

Summary
Developmental origins of dendritic cells (DCs) including conventional DCs (cDCs, comprising cDC1 and cDC2 subsets) and plasmacytoid DCs (pDCs) remain unclear. We studied DC development in unmanipulated adult mice using inducible lineage tracing combined with clonal DNA “barcoding” and single-cell transcriptome and phenotype analysis (CITE-Seq). Inducible tracing of Cx3cr1+ hematopoietic progenitors in the bone marrow showed that they simultaneously produce all DC subsets including pDCs, cDC1s and cDC2s. Clonal tracing of hematopoietic stem cells (HSCs) and of Cx3cr1+ progenitors revealed clone sharing between cDC1s and pDCs, but not between the two cDC subsets or between pDCs and B cells. Accordingly, CITE-Seq analyses of differentiating HSCs and Cx3cr1+ progenitors identified progressive stages of pDC development including Cx3cr1+ Ly-6D+ pro-pDCs that were distinct from lymphoid progenitors. These results reveal the shared origin of pDCs and cDCs, and suggest a revised scheme of DC development whereby pDCs share clonal relationship with cDC1s.
Graphical Abstract

e-TOC blurb
The origin and lineage affiliation of plasmacytoid dendritic cells (pDCs) remain controversial. Feng et al. show that pDCs originate from the same pool of bone marrow progenitors as conventional dendritic cells (cDCs), and are clonally related to the cDC1 subset of cDCs.
Introduction
Continuous differentiation of hematopoietic stem cells (HSC) in the adult bone marrow (BM) maintains the homeostasis of the immune system throughout the adult life (Pucella et al., 2020). Understanding of hematopoiesis and of homeostasis in other adult tissues requires precise delineation of differentiation trajectories from stem cells to mature cells at single-cell resolution (Jacobsen and Nerlov, 2019; Kester and van Oudenaarden, 2018; Liggett and Sankaran, 2020). Paradoxically, HSCs are proposed to be clonally uncoupled from mature hematopoietic cells and thus to comprise a reserve pool of largely inactive precursors (Sun et al., 2014). Clonal relationships between mature hematopoietic lineages have been explored using DNA “barcoding” in embryogenesis (Pei et al., 2017) or upon transplantation (Naik et al., 2013; Perie et al., 2015), whereas specific clonal labeling and tracing of adult HSCs or progenitors remains to be performed.
Dendritic cells (DCs) are key sentinel cells of the immune system that detect pathogens through pattern recognition receptors and orchestrate innate and adaptive immune responses (Steinman, 2012). Conventional or classical DCs (cDCs) efficiently present antigen to T lymphocytes and are comprised of two major types with distinct functional properties and genetic requirements, termed cDC1 and cDC2 (Guilliams et al., 2014). Plasmacytoid DCs (pDCs) specialize in the detection of pathogen-derived nucleic acids and secretion of type I interferon (IFN-I), the key mediator of antiviral immune responses (Reizis, 2019; Swiecki and Colonna, 2015). DC subsets and their core transcriptomes and functionalities are conserved between humans and mice (Crozat et al., 2010). DCs in the peripheral lymphoid organs such as the spleen are relatively short-lived (Liu et al., 2007) and their development is driven by the cytokine Flt3 ligand (Flt3L) signaling through its receptor Flt3 (Mildner and Jung, 2014). DCs have been considered a distinct branch of myeloid cells (Geissmann et al., 2010) that share an upstream progenitor with monocytes (monocyte-DC progenitor or MDP) (Fogg et al., 2006) and develop from a common progenitor of all DC subsets (common DC progenitor or CDP) (Lee et al., 2015; Naik et al., 2007; Onai et al., 2007). pDCs fully differentiate in the BM, whereas precursors of all cDCs (pre-DC) (Liu et al., 2009) and committed cDC1 or cDC2 progenitors (pre-cDC1 and pre-cDC2, respectively) (Grajales-Reyes et al., 2015; Schlitzer et al., 2015) emerge in the BM and undergo terminal differentiation in the periphery. However, these progenitors are defined primarily through transplantation, and their identity and differentiation spectrum in vivo remain unclear. Moreover, it has been recently proposed that pDCs develop predominantly from lymphoid progenitors that are distinct from DC progenitors (Dress et al., 2019; Herman et al., 2018; Rodrigues et al., 2018). Finally, clonal barcoding of transplanted murine progenitors (Naik et al., 2013) and single-cell cultures of human progenitors (Helft et al., 2017; Lee et al., 2017) suggested that commitment to individual DC subsets may occur already in HSCs or early multipotent progenitors (MPP). Thus, the origin and the hierarchy of lineage commitment during DC development remain to be elucidated at clonal resolution in naive unmanipulated animals.
Here we analyzed DC differentiation using unbiased lineage tracing based on inducible Cre recombination. We combined lineage tracing of HSC and of DC progenitors with a system for Cre-inducible clonal cell barcoding, as well as with single-cell transcriptomics and phenotyping (CITE-Seq). Our results reveal the simultaneous emergence of cDCs and pDCs from a common pool of Cx3cr1+ progenitors, which comprise a hierarchy of differentiation stages including a committed pDC progenitor (pro-pDC). They also suggest a close developmental relationship between pDCs and cDC1s, including a clonal progenitor of the two lineages.
Results
FlipJump allows Cre-inducible tracing and DNA barcoding in vivo
To analyze DC development in naive animals, we sought to develop a platform for cell type-specific clonal cell labeling. Induced mobilization of a model transposon generates a unique integration site that can be used as a clonal DNA barcode. However, the only reported system of this kind used ubiquitous doxycycline-inducible transposon mobilization (Sun et al., 2014), precluding cell type-specific labeling and tracing. We designed an all-in-one genetic construct for Cre-inducible transposon mobilization, termed FlipJump (Fig. 1A). FlipJump is based on the PiggyBac transposase, which can excise genomic fragments flanked by its cognate inverted terminal repeats and integrate them into the consensus TTAA motif anywhere in the mammalian genome (Ding et al., 2005). The FlipJump allele includes a strong CAG promoter, an engineered hyperactive PiggyBac variant and a 5’ terminal repeat (5TR) in a reverse orientation flanked by inverted LoxP sites, followed by a 3’ terminal repeat (3TR) and a promoterless green fluorescent protein (GFP) cassette. In the native FlipJump configuration, PiggyBac transposase is not expressed because of its reverse orientation. Cre recombination inverts (“flips”) the LoxP-flanked cassette and locks this position by leaving a pair of incompatible LoxP sites. This induces the expression of transposase and also restores the proper orientation of TRs, leading to mobilization (“jump”) of the transposon and its random integration. The mobilized promoterless PiggyBac cassette is removed from the CAG promoter, thereby terminating the expression of PiggyBac transposase and preventing secondary mobilizations. In addition, transposon mobilization brings the GFP cassette under the CAG promoter, leading to permanent GFP expression. The resulting transposon integration into a random TTAA site generates a unique DNA “barcode”, which can be cloned and sequenced for clonal analysis of bulk cell populations.
Figure 1. The FlipJump system for Cre-inducible transposon mobilization.
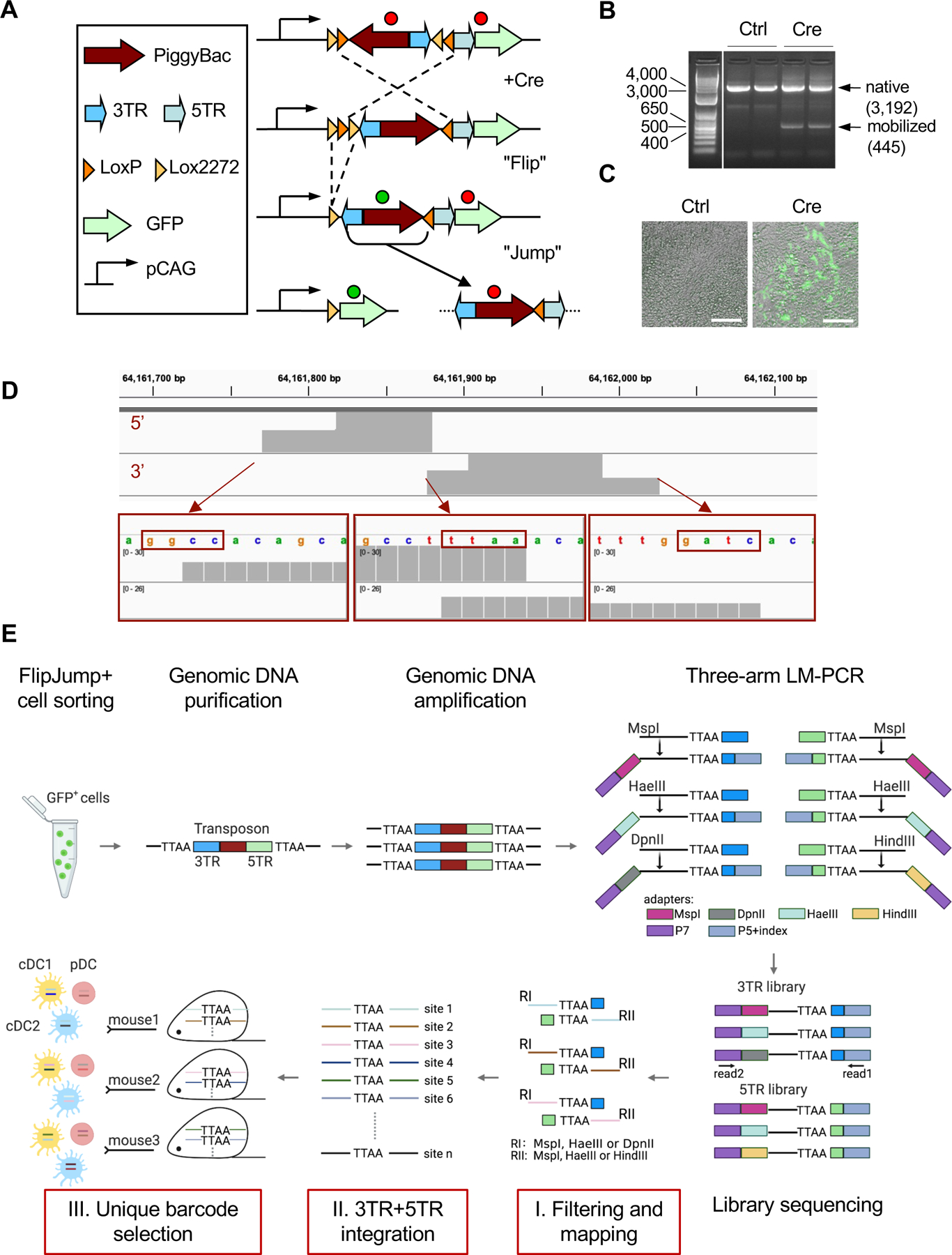
A. Scheme of the FlipJump allele and its Cre-induced inversion (“Flip”) and mobilization (“Jump”). Inactive and active transcriptional states of the PiggyBac transposase and GFP are indicated by red and green circles, respectively. 3TR and 5TR, the 3’ and 5’ terminal repeats of the PiggyBac transposon, respectively; LoxP and Lox2272, incompatible LoxP sites.
B-C. Cre-induced activation of FlipJump in murine ESC. Targeted R26FlipJump ESC were untreated (Ctrl) or treated with cell-permeable TAT-Cre recombinase protein (Cre) and examined 72 hours later.
B. Genomic PCR of two targeted ESC clones detecting the native allele configuration and the recombined configuration after PiggyBac mobilization. The PCR products with expected sizes for both alleles are indicated by arrows. Size ladder is shown on the left.
C. GFP expression by fluorescent microscopy (scale bar, 100 µm).
D. Example of a cloned PiggyBac integration site. Shown are sequencing traces from 5’ and 3’ transposon ends mapped onto the genome, with the TTAA integration site and the restriction sites used for LM-PCR highlighted.
E. Cloning and analysis of integration sites of R26FlipJump-encoded PiggyBac transposon. Following LM-PCR cloning and sequencing of integration sites at 3TR and 5TR ends of the transposon, they were filtered and mapped to the genome (step I), combined to define sites cloned from both ends (step II), and selected for sites occurring in only one animal (step III). TTAA, consensus integration site of the PiggyBac transposon; RI and RII, restriction sites used for LM-PCR.
Please also see Figure S1.
The FlipJump construct was inserted into the Gt(ROSA)26Sor (R26) locus by homologous recombination in murine embryonic stem cells (ESC), which were then injected into mouse blastocysts to establish the R26FlipJump strain. Treatment of the targeted ESC with cell-permeable TAT-Cre protein induced the mobilization of PiggyBac transposon (Fig. 1B) and the expression of GFP (Fig. 1C). Similarly, tamoxifen treatment of R26FlipJump mice crossed with the ubiquitous tamoxifen-inducible Cre recombinase (R26CreER) induced GFP expression in >50% of total splenocytes (Fig. S1A). Our initial attempts to clone transposon integration sites by ligation-mediated PCR (LM-PCR) amplification of one transposon end (Sun et al., 2014) revealed high frequency of spurious integrations. We therefore established an LM-PCR protocol to clone both 5TR and 3TR ends of transposon integrations from the same sample (Fig. 1D–E). First, we sorted and cultured single GFP+ hematopoietic progenitor cells from tamoxifen-treated R26CreER R26FlipJump mice, pooled colonies derived from 10 single cells, cloned integration sites and sequenced them by shotgun cloning in bacteria (5TR and 3TR) or by high-throughput sequencing (3TR only) (Fig. S1B). Five sites were cloned from both 5TR and 3TR in bacteria, and 3 of those were also identified by high-throughput sequencing of the 3TR cloning (Fig. S1C–D). We adopted high-throughput sequencing and implemented a computational pipeline to map and identify integration sites based on: i) the presence of the consensus PiggyBac integration site TTAA and the restriction sites used for LM-PCR; ii) independent cloning from both 5TR and 3TR ends in the same sample; iii) presence in only one induced animal or a culture derived from it (Fig. 1E).
The importance of the latter filter was demonstrated by control integration site cloning (both 5TR and 3TR) from primary BM cells of wild-type mice that did not carry FlipJump. Although the resulting products were low in abundance, they yielded 63 integration sites potentially representing PCR contamination or spurious amplification (data not shown). To filter these and other artefacts, we cloned integration sites from sorted GFP+ cells (BM monocytes) from induced FlipJump mice, including 4 samples of 104 cells and samples of different cell numbers (2×102, 2×103 and 2×104 cells). Filtering by barcode abundance (using a cutoff of 10) or by background (i.e. removing all barcodes identified in any other FlipJump sample) increased the number of unique barcodes and their correlation with cell numbers (Fig S1E and Table S1). Background filtering does not rely on an arbitrary threshold and therefore was used in this study. As a control for its efficiency, 845 unique integration sites reported in this paper include only 3 that were also cloned from FlipJump-negative DNA (Table S1). Thus, the current implementation of FlipJump is not suitable for quantitative analysis of lineage relationships, but allows qualitative description of clone sharing between primary cell populations ex vivo.
Tracing of HSCs reveals barcode sharing between pDCs and cDC1s
We crossed R26FlipJump mice to Pdzk1ip1-CreER mice that mediate tamoxifen-induced Cre recombination specifically in HSCs (Sawai et al., 2016). The resulting adult Pdzk1ip1-CreER R26FlipJump mice were treated with tamoxifen to induce FlipJump labeling in HSCs and traced to allow HSC contribution to peripheral hematopoietic cells (Fig. 2A). As expected (Sawai et al., 2016), GFP+ cells were initially absent from peripheral hematopoietic cells and slowly accrued over 20 weeks (Fig. S2A). Cloning of integration sites from major hematopoietic populations agreed with their expected relationships, including barcode sharing within myeloid and lymphoid lineages but only minimal sharing between them (data not shown). We cloned barcodes from DCs after 6 months of tracing, at which point 36% of HSCs, 31% of immature B cells in the BM and 30–33% of splenic DCs (pDC, cDC1 and cDC2) were GFP+ (Fig. 2B,C and Fig. S2B–D); these cells were sorted separately from 3 animals and used for integration site retrieval (Table S1). The identified 266 integration sites (barcodes) mapped predominantly (82%) in distal intergenic regions, gene introns or within 1–3 Kb 5’ of promoters (Fig. 2D) and were distributed evenly across the genome (Fig. 2E). Notably, 10% of HSC-derived barcodes were also detected in at least one DC subset in the same animal (Fig. 2F,G), showing that some of the originally barcoded HSCs were maintained and were contributing to mature short-lived progeny. No barcode sharing between HSC and B cells could be detected, likely reflecting bottlenecks in lymphoid commitment of HSCs and/or in the subsequent B cell selection. Within DCs, the majority of barcodes were unique to one subset; nevertheless, barcode sharing between pDCs and either cDC1s or cDC2s was detected (Fig. 2H–I). The sharing between pDCs and cDC1s was particularly prominent, representing 23% and 28% of pDC- and cDC1-derived barcodes respectively (Fig. 2J). The observed number of barcodes shared between pDCs and cDC1s (8) exceeded the number expected from a random distribution (1.5 barcodes). In contrast, only a single barcode was shared between pDCs and B cells (not exceeding a random expectation). Furthermore, barcode sharing between cDC1s and cDC2s was also minimal and not exceeding a random expectation, suggesting a closer developmental relationship between pDCs and cDC1s than between the two cDC subsets.
Figure 2. Clonal tracing of DC development from HSC in vivo.
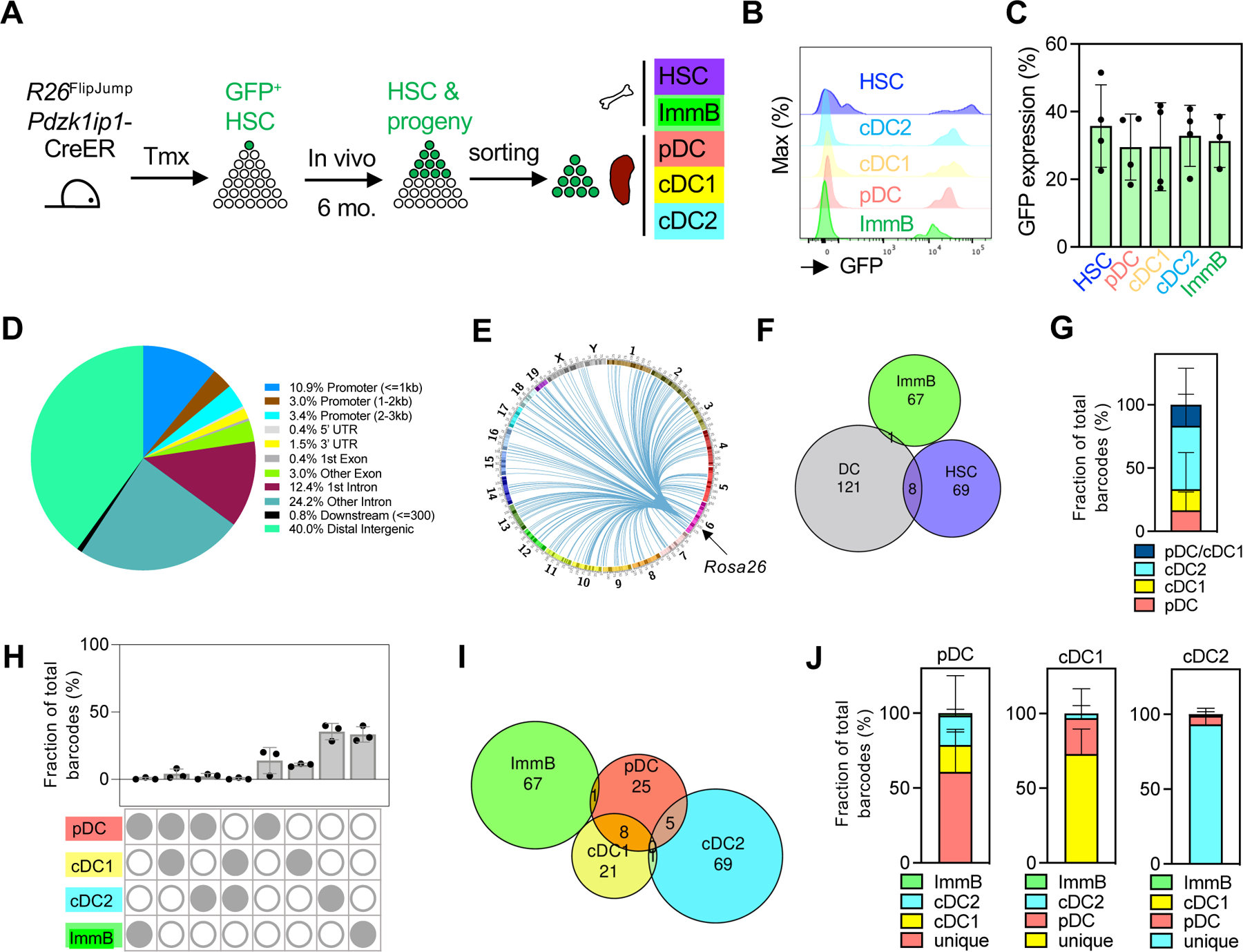
A. Clonal tracing of HSC. Pdzk1ip1-CreER R26FlipJump mice were induced with tamoxifen (Tmx) and traced for 6 months. GFP+ HSC and immature B cells (ImmB) from the BM, and DC subsets from the spleen were used to clone transposon integration sites (barcodes).
