

Abstract
A K-12 strain of Escherichia coli that overproduces methylenetetrahydrofolate reductase (MetF) has been constructed, and the enzyme has been purified to apparent homogeneity. A plasmid specifying MetF with six histidine residues added to the C terminus has been used to purify histidine-tagged MetF to homogeneity in a single step by affinity chromatography on nickel-agarose, yielding a preparation with specific activity comparable to that of the unmodified enzyme. The native protein comprises four identical 33-kDa subunits, each of which contains a molecule of noncovalently bound flavin adenine dinucleotide (FAD). No additional cofactors or metals have been detected. The purified enzyme catalyzes the reduction of methylenetetrahydrofolate to methyltetrahydrofolate, using NADH as the reductant. Kinetic parameters have been determined at 15°C and pH 7.2 in a stopped-flow spectrophotometer; the Km for NADH is 13 μM, the Km for CH2-H4folate is 0.8 μM, and the turnover number under Vmax conditions estimated for the reaction is 1,800 mol of NADH oxidized min−1 (mol of enzyme-bound FAD)−1. NADPH also serves as a reductant, but exhibits a much higher Km. MetF also catalyzes the oxidation of methyltetrahydrofolate to methylenetetrahydrofolate in the presence of menadione, which serves as an electron acceptor. The properties of MetF from E. coli differ from those of the ferredoxin-dependent methylenetetrahydrofolate reductase isolated from the homoacetogen Clostridium formicoaceticum and more closely resemble those of the NADH-dependent enzyme from Peptostreptococcus productus and the NADPH-dependent enzymes from eukaryotes.
In Escherichia coli, methylenetetrahydrofolate reductase (MetF) catalyzes the reduction of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate. This reaction commits tetrahydrofolate-bound one-carbon units to use in the methylation of homocysteine to form methionine, the terminal step in methionine biosynthesis. Hatch et al. (14) first identified the enzyme activity in crude extracts of E. coli. NADH was shown to be more effective than NADPH as the source of reducing equivalents in relatively crude preparations (4). During purification of methylenetetrahydrofolate reductase from cell extracts, the ability of the enzyme to be reduced by NADH was lost, necessitating assay of the enzyme in the presence of NADH, an NADH-flavine adenine dinucleotide (FAD) oxidoreductase, and FAD (17). Thus, it was believed that catalysis of the overall reaction shown in equation 1 required two enzymes: a methylenetetrahydrofolate reductase that catalyzed transfer of reducing equivalents from reduced FAD to CH2-H4folate, and an NADH-FAD oxidoreductase that catalyzed transfer of reducing equivalents from NADH to FAD.
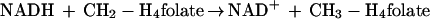 |
1 |
The metF gene specifies methylenetetrahydrofolate reductase. This gene is located in the metJBLF gene cluster, which was cloned and mapped by Zakin et al. (35). The metF gene was sequenced, and the gene product was shown to be a polypeptide of 33 kDa (24). However, we are unaware of publications reporting further characterization of the E. coli enzyme.
Methylenetetrahydrofolate reductase has previously been purified from porcine liver (7) and has been shown to contain noncovalently bound FAD and to use NADPH as a reductant. By using peptide sequences from the porcine enzyme to design oligomers, a clone for the human MTHFR gene was identified and sequenced, and the catalytic domain of the human enzyme was shown to exhibit extensive sequence similarity with MetF from E. coli (12). While two other bacterial methylenetetrahydrofolate reductases have been purified and characterized, their sequences have not been reported and they appear to differ appreciably from the human and E. coli enzymes. The enzyme from Clostridium formicoaceticum (5) is an iron-sulfur flavoprotein that catalyzes reduction of CH2-H4folate with reduced ferredoxin as an electron donor. Methylenetetrahydrofolate reductase from Peptostreptococcus productus more closely resembles the porcine and E. coli enzymes in that it lacks iron and catalyzes the reduction of methylenetetrahydrofolate with NADH as the electron donor (34). However, this enzyme appears to be associated with the cell membrane, in contrast to the E. coli and mammalian enzymes.
In this paper, we report the construction of E. coli strains for overproduction of methylenetetrahydrofolate reductase and the purification and characterization of the enzyme. The E. coli enzyme serves as a useful model for the human enzyme, mutations of which have been implicated in hyperhomocysteinemia in humans and in risk for the development of cardiovascular disease (10, 16) and neural tube defects (29). In work to be reported elsewhere, the X-ray structure of MetF from E. coli has been determined (13), and its availability will permit comparison with F420-dependent enzymes with similar functions in archaebacteria (22, 31). The ease of genetic manipulation of the E. coli metF gene will also permit further studies to define the functional properties of this important enzyme.
We have purified the MetF protein to homogeneity and shown it to be a flavoprotein capable of catalyzing the NADH-linked reduction of CH2-H4folate to CH3-H4folate without added enzymes or cofactors. The purified enzyme is a tetramer, but when diluted it readily loses its FAD cofactor, possibly accounting for the loss of catalytic function during earlier attempts at purification.
MATERIALS AND METHODS
Reagents.
Restriction enzymes and DNA-modifying enzymes were obtained from Promega, New England BioLabs, or Boehringer Mannheim. Taq polymerase and buffer were obtained from Boehringer Mannheim. Vent polymerase and buffer were purchased from New England BioLabs. Nonradioactive (6R,S)CH3-H4folate and H4folate were obtained from B. Schircks, Jona, Switzerland. NADH, menadione, FAD, and dimedone were purchased from Sigma. (6R,S)[methyl-14C]CH3-H4folate was purchased from Amersham Life Sciences as the barium salt. Luria broth was prepared as previously described (25). Formaldehyde, formic acid, hydrochloric acid, methanol, ascorbic acid, acrylamide, ampicillin, riboflavin, IPTG (isopropyl-β-d-thiogalactopyranoside), EDTA, phosphate, sodium chloride, glycerol, bovine serum albumin, nickel sulfate, imidazole, phosphate, NaCl, glycerol, and agarose were supplied by various commercial sources and were used without further purification. HiTrap chelating columns were obtained from Pharmacia, Piscataway, N.J. The DNA primers used for isolation of the metF gene were synthesized at the University of Michigan DNA Synthesis Core Facility.
Assays. (i) CH3-H4folate-menadione oxidoreductase assay.
In the presence of a high potential electron acceptor like menadione, methylenetetrahydrofolate reductase oxidizes [methyl-14C]CH3-H4folate to 14CH2O (formaldehyde) and H4folate (8, 19). Radiolabeled formaldehyde was complexed with dimedone and extracted with toluene. Formation of the formaldehyde product was measured by scintillation counting.
The 800-μl reaction mixture contained 60 μl of the radiolabeled (6R,S)[methyl-14C]CH3-H4folate cocktail (4.17 mM, 1,200 dpm/nmol), 100 μl of 1 M phosphate buffer (pH 6.3), 60 μl of 0.5 mM FAD, 200 μl of menadione stock, and 100 μl of 2% bovine serum albumin in 30 mM EDTA. This mixture was preincubated at 37°C for 5 min, and then enzyme was added to initiate the reaction. After 30 s, the assay was terminated by the addition of 300 μl of dimedone and heat treatment at 98°C for 2 min. The reaction mixtures were cooled on ice for 2 min, and 3 ml of toluene was added. The mixture was vortexed and spun in a clinical centrifuge at 4,000 rpm for 5 min. A 1-ml volume of the toluene phase was added to 10 ml of Eco-lite scintillation cocktail (ICN Biomedicals Inc.) and counted in a Beckman scintillation counter. Units for this assay are defined as micromoles of formaldehyde formed per minute.
