

Room-temperature crystallography enables researchers to resolve the conformational heterogeneity of structures. Here, the native SAD phasing of four structures at 295 K highlights the strengths of room-temperature diffraction experiments, including detailed anomalous difference maps and alternate conformations that are well supported by the electron density.
Keywords: X-ray crystallography, native SAD, room temperature, phasing, model building
Abstract
Single-wavelength anomalous diffraction (SAD) is a routine method for overcoming the phase problem when solving macromolecular structures. This technique requires the accurate measurement of intensities to determine differences between Bijvoet pairs. Although SAD experiments are commonly conducted at cryogenic temperatures to mitigate the effects of radiation damage, such temperatures can alter the conformational ensemble of the protein and may impede the merging of data from multiple crystals due to non-uniform freezing. Here, a strategy is presented to obtain high-quality data from room-temperature, single-crystal experiments. To illustrate the strengths of this approach, native SAD phasing at 6.55 keV was used to solve four structures of three model systems at 295 K. The resulting data sets allow automatic phasing and model building, and reveal alternate conformations that reflect the structure of proteins at room temperature.
1. Introduction
X-ray crystallography is the predominant experimental technique for determining the structures of macromolecules at atomic resolution. This method reconstructs the electron density of a crystal from measurements of the relative intensities of scattered X-rays, enabling researchers to build a model with atomic detail. Although the intensities of the scattered X-rays determine the amplitude of the diffracted waves, the phases of these waves are needed to reconstruct the electron density (Taylor, 2010 ▸). For the structures of novel macromolecules, this problem is often overcome using the resonance phenomenon of anomalous scattering, with single-wavelength anomalous diffraction (SAD) being the method of choice for the de novo phasing of macromolecular structures (Hendrickson, 2014 ▸; Liu & Hendrickson, 2017 ▸).
SAD phasing relies on the accurate measurement of the differences between Bijvoet pairs of reflections. An especially challenging case is native SAD phasing, which uses the weak anomalous scattering from low-Z atoms of the macromolecule or the crystallization conditions. Although the first de novo SAD structure used the anomalous scattering from native S atoms (Hendrickson & Teeter, 1981 ▸), structures solved using native SAD phasing still constitute a small fraction of those deposited in the Protein Data Bank (Rose et al., 2015 ▸).
High-redundancy data-collection strategies improve the accurate determination of Bijvoet differences and therefore increase the chance of successful phasing by the SAD method (Dauter & Adamiak, 2001 ▸; Liu et al., 2012 ▸; Weinert et al., 2015 ▸). However, cryogenic temperatures are typically used to mitigate the resulting risk of radiation damage (Hope, 1988 ▸; Pflugrath, 2015 ▸). Since the first successful S-SAD experiment (Hendrickson & Teeter, 1981 ▸), native SAD experiments have nearly exclusively been reported under cryogenic conditions or from serial crystallography applications (Nakane et al., 2015 ▸; Huang et al., 2015 ▸).
Cryocooling can alter the ensemble of conformations in a crystal (Keedy et al., 2014 ▸) and can limit the merging of multiple crystals due to imperfect isomorphism (Giordano et al., 2012 ▸). Accordingly, room-temperature X-ray crystallography is undergoing a resurgence in order to better probe the dynamics of protein structures at physiologically relevant temperatures (Fraser et al., 2009 ▸, 2011 ▸; Doukov et al., 2020 ▸). Here, we demonstrate native SAD phasing from single crystals at room temperature. We present four structures of three different model enzymes, which we solved by automatic SAD phasing and model building. The structures exhibit alternate conformations that reflect the structural heterogeneity of the crystals, and in one case the anomalous difference map identifies alternative coordination sites for manganese ions. Our high-redundancy data-collection strategy can be implemented at many macromolecular beamlines and enables native SAD phasing from single-crystal experiments at room temperature.
2. Materials and methods
2.1. Protein purification and crystallization
2.1.1. Hen egg-white lysozyme
We dissolved lyophilized hen egg-white lysozyme (HEWL; Sigma–Aldrich) in deionized water to concentrations of 15–30 mg ml−1. Using the hanging-drop vapor-diffusion method, we grew tetragonal lysozyme crystals using a well solution composed of 50 mM sodium citrate buffer pH 5.2–5.8, 20–25% glycerol, 20–25% PEG 4000. We set up drops consisting of 2 µl protein solution and 2 µl well solution over 0.5 ml wells and stored the crystallization plates at ambient temperature (approximately 22°C). Tetragonal lysozyme crystals (0.2–0.5 mm) grew after 3–7 days and could diffract to 1.2 Å resolution. The crystal used for data collection was approximately 0.3 × 0.3 × 0.1 mm in size.
2.1.2. Escherichia coli dihydrofolate reductase
We expressed wild-type E. coli dihydrofolate reductase (DHFR) from a pET-22b vector generously provided by the laboratory of James Fraser at UCSF (Keedy et al., 2014 ▸; Cameron & Benkovic, 1997 ▸). We transformed E. coli BL21(DE3) cells (Novagen) with the plasmid and grew overnight cultures in lysogeny broth (LB) with ampicillin. We diluted the overnight cultures 1:100 in 2 l Terrific broth (TB) with ampicillin, and grew the diluted cultures to an optical density of 0.6–0.8 at 600 nm. When the cultures reached the mid-log phase, we added isopropyl β-d-1-thiogalactopyranoside (IPTG; Invitrogen) to a final concentration of 1 mM, inducing DHFR expression. After 4–8 h of expression, we harvested the cells by centrifugation, flash-cooled the resulting cell pellets in liquid nitrogen and stored them at −80°C until purification.
We based our DHFR purification protocol on the thesis of Michael Sawaya (Sawaya, 1994 ▸). Briefly, we thawed, resuspended and lysed the cell pellets by sonication. We removed the cell debris by centrifugation and then used streptomycin precipitation to remove nucleotides from the supernatant. We further purified the protein fraction by ammonium sulfate precipitation at 40% saturation. The supernatant was retained and subjected to additional ammonium sulfate precipitation at 90% saturation. The pellet was resuspended and DHFR was isolated by methotrexate-affinity chromatography using methotrexate–agarose (Sigma–Aldrich) followed by anion-exchange chromatography using a HiPrep Q FF column (Cytiva). We then pooled and concentrated the eluted protein. To minimize nucleotide contamination, we only pooled fractions that had an A 280/A 260 of >1.7 based on an absorbance reading with a NanoDrop spectrophotometer (Thermo Fisher Scientific). Finally, we exchanged the purified protein into 10 mM HEPES buffer pH 7.0 containing 1 mM dithiothreitol (DTT) using an Amicon centrifugal concentrator (MilliporeSigma), flash-cooled aliquots in liquid nitrogen and stored them at −80°C.
