

Abstract
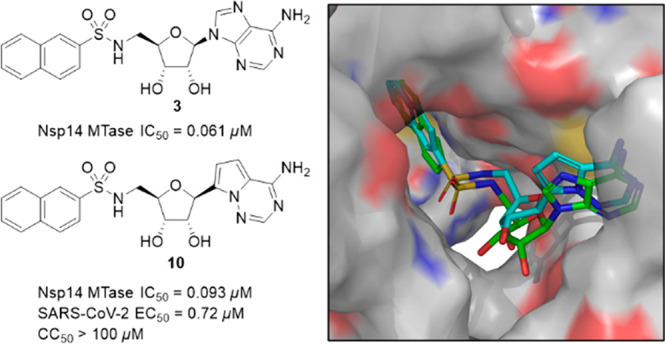
Taking advantage of the uniquely constricted active site of SARS-CoV-2 Nsp14 methyltransferase, we have designed bisubstrate inhibitors interacting with the SAM and RNA substrate binding pockets. Our efforts have led to nanomolar inhibitors including compounds 3 and 10. As a prototypic inhibitor, compound 3 also has an excellent selectivity profile over a panel of human methyltransferases. Remarkably, C-nucleoside 10 exhibits high antiviral activity and low cytotoxicity, leading to a therapeutic index (CC50/EC50) greater than 139. Furthermore, a brief metabolic profiling of these two compounds suggests that they are less likely to suffer from major metabolic liabilities. Moreover, computational docking studies point to protein–ligand interactions that can be exploited to enhance inhibitory activity. In short, discovery of inhibitor 10 clearly demonstrates that potent and selective anti-SARS-CoV-2 activity can be achieved by targeting the Nsp14 methyltransferase. Therefore, the current work strongly supports the continued pursuit of Nsp14 methyltransferase inhibitors as COVID-19 therapeutics.
Keywords: SARS-CoV-2, Nsp14, bisubstrate inhibitor, methyltransferase inhibitor, antiviral
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is a member of the genus Betacoronavirus and is an enveloped, single-stranded positive-sense RNA virus.1 As the causative agent of the current pandemic of coronavirus disease 2019 (COVID-19), SARS-CoV-2 poses a global health threat unseen in modern history. While the availability of vaccines, antibodies, and antiviral drugs has offered hope to end this devastating pandemic, recent emergence and rapid spread of more infectious variants call for continued efforts to develop effective therapeutics to combat SARS-CoV-2 infection.
Coronaviruses, such as SARS-CoV-2, possess unusually large RNA genome encoding four structural and 16 nonstructural proteins (Nsp1–16), providing ample opportunity for small molecule drug discovery.2,3 Among these viral proteins, the RNA-dependent RNA polymerase (RdRp, Nsp12) and the main protease Mpro (also known as 3CLpro, Nsp5) have been extensively pursued, leading to the FDA-approved antiviral agents remdesivir,4 molnupiravir,5 and nirmatrelvir.6
An antiviral strategy that also holds promise is targeting the S-adenosylmethionine (SAM)-dependent viral RNA cap methylation machinery.7 The RNA 5′-cap structure is methylated to ensure viral translation/replication and to evade host immune surveillance.8 In SARS-CoV-2, the RNA 5′-cap structure is sequentially methylated at the N7 position of the guanosine by the guanine N7-methyltransferase (N7-MTase, Nsp14)9 and the 2′-O position of the ribose of the first RNA nucleotide by the 2′-O-methyltransferase (2′-O-MTase, Nsp16).10 As a bifunctional protein, Nsp14 also contains an exoribonuclease domain, which is important for proofreading, mismatch excision, and drug resistance.11
Nsp14 MTase is an attractive drug discovery target because mutational analysis has shown that the MTase activity is key for SARS-CoV virus replication/transcription and viability.9,12 Furthermore, the Nsp14 N7-MTase activity of SARS-CoV-2 is required for host translational shutdown and evasion of the innate immune response.13,14 Known SARS-CoV Nsp14 MTase inhibitors are either obtained from screening10,15 or are adenine dinucleoside SAM analogs.16 Many inhibitors of SARS-CoV-2 Nsp14 MTase that have been identified through screening of commercially available libraries have been found to be relatively weak.17−19 There are potent S-adenosylhomocysteine (SAH)/SAM analogs that have been identified from a collection of other SAM-dependent MTase inhibitors20 or rationally designed.21,22 In addition, virtual screenings have been performed to identify potential Nsp14 MTase inhibitors.23 However, most of these inhibitors are highly polar and no anti-SARS-CoV-2 activity has been reported. During the preparation of this manuscript, potent and selective bisubstrate inhibitors that contained an sulfonamide functionality were reported; however, no antiviral activity was disclosed.24
Structural studies have revealed that the MTase domain of the SARS-CoV Nsp14 protein is distinct from the canonical Rossmann fold.11,25 Indeed, the MTase domain of the SARS-CoV Nsp14-Nsp10 complex (PDBs 5C8S and 5C8T) features a uniquely constricted SAM and RNA substrate binding site, in which the guanine ring of the cap structure (GpppA) is placed in a narrow pocket and positioned in close proximity to SAM for the methyl group transfer. Newly reported cryo-EM structures of SARS-CoV-2 Nsp14 MTase,26,27 albeit in an apo form, also have a similarly constrained MTase active site, while the active sites of human mRNA cap guanine-N7 methyltransferase (RNMT) (PDB 3EPP) and human mRNA cap 2′-O-Me methyltransferase (CMTR1) (PDB 4N49) are relatively more open (Figure S1).
