

Abstract
Computer-aided drug discovery methods play a major role in the development of therapeutically important small molecules, but their performance needs to be improved. Molecular dynamics simulations in mixed solvents are useful in understanding protein–ligand recognition and improving molecular docking predictions. In this work, we used ethanol as a cosolvent to find relevant interactions for ligands toward protein kinase G, an essential protein of Mycobacterium tuberculosis (Mtb). We validated the hot spots by screening a database of fragment-like compounds and another one of known kinase inhibitors. Next, we performed a pharmacophore-guided docking simulation and found three low micromolar inhibitors, including one with a novel chemical scaffold that we expanded to four derivative compounds. Binding affinities were characterized by intrinsic fluorescence quenching assays, isothermal titration calorimetry, and the analysis of melting curves. The predicted binding mode was confirmed by X-ray crystallography. Finally, the compounds significantly inhibited the viability of Mtb in infected THP-1 macrophages.
1. Introduction
Tuberculosis (TB) is currently one of the most important infectious diseases in the world. In 2019, around 10 million people fell ill with TB, and an estimated 1.4 million people died from this disease.1 Most TB cases can be treated, but the inadequate usage of antibiotics has led to the appearance of multiresistant strains that require treatments with severe secondary effects or may even be untreatable.2,3 It is therefore of utter importance to develop new effective therapies against TB.
Mycobacterium tuberculosis (Mtb), the causative agent of TB, is an obligate pathogen that mainly infects the respiratory system and spreads through the air in drops originated from an infected person.4 Once inside the lungs, Mtb is phagocytized by the macrophages, where it avoids phagolysosome fusion.5 The survival of Mtb inside the macrophages requires the presence of signal transduction systems. In this context, the family of the serine–threonine protein kinases plays a key role in the regulation of gene transcription, cellular division, and pathogen–host interactions.6−8 PknG is one of the 11 serine–threonine protein kinases identified in Mtb,9 and it regulates the tricarboxylic acid cycle by phosphorylation of protein GarA, regulates response to hypoxia, and is essential for pathogenicity.10−14
The PknG domain architecture is unique: besides the kinase domain, it has a rubredoxin domain (which may be involved in redox sensing),15 a tetratricopeptide domain (involved in protein–protein interactions),16 and a non-structured region of ∼70 residues with four auto-phosphorylatable sites. The three-dimensional structure of the PknG kinase domain has been elucidated, thus turning it into an ideal target for drug screening projects.17,18 In fact, several successful attempts have been made to discover PknG ligands; however, none of the identified compounds have advanced into clinical trials. Nonetheless, PknG still remains an important structure-based drug target.18−20
The ATP binding pocket composition differs from commonly known kinases, and it can therefore be selectively targeted by ATP competitive inhibitors.18 Indeed, the first published inhibitor (AX20017), based on an in vitro screening of a proprietary kinase library (55,000 compounds), is an excellent example of selectivity.8,18 Other in vitro approaches consisted of evaluating, as before, a library of 80 kinase inhibitory compounds,19,21 a library of triazolyl methoxy chalcones, flavanones, and 2-aminopyrimidines,21 or electrophilic unsaturated fatty acids that irreversibly react with the rubredoxin domain.22 Regarding structure-based strategies, the ATP binding pocket has once been targeted using pharmacophore-based virtual screening, followed by a biological evaluation.20 To develop this pharmacophore model, positive hits from the triazolyl methoxy aminopyrimidines were used.
Molecular docking-based virtual screening is a major tool in computer-aided drug discovery. It has been shown that docking success rates can be improved when the method is adjusted using previous knowledge,23,24 for example, using biases toward the formation of an important protein–ligand interaction. Recently, we showed that cosolvent sites, particularly ethanol sites, derived from mixed solvent molecular dynamics (MD) simulations allow us to identify (in 18 different proteins) around 70% of known protein–ligand interactions, especially those that represent the ligand-derived pharmacophore.25 We also proved in a retrospective manner that cosolvent-derived pharmacophores improve the performance of docking-based virtual screening.26
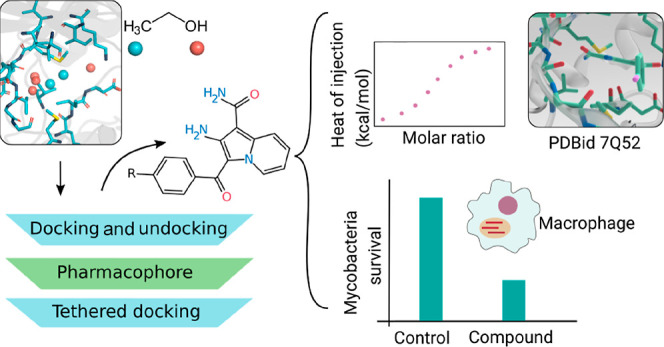
In the current work, we performed a prospective study using PknG to show that cosolvent sites are indeed a powerful tool to identify new inhibitors (Figure 1). Moreover, we solved the crystal structure of PknG bound to one of the obtained hit compounds, and we demonstrated in vitro the inhibitory activity of selected compounds on Mtb H37Rv-infected macrophages.
Figure 1.

Applied workflow that led to the discovery of new PknG inhibitors. First, ligand-binding hot spots were detected using MD in a mixture of ethanol and water. Second, hot spots were confirmed from two small screenings and used to design a scaffold and perform a tethered (structure-based) docking. Third, based on one of the hits, four derivatives were obtained, binding affinities were determined, and one inhibitor-bound crystal structure was solved. Lastly, inhibition of the survival of Mtb inside the macrophages was demonstrated.
2. Results and Discussion
2.1. Binding Site Characterization
Since their introduction in 2009, mixed solvent MD have become an important tool in structure-based drug discovery.27,28 We have previously shown that simulations in a mixture of water and ethanol allow high-specificity detection of key nonpolar and polar drug–protein interactions.25 Taking this into account, we obtained the solvent sites (SSs) inside the ATP binding pocket of PknG from the ethanol probe to find binding hot spots.
We detected several ethanol −OH-based hydrophilic sites (Figure 2). The amino acids on kinase hinges (i.e., the residues connecting the N- and C-terminal lobes of the catalytic domain) have been fully annotated from H1 to H13, where H3 corresponds to the gatekeeping residue. Kinase ATP competitive inhibitors form hydrogen bonds with these residues, and according to a survey of inhibitor-bound kinases structures, most of them engage H6 amide for hinge recognition, some present dual hydrogen bonds with the recruitment of H4 carbonyl upstream and/or H6 carbonyl downstream, and triple hydrogen bonds are rarely found.29 A detailed analysis along the MD trajectories revealed that the probe atoms from SS1 to SS3 were involved in hydrogen bonds with H4 carbonyl, H6 amide, and H6 carbonyl (Figure 2B). Therefore, the presence of SSs SS1 to SS3 suggests that, in the case of PknG, it would be better to prioritize a small chemical compound that anchors itself via three hinge hydrogen bonds. Regarding SS4, the probe atoms from this site interacted mainly with Lys181-Nζ and Asp293-Oδ1/2 (Figure 2B).
Figure 2.

MD analysis in mixed solvents reveals key interactions to design inhibitors against PknG. (A) Pipeline to detect protein hotspots. First, we carried out three MD replicates in a mixture of ethanol and water and computed the sites near the protein surface where the probability of finding a probe atom (O from the hydroxyl group or C from the methyl group) was higher than that in the bulk solvent. Then, we only kept the SSs from the active site and clustered the remaining ones to remove redundant sites. Lastly, each site was individually analyzed, and we identified the protein residues interacting with the corresponding probe atoms. For example, Val235 displayed hydrogen bond interactions with the ethanol probes from SS1, and Ile157 showed hydrophobic interactions with the ethanol probes from SS5. (B) SSs after the hot spot detection pipeline superimposed with the PknG reference structure (PDBid 4Y12). H4, H5, and H6 are, respectively, the kinase hinge residues Glu233, Tyr234, and Val235. SS1 to SS4 correspond to the ethanol −OH-based hydrophilic sites, and SS5 and SS6 correspond to the ethanol −CH3-based hydrophobic sites. (C) Superposition of the ethanol sites and two PknG known ligands, AX20017 and ATPγS.
Additionally, we obtained two ethanol −CH3-based hydrophobic sites. The probe atoms from SS5 and SS6 were close to hydrophobic residues such as Val215, Ile157, and Met283. Both of them overlap with hydrophobic moieties of two PknG known ligands (AX20017 and ATPγS), so they seem to be true positives (Figure 2C) and not the result of the usage of a dual hydrophilic–hydrophobic cosolvent molecule (probe coupling).17,18
According to a protein kinase pharmacophore model, the ATP binding site can be divided into five regions (Figure S1).30 Interestingly, the ethanol-derived SS1, SS2, SS3, SS5, and SS6 fall in the adenine region. It has been shown that more hydrogen bonds in the kinase hinge region do not necessarily translate into higher potency.29 However, the hot spots derived from the cosolvent MD suggest that PknG inhibitors will have better binding affinity if they are capable of performing the three hydrogen bonds.29 Indeed, a family of ligands derived from AX20017 has the highest binding affinity among the previously discovered inhibitors against PknG.31 This represents only one chemical scaffold; the other known ATP competitive inhibitors establish two hydrogen bonds; and a previous work using pharmacophoric features showed that their best model consisted of two hydrophobic, one donor atom, and one acceptor atom features, so whether two or three hydrogen bonds should be prioritized is still not clear for this particular system.20,21
To acquire more structural data and confirm that the three hydrogen bonds are required, we performed a docking protocol especially tailored for protein kinases where we expected to find a few binders. We docked two databases, one in-house collection of 628 fragments, and a library of 367 known kinase inhibitors (GSK Published Kinase Inhibitor Set).32 We used the rDock docking suite using a kinase hinge interaction filter, followed by dynamic undocking.33,34
The fragment collection was docked against PknG using crystal structure 2PZI18 and a pharmacophoric restrain to force the presence of a hydrogen bond between the molecules and the nitrogen atom from the backbone of Val235. This interaction was previously described and mimics the binding of the adenine moiety from the ATP (Figure 3A).35 We decided to use structure 2PZI because it has an inhibitor bound in the ATP pocket (AX20017), which has a molecular weight similar to that of the fragments.18 The 249 fragments with a binding score of less than −15 score units (AX20017) were subject to a dynamic undocking protocol.33 Briefly, the strength of the kinase hinge hydrogen bond from the kinase hinge was tested by running 12 steered MD simulations by increasing the non-hydrogen atom distance from 2.5 to 5 Å, as described by Ruiz-Carmona et al. (Figure 3A).33 The strength of the conserved interaction between the ATP and the kinases can be used as a filter to distinguish binders from non-binders after the docking runs. All fragments that contained at least one curve in which the work was not above 6 kcal/mol were discarded (Figure 3B). 53 fragments passed this filter, and 20 were finally selected by visual inspection for in vitro inhibition testing.
