

SUMMARY
Microglia are critical in brain development and Alzheimer’s disease (AD) etiology. Down syndrome (DS) is the most common genetic developmental disorder and risk factor for AD. Surprisingly, little information is available on the impact of trisomy of human chromosome 21 (Hsa21) on microglial functions during DS brain development and in AD in DS. Using induced pluripotent stem cell (iPSC)-based organoid and chimeric mouse models, we report that DS microglia exhibit enhanced synaptic pruning function, which alters neuronal synaptic functions. In response to human brain tissue-derived pathological tau, DS microglia undergo cellular senescence and exhibit elevated type I interferon signaling. Mechanistically, knockdown of Hsa21-encoded type I interferon receptors, IFNARs, rescues the DS microglial phenotypes both during brain development and in response to pathological tau. Our findings provide in vivo evidence that human microglia respond to pathological tau by exhibiting dystrophic phenotypes. Targeting IFNARs may improve DS microglial functions and prevent senescence.
Graphical Abstract
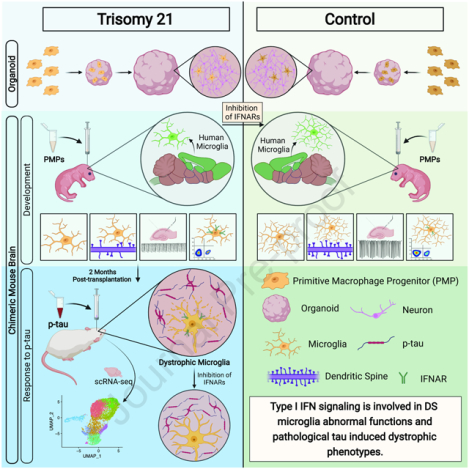
With human iPSC-based brain organoids and mouse chimeras, Jin and colleagues demonstrate that upregulated type I interferon (IFN-I) signaling in Down syndrome microglia causes elevated synaptic pruning during development and accelerated senescence, rather than massive activation/inflammation, in response to pathological tau. These phenotypes can be rescued by inhibiting IFN-I receptors.
Introduction
Down syndrome (DS), caused by trisomy of human chromosome 21 (Hsa21), is the most common genetic cause of abnormal brain development and the most common risk factor for Alzheimer’s disease (AD) (Lott and Head, 2019; Parker et al., 2010; Wiseman et al., 2015). Microglia play critical roles in maintaining CNS homeostasis and remodeling of neuronal synapses, thereby regulating synaptic plasticity, and learning and memory (Bar and Barak, 2019; Li and Barres, 2018; Wang et al., 2020). Moreover, microglial dysfunction is a central mechanism in AD etiology, and many AD risk genes are highly and sometimes exclusively expressed by microglia (Gosselin et al., 2017; Hansen et al., 2018; Holtman et al., 2017). Therefore, targeting microglia has enormous potential for improving DS brain development and treating AD. Unfortunately, the precise contributions of microglia to brain development in DS and AD in DS (henceforth referred to as DSAD) as well as underlying molecular mechanisms are poorly understood, which hampers the development of therapeutics.
Limited information is available on the influence of trisomy 21 on microglia during brain development and degeneration. A recent study using a mouse model of DS reports that microglia are activated during brain development (Pinto et al., 2020). Consistently, a study using DS postmortem human brain tissues also shows elevated microglial activation and inflammatory cytokines (Flores-Aguilar et al., 2020). Despite these characterizations at cellular levels, how trisomy of Hsa21 genes alter microglial development and functions remains largely unknown. The association between DS and AD is largely due to overexpression of the amyloid precursor protein (APP), whose gene is located on Hsa21 (Doran et al., 2017; Head et al., 2003; Lott and Head, 2019). As early as 35–40 years of age, tau pathological changes are observed in the hippocampus in DS (Head et al., 2003; Lott and Head, 2019). While the aggregation of amyloid-beta (Aβ) precedes that of tau, tau protein pathology commences in humans much sooner than was previously thought (Braak and Del Tredici, 2015). Contrary to the marked microglial activation reported in amyloidogenic mouse models (Jimenez et al., 2008; Meyer-Luehmann and Prinz, 2015), in brain tissue of AD patients, brain regions particularly relevant in AD development, such as the hippocampal formation, exhibit low and late Aβ pathology, whereas hyperphosphorylated tau (p-tau) accumulates starting early (Braak and Del Tredici, 2015; Sanchez-Mejias et al., 2016). Intriguingly, studies using AD and DSAD brain tissues showed that degenerating neuronal structures positive for p-tau invariably colocalized with severely dystrophic and senescent rather than microglial activation (Shahidehpour et al., 2021; Streit et al., 2009; Xue and Streit, 2011). In DSAD, microglial phenotypes shift with age and AD pathology, showing the presence of higher numbers of dystrophic microglia(Martini et al., 2020). Thus, the preferential accumulation of p-tau over Aβ plaques in specific brain regions could induce a different microglial response than merely activation and inflammation.
Modeling the DS-related cellular phenotypes and elucidating the molecular mechanisms underlying DS disease pathogenesis is challenging. This is because functional DS human brain tissue is relatively inaccessible, and DS mouse models often show variations and discrepancies in modeling DS-related phenotypes due to the trisomy of different subsets of Hsa21 orthologous genes (Belichenko et al., 2015; Das and Reeves, 2011; Xu et al., 2019). In addition, none of the mouse models of DS reliably reproduces Aβ or tau pathology even in aged animals (Choong et al., 2015). It is also important to note that rodent microglia cannot fully mirror the properties of human microglia (Galatro et al., 2017; Geirsdottir et al., 2019; Gosselin et al., 2017; Jiang et al., 2020). A limited overlap was also observed in microglial genes regulated during aging and neurodegeneration between mice and humans, indicating that human and mouse microglia age differently under normal and diseased conditions (Friedman et al., 2018; Galatro et al., 2017). These findings argue for using species-specific research tools to investigate microglial functions in human brain development, aging, and neurodegeneration (Smith and Dragunow, 2014).
Recent advances in stem cell technology have led to the efficient generation of microglia from human induced pluripotent stem cells (hiPSCs) (Abud et al., 2017; Brownjohn et al., 2018; Jiang et al., 2020; Pandya et al., 2017), providing an unlimited source of human microglia to study their pathophysiology. We have recently developed new hiPSC-based microglia-containing cerebral organoid and chimeric mouse brain models (Jiang et al., 2020; Xu et al., 2021; Xu et al., 2020), in which hiPSC-derived microglia undergo maturation and develop appropriate functions. In this study, we demonstrate that DS hiPSC-derived microglia show defective development and functions. Moreover, in microglial chimeric brains, DSAD human brain tissue-derived pathological tau induces senescence in DS microglia, recapitulating microglial responses in human AD and DSAD brain tissue. Importantly, inhibiting the expression of Hsa21-encoded type I interferon receptor genes improves the defective DS microglia functions during brain development and prevents DS microglial senescence in response to pathological tau.
Results
Generation and characterization of DS hiPSC-derived primitive macrophage progenitors (PMPs)
We derived PMPs from the control (Cont) and DS hiPSCs that were generated and fully characterized in our previous studies (Fig. 1A and Table S1) (Chen et al., 2014; Xu et al., 2019). Over 94% of Cont and DS hiPSC-derived PMPs expressed hematopoietic progenitor cell markers CD235 and CD43. These PMPs were highly proliferative, as indicated by expressing Ki67 (Fig. 1B–C). These DS PMPs exhibited trisomy of Hsa21, as demonstrated by fluorescence in situ hybridization (FISH) assay and gene copy number assay for Hsa21 genes, OLIG2, IFNAR1, and IFNAR2 (Fig. 1B, S1A). By performing RNA-sequencing (RNA-seq), we identified 775 differentially expressed genes (DEGs) between Cont and DS PMPs (Fig. 1D). Gene ontology (GO) analyses of the upregulated DEGs showed significant enrichment of biological processes particularly related to type I interferon (IFN) signaling pathway, neural tube development, and cell differentiation (Fig. 1E, Table S2). The downregulated DEGs were enriched in pathways relevant to immune signaling, such as innate immune response, Toll-like receptor signaling pathway, and NF- κB pathway (Fig. 1F, Table S2). Moreover, the volcano plot showed that the XAF1, IFI27, and IFI44L involved in the type I IFN (IFN-I) signaling pathway were upregulated in DS PMPs (Fig. 1G). Consistently, qPCR analysis showed that the expression IFN-I receptors (IFNARs), IFNAR1 and IFNAR2, were increased in DS PMPs compared to Cont PMPs (Fig. S1B). Furthermore, expression of IRAK4 and FCGL1 – which are involved in T cell activation (Borowski et al., 2007; Suzuki et al., 2006; Wang et al., 2019) – was also altered in DS PMPs than Cont PMPs (Fig. 1G). Interestingly, we found that 22 DEGs are Hsa21 genes. For example, the tetratricopeptide repeat domain 3 (TTC3) is an E3 ligase that interacts with Akt, and DS neural cells exhibit elevated TTC3 expression (Suizu et al., 2009), which is consistent with our observations. Taken together, these findings suggest that aberrant development of myeloid cell lineage in DS can be detected as early as the PMP stage.
Fig 1. Characterization of DS hiPSC-derived PMPs.
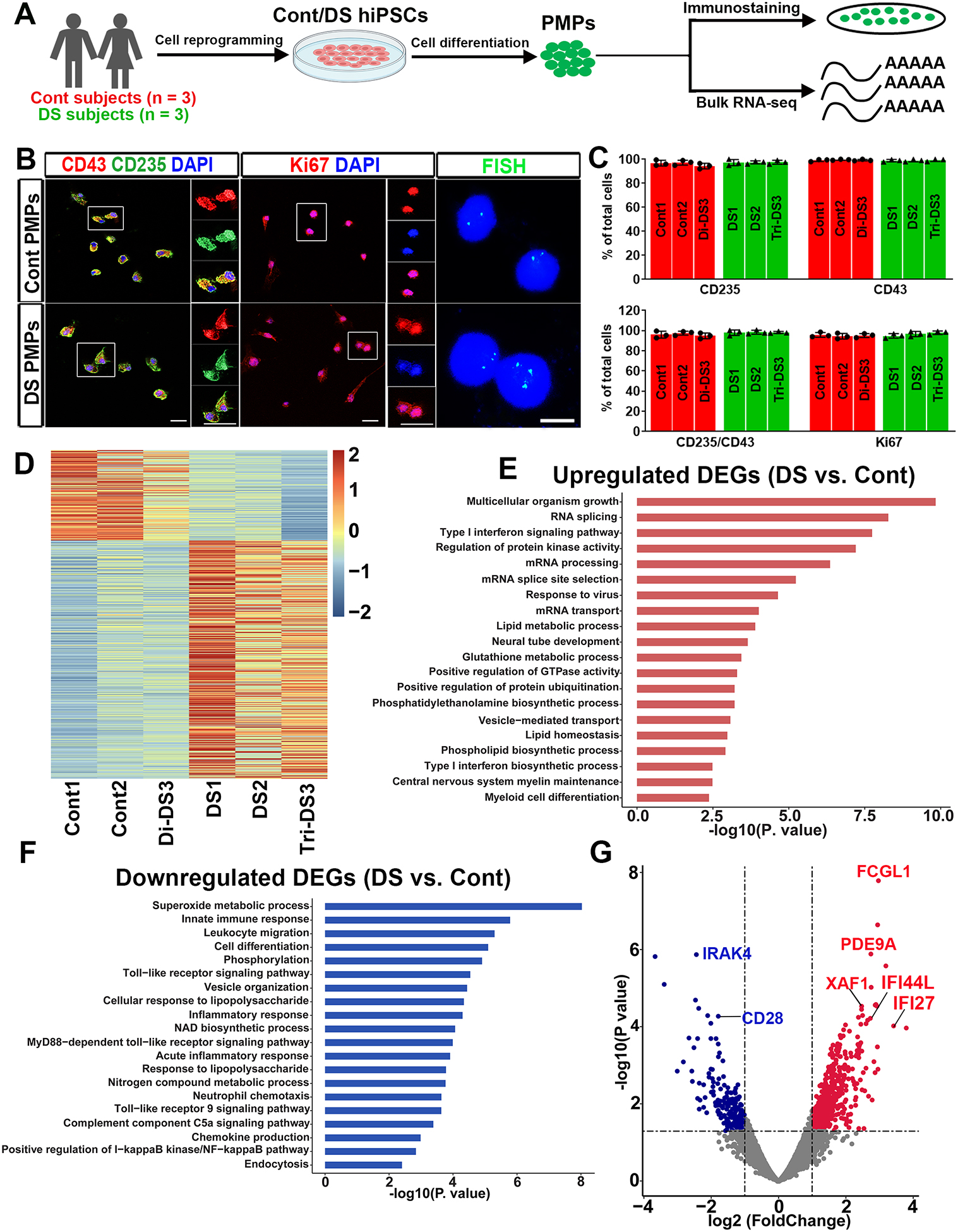
(A) Schematic representation of the generation and characterization of PMPs. This drawing was created using BioRender.com.
