

Abstract
Cx43 is the major connexin in ventricular gap junctions, and plays a pivotal role in control of electrical and metabolic communication among adjacent cardiomyocytes. We previously found that Cx43 dephosphorylation at serine 282 (pS282) caused cardiomyocyte apoptosis, which is involved in cardiac ischemia/reperfusion injury. In this study we investigated whether Cx43-S282 hyper-phosphorylation could protect cardiomyocytes against apoptosis. Adenovirus carrying rat full length Cx43 gene (Cx43-wt) or a mutant gene at S282 substituted with aspartic acid (S282D) were transfected into neonatal rat ventricular myocytes (NRVMs) or injected into rat ventricular wall. Rat abdominal aorta constriction model (AAC) was used to assess Cx43-S282 phosphorylation status. We showed that Cx43 phosphorylation at S282 was increased over 2-times compared to Cx43-wt cells at 24 h after transfection, while pS262 and pS368 were unaltered. S282D-transfected cells displayed enhanced gap junctional communication, and increased basal intracellular Ca2+ concentration and spontaneous Ca2+ transients compared to Cx43-wt cells. However, spontaneous apoptosis appeared in NRVMs transfected with S282D for 34 h. Rat ventricular myocardium transfected with S282D in vivo also exhibited apoptotic responses, including increased Bax/Bcl-xL ratio, cytochrome c release as well as caspase-3 and caspase-9 activities, while factor-associated suicide (Fas)/Fas-associated death domain expression and caspase-8 activity remained unaltered. In addition, AAC-induced hypertrophic ventricles had apoptotic injury with Cx43-S282 hyper-phosphorylation compared with Sham ventricles. In conclusion, Cx43 hyper-phosphorylation at S282, as dephosphorylation, also triggers cardiomyocyte apoptosis, but through activation of mitochondrial apoptosis pathway, providing a fine-tuned Cx43-S282 phosphorylation range required for the maintenance of cardiomyocyte function and survival.
Keywords: cardiomyocyte apoptosis, Connexin 43, serine phosphorylation, gap junction, Ca2+ signal, mitochondrial apoptosis pathway, rat abdominal aorta constriction model
Introduction
Cx43 is the major connexin in ventricular gap junctions (GJs), where it plays a pivotal role in control of electrical and metabolic communication among adjacent cardiomyocytes. In addition, Cx43 also regulates various cardiac physiological and pathological processes, including cell homeostasis, growth, differentiation and apoptosis, as well as tissue development [1, 2]. In normal myocardium Cx43 is highly phosphorylated at its carboxyl-terminal with around 21 phosphor-sites, and participates in the normal intercellular coupling and Cx43 life-cycle, including assembly, trafficking and turnover, regulations [1, 3, 4]. Under pathological conditions, dysfunctional Cx43, especially changes in its phosphorylation significantly affect cardiac function including electrical and contraction synchronization, and cardiomyocyte growth and survival, thus involved in arrythmogenesis, myocardial remodeling, apoptosis, and hypertrophy [1–7]. Until now, several important Cx43-phospho-sites have been explored and suggested to be closely related to sever cardiac diseases, such as cardiac ischemia reperfusion, hypertrophy, and heart failure across species [5–7]. In myocardial ischemia-reperfusion injury down-regulated Cx43 phosphorylation at serine 282 (S282), S325/328/330, S365, S368, and S297 have been found involved in cardiomyocyte apoptosis and myocardial injury [6–11]. Intervention with up-regulating Cx43 phosphorylation at S368 or S262 is effective to protect cardiomyocyte against apoptosis and myocardium injury [9, 11]. In another hand, upregulation of Cx43 phosphorylation for a short time is important for physiological response of cardiac adaption to environment stimulation [12, 13]. But sustained Cx43 phosphorylation has been linked to cardiac hypertrophic response, e.g. Cx43 phosphorylation at S325/328/330, S368 and S365 [2, 8, 13]. However, the mechanisms thereby Cx43 and its phosphorylation regulates cardiomyocyte survival and growth in physiological as well as pathological process remain largely unknown.
Previously, we found Cx43 dephosphorylation at S282 was closely related to cardiomyocyte apoptosis. Homozygous mice with Cx43-S282 substituted with alanine (S282A), mimicking lack of S282 phosphorylation, ceased to develop at the embryonic stage, while around 60% heterozygous mice had spontaneous arrhythmia after birth, and around 30% of these mice died before maturation, attributable to the cardiomyocyte apoptosis and cardiac arrhythmia [14]. This Cx43-S282 dephosphorylation induced myocardium damage is involved in the ischemia-reperfusion heart injury [6]. Thus, we propose that enhancing Cx43 phosphorylation at S282 could be an attractive target for protecting cardiomyocyte against apoptosis and cardiac arrhythmia due to Cx43 dephosphorylation. In this study, we found, however, that hyper Cx43 phosphorylation at S282 could increase intracellular Ca2+ concentration and intercellular communication, but cause cardiomyocyte apoptosis in normal cardiomyocytes and ventricle because of mitochondrial apoptotic pathway activation. Therefore, together with previous finings [6, 14], our results illustrate a fine-balanced Cx43-S282 phosphorylation required for the maintenance of cardiomyocyte function and survival.
Materials and methods
Animals
Wistar rats and Sprague-Dawley rats (male, 6–8 week) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd), housed at the animal care facility of the Center for Experimental Animals at Capital Medical University (Beijing, China), under specific pathogen-free conditions at 22 ± 1 °C with a 12-h light-dark cycle, and chow was provided ad libitum throughout the study. The protocols of animal experiments in the current study were approved by the Capital Medical University Animal Care and Use Committee (AEEI-2015-193 and AEEI-2016-072). All studies were conducted according to the “Guide for the Care and Use of Laboratory Animals” adopted by Beijing Municipal People’s Government, and in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 8523, revised 2011).