Figure 3.

(A) Structure of the kinase hinge binding motif with ATP illustrated by the hydrogen bond interaction between AGS and the nitrogen atom from the backbone of Val235 from PknG (PDB 4Y12). (B) Two hypothetical curves of the work required us to pull away from the active site two ATP competitive ligands using as the reaction coordinate the distance between the non-hydrogen atoms of the hydrogen bond shown in panel A. (C) AX20017-bound structure aligned with the docked pose of the two hits: the kinase inhibitor and the fragment-like compound. (D) Shared substructure between AX20017 and the two hits.
In parallel, the GSK Published Kinase Inhibitor Set32 was docked against PknG using a similar protocol to the one used for the fragments. The selected crystal structure was 4Y12,17 which has a bigger binding pocket (co-crystallized with ATP-γ-S), and the binding score threshold to perform dynamic undocking was arbitrarily set to −25 score units. This resulted in 119 compounds that were dynamically undocked from the binding site, leaving 54 possible binders, which were reduced to 21 for in vitro inhibition testing by visual inspection.
The 20 selected fragments and 21 kinase inhibitors were screened using the Kinase Glo Plus luminescent assay,36 which allows estimating the remaining ATP after incubation of the enzyme with its substrates. We performed a final point inhibition assay by incubating full-length PknG with ATP and its natural substrate, GarA. The kinase inhibitors were assayed at 40 μM and the fragments at 2 mM. We found one kinase inhibitor (GSK586581A) with ∼50% inhibition and one fragment with ∼100% inhibition (Figure S2). A structural superposition of both ligands, in addition to the known control ligand AX20017, displayed a pharmacophoric motif composed of two hydrogen bond donors and one hydrogen bond acceptor (Figure 3C,D). Interestingly, only two out of the 20 fragments and two out of the 21 kinase inhibitors were bound by the three hydrogen bonds in the docked pose. Therefore, a three-point pharmacophore filter would have theoretically improved the hit rate (two hits out of four compounds).
2.2. Scaffold-Based Docking
Based on the validated pharmacophore, we decided to exploit rDock capacity to perform a tethered docking.34 Using this methodology, the molecules are aligned with the substructure prior to docking, and then, the conformation of the free (untethered) portion is optimized. As a substructure, we used the motif shared by the known ligand AX20017 and the two hits from the fragment and kinase inhibitor (GSK set) libraries (Figure 3D). Compounds with this motif are able to establish three hydrogen bonds with the kinase hinge and should place an aromatic moiety where the ethanol −CH3-based hydrophobic site SS6 is located (Figure S3). We used rDock with a library of around 24,000 purchasable compounds having the common substructure (Figure 3D) and PknG crystal structure 4Y12.17 The 2000 best-ranked compounds were filtered by diversity (using fingerprinting and the Tanimoto diversity index), leaving 200 compounds, from which 20 were selected for experimental testing after visual inspection. The inhibition of selected compounds was assayed at 40 μM once again using the Kinase Glo luminescent assay.36 Three compounds considerably diminished kinase activity, which we named T1, T2, and T3 (Figure 4).
Figure 4.

Tethered docking of 24,000 compounds with the same motif led to the discovery of three hits (compounds T1, T2, and T3). The novel chemical scaffold from T1 was used as a starting point to synthesize/buy new compounds (R1 groups: hydrogen, fluoride, bromide, or ethoxy).
We did not benefit from the ethanol −CH3-based hydrophobic sites SS5 and SS6 during the tethered docking as this methodology requires the usage of a fixed common structure, which goes against the discovery of new chemical scaffolds. Nowadays, this interaction could be considered without that problem in Autodock Bias, which does not force any atomic position but rather modifies the energy landscape, which helps the search algorithm.37 Interestingly, we performed a cosolvent site-based biased docking after the experiment found out that the docked poses of the three active compounds were capable of establishing the important interactions without forcing any positional restraint in the ligand atoms (Figure S4).
2.3. Binding Affinity Estimations and X-ray Crystallography
Out of the three confirmed ligands, we decided to focus on T1, which had a different chemical scaffold from PknG known inhibitors.18,20,22 To validate the novel chemical scaffold, we synthesized two derivatives compounds named S1 and S2 (Scheme 1), purchased compounds B1 and B2, and measured their binding affinity using a smaller construct of PknG comprising the kinase and rubredoxin domains (Table 1 and Figure 4).17 First, we took advantage of the fact that PknG has a tryptophan near the binding site to perform intrinsic fluorescence quenching assays and estimate the equilibrium dissociation constant (Kd).38
Scheme 1. Synthesis of Compounds S1 (R=H) and S2 (R=Br).

Reagents and conditions: (a) ethanol, reflux, 5 h; (b) 2-cyanoacetamide, 1-propanol, N,N-diisopropylethylamine (DIPEA), reflux, 3 h.
Table 1. Binding Affinity Estimations from Intrinsic Tryptophan Quenching Spectroscopy, ITC, and Melting Curve Analysisa.
| name | R group | Kd—fluorescence quenching (∼21 °C) | Kd—ITC (25 °C) | KdApp—isothermal analysis (44 °C) | KdApp—TmObs (∼44 °C) |
|---|---|---|---|---|---|
| T1 | isopropyl | 1 - CI: [0.6; 1.7] | 1.5 - CI: [0.8; 2.6] | 3.8 - CI: [3.1; 4.5] | 11 - CI: [9.5; 12] |
| B1 | fluoride | 2.2 - CI: [1.3; 3.6] | 2 - CI: [1.3; 3] | 3.6 - CI: [3.1; 4.1] | 8.1 - CI: [7.1; 9.1] |
| B2 | ethoxy | 13 - CI: [11; 22] | |||
| S1 | hydrogen | 1.8 - CI: [1.2; 2.7] | |||
| S2 | bromide | 1.3 - CI: [0.7; 2.6] | |||
All Kds are in micromolar units. CI is the asymmetric confidence interval at a 95% confidenceS level.39 The fitted curves of compounds T1, B1, B2, S1 and S2 are shown in the Supporting Information (Figures S5, S7, and S8).
As a result, with the exception of B2 that had the activating substituent −OCH3CH3 (ethoxy group), we obtained Kds in the low micromolar range (Table 1 and Figures 5A and S5). Similar binding affinities were found for the control ligand AX20017 and ligand T2 (Figure S5). We also evaluated one more compound where the benzene ring was replaced by a thiophene ring (compound B3), but the affinity slightly decreased (Kd of 3.6 μM, Figure S6), suggesting that the aromatic–aromatic interaction with Tyr235 may be important. Finally, we tested one compound with a 1,2-dichloro benzene ring (compound B4), but once again, the interaction was weaker (Kd of 13 μM, Figure S6), in this case, probably due to steric effects (Figure 5E). In future studies, it would be interesting to explore compounds with hydrophilic functional groups as substituents.
Figure 5.

Binding affinity characterization and holo-structure of ligand B1. (A,B) Estimation of the equilibrium dissociation constant Kd from intrinsic fluorescence spectroscopy and ITC, respectively. (C,D) Estimation of the apparent equilibrium dissociation constant Kd from isothermal analysis of melting curves and observed melting temperature shift analysis, respectively. (E) Crystal structure of the PknG–ligand B1 complex (PDBid 7Q52).
Additionally, we performed isothermal titration calorimetry (ITC) for ligands T1 and B1. In both cases, the estimated equilibrium dissociation constant was in good agreement with the previous estimations (Table 1 and Figure 5B). To also test the binding capacity of the new scaffold, we incubated the kinase domain of PknG at different ligand concentrations and measured the change in the protein melting temperature by monitoring the intrinsic protein fluorescence at 350 nm.40,41 Then, we adjusted the data to two models that couple the unfolding and binding equilibria as described in the Experimental Methods Section.40−42 The estimated Kds turned out to be in the low micromolar range (Table 1 and Figure 5C,D).
Lastly, to confirm inhibition by direct measurement of product formation, we performed radioactive ATP (32P-γ-ATP) assays using T1. Briefly, we incubated full-length PknG with its natural substrate GarA, a mixture of cold and hot ATP and different concentrations of T1 in the micromolar range at 37 °C. We found that T1 decreased the amount of phosphorylated GarA in a concentration-dependent way in the micromolar range with a relative half inhibitory concentration (IC50) of 5.8 μM (Figure S9). Additionally, we performed the same assay with the known ligand AX20017 and found that it has similar potency with a relative IC50 of 0.9 μM, which is in high agreement with previous measurements (Figure S9).31
We tried to crystallize PknG–ligand complexes by screening at least 576 different solutions for compounds B1, T1, S1, and S2 and were successful only in the case of B1 (Figure 4E, PDBid 7Q52). A close view of the ligand inside the binding pocket reveals that B1 is performing the three hydrogen bonds derived from the pharmacophore (Figure 5E, B1 with Glu233-O, Val235-N, and Val235-O). The nonpolar part of the ligand must also be playing a crucial role in the binding affinity as it is interacting with the hydrophobic residues Val179, Ile157, Ile165, Ala158, Ile292, and Met283 (Figure 5E). The importance of the hydrogen bond interaction with Val235-N and the hydrophobic interactions with Tyr234 and Ile293 was confirmed by site-directed mutagenesis (mutants Y234L, V235P, and I292G, respectively) and thermal shift assays. Indeed, for these three mutants, there was no shift in the melting temperature at 100 μM for compounds B1 and T1 (Figure S10).
