

Abstract
Waldenström macroglobulinemia (WM) is a lymphoplasmacytic lymphoma that is characterized by the overproduction of an IgM monoclonal protein. It may cause adenopathy, hepatomegaly, splenomegaly, as well as other disease-related complications such as cold agglutinin anemia, cryoglobulinemia, hyperviscosity, and neuropathy. While light chain amyloidosis in patients with WM only occurs in about 10% of patients, it is important that advanced practitioners are able to recognize concurrent AL amyloidosis, which will affect the patient's treatment trajectory. Diagnosis of WM with AL amyloidosis is based on bone marrow biopsy and a fat pad biopsy. If AL amyloidosis is suspected, the bone marrow and fat pad biopsy should undergo Congo red staining. If it is negative, and there is a strong suspicion of AL amyloidosis, then an organ biopsy can be considered. Treatment of WM uses rituximab-based therapy in combination with a variety of other agents, including proteasome inhibitors, alkylating agents, and BTK inhibitors. Treatment of light chain amyloidosis uses bortezomib as the backbone of therapy and can be administered with cyclophosphamide, dexamethasone, and now daratumumab, which was recently approved. Waldenström macroglobulinemia and light chain amyloidosis are both rare diseases and can lead to a variety of disease-related complications. Fortunately, many options exist for both diseases. This article will highlight a case of WM with amyloidosis and a case of a patient with relapsing WM with considerations for advanced practitioners managing this patient population.
CASE STUDIES
Case Study 1: WM With Amyloidosis
A 70-year-old male presents to his primary care physician with reports of fatigue and shortness of breath. He also reports numbness and tingling to the toes that started a year ago, which has progressed to the mid-calf. He notes that he developed intermittent bruising around his eyes. His primary care physician ordered an electromyography (EMG) and complete laboratory testing. Results from the EMG were abnormal and showed moderate, generalized, axonal, sensorimotor polyneuropathy that is chronic and ongoing in the bilateral lower extremities. No classic features of a primary demyelinating process as seen in chronic inflammatory demyelinating polyneuropathy (CIDP) were found. He was found to have an elevated total protein, which led to a serum protein electrophoresis (SPEP) being ordered. He subsequently was found to have a monoclonal protein (M-protein) and was referred to an oncologist. Workup with the oncologist revealed an IgM lambda M-protein of 6.5 g/dL (Table 1).
Table 1. Initial Workup for Waldenström Macroglobulinemia.
| Serum free kappa | 1.7 mg/L |
| Serum free lambda | 70 mg/L |
| Serum free k/l ratio | 0.02 |
| IgM | 6,800 mg/dL |
| SPEP | IgM lambda M-protein 6.5 g/dL |
| Hgb | 9.3 g/dL |
| Platelets | 110 × 109/L |
| Total protein | 12 g/dL |
| LDH | 413 U/L |
| B2M | 3.7 mg/L |
| Albumin | 3.5 g/dL |
| Bone marrow biopsy | 80% lymphomplasmacytic cells with lambda light chain restriction. MYD88+, CXCR–, CD38+, CD20, CD5–, and CD19. |
| CT scan | Right axilla lymph node measuring 3 cm with mild hepatomegaly |
Note. SPEP = serum protein electrophoresis; Hgb = hemoglobin; LDH = lactate dehydrogenase; B2M = beta-2 microglobulin.
Soon after, the patient was admitted to the hospital due to worsening neuropathy, blurred vision, and shortness of breath. He was subsequently found to have an elevated blood viscosity and underwent plasma exchange. He was started on therapy with bendamustine with the addition of rituximab, which was planned to be administered at a later date due to the risk of IgM flare. In WM, patients may experience a rise in the IgM level that occurs 15 to 30 days after starting rituximab therapy and may last for several months (Dimopoulos et al., 2002). As such, this increase in the IgM level is unrelated to disease progression and so therapy should not be adjusted based on the IgM level alone. However, a patient with an already high IgM level may be at increased risk for developing hyperviscosity syndrome. In order to minimize this risk, rituximab can be delayed until after the patient receives cytotoxic therapy (Gertz, 2021).
Despite therapy, the patient continued to have symptoms of shortness of breath, worsening neuropathy, and pedal edema. Repeat lab work continued to show persistently elevated IgM level at 3,024 mg/dL. Because of the neuropathy and persistent shortness of breath, an amyloid workup was performed (Table 2). A fat pad biopsy is a cost-effective test with a sensitivity of 70% to 80% in the detection of amyloidosis (Kastritis & Dimopoulos, 2015). The biopsy is stained with Congo red, which binds with the amyloid fibrils, presenting a characteristic apple-green birefringence (Figure 1). The clinician concurrently obtained a bone marrow biopsy with Congo red staining to aid in the diagnosis of AL amyloidosis, as this may offer other key information regarding the patient's disease, including the presence of other plasma cell dyscrasias such as multiple myeloma and confirmation of WM. Typically, the level of plasma cell infiltration for AL amyloidosis is low, at 7% to 10%, and higher amounts are linked to poor prognosis (Kourelis et al., 2013). If a bone marrow biopsy should fail to detect amyloidosis, the clinician should consider obtaining an organ biopsy if organ involvement is suspected. However, the gold standard is mass spectrometry of amyloid deposits (Gertz & Zeldenrust, 2014).
Table 2. Initial Workup for Light Chain Amyloidosis.
| Echocardiogram | EF 64% with concentric thickening of the ventricle |
| Cardiac MRI | Infiltrative cardiomyopathy |
| NT-proBNP | Slightly elevated at 550 pg/mL |
| Troponin | 0.010 ng/L |
| Fat pad biopsy | Positive for Congo red stain |
| 24-hour urine | 1,500 Bence-Jones/total volume of proteinuria |
Note. EF = ejection fraction; NT-proBNP = N-terminal pro-brain natriuretic peptide.
Figure 1.