Isolation and culture of cardiomyocytes
NRVMs were isolated from 1 to 3-day-old Sprague-Dawley rats by enzymatic digestion with 0.1% trypsin and 0.03% collagenase as described previously [6]. The isolated cells at the density of 1×106/mL were used for measurements of Ca2+ transients, lucifer yellow (LY) uptake, and immunostaining, respectively. Cells at the density of 3×106/mL were used for Western blot and flow cytometry. In order to intervene factor-associated suicide (Fas) activation, soluble factor-associated suicide (sFas, 3 μg/60-mm dish) was added at the same time with virus addition and remained until harvesting.
Adenovirus construction and transfection
Recombinant plasmids and adenoviral vectors carrying rat wild-type Cx43 (Cx43-wt) or S282 substituted with aspartic acid (S282D) genes were constructed as described previously [6, 15]. The NRVMs were transfected with the adenovirus at 20 multiplicity of infection (m.o.i.) for each.
Assessment of apoptosis
Apoptosis evaluations in NRVMs and heart tissue were performed by annexin V-FITC/PI staining kit (Beyotime Biotechnology, https://www.beyotime.com/index.html) using flow cytometry and terminal deoxyribonucleotide transferase-mediated dUTP nick-end labeling (TUNEL) staining kit (Roche Applied Science, Mannheim, Germany), respectively. Caspase-3, caspase-8, and caspase-9 activities were assessed by enzyme-linked immunosorbent assay (ELISA) kit (Beyotime Biotechnology, https://www.beyotime.com/index.html). Fas, Fas-associated death domain (FADD), Bcl-2-associated X (Bax), B-cell lymphoma-extra large (Bcl-xL), and cytochrome (Cyt-c) expression was evaluated by Western blot. Cell death evaluations in NRVMs were performed by fluorescent dye YOYO-1 iodide using IncuCyte ZOOM (Essen Bioscience, Gottingen, Germany), as described previously [14].
Histological analyses and immunostaining
Myocardium morphology and ventricular structure, Cx43 distribution in NRVMs, and myocardium fibrosis were determined by immunofluorescence, hematoxylin and eosin (H&E) and MASSON staining, respectively, as described previously [6, 14].
Ca2+ transient imaging and lucifer yellow (LY) uptake assay
Cultured NRVMs were loaded with 1 μM Fluo4/AM (Invitrogen, Waltham, MA, USA), and then washed three times with Ca2+-containing HEPES-buffered saline solution (HBSS (mM)): NaCl 135, KCl 5, MgCl2 1, CaCl2 1.8, HEPES 10, and glucose 12, with pH 7.4 adjusted by NaOH for 20 min. The measurement of Ca2+ signaling was performed by Leica confocal microscopy as described previously [6, 14, 16].
The intercellular communication was evaluated in cultured NRVMs incubated with 2.5% LY (containing 1 mM EGTA) for 8 min at room temperature, and then washed three times with HBSS. The dye uptake was observed by Leica SP5 fluorescence laser scanning confocal microscopy (excitation at 405 nm and emission detection at 530 nm). Ten pictures were taken randomly from each dish for statistical analysis.
Intramyocardial injection of virus
Sprague-Dawley male rats (250 ± 20 g) were divided into three groups: vector (n = 8), Cx43-wt (n = 10), and Cx43-S282 (n = 11). After anesthetized with pentobarbital (100 mg/kg), rats were injected with adenovirus constructs of vector, Cx43-wt and S282D into the anterior wall of the left ventricle under ultrasound image guidance as previously described [6]. Briefly, four injections (25 μL for each site) were chosen in the anterior wall of rat heart, and a volume of 100 μL was injected in each heart. Virus constructs were diluted with 0.9% saline to a final concentration of 1×1010 virus particles in 1 mL. During the procedure, mice were immobilized to a warm plate, and respiration and body temperature were monitored.
Abdominal aortic construction rat model
Male Wistar rats (6–8 week, 180–220 g body weight, n = 18) were anaesthetized with a 2% sodium pentobarbital (45 mg/kg, ip). The suprarenal portion of the abdominal aorta, proximal to the left renal artery, was exposed and separated from the vena cava. A ligature (4-0 silk) was tied, using a blunt 22-G needle (the external diameter was 0.7 mm) beside the aorta between the branches of the coeliac and anterior mesenteric arteries. The needle was then removed, leaving the internal diameter of the aorta approximately 0.7 mm. The abdominal wall and skin were sutured. Sham-operated rats received the same surgical procedure without constriction. Intramuscular injection of penicillin (100,000 units/animal) was performed for 7 days after operation.
Echocardiographic assessment of cardiac dimensions and functions
Rats were intubated and anaesthetized with mechanical ventilation by inhalation of 1%–1.5% isoflurane (RWD, Batch) in 100% oxygen continuously. Transthoracic echocardiography was performed using Vevo 2100 imaging system (Visual sonics, Toronto, ON, Canada) equipped with a frequency transducer (frequency band 12–38 MHz). Two-dimensional guided M-mode tracings were recorded in both parasternal long and short axis views at the level of papillary muscles, and the measurements of cardiac function parameters were performed and calculated as described previously [17].
Preparation of junctional and non-junctional fractions
Junctional and non-junctional fractions from myocardial tissue and NRVMs were performed as described previously [6, 14]. Briefly, the myocytes were incubated in RIPA for 30 min on the ice and then centrifuged at 10,000 r/min for 5 min at 4 °C, and the supernatant was collected as nonjunctional fraction. The remaining pellet was resuspended, incubated after transient sonication in RIPA (containing 2% Triton X-100 and 0.4% SDS) for 30 min at room temperature, and then centrifuged for 15 min at 14,800 r/min at 4 °C. The supernatants were the junctional proteins. GAPDH and β-actin were used as reference for non-junctional and junctional fraction separation, respectively.
