

Abstract
In traditional Raman spectroscopy, narrow-band light is irradiated on a sample, and its inelastic scattering, i.e., Raman scattering, is detected. The energy difference between the Raman scattering and the incident light corresponds to the vibrational energy of the molecule, providing the Raman spectrum that contains rich information about the molecular-level properties of the materials. On the other hand, by using ultrashort optical pulses, it is possible to induce Raman-active coherent nuclear motion of the molecule and to observe the molecular vibration in real time. Moreover, this time-domain Raman measurement can be combined with femtosecond photoexcitation, triggering chemical changes, which enables tracking ultrafast structural dynamics in a form of “time-resolved” time-domain Raman spectroscopy, also known as time-resolved impulsive stimulated Raman spectroscopy. With the advent of stable, ultrashort laser pulse sources, time-resolved impulsive stimulated Raman spectroscopy now realizes high sensitivity and a wide detection frequency window from THz to 3000 cm–1, and has seen success in unveiling the molecular mechanisms underlying the efficient functions of complex molecular systems. In this Perspective, we overview the present status of time-domain Raman spectroscopy, particularly focusing on its application to the study of femtosecond structural dynamics. We first explain the principle and a brief history of time-domain Raman spectroscopy and then describe the apparatus and recent applications to the femtosecond dynamics of complex molecular systems, including proteins, molecular assemblies, and functional materials. We also discuss future directions for time-domain Raman spectroscopy, which has reached a status allowing a wide range of applications.
1. Introduction
Since the discovery of the Raman effect by C. V. Raman and K. S. Krishnan in 1928,1 Raman spectroscopy has extensively been utilized in a wide range of research fields from fundamental science to industrial applications, and it has become an indispensable tool for characterizing molecules, materials, biological samples, and others. In traditional Raman spectroscopy, narrow-band light is irradiated on a sample, and its inelastic scattering, i.e., Raman scattering, is detected. The energy difference between the incident light and the Raman scattering corresponds to the vibrational energy of the molecule, which provides rich information about the properties of substances at the molecular level. In this measurement, Raman scattering from the sample is collected, dispersed, and detected, directly providing a Raman spectrum in the frequency domain (Figure 1A). On the other hand, if we use ultrashort optical pulses with duration shorter than the vibrational periods of the molecule, it is possible to induce Raman-active coherent nuclear motion of the molecule and to observe molecular vibrations in real time through the temporal oscillation of the time-resolved spectroscopic signal (Figure 1B).2 This “time-domain” Raman spectroscopy has its origin in ultrafast spectroscopy,3,4 but its direct relevance to traditional frequency-domain Raman spectroscopy was recognized early on: The period and decay of the time-domain Raman signals correspond to the frequency and bandwidth of vibrational bands in the frequency-domain Raman spectrum, respectively, and they can be interconverted through Fourier transformation. While time-domain and frequency-domain Raman spectroscopy provide essentially equivalent information, time-domain Raman spectroscopy is less widely used. Nevertheless, it has been effectively utilized for studying intermolecular vibrations of liquids,5,6 collective and delocalized motions in proteins,7,8 and phonon modes of solids9,10 because it can access the low-frequency region down to THz without the disturbance of Rayleigh (elastic) scattering.
Figure 1.
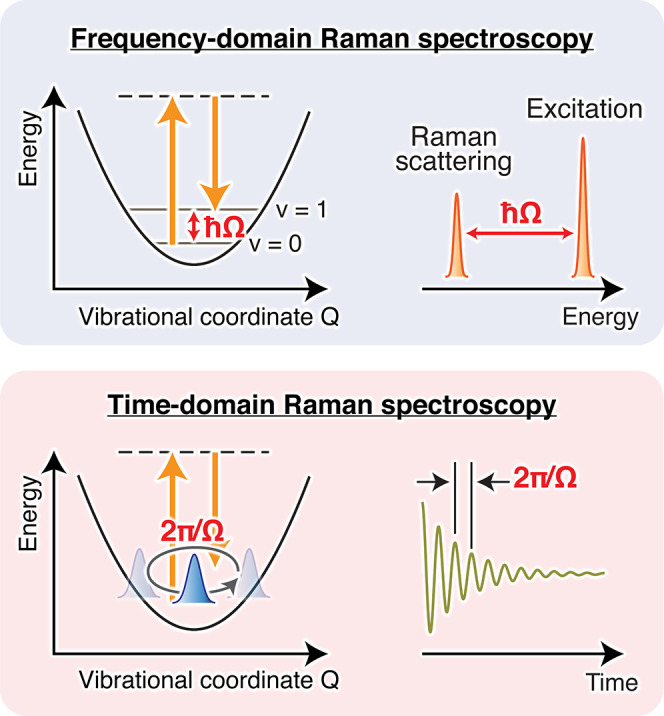
Schematic of frequency- and time-domain Raman spectroscopies. In ordinary frequency-domain Raman spectroscopy, the energy difference between the incident and scattered lights (ℏΩ) provides the vibrational energy of Raman-active modes. In time-domain Raman spectroscopy, Raman-active vibrations are observed as the temporal oscillation of the optical signal (e.g., absorption or reflection) with the period of 2π/Ω. Fourier transform of this oscillatory signal yields a frequency-domain spectrum, which carries essentially the same information as that obtained in frequency-domain Raman spectroscopy.
The uniqueness of time-domain Raman spectroscopy is that it can be performed only using femtosecond pulses. This means that we can define the timing to start the Raman process with a femtosecond accuracy in the Raman measurements. Therefore, by combining the time-domain Raman measurement with a femtosecond actinic pump pulse that initiates a chemical change, we can track ultrafast dynamics through the change of the vibrational spectrum with femtosecond time resolution. This femtosecond time-resolved Raman measurement is not possible with conventional time-resolved spontaneous Raman spectroscopy, in which a picosecond pulse is needed for the Raman probing process to maintain sufficient frequency resolution (<10 cm–1). The high potential of “time-resolved” time-domain Raman spectroscopy was first demonstrated by several studies about 2 decades ago,11−13 but its application had long been limited to a few prototypical molecular systems because of the technical difficulty of generating stable optical pulses shorter than 10 fs. Recently, with the maturation of femtosecond laser technology and ultrafast spectroscopic methods, the power of time-domain Raman spectroscopy has been successfully demonstrated in the studies of ultrafast chemical processes of complex molecular systems. These studies opened a new door to elucidate structural dynamics in a variety of molecular systems with ultrafast Raman spectroscopy.
In this Perspective, we overview the present status of time-domain Raman spectroscopy, with particular focus on its application to femtosecond time-resolved Raman measurements. We first discuss the principle and brief history of time-domain Raman spectroscopy, and then describe the apparatus and several recent applications of femtosecond time-resolved time-domain Raman spectroscopy to complex photochemical systems including proteins, molecular assemblies, and functional materials. Lastly, we comment on several possible future directions of time-resolved time-domain Raman spectroscopy.
2. Principles and Early Studies of Time-Domain Raman Spectroscopy
In time-domain Raman spectroscopy, we illuminate the sample with an ultrashort pulse having a temporal duration that is shorter than the period of the molecular vibration of interest. Because of the time–frequency relationship of the light electric field, such an ultrashort pulse possesses a broad bandwidth that exceeds the frequency of the vibration. Consequently, when interacting with the sample, this ultrashort pulse generates a coherent superposition of Raman-active vibrational states in the initial electronic state through the impulsive stimulated Raman process (Figure 2A). The generated coherent superposition state, which is often called a coherent nuclear wavepacket, is represented as
Here |S⟩ is the initial electronic state, |0⟩ and |1⟩ are its vibrational ground and first excited states, respectively, and Ω is the (angular) frequency of the vibration that corresponds to the energy spacing between the vibrational levels. Since this state is not an eigenstate, it evolves in time (Figure 2B), leading to the temporal modulation of the material properties such as the electronic transition energy, its intensity, and refractive index, with the vibrational frequency of the molecule, Ω. Therefore, the motion of the coherent nuclear wavepacket can be monitored by measuring the temporal modulation of these quantities with another ultrashort pulse that is irradiated after a delay time τ. The obtained time-domain Raman signal carries information about the period and dephasing time of the molecular vibration, which are equivalent to the information about the frequency and bandwidth of the Raman band in traditional frequency-domain Raman spectroscopy, respectively. Actually, the time-domain Raman signal in the non-resonant condition is connected to the frequency-domain Raman spectrum through Fourier transformation,14
Here, Im and 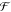 denote the
imaginary part and Fourier transform,
respectively. χ(τ), χ(ω), SS(ω), and SAS(ω)
are the time-domain molecular response (i.e., time-domain Raman signal),
susceptibility, and frequency-domain Raman spectra in the Stokes and
anti-Stokes sides, respectively. In a time-domain Raman experiment,
the first pulse, generating the coherent nuclear wavepacket, is called
the “Raman pump”, whereas the second pulse, monitoring
its motion, is called the “Raman probe”.
denote the
imaginary part and Fourier transform,
respectively. χ(τ), χ(ω), SS(ω), and SAS(ω)
are the time-domain molecular response (i.e., time-domain Raman signal),
susceptibility, and frequency-domain Raman spectra in the Stokes and
anti-Stokes sides, respectively. In a time-domain Raman experiment,
the first pulse, generating the coherent nuclear wavepacket, is called
the “Raman pump”, whereas the second pulse, monitoring
its motion, is called the “Raman probe”.
Figure 2.
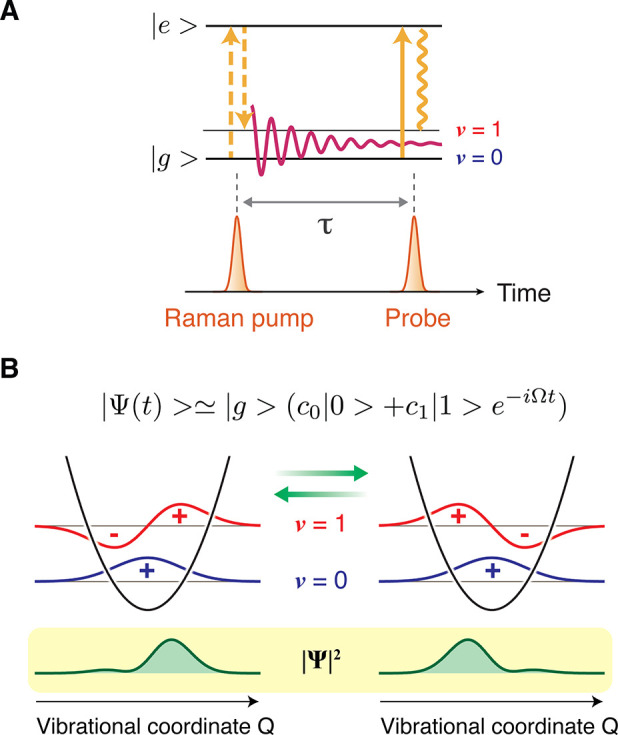
(A) Representative optical process involved in time-domain Raman spectroscopy. The ultrashort Raman pump pulse inherently possesses a bandwidth that exceeds the energy spacing between vibrational energy levels. Therefore, upon interaction with the molecule, it can induce a Raman transition within its bandwidth, creating a coherent superposition of the vibrational eigenstates. This process is called the impulsive stimulated Raman process. (B) The created superposition state evolves in time, meaning that the coherent nuclear wavepacket moves back and forth along the vibrational coordinate. This can be interpreted as a classical nuclear (vibrational) motion of the molecule.
Early time-domain Raman experiments were performed under the electronically non-resonant condition using optical configurations of transient grating (TG) spectroscopy4 or Raman-induced Kerr effect spectroscopy (RIKES),3 both of which monitor the temporal modulation of the refractive index of the sample (Figure 3A,B): In the TG configuration, the Raman pump is irradiated as two crossed beams that create spatial modulation of the refractive index in the sample with the Kerr effect (i.e., transient grating), and its temporal change is monitored by the diffraction of the Raman probe pulse (Figure 3A). The two Raman pump beams, the Raman probe beam, and the diffracted signal beam form the BOXCARS geometry. The first oscillatory time-domain Raman signal due to intramolecular vibration was measured with the TG configuration for the 173 cm–1 Raman-active Br–C–Br bend mode of CH2Br2 using 65-fs pulses.4 Although one can freely set the polarization of the Raman pump and probe pulses in TG spectroscopy for obtaining a particular tensor element of χ(τ), its drawback is that the obtained signal corresponds to the square modulus of the molecular response, i.e., |χ(τ)|2, because the total intensity of the signal diffracted in the phase-matched direction is directly detected. For measuring the relevant molecular response χ(τ) itself, heterodyne detection is required, in which another pulse (local oscillator, LO) is introduced and the interference between the signal and LO is detected. Although heterodyne detection has been realized with the TG geometry,15−18 it is more readily performed in the configuration of RIKES (Figure 3B). In RIKES, the anisotropic refractive index is induced along the polarization direction of the Raman pump pulse with the Kerr effect, and the temporal modulation of the birefringence due to the coherent molecular vibration is monitored through the rotation of the polarization of the Raman probe. The rotated component is selected and detected by placing a polarizer set perpendicular to the initial polarization direction of the Raman probe. With this configuration, heterodyne detection is achieved by making the polarization of the Raman probe slightly elliptic, thereby allowing a small portion of the Raman probe pulse to pass the polarizer and interfere with the signal to act as LO.19
Figure 3.
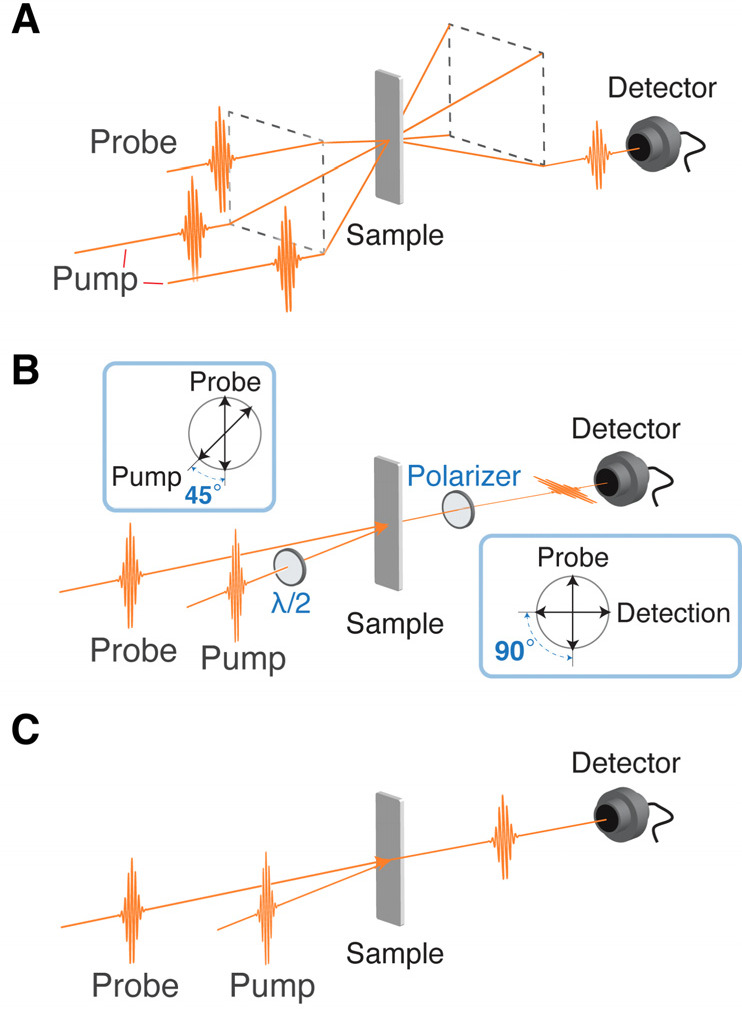
Typical geometries of the time-domain Raman measurement. (A) Transient grating (TG) spectroscopy geometry, also known as BOXCARS geometry. Spatial modulation of the refractive index is induced by the two Raman pump pulses, and its temporal oscillation is monitored by detecting the diffracted Raman probe pulse emitted in the phase-matched direction. (B) Raman-induced Kerr effect spectroscopy (RIKES) geometry. The anisotropy of the refractive index (birefringence) is induced by the Raman pump pulse, and its temporal oscillation is monitored by detecting the rotation of the polarization of the Raman probe pulse. The signal can be heterodyne-detected by making the Raman probe pulse slightly elliptic with a λ/4 plate inserted to the Raman probe path. (C) Pump–probe geometry. The Raman pump pulse impulsively excites coherent Raman vibration, and resultant temporal modulation of the refractive index or electronic transition frequency is detected through the spectral shift or the absorption intensity change, respectively, of the Raman probe pulse.
