of two‐dimensional monolayers derived from CD or non‐IBD donor biopsies. Three‐dimensional colonoids were expanded, then mechanically dissociated, cultured in Transwell plates and monitored for confluency. Once confluent, monolayers were cultured in differentiation media for 3 days (Figure 1a). There was no obvious histological difference between the two groups of monolayers (Figure 1b)—both formed visually intact monolayers, and in both cohorts, the differentiation process resulted in the development of columnar epithelium. However, monolayers from CD patients had a lower transepithelial electrical resistance (TEER) than those from non‐IBD donors. This was initially evident when the monolayers achieved confluence between days 7 and 10 but was even more evident after differentiation of the monolayers through withdrawal of Wnt 3A and R‐spondin (day 9 difference in nondifferentiation media, P < 0.001; day 12 difference in differentiation media P < 0.001; Figure 1c) as the TEER of monolayers from non‐IBD, but not CD donors, increased further during the 3 day differentiation period (non‐IBD P < 0.001, CD not significant). This raises the possibility that there are inherent differences in the epithelial barrier in CD patients, which could contribute to the development of inflammation. This finding, using individual patient‐derived monolayers from diseased and control cohorts, demonstrates the significant advantages this approach has over the traditional use of epithelial cell lines (e.g. Caco‐2) for such experiments.
Figure 1.
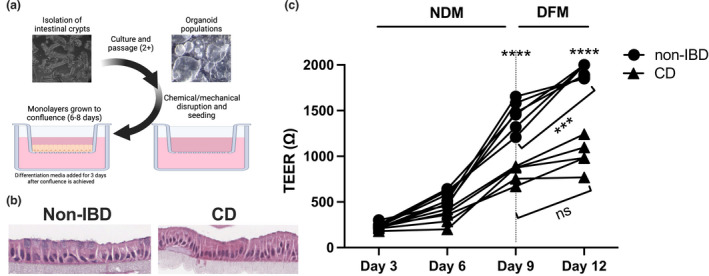
The integrity of intestinal epithelial monolayers from Crohn's disease (CD) patients is compromised compared to those from non‐inflammatory bowel disease (non‐IBD) donors. (a) Scheme of the generation of intestinal monolayers (created in Biorender). (b) Representative H&E images of non‐IBD (left) and CD donor (right) monolayers (representative of 10 non‐IBD and 8 CD). (c) Non‐IBD and CD donor intestinal monolayers were cultured for 9 days in nondifferentiation media (NDM), then for 3 days in differentiation media (DFM). Transepithelial electrical resistance (TEER) readings were taken at indicated intervals. Vertical dotted line represents the change from NDM to DFM. Non‐IBD (n = 5), CD (n = 6). Statistical analyses were calculated using a mixed‐effects model and Sidak's multiple comparisons test (***P < 0.001, ****P < 0.0001).
Peripheral T cells from CD patients have an activated, inflammatory phenotype compared to those from the non‐IBD cohort
We then investigated a second factor involved in disease pathogenesis, the immune response, in order to integrate patient‐specific immune cells into the primary cell‐derived monolayer model. Intestinal immune cells from colonoscopy biopsies are difficult to recover in sufficient numbers to test multiple parameters in these experiments. We had previously shown that the immune signatures of intestinal T cell populations were different in patients with CD compared with healthy people. 9 Therefore, we first tested whether these differences also existed in peripheral immune cells. We compared the baseline phenotype of T cells from CD patients compared with a healthy (non‐IBD) cohort to determine whether there were inherent immune differences between the two populations. Peripheral blood mononuclear cells (PBMCs) were collected from people diagnosed with CD or from non‐IBD donors (Table 1), and analysed directly ex vivo or activated with anti‐CD3 + anti‐28 antibodies (antiCD3/28). There was no significant difference in the frequency of CD4+IFN‐γ+, IL‐17+ or regulatory (CD25hiFOXP3+) T cell subsets directly ex vivo from the CD compared with the non‐IBD cohort (Figure 2a), in contrast to previous reports. 10 , 11 Quantification of gene expression of T cell inflammatory and regulatory cytokines and transcription factors directly ex vivo showed no statistically significant difference in expression of TNFA, IL23R and FOXP3 in PBMCs from CD versus non‐IBD donors (Supplementary figure 2). Upon stimulation with anti‐CD3/28, there was increased production of TNF, IL‐17 and IL‐6 in T cells from the CD versus non‐IBD cohort (Figure 2b), similar to what has been shown, 12 , 13 but there was no difference in the production of other cytokines—IL‐2, IL‐4, IL‐12p70, IL‐23 or IFN‐γ (Supplementary figure 3). To determine whether the increase in cytokine production reflected an overall increase in activation, we measured the proliferative capacity of T cells isolated from the CD or non‐IBD cohort. T cells from CD patients had a higher precursor frequency (number of cells that divided in response to stimulation) and proliferation index (extent of division) than those from the non‐IBD cohort (Figure 2c) when stimulated with anti‐CD3/28.
Table 1.
Donor information for PBMC experiments (Figure 2)
| Variable | Patients with CD (n = 25) | Health donors (non‐IBD; n = 16) |
|---|---|---|
| Age mean (SD) | 40.32 (17.1) | 30.69 (8.66) |
| Sex (%) | ||
| Female | 12 (48) | 8 (50) |
| Male | 13 (52) | 8 (50) |
| Treatment at time of appointment (%) | ||
| 6MP/azathioprine | 1 (4) | |
| Azathioprine + Pentasa + prednisone | 2 (8) | |
| Asacol | 1 (4) | |
| Humira + methotrexate | 1 (4) | |
| Pentasa | 1 (4) | |
| Azathioprine | 2 (8) | |
| Humira | 4 (16) | |
| Prednisone | 2 (8) | |
| Infliximab + azathioprine | 1 (4) | |
| Humira + azathioprine | 1 (4) | |
| Cholestyramine | 1 (4) | |
| Adalimumab + azathioprine | 1 (4) | |
| No medication | 7 (28) | |
Figure 2.