As expected by the similar results of the binding affinity measurements, the fluorine atom is not establishing any strong interaction with the protein. Interestingly, there is still space to extend the ligand in order to increase the binding affinity (Figures 5E, S1 and S11). Considering that we are only exploiting the adenine binding region, hydrophobic region II, and the sugar pocket, we could achieve higher potency by occupying the phosphate-binding region and hydrophobic pocket I (Figure S1). To test the binding mode prediction capability of the first docking protocol (used for the fragment collection and the GSK kinase inhibitor collection), we performed post hoc docking runs of compound B1. As a result, we found a strong agreement between the docked pose and the obtained crystal structure (Figure S12).
2.4. Decreased Mtb Intracellular Survival
To evaluate the effect of the compounds on mycobacterial intracellular survival, we infected THP-1 derived macrophages with the pathogenic strain Mtb H37Rv (MOI 10). Then, infected cells were cultured in the presence of compounds B1 or T1. After 24 or 48 h of treatment, viable intracellular Mtb H37Rv cells were counted by analyzing the colony forming units per milliliter (CFU/mL) (Figure 6A). We observed a significant decrease in Mtb H37Rv intracellular survival in the presence of both compounds at 1 and 10 μM for 24 h (Figure S13) and 48 h (Figure 6B). Similar inhibitory levels were also observed in a second experiment for compounds T2, T3, and AX20017 (Figure S14). In further assays, we evaluated the toxicity of B1 and T1 compounds in human-derived THP-1 macrophages (Figure 6C). The effect on cell viability was evaluated as a function of both time and compound concentration. At 3, 24, and 48 h, none of the compounds showed adverse effects at 10 μM on cell viability (Figure 6C).
Figure 6.

Compounds B1 and T1 inhibit Mtb H37Rv intracellular survival without cytotoxic effect on macrophages. (A) Experimental setup used to evaluate mycobacterial intracellular survival in the presence of compound T1/B1. (B) CFU/mL values after treatment with compound B1 or T1 at 10 or 1 μM (48 h). Rifampicin (Rif) was used as a control of inhibition (12.5 μM). Mock refers to the treatment with only the vehicle (DMSO). Statistical analysis was performed using Welch’s ANOVA, followed by Dunnett T3 post hoc test for multiple comparisons. Only the statistical significance of the comparisons against the control is shown. (C) Cytotoxicity of B1 and T1 at 10 μM. Each compound was added to differentiate THP-1 cells, and cell viability was evaluated after 3, 24, or 48 h. In both (A,B), data are shown as the mean ± SEM. NS: p > 0.05, *p ≤ 0.05, and **p ≤ 0.01 The experiments were performed in quadruplicate (A) and triplicate (B), respectively.
These results demonstrated that B1 and T1 compounds decrease Mtb H37Rv viability and had no toxic effect on host cells. To our knowledge, this is the second time that it has been proven that PknG inhibitors affect the intracellular survival of Mtb within macrophage cells.8 The previous results were based on ligand AX20017, and they showed a very similar inhibitory activity.8 Furthermore, only two other reports demonstrated inhibitory activity, but they were based on the Mycobacterium bovis BCG strain.19,20
3. Conclusions
TB is still one of the most important infectious diseases in the world. Due to the increasing cases of drug-resistant strains, there is an urgent need for the development of new treatments. PknG plays a crucial role in the pathogenesis and survival of Mtb within the host, making it an important drug target.10−13 The major strategies to target kinases consist of searching for ATP mimetic inhibitors that bind the active or inactive state, allosteric inhibitors, or inhibitors that interfere with binding of other kinase regulators/substrates.30 It has been shown that PknG is a constitutively active kinase,17,43 and there are no substrate-bound structures; thus, a suitable approach is to target the ATP binding pocket.
A common approach in kinase structure-based drug studies is to conduct biased docking runs. In this regard, hydrogen bonds are critical for potent kinase inhibition, and the majority of known kinase inhibitors anchor themselves via one or two hydrogen bonds.29 In the case of PknG, the results from the cosolvent simulation, together with the screening of fragment-like compounds and kinase inhibitors, suggest that it would be better to search compounds with a donor–acceptor–donor pattern. We have proved that compounds with this motif possess binding affinity in the low micromolar range and confirmed by X-ray crystallography that they interact through three hydrogen bonds and that they can inhibit the growth of Mtb in macrophage cells. Finally, we expect that the identified inhibitors may serve as leads for the development of potent anti-TB drugs.
4. Experimental Section
4.1. Computational Methods
4.1.1. Initial Docking of the Fragment-like and Kinase Inhibitor Libraries
To perform the docking runs of the fragment-like (molecular weight ranging from 140 to 300 Da) and GSK kinase inhibitor libraries, we used rDock software and as protein structures PDBid 2PZI (residues 73–750) and 4Y12 (residues 74–405), respectively. In both cases, up to 50 docking runs with the default parameters were performed. Cavity mapping was performed with the “reference ligand” method using ligands AX20017 and AGS, respectively. The docked compounds were constrained to interact with the kinase hinge as shown in Figure 2 (pharmacophoric restraint). As an example, the pharmacophore for PDB 4Y12 was located at point (−7.188, 7.214, −27.796). The radius of tolerance was 1 Å, and the restrain type was “Acc” (H-Bond acceptor). In both cases, the ligand libraries were prepared using LigPrep.44 An ionizer was used to generate different protonation states for groups with a pKa between 6 and 8 (-i 2 -W i, -ph 7.0, -pht 1.0). Up to eight stereoisomers (-s 8), six tautomers (-t 6), and eight ring conformers (-r 8) were generated per molecule.44
4.1.2. Molecular Dynamics in Mixed Solvents
MD simulations in a 20/80 ethanol and water mixture were carried out as described in Arcon et al., 2017.25 Structure 4Y12 was downloaded from the PDB database and all nonstructural ions and solvent and ligand molecules were removed. Missing side chains and hydrogen atoms were added using the LEaP module from the Amber16 package. The Zn2+ ion in the rubredoxin center was replaced by Fe2+, which is the metal ion typically found in rubredoxin domains.45 The standard protonation state at the physiological pH was assigned to all ionizable residues. The structure was then solvated using a truncated octahedral box of ethanol 20% v/v extending at least 10 Å from any protein atom using the pyMDMix program (http://mdmix.sourceforge.net).46 The TIP3P model was used for water molecules, and forcefield parameters for ethanol were assigned as previously reported.25 The Amber ff14SB force field was used for protein residues,47 and Rbx parameters were taken from the literature.48 Solvated systems were subjected to geometry optimization to adjust the solvent orientation and eliminate the local clashes and stereochemical inaccuracies. The subsequent equilibration protocol consisted of 0.8 ns of constant-volume MD simulation, where the temperature was slowly increased from 100 to 300 K, after which 1 ns of constant-pressure and -temperature MD simulation was performed (1 bar, 300 K) to allow the system to reach proper density. Finally, the system was subjected to three independent simulations 50 ns long at constant volume and temperature (NVT ensemble) with a quadratic positional restraint of weight 0.01 kcal/mol over all the non-hydrogen atoms of the protein to ensure that the overall 3D structure of the protein is preserved while ensuring that the binding hot spots are quantitatively correct.49 Temperature control and volume control were achieved using the Langevin thermostat (collision frequency of 4 ps–1) and Berendsen barostat, respectively. Systems were simulated using periodic boundary conditions and Ewald sums (grid spacing of 1 Å) for treating long-range electrostatic interactions with a 9 Å cutoff for direct interactions. The SHAKE algorithm was used to keep bonds involving H atoms at their equilibrium length, allowing the employment of a 2 fs time step for the integration of Newton’s equations. All simulations were performed using the PMEMD implementation of SANDER for GPU from Amber 16.50
4.1.3. SS Detection
Our protocol for the determination of the SSs from a MD simulation in a mixed solvent is inspired by the one developed by Lopez et al.(51) We focused only on the ethanol −OH-based hydrophilic sites and the ethanol −CH3-based hydrophobic sites, but the algorithm works with any probe. The procedure starts with the alignment of the MD trajectory according to a selected group of residues (i.e., binding site). Then, all snapshots are filtered to include only the probe molecule atoms (i.e., the oxygen atom from the −OH group or the C atom from the −CH3 end) close to the selected residues, leaving a 3D Cartesian data set as a result. Next, we construct a graph using as nodes each probe atom and assigning edges between nodes if their Euclidean distance is less than a user-selected parameter distThreshold. Lastly, each connected component of the graph is considered to report an SS if they have more nodes than a certain threshold watNmin. This parameter watNmin represents implicitly a time dimension. Additionally, two important parameters that describe the SSs are the probe finding probability (PFP60) and dispersion parameter (R90). The first one is the probability of finding a probe molecule in the region defined by a sphere of radius 0.6 Å, and the second one is the radius that the SS should have to contain 90% of the probe atoms that conform.
In this work, we used PknG residues 159, 167, 181, 183, 213, 233–239, 285, and 293–295 to perform the structural alignment. For each time frame of the 50 ns trajectory (500 frames), we discarded the first 1 ns prior to the analysis, the distThreshold parameter was 0.32 Å, and the watNmin parameter was 48, implying that each detected SS contained one probe atom during at least 10% of the simulation. SSs with an R90 higher than 1.2 Å and a PFP60 lower than 50 were not taken into account. Redundant SSs were removed by applying proximity-based clustering. The three production simulations and scripts to reproduce the SS detection procedure and to render the protein image from Figure 2 panel B are freely available for download at https://doi.org/10.5281/zenodo.5647767.