DHFR complexed with NADP+ and folate is a commonly used model of the Michaelis complex of the enzyme (Sawaya & Kraut, 1997 ▸). We mixed 20 mg ml−1 DHFR with a threefold molar excess of NADP+ and folate in 20 mM imidazole pH 7.0 with 2 mM DTT and then incubated the mixture on ice for 30 min. Based on the conditions of Keedy et al. (2014 ▸), we crystallized the complex using the sitting-drop vapor-diffusion method with a well solution composed of 20 mM imidazole pH 5.4–5.8, 125 mM manganese(II) chloride, 16–21%(v/v) PEG 400. The drops consisted of 0.2 µl 7–14 mg ml−1 protein solution and 0.2 µl well solution, producing rod-shaped crystals (approximately 0.1 × 0.3–1 mm) after 2–4 weeks of incubation at 4°C that could diffract to 1.1 Å resolution. The crystal used for data collection was approximately 0.1 × 0.1 mm along the short axes and 0.5 mm along the long axis.
2.1.3. Human protein tyrosine phosphatase 1B
Human protein tyrosine phosphatase 1B (PTP1B) experiments used a construct containing residues 1–321 with a C32S/C92V double mutation (Erlanson et al., 2003 ▸). We expressed PTP1B from a pET-24b vector with a kanamycin-resistance gene, which was generously provided by the laboratory of James Fraser at UCSF. We expressed and purified PTP1B following published methods (Keedy et al., 2018 ▸) with minor modifications. Briefly, we transformed E. coli Rosetta(DE3) cells (Novagen) with the PTP1B plasmid and grew overnight cultures in LB with kanamycin and chloramphenicol. We diluted the overnight cultures 1:1000 in 2 l TB with kanamycin and chloramphenicol. We incubated the final cultures at 37°C and grew them to an optical density of 0.6–0.8 at 600 nm. When the cultures reached the mid-log phase, we added IPTG (Invitrogen) to a final concentration of 1 mM, inducing PTP1B expression, and incubated them at 20°C for 12–18 h. We harvested the cells by centrifugation, flash-cooled the resulting cell pellets in liquid nitrogen and stored them at −80°C until purification.
We thawed the cell pellets, resuspended them in lysis buffer and lysed the cells by sonication. Cell debris was cleared by centrifugation and syringe filtration through a 0.45 µm filter. We purified PTP1B by cation-exchange chromatography using a HiPrep SP FF column (Cytiva). We pooled the PTP1B fractions from the elution, concentrated them using an Amicon centrifugal concentrator (MilliporeSigma) and further purified PTP1B by size-exclusion chromatography on a HiLoad Superdex 75 column (Cytiva) using a buffer consisting of 10 mM Tris pH 7.5 with 25 mM NaCl and 0.2 mM EDTA. The protein was concentrated using an Amicon centrifugal concentrator (MilliporeSigma) and was stored at −80°C until crystallization. To avoid oxidation of the catalytic Cys215 (van Montfort et al., 2003 ▸), we added fresh DTT (1–3 mM) to each buffer that was used during purification.
Before crystallization, we diluted PTP1B to 10–15 mg ml−1 using protein storage buffer. For the TCS401-bound complex, we added a 25 mM stock of TCS401 (Tocris Bioscience) dissolved in DMSO to the protein solution to a fivefold molar excess and incubated it on ice for at least 30 min. We crystallized both apo and TCS401-bound PTP1B by the hanging-drop vapor-diffusion method using a well solution composed of 100 mM HEPES pH 6.8–7.6 with 250 mM magnesium acetate, 11–15%(w/v) PEG 8000, 10%(v/v) glycerol, 6%(v/v) ethanol and 0.1%(v/v) β-mercaptoethanol. Drops were set up by mixing 1 µl protein solution and 1 µl well solution over 0.5 ml wells. Crystallization plates were incubated at 4°C and rod-shaped crystals with hexagonal faces grew to 0.2–1 mm after 1–2 weeks that could diffract to 1.7 Å resolution. The crystals used for data collection were approximately 0.1 × 0.1 mm along the short axes and 0.5 mm along the long axis.
2.2. Data collection
We collected the data sets presented in this work on the Northeastern Collaborative Access Team (NE-CAT) beamline 24-ID-C at the Advanced Photon Source, Argonne National Laboratory on 17 July 2019. We looped all crystals on-site using the MicroRT system (MiTeGen) for room-temperature data collection and conducted the diffraction experiments at ambient temperature, which was measured to be approximately 295 K. We used a beam energy of 6.55 keV to maximize the native anomalous scattering contribution within the energy range of the beamline, corresponding to anomalous scattering factors, f′′, of 3.95, 0.63 and 0.80 e− for manganese, phosphorus and sulfur, respectively (Cromer & Liberman, 1981 ▸; Brennan & Cowan, 1992 ▸).
To obtain high-redundancy data sets while minimizing radiation damage, we collected 1440-image passes (720°) from each crystal with an exposure time of 0.1 s and an oscillation angle of 0.5°. The incident X-ray intensity was attenuated to 0.2% transmission, corresponding to an estimated flux of 9.6 × 109 photons s−1. To distribute the dosage across the crystal volume, we used a beam size of approximately 5 × 30 µm (FWHM) that was collimated using a 100 µm aperture, and we used helical acquisition to continually translate along the crystal. The PILATUS 6M-F detector (Dectris) has 2463 × 2527 pixels and was positioned at the minimal distance of 150 mm. Due to the long wavelength used, the resolution was limited by the experimental geometry rather than the crystal quality for each sample. To obtain the highest resolution data that were accessible, we raised the detector by two panels during data collection. We used this experimental geometry for both passes on HEWL, both passes on PTP1B in complex with TCS401 and both crystals of apo PTP1B. For DHFR, we collected the first pass with a centered detector and the following two passes with a raised detector to record higher resolution reflections.
2.3. Data reduction, SAD phasing and structure refinement
We processed each pass from each crystal using DIALS (Winter et al., 2018 ▸). Due to the raised detector geometry, we set the beam center to (290.5, 225.2) and used a pixel mask to remove a shadow cast by the beamstop support. Our images contain leakage from a higher energy undulator harmonic. Therefore, we excluded strong reflections lower than 6 Å in resolution during indexing to avoid Bragg reflections from the secondary diffraction pattern originating from the harmonic. To index each pass on the PTP1B crystals, we used local index assignment (index.assignment.method=local), which significantly improved the percentage of indexed strong reflections by tolerating small amounts of crystal drift during the helical acquisition. This index-assignment algorithm is comparable to that used by XDS (Kabsch, 2010 ▸). After integration, we scaled and merged the multiple passes for each crystal using AIMLESS (Evans & Murshudov, 2013 ▸). For apo PTP1B, we scaled and merged one pass each from two different crystals, which we found to be necessary to improve the anomalous signal for SAD phasing. For the PTP1B–TCS401 complex, we excluded the last 360 frames from the second pass due to weaker diffraction.