In the 1990s, time-domain Raman spectroscopy was intensively utilized to study low-frequency nuclear dynamics such as relaxation of liquids and intermolecular vibrations.3,5,20 Most of the experiments were performed with (optically) heterodyne-detected RIKES (OHD-RIKES),19 and the time-domain Raman data provide high-quality depolarized Raman spectra by Fourier transformation. Scattering, which is the main obstacle in the frequency-domain Raman spectroscopy, appears as a constant (DC) offset in the temporal trace in time-domain Raman measurements, and it can be readily separated from the oscillatory components representing molecular vibrations. Therefore, time-domain Raman spectroscopy enables us to obtain high-quality Raman spectra in the low-frequency region down to THz. Castner et al. reported a representative study on the Raman spectrum of liquid water. They obtained a low-frequency Raman spectrum (0–139 cm–1) of liquid water with OHD-RIKES and connected it with a frequency-domain (depolarized) Raman spectrum in the higher-frequency region, providing a Raman spectrum in the frequency range from 0 to 1200 cm–1.21 This study nicely demonstrated the equivalence and complementarity of time- and frequency-domain Raman spectroscopies.
Time-domain Raman spectroscopy has also been performed under the electronically resonant condition, whereby intrinsically weak Raman signals can be enhanced. The first time-domain observation of coherent molecular vibration upon resonant excitation was reported by Tang and co-workers,22,23 which was soon followed by Mokhtari and co-workers and Shank and co-workers.24,25 In the electronically resonant condition, the electronic transition energy of the molecule is modulated with coherent nuclear wavepacket motion, so that the transition probability at a certain wavelength is modulated accordingly. Thus, time-domain Raman measurements can be performed with the simple pump–probe scheme shown in Figure 3C. The pump pulse acts as the Raman pump, whereas the probe pulse works as the Raman probe that monitors the temporal modulation of the absorbance at the specific wavelength. This time-domain Raman measurement with the pump–probe geometry is possible also in the non-resonant condition by monitoring the spectral shift of the probe pulse, which is induced by the temporal change of the refractive index, and it is easily realized by placing an optical filter before the detector or by dispersed detection of the probe spectrum.26−28 In addition to the simplicity of the optical configuration, this pump–probe scheme automatically realizes heterodyne detection because the Raman probe pulse also acts as LO and the time-domain Raman signal is detected as the intensity change of the Raman probe pulse as the result of the interference between the signal and LO. Therefore, the pump–probe scheme is most widely utilized for time-domain Raman measurements under the electronically resonant condition.
The term “impulsive stimulated Raman spectroscopy (ISRS)” is often used to refer to time-domain Raman spectroscopy performed in the TG configuration. However, ISRS can be used to generally refer to time-domain Raman spectroscopy, including that performed with the pump–probe configuration.
3. Time-Resolved Impulsive Stimulated Raman Spectroscopy
As described in the previous section, time-domain Raman spectroscopy is advantageous for measuring Raman spectra in the low-frequency region. However, its most distinct feature is that we can carry out Raman spectroscopy using only femtosecond pulses, which enables us to study ultrafast dynamics and short-lived transient species with “time-resolved” time-domain Raman spectroscopy.
In the extension of time-domain Raman spectroscopy to time-resolved measurements, which we call time-resolved impulsive stimulated Raman spectroscopy (TR-ISRS), an actinic pump (P1) pulse first excites the molecule to the excited state to start photochemical/photophysical processes, and the subsequent temporal changes are monitored by ISRS using Raman pump (P2) and probe (P3) pulses (Figure 4). In this measurement, two delay times, ΔT and τ, need to be controlled: ΔT is the P1–P2 delay time, which corresponds to the delay time in ordinary time-resolved spectroscopy, whereas τ is the P2–P3 delay time that is scanned for measuring time-domain Raman signals. Because both the P1 and P2 pulses are femtosecond pulses, we can define the timing to initiate the time-domain Raman measurement by the P2 pulse, i.e., ΔT, with femtosecond temporal accuracy. In other words, the temporal change of the time-resolved Raman spectra can be tracked with femtosecond time resolution. The time-resolved Raman spectrum at ΔT is obtained by Fourier transformation of the oscillatory time-domain Raman signal that is recorded with the P3 pulse by changing τ. The frequency resolution of the obtained Raman spectrum is determined by the scanning time range of τ. Therefore, when the delay time τ is scanned sufficiently long, the bandwidth of the obtained Raman band is determined only by the dephasing time of the oscillatory time-domain Raman signal, i.e., the dephasing time of the molecular vibration (a few ps). (Note that this observed dephasing may include the contribution from inhomogeneous dephasing due to the molecules in microscopically different environments.) Consequently, TR-ISRS can track the temporal change of the Raman spectra with femtosecond temporal accuracy (or femtosecond time resolution) while maintaining high frequency resolution. This is not possible with traditional time-resolved spontaneous Raman spectroscopy, in which the time and frequency resolutions are determined by the duration and bandwidth of the probe pulse, respectively. The duration and bandwidth of the optical pulse cannot be narrowed simultaneously because they are Fourier conjugates, which practically limits the time resolution of time-resolved spontaneous Raman spectroscopy at ∼2–3 ps to maintain a frequency resolution (<10 cm–1) sufficient for vibrational spectroscopy. We note that simultaneous realization of the femtosecond time resolution and high frequency resolution in TR-ISRS never violates the uncertainty principle. As explained, the time resolution of the TR-ISRS measurement is determined by the accuracy of the P1–P2 delay time ΔT, while the frequency resolution is determined by the scanning range of the P2–P3 time delay τ. Obviously, these two factors are independent of each other.
Figure 4.
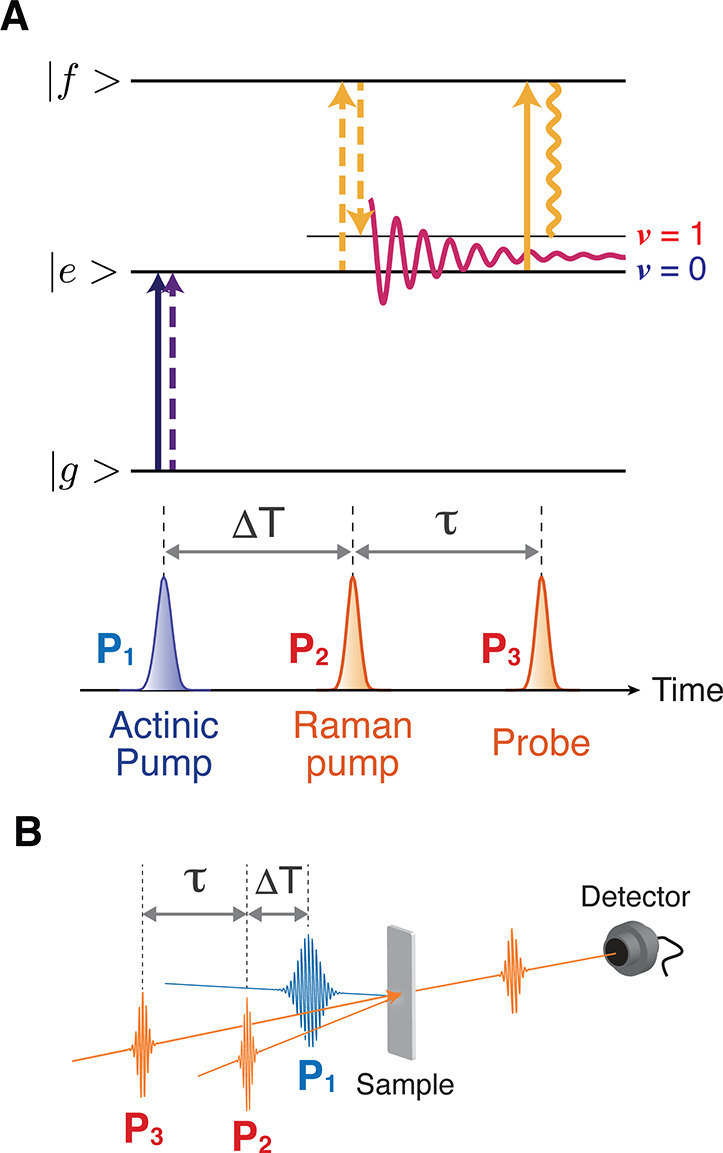
Time-resolved impulsive stimulated Raman spectroscopy (TR-ISRS). (A) Schematic diagram representing the transitions involved in TR-ISRS. The time-domain Raman measurement is performed at the arbitrary delay time ΔT, after the initiation of photoreaction by the actinic pump pulse. (B) Typical optical geometry employed in the TR-ISRS measurement. Three pulses, that is, the actinic pump, Raman pump, and Raman probe pulses, are non-collinearly introduced to the sample. The time-domain Raman signal is recorded as the oscillatory component of the Raman-pump-induced absorbance change that is monitored by the Raman probe.
The feasibility of TR-ISRS was first demonstrated by Ruhman and co-workers, who measured vibrational relaxation process of I2– that was generated by photodissociation of I3–.11 A while after this seminal work, TR-ISRS of excited-state polyatomic molecules was realized by two groups almost simultaneously. Our group carried out TR-ISRS of S1trans-stilbene,12 which is a prototypical molecule exhibiting photoisomerization,29,30 and observed in-plane bending vibration (290 cm–1) of the S1 state under the resonance condition using the Sn←S1 absorption. Cerullo, Ruhman, and co-workers reported TR-ISRS of the S1 state of an analogue of oligo(p-phenylenevinylene), which was measured under the resonance condition using the S1→S0 stimulated emission.13 Later, the true capability of TR-ISRS to investigate ultrafast structural dynamics was demonstrated by the study of the photoisomerization of cis-stilbene that proceeds with a ∼1 ps time constant.31 The obtained femtosecond time-resolved Raman spectra showed that a characteristic Raman band at 240 cm–1 exhibits a marked frequency downshift on the sub-picosecond time scale, indicating that cis-stilbene gradually changes its structure on the S1 potential energy surface on this time scale. TR-ISRS was successfully used for studying structural dynamics in solid-state materials as well. For example, Trigo and co-workers reported softening of a Raman-active phonon mode in a photoexcited perovskite, Cs2Au2I6,32 and Dexheimer and co-workers studied structural relaxation in the self-trapped exciton of the mixed-valence linear chain material [Pt(en)2][Pt(en)2Br2]·(ClO4)4.33
We note that the time-domain Raman measurements in TR-ISRS have been carried out with either two-pulse pump–probe or three-pulse TG (also known as BOXCARS)12,34 configurations. In particular, TG-type TR-ISRS has extensively been utilized by Motzkus and co-workers, which they call pump degenerate four-wave mixing (pump-DFWM).35−38 Blank and co-workers used the same geometry to study solvation dynamics by monitoring the temporal change of the non-resonant solvent Raman signal after photoexcitation of the solute dye molecule.39,40
4. Time-Resolved Time-Domain Raman Spectroscopy Using Few-Cycle Pulses
Early TR-ISRS studies successfully demonstrated the high potential of time-domain Raman spectroscopy for investigating structural dynamics of ultrafast chemical processes. However, its application had mostly been limited to simple molecules and observation of the low-frequency vibrations of <1000 cm–1 because of the technical difficulty of generating ultrashort pulses having long-term stability. In particular, because we need optical pulses with duration shorter than the period of molecular vibrations of interest, ultrashort optical pulses as short as <10 fs are required for observing all fundamental vibrations up to ∼3000 cm–1. In this regard, shortening the pulse duration from 20 to 10 fs makes an essential difference because it corresponds to doubling the observable frequency window from ∼1000 to ∼2000 cm–1, even though this factor of 2 does not make so drastic difference in the observation of ultrafast population dynamics in most cases. Thus, for utilizing TR-ISRS to track structural dynamics of complex molecular systems, we need a highly sensitive experimental setup that features very stable few-cycle, <10-fs optical pulses.
Figure 5A shows a schematic of a typical TR-ISRS setup that was developed based on a Ti:sapphire regenerative amplifier as the light source.28 The output of the regenerative amplifier is divided into two, and a half is used for generating an actinic pump pulse tunable over a broad spectral range (Figure 5B) by second/third harmonic generation or an optical parametric amplifier (OPA) followed by optional, further frequency mixing. The other half of the output is used for generating sub-10-fs pulses that are used for the Raman pump and Raman probe pulses. In order to produce such an ultrashort optical pulse for the measurements, we first need to generate a very broadband optical pulse and then temporally compress it to make its duration as short as possible. We employ an OPA with a non-collinear geometry (non-collinear OPA, NOPA41−43) and generate broadband pulses having a bandwidth that supports a few-cycle <10-fs pulse duration (Figure 5C). Because the velocities of different spectral components are different due to the material dispersion, the pulse duration of this broadband pulse is elongated after propagation in the setup, which is called “chirp”. Thus, it is necessary to temporally compress the output of the NOPA to prepare the shortest pulse possible. In this setup, the pulse compression is realized by the combination of a prism pair and a 4-f dispersion compensator equipped with a micromachined membrane deformable mirror (MMDM) at the Fourier plane. The shape of the MMDM (hence the optical path length for each color component) can be optimized using a simple genetic algorithm so that the shortest pulse can be prepared at the sample position.44−46 As shown in Figure 5D,E, nearly Fourier transform limit sub-10-fs pulses are routinely obtainable, with the shortest pulse duration available down to 5.8 fs.47 Pulse compression based on a MMDM is versatile because the pulse duration can be optimized without changing any other configurations of the setup, and very clean pulses are obtainable by compensating for higher-order dispersion. Other types of pulse shapers, e.g., those based on the spatial light modulator (SLM), acousto-optic modulator (AOM), and acousto-optic programmable dispersive filter (AOPDF), are alternatively used for fine pulse compression. Double chirped mirrors (DCM) can be also used for obtaining sub-10-fs pulses,48−51 and Kukura and co-workers carried out TR-ISRS using ultrashort pulses compressed with DCM.52−54 Compression with DCM is more economical and easy to handle, although changing the number of bounces on DCM alters the optical path and hence the delay time. In addition, the phase structure becomes complicated when the number of the bounces becomes large. If one only investigates the dynamics of low-frequency Raman modes (< ∼1000 cm–1), ∼20-fs pulses are short enough for the Raman pump and Raman probe. Such pulses are readily obtainable by traditional compression methods such as a prism pair,55 grating pair,56 or their combination.57
Figure 5.

(A) Schematic diagram of the TR-ISRS setup. Abbreviations: NOPA, non-collinear optical parametric amplifier; MMDM, micromachined membrane deformable mirror; HWP, half-wave plate; BS, beam splitter; CP, compensation plate; VND, variable neutral density filter. (B) Typical spectra of the actinic pump pulse. (C) Typical spectra of the Raman pump and Raman probe pulses, enabling transform-limited pulse duration of ∼6 fs. (D) SHG FROG trace of the compressed NOPA output, the spectrum of which is shown in panel C with a yellow solid line. (E) Retrieved intensity and phase profiles of the compressed NOPA output. Panels A–E adapted with permission from ref (28). Copyright 2016 American Institute of Physics.