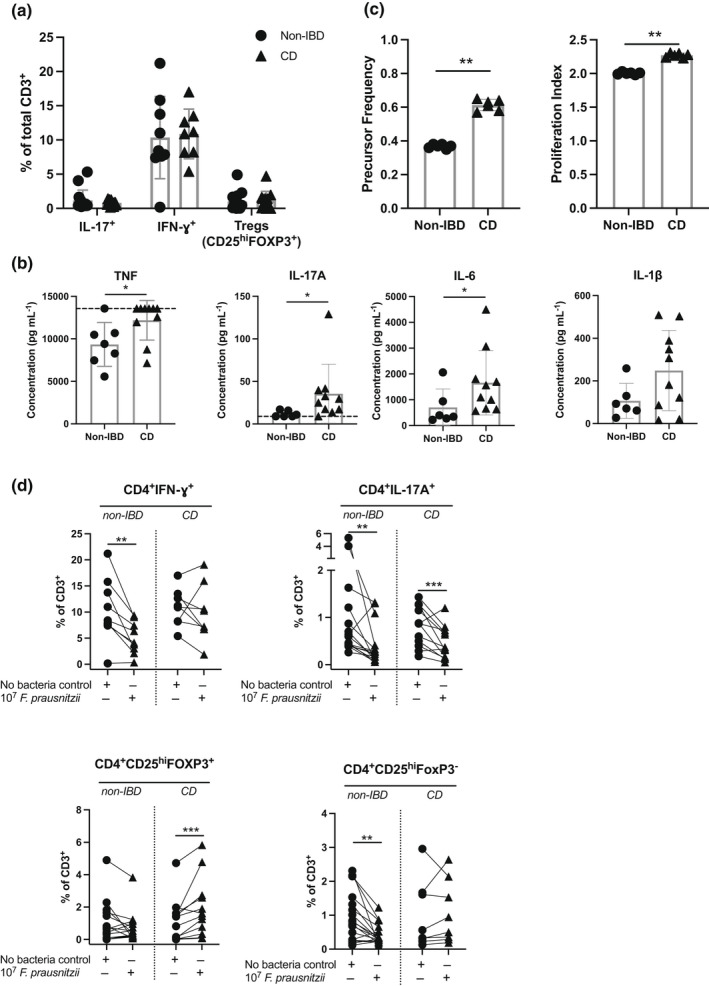
Peripheral T cells from Crohn's disease (CD) donors have a more inflammatory phenotype than those from non‐inflammatory bowel disease (non‐IBD) donors. PBMCs were isolated from non‐IBD or CD donors. (a) Frequency of PBMC CD4+IL‐17A+, CD4+IFN‐γ+ and regulatory T cells (CD4+CD25+FOXP3hi) of CD3+ cells. Data are shown as individual patients, and the bar represents the median frequency. Non‐IBD (n = 9), CD (n = 8). (b) 80 000 PBMCs were cultured with anti‐CD3/anti‐CD28 beads at a 1:1 ratio for 48 h. Cytokine supernatants were detected using a LegendPlex Human T Cell Cytokine Panel, then analysed by flow cytometry. Data are shown as individual patients, and the bar represents the median concentration (pg mL−1). Where appropriate, upper or lower limits of detection are shown with the dotted line. Non‐IBD (n = 7), CD (n = 10). (c) Tconvs were stained with Cell‐Trace Violet to allow for tracking of division events. Precursor frequency was calculated by (N/2 i )/(Total number of events/2 i ) × 100 = % P, then precursor frequency determined by the sum of % P of all peaks where division occurred (peaks 1–4). Proliferation index was calculated by ((N/2 i ) × N)/(Total number of events), where N indicates the number of events within a proliferation peak and 2 i indicates the number progeny per division stage. Non‐IBD (n = 6), CD (n = 6). Statistical analyses were calculated using the Mann–Whitney U‐test (*P < 0.05, **P < 0.01). (d) PBMCs were isolated from non‐IBD and CD donors and cultured for 72 h in the presence or absence of 107 heat‐killed Faecalibacterium prausnitzii, then analysed by flow cytometry. Left top: Frequency of CD4+IFN‐γ+ T cells. Right top: Frequency of CD4+IL‐17A+ T cells. Left bottom: Frequency of regulatory T cells (CD4+CD25+FOXP3HI). Right bottom: Frequency of CD4+CD25HIFOXP3− T cells. Statistical analyses were calculated using the Wilcoxon matched‐pairs signed‐rank test, (**P < 0.01, ***P < 0.001). Non‐IBD (n = 9), CD (n = 8). Lines represent matched‐patient PBMCs in each condition. Similar results were obtained with culture of 106 bacteria.
Differences in the peripheral T cell response from CD versus non‐IBD donors following culture with Faecalibacterium prausnitzii
Because we planned to test the effect of bacterial interactions with both immune and epithelial components in the monolayer model, we first studied the interaction between immune cells and the commensal bacterium used for these studies, F. prausnitzii. F. prausnitzii is linked to CD—its abundance is decreased in CD versus healthy patients and active versus inactive CD. 14 PBMCs were cultured in vitro directly with heat‐killed F. prausnitzii, and the changes in the frequency of T cell subsets were measured. Figure 2d shows there was no difference in the frequency of CD4+IFN‐γ+, CD4+IL‐17+ or regulatory (CD25hiFOXP3+) T cell populations between CD and non‐IBD donors. However, when stimulated with F. prausnitzii, we saw a decrease in both CD4+IFN‐γ+, CD4+IL‐17+ and CD4+CD25HIFOXP3− T cell populations in the non‐IBD cohort, but only in CD4+IL‐17+ T cells in the CD cohort (Figure 2d, top panels). We saw no difference in the median fluorescence intensity of CD25 in the FOXP3− T cells with or without bacteria (data not shown). In addition, the frequency of Tregs from CD donors but not non‐IBD donors was increased (Figure 2d, left bottom panel), indicative of a response to inflammation. These data indicate that T cells from CD patients may respond differently to commensal bacteria than T cells from non‐IBD donors.