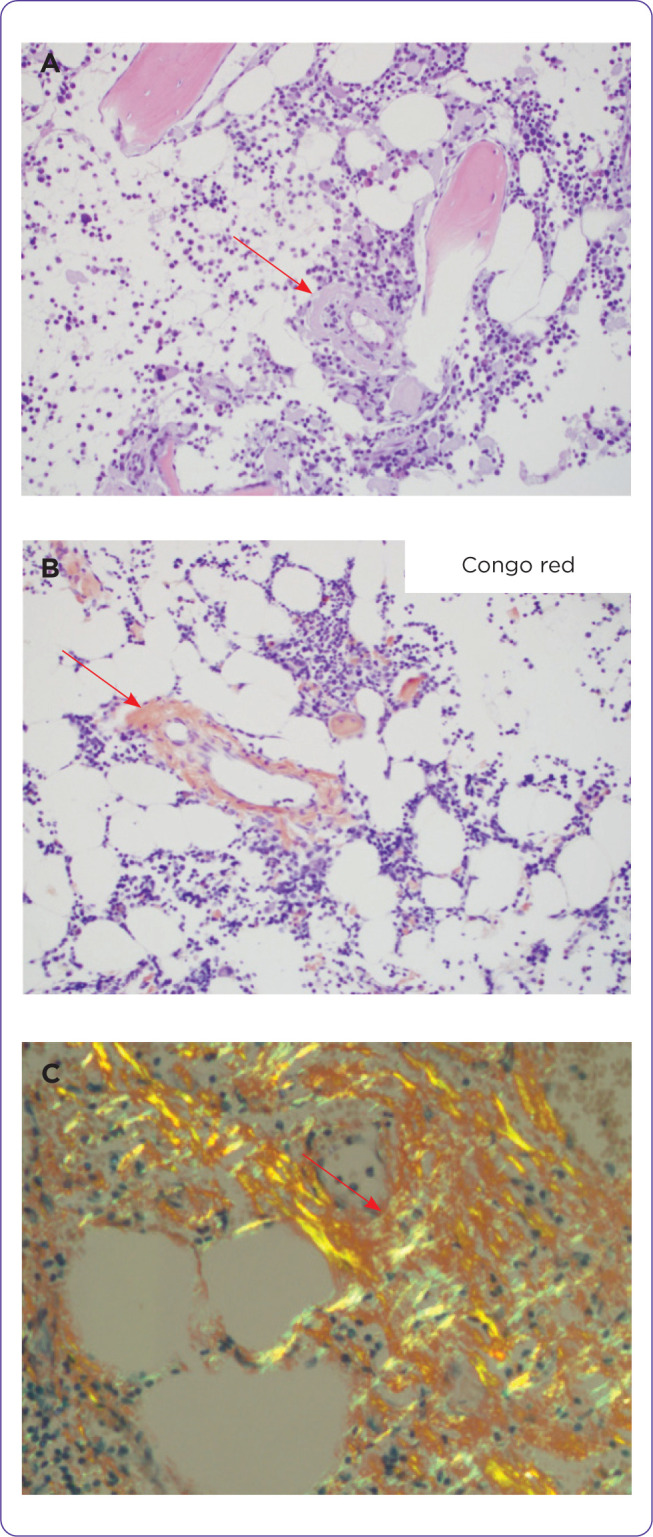
(A) H & E section showing eosinophilic amorphous materials in the wall of a vessel and some in the interstitium of a bone marrow trephine biopsy. (B) Congo red stains the vessel. (C) Apple green birefringent materials in the Congo red–stained soft tissue viewed under polarized light.
Case Study 2: Relapsing WM
A 60-year-old male presents to his local primary care provider with complaints of visual disturbances and difficulty concentrating. Labs reveal an elevated total protein of 13 g/dL. Subsequently, an SPEP was performed, which showed an IgM lambda M-protein of 6 g/dL, hemoglobin 10.3 g/dL, beta-2 microglobulin 6 mg/L, IgM 7,000 mg/dL, and viscosity of 5. He was referred to an oncologist. A bone marrow biopsy was performed, which revealed 70% involvement of lymphoplasmacytic cells, CD20+, CD138, and lambda light chain restriction. The bone marrow was positive for a MYD88 mutation and negative for CXCR. Due to symptoms of visual disturbance and an elevated IgM level, the patient was referred to the emergency room.
Upon arrival, ophthalmology was consulted, and an eye exam revealed the presence of retinal hemorrhages and retinal venous engorgement. Additionally, he was found to have purpura to the lower extremities conferring with type 1 cryoglobulinemia. He was diagnosed with symptomatic intermediate-risk Waldenström macroglobulinemia with hyperviscosity and cryoglobulinemia. He underwent plasmapheresis and then started on treatment with bendamustine and rituximab. He completed six cycles of therapy and was placed on observation. He remained in remission for several years; however, after that, his IgM levels began to increase. He returns to the clinic and the advanced practitioner notes enlarged cervical lymph nodes with palpable purpura to the lower extremities. A CT of chest, abdomen, and pelvis (CAP), labs, and a bone marrow biopsy were performed. He returns for follow-up after his workup and would like to know the treatment plan.
Waldenström macroglobulinemia (WM) is a lymphoplasmacytic lymphoma with the presence of an IgM monoclonal protein. It is characterized by bone marrow infiltration and the production of a monoclonal protein in the presence of hepatosplenomegaly and/or adenopathy. The median age at diagnosis is 71 years, with an incidence of 0.92 per 100,000 person-years for males and 0.30 per 100,000 person-years for females (Gertz, 2021). Patients under the age of 45 have a 10-year survival of 86% (Gertz, 2021).
While WM is a rare disease, several treatments have been approved in the past 5 years, and over the past decade, survival has improved from 6 to 8 years (Gertz, 2021). Light chain amyloidosis may occur in approximately 10% of patients with WM. It is a rare plasma cell dyscrasia where the responsible plasma clone will produce unstable light chains that form amyloid fibrils depositing in organs or tissues, leading to organ dysfunction. This condition is estimated to affect 10 out of 1,000,000 people per year, with the elderly population (> 65 years) at greatest risk (Ryšavá, 2018). Patients with an IgM paraproteinemia occurring concomitantly with AL amyloidosis are even rarer, accounting for 5% to 7% of light chain and/or heavy chain amyloidosis cases (Zanwar et al., 2018). This article will focus on WM and light chain amyloidosis using a case-based approach.