The radioactive methyltetrahydrofolate cocktail for this assay was prepared by adding 50 μCi of 5-[14C](6R,S)methyltetrahydrofolic acid (specific activity, 54 mCi/mmol) to 5 ml of 8 mM ascorbic acid and combining the mixture with 19 ml of ascorbate buffer containing 60.1 mg of unlabeled (6R,S)CH3-H4folate. A menadione stock solution was prepared by diluting 1 volume of a saturated solution of menadione in methanol with 4 volumes of water. The precipitate that formed was removed by filtering. The solution was stirred in the dark for 10 min and then refiltered and stored in the dark. The concentration of menadione in this stock solution was found to be 1.4 mM by measuring A340 and using a molar absorbance coefficient of 3,100 M−1 cm−1 (30).
(ii) NAD(P)H-menadione oxidoreductase assay.
The NAD(P)H-menadione oxidoreductase activity was measured at 25°C, using the method of Donaldson and Keresztesy (8) as modified by Matthews (19). In a total volume of 3 ml, the reaction mixture consisted of 1.5 ml of 100 mM potassium phosphate buffer (pH 7.2) with 0.6 mM EDTA, 300 μl of menadione stock (prepared as described above), and 125 μM NADH. The mixture was incubated at 25°C for 5 min, and then enzyme was added to initiate the reaction. Activity was monitored as a decrease in the absorbance of NADH at 343 nm, where menadione and menadiol are isosbestic. Activities are presented as the initial rate of NADH oxidation observed, using an extinction coefficient of 6,220 M−1 cm−1 for NADH at 343 nm. Activity units represent micromoles of NADH oxidized per minute.
(iii) NADH-CH2-H4folate oxidoreductase assay.
In the NADH-CH2-H4folate oxidoreductase assay, MetF oxidizes NADH and reduces CH2-H4folate to CH3-H4folate. The assay is a modification of the NADPH-CH2-H4folate oxidoreductase assay originated by Kutzbach and Stokstad (18) and described by Matthews (19). In our modification, the assay was performed in a stopped-flow spectrophotometer at 15°C. MetF (20 μM in enzyme-bound flavin) was mixed with an equal volume of buffer containing NADH and CH2-H4folate. Cycles of alternating vacuum and argon gas flow over the samples prior to mixing were used to deaerate both solutions.
To determine the Km and KiA for NADH, deaerated 20 μM enzyme was mixed anaerobically with deaerated solutions containing 60 μM (6R)CH2-H4folate and concentrations of NADH varying from 10 to 400 μM. Enzyme activity was measured as a decrease in the FAD absorbance of the enzyme by monitoring the enzyme absorbance at 447 nm. The buffer for both the enzyme and substrate solutions was 50 mM potassium phosphate (pH 7.2) (KPi) buffer containing 10% glycerol and 0.3 mM EDTA. The CH2-H4folate was added as a 6R,S mixture and was prepared anaerobically by adding 5 ml deaerated 50 mM KPi buffer, containing 10% glycerol and 0.3 mM EDTA, to 27.7 mg of H4folate under nitrogen. Reagent formaldehyde (81 μl of a 14.26 M stock) was added to generate 10 mM (6R,S)CH2-H4folate.
The Km and KiB for CH2-H4folate were also determined by the NADH CH2-H4folate oxidoreductase assay. Deaerated 20 μM enzyme was mixed anaerobically with deaerated solutions containing 200 μM NADH and concentrations of (6R)CH2-H4folate varying from 30 to 2,000 μM. The oxidation of NADH was observed as a decrease in the absorbance at 340 nm.
Subcloning of metF.
Plasmid pEJ3-1B contains the metF gene, including the 5′ upstream region regulated by an S-adenosylmethionine (AdoMet)-MetJ complex, half of Tn5 including the gene coding for neomycin phosphotransferase, and 1.5 kb of additional bacterial DNA 3′ to E. coli metF (9). To overproduce the desired protein, extraneous material from the pEJ3-1B clone was removed and the coding region of metF was subcloned into the EcoRI site of plasmid pTrc99A (Pharmacia) to generate the overexpression plasmid pCAS-5. The metF coding region in pEJ3-1B was amplified by PCR using pEJ3-1B as a template and primers 144C and 145C*, where the asterisk denotes a sequence corresponding to the reverse complement of the sense strand. Primers 144C (5′-ggaactgcagATTGATGAGGTAAGGTATGA--3′) and 145C* (5′-ggaagacgtcTTATAAACCAGGTCGAACCCC-3′) were generated to anneal to the 5′ and 3′ ends, respectively, of the E. coli metF coding sequence. The primers were designed to include the metF ribosome binding site, GAGG, the ATG translation initiation codon at the 5′ end of the gene, and a sequence complementary to the TAA stop codon, TTA, at the 3′ end of the gene. In addition, primers 144C and 145C* contain 10 bp of DNA sequence (lowercase) that are not contained within the metF gene but appear in the final construct at the 5′ and 3′ ends of the metF gene.
The 927-bp PCR product was composed of 888 bp comprising the coding region of metF, the 3-bp termination codon, 16 bp of 5′ untranslated region that contained the ribosome binding site, and 20 bp introduced by the primers that were not part of metF. Ligation of the PCR product into the Invitrogen TA cloning vector pCRII generated plasmid pCAS-3. This plasmid was then digested with the restriction enzyme EcoRI to generate a 943-bp fragment. The EcoRI site was contained within plasmid pCRII, adding 16 bp from this plasmid to the 927-bp PCR fragment containing the metF gene. This 943-bp fragment was ligated into the EcoRI site of dephosphorylated expression vector pTrc99A (Pharmacia) to generate plasmid pCAS-5.
Purification of wild-type MetF.
E. coli AB1909 (metF arg lac), which contains a mutation in metF that leads to methionine auxotrophy (32), was transformed with pCAS-5 to generate the overexpression strain CAS-9. Cells of strain CAS-9 were inoculated in LB to an optical density at 420 nm (OD420) of 0.05, grown aerobically in a rotary action shaker at 37°C to an OD420 of 1.0, and then induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and allowed to continue growing for 3 h. The cells were pelleted and resuspended in 50 mM KPi buffer (pH 7.2) containing 10% glycerol and 0.3 mM EDTA. The protease inhibitors phenylmethylsulfonyl fluoride and tosyl-l-lysine chloromethylketone were added to final concentrations of 50 μM and 100 μM, respectively, prior to sonication. The cells were sonicated with a Branson sonifier at a setting of 8. To prevent overheating, seven 1-min sonication cycles were alternated with 2-min recovery periods. The sonicate was centrifuged at 35,000 × g for 1 h at 4°C, and the supernatant was collected.