Western blot analysis
Protein extraction from myocardial tissue or NRVMs and the immunoblotting were performed as described previously [6, 14, 17].
The following primary antibodies were used: rabbit anti-Cx43 (3512, Cell Signaling Technology, Danvers, MA, USA), rabbit anti-Cx43 pS262 (sc-22267, Santa Cruz Biotechnology, CA, USA), rabbit anti-Cx43 pS368 (3511 S, Cell Signaling Technology), rabbit anti-HA (C29F4, Cell Signaling Technology), rabbit anti-Bcl-xL (ET1603-28, Huobio, http://www.huabio.cn/), rabbit anti-α-SMA (sc-53142, Santa Cruz Biotechnology, CA, USA), rabbit anti-α-actin (6487, Cell Signaling Technology), rabbit anti-FADD (sc-1023, Santa Cruz Biotechnology, CA, USA), mouse anti-Fas (ab82419, Abcam, Cambridge, UK), mouse anti-Bax (Huobio, EM1203), mouse anti-tumor necrosis factor receptor 1 (TNFR1, sc-8436, Santa Cruz Biotechnology, CA, USA), mouse anti-death receptor 5 (DR5, sc-19529, Santa Cruz Biotechnology, CA, USA), mouse anti-caspase-8 (sc-7890, Santa Cruz Biotechnology, CA, USA), mouse anti-cytochrome c (sc-13156, Santa Cruz Biotechnology, CA, USA), mouse anti-Cox-IV(sc-376731, Santa Cruz Biotechnology, CA, USA). As loading control, mouse anti-GAPDH (TA-08, ZSGB-BIO, Beijing, China) and anti-β-actin (TA-09, ZSGB-BIO, Beijing, China) were used. All antibodies were used at a ratio of 1:1000, except for anti-FADD, Bax, Bcl-xL, and anti-Fas antibody at a ratio of 1:500, respectively.
Statistical analysis
Statistical analyses were performed using the Statistical Product and Service Solutions (SPSS) program (IBM SPSS Statistics 21, NY, USA). The results were expressed as mean ± SD of at least three independent experiments. When appropriate, statistical comparisons between groups were carried out with the 2-way unpaired Student’s t-test for 2 groups, and one-way analysis of variance (ANOVA) was performed with post Tukey adjustment for multiple groups. P-value of <0.05 was considered to be statistically significant.
Results
Cx43-S282 mutation with aspartic acid increased gap communication and internal Ca2+ concentration in cardiomyocytes
To explore the effect of Cx43 phosphorylation at S282 on cardiomyocytes, we firstly performed exogenous expression of S282D (serine to aspartic acid), a phospho-mimetic mutant of Cx43, or rat wild-type Cx43 gene (Cx43-wt) in NRVMs. Both S282D and Cx43-wt NRVMs showed approximately 5 times more Cx43 expression than vector control cells, while more Cx43 phosphorylation at S282 was found in S282D than Cx43-wt cells with unaltered pS262 and pS368 abundance (Fig. 1a). Further, we evaluated the pS282 expression in junctional and non-junctional fractions isolated by Triton X-100 treatment. Higher level of pS282 in S282D cells over that in Cx43-wt cells was also found in both intercalated disk (junctional fraction) and cytoplasm (non-junctional fraction), confirming a success in manipulating hyper phospho-S282 (Fig. 1b). Immunostaining assay with anti-Cx43 antibody supports more Cx43 expression in both Cx43-gene modified cells compared with that in vector (control) cells (Fig. 1c).
Fig. 1. Cx43-S282 mutation with aspartic acid increased gap communication and Ca2+ signal in cardiomyocytes.

Neonatal rat ventricular myocytes (NRVMs) were transfected with vector, S282D or Cx43-wt gene for 24 or 34 h, and analyzed by Western blot. a Cx43 phosphorylation at S262, S368, S282 and S279 was detected using specific antibody for the respective phosphor-site, and analyzed in relative to the level of Cx43 in whole cell lysate of NRVMs transfected for 34 h. b Junctional and non-junctional fractions, referred to cell-membrane and cytosolic lysates, respectively, were used to detect Cx43 and its phosphorylation abundances distributed in both regions. No difference in Cx43 and its phosphorylation at S282 abundance from both transfected cells was found between the two fractions. c, d Confocal images of NRVMs transfected with vector control, S282D or Cx43-wt for 24 or 34 h showed Cx43 abundance and distribution using specific antibody against Cx43, and the intercellular communication in different transfected cells using lucifer yellow (LY) as indicated (c). Nucleus was stained with DAPI. Scale bar: 25 μm. The percentage of cells with LY uptake was analyzed between different groups of cells (d). e–g NRVMs were transfected with different mutants for 24 h and loaded with Fluo4 to detect the intracellular Ca2+ signal. Typical traces of Ca2+ transients (e) illustrate that S282D cells had increased spontaneous coordinated Ca2+ transients (f) and basal Ca2+ level (g). Data are expressed as mean ± SD. n values were given in each bar, and P values were obtained as indicated with a line between two groups in each panel using one-way ANOVA test.
Functional gap exchange in these cells was evaluated by the uptake of LY in NRVMs. After virus transfection for 24 or 34 h, both Cx43-wt and S282D cells showed significant more LY uptake than vector cells, and as expected, S282D transfected cells exhibited even more enhanced LY uptake than that in Cx43-wt cells (Fig. 1d), consistent with the previous observations using carboxyfluorescein (6-CF) or LY [6, 16].
We then detected whether the internal Ca2+ signal changed with the status of hyper Cx43-S282 phosphorylation in Fluo4-loaded NRVMs transfected with Cx43-wt or Cx43-S282D. After transfection for 24 h, S282D cells exhibited increased spontaneous coordinated Ca2+ transients compared to Cx43-wt cells (Fig. 1e, f), contrary to the effect of S282A (a mutate gene in Cx43-S282 with alanine) transfection in NRVMs that exhibited uncoordinated Ca2+ transients [14]. In addition, we found a higher basal intracellular Ca2+ in S282D cells than that in Cx43-wt or vector cells (Fig. 1g), indicating an activated Ca2+ signal activity in S282D NRVMs at resting state which might affect the cell function.