Collectively, these data validate previous data demonstrating greater inflammatory cytokine production in PBMCs from CD versus non‐IBD cohorts. Additionally, we show that this reflects an overall increase in baseline T cell activation, measured by proliferative capacity, and the lack of appropriate response to commensal bacterial stimulation in CD donors.
Effect of bacteria and immune cell co‐culture on epithelial integrity in the non‐IBD versus CD population
We next combined the three components involved in this model of disease: the intestinal epithelium, the immune response from the same donor, and the commensal bacteria, F. prausnitzii. We measured epithelial integrity as a readout of the effect of immune cells alone, bacteria alone, and the combination of immune cells and bacteria. We aimed to determine whether epithelial integrity in CD versus non‐IBD donors was affected (1) by the differences in immune cell phenotype between the two cohorts, (2) directly due to culture with bacteria and/or (3) by culturing immune cells in the serosal chamber and bacteria in the mucosal chamber, representing the in vivo biology.
Initially, colonic monolayers derived from CD and non‐IBD cohorts were grown to confluence, differentiated and then patient‐matched PBMCs were added to the serosal chamber and co‐cultured with the epithelial cells for a further 72 h at which point TEER was measured to assess epithelial integrity (Figure 3a). To adjust for inter‐individual variation, TEER after 72 h co‐culture is expressed as a percentage of TEER following differentiation of the monolayers through withdrawal of Wnt 3A and R‐spondin for 3 days (TEER (%) = day 3 TEER/d0 TEER × 100). Absolute TEER at the start of co‐culture (d0) varied between donors but was slightly higher in non‐IBD donors (1770 ohms, n = 4) than in CD donors (1345 ohms, n = 5; no statistically significant difference). The addition of PBMCs to the serosal chamber had no effect on the epithelial integrity of the monolayers derived from either non‐IBD or CD patients. However, in monolayers derived from both non‐IBD and CD donors, in the presence of PBMCs from the same patient activated with anti‐CD3/28 (Figure 3c), there was a dramatic reduction in TEER, suggesting complete destruction of the epithelium, confirmed by histology (Figure 3b). These data indicate that while the presence of serosal immune cells alone had no effect on the integrity of the monolayers, highly activated immune cells are sufficient to affect epithelial integrity in this model.
Figure 3.
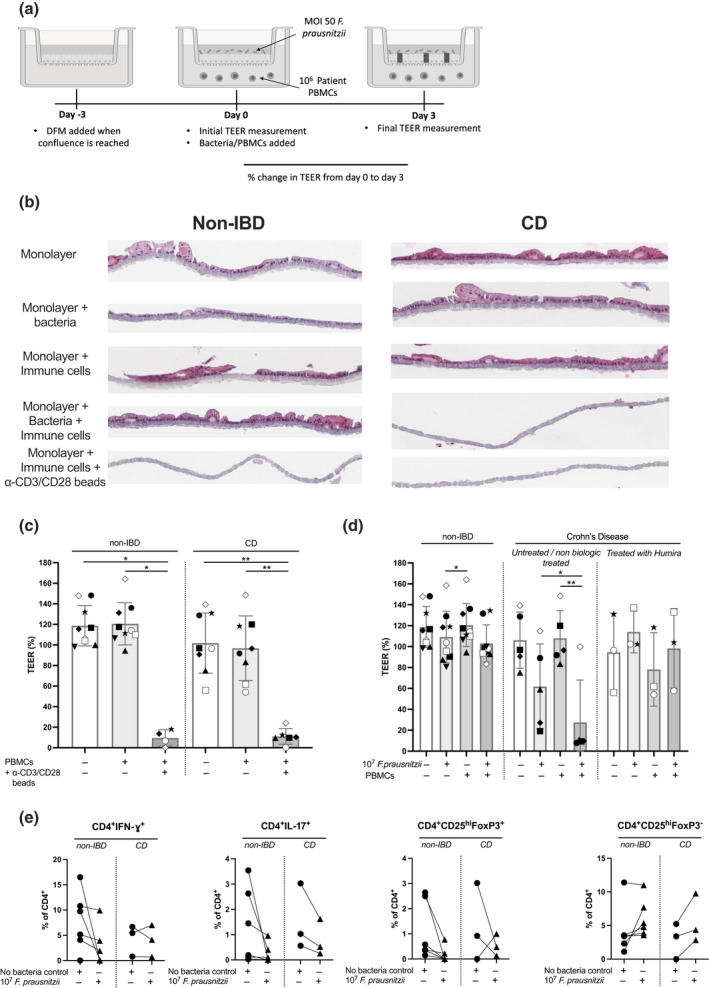
Effect of bacteria and immune cell co‐culture on epithelial integrity in non‐inflammatory bowel disease (non‐IBD) versus Crohn's disease (CD) populations. (a) Scheme of intestinal monolayers, with the addition of heat‐killed Faecalibacterium prausnitzii and matched‐patient PBMCs (created in Biorender). Differentiated intestinal monolayers were cultured alone, cultured with 106 PBMCs added to the serosal compartment, cultured with MOI 50 F. prausnitzii added to the mucosal compartment, cultured with both immune cells and MOI 50 F. prausnitzii or cultured with anti‐CD3/28 beads, for a further 72 h. (b) H&E image shows intestinal cells lining a 0.4‐μm Transwell® membrane. Images from two representative donors shown. (c) Change in transepithelial electrical resistance (TEER) from day 0 to day 3 in the presence or absence of PBMCs cultured with anti‐CD3/28 beads (non‐IBD = 9, CD = 6–8). Matched donors are shown by symbols. (d) Change in TEER from day 0 to day 3 in the presence or absence of PBMCs and/or F. prausnitzii as indicated. Left—non‐IBD donors (n = 9); middle—CD donors untreated or treated with nonbiologicals at time of biopsy (n = 6); right—CD donors treated with anti‐TNF at time of biopsy (n = 3). (e) PBMCs recovered from the monolayer co‐culture on day 3 from the monolayer + PBMCs versus monolayer + PBMCs + bacteria cultures were analysed by flow cytometry (non‐IBD donors: n = 6 HC, CD donors: n = 3). Matched donors are shown by symbols. Statistical analyses were calculated using (c) a Kruskal–Wallis test with the Dunn's multiple comparisons test separately within the non‐IBD or CD groups, or using (d) a Friedman test with the Dunn's multiple comparisons test separately within the non‐IBD, untreated CD and treated CD groups (*P < 0.05, **P < 0.01).