We solved each structure by SAD phasing using default settings in AutoSol (Terwilliger et al., 2009 ▸) in Phenix (Adams et al., 2010 ▸; Liebschner et al., 2019 ▸). We used sulfur as the anomalous scattering element for HEWL and PTP1B, and we specified manganese, phosphorus and sulfur as the anomalous scattering elements for DHFR. We successfully phased all data sets using this approach without additional intervention and constructed initial models for HEWL, DHFR and TCS401-bound PTP1B using AutoBuild (Terwilliger et al., 2008 ▸). We further refined these models by cycles of automated refinement with phenix.refine and manual model building in Coot (Emsley et al., 2010 ▸). Due to the extensive alternate conformations in the apo PTP1B electron density, we used the TCS401-bound model to initialize the apo model. We manually built the open state of the WPD loop (residues 176–193) in Coot and used group occupancies in phenix.refine to constrain mutually exclusive conformations to sum to full occupancy.
2.4. Assessing the impact of redundancy on CCanom
To characterize the effect of high-redundancy data collection, we scaled and merged each of the four data sets with increasing numbers of consecutive frames in AIMLESS (Evans & Murshudov, 2013 ▸). We computed half-data-set correlation coefficients of the anomalous differences (CCanom) for different numbers of frames using reciprocalspaceship (Greisman et al., 2021 ▸) to implement repeated twofold cross-validation. With this, we determined the mean and standard deviations of the CCanom by resolution bin for the HEWL S-SAD data set.
2.5. Assessing the impact of redundancy on real-space anomalous signal
We computed anomalous difference maps for the data sets with increasing numbers of frames using the phases from the refined models, and determined anomalous peak heights using phenix.find_peaks_and_holes (Adams et al., 2010 ▸; Liebschner et al., 2019 ▸). To analyze the anomalous peak height as a function of redundancy, we note that Terwilliger and coworkers derived an expression relating the expected anomalous difference map peak height for a SAD experiment,
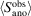 , to the useful anomalous correlation of the data set, CCano (Terwilliger et al., 2016a
▸),
, to the useful anomalous correlation of the data set, CCano (Terwilliger et al., 2016a
▸),
where N refl is the number of unique reflections in the reciprocal asymmetric unit cell, n sites is the number of sites in the anomalous substructure and f B is an average B-factor-modified anomalous atomic scattering factor. In turn, they derive CCano in terms of a normalized variance, E 2 (Terwilliger et al., 2016a ▸),
The normalized variance consists of the sum of anomalous contributions from other scatterers (for example C, N and O atoms in the protein and solvent contributions), systematic errors resulting from experimental and data-reduction imperfections not adequately compensated for in scaling, and random measurement errors. This sum is normalized by the useful anomalous signal (Terwilliger et al., 2016a ▸). The variance of the measurement errors is inversely proportional to the redundancy of observations, N, such that we can combine (1) and (2) to explicitly account for the redundancy, yielding the following functional form
where a is a coefficient representing the maximum achievable anomalous peak height and b is the variance of the measurement error relative to the sum of the variances of anomalous signal (useful and other) and of systematic errors. More pragmatically, b is the redundancy at which CCano reaches 71% [(1/2)1/2] of its maximal value. N is the average redundancy (or, equivalently, the number of frames).
For each of the data sets, we fit the anomalous peak height of each site, s, as a function of number of frames, N n , by minimizing the following least-squares objective function, based on (3), with the Levenberg–Marquardt algorithm implemented in SciPy (Virtanen et al., 2020 ▸),
where y
s, n
is the anomalous peak height of site s with N
n
frames, a
s
is a fitting parameter representing the maximal peak height for site s and b is a global fitting parameter for the data set. We also define rescaled observations as
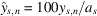 corresponding to the percentage of the maximum attainable anomalous peak height, a
s
, that was fitted for each site. This procedure ensures that the anomalous peak heights are on comparable scales among sites, and the rescaled data were compared with the normalized model given by y = 100/(1 + b/N)1/2.
corresponding to the percentage of the maximum attainable anomalous peak height, a
s
, that was fitted for each site. This procedure ensures that the anomalous peak heights are on comparable scales among sites, and the rescaled data were compared with the normalized model given by y = 100/(1 + b/N)1/2.
3. Results
3.1. Native SAD phasing of model systems at 295 K
Native SAD phasing is commonly used as a benchmark for the accuracy of new data-collection strategies and technologies (Liu et al., 2012 ▸; Nakane et al., 2015 ▸; Weinert et al., 2015 ▸; Wagner et al., 2016 ▸; Guo et al., 2019 ▸). Here, we used native SAD to solve four structures of three model systems at room temperature (295 K). Previous work by Weinert and coworkers demonstrated high-redundancy data collection from single-crystal diffraction at 6 keV and at cryogenic temperatures (Weinert et al., 2015 ▸; Basu et al., 2019 ▸). The data-collection strategy presented here for native SAD phasing at room temperature differs from their approach in several ways. They used a multi-axis goniometer to collect data at multiple sample orientations, while we instead continually translated along the crystals to distribute the X-ray dose across the crystal volume. In addition, Weinert and coworkers used fine φ-slicing to improve their data quality. Fine φ-slicing is most effective when the rotation angle is comparable to half the crystal mosaicity (Mueller et al., 2012 ▸), which in our experience is often not possible with room-temperature crystals due to their low mosaicity (0.02–0.04°; Table 1 ▸).
Table 1. Data-collection and refinement statistics.