OVERVIEW OF WM AND LIGHT CHAIN AMYLOIDOSIS
Plasma cell dyscrasias involve defective B cells, or specifically, plasma cells, which causes a monoclonal gammopathy resulting in a diverse set of problems. While multiple myeloma is easily identifiable as the most common monoclonal plasma cell proliferative disorder, WM and AL amyloidosis, although rarer, have distinct profiles and may produce multisystem challenges (Jagannath et al., 2016).
Waldenström macroglobulinemia, which is considered an indolent B-cell lymphoma, is a lymphoplasmacytic disorder caused by an IgM monoclonal gammopathy. While clinical manifestations are similar to lymphomas such as fever, anemias, adenopathy, and hepatosplenomegaly, the monoclonal IgM protein in high quantities produces a unique subset of symptoms, including hemolytic anemia, demyelinating polyneuropathy, hyperviscosity, cold agglutinin disease, and cryoglobulinemia that could lead to dermatologic and vascular conditions (Gertz, 2018; Table 3).
Table 3. Waldenström Macroglobulinemia–Related Complications.
| Complication | Pathophysiology | Exam findings | Diagnostic findings |
|---|---|---|---|
| Cold agglutin anemia (occurs in 10% of WM patients) |
|
|
|
| Cryoglobulinemia (occurs in 10%–20% of WM patients) |
|
|
|
| Hyperviscosity |
|
|
|
| Neuropathy (occurs in 30%–50% of patients with a monoclonal IgM and 20% in WM patients) |
|
|
|
Note. LDH = lactate dehydrogenase; MAG = myelin-associated glycoprotein; EMG = electromyography; VEGF = vascular endothelial growth factor. Information from Bloch & Maki (1973); D'Sa et al. (2017); Desbois et al. (2019); Dimoupolis et al. (2000); Gertz (2018, 2019).
AL amyloidosis, also known as primary amyloidosis, is a condition where the fragments of light chains (and sometimes heavy chains) form amyloid fibrils that restructure into highly organized β-pleated sheet formations that can deposit into organs or tissue, leading to organ dysfunction (Gertz & Zeldenrust, 2014). In approximately 80% of the cases, the lambda light chain is affected, which can predispose certain tissues or organs to be affected over others (Merlini, 2017). Amyloid deposition can occur virtually anywhere but will most commonly deposit in the heart and kidneys. Patients with cardiac involvement will typically present with symptoms of heart failure while patients with renal involvement may present with nephrotic range proteinuria unrelated to Bence-Jones (Quock et al., 2018)
DIAGNOSIS
Similar to myeloma and other indolent lymphomas, WM has an asymptomatic and symptomatic phase of the disease. Reasons for starting therapy include anemia/cytopenias (42%), B-symptoms (25%), hyperviscosity (17%), neuropathy (12%), amyloidosis (1.5%), symptomatic cryoglobulinemia (1.3%), and symptomatic cold agglutinin disease (0.6%; Dimopoulos & Kastritis, 2019). Table 4 lists criteria for starting treatment for WM. Diagnosing WM requires a bone marrow biopsy to confirm lymphoplasmacytic infiltration and includes labs, radiology workup, and pathology. Table 5 shows the workup for WM.
Table 4. Criteria for Starting Treatment in Waldenström Macroglobulinemia.
| Hemoglobin < 10 g/dL |
| B-symptoms |
| Platelets < 100 U/L |
| Bulky adenopathy (> 5 cm in maximum diameter or causing symptoms) |
| Symptomatic hepatomegaly or splenomegaly |
| Presence of light chain amyloidosis |
| Symptomatic hyperviscosity |
| Symptomatic cryoglobulinemia |
| Symptomatic cold agglutinin anemia |
| Neuropathy due to WM |
| Amyloidosis due to WM |
Note. Information from Dimopoulos & Kastritis (2019).
Table 5. Workup for Waldenström Macroglobulinemia.
| General workup |
| CBC |
| CMP |
| LDH |
| Disease-specific labs |
| B2M |
| SPEP |
| UPEP |
| Free light chains |
| IgG, IgA, IgM |
| Cold agglutinin titer |
| Viscosity |
| Cryoglobulin |
| Cryocrit |
| Myelin-associated glycoprotein (if neuropathy present) |
| Coagulation studies |
| Partial thromboplastin time |
| Prothrombin time |
| Von Willebrand |
| Imaging |
| CT chest, abdomen, and pelvis |
| PET-CT |
| Pathology |
| Bone marrow biopsy with flow cytometry |
| Mutation panel: MYD88 and CXCR4 |
Note. CBC = complete blood count; CMP = comprehensive metabolic panel; LDH = lactate dehydrogenase; B2M = beta-2 microglobulin; SPEP = serum protein electrophoresis; UPEP = urine protein electrophoresis.
For patients who have a concurrent diagnosis of AL amyloidosis, further workup is needed to ensure adequate treatment of the patient. The patient will require additional lab, radiology, and pathology workup (Table 6). Patients with AL amyloidosis may demonstrate key features, such as nephrotic range proteinuria in excess of 10 g/day (Gertz & Zeldenrust, 2014).
Table 6. Workup for Amyloidosis.
| General workup |
| CBC |
| CMP |
| LDH |
| Disease-specific labs |
| SPEP |
| UPEP |
| Free light chains |
| B2M |
| IgG, IgA, IgM |
| Coagulation studies |
| NT-proBNP |
| Troponin I |
| Imaging |
| Echocardiogram with strain and speckle |
| Cardiac MRI |
| Pathology |
| Fat pad biopsy |
| Bone marrow bipsy with FISH and Congo red staining; FISH: t(11:14); t(4:14), t(14:16); deletion 17p |
| Organ biopsy if fat pad biopsy negative |
| Other tests |
| PFTs |
| EMG if neuropathy present |
Note. CBC = complete blood count; CMP = comprehensive metabolic panel; LDH = lactate dehydrogenase; B2M = beta-2 microglobulin; SPEP = serum protein electrophoresis; UPEP = urine protein electrophoresis; NT-proBNP = N-terminal pro-brain natriuretic peptide; FISH = fluorescence in situ hybridization; PFT = pulmonary function test; EMG = electromyography.