B. Histograms of GFP fluorescence at the endpoint in the same animal (representative of 4 animals).
C. Fractions of GFP+ cells in HSC, immature B cells and DC subsets. Symbols represent individual mice; bars represent mean ± S.D.
D. The distribution of cloned integration sites among elements of the genome.
E. Circos plot of the integration site positions (lines) across the mouse genome relative to the transposon donor R26FlipJump allele.
F. Proportional Venn diagram of clonal barcode sharing between HSC, immature B cells and DC subsets (pooled from 3 individual mice).
G. Distribution of barcodes shared between HSC and DCs among DC subsets.
H. Cumulative fractions of barcodes in immature B cells and DC subsets. Symbols represent values from individual mice; bars represent mean ± S.D.
I. Proportional Venn diagram of clonal barcode sharing between immature B cells and DC subsets (pooled from 3 individual mice)
J. Fractions of barcodes in each DC subset that are unique or shared with immature B cells or other subsets.
Please also see Figure S2.
HSCs give rise to pDCs and cDCs independently of lymphopoiesis
In tamoxifen-treated Pdzk1ip1-CreER mice carrying a Cre-inducible red fluorescent protein tdTomato (Tom) reporter (R26LSL-Tom), nascent Tom+ myeloid cells emerge from labeled Tom+ HSCs starting at 2 weeks, whereas lymphopoiesis commences stochastically at 3–6 weeks (Upadhaya et al., 2018). To capture the earliest DC and lymphocyte progenitors emerging from HSC, we labeled HSCs with Tom in Pdzk1ip1-CreER R26LSL-Tom mice and focused on their Flt3+ progeny, which encompasses short-term HSCs (ST-HSC), MPP, early myeloid and lymphoid progenitors (here generally designated as MyP and LyP, respectively), and all DC lineage-affiliated cells. We sorted Tom+ Flt3+ cells from three independent pools of Pdzk1ip1-CreER R26LSL-Tom reporter mice 3–5 weeks post-tamoxifen and analyzed them using CITE-Seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing) (Fig. 3A). CITE-Seq combines scRNA-Seq with highly multiplexed cell surface marker detection using oligonucleotide-conjugated antibodies (Stoeckius et al., 2017); in addition, it allows cell surface labeling (“cell hashing”) and pooling of individual samples, as well as hashtag-based discrimination of cell doublets (Stoeckius et al., 2018). After the removal of contaminating mature myeloid cells (Fig. S3A), UMAP-based dimensionality reduction and clustering of the remaining 5,810 cells yielded 6 clusters (Fig. S3B). Based on differentially expressed genes (DEG) and phenotypes defined by antibody-derived tags (ADT), these clusters corresponded to MPP, myeloid and DC progenitors, and immature pDCs, cDCs and monocytes (data not shown). However, this and/or additional iterative clustering failed to identify the expected cell heterogeneity, e.g. pre-cDC1s and pre-cDC2s within the emerging DCs. We therefore used a computational method that can identify the otherwise hidden cell communities, the K-nearest-neighbor-based Network graph drawing Layout (KNetL) (Khodadadi-Jamayran and Tsirigos, 2020; Tang et al., 2021). Indeed, KNetL yielded 13 distinct clusters (Fig. 3B, Fig. S3C and Table S2), whose identity was assigned by unbiased matching to mouse immune cell transcriptome datasets from the Immunological Genome Project (ImmGen) (Heng and Painter, 2008) (Tables S3–S4). It was also supported by the analysis of top differentially expressed genes and ADT (Fig. S3D,E) and by curated analysis for characteristic transcription factors (TF), lineage-specific genes and phenotypic markers (Table S3). Thus defined clusters included: i) early progenitors including ST-HSC, MPP (2 clusters) and LyP; ii) MyP and emerging monocytes; iii) immature cDCs including pre-cDC1 and pre-cDC2; iv) pDCs (2 clusters of progressive maturity) (Fig. 3C).
Figure 3. Single-cell analysis of DC development from HSCs in vivo.
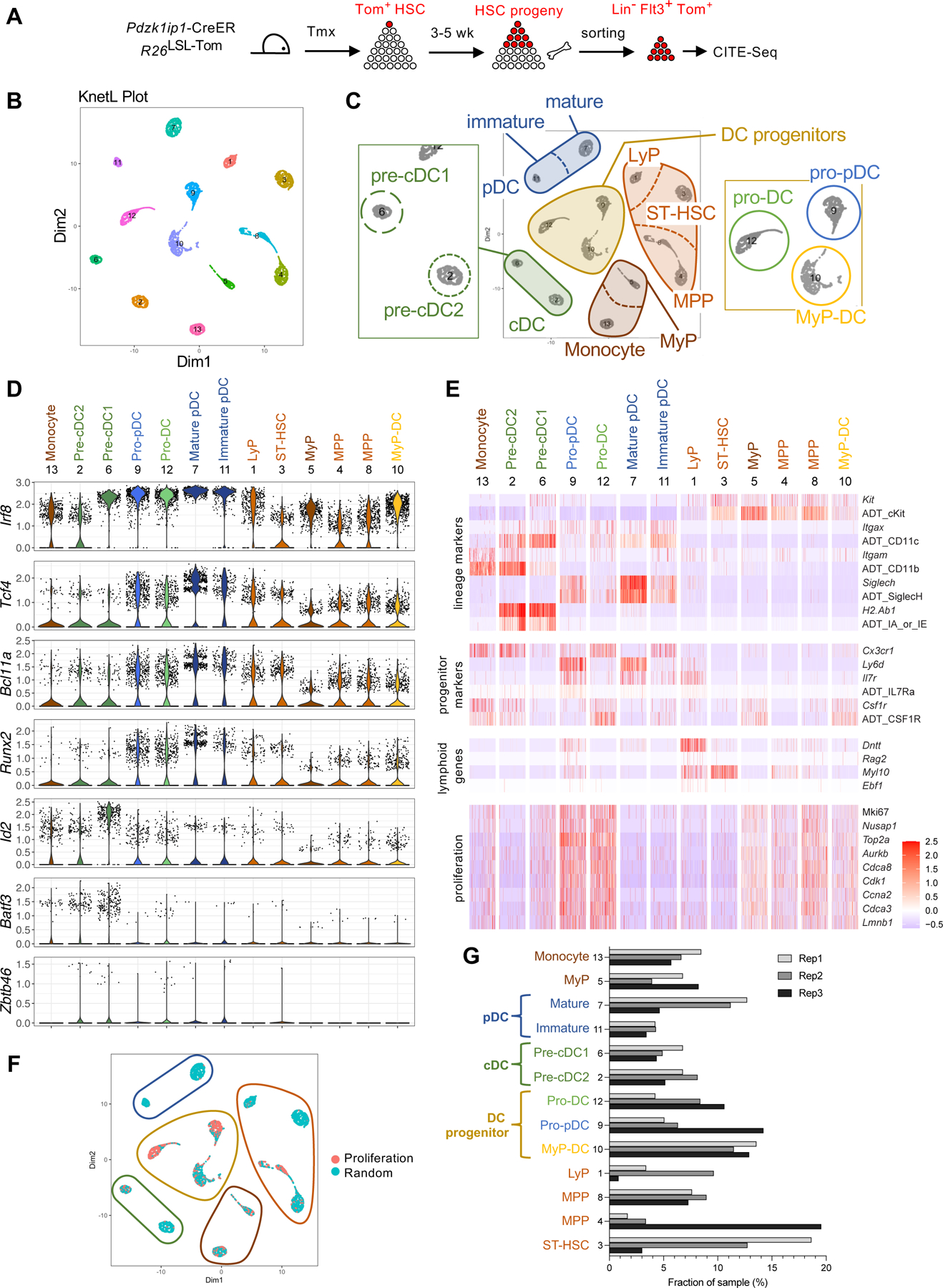
A. Pdzk1ip1-CreER R26Tom mice were induced with tamoxifen; 3–5 weeks later, Lin− Flt3+ tdTomato+ BM cells were sorted from three biological replicates and analyzed by CITESeq.
B. KNetL clustering of single cells (clusters are numbered and colored randomly).
C. Proposed identities of cell clusters.
D. Violin plots of transcript expression values for the indicated transcription factors in KNetL-defined cell clusters. The number and proposed identity of each cluster is indicated; clusters were ordered by hierarchical clustering using Euclidean distance.
E. Heat maps of transcripts for the indicated phenotypic markers, lymphocyte-specific genes and proliferation-associated genes in the cell clusters defined above. For some surface markers, expression of antibody-derived tags (ADT) is also shown.
F. Feature plot of the proliferation gene signature in KNetL-defined clusters (panel C). Cells colored red exhibited stronger correlation to proliferation signature geneset (Table S4) than to a randomly-selected gene set of equal size.
G. The fraction of cells from each biological replicate in each cluster.
Please also see Figure S3.
In addition, KNetL yielded 3 clusters whose identity was compatible with DC progenitors. These included a c-Kitlo-neg CD11c− Csf1r+ cluster without subset-specific features that was similar to MyP (designated MyP-DC); a cluster with the CD11c+ Csf1r− SiglecH+ Ly6D+ phenotype characteristic of pDCs (designated pro-pDC); and a CD11c+ Csf1r+ SiglecHlo Ly6D− cluster consistent with early progenitor of both pDCs and cDCs (designated pro-DC) (Fig. 3C). Indeed, MyP-DC matched to the ImmGen profiles of granulocyte-monocyte progenitor (GMP) and MDP, whereas pro-pDC and pro-DC matched to CDP after subtracting the gene signature of cell proliferation (Table S3). Notably, pro-DCs and pro-pDC showed prominent expression of transcription factor (TF) Irf8, which is required for cDC1 and pDC differentiation (Cytlak et al., 2020; Sichien et al., 2016) and is expressed in pre-cDC1 and emerging pDCs but is low or absent from pre-cDC2 (Fig. 3D). Pro-DCs and pro-pDCs also expressed pDC-promoting TF Bcl11a, Tcf4 and Runx2 (Reizis, 2019) together with low amounts of cDC1-promoting TCF4 inhibitor Id2; in contrast, cDC-enriched TF Zbtb46 and Batf3 were virtually absent (Fig. 3D). Furthermore, pro-DC expressed transcripts for pDC marker CD300c (Kaitani et al., 2018) and a cDC1 marker Clec9a (Schraml et al., 2013) (Table S3). Thus, the transcriptome of pro-DCs revealed potential for both pDCs and cDC1s whereas the potential for cDC2 was not evident, although it could not be excluded. Notably, clusters with general transcriptional features of cDCs could not be identified, even as subset-committed pre-cDC1 and pre-cDC2 were clearly defined. By surface markers, both pro-pDCs and pro-DCs were c-Kitlo-neg Cx3cr1+ CD11clo CD11b− MHC cl II− ; within this population, pro-pDCs were Csf1r− SiglecH+ Ly6D+ IL-7R+, whereas pro-DCs were Csf1r+ SiglecHlo Ly6D− IL-7R− (Fig. 3E). Both clusters displayed prominent proliferation-associated gene signature (Fig. 3E,F), which caused them to match to highly proliferative ImmGen reference cell populations such as pre-B cells and immature thymocytes (Table S3–S4). Notably, pro-pDCs were distinct from bona fide Ly6Dlo IL-7R+ LyP, which expressed early lymphoid markers Dntt and Myl10 but no SiglecH or Cx3cr1, and lacked proliferation-associated genes (Fig. 3D). Moreover, pro-DCs, pro-pDCs and pDCs were present in all biological replicates whereas the LyP population was nearly absent from one (Fig. 3G), suggesting that it is dispensable for pDC development. Collectively, these data reveal simultaneous development of pDCs and cDCs from HSCs through a set of progressively differentiated DC progenitors. These include proliferative progenitor populations with a pDC lineage signature (pro-pDC) and an early DC progenitor (pro-DC) with features of both pDCs and cDC1 lineage potential.
Cx3cr1+ progenitors generate all DC subsets and contain a clonal precursor of pDCs and cDC1s
Given that DC progenitors defined above expressed Cx3cr1 (Fig. 3E), we crossed R26FlipJump with mice that express tamoxifen-inducible Cre and yellow fluorescent protein (YFP) from the Cx3cr1 locus (Cx3cr1CreER-YFP)(Parkhurst et al., 2013). First, we tested the ability of Cx3cr1+ progenitors to generate DCs in Flt3L-supplemented cultures of total BM. R26FlipJump Cx3cr1CreER-YFP mice were treated with tamoxifen, and 2 days later their BM was cultured with Flt3L to generate pDC, cDC2 and cDC1-like cells (Naik et al., 2005) (Fig. 4A). Indeed, BM cultures from induced R26FlipJump Cx3cr1CreER-YFP mice yielded GFP+ cDC1, cDC2 and pDCs (Fig. 4B,C). The fraction of labeled cells was lower in pDCs than in cDC1 or cDC2, suggesting that unlabeled Cx3cr1− progenitors may contribute to pDC development in this system. We then sorted these GFP+ DCs from cultures derived from three individual mice, and analyzed the resulting barcodes (Fig. 4D). All integration sites cloned from Cx3cr1+ progenitor tracing in vitro and in vivo (see below) showed wide and unbiased genome distribution similar to HSC-derived integration sites (Fig. 4E,F). The vast majority (141/168, 84%) of Flt3L culture-derived barcodes were unique to each DC subset (Fig. 4G); however, 19 (11%) of all barcodes were shared between pDC and cDC1, representing 25% of cDC1-derived barcodes (Fig. 4H,I) and exceeding the number (8) expected from a random distribution. In contrast, only 3 barcodes (1.8%) were shared between cDC1 and cDC2, further highlighting a closer clonal relationship between pDCs and cDC1s.
Figure 4. Clonal tracing of DC development from Cx3cr1+ progenitors in vitro.

Cx3cr1CreER-YFP R26FlipJump mice were induced with tamoxifen, and 2 days later total BM was plated in Flt3L for 7 days to generated DCs.
A. Definition of pDC, cDC2 and cDC1-like cells in Flt3L-supplemented cultures by surface staining.
B. Expression of GFP in DC subsets from one culture (representative of 5 cultures from individual animals).
C. Average fraction of GFP+ cells in DC subsets. Symbols represent cultures from individual mice; bars represent mean ± S.D. The lower labeling of pDCs versus cDC1 or cDC2 is statistically significant (p=0.008 by Mann-Whitney test).
D. FlipJump was induced in Cx3cr1+ progenitors followed by Flt3L-supplemented culture, and GFP+ DC subsets were used to clone transposon integration sites (barcodes).
E The distribution among elements of the genome of the integration sites cloned in Cx3cr1+ progenitor tracing experiments in vitro (this figure) and in vivo (Fig. 5).
F. Circos plot of the integration site positions (lines) across the mouse genome relative to the transposon donor R26FlipJump allele.
G. Cumulative fractions of barcodes in DC subsets. Symbols represent values from individual mice; bars represent mean ± S.D.
H. Proportional Venn diagram of clonal barcode sharing between the DC subsets (pooled cultures from 3 individual mice).
I. Fractions of pooled barcodes in each DC subset that are unique or shared with other subsets.
Next, we used FlipJump as a fluorescent reporter to label and trace DC progenitors in vivo. A single dose of tamoxifen induced the expression of GFP in a subset of BM YFP+ cells 2 days later, consistent with the initial labeling of Cx3cr1-expressing cells (Fig. 5A). GFP+ YFP− cells progressively accumulated in the BM and in the periphery on days 6–8, representing the Cx3cr1− progeny of the initially labeled Cx3cr1+ cells. In addition to myeloid and DC progenitors, Cx3cr1 is expressed on macrophage and monocyte populations (Geissmann et al., 2003), as well as in distinct populations of cDCs such as pDC-like transitional DCs (tDCs) (Bar-On et al., 2010; Leylek et al., 2019) and monocyte-like Esam− splenic cDC2 (Lewis et al., 2011). In contrast to these populations, splenic pDC, cDC1 and Esam+ cDC2 lack Cx3cr1 expression as confirmed by their YFP− phenotype (Fig. 5B), allowing their tracing from Cx3cr1+ progenitors. Indeed, comparable fractions of GFP+ cells appeared in splenic pDC, cDC1 and cDC2 on day 6 post-tamoxifen, further increased on days 8–9 and waned by day 12 (Fig. 5C,D). Unlike in vitro cultures, the labeling of pDCs was similar to that of cDCs, suggesting that these subsets emerge from labeled Cx3cr1+ progenitors with similar efficiency and kinetics. In contrast to the bright uniform GFP expression in HSC tracing (Fig. 2B), labeled cells in R26FlipJump Cx3cr1CreER-YFP mice showed a range of GFP expression intensities (Fig. 5D). This may reflect variable accumulation of GFP during the short time frame of tracing, and/or the close compensation between the induced GFP signal and the YFP encoded by Cx3cr1. To confirm that the system does not label a distinct subset of pDCs, we performed bulk RNA sequencing of GFP+ and GFP− pDCs populations sorted from three R26FlipJump Cx3cr1CreER-YFP mice animals on day 8 post-tamoxifen (Table S5). Pairwise comparison of GFP+ and GFP− populations revealed few differentially expressed genes, none of them indicating any specific lineage affiliation (Fig. S4A). Conversely, pDC signature genes and genes shared between pDCs and lymphocytes (e.g. Ly6d, Il7r, Dntt, Ccr9, Blnk etc.) were expressed equally, whereas transcripts specific for cDCs or lymphocytes were not upregulated in either population (Fig. S4B).
Figure 5. Clonal tracing of DC development from Cx3cr1+ progenitors in vivo.

Cx3cr1CreER-YFP R26FlipJump mice were induced with tamoxifen, the expression of GFP (indicative of FlipJump activation) was examined by flow cytometry and used to clone integration sites.
A. The expression of GFP vs Cx3cr1-driven YFP among total BM or splenic cells in wildtype mice (Control) or in Cx3cr1CreER-YFP R26FlipJump mice prior to (day 0) or at days 2–8 after tamoxifen administration.
B. The expression of Cx3cr1-driven YFP in mature DC subsets. Shown are histograms of YFP fluorescence in the gated DC subsets including transitional DCs (tDCs) and EsamcDC2 (cDC2(E-)). Granulocytes (Gran) and monocytes (Mono) are included as respective negative and positive populations for YFP expression. Representative of >20 animals.