| HEWL | DHFR | Apo PTP1B | PTP1B–TCS401 | |
|---|---|---|---|---|
| PDB code | 7l84 | 7lvc | 7rin | 7mm1 |
| Anomalous scatterers | S | Mn, P, S | S | S |
| Data collection | ||||
| No. of crystals | 1 | 1 | 2 | 1 |
| Passes per crystal | 2 | 3 | 1 | 2 |
| Rotation (°) | 1440 | 2160 | 1440 | 1440 |
| Wavelength (Å) | 1.892 | 1.892 | 1.892 | 1.892 |
| Space group | P43212 | P212121 | P3121 | P3121 |
| a, b, c (Å) | 79.3, 79.3, 37.8 | 34.3, 45.6, 99.0 | 89.5, 89.5, 106.1 | 89.3, 89.3, 105.7 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 120.0 | 90.0, 90.0, 120.0 |
| Dose per crystal† (kGy) | 160 | 230 | 80 | 150 |
| Resolution (Å) | 39.67–1.71 (1.77–1.71) | 49.52–1.71 (1.77–1.71) | 62.61–1.85 (1.92–1.85) | 62.44–1.85 (1.92–1.85) |
| Total observations | 816788 (4492) | 823003 (3254) | 2413974 (111538) | 2098115 (96817) |
| Unique observations | 22855 (767) | 27967 (720) | 81017 (7932) | 79832 (7532) |
| Multiplicity | 35.7 (5.9) | 29.4 (4.5) | 29.8 (14.1) | 26.3 (12.9) |
| Completeness (%) | 90.74 (30.45) | 85.52 (22.11) | 99.68 (97.30) | 99.00 (93.15) |
| Mean I/σ(I) | 35.63 (6.70) | 30.18 (4.42) | 15.29 (1.77) | 16.60 (2.21) |
| R merge | 0.102 (0.189) | 0.101 (0.223) | 0.143 (1.466) | 0.138 (1.748) |
| R p.i.m. | 0.015 (0.084) | 0.017 (0.114) | 0.024 (0.398) | 0.026 (0.493) |
| CC1/2 | 0.999 (0.968) | 0.998 (0.945) | 0.999 (0.610) | 0.999 (0.675) |
| Mosaicity (°) | 0.021 ± 0.0002 | 0.016 ± 0.0030 | 0.043, 0.019 | 0.023 ± 0.0044 |
| Phasing | ||||
| Figure of merit‡ | 0.47 | 0.49 | 0.25 | 0.27 |
| Refinement | ||||
| R work (%) | 14.0 | 13.9 | 13.2 | 12.9 |
| R free (%) | 16.2 | 17.2 | 15.4 | 13.9 |
| No. of non-H atoms | ||||
| Total | 1209 | 1588 | 3024 | 2793 |
| Macromolecules | 1112 | 1396 | 2840 | 2592 |
| Ligands | 0 | 80 | 24 | 18 |
| Water | 97 | 107 | 160 | 159 |
| No. of protein residues | 129 | 159 | 298 | 298 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.008 | 0.009 | 0.010 | 0.010 |
| Angles (°) | 0.91 | 1.19 | 1.02 | 1.04 |
| Wilson B factor (Å2) | 17.21 | 12.78 | 29.56 | 25.49 |
| Average B factor (Å2) | ||||
| Overall | 20.12 | 16.83 | 43.45 | 34.16 |
| Macromolecules | 19.00 | 16.07 | 42.72 | 33.39 |
| Ligands | — | 13.65 | 70.00 | 21.85 |
| Water | 32.97 | 28.21 | 52.56 | 44.47 |
| Clashscore | 0.45 | 1.74 | 2.98 | 3.05 |
| Ramachandran plot | ||||
| Favored (%) | 100.0 | 99.35 | 97.64 | 98.31 |
| Allowed (%) | 0.0 | 0.65 | 2.03 | 1.35 |
| Outliers (%) | 0.0 | 0.0 | 0.34 | 0.34 |
Data were collected at 6.55 keV to maximize the anomalous scattering signal within the accessible spectrum of the APS 24-ID-C beamline. Each of the following structures was solved from multiple 1440-image (720°) passes on a single crystal, except for apo PTP1B, which was solved from passes collected from two different crystals. The data-collection statistics for these four data sets are presented in Table 1 ▸. These data sets have high redundancy (26–35× overall) for room-temperature, single-crystal experiments. The resolution was limited by the experimental geometry due to the long wavelength, leading to high mean I/σ(I) and CC1/2 values and low completeness in the high-resolution bins. Using RADDOSE-3D with our beam and crystal parameters, we determined that this strategy corresponds to an average diffraction-weighted dose of approximately 40 kGy per 360° of data collection or to a maximum dose (comparable metric to RADDOSE v2) of 0.5 MGy per 360° (Zeldin et al., 2013 ▸; Bury et al., 2018 ▸). This estimated dose is consistent with the method reported by Weinert et al. (2015 ▸), and the relative image B factors fitted by AIMLESS suggest that radiation damage did not significantly impact data reduction (Evans & Murshudov, 2013 ▸). The estimated dose per crystal is given in Table 1 ▸.
The structure of HEWL was phased using the anomalous scattering from the ten S atoms in the protein, and the anomalous difference map shows clear density for the four disulfide bonds and two methionine residues in the model (Fig. 1 ▸ a). The Michaelis complex of DHFR was solved using the anomalous scattering from the manganese ions in the crystallization solution. The refined model includes five ordered Mn2+ ions, which were supported by the anomalous difference map (Fig. 1 ▸ b). Significant anomalous density (>5σ) is also present for the three P atoms in NADP+ and six of the seven sulfur-containing side chains. PTP1B was phased by S-SAD in an apo state and in complex with the active-site inhibitor TCS401 (Iversen et al., 2000 ▸). 13 of 15 sulfur sites were resolved in the anomalous difference map of apo PTP1B at 5σ (Fig. 1 ▸ c), and 15 of 16 sulfur sites can be seen in the corresponding map of the PTP1B–TCS401 complex (Fig. 1 ▸ d). In each of these structures, the missing sulfur-containing side chains belonged to methionine residues with weak density at or near the N-terminus. The refinement statistics for the model systems are summarized in Table 1 ▸, and the peak heights from the model-phased anomalous difference maps are presented in Supplementary Tables S1–S4.
Figure 1.

High-redundancy data collection enables room-temperature, native SAD phasing of model systems. SAD solution structures for (a) hen egg-white lysozyme, (b) a model of the E. coli dihydrofolate reductase Michaelis complex, (c) apo human protein tyrosine phosphatase 1B and (d) TCS401-bound human protein tyrosine phosphatase 1B are shown with their respective anomalous difference maps (purple mesh). The maps in (a), (c) and (d) are contoured at 5σ and shown within 2 Å of modeled atoms. The DHFR anomalous difference map in (b) is contoured at 6σ and shown within 8 Å of modeled atoms due to the additional anomalous signal from partial occupancy manganese sites at crystal contacts. Images were rendered using PyMOL (version 2.4; Schrödinger).
3.2. High-redundancy data collection improves the anomalous signal
To assess the impact of redundancy on anomalous signal, we reprocessed the HEWL S-SAD data set with increasing numbers of diffraction images. The HEWL data set shows an improvement in the correlation coefficient of anomalous differences across half-data sets with increasing frame counts (Fig. 2 ▸ a). This is consistent with the expectation that the signal to noise of merged intensities should improve with redundancy (Dauter & Adamiak, 2001 ▸; Hendrickson, 2014 ▸). Furthermore, we were only able to phase the HEWL structure by the SAD method with a minimum of 720 frames. The figures of merit of the solutions improved monotonically with additional frames (Supplementary Table S5). This is in line with the expectation that the likelihood of successful phasing with the SAD method correlates with CCanom (Terwilliger et al., 2016b ▸).
Figure 2.

Redundancy improves the anomalous signal for a HEWL S-SAD data set. (a) Spearman correlation coefficient for anomalous differences, CCanom, from repeated twofold cross-validation for data sets composed of increasing numbers of frames. Error bars denote the mean ± standard deviation for ten random partitions of the frames. (b) The mean anomalous peak heights for the ten S atoms in the asymmetric unit cell after refinement using merged intensities for data sets with increasing numbers of frames (black; mean ± standard deviation). The peak heights are on an absolute scale relative to the maximal value that can be achieved. A nonlinear least-squares fit to the data is shown for y = 100/(1 + b/N)1/2 (red dashed line; b = 751 ± 76; 95% confidence interval).