(B) Representative images of CD235+, CD43+, Ki67+ cells, and FISH analysis in PMPs. Scale bars: 20 μm,10 μm, and 5 μm.
(C) Quantification of CD235+, CD43+, CD235+/CD43+, and Ki67+ PMPs derived from the three pairs of Cont and DS hiPSC lines (n=3).
(D) The heatmap showing all DEGs between Cont and DS PMPs.
(E-F) GO analyses of the upregulated and downregulated DEGs in the PMPs.
(G) A volcano plot illustrating downregulated (blue) and upregulated (red) DEGs in PMPs.
Abnormal development and function of DS microglia in cerebral organoids, human brain tissues, and human-mouse microglial chimeras
Next, we used microglia-containing cerebral organoid model (Xu et al., 2021) to examine DS microglia functions. The Cont pNPCs and DS PMPs were mixed to develop organoids. By week 4, similar percentages of CD45+ microglia were seen in both Cont and DS organoids (Fig. 2A–C). We also found cells in organoids that expressed microglia-specific marker TMEM119 (Fig. 2D). There were many MAP2+ neurons in the organoids (Fig. S1C), indicating successful differentiation in these organoids at week 8.
Fig 2. Abnormal development and function of DS microglia in cerebral organoids and human brain tissues.
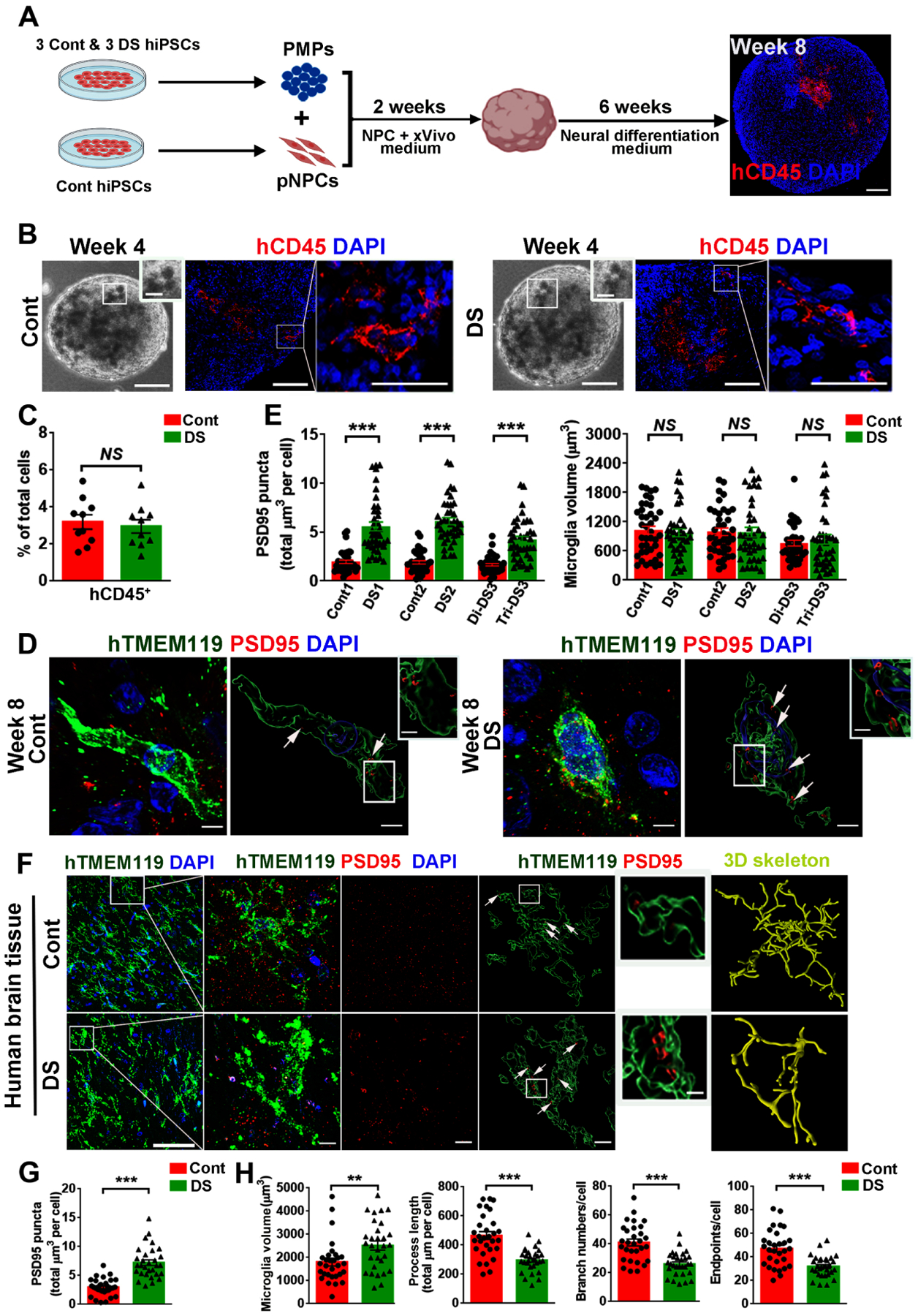
(A) A schematic representation of developing microglia-containing cerebral organoids. This drawing was created using BioRender.com. Scale bar: 100 μm.
(B) Representative images of hCD45+ microglia in Cont and DS organoids.
(C) Quantification of the percentage of hCD45+ cells in total DAPI+ cells in organoids at week 8 (n=10, the data were pooled from the three pairs of organoids).
(D) Representative raw fluorescent super-resolution and 3D surface rendered images showing colocalization of hTMEM119+ and PSD95+ staining in week 8 organoids. Scale bars: 5 μm and 1 μm.
(E) Quantification of PSD95+ puncta inside hTMEM119+ microglia and microglial volume at week 8 (n=3, the experiments were repeated 3 times, each dot represents one microglia).
(F) Representative images of hTMEM119 and PSD95 staining in hippocampal slices from a Cont and a DS individual. Arrows indicate PSD95+ puncta. Scale bar: 50 μm, 5 μm and 1 μm.
(G, H) Quantification of PSD95+ puncta inside microglia, microglial volume, process length, branch numbers, and endpoints (n=30 microglia/group).
Student’s t test, **P < 0.01 and ***P < 0.001. Data are presented as mean ± SEM.
Using super-resolution imaging techniques, we found a significantly higher number of PSD95+ puncta within hTMEM119+ DS microglia than Cont microglia (Figs. 2D–E), suggesting that DS microglia had enhanced synaptic pruning function. We also generated trisomic organoids by mixing the PMPs and pNPCs derived from DS hiPSC lines (Fig. S1D). Like DS microglia developed in a euploid environment, DS microglia in the trisomic organoids also exhibited enhanced synaptic pruning function compared to Cont microglia (Fig. S1D–E). A recent study (Pinto et al., 2020) reported that microglia in DS mice had larger soma size and branching impairment in comparison to microglia in wild-type mice. However, in the cerebral organoids, microglia extend fewer processes and do not show morphology as highly ramified as they do in vivo (Benito-Kwiecinski and Lancaster, 2020; Ormel et al., 2018; Xu et al., 2021). Consequently, we did not observe any significant morphological differences between DS vs. Cont microglia in cell size (Fig. 2D–E). We then analyzed microglia in post-mortem hippocampal tissues from DS and Cont subjects at ages of less than 1 year (Table S1). Like our brain organoid model, we observed that DS microglia in the hippocampal tissue also had a higher number of PSD95+ puncta compared to Cont microglia (Fig. 2F–G). Importantly, DS microglia also exhibited a reactive morphology as indicated by increased microglial volume, reduced numbers of branches, process lengths, and endpoints (Fig. 2F, 2H), which are consistent with the recent observations (Flores-Aguilar et al., 2020; Pinto et al., 2020).
We then transplanted PMPs derived from Cont and DS hiPSCs into the brains of postnatal day 0 (P0) Rag2−/− IL2rγ−/− hCSF1KI immunodeficient mice to generate human microglial mouse chimeras, as described in our recent study (Xu et al., 2020). At 8 weeks post-transplantation, both xenografted Cont and DS microglia labeled by human specific TMEM119 (hTMEM119) were found to migrate throughout the brain along the corpus callosum and disperse in multiple brain regions, including the cerebral cortex and hippocampus (Fig. 3A). At 3–4 months, hTMEM119+ DS microglia were widely distributed throughout the hippocampus (Fig. 3A). In both Cont and DS microglia chimeras, the vast majority of hN+ cells (>87%) were hTMEM119+ (Fig. S2A, S2C). Both Cont and DS microglia were capable of proliferating at 8 weeks, and no difference was observed (Fig. S2A, S2C). As expected, both DS and Cont microglia showed complex processes in the chimeras and developed increasingly complex morphology along with the developing mouse brain from 4 weeks to 3 months old (Fig. 3B–C). Interestingly, in the grey matter (GM), 3D skeleton analysis indicated that DS microglia had less intricate morphology than Cont microglia, as indicated by enlarged cell volume, shortened process length, fewer endpoints, and branch numbers (Fig. 3B–C, S2D). As opposed to the GM, microglia morphology is not as complex in the white matter (WM) (Fig. S2B). In the WM, the cell volume of Cont and DS microglia was similar, but the DS microglia showed significantly fewer branch numbers, reduced endpoints, and shorter process length (Fig. S2E). We also found PSD95+ puncta within both Cont and DS microglia, indicating that they were able to prune synapses (Fig. 3B). Of note, the synaptic pruning function of Cont and DS microglia is developmentally regulated, increasing along with the age of animals from 4-week to 8-week to 3 months old. Further 3D reconstructed imaging analysis showed significantly more PSD95+ puncta in DS microglia than Cont microglia in GM (Fig. 3B, 3D). In addition, we triple-stained hTMEM119, PSD95, and CD68, a marker for phagolysosomes. As shown in Fig. 3E, PSD95+ puncta were seen inside the CD68+ phagolysosomes of both hTMEM119+ Cont and DS microglia. DS microglia also showed enhanced phagocytosis, as indicated by a higher volume of CD68+ phagolysosomes and more PSD95+ puncta in CD68+ phagolysosomes (Fig. 3F). Moreover, we observed more synapsin I+ puncta within hCD45+ DS microglia than Cont microglia (Fig. 3G–H). In the WM, we also observed enhanced phagocytosis function as indicated by an increased volume of CD68+ phagolysosomes in DS microglia, compared to Cont microglia in chimeras at week 8 (Fig. S2F–S2G). The Golgi staining results showed that DS chimeras had a lower dendritic spine density than Cont chimeras (Fig. S2H), which is consistent with the findings that DS microglia exhibit excessive pruning of PSD95+ synapses. We further recorded miniature excitatory postsynaptic currents (mEPSCs) in hippocampal slices from Cont and DS chimeras at month 3. This time point was chosen because there was a wide distribution of transplanted Cont and DS microglia in the hippocampus, and a comparable level of chimerization by engrafted microglia was observed (Fig. 3A, 3I). The recorded CA1 neurons labeled by neurobiotin through recording electrodes were surrounded by engrafted hTMEM119+ microglia (Fig. 3I). Compared to Cont, DS microglial chimeric mice had mEPSCs with significantly reduced frequency and amplitude (Fig. 3J–K). Taken together, these results demonstrate that in the chimeras, DS microglia not only exhibit excessive synaptic pruning but also display less intricate morphology than Cont microglia. Excessive synaptic pruning of DS microglia may lead to impaired synaptic neurotransmission.
Fig 3. Modeling DS microglial phenotypes in human-mouse microglial chimeras.