Purification of MetF to homogeneity required a three-column purification process involving anion-exchange and hydrophobic interaction chromatography. It is similar to the procedure developed by Wohlfarth et al. (34) for the purification of methylenetetrahydrofolate reductase from P. productus. DEAE fast-flow Sepharose resin was used for the first separation. The sonicate supernatant was applied to a 2- by 50-cm DEAE column equilibrated with 50 mM KPi containing 10% glycerol and 0.3 mM EDTA. The column was washed with 50 mM KPi–10% glycerol–0.3 mM EDTA and eluted with a linear gradient from 50 to 500 mM KPi. The fraction containing MetF can be identified by absorbance at 447 nm; if sufficient enzyme is present, MetF can be visually identified by its intense yellow color. As this step is a bulk purification to remove DNA, RNA, and cellular debris, the yellow fractions were combined without regard to enzyme purity. For buffer exchange of the pooled protein fractions, an Amicon apparatus with a YM100 membrane was used. Care was taken to prevent dilution of the protein below 100 μM, which results in FAD dissociation and protein precipitation. Concentrating the protein solution and then adding buffer to restore the volume prior to concentration accomplished buffer exchange. The procedure was repeated until the protein was in 50 mM KPi (pH 7.2) with 10% glycerol and 0.3 mM EDTA.
The second purification step used fast protein liquid chromatography (FPLC) with a Pharmacia Mono Q anion-exchange resin. A Mono Q HR 16/10 column (20-ml bed volume) was used, and MetF was eluted with a salt gradient from 50 to 375 mM KPi with 10% glycerol and 0.3 mM EDTA. MetF elutes at approximately 300 mM KPi. The cleanest fractions, as determined by electrophoresis on sodium dodecyl sulfate (SDS)-polyacrylamide gels, were combined, and 1 volume of 1.6 M (NH4)2SO4 was added so that the final buffer composition of the protein was approximately 0.8 M (NH4)2SO4–100 mM KPi (pH 7.2) with 5% glycerol and 0.15 mM EDTA. For the third purification step, the protein was then loaded onto a 2- by 10-cm Phenyl-sepharose HP FPLC column (Pharmacia) preequilibrated with 0.8 M (NH4)2SO4 in 100 mM KPi (pH 7.2) containing 10% glycerol and 0.3 mM EDTA. A reverse gradient from 0.8 to 0.0 M (NH4)2SO4 in the same buffer was used to elute the enzyme. Pure E. coli MetF eluted at the very end of the gradient. The protein was exchanged into 50 mM KPi (pH 7.2) with 10% glycerol and 0.3 mM EDTA by size exclusion chromatography and stored at −80°C at a minimum concentration of 150 μM to ensure long-term protein stability.
Protein concentration determinations.
Protein determinations were performed by a Coomassie blue dye-binding method (3) implemented via the Bio-Rad protein assay (Hercules, Calif.) and were compared with standard curves obtained with bovine serum albumin. Determination of the protein concentration of purified MetF by amino acid analysis gave a value 65.2% of that obtained by the Bio-Rad protein assay. Therefore, a factor of 0.652 was used to correct values obtained by the Bio-Rad protein assay for samples of enzyme with specific activities of >2.4 μmol min−1 mg−1 in the CH3-H4folate-menadione oxidoreductase assay. Enzyme concentrations were routinely determined from the visible absorbance at 447 nm due to bound FAD, using a molar absorbance coefficient of 14,300 M−1 cm−1 (see Results).
Amino acid analysis.
Analyses were performed at the University of Michigan Protein and Carbohydrate Core Facility, using an Applied Biosystems model 420H automated amino acid analyzer, with precolumn derivatization of the protein hydrolysate with phenylisothiocyanate.
Construction of a strain producing histidine-tagged MetF.
The PCR-based primer overlap extension method (15) was used to produce a plasmid for production of histidine-tagged MetF. Generation of the histidine-tagged version of MetF required the removal of the stop codon from the metF gene and insertion of the coding sequence for the gene into the pET-23b vector (Novagen, Milwaukee, Wis.), which contains six histidine codons and a termination codon downstream of and in frame with a 6-bp XhoI site. Primers 144C (see above), 3311E* (5′-cccagtgctgcaatgataccg-3′), 3312E* (5′-gcttgcatgcctgcactcGAGTAAACCAGGTCGAAC-3′), and 3313E (5′-GTTCGACCTGGTTTACTCgagtgcaggcatgcaagc-3′) were used to generate plasmid pCAS-28 containing the wild-type coding sequence with the original stop codon removed. The asterisks indicate sequences corresponding to the reverse complement of the sense strand. Sequences contained within the coding sequence are shown in uppercase, while those from flanking regions are shown in lowercase; underlined sequences indicate changes made to introduce an XhoI site in place of the original stop codon. Separate amplification reactions were performed with the wild-type expression plasmid pCAS-5 as the DNA template and either 144C and 3312E* or 3313E and 3311E* as primers. The two amplified DNA fragments were then combined and amplified by using 144C and 3311E* as primers. The resultant 2,245-bp PCR product was digested with BglI and BspEI to generate a 1,696-bp fragment, containing the entire E. coli metF gene with a XhoI site instead of a stop codon at the 3′ end of the gene. This fragment was reintroduced between the BglI and BspEI sites in pCAS-5, replacing the original metF sequence with one that contained an XhoI restriction site at the carboxy terminus and no stop codon. The resultant plasmid, pCAS-28, was digested with XbaI and XhoI to generate a 922-bp fragment containing the full-length mutated gene. Plasmid pET-23b was digested with XbaI and XhoI, which removed a 117-bp fragment from the plasmid that specifies an unwanted amino-terminal T7 tag fusion protein, the vector-encoded ribosome binding site, and part of the multicloning region. The 922-bp XbaI-XhoI gene fragment was ligated into the 3,547-bp XbaI-XhoI fragment of pET-23b to generate the expression vector pCAS-30. This plasmid contains the metF coding sequence juxtaposed to a C-terminal histidine tag. The resulting construct specifies a protein that contains two mutated residues (Leu-Glu) at the C terminus of the original sequence, introduced by formation of the XhoI site, followed by six histidine residues.
Production of histidine-tagged MetF.
The histidine-tagged protein was produced by using E. coli EET01, which contains plasmid pCAS-30 in a BL21(DE3)recD+ (Novagen) background. Use of the BL21(DE3)recD strain results in substantially lower levels of accumulation. The BL21(DE3) background contains a chromosomal copy of the T7 RNA polymerase under control of the lacUV5 promoter. T7 RNA polymerase was required for production of the histidine-tagged proteins. Although polymerase production is induced by IPTG in this strain, we did not observe increased accumulation of MetF on addition of IPTG, and IPTG was not added to any of our cultures.
One liter of Luria broth, supplemented with ampicillin (100 μg/ml) and riboflavin (10 μM), was inoculated to an OD600 of 0.015 with strain EET01. The cells were allowed to grow in a rotary action shaker at 37°C until they reached stationary phase (OD600 of ∼5), after approximately 9 h. The cells were pelleted at 11,000 × g for 10 min, and the supernatant was removed.
Purification of histidine-tagged methylenetetrahydrofolate reductase.