In the model-phased anomalous difference maps, the mean peak height of the S atoms approaches an asymptote with increasing redundancy (Fig. 2 ▸ b). The model (equation 3) derived from the analysis of anomalous signal described by Terwilliger et al. (2016a ▸) provides an excellent fit to the observed anomalous peak heights as a function of the number of frames. The fit yields b = 751 ± 76, which corresponds to 89% of the maximum achievable anomalous signal for a 2878-frame data set at this completeness and resolution. This suggests that additional observations should have limited impact on the anomalous signal of this data set. A close correspondence between the model (equation 3) and anomalous signal is also observed for the three additional model systems used in this work (Supplementary Fig. S2).
3.3. DHFR anomalous differences highlight multiple anomalous scattering elements
Although the DHFR data set was collected with the goal of SAD phasing based on the manganese content from the crystallization conditions, we observed additional anomalous difference peaks corresponding to the native S atoms in the protein and the P atoms of the NADP+ cofactor. The active site of the refined model is shown in Fig. 3 ▸(a), which highlights sulfur and phosphorus anomalous peaks. In addition to the well ordered NADP+ cofactor, significant anomalous differences are seen for the catalytically important Met20 and Met42 side chains. Mutations of Met42 significantly impact hydride transfer in the enzyme (Wang et al., 2006 ▸), while Met20 adopts multiple rotamers in high-resolution structures of DHFR and plays a role both in hydride transfer and in regulating solvent access to promote substrate protonation (Sawaya & Kraut, 1997 ▸; Wan et al., 2014 ▸).
Figure 3.

Anomalous differences in a model of the DHFR Michaelis complex highlight active-site residues and partial occupancy ions. (a) The active site of DHFR is shown with NADP+ and folate, a substrate mimetic, in light blue and green, respectively, with nearby methionine residues shown in gray. The 2F o − F c map is displayed in blue at 1.2σ within 1.5 Å of the shown atoms. Density for the S atoms of methionine residues and the P atoms of NADP+ can be seen in the anomalous difference map (purple mesh; 5σ). (b) Multiple partial occupancy manganese sites are supported by the anomalous difference map (purple mesh; 5σ) around a phosphate group of NADP+. Only one of these sites was modeled as a manganese based on density in the 2F o − F c map (blue mesh; 1.5σ). The 2F o − F c map is displayed within 2 Å of the shown atoms and the anomalous difference map is shown within 8 Å of the P atom. Contact distances of less than 3.0 Å to manganese sites are shown as black dashed lines. Images were rendered using PyMOL (version 2.4; Schrödinger).
The anomalous difference map also contains significant density (>5σ) in regions that are not supported by strong electron density in the 2F o − F c map (Fig. 3 ▸ b). Although one manganese site is strongly supported in the 2F o − F c map in the vicinity of the phosphate group of NADP+, the additional anomalous density can be best explained by partial occupancy manganese sites around the phosphate. Each of these sites involves close contacts (<3.0 Å) to protein side chains or the phosphate group to support their coordination geometries; however, the very close distances between manganese sites (2–3 Å) suggest that occupancy at these sites is mutually exclusive. This underscores that the anomalous difference density represents an ensemble- and time-averaged view of the region. The close vicinity of Asp11 from a neighboring copy of DHFR also suggests that these sites are stabilized by crystal-packing interactions. The observation of these partial occupancy binding sites highlights the accuracy and sensitivity of these anomalous difference maps.
3.4. Room-temperature structures feature conformational heterogeneity
An advantage of room-temperature data collection is that the conformational ensemble of the macromolecule will be better represented than at cryogenic temperatures (Doukov et al., 2020 ▸; Fraser et al., 2011 ▸; Keedy et al., 2014 ▸). This is most evident in our native SAD structure of apo PTP1B, which shows a superposition of the open and closed states of the catalytically important WPD loop (Fig. 4 ▸ a). Our structure refined to 61% open and 39% closed, which is consistent with the multi-temperature refinement by Keedy and coworkers, who obtained 65% open and 35% closed at 277 K (Keedy et al., 2018 ▸). The small difference in refined occupancy may be attributable to different refinement protocols, in particular with regard to the partial occupancy waters in the vicinity of the WPD loop. In the presence of TCS401, an orthosteric inhibitor, the WPD loop fully occupies the closed state (Fig. 4 ▸ b). This is consistent with existing structures of PTP1B in complex with the inhibitor (Iversen et al., 2000 ▸; Choy et al., 2017 ▸).
Figure 4.

Room-temperature structures of PTP1B exhibit extensive conformational heterogeneity. (a) At room temperature, apo PTP1B adopts a superposition of the open and closed states of the WPD loop, which results in different positioning of important catalytic residues. (b) With a bound orthosteric inhibitor, TCS401 (green), the WPD loop is only observed in the closed state. 2F o − F c maps are shown as a blue mesh (0.5σ) within 1.5 Å of the displayed residues. (c) The F apo − F TCS401 isomorphous difference map, phased using the apo model, illustrates the sites of the protein that differ on binding TCS401. The difference map is contoured at +3.5σ (blue) and −3.5σ (red). The WPD loop is depicted in blue (open state) and red (closed state), and known allosteric sites are labeled. Most of the structural changes that occur between WPD loop states are localized to one side of the central β-sheet of PTP1B. Images were rendered using PyMOL (version 2.4; Schrödinger).
Because the apo structure exhibits a superposition of WPD loop states and the TCS401-bound complex fully populates the closed state of the WPD loop, we can visualize the regions that differ between the two WPD loop states using an isomorphous difference map, |F apo| − |F TCS401|. We used SCALEIT (Winn et al., 2011 ▸; Howell & Smith, 1992 ▸) to place the TCS401-bound data set on the same scale as the apo data set, and we then computed an isomorphous difference map using the phases from the apo model. The isomorphous difference map is shown overlaid on the apo structure in Fig. 4 ▸(c). The positive density (blue) highlights regions that are associated with the open state of the WPD loop (stronger in the apo data set) and the negative density (red) highlights those associated with the closed state of the WPD loop (stronger in the TCS401-bound data set). The isomorphous difference map illustrates that the primary sites that undergo structural changes between the two WPD loop states are localized to one side of the central β-sheet of PTP1B. Furthermore, strong difference density is observed in the vicinity of previously identified allosteric sites such as the benzbromarone binding site (BB site; Wiesmann et al., 2004 ▸), L16 site (Keedy et al., 2018 ▸; Cui et al., 2017 ▸) and 197 site (Keedy et al., 2018 ▸; Cui et al., 2017 ▸; Choy et al., 2017 ▸). The observation of these widespread structural differences within and between data sets illustrates a benefit of working near physiological temperatures (Keedy et al., 2014 ▸) and suggests that such experiments can be used to probe allosteric mechanisms in protein structures.