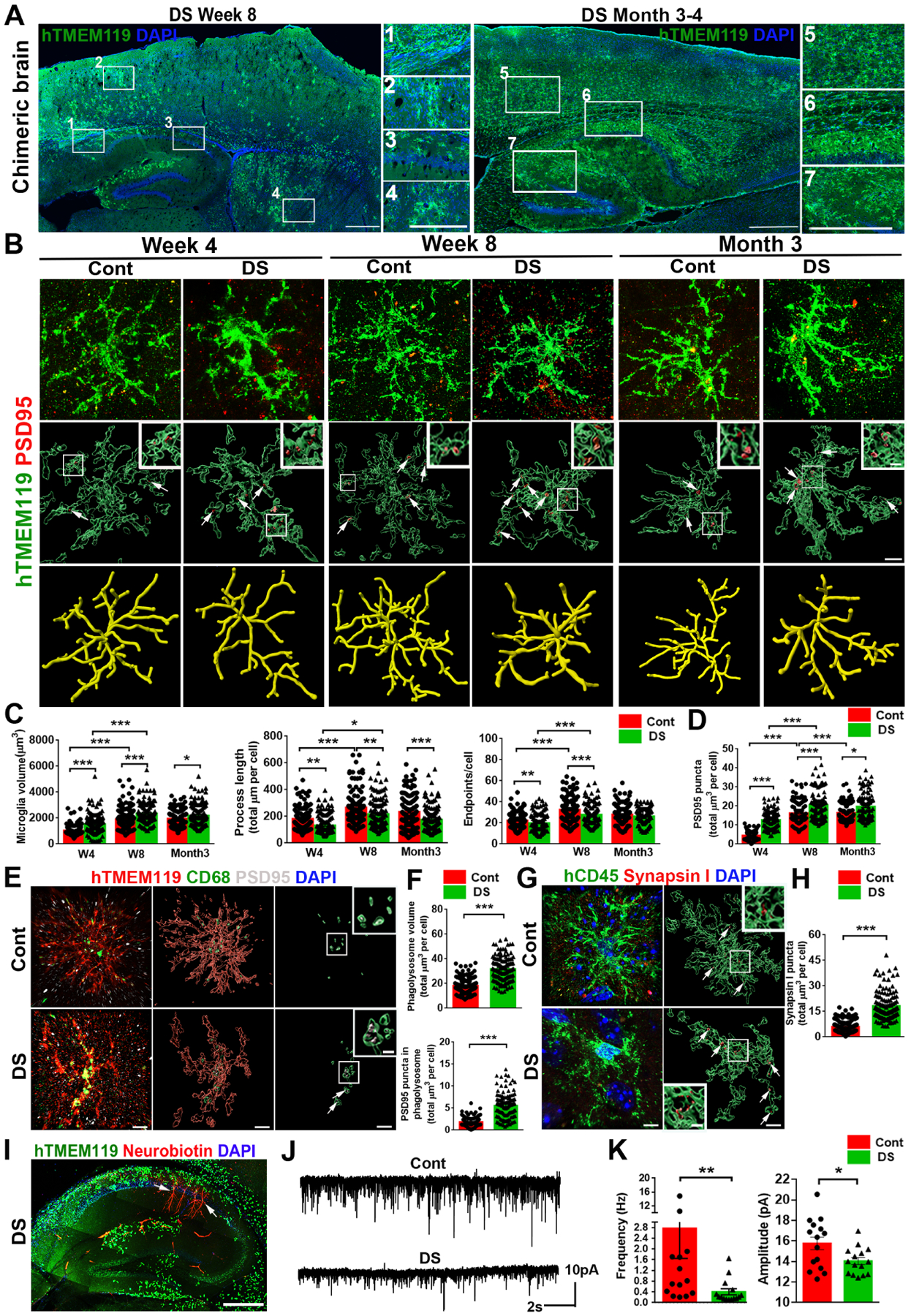
(A) Representative images from sagittal brain sections showing the distribution of transplanted DS hiPSC-derived microglia at week 8 and 3 to 4 months. Scale bar: 1 mm and 500 μm.
(B) Representative images showing colocalization of hTMEM119+ and PSD95+ staining at 4, 8 weeks, and 3 months post-transplantation. Arrows indicate PSD95+ puncta. Scale bar: 5 μm and 1μm.
(C, D) Quantification of microglial volume, process length, endpoints, and PSD95+ puncta inside microglia (n=120–160 microglia from 3–5 mice/group).
(E) Representative images showing colocalization of hTMEM119+CD68+ PSD95+ staining of week 8 chimeras. Arrows indicate PSD95+ puncta in the CD68+ phagolysosome. Scale bar: 5 μm and 1 μm.
(F) Quantification of CD68+ phagolysosome volume and PSD95+ puncta inside CD68+ phagolysosomes (n=120 microglia from 4 mice/group).
(G) Representative images of hCD45 and synapsin I staining in week 8 of chimeras. Arrows indicate synapsin I+ puncta. Scale bar: 5 μm and 1 μm.
(H) Quantification of synapsin I+ puncta in hCD45+ microglia (n=120 microglia from 4 mice/group).
(I) A representative image of hTMEM119+ microglia and Neurobiotin+ recorded neurons in the hippocampus of DS microglia chimera. Arrows indicate Neurobiotin+ recorded neurons. Scale bar: 500 μm.
(J) Representative traces of mEPSCs in CA1 hippocampal pyramidal neurons.
(K) Quantification of the frequency and amplitude of mEPSCs (n=14–15 neurons from 4–5 mice/group).
Student’s t test, *P < 0.05 and **P < 0.01. Data are presented as mean ± SEM.
Inhibiting the expression of IFNARs rescues the defective DS microglia development
We then examined the expression of Hsa21-encoded type I IFNARs in DS microglia (Fig. 4A). DS microglial chimeras had higher IFNAR1 and IFNAR2 gene transcripts expression than Cont chimeras at 8 weeks and 4 months (Fig. 4B). We also performed flow cytometry targeting hCD45+ microglia cells using 4 months old chimeras and analyzed the expression of IFNAR1 and IFNAR2 in these hCD45+ microglia. Consistently, DS chimeras showed higher levels of IFNAR1 and IFNAR2 expression than Cont chimeras (Fig. 4C–D). Therefore, these results demonstrate overexpression of IFNARs in DS microglia than in Cont microglia.
Fig 4. Knockdown of IFNARs rescues defective DS microglia in chimeras.
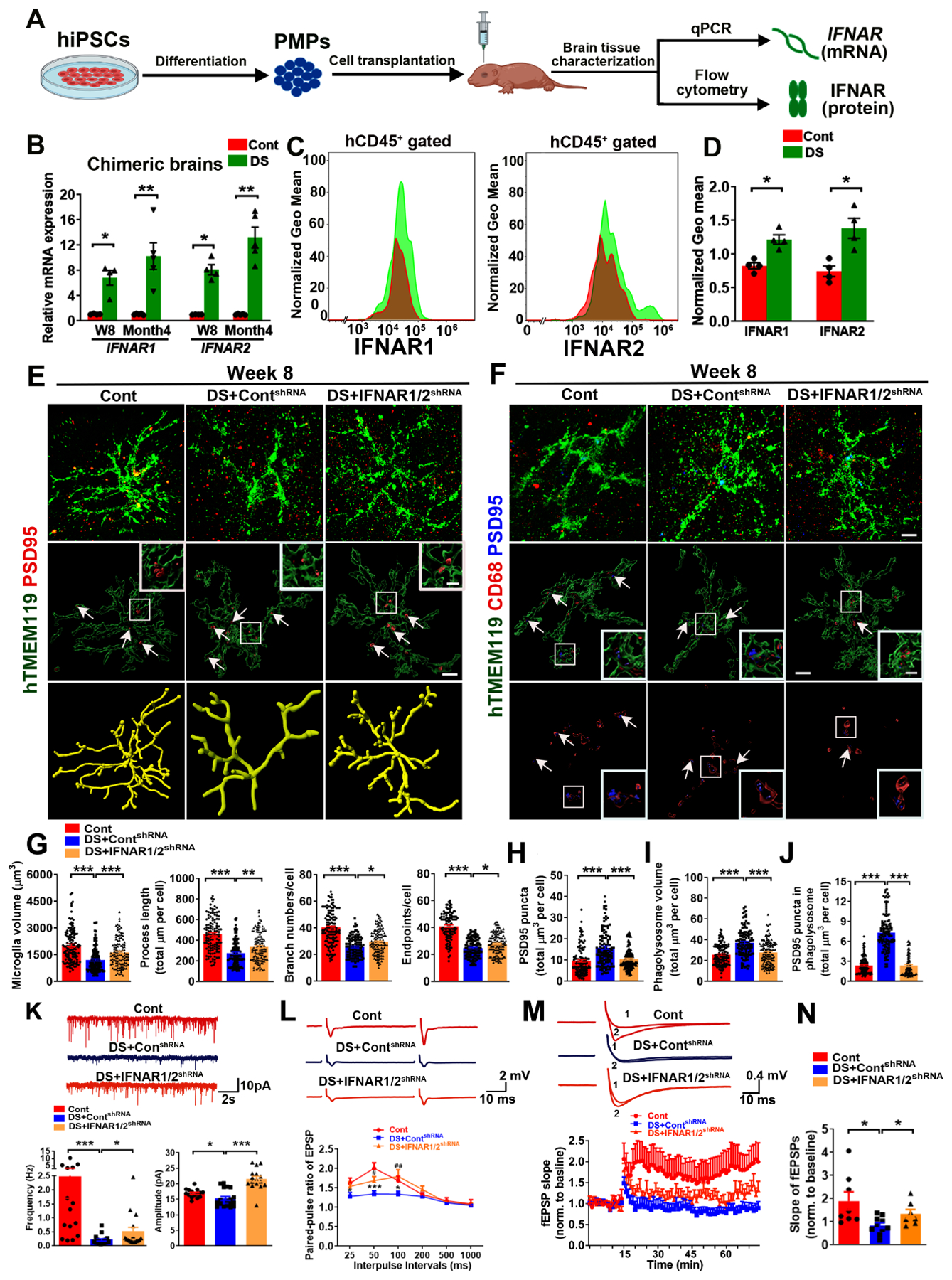
(A) A schematic diagram showing the experimental design. This drawing created using BioRender.com.
(B) qPCR analysis of IFNAR1 and IFNAR2 mRNA expression in chimeras at week 8 and month 4 (n=4–5 mice/group).
(C, D) Flow cytometry analysis and quantification of IFNAR1 and IFNAR2 expression in 4 months old chimeras (n=4).
(E–F) Representative images of hTMEM119+PSD95+ and hTMEM119+CD68+ PSD95+ staining in 8week-old chimeras (n=113–136 microglia from 3–4 mice/group). Arrows indicate PSD95+ puncta and PSD95+ puncta inside CD68+ phagolysosome. Scale bars: 5 μm and 1 μm.
(G) Quantification of microglial volume, process length, branch numbers, and endpoints (n=115–136 from 3–4 mice/group).
(H) Quantification of PSD95+ puncta in hTMEM119+microglia (n=115–133 microglia from 3–4 mice/group).
(I, J). Quantification of CD68+ phagolysosomes and PSD95+ puncta in CD68+ phagolysosomes (n=113–136 microglia from 3–4 mice/group).
(K) Representative traces of mEPSCs in CA1 hippocampal pyramidal neurons. Quantification of the frequency and amplitude of mEPSCs (n=15–18 neurons from 3 mice/group).
(L) Representative traces and quantification of PPR of EPSPs in 3–4 months old chimeras (n=9–12 slices from 3–4 mice/group). Asterisk represents Cont versus DS+ ContshRNA, and pound sign indicates DS+ContshRNA versus DS+IFNAR1/2shRNA.
(M) Representative traces of baseline (1) and last 10 min (2) fEPSP after 4×100 Hz LTP induction. Quantification of LTP after LTP induction in 3–4 months old chimeras (n=7–10 slices from 3–4 mice/group).
(N) Quantification of the last 10 min of fEPSP slope after LTP induction (n=7–10 slices from 3–4 mice/group).
Student’s t test or One-way ANOVA test, *P < 0.05, **P < 0.01 and ***P < 0.001. Data are presented as mean ± SEM.
To test the hypothesis that overexpression of IFNARs in DS microglia might be responsible for their altered development and functions, we took the RNAi knockdown approach. As observed phenotypes were highly consistent across the three DS hiPSC lines, we used two DS hiPSC lines (DS2 and Tri-DS3) to express IFNAR1/2shRNA or ContshRNA. Overexpression of IFNARs in DS could be detected at iPSC stages, as indicated by the higher expression of IFNAR1 and IFNAR2 gene transcripts in DS hiPSCs compared to Cont hiPSCs (Fig. S3A). Transduction of DS hiPSCs with lentivirus carrying IFNAR1/2shRNA, but not the ContshRNA, significantly inhibited the expression of IFNAR1 and IFNAR2 in DS hiPSCs (Fig. S3B). Then, we derived PMPs from DS hiPSCs treated with IFNAR1/2shRNA (DS+IFNAR1/2shRNA) or ContshRNA (DS+ContshRNA) and subsequently generated chimeras by engrafting these PMPs. At 8 weeks post-transplantation, we analyzed the chimeric brains and found that the expression of IFNAR1 and IFNAR2 was downregulated in DS+IFNAR1/2shRNA group than in DS+ContshRNA chimeras (Fig. S3C). Western blot analysis using an antibody specifically against human IFNAR2 confirmed the lower expression of IFNAR2 protein in DS+IFNAR1/2shRNA microglia than DS+ContshRNA microglia (Fig. S3D). In both DS+IFNAR1/2shRNA and DS+ContshRNA microglial chimeras, the vast majority of the hN+ cells were hTMEM119+(Fig. S3E–S3F). Interestingly, 3D skeleton analysis indicated that DS+IFNAR1/2shRNA microglia in the GM have more ramified morphology than DS+ContshRNA microglia (Fig. 4E, 4G). In the WM, we found that compared to DS+ContshRNA, DS+ IFNAR1/2shRNA microglia showed similar microglia volume, branch numbers, and endpoints but increased process length (Fig. S3G, S3I). Additionally, we found fewer PSD95+ puncta within DS+IFNAR1/2shRNA microglia, a smaller total volume of CD68+ phagolysosome, and fewer PSD95+ puncta inside the CD68+ phagolysosomes in the DS+IFNAR1/2shRNA microglia than in DS+ContshRNA microglia in the GM (Fig. 4F, 4H–J). The hippocampal spine density was higher in DS+IFNAR1/2shRNA chimeras than DS+ContshRNA chimeras (Fig. S4A). Similarly, in the WM, the total volume of CD68+ phagolysosomes in DS+IFNAR1/2shRNA microglia was smaller than DS+ContshRNA microglia, indicating a decrease in the phagocytic function (Fig. S3H, S3J). Taken together, inhibiting the expression of IFNARs increases the ramification of DS microglia and rescues their synaptic pruning functions.