C. Average fractions of GFP+ cells in splenic pDC, cDC1 and cDC2 at the indicated time points of tracing. Symbols represent mean ± S.D. of 10, 11 or 3 mice at days 6, 8–9 and 12, respectively. The labeling in pDCs was significantly higher than in cDC2 on day 6 (p=0.005) and lower than in cDC1 on days 8–9 (p=0.003) as determined by Mann-Whitney test; no other differences were significant.
D. Expression of GFP in splenic cell types defined above. The fractions of GFP+ cells in each subset are shown at the corresponding time points after tamoxifen administration. Representative of 3 animals on days 2–4 and 10–11 animals on days 6–8.
E. Splenic GFP+ DC subsets on day 8 post-tamoxifen were used to clone transposon integration sites (barcodes).
F. Cumulative fractions of barcodes in DC subsets. Symbols represent values from individual mice; bars represent mean ± S.D.
G. Proportional Venn diagram of clonal barcode sharing between the DC subsets (pooled from 7 individual mice).
H. Fractions of pooled barcodes in each DC subset that are unique or shared with other subsets..
Please also see Figures S4 and S5.
In contrast to Cx3cr1− DC subsets, Cx3cr1-expressing monocytes and Esam− cDC2 became labeled earlier (days 2–4), whereas granulocytes and B cells accrued little or no labeling (Fig. 5D and not shown). The analysis of FlipJump-labeled GFP+ progenitors revealed YFP+ Lin− Flt3+ c-Kitlo-neg cells consistent with DC progenitors that were present on day 2 but disappeared by day 8, concomitantly with the emergence of their DC progeny in the periphery (Fig. S5A,B). Similar results were observed in Cx3cr1CreER-YFP mice crossed with a conventional R26LSL-Tom Cre reporter: these mice showed progressive labeling of splenic DC subsets on days 4–8 post-tamoxifen, minimal labeling of granulocytes and B cells (Fig. S5C–G), and the initial labeling of Cx3cr1+ Flt3+ c-Kitlo-neg BM progenitors that waned by day 8 (Fig. S5H). The labeling of DC subsets in the R26LSL-Tom reporter mice (30–40% on day 6, Fig. S5F) was much higher than in R26FlipJump mice (6–10% on day 6, Fig. 5C) and reached >60% on day 8, revealing strong contribution of Cx3cr1+ progenitors to peripheral DCs. Thus, major fractions of pDC and cDC (but not granulocytes or lymphocytes) emerge simultaneously from a pool of Cx3cr1+ progenitors in vivo.
We analyzed transposon integrations from sorted GFP+ splenic pDC, cDC1 and cDC2 eight days after tamoxifen administration in seven independent animals (Fig. 5E). Again, the majority (389/409, 95%) of identified barcodes were subset-specific (Fig. 5F,G). However, 14 barcodes were shared between pDC and cDC1; this exceeded the number (4.4) expected from a random distribution and corresponded to 18% of cDC1- derived barcodes, whereas only 3 (4%) of those were shared with cDC2 (Fig. 5H). Across all experiments, sharing between pDCs and cDC1s exceeded the number expected from a random distribution (41 vs 15.3), whereas the sharing between other subsets did not (15 vs 15.6 for pDCs and cDC2s, and 7 vs 7.9 for cDC1s and cDC2s). Collectively, the results suggest that progenitors within the Cx3cr1+ population predominantly give rise to a single DC subset; however, they also include clonal progenitors of pDCs and cDC1s.
Cx3cr1+ progenitors give rise to pDCs via a hierarchy of differentiation stages
To characterize the real-time in vivo differentiation of Cx3cr1+ BM progenitors, we sorted Lin− GFP+ BM cells from tamoxifen-treated R26FlipJump Cx3cr1CreER-YFP mice at early (day 2) or late (day 5) time points (Fig. 6A) and analyzed them by CITE-Seq. After the removal of contaminating granulocytes (Fig. S6A), standard UMAP-based dimensionality reduction and clustering of the resulting 17,093 cells yielded 8 cell clusters corresponding to monocytes, cDCs, pDCs and progenitors thereof; however, as in the case of HSC tracing, populations such as pre-cDC1s and pre-cDC2s could not be clearly defined (Fig. S6B and data not shown). On the other hand, KNetL-based analysis revealed 22 clusters (Fig. 6B, Fig. S6C and Table S6), one of which (cl. 12) contained cells with relatively poor sequencing quality. As described for the CITE-Seq analysis in Fig. 3, we assigned cluster identities by unbiased matching to the ImmGene reference datasets after subtracting the proliferation gene signature (Table S3–S4), supported by top differentially expressed transcripts and ADT (Fig. S6D,E) and characteristic TF, lineage genes and markers (Table S3). As expected, early multipotent progenitors such as ST-HSC and MPP were absent from the differentiating Cx3cr1+ population. The resulting clusters could be designated as i) MyP (2 clusters); ii) monocytes including immature (3 clusters) and mature (3 clusters); iii) pre-cDC1 and pre-cDC2; iv) pDCs including immature (2 clusters) and mature (4 clusters). One cluster of monocytes and one cluster of pDCs manifested upregulation of IFN-I inducible genes (the interferon signature), whose origin remains to be identified.
Figure 6. Single-cell analysis of DC development from Cx3cr1+ progenitors in vivo.
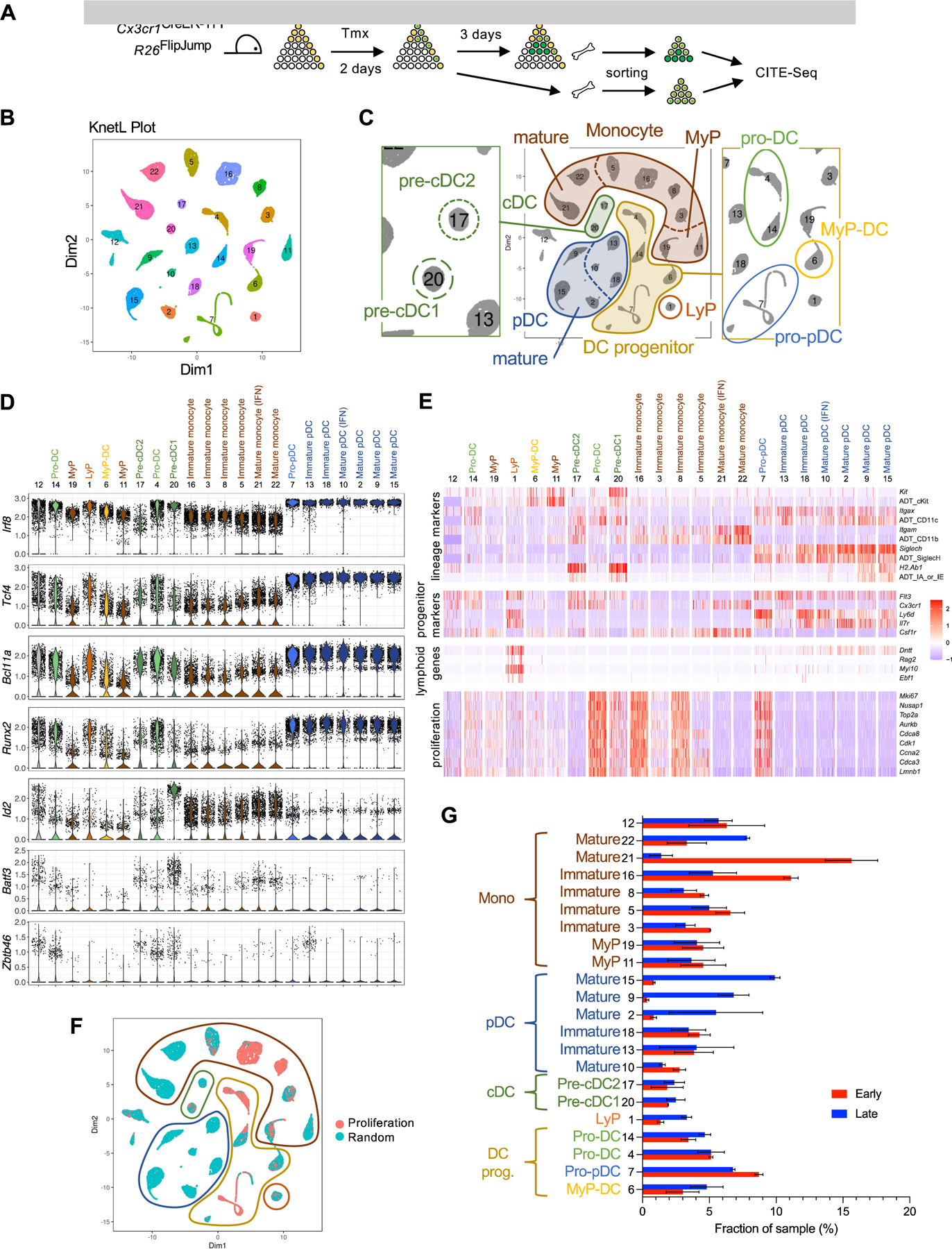
A. Cx3cr1CreER-YFP R26FlipJump mice were induced with tamoxifen; at early (2 days) or late (5 days) time points, Lin− GFP+ BM cells were sorted from two biological replicates and analyzed by CITE-Seq.
B. KNetL clustering of single cells (clusters are numbered and colored randomly).
C. Proposed identities of cell clusters (cluster 12 contains cells with poor sequencing quality).
D. Violin plots of transcript expression values for the indicated transcription factors in KNetL-defined cell clusters. The number and proposed identity of each cluster is indicated; clusters were ordered by hierarchical clustering using Euclidean distance.
E. Heat maps of transcripts for the indicated phenotypic markers, lymphocyte-specific genes and proliferation-associated genes in the cell clusters defined above. For some surface markers, expression of antibody-derived tags (ADT) is also shown.
F. Feature plot of the proliferation gene signature in KNetL-defined clusters (panel C). Cells colored red exhibited stronger correlation to proliferation signature geneset (Table S4) than to a randomly-selected gene set of equal size.
G. The fraction of cells from the early or late time point in each cluster (mean ± range of biological replicates).
Please also see Figure S6.
In addition, we defined 4 clusters of DC progenitors that were consistent with the MyP-DC, pro-pDC and pro-DC (2 clusters) identified among the HSC progeny in their TF expression (Fig. 6D), phenotype (Fig. 6E) and proliferation status (Fig. 6F). As before, the transcriptomes of pro-DC and pro-pDC matched proliferating immature lymphocyte populations but revealed similarity to CDP upon subtracting the proliferation signature (Table S3). Similar to pro-DC in the HSC progeny, pro-DCs labeled by Cx3cr1CreER showed transcriptional features of pDCs and cDC1s (Table S3). Most cell clusters including DC progenitors were present at both time points (Fig. 6G), whereas mature pDC clusters were present primarily at the late time point, revealing the emergence of pDCs from Cx3cr1+ progenitors in real time. We also detected a cluster with expression of lymphoid transcripts such as Dntt, Myl10, Ebf1 and Rag2 and the Flt3+ Csf1r− Ly-6D+ IL-7R+ phenotype of LyP (Fig. 6E). These cells were largely Cx3cr1-, had low proliferation signature and emerged primarily at the late time point (Fig. 6G), suggesting that they originate from Cx3cr1+ progenitors and do not produce pDCs within the analyzed time frame. Separate KNetL-based analysis of data from day 2 or day 5 showed that pro-DC and pro-pDC formed distinct clusters at each time point; in contrast, LyP were scarce and merged with pro-pDC on day 2 but comprised a distinct cluster on day 5 (data not shown). Altogether, these data show that Cx3cr1+ progenitors continuously give rise to all DC subsets including pDCs, and contain populations with transcriptomic features of individual DC subsets as well as of pDCs and cDC1s.
pDCs develop from unique Cx3cr1+ progenitors that express hCD2
Having characterized the hierarchy of Cx3cr1+ progenitors by CITE-Seq, we sought to confirm the results by conventional lineage tracing. First, we used Cx3cr1CreER-YFP mice to better characterize Cx3cr1+ progenitors (marked by YFP) within the Lin− c-Kitlo-neg Flt3+ population (Fig. 7A). This population comprised Cx3cr1+ cells that were either CD11c− or CD11c+; the former contained a Csf1r+ subset consistent with MyP-DC. The Cx3cr1+ CD11c+ fraction comprised cells that expressed either SiglecH or Csf1r, as well as a double-positive SiglecHlo Csf1r+ population consistent with pro-DCs. The majority of SiglecH+ Csf1r− fraction expressed Ly-6D, the phenotype corresponding to pro-pDCs. Importantly, pro-pDCs resembled pDCs in their CD11c+ SiglecH+ Ly-6D+ IL7Rlo phenotype and were distinct from canonical LyP that were Cx3cr1− CD11c− SiglecH− Ly-6D+ IL7Rhi. Similar fractions were observed among the BM Flt3+ population in wild-type mice using antibody staining for Cx3cr1 (Fig. S7A), and could even be defined without using Cx3cr1 as a marker (Fig. S7B). We then used Cx3cr1CreER-YFP R26LSL-Tom reporter mice to characterize the labeling of these populations on day 6 post-tamoxifen, close to the late time point of CITE-Seq tracing (day 5, Fig. 6) and shortly before the turnover of labeled progenitors (day 8, Fig. S5H). The initially labeled Tom+ cells on day 2 post-tamoxifen comprised almost exclusively Cx3cr1+ cells including pro-DCs and pro-pDCs (Fig. S7C). On day 6, BM pDCs were labeled comparably with pro-DCs and pro-pDCs at ~50% Tom+ cells, whereas immature B cells showed minimal labeling (Fig. 7B). Notably, a small fraction (~11%) of LyP also became labeled at that point, confirming the CITE-Seq result (Fig. 6). These data further suggest that pDC development is driven primarily by a hierarchy of Cx3cr1+ DC progenitors that are distinct from lymphoid progenitors.
Figure 7. Intersectional tracing of pDC development from Cx3cr1+ progenitors in vivo.
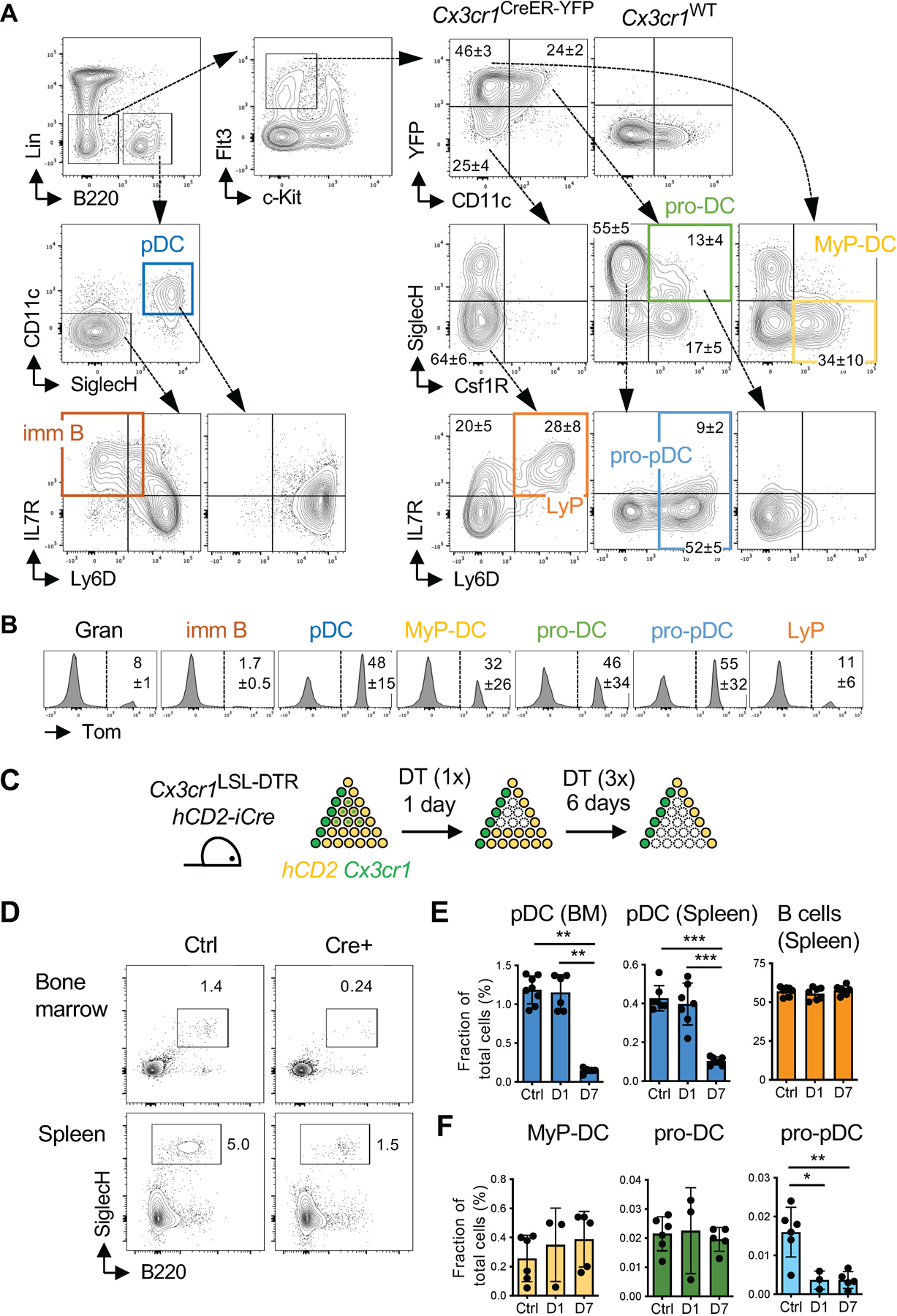
A. Definition of progenitor populations in the BM of Cx3cr1CreER-YFP mice by flow cytometry. Wild-type (Cx3cr1WT) mouse is included as a negative control for YFP expression. The fractions of select populations are indicated (mean ± S.D. of 6 mice)
B. The expression of tdTomato (Tom) in BM cell populations of Cx3cr1CreER-YFP R26LSL-Tom mice 6 days after tamoxifen induction. Populations were defined as in panel A; the fractions of Tom+ cells are indicated (mean ± S.D. of 3 mice).
C. hCD2-iCre Cx3cr1LSL-DTR mice were administered diphtheria toxin (DT) and analyzed one day later to assess the depletion of cells expressing hCD2 and Cx3cr1; or administered DT every second day during subsequent 6 days and analyzed on day 7 to assess the depletion of cells expressing hCD2 and Cx3cr1 and of their progeny. Crenegative Cx3cr1LSL-DTR mice were treated in parallel and used as controls.
D. Staining profiles of lineage (CD3,CD19,NK1.1,Ly-6G)-negative BM cells (top) or splenocytes (bottom) from hCD2-iCre Cx3cr1LSL-DTR or control mice after 7 days of continuous DT treatment. The pDC gate is indicated. Representative of 7 animals.
E-F. Fractions of indicated cell types among total splenocytes or BM cells from hCD2-iCre Cx3cr1LSL-DTR mice treated for 1 or 7 days, and in control mice treated for 7 days.Symbols represent individual mice; bars represent mean ± S.D.; statistically significant differences are indicated.
E. The fraction of pDCs and B cells.
F. The fraction of BM progenitors defined as in panel A..
Statistical significance was defined by Mann-Whitney test and shown as follows: *, p <0.05; **, p <0.01; ***, p <0.001..
Please also see Figure S7.