4.1.4. Dynamic Undocking
For each docked ligand (fragment and kinase inhibitor libraries), we computed the necessary work to increase the distance of the non-hydrogen atoms involved in the characteristic hydrogen bond from 2.5 to 5 Å (Figure 2A,B). The dynamic undocking protocol has been previously described.33 Briefly, we carried out 1000 steps of minimization (step 1), 400 ps of thermalization in the NVT ensemble to heat up the system (step 2), 1 ns of equilibration in the NPT ensemble (1 atm, 300 K) (step 3), and the production dynamics with the addition of a time-dependent potential [5 Å/ns y string constant of 50 kcal/(mol Å2)] (step 4). We performed six replicates at 300 K and six replicates at 325 K. During steps 1–3, we added a restraint of 1 kcal/(mol Å2) over all non-hydrogen atoms to avoid structural changes and a restraint to keep the hydrogen bond distance fixed [parabolic between 3 and 4 Å with a force constant of 1 kcal/(mol Å2) and linear if the distance was larger than 4 Å with a force constant of 10 kcal/(mol Å2)]. The equilibration and production steps were performed using the Langevin thermostat and a collision frequency of 4 ps–1. The cutoff for direct interactions was 9 Å, and the covalent bonds involving hydrogen atoms were kept at their equilibrium distance using the SHAKE algorithm.52 To improve speed and to avoid distal contributions not related to the hydrogen bond under investigation, we removed (and capped if necessary) all residues without at least one non-hydrogen atom at less than 10 Å from one of the non-hydrogen atoms involved in the hydrogen bond using the MOE Script provided at http://www.ub.edu/bl/undocking/.
4.1.5. Tethered Docking
For tethered docking, we used the rDock built-in functionality to define a common core and leave that part fixed {the motif is shown in Figures 2D and S3, SMARTS pattern: [NH2]C(=O)cc[#7; H1,H2]}. Compound GSK586581A docked into pknG was used as a reference against a custom library of compounds composed of various vendors from ZINC (Asinex, Chembridge, Enamine, Lifechemicals, Princeton, Specs and VitasM). The library was prepared using LigPrep (the same parameters as those explained in Section 4.1.1).44 A python script based on the RDKit package functions was used to align the library of molecules against the reference ligand.53 PDBid 4Y12 was used as the receptor. 50 runs were performed, and ligands were ranked by their interaction energy score and manually inspected to conform to the final list.
4.2. Experimental Methods
4.2.1. Expression Vectors and Mutant Generation
Cloning: Plasmids pET28a-PknG, pET28a-GarA, and pET28a-PknG-kinase-domain were a gift from Maria Natalia Lisa.54 The protein sequence was corroborated by sequencing. Mutations of PknG’s kinase domain were performed using a modified QuikChange protocol. Briefly, each PCR reaction contained the following: 21.5 μL of Autoclaved MilliQ water, 1.5 μL of the pET28a-PknG-kinase domain (100 ng/μL stock), 1 μL of forward and reverse primers (10 μM each), and 25 μL of 2x MasterMix [40 mM tris pH 8.8, 4 mM MgCl2, 120 mM KCl, 20 mM (NH4)2SO4, 0.02 mM ethylenediaminetetraacetic acid (EDTA), 0.2% tritonX-100, 8% glycerol, 0.005% xylene cyanol FF, 0.05% orange G, 0.4 mM dNTPs, and 0.04 U/μL Phu-Sso7d polymerase]. The PCR program included an initial template denaturation step at 95 °C for 5 min, 20 cycles for amplification (95 °C for 30 s, 56 °C for 60 s, and 72 °C for 4 min), and a final amplification step of 72 °C for 10 min. Each PCR product was digested using the DpnI enzyme for 2 h at 37 °C and subsequently transformed into E coli DH5α. Mutation was confirmed by Sanger sequencing of the vectors (Microsynth, Gottingen, Germany). Three mutants were utilized (Y234L, V235P, and I292G). Reverse and forward primers are provided in the Supporting Information (“Experimental methods” Section).
4.2.2. PknG and GarA Production and Purification
PknG (full-length) was overexpressed in E.coli BL21(DE3). Bacteria were grown in LB medium supplemented with 50 μg/μL kanamycin at 30 °C until OD600 = 0.6 and then for 18 h at 30 °C after the addition of 1 mM IPTG and 100 μM FeCl3. GarA (Rv1827) expression was performed in E.coli BL21(AI). Cells were grown in LB medium supplemented with 0.1% glucose and 10 μg/μL tetracycline at 37 °C until OD600 = 0.8 and then for 12 h at 18 °C after the addition of 1 mM isopropylthio-β-galactoside (IPTG) and 0.02% arabinose. The same purification protocol was used for both proteins: after cell lysis by sonication and clarification by centrifugation at 4 C and 4500 rpm, the supernatant was loaded onto a HisTrap HP column (GE Healthcare), and the His-tagged protein was purified by applying a linear imidazole gradient (50 to 400 mM). The protein-containing fractions were pooled and dialyzed against buffer D (25 mM tris–HCl, 500 mM NaCl, 5% glycerol, pH 7.6) and stored at −80 °C. Proteins were quantified by using the molar absorption coefficient predicted from the amino acid sequence using the ProtParam tool (https://web.expasy.org/protparam/).55
4.2.3. Expression of the Kinase and Rubredoxin Domains of PknG
The protein construct concerning the kinase and rubredoxin domains (PknGΔTPRΔ73) was expressed in E. coli BL21(DE3). First, transformed cells were grown overnight in 10 mL of LB medium supplemented with kanamycin (30 μg/L) at 37 °C. In the morning, cells were pelleted by centrifugation at 3500g 10′ and used to inoculate 1 L of M9 minimal medium supplemented with kanamycin (30 μg/L). After OD600 reached ∼0.8–1, 0.25 mM IPTG was added to induce protein synthesis. To achieve a redox inert protein, we also added 100 μM ZnCl2. Cells were grown for another 22 h at 15 °C, pelleted by centrifugation at 35,000g for 10 min, resuspended in tris–HCl 50 mM, NaCl 250 mM, glycerol 5%, pH 8, and lysed by using high pressure in a French Press. The cell lysate was clarified by centrifugation at 45,000g for 40 min. The supernatant with the His-tagged protein was passed through a Nickel column and eluted with imidazole. The fractions containing the protein were further purified, and the buffer was exchanged to tris–HCl 50 mM, NaCl 250 mM, glycerol 5%, pH 8, by using a molecular exclusion column HiLoad 16/600 Superdex 200 pg (SEC). Fractions with the protein were run in an SDS-PAGE to check for purity, pooled, concentrated up to 4 mg/mL, and flash-frozen.
For the expression of PknGΔTPRΔ73 mutants, the same protocol was used but using LB as the growth medium. For crystallography, the growth medium was LB, and 100 μM FeCl3 was added instead of ZnCl2. After purification, 1 mg of TEV was added to the sample to remove the HisTag. The sample was dialyzed with tris–HCl 50 mM pH 8, NaCl 250 mM, glycerol 5%, and tris(2-carboxyethyl)phosphine (TCEP) 0.5 mM. A reverse nickel column was used, and the flowthrough was concentrated and injected using the same protocol as that for the SEC.
4.2.4. Synthesis of Compounds S1 and S2
All reagents were of analytical grade and were purchased from Sigma-Aldrich Chemical Co. Solvents were purchased from local suppliers and were distilled before use. Thin-layer chromatography was carried out using precoated plates of silica gel 60 F254 from Merck, and compounds were visualized using UV detection (254 nm) and phosphomolybdic acid stain. Melting points were determined on a Fisher Johns apparatus and are uncorrected. Electrospray ionization high-resolution mass spectrometry (ESI-HRMS) spectra were measured on a Bruker micrOTOF-Q II. All NMR spectra were recorded on a Bruker Fourier-300 (300 MHz for 1H and 75 MHz for 13C). Chemical shifts (δ) are given in parts per million downfield from tetramethylsilane (TMS) as the internal standard. Coupling constant (J) values are quoted in hertz. Resonances are described as s (singlet), d (doublet), t (triplet), q (quartet), or combinations thereof. Structural determinations were confirmed by 2D NMR spectra (COSY, HSQC-DEPT, and HMBC).
4.2.5. General Procedure
4.2.5.1. Step A
To a solution of 2-bromo-l-(4-R-phenyl)-ethanone (7.5 mmol) in ethanol (10 mL) was added 2-chloropyridine (18.8 mmol, 2.5 equiv), and the reaction was heated to reflux for 5 h. The solution was left overnight at 4 °C, and the precipitate was filtered, washed with diethyl ether, and dried in vacuo to yield the title product.
4.2.5.2. Step B
4′-substituted 2-chloro-l-phenacyl-pyridinium bromide (2.9 mmol), cyanoacetamide (4.3 mmol, 1.5 equiv), and N,N-diisopropylethylamine (17.3 mmol, 6.0 equiv) in 1-propanol (12 mL) were heated to reflux for 3 h. The still-hot solution was diluted by adding dropwise 6.0 mL of water and was left overnight at 4 °C. The precipitate was filtered, washed with water/1-propanol (1:1), and dried in vacuo to yield the title product.
4.2.5.3. 2-Amino-3-benzoyl Indolizine-1-carboxamide (S1, R=H)
1H NMR (300 MHz, DMSO) δ 9.29 (1H, d, J = 6.9 Hz, Ind-4), 7.83 (1H, d, J = 8.9 Hz, Ind-7), 7.55 (5H, m, Ar), 7.39 (1H, ddd, J = 8.7, 6.9, 1.2 Hz, Ind-6), 7.07 (2H, br s, NH2), 6.88 (1H, dt, J = 6.9, 1.2 Hz, Ind-5), 5.93 (2H, br s, NH2).
13C NMR (75 MHz, DMSO) δ 182.4, 167.2, 149.4, 140.7, 137.3, 130.7, 129.2, 128.4, 128.1, 127.9, 127.0, 116.2, 112.4, 109.9, 93.9.
HRMS (ESI) m/z: (M + H)+ calcd for C16H14N3O2, 280.1081; found, 280.1073.