Cx43-S282 mutation with aspartic acid induced cardiomyocyte apoptosis through mitochondrial apoptotic pathway
As speculated, after transfected for 36 h S282D NRVMs showed observable death. By living imaging with IncuCyte ZOOM using fluorescent dye YOYO-1 iodide [14], we found that S282D transfected NRVMs exhibited a delayed dying time course compared to S282A NRVMs that began to die earlier than 24 h after transfection (Fig. 2a). The S282A transfected NRVMs demonstrated cell apoptosis mainly because of the activation of p38, followed by the activation of Fas/FADD, caspase-8 and caspase-3, the extrinsic apoptosis pathway found in previous studies [6, 14]. We then detected the two apoptosis pathways in S282D cells at 28 h after transfection. Using annexin V-FITC/PI staining, ELISA assay kit and Western blot, we found that S282D NRVMs showed 3-fold more apoptosis-positive cells, and activation of caspase-3 than those in Cx43-wt transfected cells (Fig. 2b, c), and the mitochondrial apoptosis pathway, including caspase-9, Bax and Cyt-c were significantly activated (Fig. 2d–f), whereas Fas/FADD/caspase-8, DR5 and TNFR1 pathways were inactivated (Fig. 2e–i). Then, we used the Fas inhibitor, soluble Fas (sFas), a blocker of initiating extrinsic apoptotic pathway, for further confirmation. The equal level in caspase-3 activation in both Cx43-wt and S282D cells in the presence of sFas supported the finding that Fas/FADD pathway was not involved in S282D-mediated cell death (Fig. 2c). Together with the increased basal internal Ca2+ found, all these data suggest that Cx43 hyper-phosphorylation at S282 causes cardiomyocyte apoptosis most likely through the activation of mitochondrial apoptotic pathway.
Fig. 2. Cx43-S282 mutation with aspartic acid caused cardiomyocyte apoptosis through mitochondrial apoptotic pathway.

a Time-lapse curves of NRVM death after transfection with virus carrying rat Cx43-wt, S282D or S282A gene detected by YOYO-1 probe. b–d NRVMs were transfected with virus carrying rat Cx43-wt or S282A gene, or vector for 28 h, and lysed for detection of apoptosis. Apoptotic rate was assessed by Annexin V/PI double staining and determined by a fowcytometric analysis (b). Caspase-3 (c) and caspase-9 (d) activities were detected by ELISA kits in different groups of cells. e–g The abundances of Cyt-c in mitochondria (Mt) and cytosol (e), Bax/Bcl-xL (f), and caspase-8 (g) in whole cell lysates from different groups were analyzed by Western blot. The Cyt-c fold changes in both fractions after normalized with Cox-IV or GAPDH, respectively, relative to those of control, and the fold changes in Bax, Bcl-xL and caspase-8 relative to those of Cx43-wt (after normalized with GAPDH) were thereby analyzed. h, i NRVMs were transfected with S282D or Cx43-wt gene for 24 or 34 h and the lysates were analyzed by Western blot. Fas (h), and FADD, DR5, and TNFR1 (i) were detected using specific antibody, and the fold changes in Fas and FADD relative to those of Cx43-wt (after normalized with GAPDH) were thereby analyzed. Data are expressed as mean ± SD. P values were obtained as indicated with a line between two groups in each panel using one-way ANOVA test.
Cx43-S282 mutation with aspartic acid induced myocardial apoptosis through mitochondria apoptotic pathway
To further identify the hyper phosphorylation at Cx43-S282 in cardiomyocytes, we performed an in vivo study by injecting adenovirus carrying vector, Cx43-wt or S282D mutation gene with HA tag into the anterior wall of the rat left ventricle under ultrasound guidance. Four sites in the anterior wall were selected and injected with 100 μL saline (control) or an equal volume of saline containing the adenovirus. As described previously [6], seven days after injection the positive HA-tag expression and five times increase in pS282 expression detected by Western blot indicated a successful transfection of the gene (Fig. 3d). Structural examinations showed disorganized cardiomyocytes and increased extracellular space with more interstitial fibrosis in S282D heart compared with Cx43-wt heart by H&E and MASSON staining, suggesting a myocardium injury in S282D injected ventricle (Fig. 3a). Using TUNEL staining and ELISA assay kit, S282D adenovirus infected hearts showed much more apoptosis-positive cells in the left ventricle anterior wall than that of Cx43-wt infected ventricles (Fig. 3a, b). Moreover, approximately 4-fold more caspase-3 and caspase-9 activities detected by ELISA kits were observed in S282D than Cx43-wt ventricle (Fig. 3b). In addition, significant increases in the expression of Cyt-c and Bax/Bcl-xL ratio were detected in S282D injected ventricles compared with those in Cx43-wt ventricles, while no significant changes in caspase-8 activity and Fas/FADD expression level were found (Fig. 3b, c). All these results indicate an activation of the mitochondrial apoptotic pathway involved in S282D adenovirus injected hearts, consistent with the observations found in NRVM experiment.
Fig. 3. S282D intramyocardial injection induced myocardial apoptosis through mitochondria apoptotic pathway.