We then tested whether the addition of bacterial components in the presence or absence of immune cells could affect epithelial integrity. F. prausnitzii is linked to CD—its abundance is decreased in CD versus healthy patients and active versus inactive CD. 14 Summarised data are presented in Figure 3d; data from individual patients are shown in Supplementary figure 5. As for Figure 3c, data are presented as TEER (%; = d3 TEER/d0 TEER × 100); representative H&E slides of monolayers in each tested condition from non‐IBD and CD donors are shown. Either heat‐killed F. prausnitzii was added to the mucosal chamber of confluent mature intestinal monolayers or F. prausnitzii was added to the mucosal chamber, and PBMCs from the same patient were added to the serosal chamber and TEER measured 3 days later. In monolayers from non‐IBD donors, TEER was unaffected by mucosal bacteria alone (Figure 3d, left panel), whereas in two of the six monolayers from CD donors, mucosal bacteria alone induced a marked decrease in TEER (Figure 3d, middle panel). As seen in Figure 3b, the addition of serosal PBMCs alone had no effect on TEER in the CD or non‐IBD cohort, but in the presence of mucosal bacteria and serosal PBMCs, the TEER in four of the six monolayers derived from CD patients was dramatically reduced but was not affected in monolayers from non‐IBD donors (compare Figure 3d left and middle panels). These data suggest that the epithelium of CD patients is more susceptible to disruption by the presence of F. prausnitzii than the epithelium from non‐IBD donors.
There was substantial variability in integrity in the two‐dimensional monolayer model in the CD cohort compared with the non‐IBD cohort. This was expected as the patient cohort represents a diverse and complex disease, and being heterogenous in terms of treatment. We compared patients treated at the time of biopsy with either no medication or conventional steroid medication with patients treated with the anti‐TNF antibody, Humira (adalimumab). In a very limited cohort (n = 3), those patients treated with anti‐TNF had a different pattern of results (compare Figure 3d middle and right panels). We cannot conclude an effect of anti‐TNF treatment with the data presented; however, this result highlights the need to evaluate differences in individual patients within CD cohorts.
Finally, we studied the effect of bacterial co‐culture on the phenotype of the T cells, similar to the direct culture shown in Figure 2d. There was no significant difference in the frequency of CD4+IL‐17+, CD4+IFN‐γ+, CD4+CD25HIFOXP3+ or CD4+CD25HIFOXP3− T cells (Figure 3e) or CD8+ T cell populations (Supplementary figure 4) in the presence or absence of bacteria in the monolayer model. There was also no effect on the frequency of CD4+IFN‐γ+IL‐17+ (Th17.1 cells; Supplementary figure 4).
Discussion
We have created an autologous epithelial monolayer model, integrating patient‐specific immune responses. We tested the effect of a commensal bacteria, F. prausnitzii, on both immune and epithelial functions. We demonstrated that the intestinal epithelium in monolayers derived from CD patients had less integrity than those from the non‐IBD cohort. We also showed that CD patients have T cells with a more inflammatory profile than those of the non‐IBD cohort, and responded differently from the non‐IBD cohort to exposure to the commensal bacterium, F. prausnitzii. When grown as a monolayer, epithelia from both non‐IBD and CD donors were resistant to disruption by immune cells alone from the same patient. Integrity was affected by culture with bacteria alone in monolayers from the majority of CD but not non‐IBD donors. The addition of both immune cells and bacteria led to disruption of the epithelia derived from some of the CD but none of the non‐IBD donors. There was extensive variability in results from CD patients. Our autologous patient‐specific model can therefore be used to integrate, rather than avoid, the extremely high variability in both immune responses and epithelial integrity in CD populations versus non‐IBD populations and leads us to recommend that CD should be studied per individual.
Our data highlight an important difference in the epithelial component of the disease. Monolayers derived from CD patients had impaired epithelial integrity compared to those from non‐IBD donors. The model is based on the establishment of a confluent monolayer of epithelial cells, derived from three‐dimensional organoids. At confluence, we saw reduced integrity, measured by TEER, in the CD samples compared with the non‐IBD samples. After the addition of differentiation media, the TEER increased in monolayers from non‐IBD but not CD donors. Together, these data indicate an inherent defect in the epithelial cells of patients with CD and provide a strong rationale for using patient tissue, rather than cell lines, to study this complex disease.
In the absence of bacteria or immune cells, the reduction in integrity may be a stem cell‐associated defect, with associated changes in epigenetic control of differentiation. 15 Further analysis of the monolayers from CD and non‐IBD after addition of differentiation media, including measurement of markers of stemness (Lgr5, Ephb3, Ascl2) and differentiation (Muc2, ChgA, DCLK1, NKCC1) 16 would prove maturation of the cells comprising the monolayer and identify any differences between the CD and non‐IBD monolayers. Similarly, an additional measurement of occludin, claudin‐2 and muc2, and translocation of FITC‐dextran 17 would clarify whether the differences in integrity are due to differences in the tight junctions and whether these were associated with greater permeability to bacterial components. 18 , 19
In monolayers from CD patients but not non‐IBD donors, mucosal bacteria induced a reduction in TEER in the absence of immune cells. This implies that in the monolayers from patients with CD, the interaction between the epithelial cells and the commensal bacteria results in further reduction in the integrity of the barrier. Given that interaction between the bacteria and the apical membrane of the epithelial cells is likely to be comparable in both sets of monolayers, the reduction in integrity observed in monolayers from patients with CD could be caused by direct interaction with bacteria with an already defective epithelium. The interaction of the bacteria with the epithelial cells may be modified in CD either through loss of polarity of TLRs 20 or increased translocation of bacterial components across the epithelium and interaction of the bacteria with serosal receptors. An important missing component in this model is the mucus layer van der Post et al. 21 have shown that the structure of the colonic mucus is important for the development of IBD. We saw an effect of bacteria with or without co‐culture of immune cells, therefore, it is likely that at least both of these mechanisms are involved. We also tested the addition of bacteria in the same (serosal) chamber as PBMCs to determine whether the translocation of bacteria across an impaired membrane could activate PBMCs directly. However, there was no additional reduction in integrity in CD or non‐IBD donors, although the number of samples tested with this variable was low (Supplementary figure 5; Donors 6–9). Future experiments are essential to determine whether bacterial translocation in this model is the cause of immune cell activation. Finally, it is also possible that the result described here is an artefact of the monolayer model; bacterial contact with the apical membrane of the colonic epithelium is minimal in vivo due to the epithelium structure and secretion of both mucus and antimicrobial components, 22 or that effects observed from the co‐culture of F. prausnitzii are specific to that species of commensal bacteria.