We sought to reconcile this notion with lineage tracing results from the Cre transgene driven by the human CD2 promoter (hCD2-iCre), which labels lymphocytes and pDCs but only a small fraction of cDCs (Dress et al., 2019; Siegemund et al., 2015). To this end, we crossed the hCD2-iCre transgenic strain with the Cx3cr1LSL-DTR strain that drives Cre-inducible expression of diphtheria toxin receptor (DTR) from the Cx3cr1 locus (Diehl et al., 2013). The detection of DTR expression in the resulting hCD2-iCre Cx3cr1LSL-DTR mice was limited by the low sensitivity of DTR staining (Buechler et al., 2019; Mandl et al., 2015), whereas diphtheria toxin (DT) kills DTR-expressing cells with high sensitivity. Specifically, DT administration to these mice is expected to immediately kill cells that express both the hCD2-iCre transgene and the endogenous Cx3cr1 gene (and hence DTR), and eventually to deplete their progeny. Mice were administered DT and analyzed either one day later or after continuous DT administration on day 7 (Fig. 7C). The fraction of BM and splenic pDCs on day 1 was normal, reflecting the fact that mature pDCs do not express Cx3cr1 and thus are not depleted. In contrast, on day 7 pDCs were profoundly reduced compared to similarly treated Cre-negative Cx3cr1LSL-DTR control mice, both in the BM (by >85%) and in the spleen (by >75%) (Fig. 7D, E). In contrast, neither cDCs nor B cells were affected in the same mice (Fig. 7E and data not shown). As expected given the lack of Cx3cr1 expression by lymphoid progenitors, even the earliest stages of B cell development were unaffected on day 7 (Fig S7D,E), and LyP were unaffected on day 1 or 7 (Fig. S7F). The observed delayed depletion of pDCs suggested that the progenitors of these cells uniquely express both hCD2-iCre and Cx3cr1 and were initially depleted by DT. Indeed, the analysis of BM showed that pro-pDCs were depleted on day 1 and even more so on day 7, whereas upstream progenitors including pro-DCs were not (Fig. 7F). Thus, intersectional tracing shows that the majority of pDCs are derived from unique progenitors expressing both Cx3cr1 and the “lymphoid” hCD2 transgene, and suggest pro-pDCs as such immediate pDC progenitors.
Discussion
We have developed the FlipJump system for clonal cell tracing in vivo, based on Cre-inducible mobilization of a PiggyBac transposon that creates a single integration site (“barcode”). Compared to the previously described system based on doxycycline-inducible Sleeping Beauty transposase (Sun et al., 2014), its key advantages include i) cell type specificity conferred by Cre-inducible activation; ii) irreversible self-inactivation of the transposase that reduces the possibility of secondary mobilization; iii) an all-in-one monoallelic design. We also utilized bidirectional cloning and stringent computational filtering of integration sites that guards against spurious and/or frequently occurring integrations. This yielded hundreds of unique barcodes that showed no apparent integration site bias or functional effects in either short-term (7–9 days) or long-term (~6 months) tracing. In its current implementation, FlipJump has some limitations such as a relatively cumbersome barcode retrieval and a non-quantitative readout. Although the barcoding data presented herein appear consistent between biological replicates and experimental systems, quantitative barcode readout and its robust statistical analysis still remain to be developed. Another caveat of FlipJump (and possibly of other DNA barcoding systems) is its potential sensitivity to sampling effects, which may cause the observed predominance of unique barcodes in each cell type. Nevertheless, FlipJump offers a practical opportunity for clonal lineage tracing in any tissue or cell lineage for which a Cre deleter strain is available.
We used FlipJump for clonal analysis of endogenous DC development from HSCs or from Cx3cr1+ progenitors. Our inducible labeling of HSCs or progenitors with hundreds of barcodes expands upon a previous study using several fluorescent proteins combined with Clec9aCre strain, which labels endogenous cDCs constitutively and is expressed in cDC1s throughout their development (Cabeza-Cabrerizo et al., 2019). Notably, our HSC tracing revealed that adult BM HSCs share clones with their mature short-lived progeny such as DCs. The result highlights both the long-term maintenance of barcoded HSC clones for at least 6 months, and the essential role of HSCs as the source of lifelong hematopoiesis (Pucella et al., 2020). The observed lack of barcode sharing between HSCs and B cells likely reflects the stochastic lymphoid commitment of rare individual HSCs, further emphasizing the difference between lymphopoiesis and DC development. We also found that the majority of HSC-derived barcodes were specific to one DC subset. This may reflect the high stringency of our barcode filtering or an inherent bias of transposon-mediated DNA barcoding, because a higher frequency of barcode sharing between DC subsets (including cDC1 and cDC2) was observed in lentiviral barcoding of transferred progenitors (Lin et al., 2021). The results such as the lack of barcode sharing between DCs and B cells may be further confounded by different developmental kinetics of the two lineages, undersampling of barcodes (especially from small clones) and/or exclusion of unlabeled cells. On the other hand, our data agree with transplantation- and culture-based experiments that revealed lineage commitment at the HSC stage (Carrelha et al., 2018; Lee et al., 2017).
In addition, some barcode sharing could be detected between pDCs and either cDC1s or cDC2s, but not between cDC1s or cDC2s. Given that the latter type of sharing should be as likely as the former, this result argues against an early commitment to the generic cDC fate in putative pre-DCs. This result remains to be reconciled with lineage tracing by Clec9aCre strain, which suggested the development of cDC1 and cDC2 from a common late progenitor (Schraml et al., 2013). This discrepancy may reflect technical differences between experimental systems, such as inducible versus constitutive labeling; of note, subsequent clonal tracing by Clec9aCre suggested that pre-DCs may be committed to either cDC1 or cDC2 fate prior to tissue entry (Cabeza-Cabrerizo et al., 2019). Furthermore, early commitment to the cDC1 fate has been observed by single-cell analysis of DC progenitors in culture (Balan et al., 2018) and in vivo (Bagadia et al., 2019), and by clonal barcoding of adoptively transferred progenitors during Flt3L-driven DC development in vivo (Lin et al., 2021). Accordingly, single-cell transcriptomics of traced HSC progeny identified progenitor populations with transcriptional features of individual DC subsets (e.g. pro-pDCs) or of pDCs and cDC1 (pro-DCs), although their exact function remains to be elucidated. Importantly, however, progenitors with features of “generic” cDCs but not of pDCs (i.e. the putative pre-DCs) could not be defined with certainty. Collectively, the data suggest that pDC and cDC fates are closely linked, and that the bifurcation between the two does not precede the choice between cDC subsets.
Inducible lineage tracing of Cx3cr1+ progenitors showed that this population is rapidly turning over in the BM, and is giving rise to all DC subsets with similar efficiency and kinetics. This result is consistent with the short lifespan of peripheral DCs, and with the original definition of Cx3cr1+ progenitors as the source of DCs and other mononuclear phagocytes (Fogg et al., 2006). It does not rule out the common origin of pDCs and B cells, which may bifurcate earlier in hematopoiesis; however, it reveals the continuous emergence of pDCs and cDCs from the same pool of proximal progenitors. Clonal tracing of Cx3cr1+ progenitors in Flt3L-driven cultures and in vivo showed predominance of subset-specific barcodes, consistent with clonal analysis of DC development during adoptive transfers (Naik et al., 2013) or in culture (Dursun et al., 2016; Lee et al., 2017; Lin et al., 2018). In addition, barcode sharing could be detected both in vitro and in vivo, primarily between pDCs and cDC1s (18–27% of cDC1-derived barcodes were shared with pDCs across all experiments). Thus, bi-potent progenitors of pDCs and cDC1s are present throughout the hematopoietic hierarchy from HSCs to Cx3cr1+ progenitors, revealing a close developmental connection between the two lineages. This is consistent with the detection of a Cx3cr1+ progenitor population with features of pDCs and cDC1s (pro-DC), whereas a population with features of cDC1s and cDC2s was not readily detectable. Together with the results of clonal HSC tracing, these results reinforce the developmental relationship of pDCs and cDC1s, which appears closer than the relationship between the two cDC subsets.
Our results show that pDC development shares the same kinetics and progenitor pool with cDCs. This is consistent with a common Flt3-dependent pathway of pDC and cDC differentiation, and with progressive pDC vs cDC clonal fate determination within this pathway (Lin et al., 2021; Tian et al., 2021). The observed close developmental relationship of pDCs with cDC1 likely reflects the transcriptional basis of DC differentiation: indeed, DC1s and pDCs express the highest amounts of IRF8 based on the activity of dedicated enhancers and require IRF8 for development, maintenance and/or functionality (Bagadia et al., 2019; Cytlak et al., 2020; Sichien et al., 2016). Within this IRF8-driven developmental pathway, the choice between pDC and cDC1 fates involves a cell-autonomous transcriptional circuit between E protein TCF4 and the E protein antagonist ID2 (Cisse et al., 2008; Ghosh et al., 2010; Grajkowska et al., 2017). Indeed, reduced functional ratio of TCF4 to ID2 impairs pDC development while facilitating cDC1 development (Ghosh et al., 2014; Scott et al., 2016), whereas the opposite is true following the loss of ID2 (Weigert et al., 2012). The activity of TCF4 and other E proteins (Bagadia et al., 2019) as well as of Ikaros (Allman et al., 2006), the factors that also drive lymphocyte development, likely explains the”lymphoid” features of pDCs such as the presence of immunoglobulin VH-DH rearrangements (Corcoran et al., 2003; Pelayo et al., 2005) and transcriptional similarity to immature lymphocytes (Dress et al., 2019; Herman et al., 2018). Although lymphoid progenitors can generate pDCs in vitro or upon transfer, their major role in pDC development is not consistent with their relatively small number, low proliferation and decline with age (Harman et al., 2006; Pelayo et al., 2005; Sathe et al., 2013; Shigematsu et al., 2004). Mature pDCs express lymphoid markers IL-7R and Ly-6D and are completely labeled by Il7rCre (Schlenner et al., 2010), thus committed pDC progenitors would necessarily express these markers as noticed recently (Dress et al., 2019; Rodrigues et al., 2018) and also observed in our study. Indeed, we show that IL-7R+ Ly-6D+ pDC progenitors are distinct from canonical lymphoid progenitors and instead comprise a part of Cx3cr1+ progenitor pool that rapidly generates all DCs. Importantly, the earliest Cx3cr1+ progenitors with pDC features (pro-DC) express Csf1r, consistent with a near-complete labeling of pDCs by Csf1r-Cre (Loschko et al., 2016). Finally, we used intersectional tracing to show that pDC labeling by the lymphoid-specific hCD2-Cre transgene does not necessarily indicate their lymphoid origin, because the labeled pDCs still originate from Cx3cr1+ progenitors. A minor fraction of pDCs may originate from lymphoid progenitors (Dekker et al., 2018), especially from those that are themselves derived from Cx3cr1+ progenitors as suggested by our tracing. Thus, Cx3cr1+ DC progenitors represent the major common source of both pDCs and cDCs, supporting the original definition of pDCs as a part of the DC lineage.
Collectively, our results suggest a revised model of DC development with the following key tenets. First, both pDCs and cDCs continuously differentiate from HSCs in a common Flt3-driven developmental pathway that is distinct from lymphopoiesis in its kinetics and progenitor pool. This pathway generates proliferative Cx3cr1+ progenitors that rapidly turn over to generate all DC subsets. Despite the common pool of DC progenitors, the commitment to individual DC subsets is heterogeneous and may occur at multiple stages including HSCs. At all these stages, pDC potential of individual progenitors is closely associated with the potential to produce cDC1s. In contrast, cDC2 development is clonally uncoupled from cDC1s and may not necessarily involve a common progenitor of these subsets.
Limitations of study
Technical limitations of the currently utilized clonal barcode readout are discussed above and include relatively low sensitivity and the resulting over-representation of barcodes unique to a single cell type. Furthermore, barcoding results are consistent with but not directly linked to single-cell analysis of labeled progenitors. Among the populations of putative DC progenitors described herein, only pro-pDCs have been validated for their potential by intersectional lineage tracing. For other (less differentiated) Cx3cr1+ progenitor populations such as pro-DCs, their precise position in the hierarchy and the differentiation spectrum remain to be established. Given that adoptive transfers and culture assays reveal forced potentials rather than actual developmental contributions in vivo, this task would require additional lineage tracing models. Finally, our initial analysis of DC development in the steady state remains to be tested by genetic perturbations (e.g. deletions of individual TF) and extended to inflammatory conditions.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Boris Reizis (boris.reizis@nyulangone.org).
Materials Availability
This study generated a new mouse strain, R26FlipJump. This strain will be transferred to the Jackson Laboratories repository for unrestricted distribution to research community.
Data and Code Availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. The accession number is listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-mouse CD8a (PerCP-Cy5.5 (clone 53–6.7) | eBioscience | Cat# 45–0081-82 |
| Rat anti-mouse CD11b ((eFlour 450) (clone M1/70) | eBioscience | Cat# 48–0112-82 |
| Rat anti-mouse CD11b (Biotin) (clone M1/70) | eBioscience | Cat# 13–0112-81 |
| Rat anti-mouse CD11b (APC-Cy7) (clone M1/70) | eBioscience | Cat# 47–0112-82 |
| Rat anti-mouse CD11b (PE-TR) (clone M1/70) | Invitrogen | Cat# RM2817 |
| Rat anti-mouse /human CD11b (Alexa Flour 700) (clone M1/70) | BioLegend | Cat# 101222 |
| Rat anti-mouse /human CD11b (BV 785) (clone M1/70) | BioLegend | Cat# 101243 |
| Armenian hamster anti-mouse CD11c (APC-eFlour 780) (clone N418) | eBioscience | Cat# 47–0114-80 |
| Armenian hamster anti-mouse CD11c (PerCP-Cy5.5) (clone N418) | BioLegend | Cat# 117327 |
| Armenian hamster anti-mouse CD11c (BUV 737) (clone HL3) | BD Biosciences | Cat# 612797 |
| Rat anti-mouse CD19 (Biotin) (clone 1D3) | eBioscience | Cat# 13–0193-82 |
| Anti-mouse CD19 (APC-Alexa700) (clone 1D3) | BD Biosciences | Cat #: 557958 |
| Anti-mouse CD19 (eFluor450) (clone 1D3) | eBioscience | Cat #: 48–0193-82 |
| Anti-mouse CD3 (APC-Alexa700) (clone 17A2) | BioLegend | Cat #: 100215 |
| Anti-mouse CD3 (eFluor 450) (clone 17A2) | eBioscience | Cat #: 48–0032-82 |
| Rat anti-mouse CD24 (PE) (clone M1/69) | eBioscience | Cat# 12–0242-81 |
| Rat anti-mouse CD24 (BUV 395) (clone M1/69) | BD Biosciences | Cat# 744471 |
| Rat anti-mouse CD48 (AF700) (clone HM48–1) | eBioscience | Cat# 103426 |
| (Rat anti-mouse CD93 (Biotin) (clone AA4.1) | eBioscience | Cat#13–5892-82 |
| Rat anti-mouse CD115 (c-fms) (PE) (clone AFS98) | eBioscience | Cat# 12–1152-81 |
| Rat anti-mouse CD117 (c-Kit) (APC-eFlour 780) (clone 2B8) | eBioscience | Cat# 47–1171-80 |
| Rat anti-mouse CD117 (c-Kit) (APC) (clone 2B8) | eBioscience | Cat# 17–1171-83 |
| Rat anti-mouse CD127 (BV 605) (clone A7R34) | BioLegend | Cat# 135041 |
| Rat anti-mouse CD127 (PE-Cy7) (clone A7R34) | BioLegend | Cat# 135014 |
| Rat anti-mouse CD135 (Flt3) (APC) (clone A2F10) | eBioscience | Cat# 17–1351-82 |
| Rat anti-mouse CD135 (Flt3) (APC) (clone A2F10) | eBioscience | Cat# 14–1351-82 |
| Rat anti-mouse CD135 (Flt3) (Biotin) (clone A2F10) | BioLegend | Cat# 135307 |
| Rat anti-mouse CD150 (SLAM) (PE-Cy7) (clone TC15–12F12.2) | BioLegend | Cat# 115914 |
| Rat anti-mouse CD172a (Sirp-a) APC-Fire750 (clone P84) | BioLegend | Cat # 144029 |
| Rat anti-mouse CD205 (PE-Cy7) (clone NLDC-145) | BioLegend | Cat# 138209 |
| Rat anti-mouse CD317 (PDCA-1) (PE-Cy7) (clone 927) | eBioscience | Cat# 25–3175-82 |
| Rat anti-mouse CD317 (PDCA-1) (APC) (clone 927) | Millipore Sigma | Cat# MABF2047 |
| Rat anti-mouse SiglecH (BV711) (clone 440C) | BD Biosciences | Cat#747671 |
| Rat anti-mouse SiglecH (APC) (clone 551) | BioLegend | Cat# 129612 |
| Rat anti-mouse SiglecH (PerCP-Cy5.5) (clone 551) | BioLegend | Cat# 129614 |
| Mouse anti-mouse Cx3r1 (FITC) (clone SA011F11) | BioLegend | Cat# 149019 |
| Mouse anti-mouse Cx3r1 (PE-Cy7) (clone SA011F11) | BioLegend | Cat# 149016 |
| Mouse anti-mouse Cx3r1 (BV785) (clone SA011F11) | BioLegend | Cat# 149029 |
| Rat anti-mouse/human CD45R/B220 (Alexa Flour700) (clone RA3–6B2) | BioLegend | Cat# 103232 |
| Rat anti-mouse/human CD45R/B220 (PE-Cy7) (clone RA3–6B2) | eBioscience | Cat# 25–0452-81 |
| Rat anti-mouse/human CD45R/B220 (Biotin) (clone RA3–6B2) | eBioscience | Cat# 13–0425-85 |
| Rat anti-mouse/human CD45R/B220 (eFlou 450) (clone RA3–6B2) | eBioscience | Cat# 48–0452-80 |
| Rat anti-mouse/human CD45R/B220 (BV 650) (clone RA3–6B2) | BioLegend | Cat# 103241 |
| Rat anti-mouse/human CD45R/B220 (Biotin) (clone RA3–6B2) | eBioscience | Cat# 13–0425-85 |
| Armenian hamster anti-mouse TCR beta (eFluor 450) (clone H57–597) | eBioscience | Cat# 48–5961-80 |
| Armenian hamster anti-mouse TCR beta (Biotin) (clone H57–597) | eBioscience | Cat# 13–5961-82 |
| Rat anti-mouse ESAM (PE) (clone 1G8) | eBioscience | Cat# 12–5852-81 |