In the setup shown in Figure 5A, the actinic pump, Raman pump, and Raman probe pulses are focused on the sample in a non-collinear geometry, and the total intensity of the transmitted probe pulse is detected by a photodiode (open-band detection). An alternative way of detection is that the probe pulse is dispersed with a polychromator and detected by a multichannel detector (dispersed detection).37,52 The advantage of open-band detection is the simplicity of the detection system and the high S/N readily achieved. Furthermore, open-band detection efficiently suppresses non-resonant Raman signals (e.g., those from solvent or solute in the ground state) because non-resonant signals do not noticeably change the total intensity of the Raman probe pulse.26,27 On the other hand, the advantage of dispersed detection is that it provides information about the probe wavelength dependence of the time-domain Raman signal intensity (and phase).37,52 Nevertheless, special care is needed for non-resonant Raman signals that show up as a result of the wavelength-selected monitoring in dispersed detection.52
Figure 6 shows typical non-resonant ISRS signals measured with sub-7-fs pulses. In this measurement, a colored glass filter is inserted in front of the detector for detecting non-resonant Raman signals from acetonitrile. The wavelength-selected detection is necessary in the non-resonant case because the coherent nuclear wavepacket motion modulates the spectrum of the Raman probe pulse via the Kerr effect but the change in its total intensity is negligible (Figure 6A). The Fourier transform power spectra clearly show that Raman-active vibrations of acetonitrile up to 3000 cm–1 can be easily observed with high S/N, including the CN stretching (2254 cm–1) and CH stretching (2944 cm–1) vibrations (Figure 6B). This data clearly demonstrate that, with the use of sub-7-fs pulses, TR-ISRS is capable of tracking the change of the Raman spectrum in the frequency region from THz to 3000 cm–1, covering all fundamental vibrations with femtosecond time resolution and high sensitivity.
Figure 6.
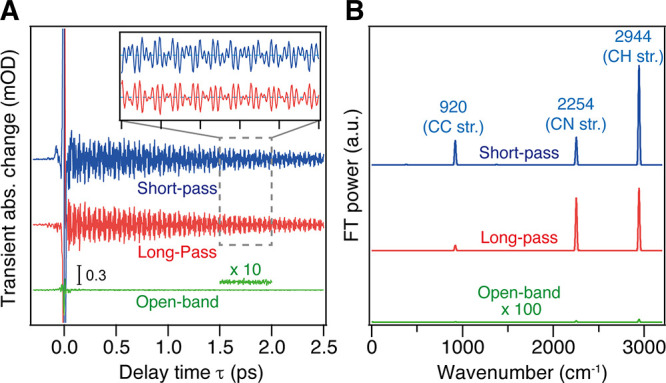
(A) Non-resonant ISRS signals of acetonitrile measured with the short-pass filter (blue), with the long-pass filter (red), and without filters (green) in front of the detector. (B) Fourier transform power spectra of the non-resonant ISRS signals of acetonitrile. Panels A and B adapted with permission from ref (28). Copyright 2016 American Institute of Physics.
5. Applications of TR-ISRS to Ultrafast Structural Dynamics in Complex Molecular Systems
The high sensitivity and wide observable frequency window (THz to 3000 cm–1) of TR-ISRS with stable sub-10-fs pulses enabled the studies on ultrafast structural dynamics of complex molecular systems. In the following, we overview several recent applications of TR-ISRS to show how the time-domain Raman approach can address the dynamics, structure, and functioning mechanism of proteins, molecular assemblies, and functional materials.
5.1. Proton Transfer in Protein: Green Fluorescent Protein
Proton transfer is one of the most important elementary processes in chemistry, and it has been extensively studied so far. In particular, recently, proton transfer in a well-organized hydrogen-bond network in proteins has attracted special attention. The high capability of TR-ISRS to track structural dynamics on the femtosecond time scale enabled addressing a controversial topic about the mechanism of the excited-state proton transfer (ESPT) in green fluorescent protein (GFP).
GFP is the prototype of fluorescent proteins that are now widely used for fluorescence imaging in biology, and the fluorescence of GFP is emitted from the autocatalytically formed chromophore, p-hydroxybenzylideneimidazolinone.58 In wild-type GFP, most of the chromophore exists in the neutral form, and it undergoes the ESPT after photoexcitation: The phenolic proton of the excited state in the neutral form (A* state) is transferred to nearby Glu222 through the hydrogen-bond network that involves a hydrogen-bonded water molecule and Ser205 (Figure 7A).59,60 The resultant anionic form of the excited-state chromophore (I* state) emits bright green fluorescence that is used for bioimaging. The photochemical reaction dynamics of GFP has attracted much attention not only for understanding the fluorescence emission mechanism to develop more useful fluorescent proteins but also for elucidating the proton-transfer dynamics that proceed in a structurally well-organized hydrogen-bond network in the protein.
Figure 7.

(A) Schematic of the excited-state proton-transfer reaction in wild-type GFP. (B) Pump–probe (black) and TR-ISRS signals (yellow area, ΔT = 0.75 ps) of wild-type GFP (pH 8.0, in H2O). The oscillatory component of the pump–probe signal is extracted (blue), and its Fourier transform power spectrum is shown in the inset. (C) Raw TR-ISRS signals at selected ΔT delay times. (D) Time-resolved Raman spectra of wild-type GFP at various ΔT delay times (ΔT = 0–1 and 40 ps), obtained as Fourier transform power spectra of the oscillatory components of the TR-ISRS signals. The corresponding 2D representation is also shown at the bottom. Panels A–D adapted with permission from ref (62). Copyright 2016 American Chemical Society.
For this ultrafast ESPT process in GFP, Mathies and co-workers carried out femtosecond stimulated Raman spectroscopy (FSRS) and succeeded in observing femtosecond time-resolved Raman spectra of the initial excited state, the A* state.61 Interestingly, it was found that the amplitude and frequency of several Raman bands of the A* state exhibit clear oscillation with ∼280 fs period, which they considered arises from the anharmonic coupling with low-frequency coherent nuclear wavepacket motion induced by photoexcitation. They assigned this low-frequency motion to the phenol-ring wagging motion of the chromophore and proposed that this coherent motion modulates the hydrogen-bond strength between the chromophore and the nearby water molecule to promote the ESPT process coherently. This ESPT mechanism, coherently driven by low-frequency nuclear motion, attracted much interest in the community.
Although this proposal was tempting, its validity remained to be examined experimentally. In fact, to prove this coherently driven ESPT mechanism, it is necessary to observe not only the coherent oscillation of the neutral reactant excited state (A* state) but also the corresponding oscillation in the growth of the anionic product excited state (I* state). This requires selective monitoring of the temporal evolution of the A* and I* states during the ultrafast ESPT process, which is difficult with femtosecond time-resolved absorption spectroscopy, because the electronic spectra are broad and not sensitive to the structure. On the other hand, Raman bands are sharp and sensitive to the structural difference, so that TR-ISRS is capable of monitoring the dynamics of the reactant and product excited states during the ESPT process, individually.62
Figure 7B shows the raw data obtained in the TR-ISRS measurement of GFP. The wavelength of the Raman probe pulse (as well as the Raman pump pulse) is tuned to the stimulated emission band of GFP, and hence the raw signal appears with a negative sign (as “negative” transient absorption). The signal obtained without the Raman pump (black) corresponds to the conventional pump–probe signal, and it exhibits a gradual growth of the stimulated emission signal on a few-picosecond time scale, representing a gradual increase in the I* state population as the ESPT proceeds. Low-frequency coherent oscillation appears with the population signal, and this oscillatory component is extracted and shown in blue. This oscillation starting at the time origin is attributed to the coherent nuclear wavepacket motion in the initial A* state, which is driven simultaneously with photoexcitation by the ∼110-fs actinic pump (P1) pulse. Upon the introduction of the Raman pump (P2) pulse at ΔT, Raman-active coherent nuclear wavepacket motion is induced. This impulsive Raman signal is accompanied by a sudden decrease of the stimulated emission signal, because the P2 pulse also dumps the I* state population to the S0 state. The difference between the signal obtained with/without the Raman pump pulse is the raw TR-ISRS signal (yellow area).
Figure 7C depicts the TR-ISRS signal of GFP obtained by introducing the Raman pump (P2) pulse at different ΔT delay times. They clearly show oscillatory features reflecting the coherent nuclear wavepacket motion of the excited-state GFP chromophore initiated at each ΔT delay time. Fourier transformation of these oscillations provides the femtosecond time-resolved Raman spectra shown in Figure 7D. In the obtained femtosecond time-resolved Raman spectra, the Raman bands due to both the reactant A* state and product I* state of the ESPT are clearly seen with high S/N, and the change of the excited-state chromophore from the neutral to anionic form is clearly traced. Notably, for the reactant A* state, a high-frequency Raman band at 1142 cm–1 (phenolic CH bend) exhibits a clear intensity modulation, confirming that the GFP chromophore certainly possesses a low-frequency mode having significant anharmonicity with high-frequency modes. Nevertheless, the actual question is whether the formation of the product I* state exhibits the corresponding oscillation or not. As clearly seen in Figure 8, no oscillation was observed within the S/N in the temporal evolution of the Raman band of the product I* state during its population growth with the ESPT. This result reveals that the coherent low-frequency motion is certainly induced in the initial A* state with photoexcitation, but it does not play a major role in the ESPT process, at least not at room temperature. The TR-ISRS data show that the ESPT in GFP at room temperature is not coherently driven solely by one single low-frequency mode of the chromophore but rather involves thermally excited motions, including those of the hydrogen-bond chain, to transfer a proton.
Figure 8.
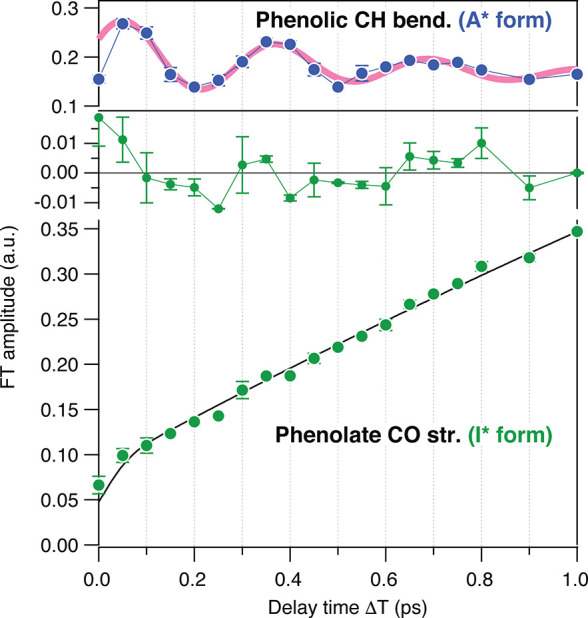
Temporal change of the Fourier transform amplitude of the phenolate CO stretch of the I* form (green, lower), with the best fit with an exponential function convoluted with the instrumental response of 110 fs (black line). The residuals are also shown (green, middle). The oscillatory feature of the phenolic CH bend of the A* form is shown for comparison (upper). Figure adapted with permission from ref (62). Copyright 2016 American Chemical Society.
This TR-ISRS study of GFP demonstrates the high capability of TR-ISRS to track femtosecond structural dynamics of not only small molecules but also complex molecules such as proteins. In fact, TR-ISRS is a very powerful method to elucidate how the ultrafast structural response triggers functional activation in photoreceptor proteins, as described in the next section.
5.2. Primary Structural Change in Photoreceptor Protein: Photoactive Yellow Protein
Photoreceptor proteins play essential roles in converting light to chemical energy for driving biological responses, and they are indispensable for living organisms.63 Functions of the photoreceptor proteins are driven by photoinduced reactions of the chromophore, followed by a chain of molecular-level changes in the protein (photocycle) that spans from femtosecond to second time scales, finally leading to macroscopic biological responses. For unraveling the molecular mechanism underlying a variety of highly efficient functions of photoreceptor proteins, various methods have been utilized to characterize the reaction intermediates in the photocycle. Nevertheless, the structural dynamics on the femto- to picosecond time scale still remain vague because of the experimental difficulties, despite its critical role in determining the fate of the subsequent chemical/biological processes. A major challenge is that not only high temporal resolution but also high sensitivity are required for clarifying the primary process in the photoreceptor proteins, because most of these proteins are not very stable and easily undergo photodegradation. The high sensitivity of TR-ISRS enables the elucidation of ultrafast structural dynamics in the primary processes of photoreceptor proteins.
Photoactive yellow protein (PYP), discovered in Halorhodospira halophila,64 is a photoreceptor protein that is responsible for the negative phototactic response of this organism.65 This function of PYP is realized by a photocycle that starts with photoinduced trans-to-cis isomerization of the chromophore, p-coumaric acid (pCA), which proceeds on the femto- to picosecond time scale (Figure 9A). Because of its relatively small size (14 kDa) and high solubility in water, PYP has been intensively studied as a model system for elucidating structure–function relationships in photoreceptor proteins.66 However, despite extensive experimental and theoretical efforts, primary structural events in PYP remained unclear. In particular, information on the structure of the short-lived excited state and the first ground-state intermediate, the I0 state, has been severely limited. TR-ISRS provided snapshot femtosecond time-resolved Raman spectra of PYP, giving new insights into the primary processes of its photoreception.67
Figure 9.

(A) Crystal structure (PDB ID: 2ZOI) and photocycle of PYP. The chromophore conformation and hydrogen-bond status (blue dotted lines) in each intermediate are indicated. pG and pG* represent the ground (S0) and excited (S1) states of PYP, respectively. (B) Oscillatory component of the TR-ISRS signal of PYP obtained at various ΔT delay times. (C) Time-resolved Raman spectra at various ΔT delay times, obtained as Fourier transform power spectra of the oscillatory components of the TR-ISRS signals. Contour representation of the Fourier transform amplitude (after interpolation) is shown at the top. The steady-state pre-resonance stimulated Raman spectrum of the S0 state (pG) obtained with 490 nm Raman excitation is also shown at the bottom. (D) Temporal profiles of the Fourier transform amplitude of selected bands, highlighting the distinct temporal behavior of the 135 cm–1 band. Broken orange lines denote the best global fits to the data using the three time constants obtained from the broadband transient absorption spectroscopy, which represent the pG* population decay. Error bars represent 95% confidence intervals. Panels A–D adapted with permission from ref (67). Copyright 2017 Nature Publishing Group.
Figure 9B shows the oscillatory component of the TR-ISRS signals measured at different ΔT delay times, and Figure 9C depicts the femtosecond time-resolved spectra obtained through Fourier transformation. The obtained data capture the vibrations of the excited-state pCA chromophore from THz to 1800 cm–1 and, notably, a marked spectral change in the low-frequency region on the femtosecond time scale: The excited-state Raman band at 135 cm–1 exhibits a drastic intensity drop within a few hundred femtoseconds after photoexcitation (Figure 9D). This low-frequency band was assigned to an intermolecular vibration that includes the motion of the hydrogen-bonded amino acid residues at the phenolic tail part of pCA, based on theoretical calculations as well as resonance Raman studies of PYP and its various mutants.68,69 The control TR-ISRS experiments for the E46Q mutant revealed that the intensity of this low-frequency 135 cm–1 band serves as the hydrogen-bond strength marker, and hence its rapid intensity drop indicates that a rapid weakening of the hydrogen bond that anchors pCA in the protein pocket occurs immediately after photoexcitation.