The peripheral immune response in patients with CD was indicative of an activated and inflammatory phenotype, similar to what we had previously shown in intestinal immune cells. 9 We had predicted that the activated phenotype of the peripheral immune cells would cause a decrease in the integrity of the intestinal epithelium in CD patients, leading to increased flux of bacterial components and a cycle of continued inflammation. The effect of an inflammatory immune response in vivo is thought to be due to modifications in tight junction proteins. 19 Addition of polyclonally activated T cells (by stimulation with antiCD3/28) led to a dramatic reduction in TEER, and almost total destruction of monolayers, from both CD and non‐IBD cohorts. We found no significant effect on epithelial integrity with the addition of patient‐matched PBMCs in monolayers from either cohort. However, we show that the epithelium is inherently compromised in CD donors. These data indicate that the activated phenotype identified in PBMCs from patients with CD was not sufficient alone to affect epithelial integrity and that an inflammatory immune response may be a consequence, rather than a cause, of disease pathology. It is possible that intestinal immune cells would respond differently than blood cells, although Rubin et al. 23 show similar phenotypes in both tissues. The frequency of central memory phenotype CD4+ T cells was higher in biopsies than in blood samples, and it is possible that these cells could induce more of a response when exposed to bacteria in our model. It is difficult to isolate sufficient cells from biopsy tissue to replicate the experiment; however, prior activation of immune cells and subsequent sorting into effector and memory subsets before use in the monolayer model may allow us to determine the role of effector status of T cells in inducing damage.
Rubin et al. 23 and van Unen et al. 24 studied the lineages and frequencies of immune cells in tissue across multiple gut diseases or inflammatory disease stages; however, there are limited data on the function of these enriched immune cells population. In particular, analysis of the production of cytokines from CD versus non‐IBD intestinal tissue is limited. 9 To determine a mechanism of immune‐mediated epithelial damage, then testing individual cytokines or cytokine‐producing immune cells in the monolayer model would be required, and an assessment of activation and proliferation of T cells. Data from a small subset of patients shown in Figure 3e did not reveal any significant differences in T cell population frequencies post culture; experiments to expand and repeat these data are currently underway.
Although T cells from CD patients were more activated in the absence of stimulation than those from non‐IBD donors, the T cells from CD patients were not sufficient alone to induce a reduction in epithelial integrity in our model. However, T cell populations expressing IFN‐γ or IL‐17 from CD patients were not decreased on exposure to F. prausnitzii in vitro unlike those from the non‐IBD cohort. We did not specifically test the activation status of T cells in response to bacterial co‐culture in the context of the epithelial monolayer, and this would be important to include in future experiments. It would be interesting to test whether immune cells from CD patients, cultured with bacteria, would induce a compromised epithelial barrier derived from a healthy donor. The published suppressive effects of F. prausnitzii may not be functional in CD patients, 25 , 26 which may prevent commensal contributions to intestinal tolerance. Previous studies have suggested that F. prausnitzii promotes tolerogenic environments by the direct activation of Treg cells via the cell‐surface protein, microbial anti‐inflammatory molecule (MAM). 25 However, our data showed no response by Tregs from non‐IBD donors to F. prausnitzii, but an increase in the frequency of Tregs from CD donors, in the presence of F. prausnitzii. The increase in Tregs from CD donors, coupled with the lack of reduction in CD4+IFN‐γ+ T cells, could suggest that Tregs from CD donors are less able to induce T cell suppression than Tregs from non‐IBD donors. It is also possible that uncontrolled expansion of Treg population in CD patients, in response to commensal stimuli, could lead to regions of excessive suppression, which could result in immune cold spots in affected regions. In addition, hyperactive Treg responses to certain commensal species may result in regions of immunosuppression, facilitating bacterial translocation into the lamina propria. Upon the initiation of an immune response to bacterial translocation, circulating PBMCs, with a high inflammatory capacity, may migrate to the intestinal site, resulting in severe inflammation.
One of the goals of the development of this autologous model was to integrate the extensive patient variability into studies of the disease. The inter‐donor variability in responses in the CD cohort, but not the non‐IBD cohort, was large. This represents the extensive heterogeneity in the CD population, encompassing past and current treatments and the type and extent of disease. 27 The model established here allows this variability to be assessed—we saw differences in responses to bacteria and immune cells across the cohort (Supplementary figure 5).
The key strength of this model is the ability to study individual patient immune responses in a patient‐specific epithelial model. 7 This self‐contained system allows the variability between immune, epithelial and microbial components within one person, therefore providing scope for personalised therapy decisions. The model described here has several advantages not only over traditional cell line‐based assays but also over three‐dimensional organoid models in terms of addressing mechanisms of disease. It allows for the study of bacteria and immune cells in the physiologically relevant locations—on the mucosal and serosal sides, respectively, of the epithelium. In this protocol three‐dimensional organoids are expanded, allowing for the generation of multiple monolayers from each individual. This allows for extensive measurements of individual samples, including immune cell phenotype and function, characterisation of genetic variability and expression of tight junction proteins. All of these can be linked to treatment, both during the generation of monolayers, which would potentially allow for optimisation of treatment and prior to sample collection.