This constricted spatial arrangement provides an ideal opportunity to design bisubstrate inhibitors of Nsp14 MTase. As a well-validated drug design strategy, bisubstrate inhibitors generally entail structural moieties from two substrates involved in a biochemical reaction. Nsp14 MTase brings together SAM and an RNA substrate and transfers a methyl group from SAM to the N7 atom of the guanine, providing a template for design of bisubstrate inhibitors targeting the Nsp14 MTase (Figure 1). In our design, we retained the adenosine moiety from SAM, albeit with a 5′-amino terminus, which served as a handle for further chemical modifications. To mimic the guanine ring moiety of the RNA substrate, a simple aromatic ring, such as 2-naphthalene, was initially used. The adenosine moiety and the aromatic ring were connected via a structurally varied linker (represented by X). Conceivably, an amino acid side chain (represented by R) could be introduced to mimic homocysteine in SAH. While such a side chain has been featured in many natural and synthetic MTase inhibitors20−22 and is believed to have major contribution to the inhibitory activity,20,22 its highly polar nature would negatively impact cell permeability and pose difficulty for further drug development. Therefore, we decided to focus on compounds without polar side chains.
Figure 1.
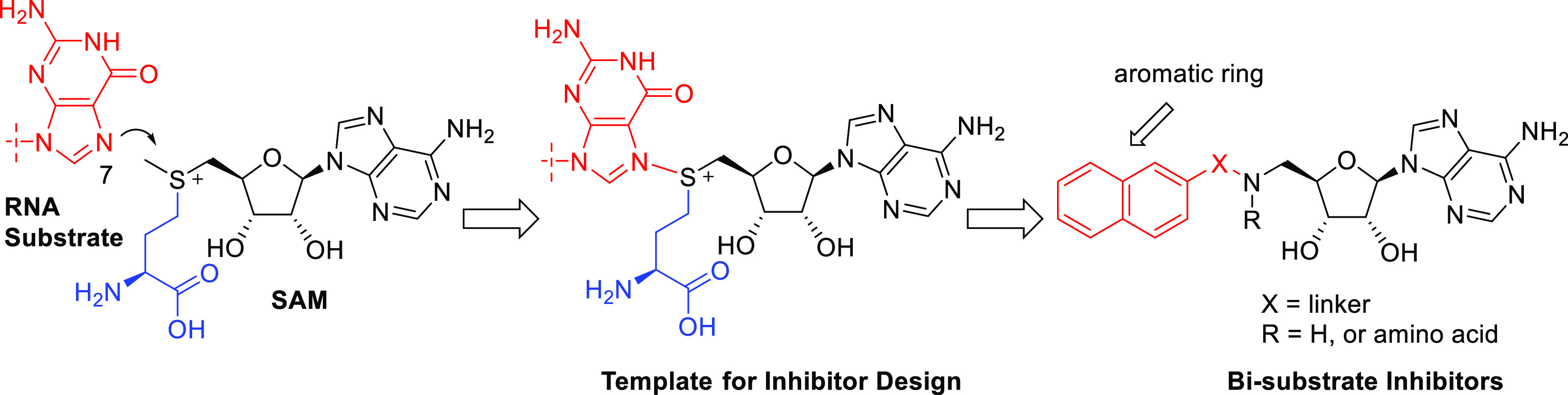
Design of bisubstrate inhibitors of Nsp14 MTase.
To guide the structure–activity relationship (SAR) studies, we established a biochemical assay in which a LC–MS/MS method was used to monitor the production of SAH, the byproduct of the methylation reaction. Typical dose–response curves for representative compounds are shown in Figure S2. This assay offered an unbiased method to evaluate a compound’s inhibitory activity. Our initial attempts to use alternative SAH detecting methods, such as an AptaFluor SAH MTase assay kit, showed difficulty in differentiating SAH from our adenosine-derived inhibitors, leading to erratic results. Sinefungin (IC50 = 260 nM, Table 1), a potent pan-MTase inhibitor, was used as a control and benchmark for MTase inhibition.
Table 1. Bisubstrate Inhibitors of SARS-CoV-2 Nsp14 MTase.


NA, not active, <15% inhibition at 50 μM.
NT, nontoxic, >95% cell viability at 50 μM. IC50, EC50 and CC50 values were determined in at least duplicate and the averages were reported.
Since the linker was a crucial element of bisubstrate inhibitors, we initiated our SAR studies by exploring several common functionalities as potential linkers (Table 1). First, when a secondary amine was used to connect the adenosine moiety and 2-naphthalene, the resulting compound 1 had an IC50 value of 9.6 μM, demonstrating the feasibility of designing bisubstrate inhibitors of Nsp14 MTase. Compound 2, which contained an amide linker, displayed enhanced inhibitory activity (IC50 = 2.7 μM). Remarkably, installation of a sulfonamide functionality as a linker led to compound 3, which was >150-fold more active than compound 1. With an IC50 value of 61 nM, compound 3 was a promising starting point for further SAR studies. To that end, we briefly examined the effect of 2-naphthalene modifications. Replacement with a 7-quinoline ring gave compound 4, which showed inhibitory activity comparable to that of compound 1, suggesting that this minor structural change was tolerated. However, use of a 6-(2-chloroquinoline) ring (5, Table 1) led to a 7-fold reduction in inhibitory activity, probably due to the relatively bulky chloro group. Furthermore, we prepared N6-acetylated compound 6, which had an IC50 value of 48 nM, suggesting that acylation at N6 was tolerated. Taken together, we proved that highly active bisubstrate inhibitors of SARS-CoV-2 Nsp14 methyltransferase could be obtained by connecting the adenosine moiety and a simply aromatic ring via a sulfonamide linker.