Pelleted cells producing histidine-tagged MetF were resuspended by adding 50 μl of ice-cold buffer (10 mM imidazole in 20 mM phosphate buffer [pH 7.4] containing 500 mM NaCl) per ml of cell culture. The cell suspensions were then sonicated in the presence of the protease inhibitors phenylmethylsulfonyl fluoride and tosyl-l-lysine chloromethyl ketone as described above. The supernatant was separated from cell debris by centrifugation at 35,000 × g for 1 h at 4°C. Purification of the histidine-tagged protein was accomplished by using a nickel affinity column. The nickel column was generated by charging the HiTrap chelating column (Pharmacia) with Ni2+, using nickel sulfate. Using a peristaltic P-1 pump (Pharmacia), the sonicate supernatant was loaded onto a 5-ml nickel column previously equilibrated with 50 ml of 10 mM imidazole in 20 mM phosphate (pH 7.4) with 500 mM NaCl. The histidine-tagged protein bound to the nickel column along with some nonspecifically bound protein. Nonspecifically bound proteins were eluted by washing with 50 ml of loading buffer followed by 50 ml of the same buffer containing 100 mM imidazole. Homogeneous histidine-tagged E. coli MetF was then eluted with the same buffer containing 300 mM imidazole. Fractions (400 μl) were collected, and glycerol was added to 10% (wt/vol). Approximately 4 μg of protein from each fraction was run on an SDS-polyacrylamide gel to identify pure fractions. The pure fractions were pooled, and the buffer was exchanged to 50 mM KPi, (pH 7.2) with 10% glycerol and 0.3 mM EDTA, using size exclusion chromatography on an FPLC Superose-12 column. The protein was stored at −80°C at a concentration of >100 μM.
Determination of the molar absorbance of enzyme-bound FAD.
Enzyme-bound flavin was released by denaturing the enzyme by addition of an equal volume of 7.5 M guanidine HCl in 1 M Tris-HCl at pH 7.2. The absorbance of the liberated FAD was compared to the absorbance of a known quantity of free FAD. The molar absorbance coefficient of FAD in aqueous solution at neutral pH is 11,300 M−1 cm−1 at 450 nm (1); the molar absorbance increases 4.85% on dilution with guanidine hydrochloride (6), yielding a molar absorbance coefficient of 11,850 M−1 cm−1.
Apparent mass of the native enzyme.
The oligomeric state of the MetF protein was determined by size exclusion chromatography using an FPLC Superose-12 HR 10/30 gel filtration column (Pharmacia). Concentrated protein was diluted to 61 μM in 50 mM KPi buffer (pH 7.2) with 10% glycerol and 0.3 mM EDTA. The protein was applied to the column and eluted with 50 mM KPi (pH 7.2) with 10% glycerol and 0.3 mM EDTA at 0.5 ml/min. The migration and elution of the protein peak was ascertained by continuous monitoring of the column eluant at 276 nm. The elution volume of the protein was compared to the elution volumes of gel filtration protein standards (Bio-Rad) dissolved in the same buffer. The elution volumes of the standards were used to generate a calibration curve for the column, and the native molecular weight for the MetF samples was calculated using this calibration curve. The subunit molecular weight of histidine-tagged MetF was determined by electrospray mass spectrometry at the University of Michigan Protein and Carbohydrate Core Facility.
RESULTS
Subcloning of metF and construction of an overproducing strain.
Plasmid pEJ3-1B, containing the E. coli metF gene specifying methylenetetrahydrofolate reductase, was obtained as a generous gift from James Johnson (9). This plasmid also contains the 5′ upstream region regulated by an AdoMet-MetJ complex, half of Tn5 including the gene coding for neomycin phosphotransferase, and 1.5 kb of additional bacterial DNA 3′ of E. coli metF. To overproduce MetF, it was desirable to remove the regulatory region, so that cells could be grown in LB medium without down-regulation of metF expression. Extraneous material from the pEJ3-1B clone was removed as described in Materials and Methods, and the coding region of metF was subcloned into pTrc99A to generate the overexpression plasmid pCAS-5. This plasmid contains a 943-bp DNA fragment of which 907 are bp 4151 to 5057 of metF (numbered according to GenBank accession no. AE000468) cloned into the EcoRI site of pTrc99A. Plasmid pCAS-5 was introduced into E. coli AB1909 (metF) to create strain CAS-9. Following growth in LB containing IPTG, approximately 20% of the total cellular protein is E. coli MetF. Thus, removing the regulatory region and placing protein production under the control of the trc promoter leads to high levels of MetF accumulation.
Purification of MetF to homogeneity.
Wild-type E. coli MetF enzyme was produced from strain CAS-9 as described in Materials and Methods. Purification of MetF to homogeneity requires a three-column purification process involving anion-exchange and hydrophobic interaction chromatography (see Materials and Methods for details). Table 1 summarizes the purification and reports specific activities measured with the CH3-H4folate-menadione oxidoreductase assay. Precautions are taken such that the enzyme is not diluted to below 100 μM to eliminate dilution-related inactivation of the protein. In particular, size exclusion chromatography was used for buffer exchange prior to storage of homogeneous enzyme. The protein was stored at −80°C in a solution of 50 mM KPi (pH 7.2) with 10% glycerol and 0.3 mM EDTA at a minimum concentration of 150 μM to ensure long-term protein stability. Protein is stable at this concentration for up to a year, provided that it does not undergo repeated freezing and thawing.
TABLE 1.
Purification of wild-type MetF from E. colia
| Purification step | Vol (ml) | Proteinb (mg) | Total activityc (U) | Sp act (U/mg) | Fold purifi-cation | Yield (%) |
|---|---|---|---|---|---|---|
| Sonication supernatant after centrifugation | 107 | 324 | 194 | 0.60 | 1.0 | 100 |
| DEAE-Sepharose chromatography | 24 | 159 | 177 | 1.11 | 1.9 | 91 |
| FPLC on Mono Q resin | 7.0 | 12.2d | 43 | 3.52 | 5.9 | 22 |
| Chromatography on phenyl-Sepharose | 9.3 | 4.0d | 18.7 | 4.67 | 7.8 | 9.6 |
Yields are reported for 1 liter of cultured cells.
Determined by the Bio-Rad protein assay (3).
Determined by using the CH3-H4folate-menadione oxidoreductase assay and 5-min incubations. We subsequently realized that the assay was not linear for 5 min, and activity values have been multiplied by 4.79, which is the ratio of activity measured in 30-s versus 5-min assays. Units are micromoles of CH3-H4folate oxidized per minute at 37°C.
Protein concentration measured by the Bio-Rad protein assay was corrected by using a correction factor of 0.652 (see Materials and Methods).
Properties of purified methylenetetrahydrofolate reductase.
The wild-type MetF protein is isolated as a flavoprotein as indicated by its yellow color. The flavin is nonfluorescent when bound to MetF apoenzyme but becomes fluorescent when released from the protein, e.g., by thermal denaturation. Since FAD is much less fluorescent than flavin mononucleotide (FMN), the two forms of flavin can be distinguished by treatment of the released flavin with snake venom phosphodiesterase, which converts FAD to FMN with an accompanying 10-fold increase in fluorescence (2). In this manner, the flavin bound to MetF was shown to be FAD, as suggested from earlier studies (14, 17). The molar absorbance of MetF was determined by comparing the absorbances of the enzyme before and after the release of FAD. FAD was released by denaturing the enzyme with guanidine hydrochloride and comparison of the absorbance of the liberated FAD with that of a known quantity of free FAD (1). The molar extinction coefficient was determined to be 14,300 M−1 cm−1 for the enzyme-bound FAD. The enzyme concentration that was determined from the absorbance at 447 nm due to enzyme-bound FAD was 1.06 times the subunit concentration determined by amino acid analysis, indicating that within experimental error, the enzyme is isolated with one equivalent of FAD bound per subunit.