The main limitation of the present study is in cohort size. Collection of samples is difficult during pandemic lockdowns, and New Zealand is limited in the total number of patients available for study. Our ongoing research is directed towards collecting donors from multiple sites and the ability to preselect donors based on treatment and timing. The variability in the CD cohort limits the ability to determine mechanisms of disease that can be applied to all patients. This includes additional clinical parameters (such as structuring and penetrating classifications) and additional medications. In particular, the response to one species of bacteria is likely to vary between individuals, and more species need to be tested. Finally, we studied epithelial integrity measured by TEER, and other assays must be incorporated, including permeability via dextran‐FITC uptake, quantification of tight junction proteins in monolayers and assessment of cellular composition. The destruction of the epithelium in both non‐IBD and CD monolayers cultured with highly and polyclonally activated PBMCs likely does not reflect the in vivo disease; we are currently developing a graded model of inflammation using partially activated PBMCs to better understand mechanisms of damage and to develop a recovery model to test therapies.
We have created an autologous colonoid monolayer co‐culture model to study bacteria and immune cell interactions and their effect on epithelial integrity in individual CD patients. Using this model, we have collected preliminary data to suggest that CD patients have impaired epithelial integrity compared with non‐IBD donors, and this integrity is worsened when the monolayers are exposed to mucosal commensal bacteria. The role of a previously inflamed immune response appears less relevant in affecting integrity.
Methods
Patient samples and ethical considerations
Patients diagnosed with Crohn's disease (CD donors) or healthy volunteers (non‐IBD donors) who provided informed consent were recruited from the Dunedin Hospital, Mercy Hospital or at the University of Otago. Collection and analysis of patient peripheral bloods and biopsies were approved by the New Zealand Health and Disability Ethics Committee (Ref# MEC/11/11/093). Ethical approval for the use of peripheral blood from healthy individuals was approved by the Human Ethics Committee of the University of Otago (Ref# 12/036, 18/088). Biopsies were obtained from macroscopically noninflamed areas of the colon from either patient with active CD or from disease‐free control participants undergoing routine screening or surveillance colonoscopy. Donor characteristics are shown in Tables 1 and 2.
Table 2.
| Variable | Patients with CD; Cohort 1 a (n = 4) | Health donors; Cohort 1 (non‐IBD; n = 5) | Patients with CD; Cohort 2 a (n = 5) | Health donors; Cohort 2 (non‐IBD; n = 4) |
|---|---|---|---|---|
| Age mean (SD) | 50.5 (7.19) | 56.2 (6.11) | 37 (14.27) | 49.27 (12.42) |
| Sex (%) | ||||
| Female | 3 (75) | 3 (60) | 1 (20) | 3 (75) |
| Male | 1 (25) | 2 (40) | 4 (80) | 1 (25) |
| Treatment at time of appointment (%) | ||||
| Pentasa + 6MP | 1 (25) | |||
| Steroids + Urosan | 1 (25) | |||
| Humira | 2 (40) | |||
| Humira + mercaptopurine | 1 (20) | |||
| Azothioprine | 1 (20) | |||
| No medication | 2 (50) | 1 (20) | ||
Organoid culture
Six biopsies were taken from macroscopically noninflamed regions of the transverse or sigmoid colon using 3 mm2 biopsy forceps and processed as a single sample. 9 Crypts were isolated following a dithiothreitol (DTT) wash and ethylenediaminetetraacetic acid (EDTA) incubation. 8 Intestinal crypts were cultured in Matrigel® (Corning, NY, USA) and specialised organoid media containing Wnt‐3a and R‐spondin. 28 , 29 Organoids were passaged weekly, when required, by mechanical disruption via a 23‐gauge needle and 1‐mL syringe. ROCK‐inhibitor was added to media for 48 h after initial crypt isolation and each passage.
Patient‐derived intestinal monolayers
Organoids were incubated in TrypLE Express (Thermo Fisher Scientific) and mechanically disrupted using a P1000 pipette. Organoid fragments were seeded on 0.4 μm pore‐size Transwell polyester (PET) membranes (Corning) in 24‐well plates, with a 6.5‐mm insert. Organoids were cultured for 8–10 days in normal organoid media (nondifferentiation media, NDM) to achieve a confluent monolayer, with media changed every 48 h. An EVOM TEER reader (World Precision Instruments, USA) was used to monitor monolayer development. Once monolayers were confluent (Ω > 800), monolayers were cultured in differentiation media (DFM, organoid media containing no Wnt‐3a or R‐spondin) for 72 h then indicated numbers of F. prausnitzii, matched‐patient immune cells, or anti‐CD3/anti‐CD28‐coated beads (Thermo Fisher Scientific) were added to mucosal (upper) or serosal (lower) Transwell compartments for a further 72 h.
Isolation of peripheral blood mononuclear cells
Human PBMCs were collected and isolated by SepMate™ Ficoll separation (50 mL tube; Stemcell Technologies, Vancouver, BC, Canada) within 6 h. PBMCs were washed in sterile phosphate‐buffered saline (PBS; Invitrogen, Carlsbad, CA, USA), then suspended in complete RPMI (with 100 μg mL−1 penicillin, 100 μg mL−1 streptomycin, 55 μm M2‐mercaptoethanol; Invitrogen), 10% fetal calf serum (PAA Laboratories, Morningside, QLD, Australia; RPMI‐10). PBMCs were stored in Corning® CoolCell® Alcohol‐free Freezing foam containers and frozen at −80°C, if required, for up to 48 h. Frozen PBMCs were then stored in liquid nitrogen.
Flow cytometry
Samples were stimulated with 10 ng mL−1 phorbol 12‐myristate 13‐acetate (PMA; Sigma‐Aldrich, St Louis, MO, USA) and 500 ng mL−1 ionomycin (Sigma‐Aldrich) and incubated at 37°C, 5% CO2 for 4 h. Brefeldin A (1 μg mL−1; Sigma‐Aldrich) was added after 2 h of incubation.