To assess the anti-SARS-CoV-2 activity of these compounds, we used A549 cells overexpressing ACE2 and TMPRSS2 (A549 AT) in an immunofluorescence (IF)-based assay. Remdesivir,28 an FDA-approved polymerase inhibitor, was used as a control. As expected, sinefungin with a polar amino acid side chain failed to show any activity. Disappointingly, compounds 1–5 at 50 μM showed no significant antiviral activity, likely due to poor cell permeability resulting from high polarity of sulfonamides and nucleosides. To mitigate high polarity and improve cell permeability, we initially prepared 7, compound 3’s di-isobutyrate prodrug (Table 1), a strategy that has been used in clinical stage HCV antiviral agents balapiravir and mericitabine as well as newly approved molnupiravir.29 While compound 7 showed antiviral activity, we had difficulty determining its EC50 value most likely due to compound 7’s low solubility in assay solutions. As a result, we synthesized the less lipophilic diacetate prodrug 8, which was more soluble and possessed moderate antiviral activity (EC50 = 53 μM). Encouraged by this result, we also prepared 9, compound 6’s diacetate prodrug, which as expected exhibited antiviral activity similar to that of compound 8. In addition, both compounds showed minimal cytotoxicity at 100 μM. Taken together, we demonstrated that we were able to elicit antiviral activity via a prodrug approach.
One drawback of nucleoside/nucleotide-based inhibitors is their metabolically labile glycosidic bond. To address this issue, we examined C-nucleosides30 in which the glycosidic bond is replaced by a metabolically stable C–C bond. To that end, we prepared compound 10, in which a pyrrolo[2,1-f][1,2,4]triazin-4-amine ring (the nucleobase of remdesivir) was used as a nucleobase. Compound 10 possessed an IC50 value comparable to that of 3, suggesting that C-nucleosides were well tolerated. Strikingly, compound 10 exhibited excellent antiviral activity (EC50 = 0.72 μM) with a therapeutic index (TI = CC50/EC50) greater than 139 (Table 1). However, compound 11, the diacetate prodrug of 10, was less potent and more toxic than 10. While there was no definite explanation to these findings, they might be linked to cell lines used in the antiviral assay, a possibility that will be investigated. Taken together, these results suggested that metabolic stability played a crucial role in antiviral activity and that the impact of the prodrug might depend on the nature of the parent nucleosides. These observations will be examined in future SAR studies.
To complement the enzymatic assay, we also performed thermal shift assays on compounds 3, 6, and 10, three nanomolar inhibitors of Nsp14 MTase, as well as sinefungin, a reference. When tested at 200 μM, sinefungin modestly increased the melting temperature (Table S1). In contrast, compounds 3, 6, and 10 drastically stabilized the Nsp14 protein judged by the remarkable shifts in melting temperature (≥8 °C). Furthermore, an inverse relationship was observed between the enzymatic IC50 values and the shifts in melting temperature (Table S1). These results confirmed the direct interactions between our inhibitors and the Nsp14 protein.
To evaluate selectivity over human MTases, we screened compound 3, the prototypic bisubstrate inhibitor, against a panel of 10 human MTases including histone lysine, protein arginine, and DNA and RNA MTases. At 10 μM, compound 3 showed minimal inhibition of this diverse set of human MTases (Table S2), indicative of an excellent selectivity profile. These preliminary results bolstered our premise that it was feasible to design selective Nsp14 MTase because of its unique structure in contrast to the canonical Rossmann fold.
To identify factors that might contribute to compound 10’s improved antiviral activity, we also assessed compounds 3 and 10 for their in vitro metabolic liabilities including plasma stability and liver S9 stability, and permeability in parallel artificial membrane permeability assay (PAMPA)31 (Table 2). Liver S9 fraction was used because the liver is the predominant site of drug metabolism and the S9 fraction contains both microsomal and cytosolic enzymes, including a wide variety of Phase I and Phase II enzymes. To facilitate metabolic transformations, nicotinamide adenine dinucleotide phosphate (NADPH) and uridine 5′-diphosphoglucuronic acid (UDPGA) were supplemented as cofactors in Phase I oxidation and II glucuronidation, respectively. Compound 3 was remarkably stable in mouse and human plasma. It was also resistant to Phase I oxidation and Phase II glucuronidation in both human and mouse liver S9 fractions. In comparison, compound 10 was similarly stable in plasma and liver S9 fractions except that it was surprisingly labile in the mouse to S9 Phase I NADPH-dependent oxidation. This metabolic instability was likely linked to compound 10’s unnatural pyrrolo[2,1-f][1,2,4]triazin-4-amine nucleobase. Nonetheless, to our best knowledge, no major metabolic liabilities associated with this nucleobase in the context of remdesivir have been reported.32 Compounds 3 and 10 were also assessed in PAMPA, which revealed that their passive diffusion rates were equally low. This finding was not unexpected because of the polar nature of these two nucleoside derivatives. This result also supported our investigation of nucleoside prodrugs to enhance cell membrane permeability. Overall, we were not able to find clues about factors that could account for the superior antiviral activity exhibited by compound 10 over compound 3. Nonetheless, our metabolic profiling indicated that neither compound suffered from major metabolic liabilities in human plasma and liver S9 fractions. In particular, compound 10 will serve as an excellent template to further improve antiviral activity and to mitigate metabolic liabilities.