In addition to the ultrafast hydrogen-bond dynamics in the excited state, TR-ISRS also captured the vibration of the first ground-state intermediate, the I0 state, which is recognized as the appearance of the 628 cm–1 band in the first few picoseconds (Figure 9C). In the I0 state, it was believed that the pCA chromophore has the cis form, but it was not directly proved, while the structure of its counterpart in the crystal phase was reported.70,71 Although the amplitude of the oscillatory signal due to the I0 state is only ∼10 μOD level (Figure 10A), it is clearly observed by virtue of the high sensitivity of the TR-ISRS measurement. As shown in Figure 10B, the Fourier transform power spectrum of the I0 state exhibits a peculiar intensity pattern that shows a markedly strong band at 628 cm–1, suggesting a unique skeletal structure of the I0 state. By comparing the obtained spectrum with a spectrum computed based on the crystal structure of the first ground-state intermediate in the crystal phase (Figure 10C),70 it was concluded that the chromophore has the cis form but is substantially twisted to maintain the hydrogen bond with the protein backbone (Figure 10D). It is noteworthy that the structure of the first ground-state intermediate in the crystal phase was controversial at that time because two groups reported two distinctly different structures, which were called pR0 and IT, from similar 100-ps time-resolved Laue diffraction experiments.70−73 The pR0 structure has the chromophore having a distorted cis form, whereas the IT structure has the chromophore having a perpendicular conformation around the isomerizing C=C bond. Although these two groups used essentially the same experimental method and analytical procedure, the determined structures were significantly different because of the difference in the constraint in the parameters to solve the structure.72,73 The Raman spectrum computed based on the pR0 structure is very similar to the spectrum of the I0 state obtained by the TR-ISRS experiment (Figure 10C). This indicates that the pR0 structure is the structure of the first ground-state intermediate in the crystal phase if we assume that the structure of the intermediate is essentially the same in the crystal and solution. Actually, later, an intermediate structure that resembles the pR0 structure was reported by femtosecond time-resolved crystallography using an X-ray free electron laser.74
Figure 10.
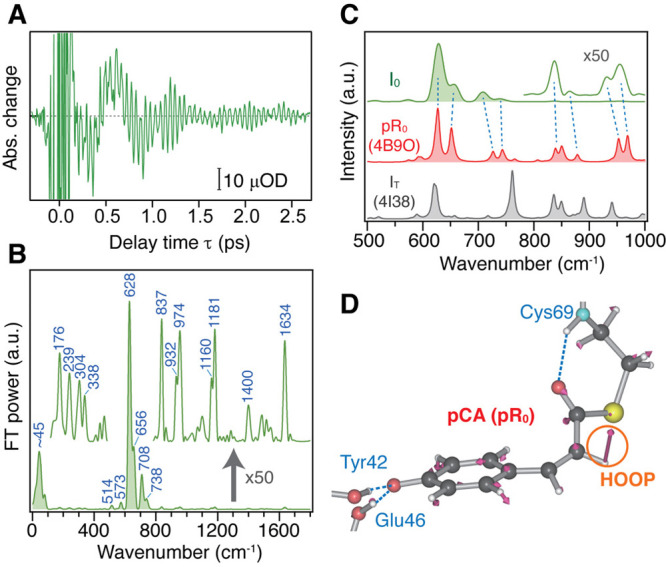
(A) Oscillatory component of the TR-ISRS signal of PYP at ΔT = 60 ps, obtained after subtracting the slowly changing population component. (B) Time-resolved Raman spectrum at ΔT = 60 ps, obtained as the Fourier transform power spectrum of the oscillatory component shown in panel A. Weak bands are shown with a magnified scale (×50). (C) Calculated Raman spectra of the two model systems based on the pR0 and IT structures (PDB ID: 4B9O and 4I38) in the fingerprint region, shown with 8 cm–1 bandwidth. The experimental I0 spectrum is also shown for comparison. (D) Optimized structure of the model PYP based on pR0. Nuclear motion associated with the calculated 628 cm–1 mode is also shown with arrows. Panels A–D adapted with permission from ref (67). Copyright 2017 Nature Publishing Group.
This study clearly demonstrates that TR-ISRS is a very powerful method for revealing ultrafast, small, but significant structural changes inside photoreceptor proteins, and it can provide unique information that even state-of-the-art X-ray crystallographic techniques are not yet able to resolve.
5.3. Chemical Bond Formation in Molecular Assembly: Gold Complex Oligomer
Formation and dissociation of chemical bonds are essential in chemical reactions, and it has been desired for a long time to elucidate how such bond rearrangement proceeds at the frontiers of reaction dynamics studies. Because bond formation/dissociation and the associated structural rearrangements proceed on the femto- to picoseconds time scale, their real-time observation is, in principle, possible using ultrafast spectroscopy.47,75,76 However, the bond formation process has been seldom investigated because triggering the bond formation at a well-defined, desired timing is difficult, although it is indispensable for applying an ultrafast spectroscopic technique. TR-ISRS has been used to acquire a detailed picture of the bond formation and subsequent structural dynamics in a unique molecular assembly system, i.e., the trimer of dicyanoaurate(I) complex ([Au(CN)2–]), in which chemical bonds are formed with photoexcitation. For this study, the advantage of time-domain Raman spectroscopy in observing low-frequency vibrations was fully utilized.
In the ground state, several [Au(CN)2–] complexes form loosely bound oligomers in solution, with the aurophilic interaction occurring between the closed-shell d10 Au atoms.77 Photoexcitation of this oligomer can promote one electron from the antibonding σ* orbital (dz2–dz2) to the bonding σ orbital (pz–pz), and consequently, tight covalent bonds are formed between Au atoms in the oligomer (Figure 11A).78 Thus, this molecular assembly serves as a good model system where one can examine ultrafast structural dynamics relevant to chemical bond formation in real time.79−81 The excited-state dynamics of the [Au(CN)2–] trimer, the most prototypical oligomer, was first studied by time-resolved absorption spectroscopy by Iwamura et al.79 On the basis of the observed spectral changes and DFT calculation, it was concluded that the trimer undergoes rapid contraction of the Au–Au bonds due to bond formation immediately after photoexcitation, which is followed by sub-picosecond intersystem crossing (ISC) and bent-to-linear structural change proceeding on the picosecond time scale. Later, Kim et al. studied this system by X-ray solution scattering82 and observed essentially the same dynamics. However, they claimed that the bent-to-linear structural change occurs first and then ISC follows, accompanied by a slight shortening of the Au–Au bonds. Since then, the structural dynamics of the [Au(CN)2–] trimer had been controversial despite its apparent simplicity in the molecular structure.80,83 To resolve this controversy, TR-ISRS was applied.84
Figure 11.

(A) Schematic of photoinduced bond formation of the [Au(CN)2–] trimer. (B) Oscillatory component of the TR-ISRS signals of 0.3 M K[Au(CN)2] aqueous solution at various ΔT delay times. (C) Time-resolved Raman spectra at various ΔT delay times, obtained as Fourier transform power spectra of the oscillatory components of the TR-ISRS signals. (D) Time-resolved Raman spectra at ΔT = 10 ps obtained for the K[Au(CN)2] aqueous solution of various concentrations. The spectra are normalized for the Au–Au breathing vibration of the T1 trimer at ∼90 cm–1, which is indicated by the asterisk. (E) Frequency change of the observed Au–Au breathing vibration. (F) Schematic of the Au–Au bond-formation dynamics of the [Au(CN)2–] trimer revealed by TR-ISRS. Panels A−F adapted with permission from ref (84). Copyright 2019 American Chemical Society.
Figure 11B shows the oscillatory component of the TR-ISRS signal obtained from the 0.3 M K[Au(CN)2] aqueous solution with photoexcitation at 310 nm, and Figure 11C depicts the femtosecond time-resolved Raman spectra obtained with Fourier transformation. The concentration employed in this study is the same as that in the previous studies using femtosecond time-resolved absorption and X-ray solution scattering.79,82 The obtained femtosecond time-resolved Raman spectra of 0.3 M K[Au(CN)2] aqueous solution clearly showed four prominent transient Raman bands, but only the ∼90 cm–1 band remained when the concentration was diluted down to 0.1 M solution (Figure 11D). This result revealed that not only the trimer but also a larger oligomer (i.e., tetramer) coexist in solution at the concentration as high as 0.3 M, and they are simultaneously excited in this experimental condition. Therefore, only the ∼90 cm–1 transient Raman band is attributed to the trimer, and it is assignable to its Au–Au breathing vibration in the lowest excited triplet (T1) state. This T1 trimer band exhibits a marked frequency upshift from 86 to 99 cm–1 with a ∼3 ps time constant (Figure 11E). This frequency change is in good agreement with the frequency shift predicted by a DFT calculation for the bent-to-linear structural change of the T1 trimer. Being supported by fluorescence up-conversion measurements conducted separately, the following ultrafast dynamics of the [Au(CN)2–] trimer has finally been clarified (Figure 11F): With photoexcitation of the loosely bound trimer in the S0 state, the tight Au–Au bonds are formed within ∼200 fs. Subsequently, the S1→T1 ISC occurs with ∼400 fs time constant, and it is followed by the bent-to-linear structural change on the T1 potential energy surface with ∼3 ps time constant. This reaction scheme is, in fact, the same as that proposed in the first time-resolved absorption study.79
The [Au(CN)2–] oligomer looks like a simple system suitable for investigating bond formation dynamics, but it turns out that it is quite complex because of the coexistence of the multiple species.79−81 The TR-ISRS study of the [Au(CN)2–] trimer demonstrated the necessity of carefully controlled experiments in studying structural dynamics of complex molecular assemblies. This study also clearly showed the power of ultrafast vibrational spectroscopy to distinguish multiple transient species that simultaneously appear in the femtosecond processes of complex systems.
5.4. Ultrafast Elementary Processes in Functional Materials
As described in the previous three sections, TR-ISRS has been successfully applied to complex molecular systems and has established its status as an exquisite tool for investigating structural dynamics of ultrafast chemical processes. Nevertheless, the application of TR-ISRS has still been limited, and there are a variety of problems left in chemistry (or in physics) that could be addressed by TR-ISRS. The subjects are not limited to samples in the solution phase, and solid-state samples such as organic/inorganic molecular crystals or semiconductors can be targets, where structural changes associated with photoinduced charge transfer and phase transitions85−89 or exciton dynamics90 are of fundamental importance. In fact, TR-ISRS has already been used as a powerful tool to study the ultrafast dynamics of functional materials.
For instance, TR-ISRS has been effectively used in the study of singlet fission. In singlet fission, two (triplet) excitons are generated from one photon, and thus it has been attracting tremendous interest as a possible way to achieve solar light–energy conversion beyond the Shockley–Queisser limit.91,92 Polyacenes have been studied as a model system of singlet fission because they exhibit triplet formation on an ultrafast time scale (∼100 fs), with the quantum yield reaching 200%.93,94 However, the detailed mechanism of this highly efficient process remained unclear.
Rao, Kukura, and co-workers carried out high time-resolution time-resolved absorption spectroscopy of a thin film of TIPS-pentacene using a 10-fs pump pulse. Resonant excitation with such an ultrashort pulse induces coherent nuclear wavepacket motion not only in the ground state (through the resonant impulsive Raman process) but also in the photoexcited state, because the bandwidth of the ultrashort pulse also exceeds the vibrational frequency of the targeted excited state. Coherent nuclear wavepacket motion launched in the excited state reports rich information on the structural change induced by photoexcitation, and thus it has been extensively used to gain insights into the coherent nuclear dynamics immediately after photoexcitation.95−100 They separately measured the Raman spectrum of the TT state after singlet fission using TR-ISRS, and compared it with the Fourier spectrum of the oscillation of transient absorption due to the excited-state nuclear wavepacket motion that was directly induced by photoexcitation (Figure 12A). This comparison led them to conclude that vibrational coherence launched in the initial photoexcited S1 state of TIPS-pentacene is transferred to the TT state, surviving the singlet fission process. In particular, vibrational coherences with periods shorter than the time constant of singlet fission (∼80 fs) were found to survive, which is difficult to explain by an incoherent singlet fission model. Based on the similarity of this observation to the ultrafast internal conversion process through a conical intersection in a model polyene system, they proposed that singlet fission in TIPS-pentacene is a vibrationally coherent process mediated by a conical intersection (Figure 12B).54 The same approach was also used to study the intramolecular singlet fission process in the pentacene dimer 13,13′-bis(mesityl)-6,6′-dipentacenyl (DP-Mes), reaching the same conclusion. For this system, the results were analyzed in more detail using a quantum-mechanical Tree Tensor Network State (TTNS) simulation, allowing the researcher group to assign the observed modes to tuning modes of the conical intersection and to further infer the crucial coupling modes.101
Figure 12.
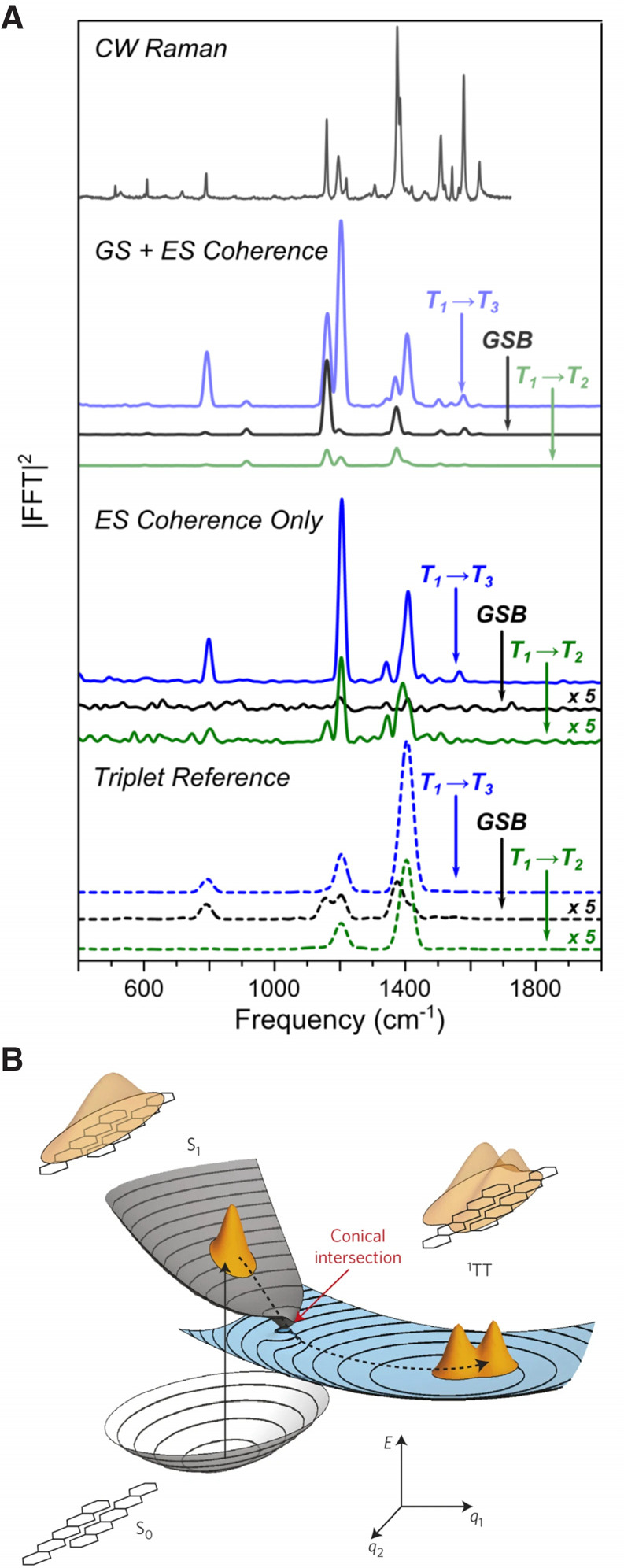
(A) Comparison of the vibrational spectra of a TIPS-pentacene thin film obtained by pump–probe spectroscopy and TR-ISRS. A spontaneous Raman spectrum is also shown for comparison. The pump–probe measurement yields a vibrational spectrum with mixed contribution from the ground and excited states because the ultrashort pump pulse induces coherent nuclear wavepacket motion in both states (GS + ES Coherence). An additional dump pulse was added to the pump–probe measurement to deplete only the excited-state population, yielding the pure excited-state vibrational spectrum as the differential signal between with and without the dump pulse (ES Coherence). The extracted excited-state vibrational spectrum agrees well with the Raman spectrum of the T1 state obtained by TR-ISRS (Triplet Reference), indicating that the vibrational coherence launched in the Franck–Condon state (S1) survives the singlet fission process and thus is transferred to the resultant triplet state. (B) Schematic of singlet fission in TIPS-pentacene via a conical intersection between the singlet- and triplet-pair manifolds. Panel A is adapted from the Supplementary Information of ref (54). Panel B is adapted with permission from ref (54). Copyright 2015 Nature Publishing Group.