The enzyme was found to lose activity upon dilution. As is evident from the data in Table 1, significant losses were observed during purification, especially during chromatographic procedures that resulted in dilution of the enzyme. We therefore decided to construct a modified MetF with a histidine tag at the C terminus, so that the protein could be purified by chromatography on nickel agarose. Strain BL21(DE3) was transformed with pCAS-30 (see Materials and Methods) and used to produce the histidine-tagged MetF protein. A summary of the purification is shown in Table 2. The protein could be purified to homogeneity in a single step with 96% yield. The specific activity of the purified protein (5.37 U/mg) is comparable to that of the wild-type protein (4.67 U/mg), but 1 liter of cell culture yielded 58 mg of purified MetF, compared to 4 mg obtained per liter of cells producing wild-type MetF.
TABLE 2.
Purification of histidine-tagged MetF from E. colia
| Purification step | Vol (ml) | Proteinb (mg) | Total activityc (U) | Sp act (U/mg) | Fold purifi-cation | Yield (%) |
|---|---|---|---|---|---|---|
| Sonication supernatant after centrifugation | 52 | 1,300 | 629 | 0.48 | 1.0 | 100 |
| Chromatography on nickel affinity column | 9 | 113d | 607 | 5.37 | 11.0 | 96 |
Two liters of cultured cells was used for this preparation.
Determined by the Bio-Rad protein assay (3).
Determined by the CH3-H4folate-menadione oxidoreductase assay using 30 s incubations. Units are micromoles of CH3-H4folate oxidized per minute at 37°C.
Protein concentration determined by the Bio-Rad protein assay (3) was corrected by using a correction factor of 0.652 (see Materials and Methods).
Histidine-tagged MetF protein has a predicted subunit molecular weight of 34,062, calculated assuming that the N-terminal methionine is removed. Determination of a peptide mass of 34,072 ± 10 by electrospray mass spectrometry was consistent with removal of the initiator methionine residue. The oligomeric state of the histidine-tagged MetF protein was determined by size exclusion chromatography as described in Materials and Methods. The elution volume of MetF is compared with those of standard proteins in Fig. 1. MetF applied to the column at an initial concentration of 61 μM enzyme-bound FAD eluted at a volume equivalent to a molecular mass of 119 ± 2 kDa, or 3.5 subunits of 34 kDa per molecule, suggesting that the protein is a tetramer. The recent determination of the structure of E. coli MetF by X-ray crystallography confirms the tetrameric structure of the native wild-type (non-histidine-tagged) enzyme (13).
FIG. 1.

Determination of the apparent molecular weight of histidine-tagged MetF by gel filtration. The oligomeric state of MetF was determined by size exclusion FPLC on a Superose-12 HR 10/30 gel filtration column. Concentrated protein was diluted to 61 μM in 50 mM KPi (pH 7.2) containing 10% glycerol and 0.3 mM EDTA, loaded on the column, and eluted with the same buffer. The elution volume is compared with those of compounds in the Bio-Rad gel filtration standard kit. These compounds and their molecular weights are as follows (unfilled circles, from left to right): bovine thyroglobulin, 670,000; bovine gamma globulin, 158,000; chicken ovalbumin, 44,000; horse myoglobin, 17,000; and vitamin B12, 1,350. The results of duplicate determinations of the standard curve are indicated. The duplicate determinations of MetF (filled circle) yielded identical elution volumes corresponding to an apparent molecular weight of 119,400. This molecular weight is consistent with the concentrated native enzyme eluting as a tetramer of expected Mr of 136,248 (4 × 34,062).
Histidine tagging did not affect any of the enzyme properties that we have examined. The unmodified enzyme also migrates as a tetramer during size exclusion chromatography. Differential scanning calorimetry indicated similar melting temperatures for the wild-type (58.6°C) and histidine-tagged (58.4°C) proteins. Thus, the introduction of additional residues at the C terminus of the protein does not appear to have altered the physical properties of the enzyme.
Catalytic properties of methylenetetrahydrofolate reductase.
Figure 2 shows the absorbance spectrum of the purified wild-type enzyme. This spectrum, with maxima at 447 and 380 nm, is characteristic of a flavoprotein. On addition of NADH, the spectrum is converted to a featureless curve lacking discrete bands above 400 nm; this spectrum is characteristic of a reduced flavoprotein, indicating that NADH is able to reduce the flavin of MetF. The initial rate of reduction of MetF by various concentrations of NADH was monitored in a stopped-flow spectrophotometer at 15°C. The results are shown in Fig. 3. A saturable hyperbolic binding curve for NADH is observed, with an apparent Kd of 37 μM and a maximum observed rate of 34 s−1, or ∼2,000 min−1.
FIG. 2.

Overlay of oxidized and NADH-reduced MetF spectra. Purified enzyme, 22 μM in potassium phosphate buffer (pH 7.2) containing 10% glycerol and 0.3 mM EDTA, was equilibrated with argon, and the spectrum was recorded. The absorbance shown (solid line) has been corrected for the subsequent twofold dilution of the enzyme and is equivalent to that of 11 μM enzyme. The enzyme was mixed with an equal volume of 200 μM NADH in the same buffer, and the spectrum of the reduced enzyme (dashed line) was recorded.
FIG. 3.

Reaction of MetF with NADH at 15°C. Under anaerobic conditions, 20 μM oxidized enzyme in potassium phosphate buffer (pH 7.2) containing 10% glycerol and 0.3 mM EDTA was mixed with various concentrations of NADH dissolved in the same buffer. The reaction was monitored at 447 nm, where the absorbance of the oxidized enzyme-bound FAD is maximal, and initial rates of reduction were determined for each concentration. The corresponding rate constants (kobx) are plotted against the NADH concentration. The data were fit to a hyperbolic equation (solid line) and yielded a Kd of 37 μM for NADH and a maximal value for kobs of 34 s−1.
The physiological reaction catalyzed by MetF is the reduction of CH2-H4folate to form CH3-H4folate. We monitored the physiological reaction at 15°C in a stopped-flow spectrophotometer by mixing enzyme with CH2-H4folate and NADH. The enzyme and substrate solutions were equilibrated with argon prior to mixing, and the reaction was conducted under rigorously anaerobic conditions. Details of the procedure are presented in Materials and Methods. Enzyme, 20 μM in bound FAD, was mixed with an equal volume of buffer containing 60 μM (6R)CH2-H4folate and 10 to 400 μM NADH, and the initial rate of flavin reduction was monitored at 447 nm. The measured initial velocity is plotted against the NADH concentration. The inset to Fig. 4 shows the initial rate of NADH oxidation measured at 340 nm when 20 μM enzyme is mixed with an equal volume of buffer containing 200 μM NADH and 30 to 2000 μM (6R)CH2-H4folate. Marked inhibition of the reaction is seen at high levels of CH2-H4folate, as is characteristic of enzymes that follow ping-pong kinetic mechanisms (11). The Vmax for this reaction must be corrected for the observed excess substrate inhibition; equation 2 has been used to determine the Km values for NADH (A) and for CH2-H4folate (B), the KiA and KiB values for substrate inhibition by NADH and CH2-H4folate respectively, and Vmax (26) by successive iterations using Kaleidograph (Abelbeck Software, Reading, Pa.) and comparison of the predicted curves with the data shown in the graph and inset.