Table 2. In Vitro Metabolic Stabilities and Permeability of Selected Compounds1.
| plasma
stability t1/2 (h), n = 3 |
human liver S9 stability t1/2 (min), n = 2 |
mouse liver S9 stability t1/2 (min), n = 2 |
|||||
|---|---|---|---|---|---|---|---|
| compound | human | mouse | phase I3 | phase II4 | phase I3 | phase II4 | PAMPA permeability Pe2 (10–6 cm/s), n = 6 |
| 3 | >245 | >245 | 674 ± 136 | >456 | 273 ± 23 | >456 | 0.2 ± 0.1 |
| 10 | >245 | >245 | 842 ± 210 | >456 | 28.9 ± 0.3 | >456 | 0.2 ± 0.1 |
| Verapamil | 19.2 ± 0.8 | 5.7 ± 0.1 | |||||
| Umbelliferone | 3.0 ± 0.05 | 0.8 ± 0.005 | |||||
Data are presented as mean ± SD.
Pe, Apparent permeability coefficient.
CYP enzyme cofactor: NADPH.
Cofactor: UDPGA.
No decrease in remaining percentage was observed at the end of incubation (24 h).
No decrease in remaining percentage was observed at the end of incubation (45 min).
To gain insight into key protein–ligand interactions that contributed to inhibitory activity, compounds were docked into the MTase domain of the SARS-CoV-2 Nsp14 structure (PDB 7EGQ).26 For compound 3, its adenine ring was sandwiched between Cys387 and Phe367, and it formed two hydrogen bonds with Tyr368 (Figure 2). Furthermore, the 3′-hydroxyl group of the ribose ring had an additional hydrogen bond with Asp352. At the same time, the sulfonamide linker projected the naphthalene ring into the guanine (of RNA substrate) binding pocket and allowed it to engage a π–π stacking with Phe426, a residue that is important for Nsp14 MTase activity.12,25 Furthermore, a similar π–π interaction was also observed for compounds 1, 4, and 5 (Figure S3). Remarkably, Phe426 formed a π–π interaction with the guanine ring of GpppA (an RNA substrate mimic) in PDB 5C8S, a SARS-CoV Nsp14 cocrystal structure in which both GpppA and SAH were present in the MTase active site. Thus, this binding mode supported our design strategy in which a simple 2-naphthalene was used to mimic the guanine ring of the RNA substrate. Docking of compound 10 revealed that its pyrrolo[2,1-f][1,2,4]triazin-4-amine nucleobase maintained the hydrogen bonds elicited by the original adenine ring (C Figure 2). However, examination of the top pose revealed no apparent π–π interaction between the naphthalene ring and Phe426 even though compound 10 displayed a binding configuration very similar to compound 3. Instead, the sulfonamide hydrogen formed a hydrogen bond with Asn386, a key residue for N7-methylation as judged by mutation analysis.12,25 Interestingly, a similar interaction (between the amide hydrogen of compound 2 and Asn386) was also identified in the top pose of compound 2 (Figure S3). Because compounds 3 and 10 possessed similar biochemical IC50 values (Table 1) and docking scores (−10.4 and −10.5 for compounds 3 and 10, respectively), our docking experiment suggested that a π–π interaction with Phe426 and a hydrogen bond with Asn386 were key contributors to the enzymatic activity and therefore will be further investigated in our future SAR efforts. In contrast to compounds 1–5, docking of compound 6 produced no pose in which its acetylated adenine ring interacted with the protein via hydrogen bonding with Tyr368. This finding was not unexpected because the narrow adenine binding groove was incapable of accommodating the bulkier acetylated adenine ring of compound 6. An alternative pose yielded a poor docking score of −6.32, which apparently could not account for the high biochemical inhibitory activity of compound 6. It is possible that protein flexibility can allow compound 6 to bind in a manner similar to that of compounds 3, which is currently being examined through crystallographic experiments. Taken together, docking studies revealed binding modes that supported our design strategy and will be used to guide the design of new bisubstrate inhibitors of Nsp14 MTase.
Figure 2.

Docking of nucleosides into the active site of SARS-CoV-2 Nsp14 MTase (PDB 7EGQ). (A) Proposed binding modes of compound 3 (green) and 10 (cyan). (B) Protein–ligand interaction diagram of compound 3. (C) Protein–ligand interaction diagram of compound 10.
The synthesis of adenosine derivatives began with commercially available 2′,3′-O-isopropylideneadenosine (12), which was converted into the corresponding azide 13 (Scheme 1).33 Reduction of azide 13 under hydrogenation conditions gave primary amine 14, which was converted into secondary amine 15, amide 16, and sulfonamides 17–19 under conventional conditions. Subsequent deprotection gave compounds 1–5 in 44–92% yield. To selectively obtain N6-acetylated adenosine derivative 6, protected azide 13 was treated with acetic anhydride to give acetamide 20 in 67% yield. Reduction of azide 20 gave primary amine 21, which was then coupled with 2-naphthalenesulfonyl chloride to give sulfonamide 22. Subsequent deprotection gave final product N6-acetylated derivative 6.