4.2.5.4. 2-Amino-3-(4-bromobenzoyl)indolizine-1-carboxamide (S2, R=Br)
1H NMR (300 MHz, DMSO) δ 9.30 (1H, d, J = 6.9 Hz, Ind-4), 7.82 (1H, d, J = 8.9 Hz, Ind-7), 7.73 (2H, d, J = 8.4 Hz, Ar-2,6), 7.52 (2H, d, J = 8.4 Hz, Ar-3,5), 7.40 (1H, ddd, J = 8.7, 7.0, 1.2 Hz, Ind-6), 7.07 (2H, br s, NH2), 6.90 (1H, dt, J = 7.0, 1.2 Hz, Ind-5), 6.03 (2H, br s, NH2).
13C NMR (75 MHz, DMSO) δ 181.0, 167.2, 149.5, 139.5, 137.4, 132.1, 129.6, 128.4, 128.3, 124.3, 116.2, 112.5, 109.8, 93.9.
HRMS (ESI) m/z: (M + H)+ calcd for C16H13BrN3O2, 358.0186; found, 358.0191.
Kinase activity was measured in a final volume of 50 μL with 25 mM tris–HCl (pH 7.4), 2 mM MnCl2, 1 μM PknG, GarA 25 μM, ATP 2 μM, and 1–2% dimethyl sulfoxide (DMSO) (reaction controls without DMSO were performed to test that the vehicle concentration was not affecting PknG activity). Reactions were started by the addition of 5 μL of ATP, and the amount of remaining ATP after 60 min of incubation at 37 °C was measured by luminescence using the commercial kit KinaseGlo Plus. The inhibition was then calculated as
| 1 |
where C2and C1are, respectively, the luminescence of the reaction without the compound (only DMSO 2% v/v) and without GarA (substrate) and X is the luminescence of the reaction under an arbitrary compound concentration. Experimental conditions were optimized to achieve a C2/C1ratio of less than 1/10. All reactions were performed in duplicate.
4.2.6. Intrinsic Tryptophan Fluorescence Spectroscopy
All tryptophan fluorescence spectroscopy experiments were performed using a TECAN SPARK microplate reader. After optimization of the conditions, the excitation wavelength was fixed to 290 nm, and the emission intensity was measured at 350 nm. The bandwidth was 5 nm in both cases. To measure PknG–ligand interactions, recombinant PknG at 2/3 μM was titrated with increasing concentrations of the ligands up to 20/40 μM at room temperature (20–22 °C). Primary inner filter effects were not present as the fluorescence intensity was determined to be linearly dependent on the protein concentration and not affected by the absorbance of the solutions used (∼0.1). For compounds T1, T2, B1, S1, S2, and AX20017, the volume of the solutions was between 85 and 95 μL (Greiner 96-well flat transparent plate). For compounds B2, B3, and B4, we used 25/28 μL (Greiner 384-well flat black plate). The fluorescence signal was fitted to a one-site binding model using the ThermoAffinity online tool.56 Asymmetric confidence intervals were calculated as suggested by Paketurytė et al., 2021.39
4.2.7. Isothermal Titration Calorimetry
ITC experiments for ligands T1 and B1 were performed in a MicroCal-PeaQ ITC instrument. To prevent possible solubility issues, the ligands were incubated in the cell, and the protein (PknG kinase domain) was injected. The injections regime consisted of one small injection of 0.4 μL and 11 injections of 3.5 μL. The injection time was 0.8 s for the first injection and 7 s for the rest. The injection time between injections was 5 min, and the stirring speed was set to 300 RPM. All ITC runs were performed at 25 °C. For T1, the protein concentration was 350 μM and the ligand concentration was 60 μM. For B1, the protein concentration was 330 μM and the ligand concentration was 35 μM. To prevent buffer mismatch, the ligands were diluted from pure DMSO to 1% v/v using the SEC buffer from the purification. DMSO was added to the protein sample to achieve the same concentration. Raw thermograms were analyzed using a custom R script provided in the Supporting Information (one-to-one binding model with subtraction of control titration). Asymmetric confidence intervals were calculated as previously mentioned.39
4.2.8. Differential Scanning Fluorimetry
Ligand binding was verified and quantified by analyzing the shift in thermal stability.41 Briefly, PknGΔTPRΔ73 was incubated at 12 μM with increasing concentrations of the ligand from 0.05 up to 500 μM, and the fluorescence at 350 nm was measured along a temperature ramp of 1 °C/min going from 20 to 95° using a Nano differential scanning fluorimetry (DSF) instrument (Prometheus NT-48, NanoTemper). In the case of PknGΔTPRΔ73 mutants (Y234L, V235P, and I292G), compounds B1 and S1 were only evaluated at 100 and 6.25 μM. Two models that assume a two-state unfolding model, where only binding to the folded state is possible, were used to estimate the apparent binding affinity against the wild-type protein.
One model is based on fitting the fraction of the unfolded protein at a fixed temperature versus the ligand concentration (isothermal analysis),40,41 while the other uses the observed melting temperatures.42,57 Data analysis was performed using the FoldAffinity online tool.40,56 For the isothermal analysis, we used 35 to 50 °C as the temperature window and the “local” option to fit the melting curves. Asymmetric confidence intervals were calculated as previously mentioned.39
4.2.9. Radioactive ATP Assay
Full-length PknG kinase activity was measured in a final volume of 50 μl with 25 mM Tris–HCl (pH 7.4), 2 mM MnCl2, GarA 25 μM, PknG 0.1 μM, 1% DMSO, ATP 2 μM, and 100 μCi/nmol ATP-32 Pγ. Reactions were started by the addition of 5 μL of ATP and stopped after 35 min of incubation at 37 °C. Then, a volume of 10–16 μL per reaction was loaded in an SDS-PAGE gel, which was run and exposed in an autoradiographic plate. The plate was revealed after 12–24 h of exposition (STORM 840 Phosphorimager), and the band corresponding to the GarA substrate was scanned using ImageJ software. Reaction conditions were optimized to achieve a linear dependence between product formation and incubation time.
4.2.10. Crystal Structure
Co-crystallization experiments were set up using the vapor diffusion method, with the 0.2 M CaCl2, 0.2 M TRIS, pH 8.5, and 20% (w/v) PEG 4000 crystallization solution with a protein concentration of 10 mg/mL and a ligand concentration of 500 μM. Initial crystals were then prepared for microseeding in a 2:1 ratio drop (2 μL protein:1 μL reservoir) using the Garnier 24-well plates using the hanging drop technique. Crystals appeared within 1 day, were harvested after 3 days, and were flash-frozen with liquid nitrogen. Cryoprotection was achieved by adding 1 μL of the precipitant solution complemented with 5% glycerol and with a 200 μM concentration of the target compound.
X-ray data sets were collected at beamline P14 operated by EMBL Hamburg at the PETRA III storage ring (DESY, Hamburg, Germany). Data sets were merged using XDS program58 and scaled using AIMLESS.59 The crystal structure was solved by the molecular replacement method, using program MOLREP60 using as a search model PDBID 4Y12. Iterative refinement and model building cycles were performed using REFMAC61 and Coot62 from the CPP4i2 suite of programs.63 Final steps of model building was also assisted with ISOLDE to correct the model for clashes.64 Data collection and refinement statistics are in Table S1. An X-ray fluorescence spectrum was obtained at 12.7 keV to determine the presence of Fe in the rubredoxin domain of pknG. The protein structure can be accessed using PDBid 7Q52. Authors will release the atomic coordinates and experimental data upon article publication.
4.2.11. In Vitro Microbicidal Activity against Mtb
4.2.11.1. Bacterial Growth Conditions
Mtb H37Rv was grown in Middlebrook 7H9 broth or on 7H10 agar with 0.5% Tween 20, 0.2% glycerol, and albumin–dextrose–catalase–oleic acid supplement. Cultures were harvested at an exponential growing phase at 37 °C. To disaggregate clumps, mycobacteria were sonicated at 2.5 W output for 4 min (Elma d-7700 Singentrans sonic) and then centrifuged for 10 min at 300g, and the was supernatant diluted in PBS. Finally, the OD at 600 nm was determined. Bacterial growth of MtbH37Rv and any experiment involving the pathogenic strain were performed in BSL3 security cabinets at the Malbran Institute, Buenos Aires, Argentina.
4.2.11.2. Cell Culture and Infection
The THP-1 (ATCC TIB-202) human monocyte cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in a RPMI-1640 medium (Gibco, 22400–071) supplemented with 10% FBS (Gibco, 10437028), L-glutamine (2 mM; Sigma, G5792), penicillin–streptomycin, and 2-mercaptoethanol (0.05 mM; Gibco) at 37 °C in a humidified atmosphere with 5% CO2. Cells were differentiated into macrophages by adding 10 ng/mL phorbol-12-myristate-13-acetate (PMA; EMD Biosciences, La Jolla, CA, USA) for 48 h in 96-well flat-bottom plates. After differentiation, THP-1 cells were infected with Mtb H37Rv (MOI 10) for 2 h. Then, cells were washed twice with warm RPMI and cultured in a complete medium without penicillin–streptomycin and in the presence of the B1 or T1 compound (1 and 10 μM) or rifampicin (12.5 and 25 μM) for 24 and 48 h.
4.2.11.3. Colony-Forming Unit Assay
THP-1-derived macrophages infected with MtbH37Rv were washed two times with warm PBS and lysed with 0.05% Triton X-100 in PBS. Serial dilution of adherent cells lysates was obtained, and 40 μL aliquots were inoculated (in quadruplicate) on Middlebrook 7H10 agar plates supplemented with oleic acid–albumin–dextrose–catalase. Plates were incubated for 3 weeks, and colonies were counted from dilutions yielding 10–100 visible colonies.
4.2.12. In Vitro Cytotoxicity Assays
For the evaluation of B1 and T1 compounds and the cytotoxic effect on THP-1-derived macrophages, cells were seeded in 96-well plates (Corning Costar, Fisher Scientific, USA) and cultured as previously described (see the Cell Culture and Infection Section).65 Then, cells were incubated with different concentrations of B1 and T1 compounds (1 and 10 μM) for 3, 5, 24, and 48 h. After incubation, cell viability was determined using the Triplan Blue criteria. Triplicates were run for each condition. Values were expressed in terms of the percent of untreated control cells set as 100%.