4. Discussion
Native SAD phasing relies on the accurate measurement of the small differences in intensity that arise due to anomalous scattering. Although single- and multi-crystal strategies have been presented for native SAD phasing at cryogenic temperatures (Liu et al., 2012 ▸; Weinert et al., 2015 ▸; Wagner et al., 2016 ▸; Guo et al., 2019 ▸; Basu et al., 2019 ▸; Cianci et al., 2019 ▸), room-temperature native SAD phasing has nearly exclusively been reported using serial crystallography experiments (Nakane et al., 2015 ▸; Huang et al., 2015 ▸) since the first S-SAD experiment in 1981 (Hendrickson & Teeter, 1981 ▸). Here, we present a strategy for room-temperature data collection that takes advantage of the capabilities of modern macromolecular beamlines to obtain high-quality, high-redundancy X-ray diffraction data sets. We used high attenuation and helical acquisition to minimize the local X-ray dose while taking advantage of the photon-counting, low-noise readout of the pixel-array detectors that are in widespread use at macromolecular beamlines. The resulting data sets enabled automatic phasing by the SAD method and the model-phased anomalous difference maps clearly identify the sites of each anomalous scattering atom (Fig. 1 ▸).
Although native SAD data sets collected at cryogenic temperatures may exceed data redundancies of 100-fold (Weinert et al., 2015 ▸), we observe that the model-phased anomalous difference peaks plateau with increasing redundancy (Fig. 2 ▸ b and Supplementary Fig. S2). This is a practical implication of equation (3) based on the analysis by Terwilliger et al. (2016a ▸). For the crystals used here, additional redundancy would have diminishing marginal improvement to the anomalous signal. Consistent with this, the HEWL and DHFR samples achieved approximately 90% of their asymptotic anomalous peak heights with 1440° of data collection.
The lower redundancy required for SAD phasing in these crystals may be explained by a previous study that found improved isomorphism in room-temperature samples (Keedy et al., 2014 ▸). Importantly, this suggests that our methodology will readily apply to multi-crystal approaches, which may be an appealing way to achieve room-temperature measurements with more radiation-sensitive crystals. This could be an important application because multi-crystal approaches have been successful for native SAD phasing from crystals that diffract to more moderate resolutions (2.3–2.8 Å; Liu et al., 2012 ▸); however, such efforts are commonly hindered by the non-isomorphism that may result from cryocooling (Giordano et al., 2012 ▸; Keedy et al., 2014 ▸). Although the crystals used in this project were fairly large (0.2–0.5 mm in length) and did not exhibit signs of radiation damage, the successful phasing of apo PTP1B required the merging of data sets from two different crystals, validating this use case. This should extend our data-collection strategy to difficult-to-freeze samples or to crystals of moderate size and resolution.
The model-phased anomalous difference map from DHFR identified multiple anomalous scattering elements (Fig. 3 ▸ a) and allowed us to resolve partial occupancy sites for manganese that were not evident in the 2F o − F c map (Fig. 3 ▸ b). This suggests that room-temperature data collection at low X-ray energies may be a valuable approach for the study of metal-coordination sites in proteins. Room-temperature anomalous signal may also be useful to study the binding poses of ligands and drug fragments containing S, P and halogen atoms. Furthermore, the room-temperature native SAD structures contain significant conformational heterogeneity, as exemplified by the superposition of loop states observed in apo PTP1B (Fig. 4 ▸ a). These features, and the resulting isomorphous difference maps (Fig. 4 ▸ c), emphasize the accuracy and sensitivity of the native SAD phasing results presented at 295 K. Since the high-redundancy strategy presented here can be implemented at many macromolecular beamlines, this work will support new investigations that use anomalous scattering to study macromolecules at physiological temperatures.
5. Data availability
All diffraction images used in this work have been deposited in SBGrid with SBGrid IDs 816, 821, 834 and 845. These depositions contain the complete processing details for DIALS, including the pixel mask that was used to exclude the beam-stop shadow on the detector. The refined structures presented here have been deposited in the Protein Data Bank (Berman et al., 2000 ▸) with PDB codes 7l84, 7lvc, 7mm1 and 7rin. The integrated intensities from DIALS and the scaled merged/unmerged data from AIMLESS have been uploaded to Zenodo for each data set at the following DOIs: https://doi.org/10.5281/zenodo.4426679, https://doi.org/10.5281/zenodo.4637114, https://doi.org/10.5281/zenodo.4767996 and https://doi.org/10.5281/zenodo.5544447.
Supplementary Material
PDB reference: hen egg-white lysozyme, 7l84
PDB reference: Escherichia coli dihydrofolate reductase, 7lvc
PDB reference: human protein tyrosine phosphatase 1B, apo, 7rin
PDB reference: complex with TCS401, 7mm1
. DOI: 10.1107/S2059798322006799/rr5221sup1.pdf
complex with TCS401, 7mm1
Diffraction data for PDB entry 7l84.: https://doi.org/10.15785/SBGRID/816
Diffraction data for PDB entry 7lvc.: https://doi.org/10.15785/SBGRID/821
Diffraction data for PDB entry 7mm1.: https://doi.org/10.15785/SBGRID/834
Diffraction data for PDB entry 7rin.: https://doi.org/10.15785/SBGRID/845
Intermediate processing results used to solve a structure of hen egg white lysozyme by native S-SAD at room temperature.: https://doi.org/10.5281/zenodo.4426679
Intermediate processing results used to solve a structure of Escherichia coli DHFR by native Mn,P,S-SAD at room temperature.: https://doi.org/10.5281/zenodo.4637114
Intermediate processing results used to solve a structure of PTP1B in complex with TCS401 by native S-SAD at room temperature.: https://doi.org/10.5281/zenodo.4767996
Intermediate processing results used to solve a structure of apo PTP1B by native S-SAD at room temperature.: https://doi.org/10.5281/zenodo.5544447
Acknowledgments
We thank the staff of the Northeastern Collaborative Access Team (NE-CAT) beamline 24-ID-C at the Advanced Photon Source for supporting our room-temperature crystallography experiments. We also thank Rachelle Gaudet (Harvard University) for helpful feedback on this manuscript. NE-CAT beamlines are supported by the National Institute of General Medical Sciences, NIH (P30 GM124165) using resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Funding Statement
This work was supported by the Searle Scholarship Program (SSP-2018-3240) and a fellowship from the George W. Merck Fund of the New York Community Trust (No. 338034). JBG is supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE1745303. KMD holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund.
References
- Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L.-W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., Richardson, J. S., Terwilliger, T. C. & Zwart, P. H. (2010). Acta Cryst. D66, 213–221. [DOI] [PMC free article] [PubMed]
- Basu, S., Finke, A., Vera, L., Wang, M. & Olieric, V. (2019). Acta Cryst. D75, 262–271. [DOI] [PMC free article] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed]
- Brennan, S. & Cowan, P. L. (1992). Rev. Sci. Instrum. 63, 850–853.