| Rat anti-mouse MHC Class II (I-A/I-E) (AF700) (clone M5/114.15.2) | eBioscience | Cat# 56–5321-82 |
| Rat anti-mouse TER-119 (eFlour 450) (clone TER-119) | eBioscience | Cat# 48–5921-82 |
| Rat anti-mouse TER119 (Biotin) (clone TER-119) | eBioscience | Cat# 13–5921-82 |
| Rat anti-mouse Ly-6A/E (Sca-1) (PerCP-Cy5.5) (clone D7) | eBioscience | Cat# 45–5981-82 |
| Rat anti-mouse Ly-6G (Gr-1) (eFlour 450) (clone RB6–8C5) | eBioscience | Cat# 48–5931-82 |
| Rat anti-mouse Ly-6G (Alexa Flour 700) (clone RB6–8C5) | BioLegend | Cat# 127621 |
| Rat anti-mouse Ly-6G (Gr-1) (Biotin) (clone RB6–8C5) | eBioscience | Cat# 13–5931-82 |
| Rat anti-mouse Ly-6C (PercP) (clone 927) | BioLegend | Cat# 128027 |
| Rat anti-mouse Ly6C BV510 (clone HK1.4) | BioLegend | Cat # 128033 |
| Rat anti-mouse Ly-6D (eFlour 450) (clone 49-H4) | eBioscience | Cat# 48–5974-80 |
| Mouse anti-mouse NK1.1 (Biotin) (clone PK136) | eBioscience | Cat# 13–5941-85 |
| Rat anti-mouse Xcr1 PerCP/Cy5.5 (clone ZET) | BioLegend | Cat # 148208 |
| Streptavidin eFluor 450 Conjugate | eBioscience | Cat# 48–4317-82 |
| PE Steptavidin | Biolegend | Cat#405203 |
| Streptavidin Alexa Fluor 647 | ThermoFisher Scientific | Cat# S21374 |
| TruStain FcX™ (anti-mouse CD16/32) Antibody | BioLegend | Cat# 101320 |
| CITE-seq antibodies, see Table S7 | ||
| Experimental Models: Organisms/Strains | ||
| Mouse strains | ||
| Pdzk1ip1-CreER | Reizis Lab (NYU) | (Sawai et al., 2016) |
| R26 FlipJump | Reizis Lab (NYU) | this study |
| Cx3cr1CreER-YFP: B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/WganJ | Jackson Laboratory, stock 021160 | (Parkhurst et al., 2013) |
| R26 CreER | Dr. Thomas Ludwig, Ohio State Univ. | (de Luca et al., 2005) |
| R26LSL-Tom: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratory, stock 007914 | (Madisen et al., 2010) |
| B6.Cg-Tg(CD2-icre)4Kio/J | Jackson Laboratory, stock 008520 | (de Boer et al., 2003) |
| B6N.129P2-Cx3cr1tm3(DTR)Litt/J | Jackson Laboratory, stock 025629 | (Diehl et al., 2013) |
| C57BL/6 | Taconic Biosciences | Black 6 |
| Cell lines | ||
| Mouse cell line: B16-FLT3L | Reizis lab (NYU) | (Mach et al., 2000) |
| Mouse cell line: 129/B6 ES cells | Dr. Victor Lin, Columbia Unversity Irving Medical Center | N/A |
| Oligonucleotides | ||
| FlipJump ES screening primer forward: GGACTAGGGCTGCGTGAGTCTCTGA | This study | N/A |
| FlipJump ES screening primer reverse: TGGCGTTACTATGGGAACATACGTC | This study | N/A |
| Linker common (long strand) GACCCGGGAGATCTGAATTCAGTGGCACAGCAGTT AGGGGGCTGGTCG | (Sun et al., 2014) | N/A |
| Blunt end linker /5Phos/CGACCAGCCC/3AmMO/ | (Sun et al., 2014) | N/A |
| CG end linker /5Phos/CGCGACCAGCCC/3AmMO/ | (Sun et al., 2014) | N/A |
| GATC end linker /5Phos/GATCCGACCAGCCC/3AmMO/ | (Sun et al., 2014) | N/A |
| AGCT end linker /5Phos/AGCTCGACCAGCCC/3AmMO/ | This study | N/A |
| Linker primer1: GACCCGGGAGATCTGAATTC | (Sun et al., 2014) | N/A |
| Linker primer3: GAATTCAGTGGCACAGCAGTTAGG | This study | N/A |
| MiSeq primer TP_P5: AATGATACGGCGACCACCGAGATCTACACA CACTCTTTCCC | This study | N/A |
| MiSeq primer TP_P7: CAAGCAGAAGACGGCATACGAGATGTGACT GGAGT | This study | N/A |
| LM_5TR_inver_A2: /5Biosg/AGATAATCATGCGTCATTTTGACTCAC | This study | N/A |
| LM_5TR_inver_B2: AGCGGCGACTGAGATGTCC | This study | N/A |
| LM_3TR_inverA: /5Biosg/TCGATATACAGACCGATAAAACACATG | This study | N/A |
| LM_3TR_inverB: ATGCGTCAATTTTACGCATGATTATC | This study | N/A |
| 5TR index primer for MiSeq sequencing: ACACTCTTTCCCTACACGACGCTCTTCCGATCTXXXXXXXCGACGGATTCGCGCTAT (Xs denote a sample-specific barcode) | This study | N/A |
| 3TR index primer for MiSeq sequencing: ACACTCTTTCCCTACACGACGCTCTTCCGATCTXXX XXXXCGTACGTCACAATATGATT(Xs denote a sample-specific barcode) | This study | N/A |
| Genotyping primer: Flipjump-F: ACGTGCTGGTTATTGTGCTGT | This study | N/A |
| Genotyping primer: Flipjump-R GTTCATGCGGAACCTGTACAT | This study | N/A |
| Genotyping primer: WT-F AAAGTCGCTCTGAGTTGTTAT | This study | N/A |
| Genotyping primer: WT-R GGAGCGGGAGAAATGGATATG | This study | N/A |
| Genotyping primer: Cre-F GGACATGTTCAGGGATCGCCAGGCG | This study | N/A |
| Genotyping primer: Cre-R GCATAACCAGTGAAACAGCATTGCTG | This study | N/A |
| CITE-Seq ADT additive primer: CCTTGGCACCCGAGAATTCC | https://cite-seq.com/protocols/ | (Stoeckius et al., 2018) |
| CITE-Seq HTO additive primer: GTGACTGGAGTTCAGACGTGTGCTC | https://cite-seq.com/protocols/ | (Stoeckius et al., 2018) |
| CITE-Seq TruSeq Small RNA RPIx primer: CAAGCAGAAGACGGCATACGAGXXXXXXXXGTGAC TGGAGTTCCTTGGCACCCGAGAATTCCA (Xs denote a sample-specific barcode) | https://cite-seq.com/protocols/ | (Stoeckius et al., 2018) |
| CITE-Seq TruSeq D7xx primer: CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGC (Xs denote a sample-specific barcode) | https://cite-seq.com/protocols/ | (Stoeckius et al., 2018) |
| CITE-Seq SI PCR primer: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTC | https://cite-seq.com/protocols/ | (Stoeckius et al., 2018) |
| Recombinant DNA | ||
| pCMV-HAhyPBase | Wellcome Trust Sanger Institute | (Yusa et al., 2011) |
| pPB-LR5 | Wellcome Trust Sanger Institute | (Cadinanos and Bradley, 2007) |
| pPB-RL5 | Wellcome Trust Sanger Institute | (Cadinanos and Bradley, 2007) |
| Rosa26 mT/mG | Addgene | Plasmid #17787 |
| pRosa26-TG | Addgene | Plasmid #40026 |
| pCMS-EGFP | Clontech | Plasmid #6101–1 |
| Software and Algorithms | ||
| FlowJo v9.9.5 and v10.1 | FlowJo, LLC | https://www.flowjo.com/ |
| Prism 8 | GraphPad | https://www.graphpad.com/ |
| samtools (v.1.3.1) | (Li and Durbin, 2009) | http://samtools.sourceforge.net/ |
| GenomicAlignments (v.1.10.1) | (Lawrence et al., 2013) | https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html |
| Bedtools2 (v2.26.0) | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| Bowtie2 (v2.2.9) | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| UCSC mm10 Known Gene Annotation Database (v.3.4) | (Speir et al., 2016) | https://bioconductor.org/packages/release/data/annotation/html/TxDb.Mmusculus.UCSC.mm10.knownGene.html |
| BLAT | (Kent, 2002) | https://genome.ucsc.edu/cgi-bin/hgBlat |
| Cutadapt (v1.18) | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| BioVenn | (Hulsen et al., 2008) | http://www.biovenn.nl |
| iCellR | (Khodadadi-Jamayran et al., 2020) | https://github.com/rezakj/iCellR |
| KNetL | (Khodadadi-Jamayran and Tsirigos, 2020) | https://github.com/rezakj/iCellR |
| ImmGen Reference Datasets | (Heng and Painter, 2008) | http://rstats.immgen.org/DataPage/ |
| NIA Array | (Sharov et al., 2005) | https://lgsun.irp.nia.nih.gov/ANOVA/) |
| HeatMapper | (Babicki et al., 2016) | http://www.heatmapper.ca/expression/ |
| STAR aligner (v2.5.0c) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| HTSeq (v 0.6.0) | (Anders et al., 2015) | https://htseq.readthedocs.io/en/master/history.html |
| Picard (v.1.126) | Picard Toolkit, the Broad Institute | http://broadinstitute.github.io/picard |
| DEseq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html |
| Reagents and Kits | ||
| REPLI-g Mini Kit (100) | QIAGEN | Cat# 150025 |
| QIAamp DNA Micro Kit (50) | QIAGEN | Cat# 56304 |
| QIAamp DNA Mini Kit (50) | QIAGEN | Cat# 51304 |
| Nucleospin Tissue XS (Takara) | Macherey-Nagel | Cat#740901 |
| QIAquick PCR Purification Kit (250) | QIAGEN | Cat# 28106 |
| TA Cloning kit | ThermoFisher Scientific | Invitrogen Cat# K2020–40 |
| Dynabeads kilobaseBINDER kit | ThermoFisher Scientific | Invitrogen Cat# 60101 |
| MspI | NEB | Cat# R0106S |
| HaeIII | NEB | Cat# R0108S |
| DpnII | NEB | Cat# R0543S |
| HindIII-HF | NEB | Cat# R3104S |
| XmaI | NEB | Cat# R0180S |
| SalI | NEB | Cat# R3138S |
| SpeI-HF | NEB | Cat# R3133S |
| BglII | NEB | Cat# R0144S |
| XhoI | NEB | Cat# R0146S |
| MluI | NEB | Cat# R0198S |
| EcoRV-HF | NEB | Cat# R3195S |
| T4 DNA Ligase | NEB | Cat# M0202S |
| Phusion High-Fidelity DNA Polymerase | NEB | Cat# M0530S |
| PfuTurbo Hotstart DNA Polymerase | Agilent Technologies | Cat# 600320 |
| Deoxynucleotide Solution Mix | NEB | Cat# N0447S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma-Aldrich | Cat# T5648–5G |
| SUNFLOWER SEED OIL | Sigma-Aldrich | Cat# S5007–250ML |
| RBC Lysis Buffer (10X) | BioLegend | Cat# 420301 |
| DMEM, high glucose, no glutamine | Thermo Fisher Scientific | Cat# 11960069 |
| Penicillin-Streptomycin (10,000 U/ml) (Gibco) | Thermo Fisher Scientific | Cat# 15140122 |
| MEM Non-Essential amino acids solution (100x) | Thermo Fisher Scientific | Cat# 11140076 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985023 |
| Sodium Pyruvate (100mM) | Thermo Fisher Scientific | Cat# 11360070 |
| L-Glutamine (200 mM) | Thermo Fisher Scientific | Cat# 25030164 |
| HyClone ADCF-Mab media | Thermo Fisher Scientific | Cat# SH3034902 |
| S-clone SF-O3 | Iwai North America | Cat# S-clone SF-O3 |
| BSA, Molecular biology grade | NEB | Cat# B9000S |
| Fixable Viability Dye eFluor 506 | eBioscience | Cat# 65–0866-14 |
| DAPI | Sigma-Aldrich | Cat# D8417 |
| ESGRO recombinant mouse LIF protein | Millipore Sigma | Cat# ESG1106 |
| TAT-CRE Recombinase | Millipore Sigma | Cat# SCR508 |
| Recombinant Murine SCF | PeproTech | Cat# 250–03 |
| Recombinant Human TPO | PeproTech | Cat# 300–18 |
| Recombinant Murine Flt3-Ligand | PeproTech | Cat# 250–31L |
| Streptavidin microbeads | Miltenyi Biotec | Cat# 130–048-101 |
| Diphtheria Toxin | Millipore Sigma | Cat# D0564 |
| Fetal Bovine Serum, charcoal stripped | Thermo Fisher Scientific | Cat# 12676029 |
| Deposited Data and Code | ||
| FlipJump transposon sequencing, CITE-seq, bulk RNA-seq | This study | GEO: GSE193733 |
| FlipJump transposon sequencing code | This study | https://doi.org/10.5281/zenodo.5879278 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Pdzk1ip1-CreER mice crossed to the homozygous R26LSL-Tom reporter have been described (Sawai et al., 2016; Upadhaya et al., 2018). Pdzk1ip1-CreER mice were crossed in a heterozygous state to heterozygous R26FlipJump mice. Cx3cr1CreER-YFP mice (Parkhurst et al., 2013) were obtained from the Jackson Laboratory and crossed in a heterozygous state to heterozygous R26FlipJump or R26LSL-Tom animals. R26CreER mice (de Luca et al., 2005) were crossed in a heterozygous state to heterozygous R26FlipJump mice. Cx3cr1LSL-DTR and hCD2-iCre mice were obtained from the Jackson Laboratory and intercrossed to obtain mice heterozygous for Cx3cr1LSL-DTR and hemizygous for hCD2-iCre.
All animal maintenance and experimentation was performed under the investigators’ protocols approved by the institutional Animal Care and Use Committees of New York University School of Medicine or Stanford University School of Medicine. For Cre recombinase induction, 50 mg tamoxifen was dissolved in 100 μL ethanol and subsequently in 900 μL sunflower seed oil. Unless indicated otherwise, mice were gavaged once with 100 μL of the resulting emulsion (5 mg tamoxifen). For CITE-Seq analysis of HSC progeny, 5 mg tamoxifen was dissolved in 1 ml as described above, and mice were gavaged once with 100 μL of the resulting emulsion (0.5 mg tamoxifen). BM biopsy was performed as described (Upadhaya et al., 2018). For DT-mediated cell depletion, Cx3cr1LSL-DTR/+ hCD2-iCre mice and Cre-negative Cx3cr1LSL-DTR/+ controls were treated with 50 ng/g body weight 100 diphtheria toxin (DT) intraperitoneally (i.p.) and analyzed 24 hr later (day 1) or inoculated with 25 ng/g DT i.p. at days 2, 4 and 6 and analyzed at day 7.
METHOD DETAILS
Generation of R26FlipJump mice
To generate the R26FlipJump strain, we have designed a cassette depicted in Fig. 1A. Plasmids encoding the hyperactive PiggyBac transposase (Yusa et al., 2011) and its cognate terminal repeats (Cadinanos and Bradley, 2007) were obtained from the Wellcome Trust Sanger Institute. The cassette was built from six DNA fragments including the LoxP/Lox2272/reversed PiggyBac (F1); 3TR (F2); reversed LoxP/reversed Lox2272/polyA (F3); reversed 5TR (F4); EGFP (F5); and polyA/FRT/Neo cassette/FRT (F6). To prevent repeat DNA sequences such as LoxP sites from inducing instability during cloning, we assembled these fragments following this specific order: F3 to F2 to F5 to F6 to F4 to F1, and then inserted into pRosa26-TG targeting vector (Addgene plasmid #40026).
Generation of targeted ES cells and chimeric mice were performed at the Genetically Modified Mouse Models Facility of the Columbia University Irving Medical Center. pRosa26-TG targeting vector containing the FlipJump construct was linearized and electroporated into ES cells, and G418- and diphtheria toxin–resistant clones were screened by PCR using forward and reverse screening primers. The PCR product for pRosa26-TG positive ES cells screening was around 1.6 kb, and was detected in ~30% of clones. Four positive clones were further expanded and confirmed for FlipJump activation using cell-permeable TAT-Cre protein. Two targeted ES clones were microinjected to generate chimeras, and the targeted allele was passed through the germline to establish the R26FlipJump strain.
Cell lines
Flt3L-secreting clone of the C57BL/6-derived B16 melanoma was cultured in DMEM medium supplemented with 10% fetal calf serum (FCS), 1% L-glutamine, 1% sodium pyruvate, 1% MEM-NEAA and 1% penicillin/streptomycin at 37°C in a humidified atmosphere at 5% CO2. FlipJump-targeted 129/B6 mice-derived ES cells were cultured in DMEM complete medium with 15% FBS and mouse leukemia inhibitory factor (LIF). For TAT-Cre administration, ES cells were washed three times with Hyclone ADCF-Mab serum-free media and resuspended in HyClone ADCF-Mab media. After prewarming ES cells and TAT-Cre at 37°C for 10 mins, ES cells were incubated with 50 ug/ml TAT-Cre at 37°C for 45 mins. ES cells were plated in a 24-well plate at a density of 1× 105 cells per well and grown for three days and harvested for further analysis.
Validation of transposon system in hematopoietic stem/progenitor cells-derived colonies
Single hematopoietic stem/progenitor cells (LSK: Lin− Sca1+ c-Kit+ GFP+) were sorted directly into individual wells of 96-well plate from R26CreERR26FlipJump mice at day 9 post tamoxifen treatment. The cells were cultured in S-clone SF-O3 medium supplemented with 10% bovine serum albumin (BSA), 50 ng/ml stem cell factor (SCF), 50 ng/ml Flt3, 50 ng/ml thrombopoietin (TPO), 50 μM 2-Mercaptoethanol and 1% penicillin/streptomycin at 37°C in a humidified atmosphere at 5% CO2. The expansion of GFP+LSK cells were visualized under a fluorescence inverted microscope. Ten wells contained expanded GFP+ LSK cells were collected together as ten LSK derived-colonies sample (10 colonies) for validation of FlipJump-based barcode system. The cells isolated from 10 colonies were performed WGA with REPLI-g Mini Kit (Qiagen) and further proceeded for three arm LM-PCR of 5TR and 3TR as described in the main method. In addition to conducting secondary PCR, half of them were prepared for Miseq with index primers and half of them were prepared for TOPO cloning in bacteria. The Miseq results were conducted using Miseq analysis pipeline described below. The TOPO cloning results were obtained from sequencing of the DNA plasmids which were extracted from bacteria. The sequencing result were aligned using BLAT search for TTAA sites.
In vivo lineage tracing of DC progenitors
Cx3cr1CreER-YFP R26FlipJump mice were administered a single dose of 5 mg tamoxifen to induce FlipJump activation and transposon mobilization in Cx3cr1+ progenitors. Animals were sacrificed at days 2 to 8 post-tamoxifen and used for the analysis of GFP expression and/or sorting of GFP+ cells for transposon integration site cloning. Cx3cr1CreER-YFP R26LSL-Tom mice were treated similarly with 5 mg tamoxifen and sacrificed at days 2 to 8 for the analysis of Tom expression by flow cytometry.
In vitro Flt3L-driven DC development
Total BM cells from Cx3cr1CreERYFPR26FlipJump mice were obtained on day 3 after tamoxifen treatment. The cells were plated in a 24-well plate at a density of 2×106 cells per well and cultured in DMEM supplemented with 10% FCS, 1% L-glutamine, 1% sodium pyruvate, 1% MEM-NEAA, 1% penicillin/streptomycin, 55 μM 2-mercaptoethanol and 10% supernatant from the cultured B16-Flt3L cell line. All DC cells were harvested after 7 days culture for immunophenotypic analysis and FlipJump-based barcode analysis.
In vivo lineage tracing of HSC
To induce recombination in the Pdzk1ip1CreER R26FlipJump mice, a single dose of 5 mg tamoxifen was administered by oral gavage. The peripheral blood was obtained by submandibular vein puncture at weeks 4, 12 and 20 post tamoxifen induction and applied for the analysis of GFP expression in different lineage. Animals were sacrificed at 20 weeks post-tamoxifen induction. The sorted GFP+ cells were analyzed for GFP expression and transposon integration site cloning.
Cell processing and flow cytometry
BM and spleen were dissected from mice which were euthanized with CO2. BM was flushed with cold PBS and pressed through a 70 μM cell strainer to yield single cell suspension. Spleen was minced and pressed through a 70 μM cell strainer. BM cells and splenocytes were treated with 1X RBC lysis buffer to remove red blood cells (BioLegend) and washed with PBS. Cells were stained with direct fluorescent antibody conjugates for 20–60 mins in PBS on ice or 15–45 mins in PBS at room temperature only for DC progenitors staining of wild-type mice. Fixable viability dye (eBioscience) or DAPI was used to identify dead cells in some of the experiments. For analysis, samples were acquired on Attune NxT (Thermo Fisher Scientific) using Attune NxT software and further analyzed with FlowJo software (Tree Star). When necessary, an optical filter set for the discrimination of GFP and YFP fluorescence on the Attune NxT was used (Thermo Fisher Scientific). Sorting was performed on BD FACSAria IIu SORP flow sorter (Becton Dickinson) at the Cytometer & Cell Sorting Laboratory, NYU School of Medicine. For sorting, GFP and YFP were excited with a single 488 nm laser, discriminating using customized filter configuration including a 530/30 nm bandpass for YFP and a 510/21 nm bandpass for GFP with the 525 nm short-pass dichroic mirror.