Scheme 1. Synthesis of Adenosine Derivatives.

Reagents and conditions: (a) i. DPPA, DBU, dioxane; ii. NaN3, TBAI, 15-crown-5, dioxane, reflux; (b) H2, Pd/C, MeOH; (c) for 15, 2-naphthaldehyde, NaBH4, MeOH, 5 h, 88%; for 16, 2-naphthoyl chloride, NEt3, CH2Cl2, rt, 18 h, 93%; for 17–19, sulfonyl chloride, NEt3, CH2Cl2, rt, 18 h, 33–93%; (d) TFA/H2O (4:1), rt, 3 h, 38–92%; (e) Ac2O, pyridine, rt, 52–67%; (f) H2, Pd/C, EtOH, 18 h, 69%; (g) 2-naphthalenesulfonyl chloride, NEt3, CH2Cl2, rt, 18 h, 41–44%; (h) isobutyric anhydride, pyridine, rt, 2 h, 29%; (i) H2, Pd/C, dioxane, 18 h.
We next synthesized di-isobutyrate and diacetate prodrugs 7 and 8, which were prepared by direct treatment of sulfonamide 3 with isobutyric anhydride and acetic anhydride, respectively (Scheme 1). Similarly, we also prepared triacetate 9, a diacetate prodrug of 6-acetamide 6, whose synthesis started with common intermediate azide 13. Deprotection followed by treatment with acetic anhydride at 50 °C gave triacetate 24 in 56% yield after a tetra-acetate byproduct was removed by column chromatography. Reduction of the azide functionality in 24 was accomplished by hydrogenation in dioxane, a solvent that was chosen to mitigate undesired deacetylation in methanol. The resulting primary amine 25 was treated with 2-naphthalenesulfonyl chloride to give triacetate derivative 9. The positions of acetylation were confirmed by the heteronuclear multiple bond correlation (HMBC) NMR experiments.
The synthesis of the pyrrolo[2,1-f][1,2,4- triazine] C-nucleosides is depicted in Scheme 2. The free amine in 7-iodopyrrolo[2,1-f][1,2,4]triazin-4-amine 27 was protected using TMSCl, followed by addition of PhMgCl, which removed acidic protons.34 A magnesium-iodide exchange promoted by iPrMgCl·LiCl followed by addition of ribonolactone 26 gave glycosylated hemiacetal 28 in 42% yield (Scheme 2). The resulting hemiacetal 26 underwent anomeric reduction with triethylsilane and BF3·OEt2 to give the stereoselective β-anomer 29.35,36 The benzyl groups were removed to give the corresponding C-nucleoside 30 in 91% yield. After protection of the hydroxyl groups, the resulting nucleoside 31 was converted into the corresponding primary amine 33 following the same synthetic sequence described for compound 12. Amine 33 was coupled with 2-naphthalenesulfonyl chloride to give sulfonamide 34, which was then deprotected to give C-nucleoside sulfonamide 10. Finally, sulfonamide 10 was treated with acetic anhydride to afford its diacetate prodrug 11.
Scheme 2. Synthesis of C-Nucleosides.
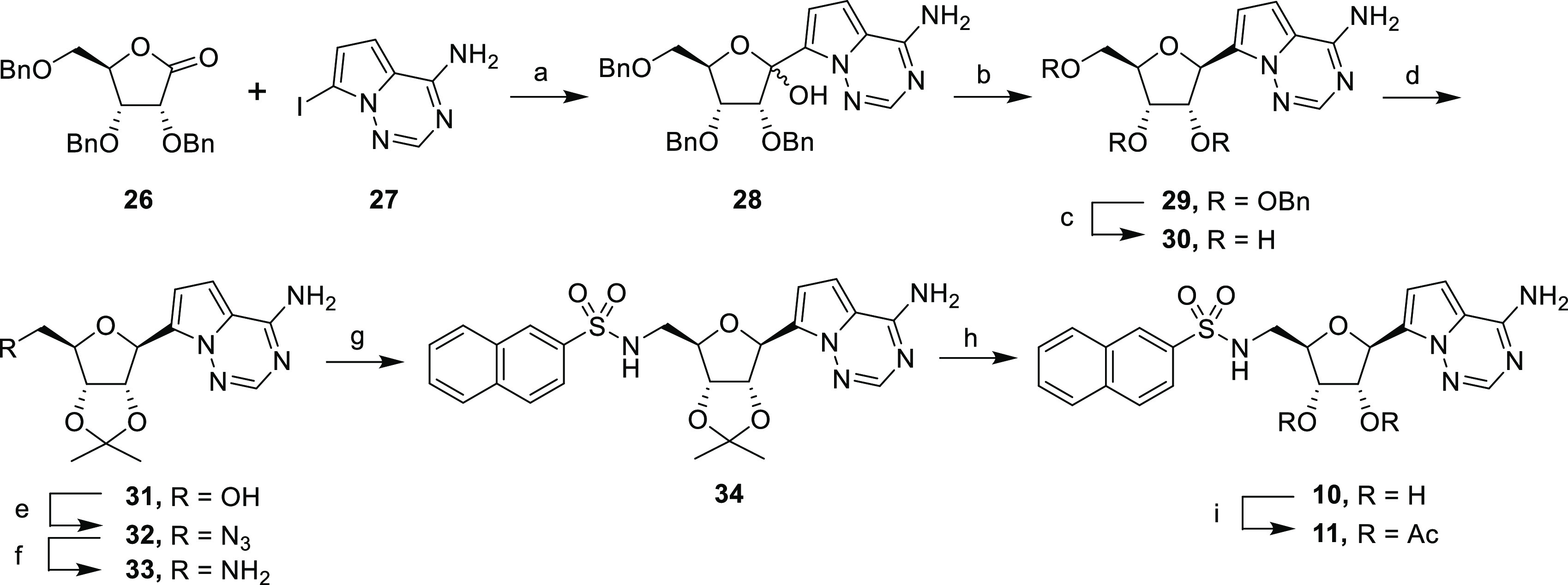
Reagents and conditions: (a) TMSCl, PhMgCl, iPrMgCl·LiCl, THF, 0 °C, 4 h, 42%; (b) BF3·OEt2, Et3SiH, CH2Cl2, 0 °C, 4 h, 87%; (c) BBr3, CH2Cl2, – 78 °C, 2 h, 91%; (d) 2,2-dimethoxypropane, H2SO4, acetone, rt, 87%; (e) i. DPPA, DBU, dioxane, rt, 16 h; ii. NaN3,15-crown-5, 110 °C, 18 h, 82% over two steps; (f) H2, Pd/C, EtOH, 18 h, 95%; (g) 2-naphthalenesulfonyl chloride, NEt3, CH2Cl2, rt, 18 h, 61%; (h) TFA/H2O (4:1), rt, 3 h, 59%; (i) Ac2O, pyridine, rt, 2 h, 35%.