4.2.12.1. Statistical Analysis
The statistical analysis for the colony-forming unit assays was performed employing Welch’s ANOVA (unequal variances) and the post hoc Dunnett T3 multiple comparison tests (low sample size and unequal variances).66,67 The p values were adjusted using the Bonferroni–Holmberg correction. Rifampicin 25 μM was left out of the analysis due to zero variance (always zero values).
4.2.13. High-Performance Liquid Chromatography
To verify the purity of the lead compounds (T1, B1, S1, and S2), of the other two hits from the tethered docking (T2 and T3), and of the control compound (AX20017), we performed high-performance liquid chromatography (HPLC) experiments with an HPLC HP series 1100 instrument and an RP-18 column (Phenomenex, description: sphereClone 5 μm ODS(2), size: 250 × 4.60 mm 5 μm). The injection volume was 20 μL, the UV wavelength for detection was 254 nm, and the mobile phase was the mixture of water/methanol 40:60 for all compounds, except for compound S2 (water/methanol 10:90). The HPLC traces are provided in the Supporting Information (Figures S15–S21). Purity of the lead compounds T1, B1, S1, and S2 and the control compound AX20017 was confirmed to be >95%. The purity values of two non-lead compounds (T2 and T3) were >96% and >84%, respectively.
5. Ancillary Information
5.1. Journal Purity Statement
All lead compounds (T1, B1, S1, and S2) were >95% pure (HPLC analysis).
Acknowledgments
We acknowledge the staff of the EMBL P14 beamline as well as the Sample Preparation and Characterization (SPC) facility of EMBL Hamburg at PETRA III (DESY, Hamburg) for assistance (Stephan Niebling, Christian Günther, Angelica Struve and David Ruiz-Carrillo). We thank Silvia Burastero for helping with the design and illustration of Figures 1, 3, and 6A.
Glossary
Abbreviations
- CFU
colony forming units.
- GarA
glycogen accumulation regulator
- MOI
multiplicity of Infection.
- Mtb
Mycobacterium tuberculosis
- PknG
protein kinase G
- Rif
rifampicin
- SS
solvent site
- TB
tuberculosis
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c02012.
Schematic representation of PknG’s active site bound to ligand B1; PknG kinase activity after incubation with the selected fragments, or the selected known kinase inhibitors; binding affinity estimations of compounds S1, S2, T1, T2, B1, B2, B3, B4, and AX20017 (kinase and rubredoxin domains of PknG); IC50 estimation of compounds T1 and AX20017 (full-length PknG and its natural substrate GarA); intracellular survival of Mtb H37Rv in human macrophages treated with B1, T1, T2, T3, or AX20017 compounds after 24 h; cosolvent site-based (AutoDock Bias) runs of the 20 compounds selected after the tethered docking; ligand B1 bound at the ATP binding site of PknG; thermal shift assay of PknGΔTPRΔ73 mutants I292G, V235P, and Y234L at different concentrations of B1 or S1; electron density map of ligand B1 inside the binding pocket of PknG; post hoc docking of compound B1; HPLC traces of compounds T1, T2, T3, B1, S1, S2, and AX20017; Data Collection and Refinement Statistics of PDBid 7Q52; forward (F) and reverse (R) primers used for mutating PknGΔTPRΔ73; quality control of the protein sample PkngΔTPRΔ73 used for crystallization and the binding affinity estimations; and purity control of compounds B2, B3, and B4 (PDF)
Raw data of fluorescence quenching, ITC, and DSF assays together with instructions to reproduce the analysis and data and analysis scripts to reproduce Figure 6 (macrophage infection) (ZIP)
Molecular Formula Strings (SMILES) of the compounds selected after the tethered docking (including T1, T2, and T3), the derivatives (B1, B2, B3, B4, S1, and S2), and the fragment and kinase inhibitor library hits (CSV)
Full wwPDB X-ray structure validation report for the crystal structure of PknG in complex (PDF)
Accession Codes
Atomic coordinates of PknG in complex with ligand B1 and associated experimental data can be accessed using PDBid 7Q52.
Author Present Address
∇ Institute for Research in Biomedicine (IRB Barcelona)—The Barcelona Institute of Science and Technology (BIST), Barcelona, Spain
Author Contributions
○ O.B. and L.A.D. contributed equally to this work.
Author Contributions
Project design was performed by M.A.M. and A.G.T. Resources, funding acquisition, and supervision were carried out by X.B., M.A.M., N.L.T., M.M.G.A., and A.G.T. The MD simulations in mixed solvents were performed by L.A.D. and J.P.A. The initial docking using rDock and dynamic undocking were performed by E.D.L. with help from X.B. The expression and purification of full-length PknG, GarA, and PknGΔTPRΔ73 was performed by O.B., L.A.D., and J.P.A. The end point kinase inhibition assays were performed by O.B. with help from J.P.A. The synthesis of ligands S1 and S2 was pefrormed by G.G. with help from J.R. Fluorescence quenching, ITC, and DSF experiments were performed by O.B. Assays with radioactive A.T.P. were performed by O.B. with help from M.D.T. and D.W. Crystals were grown by L.A.D. and O.B. Crystal shooting and data collection was performed by I.B. and model building by L.A.D. PknGΔTPRΔ73 mutants were designed, expressed, and purified by O.B. and L.A.D. Macrophage infection assays were performed by C.B.M. and N.L.T. All data analysis was mainly carried out by O.B. with help from the other authors. The first draft was written by O.B., L.A.D., and A.G.T. and further edited by all authors.
This work was supported by Agencia Nacional de Promoción Científica y Tecnológica [PICT2015-2276 and PICT-2018-02519 awarded to A.G.T. and PICT2017-1158 awarded to N.L.T.], Spanish Ministerio de Economía [RTI2018-096429-B-I00 to X.B.], and Catalan Government [2017SGR106 to X.B.]. The authors would like to thank Centro de Cómputos de Alto Rendimiento (CeCAR) for granting the use of computational resources, which allowed part of the calculations included in this work. O.B. did most of the work under a PhD fellowship from CONICET, Argentina, and then under an ARISE fellowship from EMBL. L.A.D. thanks the EMBL Interdisciplinary Postdoc Programme (EIPOD) under Marie Curie Actions COFUND 664726.
The authors declare no competing financial interest.
Notes
Protein renders from Figures 2 and 5 were performed using pymol.68 Renders from Figure 3 were performed using Rhinoceros 3D.69 Figures 2, 4, and 5 were finalized using Inkscape.70 Figures 1 and 3 were finalized using Adobe Illustrator.71 The algorithm for the calculation of the SSs requires python packages networkx, scipy, and pytraj.72−74 To load, process, analyze, and plot the data, we used the R packages, minpack.lm, tidyverse, nlstools, broom, reshape2, baseline, MESS, and plotly.75−82 MD simulations in the mixed solvent were run using AMBER 16 (https://ambermd.org/). MOE 2016.0802 was used to prepare dynamic undocking simulations. LigPrep 2017–1 was used to prepare the virtual screening libraries. REFMAC 5.8.0267, XDS VERSION Feb 5, 2021, AIMLESS 0.7.7, MOLREP 11.7.03, Coot 0.9.6, and ISOLDE 1.2 were used to solve the structure of PknG in complex with ligand B1.
Supplementary Material
References
- Global Tuberculosis Report 2020: Executive Summary. Licence: CC BY-NC-SA 3.0 IGO; Geneva; World Health Organization, 2020.