To explore the effects of inhibiting IFNARs in DS microglia on synaptic neurotransmission, we recorded mEPSCs in hippocampal CA1 neurons from DS+ContshRNA and DS+IFNAR1/2shRNA microglial chimeric mice at 3 months post-transplantation. There was a significant increase in mEPSC frequency and amplitude in DS+IFNAR1/2shRNA microglial chimeras than DS+ContshRNA group (Fig. 4K). The changes in both frequency and amplitude of mEPSCs between DS and Cont microglial chimeras suggest that in addition to the observed postsynaptic alterations (Fig. 3B, 3D–F and Fig. 4E–F, 4H–J), pre-synaptic mechanisms are involved. We thus further recorded pair-pulse ratio (PPR) to examine whether there were changes in synaptic release in the presynaptic sites. Inhibiting the IFNARs expression in DS microglia significantly enhanced the PPR, as indicated by the higher PPR at 50ms and 100ms interpulse intervals in DS+IFNAR1/2shRNA group than in DS+ContshRNA group (Fig. 4L). To assess long-term synaptic plasticity, we induced long-term potentiation (LTP) using 4 trains of 100 Hz high-frequency stimulation (HFS). As shown in Fig. 4M, the enhancement of the field excitatory postsynaptic potential (fEPSP) slope persisted for 60 min in Cont chimeras, but not in DS+ContshRNA chimeras. DS+IFNAR1/2shRNA mice exhibited a significant enhancement of fEPSP slope than in DS+ContshRNA mice (Fig. 4N). In addition, after paired-pulse facilitation (PPF) and LTP recording, we collected all the recorded hippocampal slices and performed histological analysis. All the slices similarly showed a wide distribution of xenografted, hTMEM119+ human microglia (Fig. S4B). Altogether, these data strongly demonstrate that elevated synaptic pruning function of DS microglia results in impaired neurotransmission and synaptic plasticity. Inhibiting the overexpression of IFNARs in DS microglia partially rescues the defects and improves synaptic functions.
Single-cell RNA sequencing (scRNA-seq) analysis of DS microglial chimeras following exposure to pathological tau
To examine the in vivo responses of DS microglia to pathological tau, we prepared S1 soluble protein fractions from DSAD human brain tissue with abundant tau pathology (Tangle stage 6, Table S1) qualifying for AD, as well as from age- and sex-matched Cont brain tissues. Western blot analysis showed that p-tau was not detectable in Cont brain tissue-derived soluble fractions by Tau phos Ser396/Ser404 (PHF-1) antibody that recognizes p-tau. In contrast, p-tau was highly abundant in DSAD brain tissue-derived soluble tau fractions (Fig. S5A). In addition, both Cont brain and DSAD brain tissue-derived soluble fractions had undetectable levels of Aβ content, whereas Aβ could be detected in the homogenate of DSAD cortical tissue (Fig. S5A). ELISA results also showed that both groups had a similar level of total tau (Table S3). Chimeric mice engrafted with microglia derived from two DS lines (DS2 and Tri-DS3) further received injection of either Cont or DSAD at the age of 8 to 10 weeks. At two weeks post-injection, AT8+ p-tau was distributed widely in the mouse brains that received DSAD tau, which is consistent with previous reports (Boluda et al., 2015; Nimmo et al., 2020). In the Cont tau group, very few AT8+ p-tau was detected (Fig. S5B). As shown in Fig. 5A–B, and Fig. S5C–S5D, hTMEM119+ DS microglia engulfed numerous AT8+ or PHF-1+ p-tau in the DSAD Tau group at 1–2 months post tau injection. In the Cont tau group, few p-tau was seen in DS microglia.
Fig 5. scRNA-seq analysis of DS microglial chimeras receiving injection of Cont or pathological DSAD tau.
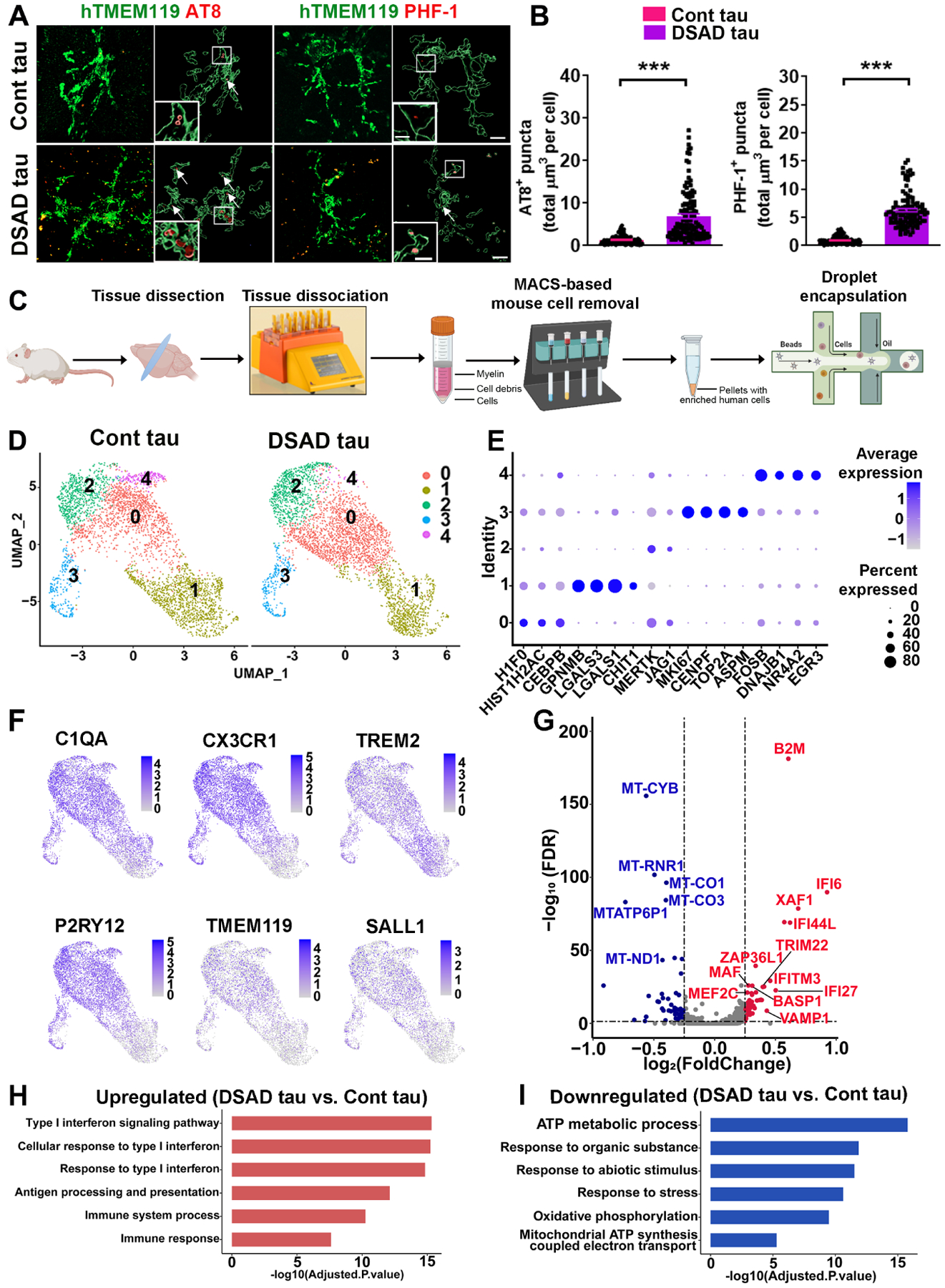
(A) Representative images of hTMEM119, AT8, and PHF-1 staining in 4 to 5-month-old chimeras receiving injection of Cont or DSAD tau at 8 weeks. Arrows indicate AT8+ or PHF-1+ p-tau. Scale bars: 7 μm and 3 μm.
(B) Quantification of AT8+ and PHF-1+ p-tau in microglia (n=110–127 from 3–4 mice/group).
(C) A schematic diagram showing the design of the scRNA-seq experiment. This drawing was created using BioRender.com.
(D) A UMAP plot showing independent subclusters (clusters 0–4) from Cont and DSAD tau groups.
(E) A dot plot showing the representative conserved markers from each subcluster.
(F) UMAP plots with dots (representing cells) colored by the expression levels of human microglial genes.
(G) A volcano plot illustrating downregulated (blue) and upregulated (red) DEGs.
(H, I) GO enrichment analyses of the upregulated and downregulated DEGs.
Student’s t test, ***P < 0.001. Data are presented as mean ± SEM.
At two months following tau injection, brain regions, including the cerebral cortex, hippocampus, corpus callosum, and olfactory bulb, were collected for scRNA-seq. To capture ample numbers of DS iPSC-derived microglia for scRNA-seq, we enriched DS microglia by using a magnetic cell sorting-based mouse cell depletion kit (Fig. 5C). A total of 7,790 human microglial cells were recovered after quality control, of which 3,919 DS microglia were from the Cont tau group and 3,871 DS microglia were from the DSAD tau group. The median gene counts were 1,041 and 1,158 per cell for Cont tau and DSAD tau groups, respectively (Fig. S5E). We identified 6 clusters, in which cluster 5 contained few cells (<0.4% of cells), expressed hematopoietic progenitor cell marker CD34 rather than microglial markers (Fig. S5F), and thus was not included for further analysis. UMAP (Uniform Manifold Approximation and Projection) and clustering of DS microglia from Cont tau and DSAD tau groups demonstrated the overall similarities in clustering of the two groups (0–4) (Fig. 5D). Clusters can be distinguished by their expression of enriched markers (Table S4), which included 5 subclusters: cluster 0 (H1F0, HIST1H2AC, and CEBPB), cluster 1 (GPNMB, LGALS3, and CHIT1), cluster 2 (MERTK and JAG1), cluster 3 (MKI67, CENPF, and ASPM), and cluster 4 (FOSB, DNAJB1, and EGR3) (Fig. 5E). All clusters highly expressed microglial markers, including C1QA, CX3CR1, TREM2, TMEM119, P2RY12, and SALL1 (Fig. 5F). We identified 122 DEGs (Fig. 5G, Table S5) in DSAD tau group than in Cont tau group. To explore the function of these DEGs, we performed GO and KEGG enrichment analyses by using g: Profiler. Interestingly, many significantly enriched DEGs that were upregulated in DSAD tau group were associated with the IFN-I signaling pathway (e.g. IFI6, XAF1, IFI44L, IFI27, and IFITM3) (Ioannidis et al., 2012) and cell senescence pathway (e.g. B2M, ZFP36L1, and XAF1) (Heo et al., 2016; Loh et al., 2020; Smith et al., 2015) (Fig. 5H, S6A). In subclusters (0 and 3), we also found upregulated IFN-I signaling (Fig. S6B). Furthermore, a considerable amount of downregulated DEGs in the DSAD tau group are involved in mitochondrial functions and ATP metabolic processes (e.g., LDHA, ALDOA, and MT-ATP). This was also seen in subclusters (e.g., 0 and 1) (Fig. 5I, S6C).
To explore the heterogeneity of these DS microglia, we compared the conserved gene markers that define each cluster with both genes that define disease-associated microglia (DAM; Table S6) (Butovsky and Weiner, 2018; Chen and Colonna, 2021; Deczkowska et al., 2018; Keren-Shaul et al., 2017; Krasemann et al., 2017; Sobue et al., 2021) and cellular senescence genes (Table S6) (Avelar et al., 2020; Zhao et al., 2016). When comparing the expression of DAM genes with the conserved markers of each cluster, we found that DAM genes were expressed by all clusters. Notably, the conserved markers of cluster 1 have the highest number of genes that overlapped with DAM genes (Fig. 6A). We found that 21 of the total upregulated DEGs detected from each cluster in DSAD tau group were DAM genes. The dot plot shown in Fig. 6B demonstrated that most of the identified DAM DEGs had higher expression levels in all clusters of DSAD tau group than Cont tau group. For senescence genes, many of these were expressed in all clusters, while cluster 3 expressed the highest number of senescence genes (Fig. 6C). 8 of the total upregulated DEGs detected from each cluster in DSAD tau group were senescence genes. Similarly, the dot plot shown in Fig. 6D demonstrated that most of the identified senescence DEGs had higher expression levels in all clusters of DSAD tau group than Cont tau group. Notably, all the 8 senescence DEGs were upregulated in clusters 1 and 3 in DSAD tau group.
Fig 6. Pathological tau induces DAM, senescence, and ISG signatures in DS microglia.