Case Study 1: Diagnosis
Based upon the workup, the patient was diagnosed with WM and light chain amyloidosis. Risk assessment in WM uses the International Prognostic Scoring System for WM (IPSSWM), which is based on age, beta-2 microglobulin (B2M), hemoglobin, platelet, and IgM level (Morel et al., 2009). More recently, an updated system was proposed that is based upon age, B2M, serum lactate dehydrogenase, and albumin (Table 7) to account for the surge of novel regimens that have improved prognosis (Kastritis et al., 2019).
Table 7. Prognostic Scoring Systems in Waldenström Macroglobulinemia.
| Categories | Range | Points | # prognostic factors | OS |
|---|---|---|---|---|
| IPSSWM | ||||
| Age | ≤ 65 years | 0 | 0–1 factors (low risk) | 142 mo |
| > 65 years | 1 | |||
| Hemoglobin | > 11.5 g/dL | 0 | Any 2 factorsor age alone (intermediate risk) | 99 mo |
| ≤ 11.5 g/dL | 1 | |||
| Platelets | > 100 × 109/L | 0 | ≥ 3 factors (high risk) | 43 mo |
| ≤ 100 × 109/L | 1 | |||
| B2M | ≤ 3 mg/L | 0 | ||
| > 3 mg/L | 1 | |||
| IgM | ≤ 7 g/dL | 0 | ||
| > 7 g/dL | 1 | |||
| Revised ISSWM a | ||||
| Age | ≤ 65 years | 0 | 0 factors | 5-yr: 95%; 10-yr: 84% |
| 66–75 years | 1 | 1 factor | 5-yr: 86%; 10-yr: 59% | |
| ≥ 76 years | 2 | 2 factors | 5-yr: 78%; 10-yr: 37% | |
| B2M | ≥ 4 mg/L | 1 | ||
| Serum albumin | < 3.5 g/dL | 1 | 3 factors | 5-yr: 47%; 10-yr: 19% |
| LDH | ≥ 250 IU/L | 1 | 4–5 factors | 5-yr: 36%; 10-yr: 9% |
Note. IPSSWM = International Prognostic Scoring System for Waldenström Macroglobulinemia; ISSWM = International Scoring System for Waldenström Macroglobulinemia; B2M = beta-2 microglobulin; LDH = lactate dehydrogenase. Adapted from Kastritis et al. (2018); Morel et al. (2009)
Survival based on number of risk prognostic factors.
The patient was found to have high-risk disease on the IPSSWM based upon age > 65, hemoglobin < 11.5, and B2M > 3. In light chain amyloidosis, the most commonly used staging systems are Mayo Clinic 2004, Mayo Clinic 2012, and the European modification (Dittrich et al., 2020). The Mayo Clinic 2012 criteria were used to score this patient. It is based on N-terminal pro-brain natriuretic peptide (NT-proBNP) level, difference between involved and uninvolved light chains, and troponin level (Table 8). The patient's prognostic score using the Mayo Clinic 2012 staging system was stage 1. There is not yet an established scoring system for IgM-related amyloidosis, although revised staging systems are currently being validated. Thus, caution should be used when considering these prognostic values (Dittrich et al., 2020). While treatment of either disease is not determined by a patient's stage, staging assists in prognostication of the patient's disease, particularly in light chain amyloidosis.
Table 8. Amyloidosis Staging System (Mayo 2012 Model).
| Stage | Troponin T (μg/L) or hs-cTnT (ng/L) | NT-proBNP (ng/L) | Difference between involved and uninvolved (mg/dL) | Prognosis in patients not undergoing AuSCT (mo) | Prognosis in patients undergoing AuSCT (mo) |
|---|---|---|---|---|---|
| I | < 0.025 μg/L | < 1,800 | < 18 | 55 | NR |
| II | Any one factor elevated | 19 | 62.8 | ||
| III | Any two factors elevated | 12 | 16.8 | ||
| IV | > 0.025 μg/L OR ≥ 40 ng/L |
> 1,800 | > 18 | 5 | 5.8 |
Note. hs-cTnT = high-sensitive cardiac troponin T; NT-proBNP = N-terminal of the prohormone brain natriuretic peptide; AuSCT = autologous stem cell transplant; OR = overall response. Adapted from Kumar et al. (2012a); Muchtar et al. (2018).
TREATMENT CONSIDERATIONS
Greater successes in the treatment of IgM-associated AL amyloidosis are seen with chemo-immunotherapy combinations rather than traditional alkylating regimens, standard treatments for myeloma, or single-agent rituximab (Wechalekar et al., 2008). While some responses have been noted with therapy, outcomes are still generally poor. The best responses are seen in patients who undergo autologous stem cell transplant, but this is often not an option for many patients due to advanced age, which is a detriment to their eligibility and ability to receive transplant. Furthermore, management of patients in the relapsed refractory setting proves even more challenging due to limited treatment options (Manwani et al., 2018). However, certain strategies have seen some promise in prolonging the progression-free survival of patients with this disease.
While there are limited data on the treatment of WM and concurrent light chain amyloidosis, the strategy has been to use agents that are known to be active in both disease states. In the management of WM, rituximab-based regimens have predominated (Table 9). Rituximab, bortezomib, and dexamethasone in the front-line setting produced response rates of 80%, while bendamustine and rituximab combination produced hematologic responses of complete response (CR) of 11%, very good partial response (VGPR) of 37%, and partial response (PR) of 11%. Rituximab maintenance thereafter contributed to even deeper responses (Manwani et al., 2018).