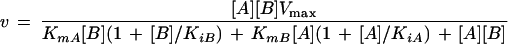 |
(2)
FIG. 4.

Measurement of turnover in the NADH-CH2-H4folate oxidoreductase reaction at 15°C. Under anaerobic conditions in a stopped-flow spectrophotometer, enzyme, 20 μM in 50 mM potassium phosphate buffer (pH 7.2) containing 10% glycerol and 0.3 mM EDTA, was mixed with an equal volume of the same buffer containing 30 μM (6R)CH2-H4folate and various concentrations of NADH. The initial velocity, measured as the rate of reduction of the enzyme at 447 nm, is plotted against the NADH concentration after mixing. The inset shows a plot of the initial velocity, measured as the rate of formation of NADH at 340 nm, determined when 20 μM enzyme was mixed with an equal volume of buffer containing 200 μM NADH and various concentrations of CH2-H4folate [added as a (6R,S)CH2-H4folate mixture]. Marked excess substrate inhibition is evident. Using equation 2, the solid-line fits to the data were determined, yielding the following values for kinetic parameters: KmA (NADH), 13 ± 2 μM; KiA, 9 ± 2 μM; KmB [(6R)CH2-H4folate], 0.8 ± 0.2 μM; KiB, 7.1 ± 1.4 μM; and Vmax/ET, 30 ± 3 s−1.
Using this equation, Vmax/ET was calculated to be 30 ± 3 s−1, KmA was 13 ± 2 μM, KiA was 9 ± 2 μM, KmB was 0.8 ± 0.2 μM, and KiB was 7.1 ± 1.4 μM. A turnover number of 1,800 min−1 can be calculated for this reaction under Vmax conditions at 15°C.
An unexpected feature of the reaction catalyzed by MetF is that the initial rate of reduction of the enzyme-bound flavin, which takes place during the approach to steady state, is the same as the observed rate of NADH oxidation during the steady state. Note that in Fig. 4, the initial rate of NADH oxidation in the presence of 30 μM CH2-H4folate is ∼160 μM s−1 (v/ET is 16 s−1) whether the reaction is monitored at 340 nm, where NADH absorbs (inset), or at 447 nm, where FAD absorbs (main graph). This agreement is seen only when the overall rate of reaction is limited by the rate of reduction of the flavin, such that kred << kox as defined by equations 3 and 4 describing the reductive and oxidative half reactions:
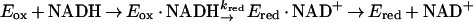 |
3 |
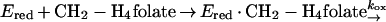 |
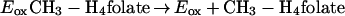 |
4 |
In agreement with this conclusion, the turnover number, estimated to be ∼1,800 min−1 after correction for excess substrate inhibition, is only slightly lower than the rate constant for reduction of the enzyme when NADH is saturating, which is 2,000 min−1 as deduced from the data in Fig. 3.
In addition to the physiological reaction, methylenetetrahydrofolate reductase also catalyzes transfer of reducing equivalents from reduced pyridine nucleotides to menadione, an artificial electron acceptor, as shown in equation 5:
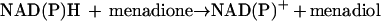 |
5 |
The Km for NADH was determined to be 30 μM in this assay at 25°C, while the Km for NADPH was >300 μM under the same conditions. Thus, NADH rather than NADPH appears to be the physiological source of reducing equivalents for the enzyme-bound flavin of MetF, as previously deduced from studies with crude extracts (14).
Neither the physiological reaction nor the NADH-menadione oxidoreductase reaction is suitable for assays of crude extracts; a CH3-H4folate-menadione oxidoreductase assay is usually used for these purposes (18, 19). When this reaction is measured at 37°C, a Km for (6S)CH3-H4folate of 75 μM is observed, and the turnover number under Vmax conditions is 190 min−1. Values approaching this turnover number are obtained only with assay incubation times of 30 s, since plots of enzyme activity versus time are linear for only 1 min.
The specific activities for the purified wild-type and histidine-tagged proteins were identical within experimental error for the NADH-menadione oxidoreductase assay at 25°C and the CH3-H4folate-menadione oxidoreductase assay at 37°C.
DISCUSSION
Properties of our enzyme preparations compared to those for enzymes from other prokaryotes.
Methylenetetrahydrofolate reductase from E. coli differs significantly from the enzymes with similar function previously purified from prokaryotes. The enzyme from C. formicoaceticum has been purified to homogeneity (5). This enzyme is an α4β4 octamer containing 26- and 35-kDa subunits. It contains 15 irons and 20 acid-labile sulfurs per octamer of 237,000, and the spectrum is characteristic of a flavoprotein containing iron-sulfur centers. The enzyme also contains 1.7 FAD per octamer and 2.3 zinc. The enzyme-bound FAD is not reduced by NADH or NADPH, but it catalyzes the reduction of CH2-H4folate with reduced ferredoxin as an electron donor.
Wohlfarth et al. (34) have purified methylenetetrahydrofolate reductase from P. productus to homogeneity. This enzyme is an α8 octamer of 32-kDa subunits and contains 4 FAD per octamer. About 40% of the enzyme is recovered in the particulate fraction, suggesting that the enzyme is associated with the cell membrane. In contrast to the clostridial enzyme, this enzyme lacks iron and catalyzes the reduction of methylenetetrahydrofolate with NADH as the electron donor. The enzyme from E. coli more closely resembles the Peptostreptococcus enzyme and eukaryotic methylenetetrahydrofolate reductases. However, it differs from these enzymes by being an α4 tetramer of identical subunits, each of which contains bound FAD if the enzyme is isolated without storage at dilute concentrations.
It is difficult to compare the specific activities of the enzymes from P. productus and E. coli. The specific activity of the P. productus enzyme in the NADH-CH2-H4folate assay was measured at pH 5.5 and 37°C and found to be 380 μmol min−1 mg of protein−1. The specific activity was reported to decrease as the pH was raised. In contrast, the specific activity of the E. coli enzyme in the same assay was measured at pH 7.2 and 15°C and found to be ∼1 μmol min−1 mg of protein−1; the variation of specific activity with pH has not yet been determined.
Comparison of properties of the enzymes from E. coli and porcine liver.
The porcine liver enzyme has been purified to homogeneity (6) and extensively characterized. By using peptide sequences obtained from the porcine enzyme, a cDNA specifying human methylenetetrahydrofolate reductase has been cloned and sequenced (10, 12). Subsequently, sequences have been obtained for putative methylenetetrahydrofolate reductase proteins from Saccharomyces cerevisiae (28), Arabidopsis thaliana (23), and Caenorhabditis elegans (33). The eukaryotic proteins are much larger (70 to 77 kDa) than the MetF protein from E. coli, and the N-terminal halves of the proteins show extensive sequence similarity with MetF. Limited proteolysis of the native porcine enzyme with trypsin cleaves the 77-kDa polypeptide into an N-terminal fragment of ∼40 kDa and a C-terminal fragment of ∼37 kDa. Cleavage does not affect the activity of the enzyme but abolishes allosteric regulation of activity by AdoMet (21). Photoaffinity labeling established that AdoMet binds to the C-terminal 37-kDa domain (27).