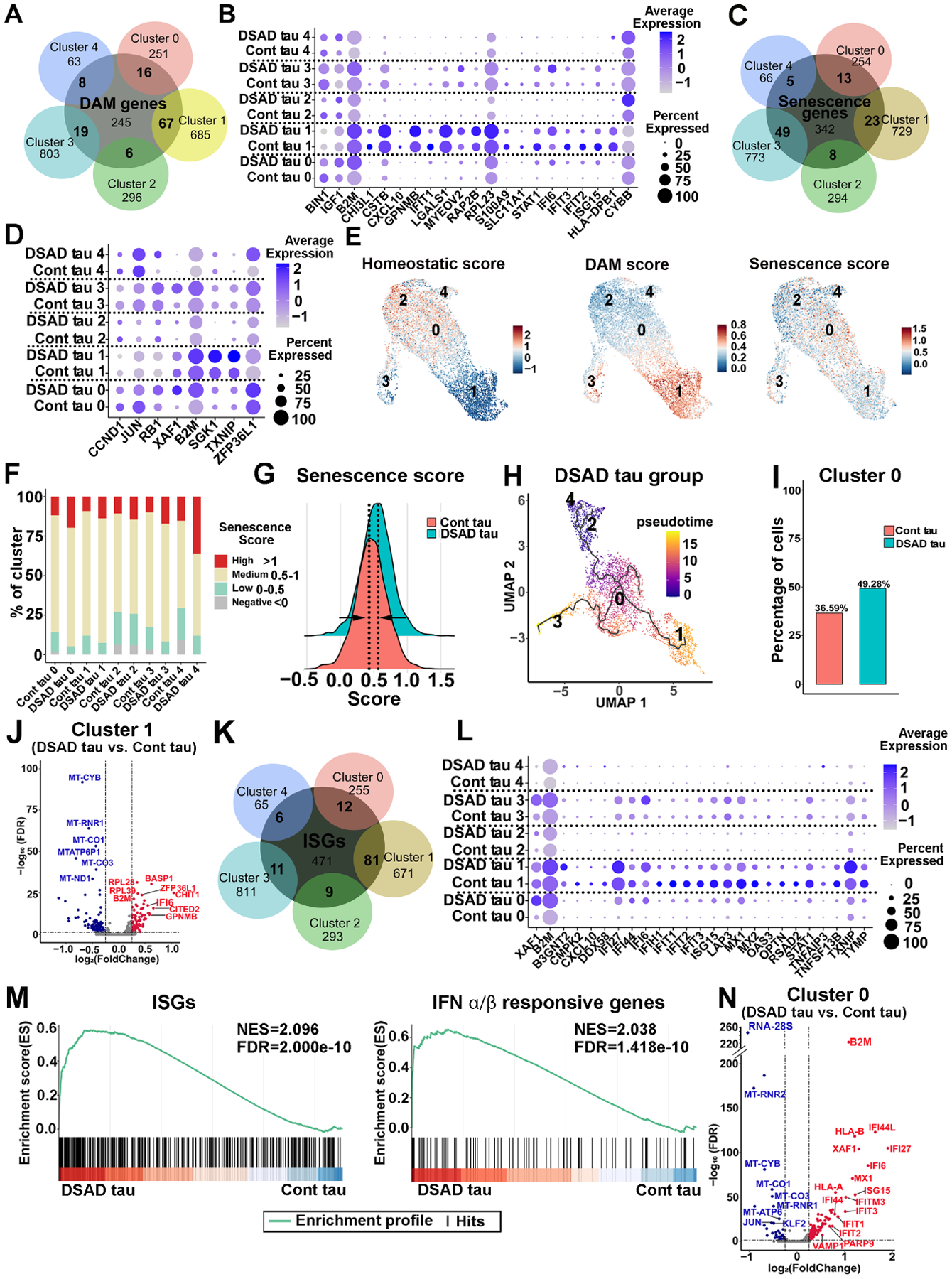
(A) A Venn diagram showing the overlap between DAM genes and the gene markers defined from each cluster.
(B) A dot plot representing the expression of DAM DEGs in each cluster from the Cont and DSAD tau groups.
(C) A Venn diagram showing the overlap between senescence genes and gene markers defined from each cluster.
(D) Dot plot showing the DEGs expression of the senescence genes signature in every subcluster from DSAD tau and Cont tau groups.
(E) Feature plots showing the scoring of homeostatic, DAM, and senescence signatures.
(F) Bar plots of the percentage of cells with a negative (<0), low (0–0.5), medium (0.5–1), or high (>1) senescence score in the custom senescence signature in each cluster.
(G) A ridge plot showing the senescence score in DSAD and Cont tau groups.
(H) UMAP representation of the trajectory of DS microglia in response to DSAD tau. Cells are colored by pseudotime.
(I) Cell ratios of cluster 0 in the Cont and DSAD tau groups.
(J) A Volcano plot illustrating the downregulated (blue) and upregulated (red) DEGs in cluster 1.
(K) A Venn diagram showing the overlap between ISGs and the gene markers defined from each cluster.
(L) A Dot plot displaying the ISG DEGs in each cluster from the Cont and DSAD tau groups.
(M) GSEA plots showing enrichment of ISGs and IFNα/β responsive genes in DSAD and Cont tau groups (NES: normalized enrichment score, FDR: false discovery rate).
(N) A Volcano plot showing the downregulated (blue) and upregulated (red) DEGs in cluster 0.
We then probed the dataset with homeostatic genes (TMEM119, P2RY12, P2RY13, CX3CR1, SELPLG, and BIN1), DAM genes (Table S6), and custom senescence signature genes (ATM, AXL, B2M, ZFP36L1, XAF1, CDKN2A, CDKN1A, CDKN2D, CASP8, IL1B, GLB1, and SERPINE1). As shown in Fig. 6E, cluster 2 expressed high levels of homeostatic genes and thus was annotated as homeostatic microglia, while cluster 1 was annotated as DAM. Interestingly, significant numbers of cells displaying senescence signature were seen in all clusters, particularly in cluster 0. As compared to Cont tau group, more cells in each subcluster in the DSAD tau group displayed greater association with senescence signature (Fig. 6F). Moreover, DSAD tau group overall showed a shift towards a higher senescence signature (Fig. 6G). Further trajectory analysis within DSAD tau group revealed a phenotypical change of DS microglia from cluster 2 (homeostatic) toward the cluster 3 (senescent state) and cluster 1 (DAM state), passing through cluster 0, an intermediate stage (Fig. 6H). Notably, cluster 0 was enriched in DSAD group (Fig. 6I). A recent study reported that DAM exhibited senescent phenotypes (Hu et al., 2021). Next, we analyzed DEGs within cluster 1, as cluster 1 had the highest DAM score. As shown in Fig. 6J (Table S5), the volcano plot showed that B2M, a pro-aging factor in neurodegenerative disease (Smith et al., 2015), was one of the top upregulated DEGs in the DSAD tau group. Moreover, ZAP36L1, whose expression was shown to promote cellular senescence (Galloway et al., 2016; Loh et al., 2020), was expressed at a much higher level in cluster 1 of DSAD tau group than Cont tau group (Fig. 6J). In addition, the expression of various mitochondria genes (e.g. MT-CYB, MT-RNR1, MT-CO1, and MT-CO3) (Lunnon et al., 2017) was decreased in cluster 1 of DSAD tau group (Fig. 6J).
We further examined the expression of interferon-stimulated genes (ISGs, Table S6) (Hubel et al., 2019; Rusinova et al., 2013). The Venn diagram shown in Fig. 6K demonstrated that ISGs were expressed by all subclusters. The DAM cluster 1 expressed the highest number of ISGs. 25 of the total upregulated DEGs detected from each cluster in DSAD tau group were ISGs (Fig. 6L). The pathway enrichment analysis also indicated increased activation of IFN-I pathway in the DSAD tau group than the Cont tau group (Fig. S6B). We performed gene set enrichment analysis (GSEA) and interestingly identified notable enrichment of ISGs and IFNα/β responsive genes in the DSAD tau group (Fig. 6M). As cluster 0 was an intermediate state (Fig. 6H), this cluster likely represented early responses of DS microglia to DSAD tau. Moreover, given the fact that cluster 0 was enriched in DSAD tau group and showed a higher senescence score (Fig. 6E), we further analyzed DEGs within cluster 0 between DSAD tau and Cont tau groups. Intriguingly, the volcano plot indicated that many IFN-I signaling-related genes (e.g., IFI44L, XAF1, IFI27, IFI44, IFIT3, and IFITM3) were significantly upregulated in cluster 0 of DSAD tau group (Fig. 6N, Table S5). Taken together, our scRNA-seq results depict that DSAD tau promotes overall senescence of DS microglia in all clusters and induces a DAM-like population of microglial clusters, specifically cluster 1. This DAM cluster 1 in DSAD tau group also shows enhanced cellular senescence. Increased cellular senescence of DS microglia subclusters in DSAD tau group largely overlaps with the elevated IFN-I signaling, suggesting that the pathological tau might work through activating IFN-I signaling to induce senescence of DS microglia.
DS microglial senescence induced by pathological tau can be rescued via inhibiting microglial IFNARs
To experimentally validate the senescent phenotype of DS microglia as indicated by scRNA-seq data, we first compared the morphology of hN+ DS microglia in Cont tau and DSAD tau groups. By double-staining Iba-1 and hN, we found that the percentage of hN+/Iba-1+ cells among total Iba-1+ cells was similar between the two groups (Fig. 7A, 7C). hN+/Iba-1+ DS microglia in DSAD tau group, but not in the Cont tau group, displayed dystrophic morphology, such as beading with shortened processes and fragmentation (Fig. 7A). Quantitative analysis showed shortened process length, enlarged soma size, and an increased ratio between soma size and process length in the DSAD tau group (Fig. 7D). Concomitantly, we found that in the same chimeras that received DSAD tau injection, the hN−negative/Iba-1+ host mouse microglia displayed hypertrophic morphology with shortened processes, decreased branch numbers, and endpoints, rather than dystrophic morphology (Fig. S6E–S6F). We next stained chimeric mouse brain tissue with hCD45 and ferritin, expression of which has been strongly linked to microglial senescence (Lopes et al., 2008; Simmons et al., 2007; Verina et al., 2011). We found that there was an increased number of ferritin+/hCD45+ DS microglia in the DSAD tau group than Cont tau group (Fig. 7B, 7E). Furthermore, scRNA-seq analysis showed that multiple inflammatory cytokines and chemokines, such as IL1B, CCL2, and CCL4, were expressed at lower levels in DS microglia in the DSAD tau group than in the Cont tau group (Fig. 7F). Consistently, qPCR analysis using human specific primers for IL1B and TNFA that respectively encode IL1β and TNFα also showed decreased expression of these inflammatory cytokines in DS microglia in DSAD tau group (Fig. 7G). We also injected either Cont or DSAD tau into Cont microglial chimeras at 8 weeks post-transplantation. As shown in Fig. S7A, we found no ferritin+/hCD45+ Cont microglia in the Cont tau group and very few ferritin+/hCD45+ Cont microglia in the DSAD tau group at two months post tau injection. Interestingly, there were many more ferritin+/hCD45+ Cont microglia at six months post DSAD tau injection, compared with that at 2 months after DSAD tau injection (Fig. S7A).
Fig 7. Knockdown of IFNARs rescues pathological tau-induced senescence in DS microglia.