Table 9. Therapies Available in the Newly Diagnosed/Relapsed Refractory Setting for Waldenström Macroglobulinemia.
| Regimen | Response rate | Time to response | PFS | OS | Author |
|---|---|---|---|---|---|
| Rituximab, cyclophosphamide, vincristine, prednisone | 50% | Wechalekar et al. (2008) | |||
| Rituximab, bortezomib, dexamethasone | 85% | 3 mo | 43 mo | 66% at 8 years | Gavriatopoulou et al. (2017) |
| Rituximab, bortezomib, dexamethasone | 96% | 1.4 mo | 57% at 5 years | 95% at 5 years | Treon et al. (2015a) |
| Bortezomib, rituximab | 88% | 3.7 mo | 37 mo | 94% at 5 years | Ghobrial et al. (2010) |
| Ixazomib, rituximab, dexamethasone | 96% | 2 mo | 73% at 22 mo | 100% at 2 years | Castillo et al. (2018) |
| Carfilzomib, rituximab, dexamethasone | 87% | 2.1 mo | 64.5% at 15 mo | NR | Treon et al. (2014) |
| Rituximab, bendamustine (subgroup analysis) | 95% | NR | 69 mo | 90.4% at 5 years | Rummel et al. (2013) |
| Rituximab, cyclophosphamide, dexamethasone | 83% | 4 mo | 36 mo | 96 mo | Kastritis et al. (2015) |
| Ibrutinib | 90.5% | 1 mo | 60% at 5 years | 87% at 5 years | Treon et al. (2015b) |
| Ibrutinib, rituximab | 92% | 1 mo | 82% at 30 mo | 94% at 30 mo | Dimopoulos et al. (2018) |
| Acalabrutinib | 79% | 4.6 mo | 90% at 24 mo | Owen et al. (2020) | |
| Zanubrutinib | 74% | 2.8 mo; time to response longer in those with CXCR mutation | 78% at 18 mo | 97% at 18 mo | Tam et al. (2020) |
| Venetoclax | 81% | 5.1 mo | 30 mo | NR | Castillo et al. (2022) |
While in WM, autologous stem cell transplant is reserved for later in the course of the disease, in patients with AL amyloidosis, autologous stem cell transplant in transplant-eligible patients is widely accepted as the standard of care post induction, with overall hematologic response rates of 100%, and with 80% achieving a VGPR or CR and organ response rates of 67% (Sissoko et al., 2015).
BTK inhibitors have also been found to be effective in both the newly diagnosed and relapsed/refractory setting, with greater than or equal to PR responses in the 90% range. However, in the patient with concurrent AL amyloidosis, response rates are poor with marked increase of adverse events. Few organ responses were seen, and distinguishing between amyloid-related complications and side effects from the drug were difficult. As such, ibrutinib is not considered for the treatment of IgM-associated amyloidosis (Pika et al., 2018).
Recently bortezomib, cyclophosphamide, dexamethasone, and daratumumab became the first US Food and Drug Administration (FDA)-approved regimen for the treatment of AL amyloidosis (Kastritis et al., 2021). This was based on a phase III randomized trial that showed excellent hematologic responses (91%) in the daratumumab arm compared with the control arm. Additionally, 53% of patients with renal involvement had a renal response, and 41% of those with cardiac involvement responded as well. The regimen was well tolerated, and the main grade 3 or 4 toxicities observed included lymphopenia (13%), pneumonia (7.8%), cardiac failure (6.2%), and diarrhea (5.7%; Kastritis et al., 2021). Other regimens in the treatment of AL amyloidosis have also been reported (Table 10). When managing patients with AL amyloidosis, they will need closer monitoring and often require dose reductions due to their organ involvement.
Table 10. Treatment of AL Amyloidosis.
| Regimen | Response rates | Organ response | Authors |
|---|---|---|---|
| Light chain amyloidosis | |||
| Bortezomib, dexamethasone | 72% | Heart (29%) Renal (19%) Liver (22%) |
– |
| Bortezomib, cyclophosphamide, dexamethasone | 94% | Renal (50%) Cardiac (71%) |
Mikhael et al. (2012) |
| Bortezomib, cyclophosphamide, dexamethasone | 60% Stage 3b: 42% |
Cardiac (71%) Renal (25%) |
Palladini et al. (2015) |
| Bortezomib, cyclophosphamide, dexamethasone, daratumumab | 91% | Renal (53%) Cardiac (41%) Hepatic (50%) |
Kastritis et al. (2021) |
| Relapsing AL amyloidosis | |||
| Ixazomib, dexamethasone | 53% | Renal (28% Cardiac (18%) |
Dispenzieri et al. (2019) |
| Bendamustine, prednisone, rituximab added for those with IgM AL amyloidosis | Non-IgM amyloid: 35% IgM amyloid: 58% Refractory to rituximab: 64% |
Renal (31%) Cardiac (12%) |
Milani et al. (2018) |
| Rituximab, bendamustine (IgM light chain amyloidosis) | Newly diagnosed: 73% Relapsed: 60% |
Both cohorts: Cardiac (17%) Renal (17%) |
Manwani et al. (2018) |
| Bendamustine, dexamethasone | 57% | Renal (46%) Cardiac (13%) |
Lentzsch et al. (2020) |
| Venetoclax | 88% | Renal (33%) Cardiac (25%) |
Sidiqi et al. (2020) |
| Isatuximab | 77% | NR | Parker et al. (2020) |
Case Study 1: Treatment
After discussion, the patient was started on bortezomib, cyclophosphamide, dexamethasone, and rituximab. After one cycle the IgM increased, which was attributed to IgM flare, so the patient continued on therapy. After three cycles of therapy, the IgM decreased to 1,500 mg/dL, and his total proteinuria decreased from 1,500 mg to 1,100 mg. He received a total of four cycles of therapy and proceeded to autologous stem cell transplant with reduced-dose melphalan. He achieved a complete hematologic response and had both cardiac and renal response in his amyloidosis. His performance status improved, as well as his neuropathy. He has been on observation since.
Case Study 2: Treatment
In patients with relapsing WM, many options are available and are well tolerated (Table 9). Since his mutational analysis revealed MYD88+ and CXCR–, treatment with a BTK inhibitor was discussed. BTK inhibitors are effective in WM both as single agents as well as in combination with rituximab. Response rates in the relapsed setting range between 74% to 90% and are well tolerated. The main side effects observed with BTK inhibitors include bleeding, hypertension, atrial fibrillation, IgM rebound, infections, neutropenia, headaches, diarrhea, fatigue, and arthralgias (Castillo et al., 2020). Acalabrutinib and zanubrutinib have a more favorable side effect profile as they do not disrupt platelet aggregation nor do they have the same incidence of atrial fibrillation. The incidence of atrial fibrillation with ibrutinib ranges from 5% to 10%, whereas with acalabrutinib is 5%, and with zanabrutinib is 2% (Owen et al., 2020; Tam et al., 2020; Treon et al., 2015b; Dimopoulos et al., 2018). In the phase III ASPEN study, investigators reported less cumulative hypertension with zanubrutinib compared with ibrutinib (Tam et al., 2020). They also observed less bleeding with zanubrutinib. In terms of myelosuppression, neutropenia was observed more often with zanubrutinib compared with ibrutinib; however, the incidence of infections was not higher in the zanubrutinib arm (Tam et al., 2020).