Cell sorting for transposon integration site cloning
For transposon integration site cloning, bone marrow cells and splenocytes were isolated from the Pdzk1ip1CreERR26FlipJump mice at weeks 20 post tamoxifen induction. HSC (Lin− c-Kit+ Sca1+ CD48− CD150+ GFP+), DC subsets (pDC, PDCA1+ B220+ CD11clow GFP+; cDC1, CD11chi MHCII+ B220− CD8+ DEC205+ GFP+; cDC2, CD11chi MHCII+ B220− CD11b+ Esam+ GFP+) and Immature B (CD11c−B220+CD93+Bst2−) were sorted from isolated bone marrow cells and splenocytes. Splenocytes from Cx3cr1CreERYFP R26FlipJump mice were obtained on days 8 post tamoxifen induction. GFP+ YFPlo/- DC subsets were sorted as follows: pDC (PDCA1+ B220+ CD11b−); cDC1 (CD11chi MHCII+ B220− SSChi CD8+ or DEC205+) and cDC2 (CD11chi MHCII+ B220− CD11b+ Esam+). GFP+ DC subsets from Flt3L cultured Cx3cr1CreERYFPR26FlipJump mouse BM cells were sorted as follows: pDC (PDCA1+ B220+ CD11b−); cDC1 (CD11chi MHCII+ B220− CD24+); and cDC2 (CD11chi MHCII+ B220− CD11b+).
For technical analysis of transposon retrieval, Pdzk1ip1CreER R26FlipJump mouse was administered a single dose of 5 mg tamoxifen and traced for 24 weeks. Four replicate samples (1×104 cells each) and three titrated samples (2×102, 2×103 and 2×104 cells) of GFP+ monocytes (Ly6Chi CD11b+ TCRb− B220−) were sorted from the BM cell suspension. Because this experiment utilized only a single FlipJump animal, BM monocytes were also sorted in duplicates (1×104 cells each) from a wild-type C57BL/6 animal and used for control amplification of integration sites.
Transposon integration site cloning and sequencing
For sorted primary DCs, ImmB and HSC (typically <15,000 cells), genomic DNA was extracted by NucleoSpin Tissue XS (Takara) and eluted DNA in 5 µl elution buffer. The total purified primary DCs, ImmB and HSC DNA was further amplified with REPLI-g Mini Kit (Qiagen) for whole genome amplification (WGA). For DCs sorted from in vitro cultures (typically 20,000–80,000 cells) genomic DNA was extracted by QIAamp Mini or Micro kit and further amplified with REPLI-g Mini Kit. Three-arm LM-PCR-based method (Sun et al., 2014) was optimized and adapted to clone FlipJump transposon integration sites from both 3TR and 5TR ends. Amplified DNA was quantified using Qubit 2.0 fluorometer (Thermo Fisher), and 1 µg was digested separately with MspI, HindIII or HaeIII for 5TR cloning, and MspI, DpnII or HaeIII for 3TR cloning. The digested DNA was purified using QIAquick PCR Purification Kit. To prepare the individual LM-PCR linker mix, the equal volume of 50 µM linker common (long strand) and 50 µM restriction enzyme (RE)-digested end-specific linker (Blunt end for HaeIII,CG end for MspI, GATC end for DpnII or AGCT end for HindIII) were mixed in a PCR tube. The linker mix was then denatured and annealed by heating it to 95° C for 5 mins and cooling slowly to room temperature. One third of purified digested DNA was ligated with the individual LM-PCR linker mix using PfuTurbo Hotstart DNA Polymerase (Agilent Technologies). The ligation reaction was then digested with EcoRV (internal RE site of PB at 5TR direction) or MluI (internal RE site of PB at 3TR direction) to remove any linker that has ligated onto the 5TR or 3TR end of DNA, respectively. The remaining DNA containing MspI, HindIII and HaeIII linkers were pooled for 5TR sampling and the remaining DNA containing MspI, DpnII and HaeIII were pooled for 3TR processing. The pooled DNA was set up for PCR amplification using linker primer 1 and biotinylated primers specific for 5TR (LM_5TR_inver_A2) or 3TR (LM_3TR_inverA). Biotinylated PCR products were pulled down using Dynabeads kilobaseBINDER kit at room temperature for 4 hours. Further nested PCR amplification was conducted using a primer that was specific for 5TR (LM_5TR_inver_B2) or 3TR (LM_3TR_inverB), and a corresponding MiSeq sequencing index primer containing a 7-base individual sample barcode (barcode sequences available upon request). The PCR products were tagged with modified P5 and P7 primers for sequencing on Miseq (Illumina) at the Genome Technology Center of NYU School of Medicine.
Transposon integration site identification and analysis
Known sequences of 5TR or 3TR and LM-PCR linker sequences were removed from the first read (R1) and the second read (R2) fastq files using cutadapt (v 1.18). To prepare the analysis-ready fastq files after trimming, only the read pairs that had TTAA at the end of R1 and a restriction site at the end of either R1 or R2 were used. All analysis-ready fastq files were then mapped to mouse genome (mm10) using Bowtie2 (v 2.3.4) end-to-end alignment method. Samtools (v 1.9) was used to sort mapped data and index the BAM files. Reads with low mapping quality (Q<30) were removed.
All analysis of the resulting reads was done in R environment (v3.5) (https://www.r-project.org/). To generate count tables for the peaks (i.e. regions with mapped reads) in each file, we first identified the regions with peaks in all the files by merging all the BAM files and converting the merged BAM file into a single BED file using BEDtools (v 2.26). Then the overlapping peaks in the BED file were merged (bedtools merge -d 500) to create an analysis-ready peaks file. This way, the following potential mapping outcomes were selected for further analysis: i) full overlap of R1 and R2; ii) partial overlap of R1 and R2; iii) no overlap with a distance between R1 and R2 <500 bp. The read count tables for every BAM file were then created using BEDtools for all mapped TTAA sites in the analysis-ready peaks file. The identified TTAA sites from 5TR and 3TR cloning for each sample were overlapped, and those present in both clonings were defined as integration sites. Finally, all integration sites that occurred more than once in unrelated mice or cultures were discarded to avoid commonly detected integration sites arising due to amplification or cloning artefacts. In the analysis of integration sites from monocytes derived from a single animal, integration sites amplified from FlipJump-negative wild-type C57BL/6 animal were used for filtering. All resulting integration sites reported in this paper (Table S1) are unique for a single animal or cell culture.
Bulk RNA-seq analysis of pDC GFP− and pDC GFP+
Cx3cr1CreERYFP R26FlipJump mice were administered a single does of 5 mg Tamoxifen and sacrificed at 8 days. The pDC (B220+ Siglech+ Bst2+) with GFP or without GFP was sorted from isolated BM cells. For RNA-seq, RNA from sorted cells was extracted using TRIzol Plus purification kit (ThermoFisher), and cDNA libraries were prepared with the Low input Clontech SMART-seq HT with Nxt HT. Sequencing was performed using Illumina NovaSeq 6000 at the Genome Technology Core at New York University School of Medicine. Sequencing reads were mapped to the reference genome (mm10) using the STAR aligner (v2.5.0c) (Dobin et al., 2013). Alignments were guided by a Gene Transfer Format (GTF) file. The mean read insert sizes and their standard deviations were calculated using Picard tools (v.1.126). The read count tables were generated using HTSeq (v0.6.0) (Anders et al., 2015), normalized based on their library size factors using DEseq2 (Love et al. 2014). Pairwise gene expression comparison was performed using NIA Array software after removing gene models (GmXXXXX).
Isolation and staining of Cx3cr1+ progenitors for CITE-Seq
To induce recombination in adult Cx3cr1CreER-YFP R26FlipJump mice, a single dose of 5 mg tamoxifen in sunflower seed oil was administered by gavage. Two or five days later, BM cells were isolated from two mice per time point and processed independently, yielding four samples. Depletion of lineage-positive cells was performed using magnetic selection as described above. Each sample was incubated with Fc blocking antibody and stained with sample-specific cell hashing reagent and a common pool of CITE-Seq antibodies as above. Cells were washed, pooled and incubated with DAPI, and Lin− GFP+ cells were sorted for CITE-Seq analysis. GFP and YFP fluorescence were discriminated, and all GFP+ cells irrespective of YFP fluorescence were included to capture the YFP− progeny of the initially labeled cells.
Isolation and staining of HSC-derived Flt3+ progenitors for CITE-Seq
To induce recombination in adult Pdzk1ip1CreER R26tdTomato mice, a single dose of 0.5 mg tamoxifen in sunflower seed oil was administered by gavage. To check labeling efficiency in the BM of induced mice, femur BM was sample by biopsy at day 2 after tamoxifen administration as previously described (Upadhaya et al., 2018). At weeks 3, 4 or 5 following tamoxifen, mice were sacrificed and BM cells were isolated from 2, 3, and 4 mice per respective timepoint. Cells were stained with biotinylated antibodies to lineage markers (Gr-1, CD19, TCR, Ter119) and negatively selected using streptavidin magnetic beads and LD MACS cell separation columns (Miltenyi Biotech). Lineage-depleted cells from all mice from each timepoint were pooled together, and pooled samples from weeks 4 and 5 were subdivided into two technical replicates, resulting in a total of 5 samples. Cells were resuspended in staining buffer (PBS with 2% BSA and 0.01% Tween) and incubated with anti-mouse CD16/32 Fc-blocking antibody (Biolegend) for 10 mins on ice. Each sample was incubated with a unique cell hashing reagent, a common pool of CITE-Seq antibodies, a fluorescently conjugated anti-CD135 antibody and Fixable Viability Dye eFluor® 506 (eBioscience) for 30 mins at 4°C. Cells were washed and resuspended in PBS and sorted using Viability Dye− tdTomato+ CD135+ as the sorting criteria.
CITE-Seq
Purified, unconjugated antibodies used in CITE-Seq were purchased from indicated providers (see Table S7) and conjugated using iEDDA click chemistry to barcode oligos as described in (van Buggenum et al., 2016). For cell hashing of HSC-derived Flt3+ progenitors, mixtures of antibodies directed against CD45 and CD29 conjugated to hashtag oligos were prepared using iEDDA click chemistry as described above. For cell hashing of Cx3cr1+ progenitors, mixtures of antibodies directed against CD45 and H-2 MHC class I conjugated to hashtag oligos were purchased from Biolegend.
CITE-Seq and cell hashing were performed as described in (Stoeckius et al., 2017; Stoeckius et al., 2018). After magnetic bead-based lineage depletion, prior to FACS sorting, each sample was stained with a pool of mouse CITE-Seq antibodies (see Table S7 for the complete CITE-Seq antibody panel for each experiment), and sample-specific hashing mixtures. Post staining and sorting, cells were run through the standard 10x Chromium (v3) protocol up until cDNA amplification, with the following modification: during cDNA PCR, 2 pmol of antibody-derived tag (ADT) additive primer and 1 pmol of hashtagging oligonucleotide (HTO) additive primer were added to the cDNA amplification master mix.
Post cDNA amplification, a 0.6X Solid Phase Reversible Immobilisation (SPRI) bbead clean-up was performed to separate the cDNA fraction (on beads) from the smaller ADT and HTO fractions (in supernatant). The cDNA fraction was converted into a 3’ tag gene expression library according to the 10x Genomics Single Cell Genomics Protocol (v3). Supernatant from cleanup was kept for ADT and HTO preparation. To the supernatant, another 1.4X SPRI was added to bring the total SPRI concentration to 2X. After washing the beads in 80% ethanol and eluting in water, a second round of 2X SPRI cleanup was performed to remove any residual primer carryover from the cDNA PCR. Post cleanup, eluate was taken into ADT PCR amplification using TruSeq Small RNA RPIx primer and SI PCR primer, and into hashtag amplification using TruSeq D7xx and SI PCR primers. Libraries were pooled and sequenced on a 100 cycle Novaseq S1 flowcell. Additional protocol details can be found for CITE-Seq and cell hashing at www.cite-seq.com
CITE-Seq Data Analysis
For quality control, we confirmed the integrity of the cDNA, quality of the libraries, number of cells sequenced and mean number of reads per cell. RNA library sequenced reads were aligned to Mus musculus reference genome (mm10/GRCm38) using CellRanger (10X Genomics). UMI correction was performed to handle PCR duplications. From this point on, analysis was done using the iCellR R package (v1.5) (https://CRAN.R-project.org/package=iCellR ) (Khodadadi-Jamayran et al., 2020). Quality control was performed by calculating the number of genes, UMIs and the proportion of mitochondrial genes for each cell. Cells were filtered out if they did not meet the following quality control thresholds:
Cx3cr1+ progenitors: ratio of reads mapping to mitochondrial/nuclear <0.05, minimum number of genes >500, maximum number of genes <7000.
HSC-derived Flt3+ progenitors: ratio of reads mapping to mitochondrial/nuclear <0.1, minimum number of genes >500, maximum number of genes <3500.
The resulting gene-cell matrix was normalized based on ranked geometric library size factor (ranked GLSF). Geometric library size factor normalization is a common normalization method used by popular tools such as DEseq2 (Love et al., 2014), however, here we use only the top ranked genes (top 500 genes sorted by base mean). This is intended to reduce the effect of dropouts (non-zero events counted as zero) in normalization by considering only the highly expressed genes. A general statistical test was then performed to calculate gene dispersion, base mean and cell coverage to build a gene model for performing Principal Component Analysis (PCA). Genes with high coverage (top 500) and high dispersion (dispersion > 1.5) were chosen and PCA analysis was performed. Principal components with high standard deviation were selected for subsequent analysis (top 13 PCs for Cx3cr1+ progenitors dataset, top 14 PCs for HSC-derived Flt3+ progenitors dataset). Dimensionality reduction was performed using either Uniform Manifold Approximation and Projection (UMAP) (Becht et al., 2018) or K-nearest-neighbor-based Network graph drawing Layout (KNetL) (Khodadadi-Jamayran and Tsirigos, 2020). KNetL borrows concepts from graph theory, utilizing force-directed graph drawing to organize cell communities in a structural visualization. KNetL finds K-nearest-neighboring cells for every cell and creates a link (edge) between the root cell and its neighbors by calculating their cell-cell distance, creating a graph/network. This graph is then used to create a force-directed layout. The coordinates of the force-compacted nodes are extracted and unpacked using secondary dimensionality reduction with UMAP. The force (resolution) of the system can be modified by adjusting the zoom parameter, the number of nearest neighbors evaluated per root. The KNetL zoom parameter was optimized over numerous iterations for maximal resolution and chosen to equal 300 for the analysis of both datasets. Clustering was then performed using the iClust function, implemented in iCellR based on PhenoGraph (Levine et al., 2015), with dist.method = “euclidean” and sensitivity=300. The latter parameter was also optimized to yield distinct and homogeneous clusters in both datasets. Numbering was assigned to identified clusters at random. The clustering shown in the manuscript was performed using both transcripts and ADT. Similar results were obtained with clustering on transcripts only.
Marker genes for each cluster were determined by unbiased differential gene expression as the average of cells within a cluster compared to the average of all other cells. Genes that achieved a minimum fold change 2 and adjusted p-value (t-test) less than 0.1 were selected as marker genes (Table S2 & S6). The predominant cell types composing each cluster were determined by unbiased cell type prediction and manual curation. Table S3 is a summary of all information that factored into final cluster cell type designations. Table S4 lists the top 25 hits of unbiased cell type prediction (the output of iCellR cell.type.pred function), which takes all of a cluster’s marker genes with minimum base mean = 0.2, calculates their cumulative expression in each population of a reference dataset as the sum of log2 normalized expression values for these genes (“CumExp-Cl. #”), and ranks them by cumulative expression. The number of marker genes (MGs) for each cluster that fit the criteria for inclusion in unbiased cell type prediction is listed in column headers of each section of Table S4. The following datasets from Immgen (https://www.immgen.org/) were used as references in unbiased cell type prediction: “microarray” = GSE15907 and “RNA-seq” = GSE109125 (http://rstats.immgen.org/DataPage/). Where indicated, genes matching to proliferation signature (Table S4, “Proliferation Signature” tab) were subtracted from marker genes list before running cell type prediction. Heatmaps were generated for top marker genes of each cluster (top 10 markers by differential expression fold change and base mean >0.2, with duplicate selection suppressed) and informative ADTs. Clusters in heatmaps were arranged by Euclidean distance.
Cellular proliferation analysis (Figure 3F & 6F) was performed using the iCellR cell.cycle function, with classification of cells into “Proliferation” or “Random” groups based on if a cell’s gene expression matches better to a proliferation signature geneset (Table S4) or an equally-sized set of random genes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Computational and statistical analysis of CITE-Seq and transposon barcoding results is described in the respective sections above. Comparison of different experimental groups (Fig. 4C, 5C and 7E–F) was done by Mann-Whitney test using Prizm 9 software (GraphPad).
Supplementary Material
Table S2. Differentially expressed genes in CITE-Seq analysis of Flt3+ HSC progeny. Related to Figure 3.
Shown are expression values of genes that were differentially expressed across cell clusters defined by KNetL analysis (Fig. 3).
Table S3. Designation of cell clusters defined by KNetL analysis of CITE-Seq datasets. Related to Figures 3 and 6.
Shown are cell clusters in the CITE-Seq datasets of Flt3+ HSC progeny (Pdzk1ip1-CreER tracing, Fig. 3) and of differentiating Cx3cr1+ progenitors (Cx3cr1CreER tracing, Fig. 6). The expression of representative transcription factors, lineage-specific genes, phenotypic markers (defined by ADT and/or transcripts) and proliferative gene signature (high, low or absent) is indicated. Also shown are two best matches to reference cell populations in the ImmGen microarray or RNA-Seq transcriptome datasets. Where indicated, the comparison to the ImmGen microarray dataset was performed after subtracting the proliferation gene signature. The raw results of matching are shown in Table S4.
Table S4. Matching of KNetL-defined cell clusters to ImmGen datasets. Related to Figures 3 and 6.
Shown are similarity scores of KNetL-defined cell clusters described in Table S3 (Pdzk1ip1-CreER and Cx3cr1CreER tracing CITE-Seq experiments) to top 25 matching populations in the ImmGen microarray or RNA-Seq transcriptome datasets. Where indicated, the comparison to the ImmGen microarray dataset was performed after subtracting the proliferation gene signature. Genes selected to comprise the proliferation signature gene set are also shown.
Table S5. Gene expression in GFP+ and GFP− fractions of pDCs traced by FlipJump activation in Cx3cr1+ progenitors. Related to Figure 5.
R26FlipJump Cx3cr1CreER-YFP mice were induced with tamoxifen, and the GFP+ and GFP− fractions of pDCs were sorted from three independent mice and analyzed by bulk RNA sequencing (RNA-Seq). Shown are normalized transcript values (excluding gene models) in individual replicates of GFP+ and GFP− fractions of pDCs (data used in Figure S4).
Table S6. Differentially expressed genes in CITE-Seq analysis of Cx3cr1+ progenitors. Related to Figure 6.
Shown are expression values of genes that were differentially expressed across cell clusters defined by KNetL analysis (Fig. 6).
Table S1. List of transposon integration sites in this study. Related to Figures 1, 2, 4 and 5.
Shown are genomic coordinates of the TTAA integration site and the presence (1) or absence (0) in the indicated cell populations from individual animals. Integration sites retrieved from experimental tracing of DCs (Fig. 2, 4 and 5) and from technical analysis of barcode retrieval from monocytes (Fig. S1E–G) are listed in separate tabs. Integration sites that were detected in control amplification from C57BL/6 DNA (n=3) are indicated with an asterisk.
Highlights.