Very recently, Kim and co-workers applied TR-ISRS to the study of the photoinduced charge-transfer process in functional π-electronic systems.102,103 They obtained femtosecond time-resolved Raman spectra of a diketopyrrolopyrrole–pyrrolopyrrole dyad using TR-ISRS and concluded that the charge-separation process proceeds in a two-step fashion, which involves the initial bright exciton state, an intermediate partial charge-transfer state, and the final charge-separated state, based on the observed spectral change with complementary quantum chemical calculation.102 For donor–acceptor–donor-type quadrupolar perylene bisimide (PBI), they observed substantial frequency shifts of characteristic high-frequency bands (i.e., C–H bend, perylene ring stretch, C=C stretch, and C=O stretch) during the photoinduced charge separation process on the femto- to picosecond time scale. This result indicates that a substantial structural change occurs on PBI upon charge separation, which they ascribed to a change in the bond-length alternation pattern on the basis of TD-DFT calculations.103
As described in this section, TR-ISRS has been used effectively to study structural dynamics in the functional materials systems that are important for device applications. These successful applications of TR-ISRS demonstrate the general versatility of time-domain Raman spectroscopy and its potential to address the role of primary structural dynamics in the device performance.
6. Discussion
6.1. TR-ISRS vs FSRS
As has been described so far, time-domain Raman spectroscopy is a powerful counterpart of traditional frequency-domain Raman spectroscopy. In particular, combined with photoexcitation to start chemical changes, it can be used for tracking structural dynamics on the femtosecond time scale as time-resolved time-domain Raman spectroscopy, i.e., TR-ISRS. As there always exist counterparts in the time domain and frequency domain, femtosecond time-resolved Raman spectroscopy can also be performed in the frequency domain by femtosecond stimulated Raman spectroscopy (FSRS).104−107 In FSRS, instead of a single ultrashort Raman pump pulse used in TR-ISRS, a pair of narrow-band picosecond pulse (Raman pump) and broadband femtosecond pulse (Raman probe) are used to induce Raman-active vibrational coherences. Then, the picosecond Raman pump pulse interacts again to generate the stimulated Raman signal, which is dispersed in a spectrograph and provides a stimulated Raman spectrum as the gain feature of the broadband Raman probe spectrum. In FSRS, the Raman probe is a femtosecond pulse, and hence it can determine the timing to start the Raman process with femtosecond accuracy, so that FSRS can also provide femtosecond time-resolved Raman spectra.108 TR-ISRS and FSRS are counterparts in the time and frequency domains, and hence they can provide essentially the same information.109 However, because of the difference in the experimental implementation, each method has advantages and disadvantages.
The biggest advantage of FSRS is the simplicity of the experimental setup. The FSRS measurements do not require an extremely short pulse (∼10 fs) and need only a narrow-band picosecond pulse and a broadband femtosecond pulse such as white-light continuum, both of which are readily prepared with the present laser technology. Actually, it is possible to perform FSRS simply by introducing a narrow-band picosecond Raman pump pulse to a conventional femtosecond time-resolved absorption setup that is implemented with a femtosecond (actinic) pump and broadband probe pulses. In addition to this easiness of the implementation, FSRS is advantageous in the acquisition time for obtaining a single spectrum because of multichannel detection in the frequency domain.
Despite this instrumental advantage, it is often difficult to obtain unambiguous time-resolved Raman spectra with FSRS for two reasons. First, FSRS needs a troublesome background subtraction. Because femtosecond time-resolved Raman spectra are usually measured under the electronically resonant condition, the Raman signals appear with a broad background that arises from the transient electronic signals (bleaching of transient absorption and/or stimulated emission) induced by the Raman pump pulse. Because the shape of these background signals is neither simple nor predictable, the background subtraction becomes somewhat arbitrary, leaving uncertainty in the obtained spectra, although some attempts at reliable background subtraction have been reported.110−113 Second, because the stimulated Raman signal is automatically heterodyned by the Raman probe pulse in FSRS, the obtained frequency-domain spectrum corresponds to the imaginary part of the relevant third-order non-linear susceptibility (Imχ(3)). This automatic heterodyning is convenient, but it can also make the bandshape of the Raman signal complicated when the Raman pump and probe pulses are in resonance with electronic transitions: The electronic resonances may be realized not only with the upper electronic excited state but also with the lower electronic ground state, which introduces additional imaginary factors to χ(3). As a result, the Raman bands do not necessarily show Lorentzian lineshapes. These two problems often complicate the femtosecond time-resolved spectra obtained by FSRS and make their interpretation difficult.
TR-ISRS does not suffer these problems of FSRS. Although subtraction of the background due to the electronic signals is also necessary in TR-ISRS, the background signal in the time domain is well fitted by exponential functions because it reflects population decays of the relevant transient species. The bandshape change depending on the resonance condition in FSRS corresponds to the change in the initial phase of the vibration in the time domain, and it can be eliminated in Fourier transform power/amplitude spectra, thus enabling the straightforward interpretation of the spectrum obtained with TR-ISRS. It is also noted again, as a common advantage of time-domain Raman measurements, that TR-ISRS can readily access the low-frequency region down to the THz region,84 because the Raman vibration is observed as the oscillatory feature in the time domain and it can easily be separated from the scattering that appears as a constant offset in the time domain.
The disadvantage of TR-ISRS is that it requires stable ∼10-fs pulses and needs a longer acquisition time for scanning the delay time τ to cover the dephasing time of the Raman-active vibrational coherence (free-induction decay time). Fortunately, the generation of ∼10-fs pulses has now become easier in the visible region thanks to the advance of ultrafast laser technology, e.g., pulse shapers and chirped mirrors, although it is still challenging in other wavelength regions. On the other hand, FSRS is more readily performed in a wide wavelength range from deep ultraviolet to near-infrared using a picosecond OPA.114−119 Therefore, at present, it may be reasonable to choose TR-ISRS for the visible region and FSRS for other regions such as ultraviolet and near-infrared for carrying out femtosecond time-resolved Raman spectroscopy.
6.2. Future Directions
Lastly, we discuss several possible future directions of time-domain Raman spectroscopy, in particular those of time-resolved time-domain Raman spectroscopy, i.e., TR-ISRS.
As described in this Perspective, TR-ISRS enables us to investigate the structural dynamics on the time scale of femto- to picoseconds. In this sense, TR-ISRS can be a very powerful tool to realize real-time tracking of the structural change of the molecules. A molecular conformational change at a relatively large scale generally proceeds on the femtosecond to early picosecond time scale, which is longer than the period of typical molecular vibrations. Therefore, the frequency of a relevant vibration can gradually change “quasi-adiabatically” while the structural change of the molecule is progressing.31 In other words, TR-ISRS allows us to track the motion of evolving structural degrees of freedom through temporal evolution of femtosecond time-resolved Raman spectra, as exemplified by the detection of the bent-to-linear structural change of the T1 [Au(CN)2–] trimer through the gradual frequency shift of the Au–Au breathing vibration.84 By extending this approach, it would be possible to map out the reaction coordinate of photochemical/photobiological reactions with the help of complementary computation. This is the distinct advantage of TR-ISRS, as well as femtosecond vibrational spectroscopy in general, over time-resolved vibrational spectroscopy on a slower time scale that monitors the spectral change corresponding to the population change from one chemical species to another. Furthermore, TR-ISRS can be an indispensable tool to examine the relevance of specific nuclear motion to the reaction, as demonstrated in the study of GFP.62 For example, recent time-resolved absorption studies on artificial light-harvesting and photovoltaics systems suggested that specific vibrations of the donor and acceptor to which the relevant electronic states are coupled play a key role in driving the charge separation, implying the importance of the vibronic coupling in the performance of these artificial, photofunctional systems.120,121 In these systems, the oscillation of the charge-transfer magnitude was deduced from the coherent oscillatory feature in the time-resolved absorption signals assigned to the relevant electronic states. However, in many molecular systems, electronic absorptions of the reactant, transients, and product are so broad and overlapped that it is very often difficult to selectively monitor the population dynamics of each species. By monitoring the temporal evolution of the species-specific Raman bands, which are sharp and distinct for each species, it will be more straightforward, reliable, and robust to address how specific nuclear motion facilitates an ultrafast chemical reaction.
From the experimental viewpoint, a natural future direction of TR-ISRS is the extension to a wider wavelength region, i.e., the ultraviolet and near-infrared regions. All the TR-ISRS experiments reported to date have been carried out in the spectral region of 450–900 nm, which is accessed with NOPA based on Ti:sapphire or Yb:KGW laser systems. Extending the tunability of the Raman pump and Raman probe pulses to the ultraviolet and near-infrared regions broadens the scope of the problems that can be studied by TR-ISRS. For example, after exciting the chromophore of photoreceptor proteins, we may be able to track the structural dynamics of the surrounding amino acid residues with TR-ISRS in the ultraviolet by taking advantage of the electronic resonance of aromatic amino acid residues (tyrosine, tryptophan, and phenylalanine).122 This experiment is expected to provide valuable information about the cooperativity of the structural dynamics of the chromophore and the surrounding protein matrix. This problem has been intensively studied on the picosecond or slower time scale using time-resolved ultraviolet resonance Raman spectroscopy123,124 but seldom investigated on the femtosecond time scale. Complementary information can be obtained also by FSRS in the ultraviolet,116,119 and FSRS in the deep ultraviolet region was recently realized to monitor the primary protein response after photoexcitation of the retinal chromophore of bacteriorhodopsin.125 Ultraviolet TR-ISRS will be advantageous for accessing the low-frequency region and hence for addressing functional roles of intermolecular vibrations and/or collective motion of the protein matrix. Near-infrared TR-ISRS will be suitable for studying structural dynamics of the excited states that have loosely bound electrons in large conjugated systems such as oligothiophenes.118 We note that the generation of tunable ultrashort pulses in the ultraviolet and near-infrared regions has been demonstrated,126−128 and hence TR-ISRS in these wavelength regions is within reach.
From the viewpoint of the light source, it is noteworthy that time-domain Raman measurements have been realized with two synchronized mode-locked lasers that oscillate with slightly different repetition rates, which achieves free-running fast scanning of the delay time τ.129 Based on this technology, TR-ISRS has also been performed recently with synchronized triple mode-locked lasers of slightly detuned repetition rates, which enables both fast data acquisition and time-resolved measurement over a wide ΔT delay time range.130 Although the applicable resonance condition is limited due to the lack of wavelength tunability, it could be an alternative to the present TR-ISRS measurements based on the amplifier systems.
It is also tempting to combine TR-ISRS with surface-enhanced Raman scattering (SERS). In SERS, the molecules are adsorbed on the rough surface of metals, and the Raman signals from these adsorbates are highly enhanced, with an apparent enhancement factor of ∼101–106 through the surface plasmon resonance.131−133 Very high SERS enhancement at a so-called hot spot has enabled the detection of the Raman signal from a single molecule,134−137 which prompts us to dream of femtosecond time-resolved Raman spectroscopy of a single molecule by the combination of SERS and TR-ISRS, that is, TR-SE-ISRS. TR-SE-ISRS will also be a powerful tool to study plasmon-induced photochemical reactions, which have been attracting extensive attention although their mechanisms have been controversial.138 In this direction, we recently reported time-domain measurements of surface-enhanced Raman signals and demonstrated that surface-enhanced ISRS (SE-ISRS) can achieve an enhancement factor as high as 106.139
Extending TR-ISRS to multi-dimensional spectroscopy is another exciting direction. In TR-ISRS, the actinic pump pulse is used primarily to populate the excited state. However, as the study on GFP demonstrated,62 TR-ISRS can monitor the vibrational coupling in the excited state by using the actinic pump pulse also to induce coherent wavepacket motion with photoexcitation. Two-dimensional Fourier transformation of such TR-ISRS signals along the two delay times, ΔT and τ, provides a two-dimensional frequency–frequency correlation map which can visualize the coupling between different vibrational motions. We recently reported the first demonstration of this two-dimensional ISRS (2D-ISRS) approach for PYP and showed its potential to characterize the multi-dimensional potential energy surface of the reactive excited-state molecules.140 Although 2D-ISRS is technically demanding at the moment, it will be a valuable tool to investigate the reactive potential energy surfaces in detail and energy flow thereon, in particular with the use of a stable, high-repetition-rate light source such as a Yb:KGW amplifier. In fact, very recently, a similar experiment was reported for GFP using a setup based on a Yb:KGW light source.141
7. Conclusion
In this Perspective, we overviewed the principles, advances, and recent applications of time-domain Raman spectroscopy, in particular those of its extension to time-resolved measurements, i.e., TR-ISRS. We discussed how time-domain Raman spectroscopy is used to probe ultrafast structural events in complex molecular systems, beyond what was considered possible in the early days of this spectroscopy. With the advent of stable, ultrashort laser pulse sources, the time-resolved time-domain Raman approach has seen success in unveiling the molecular mechanisms underlying functions of molecular systems as complex as photoreceptors, molecular assemblies, and photon-energy conversion materials, with high sensitivity and a wide detection frequency window from THz to 3000 cm–1. At present, this type of time-domain Raman spectroscopy requires an elaborate optical setup, compared to conventional ultrafast spectroscopic techniques such as transient absorption and fluorescence upconversion. However, the continuing development of laser and optical technology is eliminating technical difficulties and making this advanced spectroscopic method more accessible and versatile as a tool to investigate ultrafast structural dynamics and functioning mechanisms of complex molecular systems in a broad field ranging from biology to materials science. We envision that time-domain Raman spectroscopy will further extend the frontier of the study of ultrafast dynamics, and new insights to be obtained will facilitate a deep understanding of chemical reactions and strategic design of novel and advanced functional materials.
Acknowledgments
This work was partly supported by JST, PRESTO Grant Number JPMJPR17P4, Japan (to H.K.).
The authors declare no competing financial interest.