After discussion of the various options available, the patient elected to proceed with a BTK inhibitor as he preferred an oral regimen. The decision was made to proceed with zanubrutinib because of his history with atrial fibrillation; he is currently on oral anticoagulation. He proceeded on to therapy and achieved a VGPR with resolution of the palpable purpura and adenopathy.
CONCLUSION
Waldenström macroglobulinemia and AL amyloidosis are both distinct and rare plasma cell dyscrasias, but even more rare is the concurrence of both diagnoses. Failure to detect IgM amyloidosis with WM may have serious and possibly fatal consequences, as this diagnosis requires unique considerations than its standard, individual counterparts. Thus, early detection is imperative to obtain rapid control and suppression of the toxic clones, which may improve survival benefit while preserving organ function. While novel regimens have proven effective and have extended progression-free survival, identifying new therapeutic agents may further improve patient outcomes and give possible hope for curing this complex disease.
Footnotes
Ms. Lu has nothing to disclose. Dr. Richards has served as a consultant for Bristol Myers Squibb, GSK, Janssen/Legend Biotech, Sanofi, and Takeda.
References
- Bloch, K., & Maki, D. (1973). Hyperviscosity syndromes associated with immunoglobin abnormalities. Seminars of Hematology, 10(2), 113–124. [PubMed] [Google Scholar]
- Castillo, J. J., Advani, R. H., Branagan, A. R., Buske, C., Dimopoulos, M. A., D'Sa, S.,…Kastritis, E. (2020). Consensus treatment recommendations from the tenth International Workshop for Waldenström Macroglobulinaemia. Lancet Haematology, 7(11), e827–e837. 10.1016/s2352-3026(20)30224-6 [DOI] [PubMed] [Google Scholar]
- Castillo, J. J., Allan, J. N., Siddiqi, T., Advani, R. H., Meid, K., Leventoff, C.,…Hunter, Z. R. (2022). Venetoclax in previously treated Waldenström Macroglobulinemia. Journal of Clinical Oncology, 40(1), 63–71. 10.1200/jco.21.01194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo, J. J., Meid, K., Flynn, C. A., Chen, J., Demos, M. G., Guerrera, M. L.,…Treon, S. P. (2020). Ixazomib, dexamethasone, and rituximab in treatment-naive patients with Waldenström macroglobulinemia: Long-term follow-up. Blood Advances, 4(16), 3952–3959. 10.1182/bloodadvances.2020001963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo, J. J., Meid, K., Gustine, J. N., Dubeau, T., Severns, P., Hunter, Z. R.,…Treon, S. P. (2018). Prospective clinical trial of ixazomib, dexamethasone, and rituximab as primary therapy in Waldenström Macroglobulinemia. Clinical Cancer Research, 24(14), 3247–3252. 10.1158/1078-0432.ccr-18-0152 [DOI] [PubMed] [Google Scholar]
- D'Sa, S., Kersten, M. J., Castillo, J. J., Dimopoulos, M., Kastritis, E., Laane, E.,…Lunn, M. P. (2017). Investigation and management of IgM and Waldenström-associated peripheral neuropathies: recommendations from the IWWM-8 consensus panel. British Journal of Haematology, 176(5), 728–742. 10.1111/bjh.14492 [DOI] [PubMed] [Google Scholar]
- Desbois, A. C., Cacoub, P., & Saadoun, D. (2019). Cryoglobulinemia: An update in 2019. Joint Bone Spine, 86(6), 707–713. 10.1016/j.jbspin.2019.01.016 [DOI] [PubMed] [Google Scholar]
- Dimopoulos, M., & Kastritis, E. (2019). How I treat Waldenstroms macroglobulinemia. Blood, 134(23), 2022–2035. [DOI] [PubMed] [Google Scholar]
- Dimopoulos, M., Panayiotidis, P., Moulopoulos, L., Sfikakis, P., & Dalakas, M. (2000). Waldenström's macroglobulinemia: Clinical features, complications, and management. Journal of Clinical Oncology, 18(1), 214. 10.1200/JCO.2000.18.1.214 [DOI] [PubMed] [Google Scholar]
- Dimopoulos, M., Zervas, C., Zomas, A., Kiamouris, C., Viniou, N., Grigoraki, V.,…Anagnostopoulos, N. (2002). Treatment of Waldenstroms macroglobulinemia with rituximab. Journal of Clinical Oncology, 20(9), 2327–2333. 10.1200/JCO.2002.09.039 [DOI] [PubMed] [Google Scholar]
- Dimopoulos, M. A., Tedeschi, A., Trotman, J., García-Sanz, R., Macdonald, D., Leblond, V.,…Buske, C. (2018). Phase 3 trial of ibrutinib plus rituximab in Waldenström's macroglobulinemia. New England Journal of Medicine, 378(25), 2399–2410. 10.1056/nejmoa1802917 [DOI] [PubMed] [Google Scholar]
- Dispenzieri, A., Kastritis, E., Wechalekar, A. D., Schönland, S. O., Kim, K., Sanchorawala, V.,…Merlini, G. (2019). Primary results from the phase 3 Tourmaline-AL1 trial of ixazomib-dexamethasone versus physician's choice of therapy in patients (pts) with relapsed/refractory primary systemic AL amyloidosis (RRAL). Blood, 134(Supplement_1), 139–139. 10.1182/blood-2019-124409 [DOI] [Google Scholar]
- Dittrich, T., Kimmich, C., Hegenbart, U., & Schönland, S. O. (2020). Prognosis and staging of AL amyloidosis. Acta Haematologica, 143(4), 388–400. 10.1159/000508287 [DOI] [PubMed] [Google Scholar]