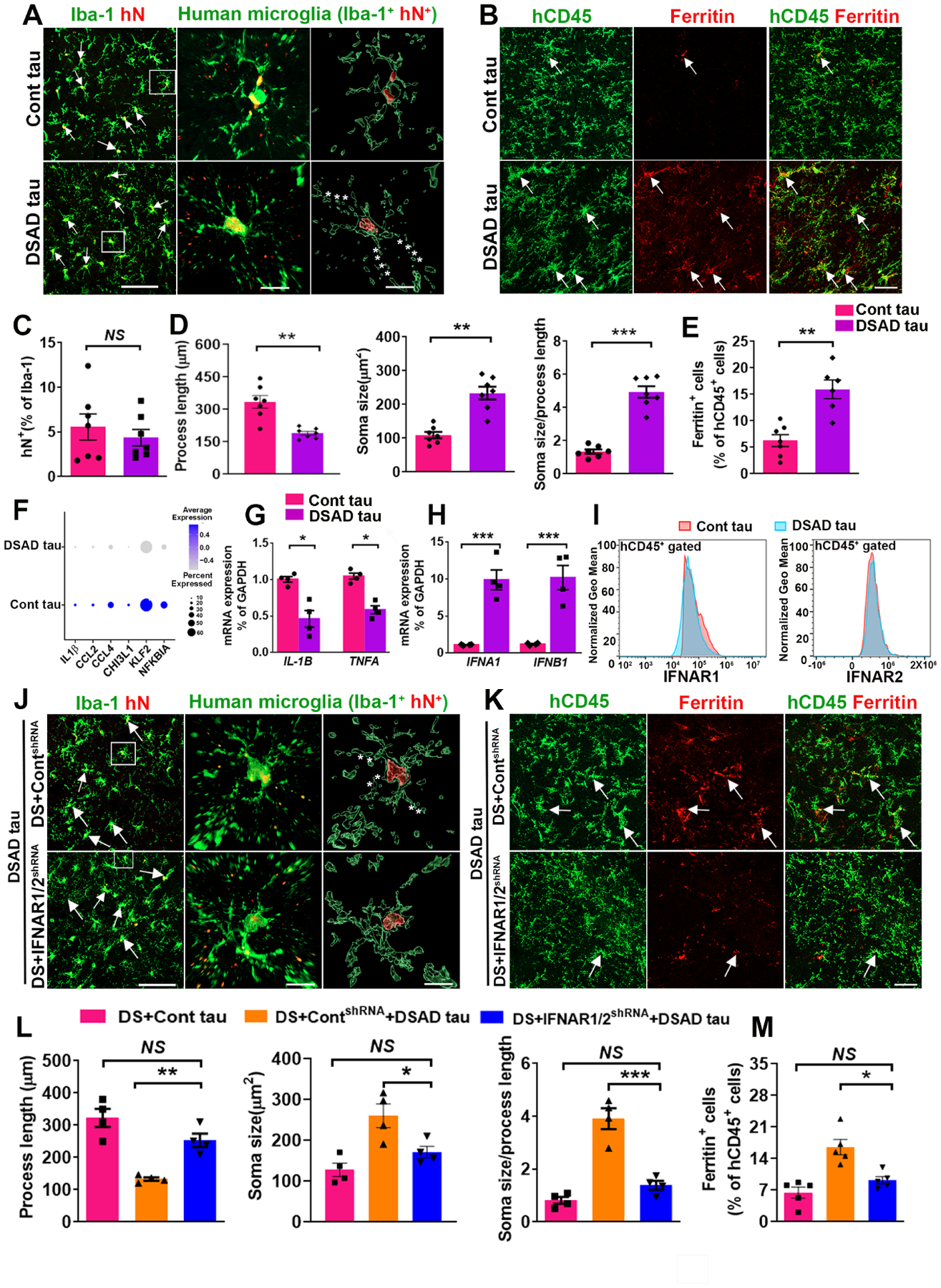
(A) Representative images of Iba+/hN+ human microglia in Cont and DSAD tau groups. Arrows indicate Iba+/hN+ human microglia. Asterisks indicate fragmented processes. Scale bars: 50 μm and 10 μm.
(B) Representative images showing colocalization of hCD45+ and Ferritin+ staining in Cont and DSAD tau groups. Arrows indicate Ferritin+ and/or hCD45+ staining. Scale bar: 50 μm.
(C) Quantification of the percentage of hN+ in Iba-1+ cells (n=7 mice/group).
(D) Quantification of the process length, soma size, and soma size/process length cells (n=7 mice/group).
(E) Quantification of the percentage of Ferritin in hCD45+cells (n=6–7 mice/group).
(F) A Dot plot representing the expression of the inflammation-related genes IL1B, CCL2, CCL4, CHI3L1, KLF2, NFKBIA identified from scRNA-seq.
(G) qPCR analysis of IL-1B and TNFA mRNA expression (n=4 mice/group).
(H) qPCR analysis of IFNA1 and IFNB1 mRNA expression (n=4 mice/group).
(I) Flow cytometry analysis showing the expression of IFNAR1 and IFNAR2 in Cont tau and DSAD tau group (n=2 mice/group).
(J) Representative images of human microglia (Iba+hN+) in chimeras. Arrows indicate Iba+/hN+ human microglia. Asterisks indicate fragmented processes. Scale bar: 50 μm and 10 μm.
(K) Representative image showing colocalization of hCD45+ and Ferritin+ staining in chimeras. Arrows indicate Ferritin+ and/or hCD45+ staining. Scale bar: 50 μm.
(L) Quantification of the process length, soma size, and soma size/process length (n=4 mice/group).
(M) Quantification of the percentage of Ferritin+ in hCD45+ cells (n=5 mice/group).
Student’s t test or One-way ANOVA test, *P < 0.05, **P < 0.01 and ***P < 0.001. Data are presented as mean ± SEM.
The IFNA1 and IFNB1 gene transcripts, which respectively encode IFNα and IFNβ, were significantly upregulated in the DSAD tau group compared to the Cont group (Fig. 7H), but no differences in the expression of IFNARs (Fig. S6G). In addition, flow cytometry analysis also showed that the expression of IFNARs was similar in both groups (Fig. 7I). These results suggest that pathological tau increases the production of IFN-I but is not able to further increase the already overexpressed IFNARs in DS microglia (relative to Cont microglia as shown in Fig. 7I, S6G). We then proposed that cell-type specific inhibition of IFNARs in DS microglia might prevent their senescence in response to pathological tau. We took the RNAi knockdown approach again and used the two DS hiPSC lines (DS2 and Tri-DS3) expressing IFNAR1/2shRNA or ContshRNA. We found that the expression of IFNA1 and IFNB1 remained higher in the DS IFNAR1/2shRNA+DSAD tau group compared to the DS IFNAR1/2shRNA+Cont tau group (Fig. S6H). Compared to DS microglia in the DS ContshRNA+DSAD tau, DS microglia in the DS IFNAR1/2shRNA+DSAD tau showed less fragmented processes, longer process length, smaller soma size, and decreased soma size/process length ratio (Fig. 7J, 7L). DS microglia in the DS IFNAR1/2shRNA+DSAD tau group showed similar process length and soma size than in the DS+Cont tau group (Fig. 7L). Moreover, there were fewer ferritin+/hCD45+ microglia in the DS IFNAR1/2shRNA+DSAD tau group than in the DS ContshRNA+DSAD tau group (Fig. 7K, 7M). Taken together, these results demonstrate that pathological tau induces cellular senescence of DS human microglia rather than causing massive activation. Inhibiting the expression of IFNARs in DS microglia rescues their senescent phenotypes following DSAD tau injection.
Discussion
We find that the human microglia chimeric mice and cerebral organoids faithfully recapitulate the defective phenotypes of DS microglia. Of note, as compared to Cont microglial chimeras, DS microglial chimeras showed impaired synaptic functions, corroborating the similar observations in Dp (16) mice (Pinto et al., 2020). Mechanistically, we focus on IFN-I signaling, because the roles of IFN-I signaling are not only limited to antiviral and immunomodulation, but are also involved in regulating homeostatic processes in the CNS (Blank and Prinz, 2017). It is known that under healthy conditions, baseline levels of constant IFN-I signaling play critical roles in brain development and functions, such as synaptic plasticity (Blank and Prinz, 2017; Ejlerskov et al., 2015; Hosseini et al., 2020). Moreover, trisomy 21 consistently activates IFN-I responses (Araya et al., 2019; Sullivan et al., 2016; Waugh et al., 2019), because of the increased gene dosage of IFNARs (Araya et al., 2019; Sullivan et al., 2016; Waugh et al., 2019). We show that inhibition of the IFNAR expression improves the morphological complexity of DS microglia, corrects its synaptic pruning functions, and rescues the hippocampal synaptic functions, suggesting that IFN-I signaling could be targeted to improve microglial functions in DS brain development.
Although the amyloid cascade/neuroinflammation theory of AD has been widely accepted, several caveats have arisen. Based on postmortem neuropathological examinations of human AD and DSAD brain tissues, a paradigm-shifting tau/microglial senescence hypothesis has emerged, in which human microglia respond to pathological tau by exhibiting accelerated senescence and dystrophic phenotypes rather than massive activation as previously thought (Guerrero et al., 2021; Navarro et al., 2018; Sanchez-Mejias et al., 2016; Streit et al., 2020; Streit et al., 2004). The supporting evidence for tau/microglial senescence hypothesis from live, functional brain tissue that contains human microglia as well as knowledge of underlying molecular mechanisms have so far been out of reach. Both clinical manifestations and biological evidence demonstrate accelerated aging of the brain and the immune system in individuals with DS (Cuadrado and Barrena, 1996; Horvath et al., 2015; Lott and Head, 2005; Teipel and Hampel, 2006). Recent work has also shown higher numbers of dystrophic/senescent microglia in the brains of people with DS (Martini et al., 2020). Moreover, in human microglial chimeric mouse brains, maturation and aging of donor-derived human microglia appears to be accelerated in the much faster-developing mouse brain relative to human brain (Jiang et al., 2020). Therefore, the combination of DS and hiPSC microglial chimeric mouse model presents an unprecedented opportunity to examine the novel tau/microglial senescence hypothesis and reveal aging-related responses of human microglia to pathological tau.
Here we provide in vivo evidence demonstrating that human microglia respond to pathological tau with accelerated senescence. First, scRNA-seq data clearly show that DS microglia in DSAD tau group exhibit enhanced senescence compared to DS microglia in Cont tau group. Senescence scoring (Fig. 6F,6G) and KEGG enrichment analysis of DEGs (Fig. S6A) consistently indicate enhanced microglial senescence in DSAD tau group. Some senescence-associated genes, such as B2M, XAF1, and ZFP36L1, are significantly upregulated in multiple clusters in DSAD tau group, particularly the intermediate, transition cluster 0 (Fig. 6N). Many DEGs that are downregulated in DSAD tau group are involved in mitochondrial functions and cell metabolism, indicating decreased metabolic processes in DS microglia in DSAD tau group. Reduced metabolic processes also suggest cellular senescence (Lopez-Otin et al., 2013; Vasileiou et al., 2019). Moreover, our scRNA-seq and qPCR analyses show that DS microglia in DSAD tau group express much lower levels of proinflammatory cytokines and chemokines than DS microglia in the Cont tau group, suggesting that pathological tau may not induce massive activation of DS microglia. Interestingly, as opposed to human DS microglia, the host mouse microglia in the same chimeric mouse brains in DSAD tau group exhibit hypertrophic morphology, suggestive of activation. Second, histological evidence also points to enhanced senescence in DSAD tau group. As compared to DS microglia in Cont tau group, DS microglia in DSAD tau group show increased expression of ferritin and dystrophic morphology, characteristics of senescent microglia (Streit et al., 2020). Lastly, our mechanistic studies also reveal enhanced microglial senescence in DSAD tau group. Our scRNA-seq analysis of DS microglia chimeric mouse brains show that IFN-I signaling is significantly upregulated in DS microglia in DSAD tau group, as compared to Cont tau group. Previous studies in human AD brain tissues report increased production of IFN-I (DiPatre and Gelman, 1997; Overmyer et al., 1999; Sheng et al., 1997; Taylor et al., 2014; Taylor et al., 2018). The receptors for IFN-I are expressed in microglia in the brain tissue from AD patients (Yamada and Yamanaka, 1995). Importantly, elevated IFN-I signaling has been shown to promote cellular senescence and aging (Frisch and MacFawn, 2020; Yu et al., 2015). By inhibiting IFNARs in xenografted microglia, we demonstrate that inhibition of IFNARs in DS microglia prevents their senescence after DSAD tau injection. A previous study (Bussian et al., 2018) has demonstrated a causal link between the accumulation of tau-pathology-induced senescent cells and cognition-associated neuronal loss. Clearance of senescent glial cells, including senescent microglia, prevents tau-dependent pathology and cognitive decline. Our findings suggest that new therapeutic strategies targeting IFNARs could prevent human microglial senescence to potentially slow the progression of AD in DS.
Accumulation of Aβ in individuals with DS starts as early as childhood and lasts lifelong (Lott and Head, 2005, 2019). Soluble Aβ proteins can induce microglia to prune synapses in early AD (Hong et al., 2016). A recent study further reported that Aβ pathology induces activation of type 1 IFN signaling in microglia, which drives microglia activation and microglia-mediated synaptic loss at early stages of β-amyloidosis (Roy et al., 2022). In our study, we find that DS microglia exhibit enhanced synaptic pruning functions in the developing chimeric mouse brains, due to their overexpression of IFNARs and elevated IFN-I signaling. Thus, the dysfunctional DS microglia in the developing brain likely contribute to the progression of AD and dementia later in life in people with DS. In addition, our microglial chimeras challenged by pathological tau are more of modeling AD at later stages when p-tau proteins are produced or in brain regions where p-tau is preferentially accumulated over Aβ (Braak and Del Tredici, 2015; Sanchez-Mejias et al., 2016). Recent RNA-seq analysis of human AD microglia reports aging profiles of AD microglia, which is not a direct response to amyloid pathology (Srinivasan et al., 2020). Here we find that pathological tau can directly induce aging of human microglia in vivo in chimeric mouse brains. Nevertheless, we support the notion that parenchymal deposition of Aβ and microglia activation is likely necessary for triggering and driving tau pathology (Edwards, 2019; Hopp et al., 2018; Ising et al., 2019; Long and Holtzman, 2019; Pascoal et al., 2021). In parallel with Aβ-pathology-induced microglial activation, microglia show rapid and proliferative response (Condello et al., 2015), which may indirectly promote senescence (Hu et al., 2021). Therefore, pathological Aβ and tau likely collude to promote microglial senescence. Importantly, in future studies, our human microglial chimeric mouse brain model will provide new opportunities to investigate how pathological tau-induced human microglial senescence further contributes to tau spread, neurodegeneration, and dementia.