- de Jonge M. R.; Koymans L. H. M.; Guillemont J. E. G.; Koul A.; Andries K.; Andries K. A Computational Model of the Inhibition of Mycobacterium Tuberculosis ATPase by a New Drug Candidate R207910. Proteins: Struct., Funct., Bioinf. 2007, 67, 971–980. 10.1002/prot.21376. [DOI] [PubMed] [Google Scholar]
- Diacon A. H.; Pym A.; Grobusch M.; Patientia R.; Rustomjee R.; Page-Shipp L.; Pistorius C.; Krause R.; Bogoshi M.; Churchyard G.; Venter A.; Allen J.; Palomino J. C.; De Marez T.; van Heeswijk R. P. G.; Lounis N.; Meyvisch P.; Verbeeck J.; Parys W.; de Beule K.; Andries K.; Neeley D. F. M. The Diarylquinoline TMC207 for Multidrug-Resistant Tuberculosis. N. Engl. J. Med. 2009, 360, 2397–2405. 10.1056/nejmoa0808427. [DOI] [PubMed] [Google Scholar]
- Martinez A.; Torello S.; Kolter R. Sliding Motility in Mycobacteria. J. Bacteriol. 1999, 181, 7331–7338. 10.1128/jb.181.23.7331-7338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane J.; Balcewicz-Sablinska M. K.; Remold H. G.; Chupp G. L.; Meek B. B.; Fenton M. J.; Kornfeld H. Infection by Mycobacterium Tuberculosis Promotes Human Alveolar Macrophage Apoptosis. Infect. Immun. 1997, 65, 298–304. 10.1128/iai.65.1.298-304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Av-Gay Y.; Everett M. The Eukaryotic-like Ser/Thr Protein Kinases of Mycobacterium Tuberculosis. Trends Microbiol. 2000, 8, 238–244. 10.1016/s0966-842x(00)01734-0. [DOI] [PubMed] [Google Scholar]
- Sassetti C. M.; Rubin E. J. Genetic Requirements for Mycobacterial Survival during Infection. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 12989–12994. 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walburger A.; Koul A.; Ferrari G.; Nguyen L.; Prescianotto-Baschong C.; Huygen K.; Klebl B.; Thompson C.; Bacher G.; Pieters J. Protein Kinase G from Pathogenic Mycobacteria Promotes Survival Within Macrophages. Science 2004, 304, 1800–1804. 10.1126/science.1099384. [DOI] [PubMed] [Google Scholar]
- Johnson L. N.; Lewis R. J. Structural Basis for Control by Phosphorylation. Chem. Rev. 2001, 101, 2209–2242. 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- Cowley S.; Ko M.; Pick N.; Chow R.; Downing K. J.; Gordhan B. G.; Betts J. C.; Mizrahi V.; Smith D. A.; Stokes R. W.; Av-Gay Y. The Mycobacterium Tuberculosis Protein Serine/threonine Kinase PknG Is Linked to Cellular Glutamate/glutamine Levels and Is Important for Growth in Vivo. Mol. Microbiol. 2004, 52, 1691–1702. 10.1111/j.1365-2958.2004.04085.x. [DOI] [PubMed] [Google Scholar]
- Ventura M.; Rieck B.; Boldrin F.; Degiacomi G.; Bellinzoni M.; Barilone N.; Alzaidi F.; Alzari P. M.; Manganelli R.; O’Hare H. M. GarA Is an Essential Regulator of Metabolism inMycobacterium Tuberculosis. Mol. Microbiol. 2013, 90, 356. 10.1111/mmi.12368. [DOI] [PubMed] [Google Scholar]
- Wolff K. A.; de la Peña A. H.; Nguyen H. T.; Pham T. H.; Amzel L. M.; Gabelli S. B.; Nguyen L. A Redox Regulatory System Critical for Mycobacterial Survival in Macrophages and Biofilm Development. PLoS Pathog. 2015, 11, e1004839 10.1371/journal.ppat.1004839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff K. A.; Nguyen H. T.; Cartabuke R. H.; Singh A.; Ogwang S.; Nguyen L. Protein Kinase G Is Required for Intrinsic Antibiotic Resistance in Mycobacteria. Antimicrob. Agents Chemother. 2009, 53, 3515–3519. 10.1128/aac.00012-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima A.; Leyva A.; Rivera B.; Portela M. M.; Gil M.; Cascioferro A.; Lisa M. N.; Wehenkel A.; Bellinzoni M.; Carvalho P. C.; Batthyány C.; Alvarez M. N.; Brosch R.; Alzari P. M.; Durán R. Proteome Remodeling in the Mycobacterium Tuberculosis PknG Knockout: Molecular Evidence for the Role of This Kinase in Cell Envelope Biogenesis and Hypoxia Response. J. Proteonomics 2021, 244, 104276. 10.1016/j.jprot.2021.104276. [DOI] [PubMed] [Google Scholar]
- Wittwer M.; Luo Q.; Kaila V. R. I.; Dames S. A. Oxidative Unfolding of the Rubredoxin Domain and the Natively Disordered N-Terminal Region Regulate the Catalytic Activity ofMycobacterium tuberculosisProtein Kinase G. J. Biol. Chem. 2016, 291, 27062–27072. 10.1074/jbc.m116.747089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisa M.-N.; Sogues A.; Barilone N.; Baumgart M.; Gil M.; Graña M.; Durán R.; Biondi R. M.; Bellinzoni M.; Bott M.; Alzari P. M.. A TPR Scaffold Couples Signal Detection to OdhI Phosphorylation in Metabolic Control by the Protein Kinase PknG, 2021. [DOI] [PMC free article] [PubMed]
- Lisa M.-N.; Gil M.; André-Leroux G.; Barilone N.; Durán R.; Biondi R. M.; Alzari P. M. Molecular Basis of the Activity and the Regulation of the Eukaryotic-like S/T Protein Kinase PknG from. Mycobacterium Tuberculosis. Structure 2015, 23, 1039–1048. 10.1016/j.str.2015.04.001. [DOI] [PubMed] [Google Scholar]
- Scherr N.; Honnappa S.; Kunz G.; Mueller P.; Jayachandran R.; Winkler F.; Pieters J.; Steinmetz M. O. Structural Basis for the Specific Inhibition of Protein Kinase G, a Virulence Factor of Mycobacterium Tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 12151–12156. 10.1073/pnas.0702842104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehiro Y.; Tomioka H.; Pieters J.; Tatano Y.; Kim H.; Iizasa H.; Yoshiyama H. Identification of Novel Mycobacterial Inhibitors Against Mycobacterial Protein Kinase G. Front. Microbiol. 2018, 9, 1517. 10.3389/fmicb.2018.01517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N.; Tiwari S.; Srivastava K. K.; Siddiqi M. I. Identification of Novel Inhibitors of Mycobacterium Tuberculosis PknG Using Pharmacophore Based Virtual Screening, Docking, Molecular Dynamics Simulation, and Their Biological Evaluation. J. Chem. Inf. Model. 2015, 55, 1120–1129. 10.1021/acs.jcim.5b00150. [DOI] [PubMed] [Google Scholar]
- Anand N.; Singh P.; Sharma A.; Tiwari S.; Singh V.; Singh D. K.; Srivastava K. K.; Singh B. N.; Tripathi R. P. Synthesis and Evaluation of Small Libraries of Triazolylmethoxy Chalcones, Flavanones and 2-Aminopyrimidines as Inhibitors of Mycobacterial FAS-II and PknG. Bioorg. Med. Chem. 2012, 20, 5150–5163. 10.1016/j.bmc.2012.07.009. [DOI] [PubMed] [Google Scholar]
- Gil M.; Graña M.; Schopfer F. J.; Wagner T.; Denicola A.; Freeman B. A.; Alzari P. M.; Batthyány C.; Durán R. Inhibition of Mycobacterium Tuberculosis PknG by Non-Catalytic Rubredoxin Domain Specific Modification: Reaction of an Electrophilic Nitro-Fatty Acid with the Fe–S Center. Free Radic. Biol. Med. 2013, 65, 150–161. 10.1016/j.freeradbiomed.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B.; Lill M. A. PharmDock: A Pharmacophore-Based Docking Program. J. Cheminf. 2014, 6, 14. 10.1186/1758-2946-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleves A. E.; Jain A. N. Knowledge-Guided Docking: Accurate Prospective Prediction of Bound Configurations of Novel Ligands Using Surflex-Dock. J. Comput. Aided Mol. Des. 2015, 29, 485–509. 10.1007/s10822-015-9846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcon J. P.; Defelipe L. A.; Modenutti C. P.; López E. D.; Alvarez-Garcia D.; Barril X.; Turjanski A. G.; Martí M. A. Molecular Dynamics in Mixed Solvents Reveals Protein–Ligand Interactions, Improves Docking, and Allows Accurate Binding Free Energy Predictions. J. Chem. Inf. Model. 2017, 57, 846–863. 10.1021/acs.jcim.6b00678. [DOI] [PubMed] [Google Scholar]
- Arcon J. P.; Defelipe L. A.; Lopez E. D.; Burastero O.; Modenutti C. P.; Barril X.; Marti M. A.; Turjanski A. G. Cosolvent-Based Protein Pharmacophore for Ligand Enrichment in Virtual Screening. J. Chem. Inf. Model. 2019, 59, 3572–3583. 10.1021/acs.jcim.9b00371. [DOI] [PubMed] [Google Scholar]
- Ghanakota P.; Carlson H. A. Driving Structure-Based Drug Discovery through Cosolvent Molecular Dynamics. J. Med. Chem. 2016, 59, 10383–10399. 10.1021/acs.jmedchem.6b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defelipe L.; Arcon J.; Modenutti C.; Marti M.; Turjanski A.; Barril X. Solvents to Fragments to Drugs: MD Applications in Drug Design. Molecules 2018, 23, 3269. 10.3390/molecules23123269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L.; Klug-Mcleod J.; Rai B.; Lunney E. A. Kinase Hinge Binding Scaffolds and Their Hydrogen Bond Patterns. Bioorg. Med. Chem. 2015, 23, 6520–6527. 10.1016/j.bmc.2015.08.006. [DOI] [PubMed] [Google Scholar]
- Traxler P.; Furet P. Strategies toward the Design of Novel and Selective Protein Tyrosine Kinase Inhibitors. Pharmacol. Ther. 1999, 82, 195–206. 10.1016/s0163-7258(98)00044-8. [DOI] [PubMed] [Google Scholar]
- Székely R.; Wáczek F.; Szabadkai I.; Németh G.; Hegymegi-Barakonyi B.; Erős D.; Szokol B.; Pató J.; Hafenbradl D.; Satchell J.; Saint-Joanis B.; Cole S. T.; Őrfi L.; Klebl B. M.; Kéri G. A Novel Drug Discovery Concept for Tuberculosis: Inhibition of Bacterial and Host Cell Signalling. Immunol. Lett. 2008, 116, 225–231. 10.1016/j.imlet.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Dranchak P.; MacArthur R.; Guha R.; Zuercher W. J.; Drewry D. H.; Auld D. S.; Inglese J. Profile of the GSK Published Protein Kinase Inhibitor Set across ATP-Dependent and-Independent Luciferases: Implications for Reporter-Gene Assays. PLoS One 2013, 8, e57888 10.1371/journal.pone.0057888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Carmona S.; Schmidtke P.; Luque F. J.; Baker L.; Matassova N.; Davis B.; Roughley S.; Murray J.; Hubbard R.; Barril X. Dynamic Undocking and the Quasi-Bound State as Tools for Drug Discovery. Nat. Chem. 2017, 9, 201–206. 10.1038/nchem.2660. [DOI] [PubMed] [Google Scholar]
- Ruiz-Carmona S.; Alvarez-Garcia D.; Foloppe N.; Garmendia-Doval A. B.; Juhos S.; Schmidtke P.; Barril X.; Hubbard R. E.; Morley S. D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571 10.1371/journal.pcbi.1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble M. E.; Endicott J. A.; Johnson L. N. Protein Kinase Inhibitors: Insights into Drug Design from Structure. Science 2004, 303, 1800–1805. 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- Koresawa M.; Okabe T. High-Throughput Screening with Quantitation of ATP Consumption: A Universal Non-Radioisotope, Homogeneous Assay for Protein Kinase. Assay Drug Dev. Technol. 2004, 2, 153–160. 10.1089/154065804323056495. [DOI] [PubMed] [Google Scholar]
- Arcon J. P.; Modenutti C. P.; Avendaño D.; Lopez E. D.; Defelipe L. A.; Ambrosio F. A.; Turjanski A. G.; Forli S.; Marti M. A. AutoDock Bias: Improving Binding Mode Prediction and Virtual Screening Using Known Protein–ligand Interactions. Bioinformatics 2019, 35, 3836. 10.1093/bioinformatics/btz152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindrewicz P.; Li X.; Yates E. A.; Turnbull J. E.; Lian L.-Y.; Yu L.-G. Intrinsic Tryptophan Fluorescence Spectroscopy Reliably Determines Galectin-Ligand Interactions. Sci. Rep. 2019, 9, 11851. 10.1038/s41598-019-47658-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paketurytė V.; Petrauskas V.; Zubrienė A.; Abian O.; Bastos M.; Chen W.-Y.; Moreno M. J.; Krainer G.; Linkuvienė V.; Sedivy A.; Velazquez-Campoy A.; Williams M. A.; Matulis D. Uncertainty in Protein-Ligand Binding Constants: Asymmetric Confidence Intervals versus Standard Errors. Eur. Biophys. J. 2021, 50, 661–670. 10.1007/s00249-021-01518-4. [DOI] [PubMed] [Google Scholar]
- Niebling S.; Burastero O.; Bürgi J.; Günther C.; Defelipe L. A.; Sander S.; Gattkowski E.; Anjanappa R.; Wilmanns M.; Springer S.; Tidow H.; García-Alai M. FoldAffinity: Binding Affinities from nDSF Experiments. Sci. Rep. 2021, 11, 9572. 10.1038/s41598-021-88985-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai N.; Roder H.; Dickson A.; Karanicolas J. Isothermal Analysis of ThermoFluor Data Can Readily Provide Quantitative Binding Affinities. Sci. Rep. 2019, 9, 2650. 10.1038/s41598-018-37072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellman J. A. Macromolecular Binding. Biopolymers 1975, 14, 999–1018. 10.1002/bip.1975.360140509. [DOI] [Google Scholar]
- Specificity and reactivity of Mycobacterium Tuberculosis serine/threonine Kinases PknG and PknB.