The anterior walls of rat left ventricles were injected with vector or adenovirus carrying Cx43-wt or S282D gene. After seven days, the hearts were isolated for investigation. a, b Representative images of hearts (upper panel) and section of left ventricles stained with H&E, MASSON or TUNEL as indicated (a), and the percentages of fibrosis area and apoptosis positive cells as well as caspase-8/-9/-3 activities detected using ELISA kits were analyzed (b). c The levels of Bax, Bcl-xL, Cyt-c, Fas and FADD were determined by Western blot, and the fold changes in their abundances relative to those of control (after normalized with GAPDH) were thereby analyzed. d Cx43 expression and its phosphorylation at S282 were detected by Western blot using specific antibody, and fold changes in different groups of injected left ventricles were thereby analyzed. Data are expressed as mean ± SD. P values were obtained as indicated with a line between two groups in each panel using one-way ANOVA test.
Hyper-phosphorylation at Cx43-S282 associated with cardiomyocyte apoptosis in hypertrophy model
Cardiac hypertrophy and failure have been characterized by abnormal Ca2+ handling and apoptosis occurred in myocardium [18, 19]. Here, this study demonstrated a basal internal Ca2+ increase in vitro and cardiomyocyte apoptosis in vitro and in vivo. We thus further investigated the relationship between the status of S282 phosphorylation and the cardiomyocyte hypertrophy and apoptosis occurred in AAC rat heart.
AAC rats at 4 weeks after surgery were evaluated by echocardiography. An increased left ventricular wall thickness compared with that in Sham rats was found. Systolic function, as represented by fractional shortening (FS), was similar between the groups, indicating that the heart had not reached the stage of heart failure (Fig. 4a, b). The approximately 2-fold larger cardiomyocytes than those in Sham group detected by FITC labeled wheat germ agglutinin (WGA) (Fig. 4c) and together with the echocardiographic evaluation indicated these AAC rats on the stage of cardiac hypertrophy.
Fig. 4. Abdominal aortic construction induced cardiac hypertrophy in rats.

Aortic construction was performed in rat to induce cardiac hypertrophy (AAC, see Materials and Methods). a Representative images of M-mode echocardiography and the parameters obtained from each group of rats. b, c The cardiac hypertrophy was evaluated by examining the ratios of heart and left ventricle with body weights (HW/BW) and (LVW/BW) (b), and H&E and WGA staining, respectively (c). Representative images of H&E or WGA staining of the left ventricle from each group, and the areas of ventricular cardiomyocyte were compared between Sham and ACC rats. d, e Representative images of the left ventricle stained with Sirius Red, MASSON or TUNEL as indicated (d), and the percentages of fibrosis and apoptosis positive cells as well as caspase-8/-9/-3 activities using ELISA kits were analyzed and compared with those of Sham rats (e). f, g Apoptosis in the hypertrophic rat myocardium was determined by measuring the levels of Bcl-xL, Bax, and Cyt-c (e), and Fas/FADD (g), expression by Western blot, and the fold increases in their abundances relative to those of control (after normalized with GAPDH) were thereby obtained and compared between the two groups. Data are expressed as mean ± SD. P values were obtained as indicated with a line between two groups in each panel using unpaired two-tailed Student’s t test.
To investigate whether apoptosis occurred in the cardiac hypertrophy model, AAC left ventricles were detected by TUNEL staining and ELISA assay kit. AAC ventricles showed approximately 4-fold more apoptosis-positive cells, and approximately 2-, 4- and 7-fold higher caspase-8, -9 and -3 activity than those in Sham ventricles. A significant increased interstitial fibrosis was also observed in the AAC heart by MASSON and Sirius staining compared with those in Sham hearts, respectively (Fig. 4d, e). In addition, increased Cyt-c, Bax/Bcl-xL ratio as well as Fas and FADD expression was found in AAC ventricles, indicating apoptosis occurred in the stage of hypertrophic heart through the activation of mitochondrial and extrinsic apoptotic pathways in AAC rat model (Fig. 4f, g) as reported [20, 21].
Further, Cx43 phosphorylation was examined by Western blot, and approximately 2-fold higher expression was found in pS282, pS262 and pS368 in AAC ventricles than those in Sham ventricles (Fig. 5a). The Cx43 phosphorylation at pS282 in junctional and non-junctional fractions isolated by Triton X-100 treatment also showed approximately 2 times more than those in Sham ventricles (Fig. 5b).
Fig. 5. Hypertrophic myocardium exhibited increased Cx43 phosphorylation at S282 in AAC rat model.

a The levels of phospho-S262, -S282, -S368, and Cx43 expression in Sham and AAC rats were determined by Western blot using specific antibody, and fold increases in their abundances relative to those of Sham control (after normalized with GAPDH) were thereby obtained and compared between the Sham and AAC rats. b Junctional and non-junctional fractions were used to detect phospho-S282 and Cx43 abundance distributed in both regions. Data are expressed as mean ± SD. P values obtained as indicated with a line between two groups in each panel using unpaired two-tailed Student’s t test.
Discussion
Cx43 plays a pivotal role in regulating heart development and cardiomyocyte survival in addition to the regulation of electrical/contraction activities. In this regard, Cx43 phosphorylation modification performs mostly effective for acute and chronic regulation. Cx43 phosphorylation is dynamic and changes in response to activation of multiple different kinases, including protein kinase A, protein kinase C, Akt, extracellular regulated protein kinase, p34cdc2/cyclin B kinase, protein kinase CK1, mitogen-activated protein kinase, and pp60Src kinase [22–24]. One kinase activation may phosphorylate several Cx43 residues, and one residue can response to different kinase activation, rendering a much complicated crosslinking network and uncertainty in seeking the net effect of one phosphor-site in the regulation of gap communication in heart in vivo [4, 23–26].