Limitations of the Study
Some limitations of this study include: i) There is a lack of peripheral adaptive immune system in the chimeric mice, due to the use of immunodeficient mice. We recently discussed potential approaches to developing chimeric mice with a chimeric brain and humanized peripheral adaptive immune system derived from the same human donor (Jiang et al., 2020). ii) it is important to use behavioral tests to evaluate cognitive functions of the chimeric mice. We examined the behavioral performance of chimeric mice, including open field, novel object recognition, and elevated plus maze tests, but found no significant difference among the tested groups (Fig. S7B–S7D). As previously reported, Rag2 is necessary for proper retinal development, and Rag2−/− mice are blind (Alvarez-Lindo et al., 2019; Han et al., 2013). Thus, these Rag2−/− immunodeficient mice are likely not ideal for behavioral tests that require visual input. This issue could be potentially circumvented by using vision independent tests or using other immunodeficient mouse strains expressing human IL-34 or CSF-1 that can support the survival of engrafted human microglia (Mathews et al., 2019; Svoboda et al., 2019). Moreover, DS is associated with a significantly increased risk of autism spectrum disorder (ASD) (Moss et al., 2013; Warner et al., 2014). Accumulating evidence has indicated that defects in synaptic pruning mediated by microglia are closely linked to the development of ASD (Lukens and Eyo, 2022). Performing behavioral tests concerning ASD-related social interactions may also help reveal differences in cognitive functions between Cont and DS microglial chimeras.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peng Jiang (peng.jiang@rutgers.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The RNA-seq and scRNA-seq datasets generated in this study have been deposited at NCBI GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Mouse anti-β-amyloid | Biolegend | RRID:AB_2715854 |
| Goat anti-PSD95 | Abcam | RRID:AB_298846 |
| AF700 anti-human IFNAR1 | R&D Systems | RRID:AB_2920857 |
| FITC anti-human CD45 | BioLegend | RRID:AB_2566368 |
| PE anti-human IFNAR2 | Miltenyi Biotec | RRID:AB_2652221 |
| Mouse anti-AT8 | Invitrogen | RRID:AB_223647 |
| Mouse anti-CD43 | Invitrogen | RRID:AB_763493 |
| Mouse anti-CD45 | Invitrogen | RRID:AB_11063696 |
| Mouse anti-CD68 | Invitrogen | RRID:AB_10987212 |
| Mouse anti-GAPDH | Santa Cruz Biotechnologies | RRID:AB_627679 |
| Mouse anti-human nuclei | Millipore | RRID:AB_827439 |
| Mouse anti-Ki67 | Thermo Fisher Scientific | RRID:AB_2341197 |
| Mouse anti-MAP2 | Santa Cruz Biotechnologies | RRID:AB_1126219 |
| Mouse anti-PHF1 | Dr. Peter Davies, Albert Einstein College of Medicine | N/A |
| Rabbit anti-Synapsin 1 | Millipore | RRID:AB_90757 |
| Rabbit anti-CD235 | Invitrogen | RRID: AB_2544630 |
| Rabbit anti-Ferritin | Sigma-Aldrich | RRID:AB_259684 |
| Rabbit anti-Iba1 | Wako | RRID:AB_839504 |
| Rabbit anti-IFNAR2 | Invitrogen | RRID:AB_2545779 |
| Rabbit anti-Ki67 | Cell signaling | RRID:AB_2687446 |
| Rabbit anti-TMEM119 | Invitrogen | RRID:AB_2648507 |
| Donkey anti-goat 488 | Invitrogen | RRID:AB_2534102 |
| Donkey anti-goat 594 | Invitrogen | RRID:AB_142540 |
| Donkey anti-goat 647 | Invitrogen | RRID:AB_2535864 |
| Goat anti-mouse 488 | Invitrogen | RRID:AB_2534088 |
| Goat anti-mouse 594 | Invitrogen | RRID:AB_2534091 |
| Goat anti-mouse 647 | Invitrogen | RRID:AB_2535804 |
| Goat anti-rabbit 488 | Invitrogen | RRID:AB_2536097 |
| Goat anti-rabbit 594 | Invitrogen | RRID:AB_2534095 |
| Goat anti-rabbit 647 | Invitrogen | RRID:AB_2535813 |
| Bacterial and Virus Strains | ||
| IFNAR1 shRNA | Santa Cruz | sc-35637-V |
| IFNAR2 shRNA | Santa Cruz | sc-40091-V |
| Control shRNA | Santa Cruz | sc-108080 |
| Biological samples | ||
| Human brain tissue samples | NIH NeuroBioBank | See Table S3 for detailed information |
| Human brain tissue samples | UCI-ADRC | See Table S4 for detailed information |
| Chemicals, peptides, and recombinant proteins | ||
| 2-Mercaptoethanol | Gibco | Catalog# 21985-023 |
| 4% paraformaldehyde | Thermo Fisher Scientific | Catalog# J19943-K2 |
| Adult brain dissociation kit | Miltenyi Biotec | Catalog# 130-107-677 |
| Ascorbic acid | Sigma | Catalog# A4403 |
| B27-RA | Thermo Fisher Scientific | Catalog# 12587010 |
| BDNF | Peprotech | Catalog# 450-02 |
| BMP4 | Peprotech | Catalog# 120-05ET |
| CHIR99021 | Biogems | Catalog# 2520691 |
| Compensation beads | Miltenyi Biotec | Catalog# 01-2222-41 |
| Dibutyryl cAMP | Sigma | Catalog# D0260 |
| DMEM/F12 | HyClone | Catalog# SH3002201 |
| D-Sucrose | Fisher Bioreagents | Catalog# BP220-212 |
| FGF-2 | Peprotech | Catalog# 100-18B |
| GDNF | Peprotech | Catalog# 450-10 |
| GlutaMAX | Gibco | Catalog# 35050-061 |
| GM-CSF | Peprotech | Catalog# 300-03 |
| Growth factor reduced matrigel | Corning | Catalog# 354230 |
| hLIF | Millipore | Catalog# LIF1005 |
| IL-3 | Peprotech | Catalog# 200-03 |
| IL-34 | Peprotech | Catalog# 200-34 |
| Matrigel | Corning | Catalog# 354277 |
| M-CSF | Invitrogen | Catalog# PHC9501 |
| Mini-PROTEAN TGX Gels | Bio-rad | Catalog# 4561043 |
| Mouse cell depletion cocktail | Miltenyi Biotec | Catalog# 130-104-694 |
| M-PER Mammalian protein extraction reagent | Thermo Fisher Scientific | Catalog# 78501 |
| mTeSR 1 | Stemcell Technologies | Catalog# 85850 |
| mTeSR plus | Stemcell Technologies | Catalog# 100-0276 |
| N2 | Thermo Fisher Scientific | Catalog# 17502048 |
| Neurobasal | Thermo Fisher Scientific | Catalog# 21103-049 |
| Pierce BCA Protein assay kit | Thermo Fisher Scientific | Catalog# 23225 |
| Pierce ECL Western blotting substrate | Thermo Fisher Scientific | Catalog# 32106 |
| ReLeSR | Stemcell Technologies | Catalog# 100-0484 |
| RNeasy Kit | Qiagen | Catalog# 74004 |
| SB431542 | Stemgent | Catalog# 04-0010-05 |
| SCF | Peprotech | Catalog# 300-07 |
| Taqman Gene expression Master Mix | Applied Biosystem | Catalog# 4369016 |
| Tau (total) human ELISA kit | Invitrogen | Catalog# KHB0041 |
| Triton X-100 | Sigma | Catalog# T9284 |
| TRIzol | Thermo Fisher Scientific | Catalog# 15596026 |
| TrypLE Express | Thermo Fisher Scientific | Catalog# 12605028 |
| Tween 20 | Fisher Bioreagents | Catalog# BP337-100 |
| VEGF | Peprotech | Catalog# 100-20 |
| X-Vivo 15 | Lonza | Catalog# 04-418Q |
| Y-27632 | Tocris | Catalog# 1254 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3' GEM, Library & Gel Bead Kit v3.1 | 10x Genomics | Catalog# 1000128 |
| Chromium Next GEM Chip G Single Cell Kit | 10x Genomics | Catalog#1000127 |
| Deposited data | ||
| RNA-seq data | This paper | NCBI GEO: GSE189227 |
| scRNA-seq data | This paper | NCBI GEO: GSE189227 |
| Single Index Kit T Set A | 10x Genomics | Catalog#1000213 |
| Experimental Model | ||
| Cell lines | Coriell Institute for Medical Research | See Table S1 for detailed information |
| Mice: Rag2-/-hCSF1 | The Jackson Laboratory | Stock no:017708 |
| Oligonucleotides | ||
| qPCR primers | Thermo Fisher Scientific | See Table S11 for detailed information |
| Software and algorithms | ||
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| Adobe Photoshop | Adobe | https://www.adobe.com/products/photoshop.html |
| BioRender | BioRender.Inc | https://biorender.com/ |
| Cell Ranger v6.1.1 | 10x Genomics | https://github.com/10XGenomics/cellranger |
| Cluster Profiler 4.0.5 | https://guangchuangyu.github.io/software/clusterProfiler/ | |
| Clampfit 10.5 | Molecular Devices | https://support.moleculardevices.com/s/ |
| FlowJo_V10 | TreeStar | https://www.flowjo.com/solutions/flowjo/ |
| g:Profiler | e104_eg51_p15_3922dba | https://biit.cs.ut.ee/gprofiler/gost |
| GraphPad Prism 9.2.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.net/software/fiji/ |
| Imaris 9.5.1 | Bitplane | https://imaris.oxinst.com |
| Monocle v 1.0.0 | https://cole-trapnell-lab.github.io/monocle3/ | |
| R 4.1.0 | https://www.r-project.org | |
| RStudio | RStudio | https://www.rstudio.com/products/rstudio/download/ |
| Seurat v4.0.4 | https://satijalab.org/seurat/ | |
| Zen 2.3 | Carl Zeiss | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal work was performed without gender bias with the approval of the Rutgers University Institutional Animal Care and Use Committee. The Rag2−/−hCSF1 immunodeficient mice (C;129S4-Rag2tm1.1Flv Csf1tm1(CSF1)Flv Il2rgtm1.1Flv/J, The Jackson Laboratory) were used in this study.
Human samples
The post-mortem hippocampal tissues were obtained from the NIH NeuroBioBank. Soluble S1 fractions from human samples (provided by the University of California Alzheimer’s Disease Research Center (UCI-ADRC) and the Institute for Memory Impairments and Neurological Disorders). All experiments involving human samples were conducted in accordance with regulations and guidelines by the ethics committee of the Rutgers University.
Human iPSC lines
A total of six hiPSC lines were used in this study: three healthy control (Cont) and DS iPSC cell lines (Table S1). The DS hiPSC lines include two DS hiPSC lines (DS1, female; and DS2, male) and isogenic Di-DS3 and Tri-DS3 hiPSCs that were generated from a single female patient.
METHOD DETAILS
Human iPSC lines generation, culture, and quality control
The hiPSC lines were fully characterized and completely de-identified (Chen et al., 2014; Xu et al., 2019). All hiPSCs were cultured on dishes coated with hESC-qualified Matrigel (Corning) in mTeSR plus media (STEMCELL Technologies) under a feeder-free condition. The hiPSCs were passaged with ReLeSR media (STEMCELL Technologies) once per week.
Differentiation and culture of PMPs and pNPCs
PMPs were generated from the three pairs of Cont and DS hiPSC cell lines using a previously established protocol (Haenseler et al., 2017). The yolk sac embryoid bodies (YS-EBs) were generated by treating the YS-EBs with mTeSR 1 media (STEMCELL Technologies) supplemented with bone morphogenetic protein 4 (BMP4, 50 ng/ml), vascular endothelial growth factor (VEGF, 50 ng/ml), and stem cell factor (SCF, 20 ng/ml) for 6 days. To stimulate myeloid differentiation, the YS-EBs were plated on dishes with X-VIVO 15 medium (Lonza) supplemented with interleukin-3 (IL-3, 25 ng/ml) and macrophage colony-stimulating factor (M-CSF, 100 ng/ml). At 4–6 weeks after plating, human PMPs emerged into the supernatant and were continuously produced for more than 3 months. In this study, the fluorescence in situ hybridization (FISH) analyses were performed with a chromosome 21-specific probe (Vysis LSI 21 probe; Abbott Molecular) to examine the copy number of Hsa21 in the DS and control hiPSC-derived PMPs (Chen et al., 2014).