- Bury, C. S., Brooks-Bartlett, J. C., Walsh, S. P. & Garman, E. F. (2018). Protein Sci. 27, 217–228. [DOI] [PMC free article] [PubMed]
- Cameron, C. E. & Benkovic, S. J. (1997). Biochemistry, 36, 15792–15800. [DOI] [PubMed]
- Choy, M. S., Li, Y., Machado, L. E. S. F., Kunze, M. B. A., Connors, C. R., Wei, X., Lindorff-Larsen, K., Page, R. & Peti, W. (2017). Mol. Cell, 65, 644–658. [DOI] [PMC free article] [PubMed]
- Cianci, M., Nanao, M. & Schneider, T. R. (2019). Acta Cryst. D75, 192–199. [DOI] [PMC free article] [PubMed]
- Cromer, D. T. & Liberman, D. A. (1981). Acta Cryst. A37, 267–268.
- Cui, D. S., Beaumont, V., Ginther, P. S., Lipchock, J. M. & Loria, J. P. (2017). J. Mol. Biol. 429, 2360–2372. [DOI] [PMC free article] [PubMed]
- Dauter, Z. & Adamiak, D. A. (2001). Acta Cryst. D57, 990–995. [DOI] [PubMed]
- Doukov, T., Herschlag, D. & Yabukarski, F. (2020). J. Appl. Cryst. 53, 1493–1501. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Erlanson, D. A., McDowell, R. S., He, M. M., Randal, M., Simmons, R. L., Kung, J., Waight, A. & Hansen, S. K. (2003). J. Am. Chem. Soc. 125, 5602–5603. [DOI] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Fraser, J. S., Clarkson, M. W., Degnan, S. C., Erion, R., Kern, D. & Alber, T. (2009). Nature, 462, 669–673. [DOI] [PMC free article] [PubMed]
- Fraser, J. S., van den Bedem, H., Samelson, A. J., Lang, P. T., Holton, J. M., Echols, N. & Alber, T. (2011). Proc. Natl Acad. Sci. USA, 108, 16247–16252. [DOI] [PMC free article] [PubMed]
- Giordano, R., Leal, R. M. F., Bourenkov, G. P., McSweeney, S. & Popov, A. N. (2012). Acta Cryst. D68, 649–658. [DOI] [PubMed]
- Greisman, J. B., Dalton, K. M. & Hekstra, D. R. (2021). J. Appl. Cryst. 54, 1521–1529. [DOI] [PMC free article] [PubMed]
- Guo, G., Zhu, P., Fuchs, M. R., Shi, W., Andi, B., Gao, Y., Hendrickson, W. A., McSweeney, S. & Liu, Q. (2019). IUCrJ, 6, 532–542. [DOI] [PMC free article] [PubMed]
- Hendrickson, W. A. (2014). Q. Rev. Biophys. 47, 49–93. [DOI] [PMC free article] [PubMed]
- Hendrickson, W. A. & Teeter, M. M. (1981). Nature, 290, 107–113. [DOI] [PMC free article] [PubMed]
- Hope, H. (1988). Acta Cryst. B44, 22–26. [DOI] [PubMed]
- Howell, P. L. & Smith, G. D. (1992). J. Appl. Cryst. 25, 81–86.
- Huang, C.-Y., Olieric, V., Ma, P., Panepucci, E., Diederichs, K., Wang, M. & Caffrey, M. (2015). Acta Cryst. D71, 1238–1256. [DOI] [PMC free article] [PubMed]
- Iversen, L. F., Andersen, H. S., Branner, S., Mortensen, S. B., Peters, G. H., Norris, K., Olsen, O. H., Jeppesen, C. B., Lundt, B. F., Ripka, W., Møller, K. B. & Møller, N. P. H. (2000). J. Biol. Chem. 275, 10300–10307. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Keedy, D. A., Hill, Z. B., Biel, J. T., Kang, E., Rettenmaier, T. J., Brandão-Neto, J., Pearce, N. M., von Delft, F., Wells, J. A. & Fraser, J. S. (2018). eLife, 7, e36307. [DOI] [PMC free article] [PubMed]
- Keedy, D. A., van den Bedem, H., Sivak, D. A., Petsko, G. A., Ringe, D., Wilson, M. A. & Fraser, J. S. (2014). Structure, 22, 899–910. [DOI] [PMC free article] [PubMed]
- Liebschner, D., Afonine, P. V., Baker, M. L., Bunkóczi, G., Chen, V. B., Croll, T. I., Hintze, B., Hung, L.-W., Jain, S., McCoy, A. J., Moriarty, N. W., Oeffner, R. D., Poon, B. K., Prisant, M. G., Read, R. J., Richardson, J. S., Richardson, D. C., Sammito, M. D., Sobolev, O. V., Stockwell, D. H., Terwilliger, T. C., Urzhumtsev, A. G., Videau, L. L., Williams, C. J. & Adams, P. D. (2019). Acta Cryst. D75, 861–877.
- Liu, Q., Dahmane, T., Zhang, Z., Assur, Z., Brasch, J., Shapiro, L., Mancia, F. & Hendrickson, W. A. (2012). Science, 336, 1033–1037. [DOI] [PMC free article] [PubMed]
- Liu, Q. & Hendrickson, W. A. (2017). Methods Mol. Biol. 1607, 377–399. [DOI] [PMC free article] [PubMed]
- Montfort, R. L. M. van, Congreve, M., Tisi, D., Carr, R. & Jhoti, H. (2003). Nature, 423, 773–777. [DOI] [PubMed]
- Mueller, M., Wang, M. & Schulze-Briese, C. (2012). Acta Cryst. D68, 42–56. [DOI] [PMC free article] [PubMed]
- Nakane, T., Song, C., Suzuki, M., Nango, E., Kobayashi, J., Masuda, T., Inoue, S., Mizohata, E., Nakatsu, T., Tanaka, T., Tanaka, R., Shimamura, T., Tono, K., Joti, Y., Kameshima, T., Hatsui, T., Yabashi, M., Nureki, O., Iwata, S. & Sugahara, M. (2015). Acta Cryst. D71, 2519–2525. [DOI] [PMC free article] [PubMed]
- Pflugrath, J. W. (2015). Acta Cryst. F71, 622–642. [DOI] [PMC free article] [PubMed]
- Rose, J. P., Wang, B.-C. & Weiss, M. S. (2015). IUCrJ, 2, 431–440. [DOI] [PMC free article] [PubMed]
- Sawaya, M. R. (1994). PhD thesis. University of California, San Diego, USA.