References
- Raman C. V.; Krishnan K. S. A New Type of Secondary Radiation. Nature 1928, 121 (3048), 501–502. 10.1038/121501c0. [DOI] [Google Scholar]
- Dhar L.; Rogers J. A.; Nelson K. A. Time-Resolved Vibrational Spectroscopy in the Impulsive Limit. Chem. Rev. 1994, 94 (1), 157–193. 10.1021/cr00025a006. [DOI] [Google Scholar]
- Kalpouzos C.; Lotshaw W. T.; McMorrow D.; Kenney-Wallace G. A. Femtosecond Laser-Induced Kerr Responses in Liquid Carbon Disulfide. J. Phys. Chem. 1987, 91 (8), 2028–2030. 10.1021/j100292a008. [DOI] [Google Scholar]
- Ruhman S.; Joly A. G.; Nelson K. A. Time-Resolved Observations of Coherent Molecular Vibrational Motion and the General Occurrence of Impulsive Stimulated Scattering. J. Chem. Phys. 1987, 86 (11), 6563–6565. 10.1063/1.452400. [DOI] [Google Scholar]
- Chang Y. J.; Castner E. W. Fast Responses from ‘Slowly Relaxing’ Liquids: A Comparative Study of the Femtosecond Dynamics of Triacetin, Ethylene Glycol, and Water. J. Chem. Phys. 1993, 99 (10), 7289–7299. 10.1063/1.465710. [DOI] [Google Scholar]
- Heisler I. A.; Meech S. R. Low-Frequency Modes of Aqueous Alkali Halide Solutions: Glimpsing the Hydrogen Bonding Vibration. Science 2010, 327 (5967), 857–860. 10.1126/science.1183799. [DOI] [PubMed] [Google Scholar]
- Turton D. A.; Senn H. M.; Harwood T.; Lapthorn A. J.; Ellis E. M.; Wynne K. Terahertz Underdamped Vibrational Motion Governs Protein-Ligand Binding in Solution. Nat. Commun. 2014, 5, 3999. 10.1038/ncomms4999. [DOI] [PubMed] [Google Scholar]
- Giraud G.; Karolin J.; Wynne K. Low-Frequency Modes of Peptides and Globular Proteins in Solution Observed by Ultrafast OHD-RIKES Spectroscopy. Biophys. J. 2003, 85 (3), 1903–1913. 10.1016/S0006-3495(03)74618-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portuondo-Campa E.; Tortschanoff A.; van Mourik F.; Chergui M. Ultrafast Nonresonant Response of TiO2 Nanostructured Films. J. Chem. Phys. 2008, 128 (24), 244718. 10.1063/1.2949517. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Miyata K.; Fu Y.; Wang J.; Joshi P. P.; Niesner D.; Williams K. W.; Jin S.; Zhu X. Y. Screening in Crystalline Liquids Protects Energetic Carriers in Hybrid Perovskites. Science 2016, 353 (6306), 1409. 10.1126/science.aaf9570. [DOI] [PubMed] [Google Scholar]
- Banin U.; Ruhman S. Ultrafast Vibrational Dynamics of Nascent Diiodide Fragments Studied by Femtosecond Transient Resonance Impulsive Stimulated Raman Scattering. J. Chem. Phys. 1993, 99 (11), 9318–9321. 10.1063/1.465501. [DOI] [Google Scholar]
- Fujiyoshi S.; Takeuchi S.; Tahara T. Time-Resolved Impulsive Stimulated Raman Scattering from Excited-State Polyatomic Molecules in Solution. J. Phys. Chem. A 2003, 107 (4), 494–500. 10.1021/jp0270856. [DOI] [Google Scholar]
- Cerullo G.; Lüer L.; Manzoni C.; De Silvestri S.; Shoshana O.; Ruhman S. Time Domain Investigation of Excited-State Vibrational Motion in Organic Molecules by Stimulated Emission Pumping. J. Phys. Chem. A 2003, 107 (40), 8339–8344. 10.1021/jp022696j. [DOI] [Google Scholar]
- Cho M.; Du M.; Scherer N. F.; Fleming G. R.; Mukamel S. Off-Resonant Transient Birefringence in Liquids. J. Chem. Phys. 1993, 99 (4), 2410–2428. 10.1063/1.465256. [DOI] [Google Scholar]
- Vöhringer P.; Scherer N. F. Transient Grating Optical Heterodyne Detected Impulsive Stimulated Raman Scattering in Simple Liquids. J. Phys. Chem. 1995, 99 (9), 2684–2695. 10.1021/j100009a027. [DOI] [Google Scholar]
- Matsuo S.; Tahara T. Phase-Stabilized Optical Heterodyne Detection of Impulsive Stimulated Raman Scattering. Chem. Phys. Lett. 1997, 264 (6), 636–642. 10.1016/S0009-2614(96)01399-1. [DOI] [Google Scholar]
- Goodno G. D.; Dadusc G.; Miller R. J. D. Ultrafast Heterodyne-Detected Transient-Grating Spectroscopy Using Diffractive Optics. J. Opt. Soc. Am. B 1998, 15 (6), 1791–1794. 10.1364/JOSAB.15.001791. [DOI] [Google Scholar]
- Maznev A. A.; Nelson K. A.; Rogers J. A. Optical Heterodyne Detection of Laser-Induced Gratings. Opt. Lett. 1998, 23 (16), 1319–1321. 10.1364/OL.23.001319. [DOI] [PubMed] [Google Scholar]
- Smith N. A.; Meech S. R. Optically-Heterodyne-Detected Optical Kerr Effect (OHD-OKE): Applications in Condensed Phase Dynamics. Int. Rev. Phys. Chem. 2002, 21 (1), 75–100. 10.1080/01442350110092701. [DOI] [Google Scholar]
- Righini R. Ultrafast Optical Kerr Effect in Liquids and Solids. Science 1993, 262 (5138), 1386–1390. 10.1126/science.262.5138.1386. [DOI] [PubMed] [Google Scholar]
- Castner E. W.; Chang Y. J.; Chu Y. C.; Walrafen G. E. The Intermolecular Dynamics of Liquid Water. J. Chem. Phys. 1995, 102 (2), 653–659. 10.1063/1.469177. [DOI] [Google Scholar]
- Rosker M. J.; Wise F. W.; Tang C. L. Femtosecond Relaxation Dynamics of Large Molecules. Phys. Rev. Lett. 1986, 57 (3), 321–324. 10.1103/PhysRevLett.57.321. [DOI] [PubMed] [Google Scholar]
- Wise F. W.; Rosker M. J.; Tang C. L. Oscillatory Femtosecond Relaxation of Photoexcited Organic Molecules. J. Chem. Phys. 1987, 86 (5), 2827–2832. 10.1063/1.452032. [DOI] [Google Scholar]
- Chesnoy J.; Mokhtari A. Resonant Impulsive-Stimulated Raman Scattering on Malachite Green. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38 (7), 3566–3576. 10.1103/PhysRevA.38.3566. [DOI] [PubMed] [Google Scholar]
- Fragnito H. L.; Bigot J. Y.; Becker P. C.; Shank C. V. Evolution of the Vibronic Absorption Spectrum in a Molecule Following Impulsive Excitation with a 6 fs Optical Pulse. Chem. Phys. Lett. 1989, 160 (2), 101–104. 10.1016/0009-2614(89)87564-5. [DOI] [Google Scholar]
- Constantine S.; Zhou Y.; Morais J.; Ziegler L. D. Dispersed Optical Heterodyne Detected Birefringence and Dichroism of Transparent Liquids. J. Phys. Chem. A 1997, 101 (30), 5456–5462. 10.1021/jp971227s. [DOI] [Google Scholar]
- Rosca F.; Ionascu D.; Kumar A. T. N.; Demidov A. A.; Champion P. M. Femtosecond Coherence Spectroscopy Using Spectrally Selective Differential Photodetection. Chem. Phys. Lett. 2001, 337 (1–3), 107–116. 10.1016/S0009-2614(01)00200-7. [DOI] [Google Scholar]
- Kuramochi H.; Takeuchi S.; Tahara T. Femtosecond Time-Resolved Impulsive Stimulated Raman Spectroscopy Using Sub-7-fs Pulses: Apparatus and Applications. Rev. Sci. Instrum. 2016, 87 (4), 043107. 10.1063/1.4945259. [DOI] [PubMed] [Google Scholar]
- Iwata K.; Hamaguchi H. Picosecond Structural Relaxation of S1 Trans-Stilbene in Solution as Revealed by Time-Resolved Raman Spectroscopy. Chem. Phys. Lett. 1992, 196 (5), 462–468. 10.1016/0009-2614(92)85721-L. [DOI] [Google Scholar]
- Hamaguchi H.; Iwata K. Exchange Model of Vibrational Dephasing in S1 Trans-Stilbene in Solution and Its Possible Correlation with the Isomerization Reaction. Chem. Phys. Lett. 1993, 208 (5–6), 465–470. 10.1016/0009-2614(93)87174-2. [DOI] [Google Scholar]
- Takeuchi S.; Ruhman S.; Tsuneda T.; Chiba M.; Taketsugu T.; Tahara T. Spectroscopic Tracking of Structural Evolution in Ultrafast Stilbene Photoisomerization. Science 2008, 322 (5904), 1073–7. 10.1126/science.1160902. [DOI] [PubMed] [Google Scholar]
- Trigo M.; Chen J.; Jiang M. P.; Mao W. L.; Riggs S. C.; Shapiro M. C.; Fisher I. R.; Reis D. A. Ultrafast Pump-Probe Measurements of Short Small-Polaron Lifetimes in the Mixed-Valence Perovskite Cs2Au2I6 under High Pressures. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 85 (8), 081102. 10.1103/PhysRevB.85.081102. [DOI] [Google Scholar]
- Morrissey F. X.; Dexheimer S. L. Vibrational Spectroscopy of Structurally Relaxed Self-Trapped Excitons Via Excited-State Resonant Impulsive Stimulated Raman Spectroscopy. J. Phys. Chem. B 2012, 116 (35), 10582–10589. 10.1021/jp303288s. [DOI] [PubMed] [Google Scholar]
- Fujiyoshi S.; Takeuchi S.; Tahara T. Time-Resolved Impulsive Stimulated Raman Studies of 1,1′-Binaphthyl in the Excited State: Low-Frequency Vibrations and Conformational Relaxation. J. Phys. Chem. A 2004, 108 (28), 5938–5943. 10.1021/jp048968v. [DOI] [Google Scholar]
- Hornung T.; Skenderović H.; Motzkus M. Observation of All-Trans-β-Carotene Wavepacket Motion on the Electronic Ground and Excited Dark State Using Degenerate Four-Wave Mixing (DFWM) and Pump-DFWM. Chem. Phys. Lett. 2005, 402 (4–6), 283–288. 10.1016/j.cplett.2004.11.135. [DOI] [Google Scholar]
- Kraack J. P.; Buckup T.; Motzkus M. Evidence for the Two-State-Two-Mode Model in Retinal Protonated Schiff-Bases from Pump Degenerate Four-Wave-Mixing Experiments. Phys. Chem. Chem. Phys. 2012, 14 (40), 13979–13988. 10.1039/c2cp42248d. [DOI] [PubMed] [Google Scholar]
- Kraack J. P.; Wand A.; Buckup T.; Motzkus M.; Ruhman S. Mapping Multidimensional Excited State Dynamics Using Pump-Impulsive-Vibrational-Spectroscopy and Pump-Degenerate-Four-Wave-Mixing. Phys. Chem. Chem. Phys. 2013, 15 (34), 14487–14501. 10.1039/c3cp50871d. [DOI] [PubMed] [Google Scholar]
- Kraack J. P.; Motzkus M.; Buckup T. Excited State Vibrational Spectra of All-Trans Retinal Derivatives in Solution Revealed by Pump-DFWM Experiments. J. Phys. Chem. B 2018, 122 (51), 12271. 10.1021/acs.jpcb.8b08495. [DOI] [PubMed] [Google Scholar]
- Underwood D. F.; Blank D. A. Measuring the Change in the Intermolecular Raman Spectrum During Dipolar Solvation. J. Phys. Chem. A 2005, 109 (15), 3295–3306. 10.1021/jp044187i. [DOI] [PubMed] [Google Scholar]
- Schmidtke S. J.; Underwood D. F.; Blank D. A. Following the Solvent Directly During Ultrafast Excited State Proton Transfer. J. Am. Chem. Soc. 2004, 126 (28), 8620–8621. 10.1021/ja048639g. [DOI] [PubMed] [Google Scholar]
- Wilhelm T.; Piel J.; Riedle E. Sub-20-fs Pulses Tunable across the Visible from a Blue-Pumped Single-Pass Noncollinear Parametric Converter. Opt. Lett. 1997, 22 (19), 1494–1496. 10.1364/OL.22.001494. [DOI] [PubMed] [Google Scholar]
- Cerullo G.; Nisoli M.; De Silvestri S. Generation of 11 fs Pulses Tunable across the Visible by Optical Parametric Amplification. Appl. Phys. Lett. 1997, 71 (25), 3616–3618. 10.1063/1.120458. [DOI] [Google Scholar]
- Shirakawa A.; Kobayashi T. Noncollinearly Phase-Matched Femtosecond Optical Parametric Amplification with a 2000 cm–1 Bandwidth. Appl. Phys. Lett. 1998, 72 (2), 147–149. 10.1063/1.120670. [DOI] [Google Scholar]
- Zeek E.; Maginnis K.; Backus S.; Russek U.; Murnane M.; Mourou G.; Kapteyn H.; Vdovin G. Pulse Compression by Use of Deformable Mirrors. Opt. Lett. 1999, 24 (7), 493–495. 10.1364/OL.24.000493. [DOI] [PubMed] [Google Scholar]
- Armstrong M. R.; Plachta P.; Ponomarev E. A.; Miller R. J. D. Versatile 7-fs Optical Parametric Pulse Generation and Compression by Use of Adaptive Optics. Opt. Lett. 2001, 26 (15), 1152–1154. 10.1364/OL.26.001152. [DOI] [PubMed] [Google Scholar]
- Baltuška A.; Fuji T.; Kobayashi T. Visible Pulse Compression to 4 fs by Optical Parametric Amplification and Programmable Dispersion Control. Opt. Lett. 2002, 27 (5), 306–308. 10.1364/OL.27.000306. [DOI] [PubMed] [Google Scholar]
- Kuramochi H.; Takeuchi S.; Tahara T. Ultrafast Photodissociation Dynamics of Diphenylcyclopropenone Studied by Time-Resolved Impulsive Stimulated Raman Spectroscopy. Chem. Phys. 2018, 512, 88–92. 10.1016/j.chemphys.2018.02.023. [DOI] [Google Scholar]
- Shirakawa A.; Sakane I.; Takasaka M.; Kobayashi T. Sub-5-fs Visible Pulse Generation by Pulse-Front-Matched Noncollinear Optical Parametric Amplification. Appl. Phys. Lett. 1999, 74 (16), 2268–2270. 10.1063/1.123820. [DOI] [Google Scholar]
- Zavelani-Rossi M.; Polli D.; Cerullo G.; De Silvestri S.; Gallmann L.; Steinmeyer G.; Keller U. Few-Optical-Cycle Laser Pulses by OPA: Broadband Chirped Mirror Compression and SPIDER Characterization. Appl. Phys. B: Lasers Opt. 2002, 74 (S1), s245–s251. 10.1007/s00340-002-0892-x. [DOI] [Google Scholar]
- Pervak V.; Ahmad I.; Trubetskov M. K.; Tikhonravov A. V.; Krausz F. Double-Angle Multilayer Mirrors with Smooth Dispersion Characteristics. Opt. Express 2009, 17 (10), 7943–7951. 10.1364/OE.17.007943. [DOI] [PubMed] [Google Scholar]
- Liebel M.; Schnedermann C.; Kukura P. Sub-10-fs Pulses Tunable from 480 to 980 nm from a NOPA Pumped by an Yb:KGW Source. Opt. Lett. 2014, 39 (14), 4112–4115. 10.1364/OL.39.004112. [DOI] [PubMed] [Google Scholar]
- Liebel M.; Kukura P. Broad-Band Impulsive Vibrational Spectroscopy of Excited Electronic States in the Time Domain. J. Phys. Chem. Lett. 2013, 4 (8), 1358–1364. 10.1021/jz4004203. [DOI] [PubMed] [Google Scholar]