The pathological role of Cx43 phosphorylation has been investigated more than its physiological regulation, particularly in the participation of cardiac arrhythmia and ischemic injury. Cx43 is dephosphorylated by mostly undefined mechanisms in ischemia and nonischemic heart diseases across species, including myocardial ischemia, arrhythmia, diabetes, and aging and failing hearts [3, 5, 22, 23, 27, 28]. Specific dephosphorylated serines, including S325/328/330, S306, S365, S368 and S296/297, are found in ischemic and ischemic/reperfusion heart, while increased phosphorylated serines, including total phosphorylation has been found in different hypertrophy heart model [6–9, 29–36]. We previously found that Cx43 phosphorylation at S282 can regulate cardiomyocyte survival and electrical stability. Dephosphorylated Cx43-S282 caused cardiomyocyte apoptosis, which is involved in the ischemia-reperfusion heart injury [6, 14]. In this study we found that hyper-phosphorylated S282 also triggered cardiomyocyte apoptosis in vivo and in vitro through the activation of mitochondrial apoptosis pathway (Figs. 1, 2, 3), which was likely involved in the apoptosis process in AAC-induced heart hypertrophy (Figs. 4, 5). Thus, together with previous results, this study illustrate a picture of limited range required for Cx43-S282 phosphorylation in the regulation of cardiomyocyte survival, either lower or higher than the limitation deems to cause cardiomyocyte death, which may contribute to several cardiac pathological injuries (Fig. 6).
Fig. 6. A cartoon illustrates Cx43 phosphorylation at S282 regulates cardiomyocyte survival in physiological and pathological status.
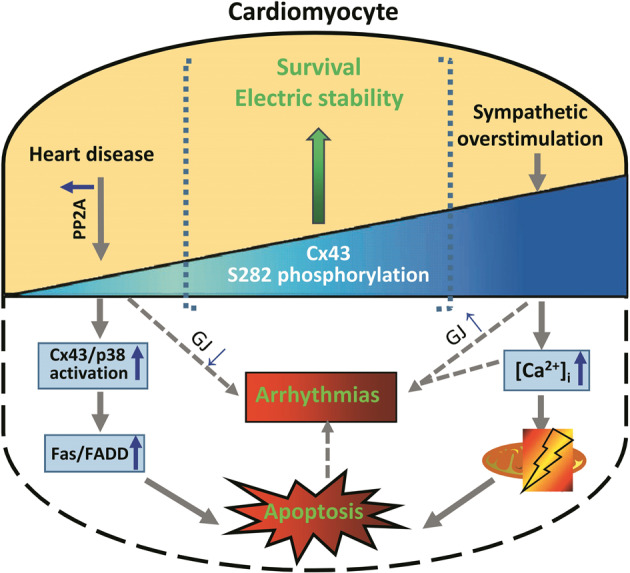
In cardiomyocyte, basal Cx43 phosphorylation on S282 plays a critical role in the regulations of cell survival and electrical stability. Either upregulation or downregulation of phosphor-S282 is highly risky in inducing arrhythmia and cardiomyocyte apoptosis because of the activation of mitochondrial or extrinsic apoptotic pathway, a situation that may occur in pathological conditions of ischemia or hypertrophy. Fas indicates factor-associated suicide; FADD, Fas-associated death domain; GJ, gap junction communication; Mt, mitochondria; [Ca2+]i, intracellular free Ca2+ ion concentration.
Myocardial hypertrophy is a reversible pathological process. When all kinds of pathological stimuli are removed, the hypertrophic myocardium can be improved [4]. However, when pathological stimuli are persistently active, the cardiac hypertrophy often proceeds to heart failure, and at this stage cardiomyocyte apoptosis plays an important role in the transition of the hypertrophic heart to failure heart [20, 37]. Enhanced internal Ca2+ activity is highly linked to cardiac hypertrophy though the activation of carcineurin [18, 19]. It is also well accepted that Ca2+ overload directly causes myocyte apoptosis through the mitochondrial apoptotic pathway [38, 39]. In the present study, the basal internal Ca2+ increase and enhanced spontaneous Ca2+ transients indicate a highly activated Ca2+ signal in S282D transfected cardiomyocytes (Fig. 2), which should be mechanically leading to cardiac hypertrophy. However, hypertrophic response was unable to be detected in S282D NRVMs because apoptotic phenotype also existed. In another hand, the overloaded Ca2+may result in excessive Ca2+ uptake by mitochondria [38], and the increased Ca2+ in the mitochondrial matrix interacts with cyclophilin D to induce opening of the mitochondrial permeability transition pore, giving rise to release of the Cyt-c and cell apoptosis, while the extrinsic apoptotic pathway remains inactivated (Fig. 2). The hypertrophic heart in AAC rat model also exhibited an increased Cx43 phosphorylation at S282 and occurrence of cardiomyocyte apoptosis (Figs. 4, 5). The phosphor-S368 and -S262 were upregulated too in AAC heart (Fig. 5), in agreement with the Cx43 phosphorylation on several serine sites by hypertrophic stimuli [23, 33, 34]. Concerning the protective effect of enhanced S262 or S368 phosphorylation against myocardium apoptosis and injury [10, 11, 40], and the resistance to arrhythmia by phosphorylation on S325/328/330 [7], the increased Cx43-S282 phosphorylation, in another hand, likely contributes to the cardiomyocyte apoptosis in AAC heart, which is strongly supported by the spontaneous cardiomyocyte apoptosis because of exogenous expression of S282D gene in vivo and in vitro. Moreover, the totally different apoptotic pathway between S282D- and S282A-induced cardiomyocyte death demonstrates that the phosphorylation status, rather than the substituted residual in Cx43-S282, is associated with the regulation of cardiomyocyte survival. This proposal is further supported by the multiple lines of pharmacological evidence that the internal Ca2+ augmentation and cell hypertrophy induced by phenylephrine, a typical myocyte hypertrophic model, which is inhibited by 2-aminoethoxydiphenyl borate, a gap junction uncoupler, are correlated well with the increased and inhibited Cx43-S282 phosphorylation in cardiomyocytes [16, 41]. There are, however, several issues need to be further investigated in future, including how the increased Cx43-S282 phosphorylation enhances internal Ca2+ signal, and at which level of Cx43-S282 phosphorylation initiates cardiomyocyte apoptosis.