- Schrödinger Release 2021-1: LigPrep; Schrödinger, LLC: New York, NY, 2021.
- Beinert H.; Holm R. H.; Münck E. Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science 1997, 277, 653–659. 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- Alvarez-Garcia D.; Barril X. Molecular Simulations with Solvent Competition Quantify Water Displaceability and Provide Accurate Interaction Maps of Protein Binding Sites. J. Med. Chem. 2014, 57, 8530–8539. 10.1021/jm5010418. [DOI] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho A. T. P.; Teixeira A. F. S.; Ramos M. J. Parameters for Molecular Dynamics Simulations of Iron-Sulfur Proteins. J. Comput. Chem. 2013, 34, 1540–1548. 10.1002/jcc.23287. [DOI] [PubMed] [Google Scholar]
- Alvarez-Garcia D.; Barril X. Relationship between Protein Flexibility and Binding: Lessons for Structure-Based Drug Design. J. Chem. Theory Comput. 2014, 10, 2608–2614. 10.1021/ct500182z. [DOI] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Götz A. W.; Poole D.; Le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- López E. D.; Arcon J. P.; Gauto D. F.; Petruk A. A.; Modenutti C. P.; Dumas V. G.; Marti M. A.; Turjanski A. G. WATCLUST: A Tool for Improving the Design of Drugs Based on Protein-Water Interactions. Bioinformatics 2015, 31, 3697–3699. 10.1093/bioinformatics/btv411. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- RDKit. Open-source cheminformatics. http://www.rdkit.org, accessed 2017.
- O’Hare H. M.; Durán R.; Cerveñansky C.; Bellinzoni M.; Wehenkel A. M.; Pritsch O.; Obal G.; Baumgartner J.; Vialaret J.; Johnsson K.; Alzari P. M. Regulation of Glutamate Metabolism by Protein Kinases in Mycobacteria. Mol. Microbiol. 2008, 70, 1408–1423. 10.1111/j.1365-2958.2008.06489.x. [DOI] [PubMed] [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud S. e.; Wilkins M. R.; Appel R. D.; Bairoch A.. Protein Identification and Analysis Tools on the ExPASy Server. The Proteomics Protocols Handbook; Humana Press, 2005; pp 571–607. [Google Scholar]
- Burastero O.; Niebling S.; Defelipe L. A.; Günther C.; Struve A.; Garcia Alai M. M. eSPC: An Online Data-Analysis Platform for Molecular Biophysics. Acta Crystallogr., Sect. D: Struct. Biol. 2021, 77, 1241–1250. 10.1107/s2059798321008998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T.; Yasui N.; Kusakabe Y.; Ito C.; Akamatsu M.; Yamashita A.. Differential Scanning Fluorimetric Analysis of the Amino-Acid Binding to Taste Receptor Using a Model Receptor Protein, the Ligand-Binding Domain of Fish T1r2a/T1r, 2019. [DOI] [PMC free article] [PubMed]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/s0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R. An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 282–292. 10.1107/s090744491003982x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. 10.1107/s0021889897006766. [DOI] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/s0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- Potterton L.; Agirre J.; Ballard C.; Cowtan K.; Dodson E.; Evans P. R.; Jenkins H. T.; Keegan R.; Krissinel E.; Stevenson K.; Lebedev A.; McNicholas S. J.; Nicholls R. A.; Noble M.; Pannu N. S.; Roth C.; Sheldrick G.; Skubak P.; Turkenburg J.; Uski V.; von Delft F.; Waterman D.; Wilson K.; Winn M.; Wojdyr M. CCP4i2: The New Graphical User Interface to the CCP4 Program Suite. Acta Crystallogr., Sect. D: Struct. Biol. 2018, 74, 68–84. 10.1107/s2059798317016035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll T. I. ISOLDE A Physically Realistic Environment for Model Building into Low-Resolution Electron-Density Maps. Acta Crystallogr., Sect. D: Struct. Biol. 2018, 74, 519–530. 10.1107/s2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotz E.; Tateosian N. L.; Salgueiro J.; Bernabeu E.; Gonzalez L.; Manca M. L.; Amiano N.; Valenti D.; Manconi M.; García V.; Moretton M. A. Pulmonary Delivery of Rifampicin-Loaded Soluplus Micelles against Mycobacterium Tuberculosis. J. Drug Deliv. Sci. Technol. 2019, 53, 101170. 10.1016/j.jddst.2019.101170. [DOI] [Google Scholar]
- Welch B. L. On the Comparison of Several Mean Values: an Alternative Approach. Biometrika 1951, 38, 330–336. 10.1093/biomet/38.3-4.330. [DOI] [Google Scholar]
- Dunnett C. W. Pairwise Multiple Comparisons in the Unequal Variance Case. J. Am. Stat. Assoc. 1980, 75, 796–800. 10.1080/01621459.1980.10477552. [DOI] [Google Scholar]
- Schrödinger, LLC . The PyMOL Molecular Graphics System Version 1.8, November, 2015..
- McNeel R. Others Rhinoceros 3D, Version 6.0; Robert McNeel & Associates: Seattle, WA, 2010..
- Harrington B. Others Inkscape 0.92.3 (2405546, 2018-03-11), accessed 2018. http://www.inkscape.org.
- Adobe Inc . Adobe Illustrator April 1, 2019.
- Hagberg A.; Swart P.; Chult D.. Exploring Network Structure, Dynamics, and Function Using Networkx, 2008.
- Virtanen P.; Gommers R.; Oliphant T. E.; Haberland M.; Reddy T.; Cournapeau D.; Burovski E.; Peterson P.; Weckesser W.; Bright J.; van der Walt S. J.; Brett M.; Wilson J.; Millman K. J.; Mayorov N.; Nelson A. R. J.; Jones E.; Kern R.; Larson E.; Carey C. J.; Polat İ.; Feng Y.; Moore E. W.; VanderPlas J.; Laxalde D.; Perktold J.; Cimrman R.; Henriksen I.; Quintero E. A.; Harris C. R.; Archibald A. M.; Ribeiro A. H.; Pedregosa F.; van Mulbregt P. SciPy 1.0 Contributors. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. 3rd PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Elzhov T. V.; Mullen K. M.; Spiess A.-N.; Bolker B.. Minpack. Lm: R Interface to the Levenberg-Marquardt Nonlinear Least-Squares Algorithm Found in MINPACK, plus Support for Bounds. R Package Version 1.2-1, 2016.
- Wickham H.; Averick M.; Bryan J.; Chang W.; McGowan L.; François R.; Grolemund G.; Hayes A.; Henry L.; Hester J.; Kuhn M.; Pedersen T.; Miller E.; Bache S.; Müller K.; Ooms J.; Robinson D.; Seidel D.; Spinu V.; Takahashi K.; Vaughan D.; Wilke C.; Woo K.; Yutani H. Welcome to the Tidyverse. J. Open Source Software 2019, 4, 1686. 10.21105/joss.01686. [DOI] [Google Scholar]
- Baty F.; Ritz C.; Charles S.; Brutsche M.; Flandrois J.-P.; Delignette-Muller M.-L. A Toolbox for Nonlinear Regression in R: The Package Nlstools. J. Stat. Software 2015, 66, 1–21. 10.18637/jss.v066.i05. [DOI] [Google Scholar]
- Robinson D.; Hayes A.; Couch S.. Broom Convert Statistical Objects into Tidy Tibbles, 2021.
- Wickham H. Reshaping Data with the Reshape Package. J. Stat. Software 2007, 21, 1–20. 10.18637/jss.v021.i12. [DOI] [Google Scholar]
- Liland K. H.; Almøy T.; Mevik B.-H. Optimal Choice of Baseline Correction for Multivariate Calibration of Spectra. Appl. Spectrosc. 2010, 64, 1007–1016. 10.1366/000370210792434350. [DOI] [PubMed] [Google Scholar]
- Ekstrøm C. T.MESS: Miscellaneous Esoteric Statistical Scripts, 2020.
- Sievert C.Interactive Web-Based Data Visualization with R, Plotly, and Shiny; CRC Press, 2020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.