- Wende T.; Liebel M.; Schnedermann C.; Pethick R. J.; Kukura P. Population-Controlled Impulsive Vibrational Spectroscopy: Background- and Baseline-Free Raman Spectroscopy of Excited Electronic States. J. Phys. Chem. A 2014, 118 (43), 9976–9984. 10.1021/jp5075863. [DOI] [PubMed] [Google Scholar]
- Musser A. J.; Liebel M.; Schnedermann C.; Wende T.; Kehoe T. B.; Rao A.; Kukura P. Evidence for Conical Intersection Dynamics Mediating Ultrafast Singlet Exciton Fission. Nat. Phys. 2015, 11 (4), 352–357. 10.1038/nphys3241. [DOI] [Google Scholar]
- Fork R. L.; Martinez O. E.; Gordon J. P. Negative Dispersion Using Pairs of Prisms. Opt. Lett. 1984, 9 (5), 150–152. 10.1364/OL.9.000150. [DOI] [PubMed] [Google Scholar]
- Treacy E. Optical Pulse Compression with Diffraction Gratings. IEEE J. Quantum Electron. 1969, 5 (9), 454–458. 10.1109/JQE.1969.1076303. [DOI] [Google Scholar]
- Fork R. L.; Cruz C. H. B.; Becker P. C.; Shank C. V. Compression of Optical Pulses to Six Femtoseconds by Using Cubic Phase Compensation. Opt. Lett. 1987, 12 (7), 483–485. 10.1364/OL.12.000483. [DOI] [PubMed] [Google Scholar]
- Tsien R. Y. The Green Fluorescent Protein. Annu. Rev. Biochem. 1998, 67 (1), 509–544. 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- van Thor J. J. Photoreactions and Dynamics of the Green Fluorescent Protein. Chem. Soc. Rev. 2009, 38, 2935–2950. 10.1039/b820275n. [DOI] [PubMed] [Google Scholar]
- Meech S. R. Excited State Reactions in Fluorescent Proteins. Chem. Soc. Rev. 2009, 38, 2922–2934. 10.1039/b820168b. [DOI] [PubMed] [Google Scholar]
- Fang C.; Frontiera R. R.; Tran R.; Mathies R. A. Mapping GFP Structure Evolution During Proton Transfer with Femtosecond Raman Spectroscopy. Nature 2009, 462 (7270), 200–204. 10.1038/nature08527. [DOI] [PubMed] [Google Scholar]
- Fujisawa T.; Kuramochi H.; Hosoi H.; Takeuchi S.; Tahara T. Role of Coherent Low-Frequency Motion in Excited-State Proton Transfer of Green Fluorescent Protein Studied by Time-Resolved Impulsive Stimulated Raman Spectroscopy. J. Am. Chem. Soc. 2016, 138 (12), 3942–3945. 10.1021/jacs.5b11038. [DOI] [PubMed] [Google Scholar]
- Briggs W. R.; Spudich J. L.. Handbook of Photosensory Receptors; John Wiley & Sons, 2005. [Google Scholar]
- Meyer T. E. Isolation and Characterization of Soluble Cytochromes, Ferredoxins and Other Chromophoric Proteins from the Halophilic Phototrophic Bacterium Ectothiorhodospira Halophila. Biochim. Biophys. Acta, Bioenerg. 1985, 806 (1), 175–183. 10.1016/0005-2728(85)90094-5. [DOI] [PubMed] [Google Scholar]
- Sprenger W. W.; Hoff W. D.; Armitage J. P.; Hellingwerf K. J. The Eubacterium Ectothiorhodospira Halophila Is Negatively Phototactic, with a Wavelength Dependence That Fits the Absorption Spectrum of the Photoactive Yellow Protein. J. Bacteriol. 1993, 175 (10), 3096–3104. 10.1128/JB.175.10.3096-3104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellingwerf K. J.; Hendriks J.; Gensch T. Photoactive Yellow Protein, a New Type of Photoreceptor Protein: Will This “Yellow Lab” Bring Us Where We Want to Go?. J. Phys. Chem. A 2003, 107 (8), 1082–1094. 10.1021/jp027005y. [DOI] [Google Scholar]
- Kuramochi H.; Takeuchi S.; Yonezawa K.; Kamikubo H.; Kataoka M.; Tahara T. Probing the Early Stages of Photoreception in Photoactive Yellow Protein with Ultrafast Time-Domain Raman Spectroscopy. Nat. Chem. 2017, 9 (7), 660–666. 10.1038/nchem.2717. [DOI] [PubMed] [Google Scholar]
- Chosrowjan H.; Taniguchi S.; Mataga N.; Unno M.; Yamauchi S.; Hamada N.; Kumauchi M.; Tokunaga F. Low-Frequency Vibrations and Their Role in Ultrafast Photoisomerization Reaction Dynamics of Photoactive Yellow Protein. J. Phys. Chem. B 2004, 108, 2686–2698. 10.1021/jp031126w. [DOI] [Google Scholar]
- Adesokan A. A.; Pan D.; Fredj E.; Mathies R. A.; Gerber R. B. Anharmonic Vibrational Calculations Modeling the Raman Spectra of Intermediates in the Photoactive Yellow Protein (PYP) Photocycle. J. Am. Chem. Soc. 2007, 129 (15), 4584–4594. 10.1021/ja066903v. [DOI] [PubMed] [Google Scholar]
- Schotte F.; Cho H. S.; Kaila V. R. I.; Kamikubo H.; Dashdorj N.; Henry E. R.; Graber T. J.; Henning R.; Wulff M.; Hummer G.; Kataoka M.; Anfinrud P. A. Watching a Signaling Protein Function in Real Time Via 100-ps Time-Resolved Laue Crystallography. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (47), 19256–19261. 10.1073/pnas.1210938109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y. O.; Lee J. H.; Kim J.; Schmidt M.; Moffat K.; Šrajer V.; Ihee H. Volume-Conserving Trans-Cis Isomerization Pathways in Photoactive Yellow Protein Visualized by Picosecond X-Ray Crystallography. Nat. Chem. 2013, 5 (3), 212–220. 10.1038/nchem.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila V. R. I.; Schotte F.; Cho H. S.; Hummer G.; Anfinrud P. A. Contradictions in X-Ray Structures of Intermediates in the Photocycle of Photoactive Yellow Protein. Nat. Chem. 2014, 6 (4), 258–259. 10.1038/nchem.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y. O.; Lee J. H.; Kim J.; Schmidt M.; Moffat K.; Srajer V.; Ihee H. Reply to ‘Contradictions in X-Ray Structures of Intermediates in the Photocycle of Photoactive Yellow Protein’. Nat. Chem. 2014, 6 (4), 259–260. 10.1038/nchem.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande K.; Hutchison C. D. M.; Groenhof G.; Aquila A.; Robinson J. S.; Tenboer J.; Basu S.; Boutet S.; DePonte D. P.; Liang M.; White T. A.; Zatsepin N. A.; Yefanov O.; Morozov D.; Oberthuer D.; Gati C.; Subramanian G.; James D.; Zhao Y.; Koralek J.; Brayshaw J.; Kupitz C.; Conrad C.; Roy-Chowdhury S.; Coe J. D.; Metz M.; Xavier P. L.; Grant T. D.; Koglin J. E.; Ketawala G.; Fromme R.; Šrajer V.; Henning R.; Spence J. C. H.; Ourmazd A.; Schwander P.; Weierstall U.; Frank M.; Fromme P.; Barty A.; Chapman H. N.; Moffat K.; van Thor J. J.; Schmidt M. Femtosecond Structural Dynamics Drives the Trans/Cis Isomerization in Photoactive Yellow Protein. Science 2016, 352 (6286), 725–729. 10.1126/science.aad5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian R.; Corthey G.; Rogers D. M.; Morrison C. A.; Prokhorenko V. I.; Hayes S. A.; Miller R. J. D. Coherent Ultrafast Lattice-Directed Reaction Dynamics of Triiodide Anion Photodissociation. Nat. Chem. 2017, 9 (6), 516–522. 10.1038/nchem.2751. [DOI] [PubMed] [Google Scholar]
- Wolf T. J. A.; Sanchez D. M.; Yang J.; Parrish R. M.; Nunes J. P. F.; Centurion M.; Coffee R.; Cryan J. P.; Gühr M.; Hegazy K.; Kirrander A.; Li R. K.; Ruddock J.; Shen X.; Vecchione T.; Weathersby S. P.; Weber P. M.; Wilkin K.; Yong H.; Zheng Q.; Wang X. J.; Minitti M. P.; Martínez T. J. The Photochemical Ring-Opening of 1,3-Cyclohexadiene Imaged by Ultrafast Electron Diffraction. Nat. Chem. 2019, 11 (6), 504–509. 10.1038/s41557-019-0252-7. [DOI] [PubMed] [Google Scholar]
- Rawashdeh-Omary M. A.; Omary M. A.; Patterson H. H. Oligomerization of Au(CN)2– and Ag(CN)2– Ions in Solution Via Ground-State Aurophilic and Argentophilic Bonding. J. Am. Chem. Soc. 2000, 122 (42), 10371–10380. 10.1021/ja001545w. [DOI] [Google Scholar]
- Rawashdeh-Omary M. A.; Omary M. A.; Patterson H. H.; Fackler J. P. Excited-State Interactions for [Au(CN)2–]n and [Ag(CN)2–]n Oligomers in Solution. Formation of Luminescent Gold-Gold Bonded Excimers and Exciplexes. J. Am. Chem. Soc. 2001, 123 (45), 11237–11247. 10.1021/ja011176j. [DOI] [PubMed] [Google Scholar]
- Iwamura M.; Nozaki K.; Takeuchi S.; Tahara T. Real-Time Observation of Tight Au-Au Bond Formation and Relevant Coherent Motion Upon Photoexcitation of [Au(CN)2–] Oligomers. J. Am. Chem. Soc. 2013, 135 (2), 538–541. 10.1021/ja310004z. [DOI] [PubMed] [Google Scholar]
- Iwamura M.; Wakabayashi R.; Maeba J.; Nozaki K.; Takeuchi S.; Tahara T. Coherent Vibration and Ultrafast Dynamics Upon Bond Formation in Excited Dimers of an Au(I) Complex. Phys. Chem. Chem. Phys. 2016, 18 (7), 5103–5107. 10.1039/C5CP06651D. [DOI] [PubMed] [Google Scholar]
- Iwamura M.; Kimoto K.; Nozaki K.; Kuramochi H.; Takeuchi S.; Tahara T. Metal-Metal Bond Formations in [Au(CN)2–]n (n = 3–5) Oligomers in Water Identified by Coherent Nuclear Wavepacket Motions. J. Phys. Chem. Lett. 2018, 9, 7085–7089. 10.1021/acs.jpclett.8b03139. [DOI] [PubMed] [Google Scholar]
- Kim K. H.; Kim J. G.; Nozawa S.; Sato T.; Oang K. Y.; Kim T. W.; Ki H.; Jo J.; Park S.; Song C.; Sato T.; Ogawa K.; Togashi T.; Tono K.; Yabashi M.; Ishikawa T.; Kim J.; Ryoo R.; Kim J.; Ihee H.; Adachi S.-i. Direct Observation of Bond Formation in Solution with Femtosecond X-Ray Scattering. Nature 2015, 518 (7539), 385–389. 10.1038/nature14163. [DOI] [PubMed] [Google Scholar]
- Kim K. H.; Kim J. G.; Oang K. Y.; Kim T. W.; Ki H.; Jo J.; Kim J.; Sato T.; Nozawa S.; Adachi S.-i.; Ihee H. Femtosecond X-Ray Solution Scattering Reveals That Bond Formation Mechanism of a Gold Trimer Complex Is Independent of Excitation Wavelength. Struct. Dyn. 2016, 3 (4), 043209. 10.1063/1.4948516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi H.; Takeuchi S.; Iwamura M.; Nozaki K.; Tahara T. Tracking Photoinduced Au-Au Bond Formation through Transient Terahertz Vibrations Observed by Femtosecond Time-Domain Raman Spectroscopy. J. Am. Chem. Soc. 2019, 141 (49), 19296–19303. 10.1021/jacs.9b06950. [DOI] [PubMed] [Google Scholar]
- Gao M.; Lu C.; Jean-Ruel H.; Liu L. C.; Marx A.; Onda K.; Koshihara S.-y.; Nakano Y.; Shao X.; Hiramatsu T.; Saito G.; Yamochi H.; Cooney R. R.; Moriena G.; Sciaini G.; Miller R. J. D. Mapping Molecular Motions Leading to Charge Delocalization with Ultrabright Electrons. Nature 2013, 496 (7445), 343–346. 10.1038/nature12044. [DOI] [PubMed] [Google Scholar]
- Cammarata M.; Bertoni R.; Lorenc M.; Cailleau H.; Di Matteo S.; Mauriac C.; Matar S. F.; Lemke H.; Chollet M.; Ravy S.; Laulhé C.; Létard J.-F.; Collet E. Sequential Activation of Molecular Breathing and Bending During Spin-Crossover Photoswitching Revealed by Femtosecond Optical and X-Ray Absorption Spectroscopy. Phys. Rev. Lett. 2014, 113 (22), 227402. 10.1103/PhysRevLett.113.227402. [DOI] [PubMed] [Google Scholar]
- Ishikawa T.; Hayes S. A.; Keskin S.; Corthey G.; Hada M.; Pichugin K.; Marx A.; Hirscht J.; Shionuma K.; Onda K.; Okimoto Y.; Koshihara S.-y.; Yamamoto T.; Cui H.; Nomura M.; Oshima Y.; Abdel-Jawad M.; Kato R.; Miller R. J. D. Direct Observation of Collective Modes Coupled to Molecular Orbital-Driven Charge Transfer. Science 2015, 350 (6267), 1501–1505. 10.1126/science.aab3480. [DOI] [PubMed] [Google Scholar]
- Iwano K.; Shimoi Y.; Miyamoto T.; Hata D.; Sotome M.; Kida N.; Horiuchi S.; Okamoto H. Ultrafast Photoinduced Electric-Polarization Switching in a Hydrogen-Bonded Ferroelectric Crystal. Phys. Rev. Lett. 2017, 118 (10), 107404. 10.1103/PhysRevLett.118.107404. [DOI] [PubMed] [Google Scholar]
- Kuramochi H.; Aoyama G.; Okajima H.; Sakamoto A.; Kanegawa S.; Sato O.; Takeuchi S.; Tahara T. Femtosecond Polarization Switching in the Crystal of a [CrCo] Dinuclear Complex. Angew. Chem., Int. Ed. 2020, 59 (37), 15865–15869. 10.1002/anie.202004583. [DOI] [PubMed] [Google Scholar]
- Miyata K.; Atallah T. L.; Zhu X. Y. Lead Halide Perovskites: Crystal-Liquid Duality, Phonon Glass Electron Crystals, and Large Polaron Formation. Sci. Adv. 2017, 3 (10), e1701469. 10.1126/sciadv.1701469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. B.; Michl J. Singlet Fission. Chem. Rev. 2010, 110 (11), 6891–6936. 10.1021/cr1002613. [DOI] [PubMed] [Google Scholar]
- Rao A.; Friend R. H. Harnessing Singlet Exciton Fission to Break the Shockley-Queisser Limit. Nat. Rev. Mater. 2017, 2 (11), 17063. 10.1038/natrevmats.2017.63. [DOI] [Google Scholar]
- Wilson M. W. B.; Rao A.; Clark J.; Kumar R. S. S.; Brida D.; Cerullo G.; Friend R. H. Ultrafast Dynamics of Exciton Fission in Polycrystalline Pentacene. J. Am. Chem. Soc. 2011, 133 (31), 11830–11833. 10.1021/ja201688h. [DOI] [PubMed] [Google Scholar]
- Yost S. R.; Lee J.; Wilson M. W. B.; Wu T.; McMahon D. P.; Parkhurst R. R.; Thompson N. J.; Congreve D. N.; Rao A.; Johnson K.; Sfeir M. Y.; Bawendi M. G.; Swager T. M.; Friend R. H.; Baldo M. A.; Van Voorhis T. A Transferable Model for Singlet-Fission Kinetics. Nat. Chem. 2014, 6 (6), 492–497. 10.1038/nchem.1945. [DOI] [PubMed] [Google Scholar]
- Takeuchi S.; Tahara T. Vibrational Coherence of S1 Trans-Stilbene in Solution Observed by 40-fs-Resolved Absorption Spectroscopy: Comparison of the Low-Frequency Vibration Appearing in the Frequency-Domain and Time-Domain Spectroscopies. Chem. Phys. Lett. 2000, 326 (5–6), 430–438. 10.1016/S0009-2614(00)00822-8. [DOI] [Google Scholar]