In conclusion, this study demonstrates that a sustained hyper Cx43 phosphorylation at S282 can increase internal Ca2+ concentration and trigger cardiomyocyte apoptosis through the activation of mitochondrial apoptotic pathway. This mode of cardiomyocyte death is likely involved in cardiac hypertrophy progress. In addition, this study also demonstrates additional data on an insight into a well-arranged Cx43-S282 phosphorylation for the maintenance of cardiomyocyte function and survival physiologically as well as pathologically, thus providing cardiac Cx43 as a crossroads between a variety of pathological stimuli and cell apoptosis.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81570206, 81970197 and 82170319), and the Scientific Research Key Program of Beijing Municipal Commission of Education (KZ201710025023).
Author contributions
ZPF and LLW performed validation assay involving animal models and cardiomyocytes; JYX, LEZ, CL, and HJY helped in data collection and analysis in Western blot assay; DLL designed project, wrote the manuscript, and supervised the whole project.
Competing interests
The authors declare no conflict of interest.
References
- 1.Solan JL, Lampe PD. Connexin 43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261–72. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bupha-Intr T, Haizlip KM, Janssen PM. Temporal changes in expression of connexin 43 after load-induced hypertrophy in vitro. Am J Physi Heart Circ Physiol. 2009;296:H806–14. doi: 10.1152/ajpheart.01058.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martins-Marques T, Catarino S, Gonalves A, Miranda-Silva D, Girao H. EHD1 modulates Cx43 gap junction remodeling associated with cardiac diseases. Circ Res. 2020;126:e97–e113. doi: 10.1161/CIRCRESAHA.119.316502. [DOI] [PubMed] [Google Scholar]
- 4.Falk MM, Bell CL, Kells Andrews RM, Murray SA. Molecular mechanisms regulating formation, trafficking and processing of annular gap junctions. BMC Cell Biol. 2016;17:22–45. doi: 10.1186/s12860-016-0087-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ai X, Yan J, Pogwizd SM. Serine-threonine protein phosphatase regulation of Cx43 dephosphorylation in arrhythmogenic disorders. Cell Signal. 2021;86:110070. doi: 10.1016/j.cellsig.2021.110070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xue J, Yan X, Yang Y, Chen M, Wu L, Gou Z, et al. Connexin 43 dephosphorylation contributes to arrhythmias and cardiomyocyte apoptosis in ischemia/reperfusion hearts. Basic Res Cardiol. 2019;114:40–56. doi: 10.1007/s00395-019-0748-8. [DOI] [PubMed] [Google Scholar]
- 7.Remo BF, Qu J, Volpicelli FM, Giovannone S, Shin D, Lader J, et al. Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ Res. 2011;108:1459–66. doi: 10.1161/CIRCRESAHA.111.244046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of Connexin 43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119:3435–42. doi: 10.1242/jcs.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation of Cx43 at S365 is a gatekeeper event that changes the structure of Cx43 and prevents downregulation by PKC. J Cell Biol. 2007;179:1301–9. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cotter ML, Boitano S, Lampe PD, Solan JL, Vagner J, Ek-Vitorin JF, et al. The lipidated connexin mimetic peptide srptekt-hdc is a potent inhibitor of Cx43 channels with specificity for the pS368 phospho-isoform. Am J Physiol Cell Physiol. 2019;317:C825–C42. doi: 10.1152/ajpcell.00160.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srisakuldee W, Makazan Z, Nickel BE, Zhang F, Thliveris JA, Pasumarthi KB, et al. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc Res. 2014;103:72–80. doi: 10.1093/cvr/cvu066. [DOI] [PubMed] [Google Scholar]
- 12.Crassous PA, Shu P, Huang C, Gordan R, Brouckaert P, Lampe PD, et al. Newly identified no-sensor guanylyl cyclase/connexin 43 association is involved in cardiac electrical function. J Am Heart Assoc. 2017;6:e006397. doi: 10.1161/JAHA.117.006397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lei Y, Peng X, Li T, Liu L, Yang G. Erk and miRNA-1 target Cx43 expression and phosphorylation to modulate the vascular protective effect of angiotensin II. Life Sci. 2019;216:59–66. doi: 10.1016/j.lfs.2018.11.019. [DOI] [PubMed] [Google Scholar]
- 14.Yang Y, Yan X, Xue J, Zheng Y, Chen M, Sun Z, et al. Connexin43 dephosphorylation at serine 282 is associated with connexin 43-mediated cardiomyocyte apoptosis. Cell Death Differ. 2019;26:1332–45. doi: 10.1038/s41418-019-0277-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun Z, Yang Y, Wu L, Talabieke S, You H, Zheng Y, et al. Connexin 43-serine 282 modulates serine 279 phosphorylation in cardiomyocytes. Biochem BiophysRes Commun. 2019;513:567–72. doi: 10.1016/j.bbrc.2019.04.032. [DOI] [PubMed] [Google Scholar]
- 16.Kang M, Lin N, Li C, Meng Q, Zheng Y, Yan X, et al. Cx43 phosphorylation on S279/282 and intercellular communication are regulated by IP3/IP3 receptor signaling. Cell Commun Signal. 2014;12:58–70. doi: 10.1186/s12964-014-0058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Z, Wang L, Han L, Wang Y, Zhou Y, Li Q, et al. Functional calsequestrin-1 is expressed in the heart and its deficiency is causally related to malignant hyperthermia-like arrhythmia. Circulation. 2021;144:788–804. doi: 10.1161/CIRCULATIONAHA.121.053255. [DOI] [PubMed] [Google Scholar]