FlipJump allows Cre-inducible DNA barcoding and clonal cell tracing in naive mice
Lineage tracing reveals cDC and pDC development from Cx3cr1+ progenitors
DNA barcoding suggests shared clonal origin of pDC and cDC1 subsets
CITE-Seq identifies a hierarchy of pDC development from Cx3cr1+ DC progenitors
Acknowledgments
Supported by the NIH grants AI072571, AG049074, AI115382 and AI128730 (B.R), AI158808 (J.I.), HL145997 and CA009161 (J.N.P) and AI100853 (C.M.L.), and by Damon Runyon postdoctoral fellowship DRG 2408–20 (N.M.A.). We acknowledge the use of resources provided by NYU Genome Technology Center (GTC), NYU Applied Bioinformatics Facility Laboratories (ABL) and the NYU High Performance Computing Facility (HPCF). GTC and ABL are Shared Resources partially supported by NIH grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
M.S. and P.S. are inventors on a patent related to the CITE-Seq method. They are current employees of 10x Genomics, Inc., but were not affiliated with it or any other commercial entity at the time of the work described in this manuscript. B.R. is an advisor for Related Sciences and a co-founder of Danger Bio, which are not related to this work. Other authors declare no competing interests.
References
- Allman D, Dalod M, Asselin-Paturel C, Delale T, Robbins SH, Trinchieri G, Biron CA, Kastner P, and Chan S (2006). Ikaros is required for plasmacytoid dendritic cell differentiation. Blood 108, 4025–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, and Wishart DS (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44, W147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagadia P, Huang X, Liu TT, Durai V, Grajales-Reyes GE, Nitschke M, Modrusan Z, Granja JM, Satpathy AT, Briseno CG, et al. (2019). An Nfil3-Zeb2-Id2 pathway imposes Irf8 enhancer switching during cDC1 development. Nat Immunol 20, 1174–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balan S, Arnold-Schrauf C, Abbas A, Couespel N, Savoret J, Imperatore F, Villani AC, Vu Manh TP, Bhardwaj N, and Dalod M (2018). Large-Scale Human Dendritic Cell Differentiation Revealing Notch-Dependent Lineage Bifurcation and Heterogeneity. Cell Rep 24, 1902–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On L, Birnberg T, Lewis KL, Edelson BT, Bruder D, Hildner K, Buer J, Murphy KM, Reizis B, and Jung S (2010). CX3CR1+ CD8alpha+ dendritic cells are a steady-state population related to plasmacytoid dendritic cells. Proc Natl Acad Sci U S A 107, 14745–14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol [DOI] [PubMed]
- Buechler MB, Kim KW, Onufer EJ, Williams JW, Little CC, Dominguez CX, Li Q, Sandoval W, Cooper JE, Harris CA, et al. (2019). A Stromal Niche Defined by Expression of the Transcription Factor WT1 Mediates Programming and Homeostasis of Cavity-Resident Macrophages. Immunity 51, 119–130 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabeza-Cabrerizo M, van Blijswijk J, Wienert S, Heim D, Jenkins RP, Chakravarty P, Rogers N, Frederico B, Acton S, Beerling E, et al. (2019). Tissue clonality of dendritic cell subsets and emergency DCpoiesis revealed by multicolor fate mapping of DC progenitors. Sci Immunol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadinanos J, and Bradley A (2007). Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res 35, e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrelha J, Meng Y, Kettyle LM, Luis TC, Norfo R, Alcolea V, Boukarabila H, Grasso F, Gambardella A, Grover A, et al. (2018). Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 554, 106–111. [DOI] [PubMed] [Google Scholar]
- Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG, et al. (2008). Transcription factor E2–2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 135, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran L, Ferrero I, Vremec D, Lucas K, Waithman J, O’Keeffe M, Wu L, Wilson A, and Shortman K (2003). The lymphoid past of mouse plasmacytoid cells and thymic dendritic cells. J Immunol 170, 4926–4932. [DOI] [PubMed] [Google Scholar]
- Crozat K, Guiton R, Guilliams M, Henri S, Baranek T, Schwartz-Cornil I, Malissen B, and Dalod M (2010). Comparative genomics as a tool to reveal functional equivalences between human and mouse dendritic cell subsets. Immunol Rev 234, 177–198. [DOI] [PubMed] [Google Scholar]
- Cytlak U, Resteu A, Pagan S, Green K, Milne P, Maisuria S, McDonald D, Hulme G, Filby A, Carpenter B, et al. (2020). Differential IRF8 Transcription Factor Requirement Defines Two Pathways of Dendritic Cell Development in Humans. Immunity 53, 353–370 e358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, and Kioussis D (2003). Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol 33, 314–325. [DOI] [PubMed] [Google Scholar]
- de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, and Chua SC Jr. (2005). Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest 115, 3484–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker JD, Rhee C, Hu Z, Lee BK, Lee J, Iyer VR, Ehrlich LIR, Georgiou G, Tucker HO, and Ippolito GC (2018). Lymphoid origin of a lineage of intrinsically activated plasmacytoid dendritic cell in mice and humans. BioRxiv
- Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, and Littman DR (2013). Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature 494, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Wu X, Li G, Han M, Zhuang Y, and Xu T (2005). Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122, 473–483. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dress RJ, Dutertre CA, Giladi A, Schlitzer A, Low I, Shadan NB, Tay A, Lum J, Kairi M, Hwang YY, et al. (2019). Plasmacytoid dendritic cells develop from Ly6D(+) lymphoid progenitors distinct from the myeloid lineage. Nat Immunol 20, 852–864. [DOI] [PubMed] [Google Scholar]
- Dursun E, Endele M, Musumeci A, Failmezger H, Wang SH, Tresch A, Schroeder T, and Krug AB (2016). Continuous single cell imaging reveals sequential steps of plasmacytoid dendritic cell development from common dendritic cell progenitors. Sci Rep 6, 37462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, Cumano A, and Geissmann F (2006). A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311, 83–87. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Jung S, and Littman DR (2003). Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, and Ley K (2010). Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh HS, Ceribelli M, Matos I, Lazarovici A, Bussemaker HJ, Lasorella A, Hiebert SW, Liu K, Staudt LM, and Reizis B (2014). ETO family protein Mtg16 regulates the balance of dendritic cell subsets by repressing Id2. J Exp Med 211, 1623–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh HS, Cisse B, Bunin A, Lewis KL, and Reizis B (2010). Continuous expression of the transcription factor e2–2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity 33, 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, Kretzer NM, Briseno CG, Durai V, Bagadia P, et al. (2015). Batf3 maintains autoactivation of Irf8 for commitment of a CD8alpha(+) conventional DC clonogenic progenitor. Nat Immunol 16, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grajkowska LT, Ceribelli M, Lau CM, Warren ME, Tiniakou I, Nakandakari Higa S, Bunin A, Haecker H, Mirny LA, Staudt LM, and Reizis B (2017). Isoform-Specific Expression and Feedback Regulation of E Protein TCF4 Control Dendritic Cell Lineage Specification. Immunity 46, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, and Yona S (2014). Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman BC, Miller JP, Nikbakht N, Gerstein R, and Allman D (2006). Mouse plasmacytoid dendritic cells derive exclusively from estrogen-resistant myeloid progenitors. Blood 108, 878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helft J, Anjos-Afonso F, van der Veen AG, Chakravarty P, Bonnet D, and Reis e Sousa C (2017). Dendritic Cell Lineage Potential in Human Early Hematopoietic Progenitors. Cell Rep 20, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TS, and Painter MW (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Herman JS, Sagar, and Grun D (2018). FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nature methods 15, 379–386. [DOI] [PubMed] [Google Scholar]
- Hulsen T, de Vlieg J, and Alkema W (2008). BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen SEW, and Nerlov C (2019). Haematopoiesis in the era of advanced single-cell technologies. Nat Cell Biol 21, 2–8. [DOI] [PubMed] [Google Scholar]
- Kaitani A, Izawa K, Maehara A, Isobe M, Takamori A, Matsukawa T, Takahashi M, Yamanishi Y, Oki T, Yamada H, et al. (2018). Leukocyte mono-immunoglobulin-like receptor 8 (LMIR8)/CLM-6 is an FcRgamma-coupled receptor selectively expressed in mouse tissue plasmacytoid dendritic cells. Sci Rep 8, 8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ (2002). BLAT--the BLAST-like alignment tool. Genome Res 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kester L, and van Oudenaarden A (2018). Single-Cell Transcriptomics Meets Lineage Tracing. Cell Stem Cell 23, 166–179. [DOI] [PubMed] [Google Scholar]
- Khodadadi-Jamayran A, Pucella JN, Zhou H, Doudican N, Carucci J, Heguy A, Reizis B, and Tsirigos A (2020). iCellR: Combined Coverage Correction and Principal Component Alignment for Batch Alignment in Single-Cell Sequencing Analysis. BioRxiv
- Khodadadi-Jamayran A, and Tsirigos A (2020). Graph Drawing-based Dimensionality Reduction to Identify Hidden Communities in Single-Cell Sequencing Spatial Representation. BioRxiv
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Breton G, Oliveira TY, Zhou YJ, Aljoufi A, Puhr S, Cameron MJ, Sekaly RP, Nussenzweig MC, and Liu K (2015). Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J Exp Med 212, 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Zhou YJ, Ma W, Zhang W, Aljoufi A, Luh T, Lucero K, Liang D, Thomsen M, Bhagat G, et al. (2017). Lineage specification of human dendritic cells is marked by IRF8 expression in hematopoietic stem cells and multipotent progenitors. Nat Immunol 18, 877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015). Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, Klinakis A, Charo IF, Jung S, Gommerman JL, et al. (2011). Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leylek R, Alcantara-Hernandez M, Lanzar Z, Ludtke A, Perez OA, Reizis B, and Idoyaga J (2019). Integrated Cross-Species Analysis Identifies a Conserved Transitional Dendritic Cell Population. Cell Rep 29, 3736–3750 e3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liggett LA, and Sankaran VG (2020). Unraveling Hematopoiesis through the Lens of Genomics. Cell 182, 1384–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DS, Kan A, Gao J, Crampin EJ, Hodgkin PD, and Naik SH (2018). DiSNE Movie Visualization and Assessment of Clonal Kinetics Reveal Multiple Trajectories of Dendritic Cell Development. Cell Rep 22, 2557–2566. [DOI] [PubMed] [Google Scholar]
- Lin DS, Tian L, Tomei S, Amann-Zalcenstein D, Baldwin TM, Weber TS, Schreuder J, Stonehouse OJ, Rautela J, Huntington ND, et al. (2021). Single-cell analyses reveal the clonal and molecular aetiology of Flt3L-induced emergency dendritic cell development. Nat Cell Biol 23, 219–231. [DOI] [PubMed] [Google Scholar]
- Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, Chu FF, Randolph GJ, Rudensky AY, and Nussenzweig M (2009). In vivo analysis of dendritic cell development and homeostasis. Science 324, 392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Waskow C, Liu X, Yao K, Hoh J, and Nussenzweig M (2007). Origin of dendritic cells in peripheral lymphoid organs of mice. Nat Immunol 8, 578–583. [DOI] [PubMed] [Google Scholar]
- Loschko J, Rieke GJ, Schreiber HA, Meredith MM, Yao KH, Guermonprez P, and Nussenzweig MC (2016). Inducible targeting of cDCs and their subsets in vivo. J Immunol Methods 434, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach N, Gillessen S, Wilson SB, Sheehan C, Mihm M, and Dranoff G (2000). Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res 60, 3239–3246. [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl M, Drechsler M, Jansen Y, Neideck C, Noels H, Faussner A, Soehnlein O, Weber C, and Doring Y (2015). Evaluation of the BDCA2-DTR Transgenic Mouse Model in Chronic and Acute Inflammation. PLoS One 10, e0134176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. [Google Scholar]
- Mildner A, and Jung S (2014). Development and function of dendritic cell subsets. Immunity 40, 642–656. [DOI] [PubMed] [Google Scholar]
- Naik SH, Perie L, Swart E, Gerlach C, van Rooij N, de Boer RJ, and Schumacher TN (2013). Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature 496, 229–232. [DOI] [PubMed] [Google Scholar]
- Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, Lahoud MH, O’Keeffe M, Shao QX, Chen WF, et al. (2005). Cutting edge: generation of splenic CD8+ and CD8- dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol 174, 6592–6597. [DOI] [PubMed] [Google Scholar]
- Naik SH, Sathe P, Park HY, Metcalf D, Proietto AI, Dakic A, Carotta S, O’Keeffe M, Bahlo M, Papenfuss A, et al. (2007). Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol 8, 1217–1226. [DOI] [PubMed] [Google Scholar]
- Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, and Manz MG (2007). Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol 8, 1207–1216. [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, and Gan WB (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei W, Feyerabend TB, Rossler J, Wang X, Postrach D, Busch K, Rode I, Klapproth K, Dietlein N, Quedenau C, et al. (2017). Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 548, 456–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelayo R, Hirose J, Huang J, Garrett KP, Delogu A, Busslinger M, and Kincade PW (2005). Derivation of 2 categories of plasmacytoid dendritic cells in murine bone marrow. Blood 105, 4407–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perie L, Duffy KR, Kok L, de Boer RJ, and Schumacher TN (2015). The Branching Point in Erythro-Myeloid Differentiation. Cell 163, 1655–1662. [DOI] [PubMed] [Google Scholar]
- Pucella JN, Upadhaya S, and Reizis B (2020). The Source and Dynamics of Adult Hematopoiesis: Insights from Lineage Tracing. Annu Rev Cell Dev Biol 36, 529–550. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizis B (2019). Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 50, 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues PF, Alberti-Servera L, Eremin A, Grajales-Reyes GE, Ivanek R, and Tussiwand R (2018). Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat Immunol 19, 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathe P, Vremec D, Wu L, Corcoran L, and Shortman K (2013). Convergent differentiation: myeloid and lymphoid pathways to murine plasmacytoid dendritic cells. Blood 121, 11–19. [DOI] [PubMed] [Google Scholar]
- Sawai CM, Babovic S, Upadhaya S, Knapp DJ, Lavin Y, Lau CM, Goloborodko A, Feng J, Fujisaki J, Ding L, et al. (2016). Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity 45, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenner SM, Madan V, Busch K, Tietz A, Laufle C, Costa C, Blum C, Fehling HJ, and Rodewald HR (2010). Fate mapping reveals separate origins of T cells and myeloid lineages in the thymus. Immunity 32, 426–436. [DOI] [PubMed] [Google Scholar]
- Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HR, Schreuder J, Lum J, Malleret B, Zhang S, Larbi A, Zolezzi F, et al. (2015). Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol 16, 718–728. [DOI] [PubMed] [Google Scholar]
- Schraml BU, van Blijswijk J, Zelenay S, Whitney PG, Filby A, Acton SE, Rogers NC, Moncaut N, Carvajal JJ, and Reis e Sousa C (2013). Genetic tracing via DNGR-1 expression history defines dendritic cells as a hematopoietic lineage. Cell 154, 843–858. [DOI] [PubMed] [Google Scholar]
- Scott CL, Soen B, Martens L, Skrypek N, Saelens W, Taminau J, Blancke G, Van Isterdael G, Huylebroeck D, Haigh J, et al. (2016). The transcription factor Zeb2 regulates development of conventional and plasmacytoid DCs by repressing Id2. J Exp Med 213, 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov AA, Dudekula DB, and Ko MS (2005). A web-based tool for principal component and significance analysis of microarray data. Bioinformatics 21, 2548–2549. [DOI] [PubMed] [Google Scholar]
- Shigematsu H, Reizis B, Iwasaki H, Mizuno S, Hu D, Traver D, Leder P, Sakaguchi N, and Akashi K (2004). Plasmacytoid dendritic cells activate lymphoid-specific genetic programs irrespective of their cellular origin. Immunity 21, 43–53. [DOI] [PubMed] [Google Scholar]
- Sichien D, Scott CL, Martens L, Vanderkerken M, Van Gassen S, Plantinga M, Joeris T, De Prijck S, Vanhoutte L, Vanheerswynghels M, et al. (2016). IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity 45, 626–640. [DOI] [PubMed] [Google Scholar]
- Siegemund S, Shepherd J, Xiao C, and Sauer K (2015). hCD2-iCre and Vav-iCre mediated gene recombination patterns in murine hematopoietic cells. PLoS One 10, e0124661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, et al. (2016). The UCSC Genome Browser database: 2016 update. Nucleic Acids Res 44, D717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM (2012). Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 30, 1–22. [DOI] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P (2017). Simultaneous epitope and transcriptome measurement in single cells. Nature methods 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, Smibert P, and Satija R (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, and Camargo FD (2014). Clonal dynamics of native haematopoiesis. Nature 514, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, and Colonna M (2015). The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 15, 471–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang KH, Li S, Khodadadi-Jamayran A, Jen J, Han H, Guidry K, Chen T, Hao Y, Fedele C, Zebala JA, et al. (2021). Combined Inhibition of SHP2 and CXCR1/2 Promotes Antitumor T-cell Response in NSCLC. Cancer Discov [DOI] [PMC free article] [PubMed]
- Tian L, Tomei S, Schreuder J, Weber TS, Amann-Zalcenstein D, Lin DS, Tran J, Audiger C, Chu M, Jarratt A, et al. (2021). Clonal multi-omics reveals Bcor as a negative regulator of emergency dendritic cell development. Immunity 54, 1338–1351 e1339. [DOI] [PubMed] [Google Scholar]
- Upadhaya S, Sawai CM, Papalexi E, Rashidfarrokhi A, Jang G, Chattopadhyay P, Satija R, and Reizis B (2018). Kinetics of adult hematopoietic stem cell differentiation in vivo. J Exp Med 215, 2815–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Buggenum JA, Gerlach JP, Eising S, Schoonen L, van Eijl RA, Tanis SE, Hogeweg M, Hubner NC, van Hest JC, Bonger KM, and Mulder KW (2016). A covalent and cleavable antibody-DNA conjugation strategy for sensitive protein detection via immuno-PCR. Sci Rep 6, 22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert A, Weichand B, Sekar D, Sha W, Hahn C, Mora J, Ley S, Essler S, Dehne N, and Brune B (2012). HIF-1alpha is a negative regulator of plasmacytoid DC development in vitro and in vivo. Blood 120, 3001–3006. [DOI] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, and Craig NL (2011). A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A 108, 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Differentially expressed genes in CITE-Seq analysis of Flt3+ HSC progeny. Related to Figure 3.
Shown are expression values of genes that were differentially expressed across cell clusters defined by KNetL analysis (Fig. 3).
Table S3. Designation of cell clusters defined by KNetL analysis of CITE-Seq datasets. Related to Figures 3 and 6.
Shown are cell clusters in the CITE-Seq datasets of Flt3+ HSC progeny (Pdzk1ip1-CreER tracing, Fig. 3) and of differentiating Cx3cr1+ progenitors (Cx3cr1CreER tracing, Fig. 6). The expression of representative transcription factors, lineage-specific genes, phenotypic markers (defined by ADT and/or transcripts) and proliferative gene signature (high, low or absent) is indicated. Also shown are two best matches to reference cell populations in the ImmGen microarray or RNA-Seq transcriptome datasets. Where indicated, the comparison to the ImmGen microarray dataset was performed after subtracting the proliferation gene signature. The raw results of matching are shown in Table S4.
Table S4. Matching of KNetL-defined cell clusters to ImmGen datasets. Related to Figures 3 and 6.
Shown are similarity scores of KNetL-defined cell clusters described in Table S3 (Pdzk1ip1-CreER and Cx3cr1CreER tracing CITE-Seq experiments) to top 25 matching populations in the ImmGen microarray or RNA-Seq transcriptome datasets. Where indicated, the comparison to the ImmGen microarray dataset was performed after subtracting the proliferation gene signature. Genes selected to comprise the proliferation signature gene set are also shown.
Table S5. Gene expression in GFP+ and GFP− fractions of pDCs traced by FlipJump activation in Cx3cr1+ progenitors. Related to Figure 5.
R26FlipJump Cx3cr1CreER-YFP mice were induced with tamoxifen, and the GFP+ and GFP− fractions of pDCs were sorted from three independent mice and analyzed by bulk RNA sequencing (RNA-Seq). Shown are normalized transcript values (excluding gene models) in individual replicates of GFP+ and GFP− fractions of pDCs (data used in Figure S4).