The porcine enzyme is a dimer, but scanning transmission electron microscopy of preparations of enzyme that have been shadowed with uranyl sulfate reveals the appearance of tetrameric planar rosettes, suggesting that each subunit consists of two separable domains (21). Sequence comparisons identify the N-terminal domain as the catalytic domain, presumably responsible for binding of FAD and substrates, and the C-terminal domain is thought to be involved in allosteric regulation of enzyme activity. In contrast, the bacterial enzyme is a tetramer of identical subunits, also arranged in a planar rosette with only one twofold axis of symmetry (13).
Table 3 compares the properties of the methylenetetrahydrofolate reductase enzymes from E. coli and porcine liver. Both the prokaryotic and eukaryotic enzymes are capable of transferring reducing equivalents from reduced pyridine nucleotides to either CH2-H4folate or menadione, and they can also transfer reducing equivalents from CH3-H4folate to menadione. As mentioned above, the enzyme from E. coli uses NADH in preference to NADPH, while the porcine enzyme uses NADPH preferentially. However, the porcine enzyme can use NADH as a reductant, provided that phosphate ions are present in the assay buffer (20).
TABLE 3.
Comparison of properties of enzymes from E. coli and pig liver
| Property | E. coli MetF | Porcine liver methylenetetra-hydrofolate reductase |
|---|---|---|
| Km for CH2-H4folate (μM) | 0.8 (pH 7.2) | 88 (pH 7.2) |
| Ki for CH2-H4folate (μM) | 7.1 | Not determined |
| Km for NAD(P)H (μM) | 13 (NADH) | 16 (NADPH) |
| Ki for NAD(P)H (μM) | 9 (NADH) | Not determined |
| Maximum turnover in NAD(P)H-CH2-H4folate oxidoreductase assay (min−1) | 1,800 (15°C) | 1,700 (25°C) (19) |
| Km for (6S)CH3-H4folate (μM) | 75 (pH 6.3) | 25 (pH 6.7) |
| Maximum turnover in CH3-H4folate-menadione oxidoreductase assay (min−1) | 190 (37°C) | 320 (25°C) (30) |
| Km for NADH (μM) | 30 (pH 7.2) | Not determined |
| Km for NADPH (μM) | >300 | 28 (pH 7.2) |
| Turnover in NAD(P)H-menadione oxidoreductase assay (min−1) | 2,600 (25°C) | 6,700 (25°C) (30) |
| Kd for NAD(P)Ha (μM) | 37 (NADH) | 28 (NADPH) |
| Extinction coefficient (mM−1 cm−1) | 14.3 | 12.1 |
| Oligomeric state | Tetramer | Dimer |
| Subunit mol wt | 33,100b | 77,300c |
Determined by measuring the rate of reduction of enzyme in a stopped-flow spectrophotometer as a function of the concentration of reduced pyridine nucleotide.
Determined from the sequence of the metF open reading frame.
Determined by polyacrylamide gel electrophoresis in the presence of SDS (6).
The enzyme from E. coli is markedly less stable than its eukaryotic counterpart and readily loses its activity and flavin cofactor when present in dilute solution. In a study to be reported elsewhere (13), we have shown that activity loss on dilution results when the tetramer dissociates into dimers and is associated with release of the flavin cofactor. Addition of FAD to the buffer does not completely prevent dissociation of the tetramer into dimers when the enzyme is dilute, although it slows the net rate of dissociation. These properties pose difficulties both for purification and for assay and led us to construct a vector for overproduction of a histidine-tagged MetF protein that can be purified in a single step and maintained at high concentration throughout the purification.
ACKNOWLEDGMENTS
This work was supported in part by NIH grant GM24908, by NIH predoctoral fellowship DK49201 (C.A.S.), and by an NIH Cellular & Molecular Biology training grant to the University of Michigan (C.A.S.).
We thank Vincent Massey, David Ballou, and Bruce Palfey for help with the performance and analysis of the stopped-flow experiments. Plasmid pEJ3-1B was provided by James Johnson.
REFERENCES
- 1.Beinert H. Flavin coenzymes. In: Boyer P D, Lardy H, Myrback K, editors. The enzymes. 2nd ed. Vol. 2. New York, N.Y: Academic Press; 1960. pp. 339–416. [Google Scholar]
- 2.Bessey O A, Lowrey O, Love R. The fluorometric measurement of the nucleotides of riboflavin and their concentration in tissues. J Biol Chem. 1949;180:755–769. [PubMed] [Google Scholar]
- 3.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 4.Cathou R E, Buchanan J M. Enzymatic synthesis of the methyl group of methionine. V. Studies with 5,10-methylenetetrahydrofolate reductase from Escherichia coli. J Biol Chem. 1963;238:1746–1751. [PubMed] [Google Scholar]
- 5.Clark J E, Ljungdahl L G. Purification and properties of 5,10-methylenetetrahydrofolate reductase, an iron-sulfur flavoprotein from Clostridium formicoaceticum. J Biol Chem. 1984;259:10845–10849. [PubMed] [Google Scholar]
- 6.Daubner S C, Matthews R G. Purification and properties of methylenetetrahydrofolate reductase from pig liver. J Biol Chem. 1982;257:140–145. [PubMed] [Google Scholar]
- 7.Daubner S C, Matthews R G. Purification and properties of methylenetetrahydrofolate reductase from pig liver. In: Massey V, Williams C H, editors. Flavins and flavoproteins. New York, N.Y: Elsevier; 1982. pp. 165–172. [PubMed] [Google Scholar]
- 8.Donaldson K O, Keresztesy J C. Naturally occurring forms of folic acid. II. Enzymatic conversion of methylenetetrahydrofolic acid to prefolic A-methyltetrahydrofolate. J Biol Chem. 1962;237:1298–1304. [PubMed] [Google Scholar]
- 9.Emmett M R, Johnson J R. Control of metF gene expression in maxicell preparations of Escherichia coli K-12: reversible action of the MetJ protein and effect of vitamin B12. J Bacteriol. 1986;168:1491–1494. doi: 10.1128/jb.168.3.1491-1494.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frosst P, Blom H J, Milos R, Goyette P, Sheppard C A, Matthews R G, Boers G J H, den Heijer M, Kluijtmans L A J, van den Heuvel L P, Rozen R. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 11.Garces E, Cleland W W. Kinetic studies of yeast nucleoside diphosphate kinase. Biochemistry. 1969;8:633–640. doi: 10.1021/bi00830a026. [DOI] [PubMed] [Google Scholar]
- 12.Goyette P, Sumner J S, Milos R, Duncan A M V, Rosenblatt D S, Matthews R G, Rozen R. Human methylenetetrahydrofolate reductase: isolation of cDNA, mapping and mutation identification. Nat Genet. 1994;7:195–200. doi: 10.1038/ng0694-195. [DOI] [PubMed] [Google Scholar]