In summary, the uniquely constricted active site of SARS-CoV-2 Nsp14 MTase has allowed us to design bisubstrate inhibitors, which simultaneously engage residues in the SAM and RNA substrate binding pockets. Our preliminary effort has led to nanomolar inhibitors including 3 and 10, validating our bisubstrate drug design. Successful identification of potent inhibitors has also demonstrated that it is feasible to obtain inhibitors devoid of a polar amino acid moiety. Furthermore, a brief metabolic profiling of these two compounds suggests that they are less likely to have major metabolic liabilities. Moreover, compound 3, as a representative of these bisubstrate inhibitors, displays an excellent selectivity profile over a panel of human MTases. Remarkably, use of prodrugs has led to compound 8, which showed significant, albeit moderate, antiviral activity. More importantly, we have discovered C-nucleoside 10, which exhibits high antiviral activity and low cytotoxicity, clearly proving that superior anti-SARS-CoV-2 inhibition can be accomplished by targeting the Nsp14 MTase. To our best knowledge, compound 10 is the first designed SARS-CoV-2 Nsp14 MTase inhibitor that has achieved submicromolar antiviral activity without significant cytotoxicity. Therefore, while many questions remain unanswered, identification of compound 10 represents a substantial advancement in Nsp14 MTase drug discovery and strongly supports the continued pursuit of Nsp14 MTase inhibitors as COVID-19 therapeutics.
Acknowledgments
The reported work was supported by the Center of Drug Design at the University of Minnesota. The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281.
Glossary
Abbreviations
- SARS-CoV-2
severe acute respiratory syndrome coronavirus-2
- COVID-19
coronavirus disease 2019
- Nsp
nonstructural protein
- RdRp
RNA-dependent RNA polymerase
- SAM
S-adenosylmethionine
- MTase
methyltransferase
- N7-MTase
guanine N7-methyltransferase
- 2′-O-MTase
2′-O-methyltransferase
- SAH
S-adenosylhomocysteine
- SAR
structure–activity relationship
- IF
immunofluorescence
- TI
therapeutic index
- PAMPA
parallel artificial membrane permeability assay
- NADPH
nicotinamide adenine dinucleotide phosphate
- UDPGA
uridine 5′-diphosphoglucuronic acid
- HMBC
heteronuclear multiple bond correlation.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00265.
Thermal shift assay data, human MTases counter-screening data, overall geometry of MTase active sites, dose–response curves of representative compounds, protein–ligand interaction diagrams of selected nucleosides, experimental procedures, NMR spectra of selected compounds (PDF)
The authors declare no competing financial interest.
This paper was published ASAP on July 22, 2022, with a typographical error in the title. The corrected version was reposted on August 3, 2022.
This article is made available via the ACS COVID-19 subset for unrestricted RESEARCH re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for the duration of the World Health Organization (WHO) declaration of COVID-19 as a global pandemic.