- Gavriatopoulou, M., García-Sanz, R., Kastritis, E., Morel, P., Kyrtsonis, M.-C., Michalis, E.,…Dimopoulos, M. A. (2017). BDR in newly diagnosed patients with WM: Final analysis of a phase 2 study after a minimum follow-up of 6 years. Blood, 129(4), 456–459. 10.1182/blood-2016-09-742411 [DOI] [PubMed] [Google Scholar]
- Gertz, M. (2019). Waldenstrom macroglobulinemia: 2019 update on diagnosis, risk stratification, and managmenet. Annual Clinical Updates in Hematological Malignancies, 94(2), 266–276. 10.1002/ajh.25292 [DOI] [PubMed] [Google Scholar]
- Gertz, M. A. (2018). Waldenström macroglobulinemia: 2019 update on diagnosis, risk stratification, and management. American Journal of Hematology, 94(2), 266–276. 10.1002/ajh.25292 [DOI] [PubMed] [Google Scholar]
- Gertz, M. A. (2021). Waldenström macroglobulinemia: 2021 update on diagnosis, risk stratification, and management. American Journal of Hematology, 96(2), 258–269. 10.1002/ajh.26082 [DOI] [PubMed] [Google Scholar]
- Gertz, M. A., & Zeldenrust, S. R. (2014). Amyloidosis. In Gertz M.A. & Rajkumar S.V. (Eds.), Multiple myeloma: Diagnosis and treatment (pp. 265–282). http://citeseerx.ist.psu.edu/viewdoc/download;jsessionid=518EFA3AAC2E74E3078036EF60F11F78?doi=10.1.1.655.353&rep=rep1&type=pdf
- (2016). IgM amyloidosis. In Leblond V.., Treon S.., & Dimoploulos M. (Ed.). Waldenstrom's Macroglobulinemia (pp. 195–207). Switzerland: Springer. [Google Scholar]
- Ghobrial, I. M., Xie, W., Padmanabhan, S., Badros, A., Rourke, M., Leduc, R.,…Matous, J. (2010). Phase II trial of weekly bortezomib in combination with rituximab in untreated patients with Waldenström Macroglobulinemia. American Journal of Hematology, 85(9), 670–674. 10.1002/ajh.21788 [DOI] [PubMed] [Google Scholar]
- Jagannath, S., Munshi, N. C., & Richardson, P. G. (2016). Multiple myeloma and other plasma cell dyscrasias. https://www.cancernetwork.com/view/multiple-myeloma-and-other-plasma-cell-dyscrasias
- Kastritis, E., & Dimopoulos, M. A. (2015). Recent advances in the management of AL Amyloidosis. British Journal of Haematology, 172(2), 170–186. 10.1111/bjh.13805 [DOI] [PubMed] [Google Scholar]
- Kastritis, E., Gavriatopoulou, M., Kyrtsonis, M.-C., Roussou, M., Hadjiharissi, E., Symeonidis, A.,…Dimopoulos, M. A. (2015). Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenström macroglobulinemia: Final analysis of a phase 2 study. Blood, 126(11), 1392–1394. 10.1182/blood-2015-05-647420 [DOI] [PubMed] [Google Scholar]
- Kastritis, E., Gavriatopoulou, M., Roussou, M., Bagratuni, T., Migkou, M., Fotiou, D., Ziogas, D.,…Dimopoulos, M. (2018). Efficacy of lenalidomide as salvage therapy for patients with AL amyloidosis. Amyloid, 25(4), 234–241. 10.1080/13506129.2018.1540410 [DOI] [PubMed] [Google Scholar]
- Kastritis, E., Morel, P., Duhamel, A., Gavriatopoulou, M., Kyrtsonis, M. C., Durot, E.,…Terpos, E. (2019). A revised international prognostic score system for Waldenström's macroglobulinemia. Leukemia, 33(11), 2654–2661. 10.1038/s41375-019-0431-y [DOI] [PubMed] [Google Scholar]
- Kastritis, E., Palladini, G., Minnema, M. C., Wechalekar, A. D., Jaccard, A., Lee, H. C.,…Hungria, V. (2021). Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. New England Journal of Medicine, 385(1), 46–58. 10.1056/nejmoa2028631 [DOI] [PubMed] [Google Scholar]
- Kourelis, T. V., Kumar, S. K., Gertz, M. A., Lacy, M. Q., Buadi, F. K., Hayman, S. R.,…Dispenzieri, A. (2013). Coexistent multiple myeloma or increased bone marrow plasma cells define equally high-risk populations in patients with immunoglobulin light chain amyloidosis. Journal of Clinical Oncology, 31(34), 4319–4324. 10.1200/jco.2013.50.8499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentzsch, S., Lagos, G., Comenzo, R., Zonder, J., Osman, K., Pan, S.,…Landau, H. (2020). Bendamustine with dexamethasone in relapsed/refractory systemic light chain amyloidosis:results of a phase II study. Journal of Clinical Oncology, 38(13), 1455–1462. 10.1200/JCO.19.01721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, R., & Richards, T. (2019). AL amyloidosis: Unfolding a complex disease. Journal of the Advanced Practitioner in Oncology, 10(8), 813–825. 10.6004/jadpro.2019.10.8.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manwani, R., Sachchithanantham, S., Mahmood, S., Foard, D., Sharpley, F., Rezk, T.,…Wechalekar, A. D. (2018). Treatment of IgM-associated immunoglobulin light-chain amyloidosis with rituximab-bendamustine. Blood, 132(7), 761–764. 10.1182/blood-2018-04-846493 [DOI] [PubMed] [Google Scholar]
- Merlini, G. (2017). AL amyloidosis: From molecular mechanisms to targeted therapies. Hematology, 2017(1), 1–12. 10.1182/asheducation-2017.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhael, J. R., Schuster, S. R., Jimenez-Zepeda, V. H., Bello, N., Spong, J., Reeder, C. B.,…Fonseca, R. (2012). Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood, 119(19), 4391–4394. 10.1182/blood-2011-11-390930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani, P., Schönland, S., Merlini, G., Kimmich, C., Foli, A., Dittrich, T.,…Hegenbart, U. (2018). Treatment of AL amyloidosis with bendamustine: A study of 122 patients. Blood, 132(18), 1988–1991. 10.1182/blood-2018-04-845396 [DOI] [PubMed] [Google Scholar]