Cells were resuspended in sterile PBS and incubated with Zombie NIR live/dead dye (eBioScience, San Diego, CA, USA) in the dark, on ice, for 30 min. Cells were incubated with a surface antibody cocktail [AF700‐anti‐CD3 (clone WCHT1; BioLegend, San Diego, CA, USA), V500‐anti‐CD8 (clone RPAT8; BD Biosciences) and PE/Cy7‐anti‐CD25 (clone BC96; BioLegend)], then incubated in the dark, on ice, for 30 min. Cells were washed in fluorescence‐activated cell sorting (FACS) buffer, then fixed in 1% paraformaldehyde (Sigma‐Aldrich) for 1 h. Fluorescence minus one control was used as gating controls.
Cells were fixed using FOXP3 Fix/Perm Buffer Set (BioLegend) and incubated as per the manufacturer's instructions. Cells were incubated with antibody cocktail [Pacific Blue‐anti‐CD4 (clone SK3; BioLegend), BV605‐anti‐IL‐2 (clone MQ1‐17H12; BioLegend), PE/DAZZLE‐anti‐IL‐17A (clone BL168; BD Biosciences), PE‐anti‐IL‐22 (clone BG/IL22; BioLegend), PerCP/Cy5.5‐anti‐IFN‐γ (clone 45.B3; BioLegend) and AF488‐anti‐FOXP3 (clone 150D/E4; BioLegend)], and incubated in the dark, on ice, for 90 min. Cells were then washed twice in permeabilization buffer, then once in FACS buffer, then resuspended in FACS buffer for analysis.
eBioscience OneComp eBeads (eBioscience) single‐stain bead controls were used to adjust voltages and calculate compensation. Data were acquired using an LSR‐FORTESSA (Becton Dickinson, Franklin Lakes, NJ, USA) using FACSDiva software (Becton Dickinson). Data were analysed using FlowJo software (FlowJo X 10.6.0, BD Life Sciences, Ashland, OR, USA). Representative gating is shown in Supplementary figure 1.
Cytokine analysis
8 × 104 human PBMCs were added to individual wells of a 96‐well plate (Corning), in 200 μL RPMI‐10. Cells were stimulated with Dynabeads™ Human T‐Activator CD3/28 beads (Thermo Fisher Scientific) at a 1:1 (cell:bead) ratio, or remained unstimulated, and incubated at 37°C, 5% CO2, for 48 h. Twenty‐five microlitres of cell supernatant was collected and then stained with fluorescent beads as per the LegendPlex Human Inflammation Cytokine Analysis Kit (BioLegend). Data were acquired using FACSDiva software (Becton Dickinson) and analysed using LegendPlex software (LegendPlex, v 8.0; BioLegend).
CD4 T cell proliferation studies
Conventional T cells (CD3+CD4+CD127+CD25lo‐int) from isolated PBMCs were sorted using a FACSAria II (BD Biosciences), and Tregs (CD3+CD4+CD127‐CD25hi) were excluded (APC/Cy7‐anti‐CD3 (clone HIT3a), BV605‐anti‐CD4 (clone OKT4), PE/Cy7‐anti‐CD25 (clone BC96) and APC‐anti‐CD127 (clone A019D5), all from Biolegend). T cells were stained with Cell‐Trace Violet (Thermo Fisher Scientific) and Dynabeads™ Human T‐Activator CD3/28 beads (Thermo Fisher Scientific) were added at a 10:1 cell:bead ratio, and incubated for 80 h at 37°C in 5% CO2, then stained again for CD3 and CD4 before acquisition on a BD LSR‐FORTESSA.
Bacterial co‐culture
Isolated PBMCs were co‐cultured with indicated dosages of heat‐killed F. prausnitzii DSM 17677 (kindly provided by Professor G Tannock, University of Otago), 30 in RPMI‐10, and incubated at 37°C in 5% CO2 for 72 h.
Statistical analyses
Statistical analysis was performed using GraphPad Prism 8 (CA, USA) and tests are outlined in figure captions. PCR data were analysed using the unpaired Student's t‐test. ANOVA was used for statistical testing for cell experiments and where pair‐wise comparison was made, the Mann–Whitney rank test or Wilcoxon matched‐pairs signed rank test was used. For two‐way analysis, a mixed‐effects model was used followed by the Sidak's multiple comparisons test. For monolayer experiments, the Friedman with the Dunn's post hoc test was used for matched data; the Kruskal–Wallis with the Dunn's post hoc test was used when there was not a full matching data set. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Hamish CK Angus: Conceptualization; data curation; formal analysis; investigation; methodology; writing – original draft; writing – review and editing. Paulo CM Urbano: Data curation; formal analysis; investigation; supervision; validation. Gemma A Laws: Formal analysis; methodology. Shijun Fan: Methodology; validation. Safina Gadeock: Methodology; resources. Michael Schultz: Conceptualization; funding acquisition; writing – review and editing. Grant Butt: Conceptualization; formal analysis; investigation; methodology; supervision; writing – original draft; writing – review and editing. Andrew J Highton: Formal analysis; writing – review and editing.
Supporting information
Supplementary figures 1–5
Supplementary table 1
Acknowledgments
We thank the patients and donors for their kind participation. We also thank the gastroenterology nurses for their contributions to this work. We thank Michelle Wilson and Joanna Roberts for flow cytometry support, and Murray Barclay and Jake Begun for input into the project. This work was funded by the Otago Medical Research Foundation, the University of Otago and the Healthcare Otago Trust. HCKA was the recipient of a University of Otago PhD scholarship. GAL was the recipient of a Department of Medicine PhD scholarship.
Contributor Information
Andrew J Highton, Email: andrew.highton@otago.ac.nz.
Roslyn A Kemp, Email: roslyn.kemp@otago.ac.nz.