Supplementary Material
References
- Li J.; Lai S.; Gao G. F.; Shi W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600 (7889), 408–418. 10.1038/s41586-021-04188-6. [DOI] [PubMed] [Google Scholar]
- Su H.; Zhou F.; Huang Z.; Ma X.; Natarajan K.; Zhang M.; Huang Y.; Su H. Molecular Insights into Small-Molecule Drug Discovery for SARS-CoV-2. Angew. Chem., Int. Ed. Engl. 2021, 60 (18), 9789–9802. 10.1002/anie.202008835. [DOI] [PubMed] [Google Scholar]
- Pillaiyar T.; Wendt L. L.; Manickam M.; Easwaran M. The recent outbreaks of human coronaviruses: A medicinal chemistry perspective. Med. Res. Rev. 2021, 41 (1), 72–135. 10.1002/med.21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb Y. N. Remdesivir: First Approval. Drugs 2020, 80 (13), 1355–1363. 10.1007/s40265-020-01378-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed Y. Y. Molnupiravir: First Approval. Drugs 2022, 82 (4), 455–460. 10.1007/s40265-022-01684-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb Y. N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82 (5), 585–591. 10.1007/s40265-022-01692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decroly E.; Ferron F.; Lescar J.; Canard B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2012, 10 (1), 51–65. 10.1038/nrmicro2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron F.; Decroly E.; Selisko B.; Canard B. The viral RNA capping machinery as a target for antiviral drugs. Antiviral Res. 2012, 96 (1), 21–31. 10.1016/j.antiviral.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Cai H.; Pan J.; Xiang N.; Tien P.; Ahola T.; Guo D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (9), 3484–9. 10.1073/pnas.0808790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet M.; Debarnot C.; Imbert I.; Selisko B.; Snijder E. J.; Canard B.; Decroly E. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010, 6 (4), e1000863 10.1371/journal.ppat.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron F.; Subissi L.; Silveira De Morais A. T.; Le N. T. T.; Sevajol M.; Gluais L.; Decroly E.; Vonrhein C.; Bricogne G.; Canard B.; Imbert I. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (2), E162–E171. 10.1073/pnas.1718806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogando N. S.; El Kazzi P.; Zevenhoven-Dobbe J. C.; Bontes B. W.; Decombe A.; Posthuma C. C.; Thiel V.; Canard B.; Ferron F.; Decroly E.; Snijder E. J. Structure-function analysis of the nsp14 N7-guanine methyltransferase reveals an essential role in Betacoronavirus replication. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (49), e2108709118 10.1073/pnas.2108709118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J. C.; Laurent-Rolle M.; Pawlak J. B.; Wilen C. B.; Cresswell P. Translational shutdown and evasion of the innate immune response by SARS-CoV-2 NSP14 protein. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (24), e2101161118 10.1073/pnas.2101161118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan R.; Kindler E.; Cao L.; Zhou Y.; Zhang Z.; Liu Q.; Ebert N.; Zust R.; Sun Y.; Gorbalenya A. E.; Perlman S.; Thiel V.; Chen Y.; Guo D. N7-Methylation of the Coronavirus RNA Cap Is Required for Maximal Virulence by Preventing Innate Immune Recognition. mBio 2022, 13, e0366221 10.1128/mbio.03662-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouadi W.; Eydoux C.; Coutard B.; Martin B.; Debart F.; Vasseur J. J.; Contreras J. M.; Morice C.; Querat G.; Jung M. L.; Canard B.; Guillemot J. C.; Decroly E. Toward the identification of viral cap-methyltransferase inhibitors by fluorescence screening assay. Antiviral Res. 2017, 144, 330–339. 10.1016/j.antiviral.2017.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed-Belkacem R.; Sutto-Ortiz P.; Guiraud M.; Canard B.; Vasseur J. J.; Decroly E.; Debart F. Synthesis of adenine dinucleosides SAM analogs as specific inhibitors of SARS-CoV nsp14 RNA cap guanine-N7-methyltransferase. Eur. J. Med. Chem. 2020, 201, 112557. 10.1016/j.ejmech.2020.112557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S.; Mak T.; Ulferts R.; Wu M.; Deegan T.; Fujisawa R.; Tan K. W.; Lim C. T.; Basier C.; Canal B.; Curran J. F.; Drury L. S.; McClure A. W.; Roberts E. L.; Weissmann F.; Zeisner T. U.; Beale R.; Cowling V. H.; Howell M.; Labib K.; Diffley J. F. X. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of Nsp14 RNA cap methyltransferase. Biochem. J. 2021, 478 (13), 2481–2497. 10.1042/BCJ20210219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasprzyk R.; Spiewla T. J.; Smietanski M.; Golojuch S.; Vangeel L.; De Jonghe S.; Jochmans D.; Neyts J.; Kowalska J.; Jemielity J. Identification and evaluation of potential SARS-CoV-2 antiviral agents targeting mRNA cap guanine N7-Methyltransferase. Antiviral Res. 2021, 193, 105142. 10.1016/j.antiviral.2021.105142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson L. A.; Green C. J.; Lin; Petit A. P.; Gray D. W.; Cowling V. H.; Fordyce E. A. F. Development of a High-Throughput Screening Assay to Identify Inhibitors of the SARS-CoV-2 Guanine-N7-Methyltransferase Using RapidFire Mass Spectrometry. SLAS Discovery 2021, 26 (6), 749–756. 