- 18.Nakayama H, Bodi I, Correll RN, Chen X, Lorenz J, Houser SR, et al. Alpha1g-dependent t-type Ca2+ current antagonizes cardiac hypertrophy through a NOS3-dependent mechanism in mice. J Clin Invest. 2009;119:3787–96. doi: 10.1172/JCI39724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subedi KP, Son MJ, Chidipi B, Kim SW, Wang J, Kim KH, et al. Signaling pathway for endothelin-1- and phenylephrine-induced camp response element binding protein activation in rat ventricular myocytes: role of inositol 1,4,5-trisphosphate receptors and CaMKII. Cell Physiol Biochem. 2017;41:399–412. doi: 10.1159/000456422. [DOI] [PubMed] [Google Scholar]
- 20.Oldfield CJ, Duhamel TA, Dhalla NS. Mechanisms for the transition from physiological to pathological cardiac hypertrophy. Can J Physiol Pharmacol. 2020;98:74–84. doi: 10.1139/cjpp-2019-0566. [DOI] [PubMed] [Google Scholar]
- 21.Qu J, Volpicelli FM, Garcia LI, Sandeep N, Zhang J, Márquez-Rosado L, et al. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ Res. 2009;104:365–7. doi: 10.1161/CIRCRESAHA.108.184044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leithe E, Mesnil M, Aasen T. The connexin 43 C-terminus: a tail of many tales. Biochim Biophys Acta Biomembr. 2018;1860:48–64. doi: 10.1016/j.bbamem.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Jeyaraman MM, Srisakuldee W, Nickel BE, Kardami E. Connexin 43 phosphorylation and cytoprotection in the heart. Biochim Biophys Acta. 2012;1818:2009–13. doi: 10.1016/j.bbamem.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 24.Wang G, Bi Y, Liu X, Wei M, Zhang Q. Upregulation of connexin43 by glucose deprivation in H9C2 cells via the extracellular signal-regulated kinase/mitogen-activated protein kinase signaling pathway. Mol Med Rep. 2018;17:729–34. doi: 10.3892/mmr.2017.7967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugita J, Fujiu K, Nakayama Y, Matsubara T, Komuro I. Cardiac macrophages prevent sudden death during heart stress. Nat Commun. 2021;12:1910. doi: 10.1038/s41467-021-22178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fong JT, Nimlamool W, Falk MM. Egf induces efficient Cx43 gap junction endocytosis in mouse embryonic stem cell colonies via phosphorylation of Ser262, Ser279/282, and Ser368. FEBS Lett. 2014;588:836–44. doi: 10.1016/j.febslet.2014.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly JJ, Esseltine JL, Shao Q, Jabs EW, Sampson J, Auranen M, et al. Specific functional pathologies of Cx43 mutations associated with oculodentodigital dysplasia. Mol Biol Cell. 2016;27:2172–85. doi: 10.1091/mbc.E16-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Himelman E, Lillo MA, Nouet J, Gonzalez JP, Fraidenraich D. Prevention of connexin 43 remodeling protects against duchenne muscular dystrophy cardiomyopathy. J Clin Invest. 2020;130:1713–27. doi: 10.1172/JCI128190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of connexin 43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119:3435–42. doi: 10.1242/jcs.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol. 2007;179:1301–9. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang J, Hoagland D, Palatinus JA, He H, Iyyathurai J, Jourdan LJ, et al. Interaction of α carboxyl terminus 1 peptide with the connexin 43 carboxyl terminus preserves left ventricular function after ischemia-reperfusion injury. J Am Heart Assoc. 2019;8:e012385. doi: 10.1161/JAHA.119.012385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Procida K, Jørgensen L, Schmitt N, Delmar M, Taffet SM, Holstein-Rathlou NH, et al. Phosphorylation of connexin 43 on serine 306 regulates electrical coupling. Heart Rhythm. 2009;6:1632–8. doi: 10.1016/j.hrthm.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solan JL, Lampe PD. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014;588:1423–9. doi: 10.1016/j.febslet.2014.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solan JL, Márquez-Rosado L, Lampe PD. Cx43 phosphorylation-mediated effects on ERK and Akt protect against ischemia reperfusion injury and alter the stability of the stress-inducible protein NDRG1. J Biol Chem. 2019;294:11762–71. doi: 10.1074/jbc.RA119.009162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hood AR, Xun A, Pogwizd SM. Regulation of cardiac gap junctions by protein phosphatases. J Mol Cell Cardiol. 2017;107:52–7. doi: 10.1016/j.yjmcc.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schulz R, Görge PM, Görbe A, Ferdinandy P, Lampe PD, Leybaert L. Connexin 43 is an emerging therapeutic target in ischemia/reperfusion injury, cardioprotection and neuroprotection. Pharmacol Ther. 2015;153:90–106. doi: 10.1016/j.pharmthera.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang L, Deng J, Ma W, Qiao A, Xu S, Yu Y, et al. Ablation of lncrna miat attenuates pathological hypertrophy and heart failure. Theranostics. 2021;11:7995–8007. doi: 10.7150/thno.50990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiao L, Li M, Shao Y, Zhang Y, Gong M, Yang X, et al. Lncrna-zfas1 induces mitochondria-mediated apoptosis by causing cytosolic Ca2+ overload in myocardial infarction mice model. Cell Death Dis. 2019;10:942. doi: 10.1038/s41419-019-2136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–57. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmad Waza A, Andrabi K, Ul Hussain M. Adenosine-triphosphate-sensitive K+ channel (kir6.1): A novel phosphospecific interaction partner of connexin 43 (Cx43) Exp Cell Res. 2012;318:2559–66. doi: 10.1016/j.yexcr.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 41.Luo D, Lan X, Wang G, Wei S, Xiao RP, Han Q. Role of inositol 1,4,5-trisphosphate receptors in α1-adrenergic receptorinduced cardiomyocyte hypertrophy. Acta Pharmacol Sin. 2006;27:895–900. doi: 10.1111/j.1745-7254.2006.00382.x. [DOI] [PubMed] [Google Scholar]