- Sawaya, M. R. & Kraut, J. (1997). Biochemistry, 36, 586–603. [DOI] [PubMed]
- Taylor, G. L. (2010). Acta Cryst. D66, 325–338. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C., Adams, P. D., Read, R. J., McCoy, A. J., Moriarty, N. W., Grosse-Kunstleve, R. W., Afonine, P. V., Zwart, P. H. & Hung, L.-W. (2009). Acta Cryst. D65, 582–601. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C., Bunkóczi, G., Hung, L.-W., Zwart, P. H., Smith, J. L., Akey, D. L. & Adams, P. D. (2016a). Acta Cryst. D72, 346–358. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C., Bunkóczi, G., Hung, L.-W., Zwart, P. H., Smith, J. L., Akey, D. L. & Adams, P. D. (2016b). Acta Cryst. D72, 359–374. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C., Grosse-Kunstleve, R. W., Afonine, P. V., Moriarty, N. W., Zwart, P. H., Hung, L.-W., Read, R. J. & Adams, P. D. (2008). Acta Cryst. D64, 61–69. [DOI] [PMC free article] [PubMed]
- Virtanen, P., Gommers, R., Oliphant, T. E., Haberland, M., Reddy, T., Cournapeau, D., Burovski, E., Peterson, P., Weckesser, W., Bright, J., van der Walt, S. J., Brett, M., Wilson, J., Millman, K. J., Mayorov, N., Nelson, A. R. J., Jones, E., Kern, R., Larson, E., Carey, C. J., Polat, İ., Feng, Y., Moore, E. W., VanderPlas, J., Laxalde, D., Perktold, J., Cimrman, R., Henriksen, I., Quintero, E. A., Harris, C. R., Archibald, A. M., Ribeiro, A. H., Pedregosa, F., van Mulbregt, P., Vijaykumar, A., Bardelli, A. P., Rothberg, A., Hilboll, A., Kloeckner, A., Scopatz, A., Lee, A., Rokem, A., Woods, C. N., Fulton, C., Masson, C., Häggström, C., Fitzgerald, C., Nicholson, D. A., Hagen, D. R., Pasechnik, D. V., Olivetti, E., Martin, E., Wieser, E., Silva, F., Lenders, F., Wilhelm, F., Young, G., Price, G. A., Ingold, G., Allen, G. E., Lee, G. R., Audren, H., Probst, I., Dietrich, J. P., Silterra, J., Webber, J. T., Slavič, J., Nothman, J., Buchner, J., Kulick, J., Schönberger, J. L., de Miranda Cardoso, J. V., Reimer, J., Harrington, J., Rodríguez, J. L. C., Nunez-Iglesias, J., Kuczynski, J., Tritz, K., Thoma, M., Newville, M., Kümmerer, M., Bolingbroke, M., Tartre, M., Pak, M., Smith, N. J., Nowaczyk, N., Shebanov, N., Pavlyk, O., Brodtkorb, P. A., Lee, P., McGibbon, R. T., Feldbauer, R., Lewis, S., Tygier, S., Sievert, S., Vigna, S., Peterson, S., More, S., Pudlik, T., Oshima, T., Pingel, T. J., Robitaille, T. P., Spura, T., Jones, T. R., Cera, T., Leslie, T., Zito, T., Krauss, T., Upadhyay, U., Halchenko, Y. O. & Vázquez-Baeza, Y. (2020). Nat. Methods, 17, 261–272.
- Wagner, A., Duman, R., Henderson, K. & Mykhaylyk, V. (2016). Acta Cryst. D72, 430–439. [DOI] [PMC free article] [PubMed]
- Wan, Q., Bennett, B. C., Wilson, M. A., Kovalevsky, A., Langan, P., Howell, E. E. & Dealwis, C. (2014). Proc. Natl Acad. Sci. USA, 111, 18225–18230. [DOI] [PMC free article] [PubMed]
- Wang, L., Goodey, N. M., Benkovic, S. J. & Kohen, A. (2006). Proc. Natl Acad. Sci. USA, 103, 15753–15758. [DOI] [PMC free article] [PubMed]
- Weinert, T., Olieric, V., Waltersperger, S., Panepucci, E., Chen, L., Zhang, H., Zhou, D., Rose, J., Ebihara, A., Kuramitsu, S., Li, D., Howe, N., Schnapp, G., Pautsch, A., Bargsten, K., Prota, A. E., Surana, P., Kottur, J., Nair, D. T., Basilico, F., Cecatiello, V., Pasqualato, S., Boland, A., Weichenrieder, O., Wang, B.-C., Steinmetz, M. O., Caffrey, M. & Wang, M. (2015). Nat. Methods, 12, 131–133. [DOI] [PubMed]
- Wiesmann, C., Barr, K. J., Kung, J., Zhu, J., Erlanson, D. A., Shen, W., Fahr, B. J., Zhong, M., Taylor, L., Randal, M., McDowell, R. S. & Hansen, S. K. (2004). Nat. Struct. Mol. Biol. 11, 730–737. [DOI] [PubMed]
- Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G. W., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A. & Wilson, K. S. (2011). Acta Cryst. D67, 235–242. [DOI] [PMC free article] [PubMed]
- Winter, G., Waterman, D. G., Parkhurst, J. M., Brewster, A. S., Gildea, R. J., Gerstel, M., Fuentes-Montero, L., Vollmar, M., Michels-Clark, T., Young, I. D., Sauter, N. K. & Evans, G. (2018). Acta Cryst. D74, 85–97. [DOI] [PMC free article] [PubMed]
- Zeldin, O. B., Gerstel, M. & Garman, E. F. (2013). J. Appl. Cryst. 46, 1225–1230.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: hen egg-white lysozyme, 7l84
PDB reference: Escherichia coli dihydrofolate reductase, 7lvc
PDB reference: human protein tyrosine phosphatase 1B, apo, 7rin
PDB reference: complex with TCS401, 7mm1
. DOI: 10.1107/S2059798322006799/rr5221sup1.pdf
complex with TCS401, 7mm1
Diffraction data for PDB entry 7l84.: https://doi.org/10.15785/SBGRID/816
Diffraction data for PDB entry 7lvc.: https://doi.org/10.15785/SBGRID/821
Diffraction data for PDB entry 7mm1.: https://doi.org/10.15785/SBGRID/834
Diffraction data for PDB entry 7rin.: https://doi.org/10.15785/SBGRID/845
Intermediate processing results used to solve a structure of hen egg white lysozyme by native S-SAD at room temperature.: https://doi.org/10.5281/zenodo.4426679
Intermediate processing results used to solve a structure of Escherichia coli DHFR by native Mn,P,S-SAD at room temperature.: https://doi.org/10.5281/zenodo.4637114
Intermediate processing results used to solve a structure of PTP1B in complex with TCS401 by native S-SAD at room temperature.: https://doi.org/10.5281/zenodo.4767996
Intermediate processing results used to solve a structure of apo PTP1B by native S-SAD at room temperature.: https://doi.org/10.5281/zenodo.5544447
Data Availability Statement
All diffraction images used in this work have been deposited in SBGrid with SBGrid IDs 816, 821, 834 and 845. These depositions contain the complete processing details for DIALS, including the pixel mask that was used to exclude the beam-stop shadow on the detector. The refined structures presented here have been deposited in the Protein Data Bank (Berman et al., 2000 ▸) with PDB codes 7l84, 7lvc, 7mm1 and 7rin. The integrated intensities from DIALS and the scaled merged/unmerged data from AIMLESS have been uploaded to Zenodo for each data set at the following DOIs: https://doi.org/10.5281/zenodo.4426679, https://doi.org/10.5281/zenodo.4637114, https://doi.org/10.5281/zenodo.4767996 and https://doi.org/10.5281/zenodo.5544447.