- 13.Guenther, B., C. Sheppard, P. Tran, R. Rozen, R. G. Matthews, and M. L. Ludwig. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli: a model for an enzyme linked to hyperhomocysteinemia. Nat. Struct. Biol., in press. [DOI] [PubMed]
- 14.Hatch F T, Larrabee A R, Cathou R E, Buchanan J M. Enzymatic synthesis of the methyl group of methionine. I. Identification of the enzymes and cofactors involved in the system isolated from Escherichia coli. J Biol Chem. 1961;236:1095–1101. [PubMed] [Google Scholar]
- 15.Horton R M, Ho S N, Pullen J K, Hunt H D, Cai Z, Pease L R. Gene splicing by overlap extension. Methods Enzymol. 1993;217:270–279. doi: 10.1016/0076-6879(93)17067-f. [DOI] [PubMed] [Google Scholar]
- 16.Kang S-S, Wong P W K, Zhou J, Sora J, Lessick M, Ruggie N, Grcevich G. Thermolabile methylenetetrahydrofolate reductase in patients with coronary artery disease. Metabolism. 1988;37:611–613. doi: 10.1016/0026-0495(88)90076-5. [DOI] [PubMed] [Google Scholar]
- 17.Katzen H M, Buchanan J M. Enzymatic synthesis of the methyl group of methionine. VIII. Repression, derepression, purification, and properties of 5,10-methylenetetrahydrofolate reductase from Escherichia coli. J Biol Chem. 1965;240:825–835. [PubMed] [Google Scholar]
- 18.Kutzbach C, Stokstad E L R. Mammalian methylenetetrahydrofolate reductase: partial purification, properties, and inhibition by S-adenosylmethionine. Biochim Biophys Acta. 1971;250:459–477. doi: 10.1016/0005-2744(71)90247-6. [DOI] [PubMed] [Google Scholar]
- 19.Matthews R G. Methylenetetrahydrofolate reductase from pig liver. Methods Enzymol. 1986;122:372–381. doi: 10.1016/0076-6879(86)22196-5. [DOI] [PubMed] [Google Scholar]
- 20.Matthews R G, Kaufman S. Characterization of the dihydropterin reductase activity of pig liver methylenetetrahydrofolate reductase. J Biol Chem. 1980;255:6014–6017. [PubMed] [Google Scholar]
- 21.Matthews R G, Vanoni M A, Hainfeld J A, Wall J. Methylenetetrahydrofolate reductase: evidence for spatially distinct subunit domains obtained by scanning transmission electron microscopy and limited proteolysis. J Biol Chem. 1984;259:11647–11650. [PubMed] [Google Scholar]
- 22.Nölling J, Pihl T D, Reeve J N. Cloning, sequencing, and growth phase-dependent transcription of the coenzyme F420-dependent N5,N10-methylenetetrahydromethanopterin reductase-encoding genes in two regions of the Methanobacterium thermoautotrophicum genome. J Bacteriol. 1995;177:7238–7244. doi: 10.1128/jb.177.24.7238-7244.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rounsley S D, Kaul S, Lin X, Ketchum K A, Crosby M L, Brandon R C, Sykes S M, Mason T M, Kerlavage A R, Adams M D, Somerville C R, Venter J C. Arabidopsis thaliana chromosome II BAC F6E13 genomic sequence. 1998. GenBank accession no. AC004005. [Google Scholar]
- 24.Saint-Girons I, Duchange N, Zakin M M, Park I, Margarita D, Ferrara P, Cohen G N. Nucleotide sequence of metF, the E. coli structural gene for 5,10-methylenetetrahydrofolate reductase and of its control region. Nucleic Acids Res. 1983;11:6723–6732. doi: 10.1093/nar/11.19.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 26.Segel I. Enzyme kinetics. New York, N.Y: Wiley-Interscience; 1975. [Google Scholar]
- 27.Sumner J, Jencks D A, Khani S, Matthews R G. Photoaffinity labeling of methylenetetrahydrofolate reductase with 8-azido-S-adenosylmethionine. J Biol Chem. 1986;261:7697–7700. [PubMed] [Google Scholar]
- 28.Tizon B, Rodriguez-Torres M, Rodriguez-Belmonte E, Cadahia J L, Cerdan E. Identification of a putative methylenetetrahydrofolate reductase by sequence analysis of a 6.8 kb DNA fragment of yeast chromosome VII. Yeast. 1996;12:1047–1051. doi: 10.1002/(SICI)1097-0061(199609)12:10B%3C1047::AID-YEA991%3E3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 29.van der Put N M J, Eskes T K A B, Blom H J. Is the common 677C→T mutation in the methylenetetrahydrofolate reductase gene a risk factor for neural tube defects? A meta-analysis. Q J Med. 1997;90:111–115. doi: 10.1093/qjmed/90.2.111. [DOI] [PubMed] [Google Scholar]
- 30.Vanoni M A, Ballou D P, Matthews R G. Methylenetetrahydrofolate reductase: steady state and rapid reaction studies on the NADPH-methylenetetrahydrofolate, NADPH-menadione, and methyltetrahydrofolate-menadione oxidoreductase activities of the enzyme. J Biol Chem. 1983;258:11510–11514. [PubMed] [Google Scholar]
- 31.Vaupel M, Thauer R K. Coenzyme F420-dependent N5,N10-methylenetetrahydromethanopterin reductase (Mer) from Methanobacterium thermoautotrophicum strain Marburg. Cloning, sequencing, transcriptional analysis and functional expression in Escherichia coli of the mer gene. Eur J Biochem. 1995;231:773–778. doi: 10.1111/j.1432-1033.1995.0773d.x. [DOI] [PubMed] [Google Scholar]
- 32.Whitfield C, Steers E J, Weisbach H. Purification and properties of 5-methyltetrahydropteroyltriglutamate-homocysteine transmethylase. J Biol Chem. 1970;245:390–401. [PubMed] [Google Scholar]
- 33.Wilson R, Ainscough R, Anderson K, Baynes C, Berks M, Bonfield J, Burton J, Connell M, Copsey T, Cooper J, Coulson A, Craxton M, Dear S, Du Z, Durbin R, Favello A, Fulton L, Gardner A, Green P, Hawkins T, Hillier L, Jier M, Johnston L, Jones M, Kershaw J, Kirsten J, Laister N, Latreille P, Lightning J, Lloyd C, McMurray A, Mortimore B, O’Callaghan M, Parsons J, Percy C, Rifken L, Roopra A, Saunders D, Shownkeen R, Smaldon N, Smith A, Sonnhammer E, Staden R, Sulston J, Thierry-Mieg J, Thomas K, Vaudin M, Vaughan K, Waterston R, Watson A, Weinstock L, Wilkinson-Sproat J, Wohldman P. 2.2 Mb of contiguous nucleotide sequence from chromosome III of C. elegans. Nature. 1994;368:32–38. doi: 10.1038/368032a0. [DOI] [PubMed] [Google Scholar]
- 34.Wohlfarth G, Geerligs G, Diekert G. Purification and properties of an NADH-dependent 5,10-methylenetetrahydrofolate reductase from Peptostreptococcus productus. Eur J Biochem. 1990;192:411–417. doi: 10.1111/j.1432-1033.1990.tb19242.x. [DOI] [PubMed] [Google Scholar]
- 35.Zakin M M, Greene R C, Dautry-Varsat A, Duchange N, Ferrara P, Py M-C, Margarita D, Cohen G M. Construction and physical mapping of plasmids containing the metJBLF gene cluster of E. coli K-12. Mol Gen Genet. 1982;187:101–106. doi: 10.1007/BF00384390. [DOI] [PubMed] [Google Scholar]