References
- 1. Uhlig HH, Powrie F. Translating immunology into therapeutic concepts for inflammatory bowel disease. Annu Rev Immunol 2018; 36: 755–781. [DOI] [PubMed] [Google Scholar]
- 2. Roda G, Jharap B, Neeraj N, Colombel J‐F. Loss of response to anti‐TNFs: definition, epidemiology, and management. Clin Transl Gastroenterol 2016; 7: e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kemp R, Dunn E, Schultz M. Immunomodulators in inflammatory bowel disease: an emerging role for biologic agents. BioDrugs 2013; 27: 585–590. [DOI] [PubMed] [Google Scholar]
- 4. de Souza HSP, Fiocchi C, Iliopoulos D. The IBD interactome: an integrated view of aetiology, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol 2017; 14: 739–749. [DOI] [PubMed] [Google Scholar]
- 5. Ben‐Horin S, Chowers Y. Loss of response to anti‐TNF treatments in Crohn's disease. Aliment Pharmacol Ther 2011; 33: 987–995. [DOI] [PubMed] [Google Scholar]
- 6. Giri R, Hoedt EC, Khushi S et al. Secreted NF‐κB suppressive microbial metabolites modulate gut inflammation. Cell Rep 2022; 39: 110646. [DOI] [PubMed] [Google Scholar]
- 7. Noel G, Baetz NW, Staab JF et al. A primary human macrophage‐enteroid co‐culture model to investigate mucosal gut physiology and host‐pathogen interactions. Sci Rep 2017; 7: 45270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sato T, Stange DE, Ferrante M et al. Long‐term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 2011; 141: 1762–1772. [DOI] [PubMed] [Google Scholar]
- 9. Dunn ETJ, Taylor ES, Stebbings S, Schultz M, Butt AG, Kemp RA. Distinct immune signatures in the colon of Crohn's disease and ankylosing spondylitis patients in the absence of inflammation. Immunol Cell Biol 2016; 94: 421–429. [DOI] [PubMed] [Google Scholar]
- 10. Eastaff‐Leung N, Mabarrack N, Barbour A, Cummins A, Barry S. Foxp3+ regulatory T cells, Th17 effector cells, and cytokine environment in inflammatory bowel disease. J Clin Immunol 2010; 30: 80–89. [DOI] [PubMed] [Google Scholar]
- 11. Maul J, Loddenkemper C, Mundt P et al. Peripheral and intestinal regulatory CD4+CD25high T cells in inflammatory bowel disease. Gastroenterology 2005; 128: 1868–1878. [DOI] [PubMed] [Google Scholar]
- 12. Nemeth ZH, Bogdanovski DA, Barratt‐Stopper P, Paglinco SR, Antonioli L, Rolandelli RH. Crohn's disease and ulcerative colitis show unique cytokine profiles. Cureus 2017; 9: e1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hegazy AN, West NR, Stubbington MJT et al. Circulating and tissue‐resident CD4+ T cells with reactivity to intestinal microbiota are abundant in healthy individuals and function is altered during inflammation. Gastroenterology 2017; 153: 1320–1337.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao H, Xu H, Chen S, He J, Zhou Y, Nie Y. Systematic review and meta‐analysis of the role of Faecalibacterium prausnitzii alteration in inflammatory bowel disease. J Gastroenterol Hepatol 2021; 36: 320–328. [DOI] [PubMed] [Google Scholar]
- 15. Verzi MP, Shivdasani RA. Epigenetic regulation of intestinal stem cell differentiation. Am J Physiol Gastrointest Liver Physiol 2020; 319: G189–G196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee J, Wasinger V, Yau Y, Chuang E, Yajnik V, Leong R. Molecular pathophysiology of epithelial barrier dysfunction in inflammatory bowel diseases. Proteomes 2018; 6: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schoultz I, Keita ÅV. The intestinal barrier and current techniques for the assessment of gut permeability. Cell 2020; 9: 1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol 2009; 1: a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol 2011; 73: 283–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee J, Mo JH, Katakura K et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol 2006; 8: 1327–1336. [DOI] [PubMed] [Google Scholar]
- 21. van der Post S, Jabbar KS, Birchenough G et al. Structural weakening of the colonic mucus barrier is an early event in ulcerative colitis pathogenesis. Gut 2019; 68: 2142–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coleman OI, Haller D. Microbe–mucus interface in the pathogenesis of colorectal cancer. Cancers (Basel) 2021; 13: 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rubin SJS, Bai L, Haileselassie Y et al. Mass cytometry reveals systemic and local immune signatures that distinguish inflammatory bowel diseases. Nat Commun 2019; 10: 2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Unen V, Li N, Molendijk I et al. Mass cytometry of the human mucosal immune system identifies tissue‐ and disease‐associated immune subsets. Immunity 2016; 44: 1227–1239. [DOI] [PubMed] [Google Scholar]
- 25. Breyner NM, Michon C, de Sousa CS et al. Microbial anti‐inflammatory molecule (MAM) from Faecalibacterium prausnitzii shows a protective effect on DNBS and DSS‐induced colitis model in mice through inhibition of NF‐κB pathway. Front Microbiol 2017; 8: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lopez‐Siles M, Duncan SH, Garcia‐Gil LJ, Martinez‐Medina M. Faecalibacterium prausnitzii: from microbiology to diagnostics and prognostics. ISME J 2017; 11: 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feakins RM. Ulcerative colitis or Crohn's disease? Pitfalls and problems. Histopathology 2014; 64: 317–335. [DOI] [PubMed] [Google Scholar]
- 28. Sayoc‐Becerra A, Krishnan M, Fan S et al. The JAK‐inhibitor tofacitinib rescues human intestinal epithelial cells and colonoids from cytokine‐induced barrier dysfunction. Inflamm Bowel Dis 2020; 26: 407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Angus HCK, Butt AG, Schultz M, Kemp RA. Intestinal organoids as a tool for inflammatory bowel disease research. Front Med 2020; 6: 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Roock S, van Elk M, Hoekstra MO, Prakken BJ, Rijkers GT, de Kleer IM. Gut derived lactic acid bacteria induce strain specific CD4+ T cell responses in human PBMC. Clin Nutr 2011; 30: 845–851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1–5
Supplementary table 1