10.1177/24725552211000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota K.; Schapira M.; Perveen S.; Khalili Yazdi A.; Li F.; Chau I.; Ghiabi P.; Hajian T.; Loppnau P.; Bolotokova A.; Satchell K. J. F.; Wang K.; Li D.; Liu J.; Smil D.; Luo M.; Jin J.; Fish P. V.; Brown P. J.; Vedadi M. Probing the SAM Binding Site of SARS-CoV-2 Nsp14 In Vitro Using SAM Competitive Inhibitors Guides Developing Selective Bisubstrate Inhibitors. SLAS Discovery 2021, 26 (9), 1200–1211. 10.1177/24725552211026261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otava T.; Sala M.; Li F.; Fanfrlik J.; Devkota K.; Perveen S.; Chau I.; Pakarian P.; Hobza P.; Vedadi M.; Boura E.; Nencka R. The Structure-Based Design of SARS-CoV-2 nsp14 Methyltransferase Ligands Yields Nanomolar Inhibitors. ACS Infect. Dis. 2021, 7 (8), 2214–2220. 10.1021/acsinfecdis.1c00131. [DOI] [PubMed] [Google Scholar]
- Bobileva O.; Bobrovs R.; Kanepe I.; Patetko L.; Kalnins G.; Sisovs M.; Bula A. L.; Gri Nberga S.; Boroduskis M. R.; Ramata-Stunda A.; Rostoks N.; Jirgensons A.; Ta Rs K.; Jaudzems K. Potent SARS-CoV-2 mRNA Cap Methyltransferase Inhibitors by Bioisosteric Replacement of Methionine in SAM Cosubstrate. ACS Med. Chem. Lett. 2021, 12 (7), 1102–1107. 10.1021/acsmedchemlett.1c00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobrovs R.; Kanepe I.; Narvaiss N.; Patetko L.; Kalnins G.; Sisovs M.; Bula A. L.; Grinberga S.; Boroduskis M.; Ramata-Stunda A.; Rostoks N.; Jirgensons A.; Tars K.; Jaudzems K. Discovery of SARS-CoV-2 Nsp14 and Nsp16 Methyltransferase Inhibitors by High-Throughput Virtual Screening. Pharmaceuticals 2021, 14 (12), 1243. 10.3390/ph14121243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed-Belkacem R.; Hausdorff M.; Delpal A.; Sutto-Ortiz P.; Colmant A. M. G.; Touret F.; Ogando N. S.; Snijder E. J.; Canard B.; Coutard B.; Vasseur J. J.; Decroly E.; Debart F. Potent Inhibition of SARS-CoV-2 nsp14 N7-Methyltransferase by Sulfonamide-Based Bisubstrate Analogues. J. Med. Chem. 2022, 65 (8), 6231–6249. 10.1021/acs.jmedchem.2c00120. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Wu L.; Shaw N.; Gao Y.; Wang J.; Sun Y.; Lou Z.; Yan L.; Zhang R.; Rao Z. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (30), 9436–41. 10.1073/pnas.1508686112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L.; Yang Y.; Li M.; Zhang Y.; Zheng L.; Ge J.; Huang Y. C.; Liu Z.; Wang T.; Gao S.; Zhang R.; Huang Y. Y.; Guddat L. W.; Gao Y.; Rao Z.; Lou Z. Coupling of N7-methyltransferase and 3′-5′ exoribonuclease with SARS-CoV-2 polymerase reveals mechanisms for capping and proofreading. Cell 2021, 184 (13), 3474–3485. 10.1016/j.cell.2021.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Shi W.; Becker S. T.; Schatz D. G.; Liu B.; Yang Y. Structural basis of mismatch recognition by a SARS-CoV-2 proofreading enzyme. Science 2021, 373 (6559), 1142–1146. 10.1126/science.abi9310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Cao R.; Zhang L.; Yang X.; Liu J.; Xu M.; Shi Z.; Hu Z.; Zhong W.; Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30 (3), 269–271. 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dousson C. B. Current and future use of nucleo(s)tide prodrugs in the treatment of hepatitis C virus infection. Antivir. Chem. Chemother. 2018, 26, 2040206618756430. 10.1177/2040206618756430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambasky J.; Hocek M.; Kocovsky P. C-nucleosides: synthetic strategies and biological applications. Chem. Rev. 2009, 109 (12), 6729–64. 10.1021/cr9002165. [DOI] [PubMed] [Google Scholar]
- Chen X.; Murawski A.; Patel K.; Crespi C. L.; Balimane P. V. A novel design of artificial membrane for improving the PAMPA model. Pharm. Res. 2008, 25 (7), 1511–20. 10.1007/s11095-007-9517-8. [DOI] [PubMed] [Google Scholar]
- Xie J.; Wang Z. Can remdesivir and its parent nucleoside GS-441524 be potential oral drugs? An in vitro and in vivo DMPK assessment. Acta Pharm. Sin. B 2021, 11 (6), 1607–1616. 10.1016/j.apsb.2021.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Austin D. J. Synthesis of 5′-functionalized adenosine: suppression of cyclonucleoside formation. Tetrahedron Lett. 2001, 42 (18), 3153–3154. 10.1016/S0040-4039(01)00395-1. [DOI] [Google Scholar]
- von Keutz T.; Williams J. D.; Kappe C. O. Continuous Flow C-Glycosylation via Metal-Halogen Exchange: Process Understanding and Improvements toward Efficient Manufacturing of Remdesivir. Org. Process Res. Dev. 2020, 24 (10), 2362–2368. 10.1021/acs.oprd.0c00370. [DOI] [Google Scholar]
- Metobo S. E.; Xu J.; Saunders O. L.; Butler T.; Aktoudianakis E.; Cho A.; Kim C. U. Practical synthesis of 1 ’-substituted Tubercidin C-nucleoside analogs. Tetrahedron Lett. 2012, 53 (5), 484–486. 10.1016/j.tetlet.2011.11.055. [DOI] [Google Scholar]
- Li Q.; Lescrinier E.; Groaz E.; Persoons L.; Daelemans D.; Herdewijn P.; De Jonghe S. Synthesis and Biological Evaluation of Pyrrolo[2,1-f][1,2,4]triazine C-Nucleosides with a Ribose, 2’-Deoxyribose, and 2’,3′-Dideoxyribose Sugar Moiety. ChemMedChem. 2018, 13 (1), 97–104. 10.1002/cmdc.201700657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.