Table S6. Differentially expressed genes in CITE-Seq analysis of Cx3cr1+ progenitors. Related to Figure 6.
Shown are expression values of genes that were differentially expressed across cell clusters defined by KNetL analysis (Fig. 6).
Table S1. List of transposon integration sites in this study. Related to Figures 1, 2, 4 and 5.
Shown are genomic coordinates of the TTAA integration site and the presence (1) or absence (0) in the indicated cell populations from individual animals. Integration sites retrieved from experimental tracing of DCs (Fig. 2, 4 and 5) and from technical analysis of barcode retrieval from monocytes (Fig. S1E–G) are listed in separate tabs. Integration sites that were detected in control amplification from C57BL/6 DNA (n=3) are indicated with an asterisk.
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. The accession number is listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-mouse CD8a (PerCP-Cy5.5 (clone 53–6.7) | eBioscience | Cat# 45–0081-82 |
| Rat anti-mouse CD11b ((eFlour 450) (clone M1/70) | eBioscience | Cat# 48–0112-82 |
| Rat anti-mouse CD11b (Biotin) (clone M1/70) | eBioscience | Cat# 13–0112-81 |
| Rat anti-mouse CD11b (APC-Cy7) (clone M1/70) | eBioscience | Cat# 47–0112-82 |
| Rat anti-mouse CD11b (PE-TR) (clone M1/70) | Invitrogen | Cat# RM2817 |
| Rat anti-mouse /human CD11b (Alexa Flour 700) (clone M1/70) | BioLegend | Cat# 101222 |
| Rat anti-mouse /human CD11b (BV 785) (clone M1/70) | BioLegend | Cat# 101243 |
| Armenian hamster anti-mouse CD11c (APC-eFlour 780) (clone N418) | eBioscience | Cat# 47–0114-80 |
| Armenian hamster anti-mouse CD11c (PerCP-Cy5.5) (clone N418) | BioLegend | Cat# 117327 |
| Armenian hamster anti-mouse CD11c (BUV 737) (clone HL3) | BD Biosciences | Cat# 612797 |
| Rat anti-mouse CD19 (Biotin) (clone 1D3) | eBioscience | Cat# 13–0193-82 |
| Anti-mouse CD19 (APC-Alexa700) (clone 1D3) | BD Biosciences | Cat #: 557958 |
| Anti-mouse CD19 (eFluor450) (clone 1D3) | eBioscience | Cat #: 48–0193-82 |
| Anti-mouse CD3 (APC-Alexa700) (clone 17A2) | BioLegend | Cat #: 100215 |
| Anti-mouse CD3 (eFluor 450) (clone 17A2) | eBioscience | Cat #: 48–0032-82 |
| Rat anti-mouse CD24 (PE) (clone M1/69) | eBioscience | Cat# 12–0242-81 |
| Rat anti-mouse CD24 (BUV 395) (clone M1/69) | BD Biosciences | Cat# 744471 |
| Rat anti-mouse CD48 (AF700) (clone HM48–1) | eBioscience | Cat# 103426 |
| (Rat anti-mouse CD93 (Biotin) (clone AA4.1) | eBioscience | Cat#13–5892-82 |
| Rat anti-mouse CD115 (c-fms) (PE) (clone AFS98) | eBioscience | Cat# 12–1152-81 |
| Rat anti-mouse CD117 (c-Kit) (APC-eFlour 780) (clone 2B8) | eBioscience | Cat# 47–1171-80 |
| Rat anti-mouse CD117 (c-Kit) (APC) (clone 2B8) | eBioscience | Cat# 17–1171-83 |
| Rat anti-mouse CD127 (BV 605) (clone A7R34) | BioLegend | Cat# 135041 |
| Rat anti-mouse CD127 (PE-Cy7) (clone A7R34) | BioLegend | Cat# 135014 |
| Rat anti-mouse CD135 (Flt3) (APC) (clone A2F10) | eBioscience | Cat# 17–1351-82 |
| Rat anti-mouse CD135 (Flt3) (APC) (clone A2F10) | eBioscience | Cat# 14–1351-82 |
| Rat anti-mouse CD135 (Flt3) (Biotin) (clone A2F10) | BioLegend | Cat# 135307 |
| Rat anti-mouse CD150 (SLAM) (PE-Cy7) (clone TC15–12F12.2) | BioLegend | Cat# 115914 |
| Rat anti-mouse CD172a (Sirp-a) APC-Fire750 (clone P84) | BioLegend | Cat # 144029 |
| Rat anti-mouse CD205 (PE-Cy7) (clone NLDC-145) | BioLegend | Cat# 138209 |
| Rat anti-mouse CD317 (PDCA-1) (PE-Cy7) (clone 927) | eBioscience | Cat# 25–3175-82 |
| Rat anti-mouse CD317 (PDCA-1) (APC) (clone 927) | Millipore Sigma | Cat# MABF2047 |
| Rat anti-mouse SiglecH (BV711) (clone 440C) | BD Biosciences | Cat#747671 |
| Rat anti-mouse SiglecH (APC) (clone 551) | BioLegend | Cat# 129612 |
| Rat anti-mouse SiglecH (PerCP-Cy5.5) (clone 551) | BioLegend | Cat# 129614 |
| Mouse anti-mouse Cx3r1 (FITC) (clone SA011F11) | BioLegend | Cat# 149019 |
| Mouse anti-mouse Cx3r1 (PE-Cy7) (clone SA011F11) | BioLegend | Cat# 149016 |
| Mouse anti-mouse Cx3r1 (BV785) (clone SA011F11) | BioLegend | Cat# 149029 |
| Rat anti-mouse/human CD45R/B220 (Alexa Flour700) (clone RA3–6B2) | BioLegend | Cat# 103232 |
| Rat anti-mouse/human CD45R/B220 (PE-Cy7) (clone RA3–6B2) | eBioscience | Cat# 25–0452-81 |
| Rat anti-mouse/human CD45R/B220 (Biotin) (clone RA3–6B2) | eBioscience | Cat# 13–0425-85 |
| Rat anti-mouse/human CD45R/B220 (eFlou 450) (clone RA3–6B2) | eBioscience | Cat# 48–0452-80 |
| Rat anti-mouse/human CD45R/B220 (BV 650) (clone RA3–6B2) | BioLegend | Cat# 103241 |
| Rat anti-mouse/human CD45R/B220 (Biotin) (clone RA3–6B2) | eBioscience | Cat# 13–0425-85 |
| Armenian hamster anti-mouse TCR beta (eFluor 450) (clone H57–597) | eBioscience | Cat# 48–5961-80 |
| Armenian hamster anti-mouse TCR beta (Biotin) (clone H57–597) | eBioscience | Cat# 13–5961-82 |
| Rat anti-mouse ESAM (PE) (clone 1G8) | eBioscience | Cat# 12–5852-81 |
| Rat anti-mouse MHC Class II (I-A/I-E) (AF700) (clone M5/114.15.2) | eBioscience | Cat# 56–5321-82 |
| Rat anti-mouse TER-119 (eFlour 450) (clone TER-119) | eBioscience | Cat# 48–5921-82 |
| Rat anti-mouse TER119 (Biotin) (clone TER-119) | eBioscience | Cat# 13–5921-82 |
| Rat anti-mouse Ly-6A/E (Sca-1) (PerCP-Cy5.5) (clone D7) | eBioscience | Cat# 45–5981-82 |
| Rat anti-mouse Ly-6G (Gr-1) (eFlour 450) (clone RB6–8C5) | eBioscience | Cat# 48–5931-82 |
| Rat anti-mouse Ly-6G (Alexa Flour 700) (clone RB6–8C5) | BioLegend | Cat# 127621 |
| Rat anti-mouse Ly-6G (Gr-1) (Biotin) (clone RB6–8C5) | eBioscience | Cat# 13–5931-82 |
| Rat anti-mouse Ly-6C (PercP) (clone 927) | BioLegend | Cat# 128027 |
| Rat anti-mouse Ly6C BV510 (clone HK1.4) | BioLegend | Cat # 128033 |
| Rat anti-mouse Ly-6D (eFlour 450) (clone 49-H4) | eBioscience | Cat# 48–5974-80 |
| Mouse anti-mouse NK1.1 (Biotin) (clone PK136) | eBioscience | Cat# 13–5941-85 |
| Rat anti-mouse Xcr1 PerCP/Cy5.5 (clone ZET) | BioLegend | Cat # 148208 |
| Streptavidin eFluor 450 Conjugate | eBioscience | Cat# 48–4317-82 |
| PE Steptavidin | Biolegend | Cat#405203 |
| Streptavidin Alexa Fluor 647 | ThermoFisher Scientific | Cat# S21374 |
| TruStain FcX™ (anti-mouse CD16/32) Antibody | BioLegend | Cat# 101320 |
| CITE-seq antibodies, see Table S7 | ||
| Experimental Models: Organisms/Strains | ||
| Mouse strains | ||
| Pdzk1ip1-CreER | Reizis Lab (NYU) | (Sawai et al., 2016) |
| R26 FlipJump | Reizis Lab (NYU) | this study |
| Cx3cr1CreER-YFP: B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/WganJ | Jackson Laboratory, stock 021160 | (Parkhurst et al., 2013) |
| R26 CreER | Dr. Thomas Ludwig, Ohio State Univ. | (de Luca et al., 2005) |
| R26LSL-Tom: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratory, stock 007914 | (Madisen et al., 2010) |
| B6.Cg-Tg(CD2-icre)4Kio/J | Jackson Laboratory, stock 008520 | (de Boer et al., 2003) |
| B6N.129P2-Cx3cr1tm3(DTR)Litt/J | Jackson Laboratory, stock 025629 | (Diehl et al., 2013) |
| C57BL/6 | Taconic Biosciences | Black 6 |
| Cell lines | ||
| Mouse cell line: B16-FLT3L | Reizis lab (NYU) | (Mach et al., 2000) |
| Mouse cell line: 129/B6 ES cells | Dr. Victor Lin, Columbia Unversity Irving Medical Center | N/A |
| Oligonucleotides | ||
| FlipJump ES screening primer forward: GGACTAGGGCTGCGTGAGTCTCTGA | This study | N/A |
| FlipJump ES screening primer reverse: TGGCGTTACTATGGGAACATACGTC | This study | N/A |
| Linker common (long strand) GACCCGGGAGATCTGAATTCAGTGGCACAGCAGTT AGGGGGCTGGTCG | (Sun et al., 2014) | N/A |
| Blunt end linker /5Phos/CGACCAGCCC/3AmMO/ | (Sun et al., 2014) | N/A |
| CG end linker /5Phos/CGCGACCAGCCC/3AmMO/ | (Sun et al., 2014) | N/A |
| GATC end linker /5Phos/GATCCGACCAGCCC/3AmMO/ | (Sun et al., 2014) | N/A |
| AGCT end linker /5Phos/AGCTCGACCAGCCC/3AmMO/ | This study | N/A |
| Linker primer1: GACCCGGGAGATCTGAATTC | (Sun et al., 2014) | N/A |
| Linker primer3: GAATTCAGTGGCACAGCAGTTAGG | This study | N/A |
| MiSeq primer TP_P5: AATGATACGGCGACCACCGAGATCTACACA CACTCTTTCCC | This study | N/A |
| MiSeq primer TP_P7: CAAGCAGAAGACGGCATACGAGATGTGACT GGAGT | This study | N/A |
| LM_5TR_inver_A2: /5Biosg/AGATAATCATGCGTCATTTTGACTCAC | This study | N/A |
| LM_5TR_inver_B2: AGCGGCGACTGAGATGTCC | This study | N/A |
| LM_3TR_inverA: /5Biosg/TCGATATACAGACCGATAAAACACATG | This study | N/A |
| LM_3TR_inverB: ATGCGTCAATTTTACGCATGATTATC | This study | N/A |
| 5TR index primer for MiSeq sequencing: ACACTCTTTCCCTACACGACGCTCTTCCGATCTXXXXXXXCGACGGATTCGCGCTAT (Xs denote a sample-specific barcode) | This study | N/A |
| 3TR index primer for MiSeq sequencing: ACACTCTTTCCCTACACGACGCTCTTCCGATCTXXX XXXXCGTACGTCACAATATGATT(Xs denote a sample-specific barcode) | This study | N/A |
| Genotyping primer: Flipjump-F: ACGTGCTGGTTATTGTGCTGT | This study | N/A |
| Genotyping primer: Flipjump-R GTTCATGCGGAACCTGTACAT | This study | N/A |
| Genotyping primer: WT-F AAAGTCGCTCTGAGTTGTTAT | This study | N/A |
| Genotyping primer: WT-R GGAGCGGGAGAAATGGATATG | This study | N/A |
| Genotyping primer: Cre-F GGACATGTTCAGGGATCGCCAGGCG | This study | N/A |
| Genotyping primer: Cre-R GCATAACCAGTGAAACAGCATTGCTG | This study | N/A |
| CITE-Seq ADT additive primer: CCTTGGCACCCGAGAATTCC | https://cite-seq.com/protocols/ | (Stoeckius et al., 2018) |
| CITE-Seq HTO additive primer: GTGACTGGAGTTCAGACGTGTGCTC | https://cite-seq.com/protocols/ | (Stoeckius et al., 2018) |
| CITE-Seq TruSeq Small RNA RPIx primer: CAAGCAGAAGACGGCATACGAGXXXXXXXXGTGAC TGGAGTTCCTTGGCACCCGAGAATTCCA (Xs denote a sample-specific barcode) | https://cite-seq.com/protocols/ | (Stoeckius et al., 2018) |
| CITE-Seq TruSeq D7xx primer: CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGC (Xs denote a sample-specific barcode) | https://cite-seq.com/protocols/ | (Stoeckius et al., 2018) |
| CITE-Seq SI PCR primer: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTC | https://cite-seq.com/protocols/ | (Stoeckius et al., 2018) |
| Recombinant DNA | ||
| pCMV-HAhyPBase | Wellcome Trust Sanger Institute | (Yusa et al., 2011) |
| pPB-LR5 | Wellcome Trust Sanger Institute | (Cadinanos and Bradley, 2007) |
| pPB-RL5 | Wellcome Trust Sanger Institute | (Cadinanos and Bradley, 2007) |
| Rosa26 mT/mG | Addgene | Plasmid #17787 |
| pRosa26-TG | Addgene | Plasmid #40026 |
| pCMS-EGFP | Clontech | Plasmid #6101–1 |
| Software and Algorithms | ||
| FlowJo v9.9.5 and v10.1 | FlowJo, LLC | https://www.flowjo.com/ |
| Prism 8 | GraphPad | https://www.graphpad.com/ |
| samtools (v.1.3.1) | (Li and Durbin, 2009) | http://samtools.sourceforge.net/ |
| GenomicAlignments (v.1.10.1) | (Lawrence et al., 2013) | https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html |
| Bedtools2 (v2.26.0) | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| Bowtie2 (v2.2.9) | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| UCSC mm10 Known Gene Annotation Database (v.3.4) | (Speir et al., 2016) | https://bioconductor.org/packages/release/data/annotation/html/TxDb.Mmusculus.UCSC.mm10.knownGene.html |
| BLAT | (Kent, 2002) | https://genome.ucsc.edu/cgi-bin/hgBlat |
| Cutadapt (v1.18) | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| BioVenn | (Hulsen et al., 2008) | http://www.biovenn.nl |
| iCellR | (Khodadadi-Jamayran et al., 2020) | https://github.com/rezakj/iCellR |
| KNetL | (Khodadadi-Jamayran and Tsirigos, 2020) | https://github.com/rezakj/iCellR |
| ImmGen Reference Datasets | (Heng and Painter, 2008) | http://rstats.immgen.org/DataPage/ |
| NIA Array | (Sharov et al., 2005) | https://lgsun.irp.nia.nih.gov/ANOVA/) |
| HeatMapper | (Babicki et al., 2016) | http://www.heatmapper.ca/expression/ |
| STAR aligner (v2.5.0c) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| HTSeq (v 0.6.0) | (Anders et al., 2015) | https://htseq.readthedocs.io/en/master/history.html |
| Picard (v.1.126) | Picard Toolkit, the Broad Institute | http://broadinstitute.github.io/picard |
| DEseq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html |
| Reagents and Kits | ||
| REPLI-g Mini Kit (100) | QIAGEN | Cat# 150025 |
| QIAamp DNA Micro Kit (50) | QIAGEN | Cat# 56304 |
| QIAamp DNA Mini Kit (50) | QIAGEN | Cat# 51304 |
| Nucleospin Tissue XS (Takara) | Macherey-Nagel | Cat#740901 |
| QIAquick PCR Purification Kit (250) | QIAGEN | Cat# 28106 |
| TA Cloning kit | ThermoFisher Scientific | Invitrogen Cat# K2020–40 |
| Dynabeads kilobaseBINDER kit | ThermoFisher Scientific | Invitrogen Cat# 60101 |
| MspI | NEB | Cat# R0106S |
| HaeIII | NEB | Cat# R0108S |
| DpnII | NEB | Cat# R0543S |
| HindIII-HF | NEB | Cat# R3104S |
| XmaI | NEB | Cat# R0180S |
| SalI | NEB | Cat# R3138S |
| SpeI-HF | NEB | Cat# R3133S |
| BglII | NEB | Cat# R0144S |
| XhoI | NEB | Cat# R0146S |
| MluI | NEB | Cat# R0198S |
| EcoRV-HF | NEB | Cat# R3195S |
| T4 DNA Ligase | NEB | Cat# M0202S |
| Phusion High-Fidelity DNA Polymerase | NEB | Cat# M0530S |
| PfuTurbo Hotstart DNA Polymerase | Agilent Technologies | Cat# 600320 |
| Deoxynucleotide Solution Mix | NEB | Cat# N0447S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma-Aldrich | Cat# T5648–5G |
| SUNFLOWER SEED OIL | Sigma-Aldrich | Cat# S5007–250ML |
| RBC Lysis Buffer (10X) | BioLegend | Cat# 420301 |
| DMEM, high glucose, no glutamine | Thermo Fisher Scientific | Cat# 11960069 |
| Penicillin-Streptomycin (10,000 U/ml) (Gibco) | Thermo Fisher Scientific | Cat# 15140122 |
| MEM Non-Essential amino acids solution (100x) | Thermo Fisher Scientific | Cat# 11140076 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985023 |
| Sodium Pyruvate (100mM) | Thermo Fisher Scientific | Cat# 11360070 |
| L-Glutamine (200 mM) | Thermo Fisher Scientific | Cat# 25030164 |
| HyClone ADCF-Mab media | Thermo Fisher Scientific | Cat# SH3034902 |
| S-clone SF-O3 | Iwai North America | Cat# S-clone SF-O3 |
| BSA, Molecular biology grade | NEB | Cat# B9000S |
| Fixable Viability Dye eFluor 506 | eBioscience | Cat# 65–0866-14 |
| DAPI | Sigma-Aldrich | Cat# D8417 |
| ESGRO recombinant mouse LIF protein | Millipore Sigma | Cat# ESG1106 |
| TAT-CRE Recombinase | Millipore Sigma | Cat# SCR508 |
| Recombinant Murine SCF | PeproTech | Cat# 250–03 |
| Recombinant Human TPO | PeproTech | Cat# 300–18 |
| Recombinant Murine Flt3-Ligand | PeproTech | Cat# 250–31L |
| Streptavidin microbeads | Miltenyi Biotec | Cat# 130–048-101 |
| Diphtheria Toxin | Millipore Sigma | Cat# D0564 |
| Fetal Bovine Serum, charcoal stripped | Thermo Fisher Scientific | Cat# 12676029 |
| Deposited Data and Code | ||
| FlipJump transposon sequencing, CITE-seq, bulk RNA-seq | This study | GEO: GSE193733 |
| FlipJump transposon sequencing code | This study | https://doi.org/10.5281/zenodo.5879278 |