- Morel, P., Duhamel, A., Gobbi, P., Dimopoulos, M. A., Dhodapkar, M. V., McCoy, J.,…Merlini, G. (2009). International prognostic scoring system for Waldenström macroglobulinemia. Blood, 113(18), 4163–4170. 10.1182/blood-2008-08-174961 [DOI] [PubMed] [Google Scholar]
- Owen, R. G., McCarthy, H., Rule, S., D'Sa, S., Thomas, S. K., Tournilhac, O.,…Hamdy, A. (2020). Acalabrutinib monotherapy in patients with Waldenström macroglobulinemia: A single-arm, multicentre, phase 2 study. Lancet Haematology, 7(2), e112–e121. 10.1016/s2352-3026(19)30210-8 [DOI] [PubMed] [Google Scholar]
- Palladini, G., Sachchithanantham, S., Milani, P., Gillmore, J., Foli, A., Lachmann, H.,…Wechalekar, A. D. (2015). A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood, 126(5), 612–615. 10.1182/blood-2015-01-620302 [DOI] [PubMed] [Google Scholar]
- Parker, T. L., Rosenthal, A., Sanchorawala, V., Landau, H. J., Campagnaro, E., Kapoor, P.,…Orlowski, R. Z. (2020). A phase II study of isatuximab (SAR650984) (NSC-795145) for patients with previously treated AL amyloidosis (SWOG S1702; NCT#03499808). Blood, 136(Supplement 1), 20–21. 10.1182/blood-2020-143180 [DOI] [Google Scholar]
- Pika, T., Hegenbart, U., Flodrova, P., Maier, B., Kimmich, C., & Schönland, S. O. (2018). First report of ibrutinib in IgM-related amyloidosis: Few responses, poor tolerability, and short survival. Blood, 131(3), 368–371. 10.1182/blood-2017-09-806463 [DOI] [PubMed] [Google Scholar]
- Quock, T. P., Yan, T., Chang, E., Guthrie, S., & Broder, M. S. (2018). Epidemiology of AL amyloidosis: A real-world study using US claims data. Blood Advances, 2(10), 1046–1053. 10.1182/bloodadvances.2018016402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rummel, M. J., Niederle, N., Maschmeyer, G., Banat, G. A., von Grünhagen, U., Losem, C.,…Brugger, W. (2013). Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. The Lancet, 381(9873), 1203–1210. 10.1016/s01406736(12)61763-2 [DOI] [PubMed] [Google Scholar]
- Ryšavá, R. (2018). AL amyloidosis: Advances in diagnostics and treatment. Nephrology Dialysis Transplantation, 34(9), 1460–1466. 10.1093/ndt/gfy291 [DOI] [PubMed] [Google Scholar]
- Sidiqi, M. H., Al Saleh, A. S., Leung, N., Jevremovic, D., Aljama, M. A., Gonsalves, W. I.,…Fonseca, R. (2020). Venetoclax for the treatment of translocation (11;14) AL amyloidosis. Blood Cancer Journal, 10(5), 55. 10.1038/s41408-020-0321-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sissoko, M., Sanchorawala, V., Seldin, D., Sworder, B., Angelino, K., Broce, M.,…Sloan, J. M. (2015). Clinical presentation and treatment responses in IGM-related al amyloidosis. Amyloid, 22(4), 229–235. 10.3109/13506129.2015.1092433 [DOI] [PubMed] [Google Scholar]
- Stone, M. J., & Pascual, V. (2010). Pathophysiology of Waldenstrom's macroglobulinemia. Haematologica, 95(3), 359–364. 10.3324/haematol.2009.017251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam, C. S., Opat, S., D'Sa, S., Jurczak, W., Lee, H.-P., Cull, G.,… Motta, M. (2020). A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: The ASPEN study. Blood, 136(18), 2038–2050. 10.1182/blood.2020006844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treon, S. P., Meid, K., Gustine, J., Patterson, C. J., Matous, J. V., Ghobrial, I. M., & Castillo, J. J. (2015a). Long-term outcome of a prospective study of bortezomib, dexamethasone and rituximab (BDR) in previously untreated, symptomatic patients with Waldenstrom's Macroglobulinemia. Blood, 126(23), 1833–1833. 10.1182/blood.v126.23.1833.1833 [DOI] [Google Scholar]
- Treon, S. P., Tripsas, C. K., Meid, K., Kanan, S., Sheehy, P., Chuma, S., … Castillo, J. J. (2014). Carfilzomib, rituximab, and dexamethasone (CaRD) treatment offers a neuropathy-sparing approach for treating Waldenström's macroglobulinemia. Blood, 124(4), 503–510. 10.1182/blood-2014-03-566273 [DOI] [PubMed] [Google Scholar]
- Treon, S. P., Tripsas, C. K., Meid, K., Warren, D., Varma, G., Green, R.,…Hunter, Z. R. (2015b). Ibrutinib in previously treated Waldenström's Macroglobulinemia. New England Journal of Medicine, 372(15), 1430–1440. 10.1056/nejmoa1501548 [DOI] [PubMed] [Google Scholar]
- Wechalekar, A. D., Lachmann, H. J., Goodman, H. J. B., Bradwell, A., Hawkins, P. N., & Gillmore, J. D. (2008). AL amyloidosis associated with IgM paraproteinemia: clinical profile and treatment outcome. Blood, 112(10), 4009–4016. 10.1182/blood-2008-02-138156 [DOI] [PubMed] [Google Scholar]
- Zanwar, S., Abeykoon, J. P., Ansell, S. M., Gertz, M. A., Dispenzieri, A., Muchtar, E.,…Nowakowski, G. (2018). Primary systemic amyloidosis in patients with Waldenström macroglobulinemia. Leukemia, 33(3), 790–794. 10.1038/s41375-018-0286-7 [DOI] [PubMed] [Google Scholar]