- Ishii K.; Takeuchi S.; Tahara T. A 40-fs Time-Resolved Absorption Study on Cis-Stilbene in Solution: Observation of Wavepacket Motion on the Reactive Excited State. Chem. Phys. Lett. 2004, 398 (4–6), 400–406. 10.1016/j.cplett.2004.09.075. [DOI] [Google Scholar]
- Takeuchi S.; Tahara T. Coherent Nuclear Wavepacket Motions in Ultrafast Excited-State Intramolecular Proton Transfer: Sub-30-fs Resolved Pump-Probe Absorption Spectroscopy of 10-Hydroxybenzo[h]quinoline in Solution. J. Phys. Chem. A 2005, 109 (45), 10199–207. 10.1021/jp0519013. [DOI] [PubMed] [Google Scholar]
- Iwamura M.; Watanabe H.; Ishii K.; Takeuchi S.; Tahara T. Coherent Nuclear Dynamics in Ultrafast Photoinduced Structural Change of Bis(diimine)copper(I) Complex. J. Am. Chem. Soc. 2011, 133 (20), 7728–7736. 10.1021/ja108645x. [DOI] [PubMed] [Google Scholar]
- Liebel M.; Schnedermann C.; Kukura P. Vibrationally Coherent Crossing and Coupling of Electronic States During Internal Conversion in β-Carotene. Phys. Rev. Lett. 2014, 112 (19), 198302. 10.1103/PhysRevLett.112.198302. [DOI] [PubMed] [Google Scholar]
- Liebel M.; Schnedermann C.; Bassolino G.; Taylor G.; Watts A.; Kukura P. Direct Observation of the Coherent Nuclear Response after the Absorption of a Photon. Phys. Rev. Lett. 2014, 112 (23), 238301. 10.1103/PhysRevLett.112.238301. [DOI] [PubMed] [Google Scholar]
- Schnedermann C.; Alvertis A. M.; Wende T.; Lukman S.; Feng J.; Schröder F. A. Y. N.; Turban D. H. P.; Wu J.; Hine N. D. M.; Greenham N. C.; Chin A. W.; Rao A.; Kukura P.; Musser A. J. A Molecular Movie of Ultrafast Singlet Fission. Nat. Commun. 2019, 10 (1), 4207. 10.1038/s41467-019-12220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.; Kim W.; Vakuliuk O.; Gryko D. T.; Kim D. Two-Step Charge Separation Passing through the Partial Charge-Transfer State in a Molecular Dyad. J. Am. Chem. Soc. 2020, 142 (3), 1564–1573. 10.1021/jacs.9b12016. [DOI] [PubMed] [Google Scholar]
- Kim W.; Kim T.; Kang S.; Hong Y.; Würthner F.; Kim D. Tracking Structural Evolution During Symmetry-Breaking Charge Separation in Quadrupolar Perylene Bisimide with Time-Resolved Impulsive Stimulated Raman Spectroscopy. Angew. Chem., Int. Ed. 2020, 59 (22), 8571–8578. 10.1002/anie.202002733. [DOI] [PubMed] [Google Scholar]
- Yoshizawa M.; Kurosawa M. Femtosecond Time-Resolved Raman Spectroscopy Using Stimulated Raman Scattering. Phys. Rev. A: At., Mol., Opt. Phys. 1999, 61 (1), 013808. 10.1103/PhysRevA.61.013808. [DOI] [Google Scholar]
- Kukura P.; McCamant D. W.; Mathies R. A. Femtosecond Stimulated Raman Spectroscopy. Annu. Rev. Phys. Chem. 2007, 58 (1), 461–488. 10.1146/annurev.physchem.58.032806.104456. [DOI] [PubMed] [Google Scholar]
- Dietze D. R.; Mathies R. A. Femtosecond Stimulated Raman Spectroscopy. ChemPhysChem 2016, 17 (9), 1224–1251. 10.1002/cphc.201600104. [DOI] [PubMed] [Google Scholar]
- Fang C.; Tang L.; Oscar B. G.; Chen C. Capturing Structural Snapshots During Photochemical Reactions with Ultrafast Raman Spectroscopy: From Materials Transformation to Biosensor Responses. J. Phys. Chem. Lett. 2018, 9 (12), 3253–3263. 10.1021/acs.jpclett.8b00373. [DOI] [PubMed] [Google Scholar]
- The Raman scattering process is a third-order optical process in which the light electric field interacts with the materials three times. In TR-ISRS, the first two interactions are provided by the Raman pump pulse, which creates the Raman-active vibrational coherence, and the third interaction occurs with the Raman probe pulse. On the other hand, in FSRS, the first two interactions creating the Raman-active vibrational coherence are provided with the picosecond Raman pump and femtosecond Raman probe pulses, and the third interaction is provided again with the picosecond Raman pump while the vibrational coherence persists.
- Dorfman K. E.; Fingerhut B. P.; Mukamel S. Time-Resolved Broadband Raman Spectroscopies: A Unified Six-Wave-Mixing Representation. J. Chem. Phys. 2013, 139 (12), 124113–17. 10.1063/1.4821228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloz M.; Grondelle R. v.; Kennis J. T. M. Wavelength-Modulated Femtosecond Stimulated Raman Spectroscopy-Approach Towards Automatic Data Processing. Phys. Chem. Chem. Phys. 2011, 13 (40), 18123–18133. 10.1039/c1cp21650c. [DOI] [PubMed] [Google Scholar]
- Grumstrup E. M.; Chen Z.; Vary R. P.; Moran A. M.; Schanze K. S.; Papanikolas J. M. Frequency Modulated Femtosecond Stimulated Raman Spectroscopy of Ultrafast Energy Transfer in a Donor-Acceptor Copolymer. J. Phys. Chem. B 2013, 117 (27), 8245–8255. 10.1021/jp404498u. [DOI] [PubMed] [Google Scholar]
- Kloz M.; Weißenborn J.; Polívka T.; Frank H. A.; Kennis J. T. M. Spectral Watermarking in Femtosecond Stimulated Raman Spectroscopy: Resolving the Nature of the Carotenoid S* State. Phys. Chem. Chem. Phys. 2016, 18 (21), 14619–14628. 10.1039/C6CP01464J. [DOI] [PubMed] [Google Scholar]
- Bera K.; Kwang S. Y.; Cassabaum A. A.; Rich C. C.; Frontiera R. R. Facile Background Discrimination in Femtosecond Stimulated Raman Spectroscopy Using a Dual-Frequency Raman Pump Technique. J. Phys. Chem. A 2019, 123 (37), 7932–7939. 10.1021/acs.jpca.9b02473. [DOI] [PubMed] [Google Scholar]
- Co D. T.; Lockard J. V.; McCamant D. W.; Wasielewski M. R. Narrow-Bandwidth Tunable Picosecond Pulses in the Visible Produced by Noncollinear Optical Parametric Amplification with a Chirped Blue Pump. Appl. Opt. 2010, 49 (10), 1880–1885. 10.1364/AO.49.001880. [DOI] [PubMed] [Google Scholar]
- Kovalenko S. A.; Dobryakov A. L.; Ernsting N. P. An Efficient Setup for Femtosecond Stimulated Raman Spectroscopy. Rev. Sci. Instrum. 2011, 82 (6), 063102–9. 10.1063/1.3596453. [DOI] [PubMed] [Google Scholar]
- Kuramochi H.; Takeuchi S.; Tahara T. Ultrafast Structural Evolution of Photoactive Yellow Protein Chromophore Revealed by Ultraviolet Resonance Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. Lett. 2012, 3 (15), 2025–2029. 10.1021/jz300542f. [DOI] [Google Scholar]
- Zhu L.; Liu W.; Fang C. A Versatile Femtosecond Stimulated Raman Spectroscopy Setup with Tunable Pulses in the Visible to near Infrared. Appl. Phys. Lett. 2014, 105 (4), 041106. 10.1063/1.4891766. [DOI] [Google Scholar]
- Takaya T.; Iwata K. Development of a Femtosecond Time-Resolved near-IR Multiplex Stimulated Raman Spectrometer in Resonance with Transitions in the 900–1550 nm Region. Analyst 2016, 141 (14), 4283–4292. 10.1039/C6AN01051B. [DOI] [PubMed] [Google Scholar]
- Kuramochi H.; Fujisawa T.; Takeuchi S.; Tahara T. Broadband Stimulated Raman Spectroscopy in the Deep Ultraviolet Region. Chem. Phys. Lett. 2017, 683, 543–546. 10.1016/j.cplett.2017.02.015. [DOI] [Google Scholar]
- Andrea Rozzi C.; Maria Falke S.; Spallanzani N.; Rubio A.; Molinari E.; Brida D.; Maiuri M.; Cerullo G.; Schramm H.; Christoffers J.; Lienau C. Quantum Coherence Controls the Charge Separation in a Prototypical Artificial Light-Harvesting System. Nat. Commun. 2013, 4 (1), 1602. 10.1038/ncomms2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falke S. M.; Rozzi C. A.; Brida D.; Maiuri M.; Amato M.; Sommer E.; De Sio A.; Rubio A.; Cerullo G.; Molinari E.; Lienau C. Coherent Ultrafast Charge Transfer in an Organic Photovoltaic Blend. Science 2014, 344 (6187), 1001. 10.1126/science.1249771. [DOI] [PubMed] [Google Scholar]
- Harada I.; Takeuchi H.. Raman and Ultraviolet Resonance Raman Spectra of Proteins and Related Compounds. Advances in Infrared and Raman Spectroscopy; Wiley, 1986; Vol. 13, pp 113–175. [Google Scholar]
- Sato A.; Gao Y.; Kitagawa T.; Mizutani Y. Primary Protein Response after Ligand Photodissociation in Carbonmonoxy Myoglobin. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (23), 9627–9632. 10.1073/pnas.0611560104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno M.; Kamikubo H.; Kataoka M.; Mizutani Y. Changes in the Hydrogen-Bond Network around the Chromophore of Photoactive Yellow Protein in the Ground and Excited States. J. Phys. Chem. B 2011, 115, 9306–9310. 10.1021/jp2029399. [DOI] [PubMed] [Google Scholar]
- Tahara S.; Kuramochi H.; Takeuchi S.; Tahara T. Protein Dynamics Preceding Photoisomerization of the Retinal Chromophore in Bacteriorhodopsin Revealed by Deep-UV Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. Lett. 2019, 10 (18), 5422–5427. 10.1021/acs.jpclett.9b02283. [DOI] [PubMed] [Google Scholar]
- Adachi S.; Kumbhakar P.; Kobayashi T. Quasi-Monocyclic near-Infrared Pulses with a Stabilized Carrier-Envelope Phase Characterized by Noncollinear Cross-Correlation Frequency-Resolved Optical Gating. Opt. Lett. 2004, 29 (10), 1150–1152. 10.1364/OL.29.001150. [DOI] [PubMed] [Google Scholar]
- Brida D.; Bonora S.; Manzoni C.; Marangoni M.; Villoresi P.; De Silvestri S.; Cerullo G. Generation of 8.5-fs Pulses at 1.3 μm for Ultrabroadband Pump-Probe Spectroscopy. Opt. Express 2009, 17 (15), 12510–12515. 10.1364/OE.17.012510. [DOI] [PubMed] [Google Scholar]
- Varillas R. B.; Candeo A.; Viola D.; Garavelli M.; De Silvestri S.; Cerullo G.; Manzoni C. Microjoule-Level, Tunable Sub-10 fs UV Pulses by Broadband Sum-Frequency Generation. Opt. Lett. 2014, 39 (13), 3849–3852. 10.1364/OL.39.003849. [DOI] [PubMed] [Google Scholar]
- Ideguchi T.; Holzner S.; Bernhardt B.; Guelachvili G.; Picque N.; Hansch T. W. Coherent Raman Spectro-Imaging with Laser Frequency Combs. Nature 2013, 502 (7471), 355–358. 10.1038/nature12607. [DOI] [PubMed] [Google Scholar]
- Kim J.; Yoon T. H.; Cho M. Time-Resolved Impulsive Stimulated Raman Spectroscopy with Synchronized Triple Mode-Locked Lasers. J. Phys. Chem. Lett. 2020, 11 (8), 2864–2869. 10.1021/acs.jpclett.0c00596. [DOI] [PubMed] [Google Scholar]
- Stiles P. L.; Dieringer J. A.; Shah N. C.; Van Duyne R. P. Surface-Enhanced Raman Spectroscopy. Annu. Rev. Anal. Chem. 2008, 1 (1), 601–626. 10.1146/annurev.anchem.1.031207.112814. [DOI] [PubMed] [Google Scholar]
- Sharma B.; Frontiera R. R.; Henry A.-I.; Ringe E.; Van Duyne R. P. SERS: Materials, Applications, and the Future. Mater. Today 2012, 15 (1), 16–25. 10.1016/S1369-7021(12)70017-2. [DOI] [Google Scholar]
- Zong C.; Xu M.; Xu L.-J.; Wei T.; Ma X.; Zheng X.-S.; Hu R.; Ren B. Surface-Enhanced Raman Spectroscopy for Bioanalysis: Reliability and Challenges. Chem. Rev. 2018, 118 (10), 4946–4980. 10.1021/acs.chemrev.7b00668. [DOI] [PubMed] [Google Scholar]
- Nie S.; Emory S. R. Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering. Science 1997, 275 (5303), 1102. 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- Kneipp K.; Wang Y.; Kneipp H.; Perelman L. T.; Itzkan I.; Dasari R. R.; Feld M. S. Single Molecule Detection Using Surface-Enhanced Raman Scattering (SERS). Phys. Rev. Lett. 1997, 78 (9), 1667–1670. 10.1103/PhysRevLett.78.1667. [DOI] [Google Scholar]
- Dieringer J. A.; Lettan R. B.; Scheidt K. A.; Van Duyne R. P. A Frequency Domain Existence Proof of Single-Molecule Surface-Enhanced Raman Spectroscopy. J. Am. Chem. Soc. 2007, 129 (51), 16249–16256. 10.1021/ja077243c. [DOI] [PubMed] [Google Scholar]
- Blackie E. J.; Le Ru E. C.; Etchegoin P. G. Single-Molecule Surface-Enhanced Raman Spectroscopy of Nonresonant Molecules. J. Am. Chem. Soc. 2009, 131 (40), 14466–14472. 10.1021/ja905319w. [DOI] [PubMed] [Google Scholar]
- Kazuma E.; Kim Y. Mechanistic Studies of Plasmon Chemistry on Metal Catalysts. Angew. Chem., Int. Ed. 2019, 58 (15), 4800–4808. 10.1002/anie.201811234. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Kuramochi H.; Takeuchi S.; Tahara T. Time-Domain Observation of Surface-Enhanced Coherent Raman Scattering with 105-106 Enhancement. J. Phys. Chem. Lett. 2020, 11, 6305–6311. 10.1021/acs.jpclett.0c01411. [DOI] [PubMed] [Google Scholar]
- Kuramochi H.; Takeuchi S.; Kamikubo H.; Kataoka M.; Tahara T. Fifth-Order Time-Domain Raman Spectroscopy of Photoactive Yellow Protein for Visualizing Vibrational Coupling in Its Excited State. Sci. Adv. 2019, 5 (6), eaau4490. 10.1126/sciadv.aau4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumero G.; Schnedermann C.; Batignani G.; Wende T.; Liebel M.; Bassolino G.; Ferrante C.; Mukamel S.; Kukura P.; Scopigno T. Two-Dimensional Impulsively Stimulated Resonant Raman Spectroscopy of Molecular Excited States. Phys. Rev. X 2020, 10 (1), 011051. 10.1103/PhysRevX.10.011051. [DOI] [Google Scholar]