Human pNPCs were generated from Cont2 hiPSCs (Chen et al., 2016; Xu et al., 2019). The pNPCs were cultured in a medium, which is composed of a 1:1 mixture of Neurobasal (Thermo Fisher Scientific) and DMEM/F12 (Hyclone), supplemented with 1x N2, 1x B27-RA (Thermo Fisher Scientific), FGF2 (20 ng/ml, Peprotech), CHIR99021 (3 mM, Biogems), human leukemia inhibitory factor (hLIF, 10 ng/ml, Millipore), SB431542 (2 mM), and ROCK inhibitor Y-27632 (10 mM, Tocris). The pNPCs were passaged with TrypLE Express (Thermo Fisher Scientific) once per week. pNPCs within 6 passages were used for organoid generation.
Brain organoid culture
Each brain organoid was generated from a total of 10,000 cells (7,000 pNPCs and 3,000 PMPs), in each well of ultra-low-attachment 96-well plates in the presence of ROCK inhibitor Y-27632 (10 mM) (Xu et al., 2021). The culture medium was composed of a 1:1 mixture of PMP medium and NPC medium (1:1 mixture of Neurobasal and DMEM/F12, supplemented with 1x N2, 1x B27-RA, and FGF2 (20 ng/ml, Peprotech) for three days (day 3). Then, the organoids were transferred to ultra-low-attachment 6-well plates and cultured with 1:1 mixture of PMP medium and NPC medium for another 11 days (2 weeks). The medium was replenished every two days, and the cell culture plates were kept on an orbital shaker at a speed of 80 rpm/min starting from day 8. To promote neural and microglial differentiation, organoids were cultured in differentiation media, comprised of a 1:1 mixture of Neurobasal and DMEM/F12, supplemented with 1x N2 (Thermo Fisher Scientific), BDNF (20 ng/ml, Peprotech), GDNF (20 ng/ml, Peprotech), dibutyryl-cyclic AMP (1mM, Sigma), ascorbic acid (200 nM, Sigma), IL-34 (100 ng/ml, Peprotech), and Granulocyte-macrophage colony-stimulating factor (GM-CSF, 10 ng/ml, Peprotech) from day 15 onwards. As shown in our recent study (Xu et al., 2021), cells are highly proliferative in the early stages of organoid development. Ventricular zone-like structures containing proliferative neural stem cells are seen in organoids even after long-term culture, whereas PMPs differentiate into microglia that are not actively proliferating. Thus, the different proliferation rates between pNPCs vs. PMPs as well as protracted proliferation of human neural stem cells likely lead to a lower ratio of microglia cells in organoids than the starting 7:3 ratio. The medium was replenished every other day. After 4 weeks, the organoids were used for further experimentation.
In vitro differentiation of PMPs to microglia
PMPs were differentiated in the medium composed of DMEM/ F12 supplemented with N2, 2 mM Glutamax, 100 U/mL penicillin and 100 mg/mL streptomycin, 100 ng/mL M-CSF (Peprotech), 100 ng/mL IL-34 (Peprotech), and 10 ng/mL GM-CSF (Peprotech) for two weeks (Haenseler et al., 2017). The medium was changed once a week. After two weeks, cells were collected for Western blotting.
Cell transplantation
PMPs were collected from the supernatant and suspended at a concentration of 100,000 cells/μl in PBS. Cells were then injected into the brains of P0 immunodeficient mice (C;129S4-Rag2tm1.1Flv Csf1tm1(CSF1)Flv Il2rgtm1.1Flv/J, The Jackson Laboratory). The transplantation sites were bilateral from the midline = ±1.0 mm, posterior from bregma = −2.0 mm, and dorsoventral depths = −1.5 and −1.2 mm (Xu et al., 2020). All pups were placed in ice for 4–5 mins to anesthetize. The pups were then injected with 0.5 μl of cells into each site (four sites total), using a digital stereotaxic device (David KOPF Instruments) that was equipped with a neonatal mouse adapter (Stoelting). The pups were weaned at three weeks and kept for further experimentation at different time points.
Preparation of soluble S1 fractions
Soluble S1 fractions from human samples (provided by the University of California Alzheimer’s Disease Research Center (UCI-ADRC) and the Institute for Memory Impairments and Neurological Disorders) were prepared as described before (Sanchez-Mejias et al., 2016). The human tissues were homogenized in TBS (20 mM Tris-HCl, 140 mM NaCl, pH 7.5) containing protease and phosphatase inhibitors (Roche). Homogenates were ultracentrifuged (4 °C for 60 min) at 100,000×g (Optima MAX Preparative Ultracentrifuge, Beckman Coulter). Supernatants, S1 fractions, were aliquoted and stored at −80 °C.
Tau quantification by ELISA
The total amount of soluble tau in S1 fractions was determined by using an ELISA kit (human Tau, Invitrogen), according to the manufacturer’s protocol. The ELISA experiments were repeated in three independent experiments using triplicate replicas. The results were summarized in Table S3.
Intracerebral adult brain injection
All adult brain injections were performed using a Kopf stereotaxic apparatus (David Kopf, Tujunga, CA). Stereotaxic surgery was performed on two months old transplanted immunodeficient mice. The mice were aseptically injected with human brain extracts (Cont tau or DSAD tau) in the dorsal hippocampus and the overlying cortex (bregma: −2.5 mm; lateral: +2 mm; depth: −2.4 mm and −1.8 mm from the skull). A dose of tau at 3.2 μg, which was previously shown to be able to induce tau pathology in tau transgenic mice (Boluda et al., 2015; Iba et al., 2013; Peeraer et al., 2015), was injected into each human microglial chimeric mouse. Concentrations of tau per injection site were 0.8 μg/μl each site for both DSAD tau and Cont tau. The mice were injected with 1 μl of Cont or DSAD tau into each site (four sites total). After the injection, the mice were kept for further experiments.
Flow cytometric analysis
Single-cell suspensions from the chimeric brains were washed and suspended in PBS with 1% BSA and 1 mg/ml of anti-FcR to block FcR binding. After 10 min of incubation on ice, the appropriate primary Abs, unconjugated or conjugated to different fluorescent markers, were added to the cells at a concentration of 1–10 μg/ml and incubated for 30 mins on ice. Then, the cells were washed twice in PBS+1% BSA and fixed in PBS+1% paraformaldehyde. Flow cytometric analysis was performed on a FACS Calibur or Cytek Aurora cytometer, and results were analyzed with FlowJo software (TreeStar).
RNA isolation and quantitative reverse transcription PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, 15596026), and 600 μg RNA was reverse transcribed into complementary DNA (cDNA) using TaqMan™ Reverse Transcription Reagents (Thermo Fisher Scientific; N8080234). Total DNA was prepared with Superscript III First-Strand kit (Invitrogen). Real-time PCR was performed on the ABI 7500 Real-Time PCR System using the TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific). All primers are listed in Table S12. The 2–ΔΔCt method was used to calculate relative gene expression after normalization to the GAPDH internal control.
Bulk RNA-seq
PMPs generated from the three pairs of Cont and DS hiPSC lines were used for RNA extraction and RNA sequencing sample preparation. Total RNA was prepared with an RNAeasy kit (QIAGEN) (Chen et al., 2014) and libraries were constructed by using 600 ng of total RNA from each sample and utilizing a TruSeqV2 kit from Illumina (Illumina, San Diego, CA) following the manufacturer’s suggested protocol. The libraries were subjected to 75 bp paired read sequencing using a NextSeq500 Illumina sequencer to generate approximately 30 to 35 million paired-reads per sample. Fastq files were generated using the Bcl2Fastq software, version 1.8.4. The genome sequence was then indexed using the rsem-prepare-reference command. Each fastq file was trimmed using the fastp program (v0.12.2) and then aligned to the human genome (GRCh38/hg38) using HISAT2 (v.2.2.0). Gene counts were extracted from bam files using Rsubread/featureCounts in R with a UCSC transcript map. To analyze the transcripts, CPM > 1 was set as a cutoff to filter transcripts. Fold change > 2 was set as criteria to filter differential expressed genes (DEGs). The raw gene counts were processed with R package edgeR for differential expression analysis, exact Test hypothesis testing was used for each pairwise analysis. Differentially expressed genes were defined with |log2FC| > 1 between control and DS groups, and p-value < 0.05. Gene ontology enrichment analysis was performed with DAVID version 6.8, upregulated and downregulated genes were enriched separately. Data shown in the paper were from terms enriched in GOTERM_BP_DIRECT.
Tissue dissociation for scRNA-seq
After perfusion with cold PBS, whole brains were dissected and stored briefly in 1x DPBS. The tissue dissociation was then performed utilizing the Adult Brain Dissociation Kit (Miltenyi) according to the manufacturer’s instructions. Following tissue dissociation, the tissue was dissected into 1xmm3 pieces and placed into the C-tubes equipped with enzymes. These samples were further dissociated in gentleMACS OctoDissociator with heaters (Miltenyi) using the established preprogrammed protocol. Following enzymatic digestion, samples were isolated using a 70 μm cell strainer and pelleted by centrifugation. Myelin and debris byproducts were removed by debris removal solution, overlaid with 6mL of 1X DPBS, and spun at 3000xg for 10 min at 4°C. The supernatant was discarded, and the cell pellet was processed for Magnetic Isolation.
Magnetic Isolation of human microglia for scRNA-seq
Dissociated cell pellets were resuspended in 160 μL FACS buffer (0.5% BSA in 1X DPBS) + 40uL Mouse cell removal beads (Miltenyi) and incubated at 4°C for 15 min. The resulting samples were then isolated using LS columns and the MidiMACs separator (Miltenyi), the human cells were collected in the flow through. Using centrifugation (10 min, 400xg), the cells were pelleted and resuspended to 1,000 cells per microliter in FACS buffer for scRNA-seq library preparation, according to the previous study (Hasselmann et al., 2019).
ScRNA-seq
Following magnetic isolation, we used Chromium™ Single Cell 3′ Library and Gel Bead Kit v3.1, Chromium™ Single Cell Chip G Kit, and Chromium™ i7 Multiplex Kit, 96 rxns for capture and library preparation. The libraries were analyzed on Agilent 4200 TapeStation System using High Sensitivity D1000 ScreenTape Assay and quantified using KAPA qPCR. Libraries were then normalized to 10 nM before being pooled together. Next, the pooled library was clustered and sequenced on Illumina NovaSeq 6000 S4 flowcell for 150bp paired-end sequencing, Read 1 and Read 2 are sequenced from both ends of the fragment. For each individual library, the sequencing data from four unique indexes were combined before further analysis.
Raw data pre-processing
The sequencing data was aligned with the pooled mouse (mm10, Ensembl 93) and human (hg19, Ensembl 87) reference genomes (10x Genomics pre-built human and mouse reference genome v3.0.0) and interpreted via barcodes analyzed with Cell Ranger software (10x Genomics, v.6.0.2). The resulting matrices of gene count × barcodes were coded by individual sample identifiers and loaded into Seurat (v.4.0.3) software in R/Bioconductor. An initial analysis revealed a distinct cluster of human-expressing cells. Cells with gene number <200 or >10,000, and mitochondria gene >10% were excluded. Leaving a total number of 47,826 cells from four samples. Cells were summarized for all genes by species and those with >75% of reads aligning with hg19 were selected as human. Raw hg19 counts from these cells were loaded into a new Seurat object, normalized and clustered.
Data processing and quality control
The separated fastq files were re-aligned to the human hg19 reference genome using the same version of Cell Ranger as described above. Seurat R package v4.0.4 was used for quality control and dimensional reduction analysis. Gene-barcode matrices were merged after inputting into R as Seurat objects. Cells with gene number <200 or >4,000, and mitochondria gene >10% were excluded. Leaving a total number of 7,790 cells.
Dimensional reduction and differential expression analysis
Data were normalized by RPM following log transformation and top 2,000 highly variable genes were selected for scaling and principal component analysis (PCA). The top 15 principal components were used for downstream uniform manifold approximation and projection (UMAP) visualization and clustering. Louvain algorithm with resolution 0.2 was used to cluster cells, which resulted in 6 distinct cell clusters. Cluster 5 was removed due to the size of the cluster (15 cells). Differential expression analysis was performed by using Wilcoxon rank sum test embedded in FindMarkers function from Seurat. A gene was considered to be differentially expressed if it was detected in at least 25% of one group and with at least 0.25 log fold change between two groups and the significant level of Benjamini–Hochberg (BH) adjusted p-value < 0.05. Of note, cluster 4 accounted for a very small proportion in the DSAD tau group (0.64%), whereas in the Cont tau group, cluster 4 accounted for 6.66% (Fig.S5F). Thus, a small number of downregulated DEGs in cluster 4 of DSAD tau group were found, which were not enriched for any significant pathways in our GO analysis of cluster 4 (Fig.S6C).
Gene ontology (GO) and gene set enrichment analysis (GSEA)
GO analysis used the g:Profiler website (https://biit.cs.ut.ee/gprofiler/gost). All genes were calculated between Control and DSAD group before logFC, and adjusted p-value filtration were ranked by logFC value. Pre-defined gene sets were used to test their distribution on the ranked gene list, GSEA analysis and visualization were performed by gseaplot function from clusterProfiler v4.0.5 R package.
Pseudotime trajectory inference analysis
Single cell pseudotime trajectory was predicted by Monocle 3 v1.0.0. The gene-cell matrix was exported from Seurat object as input into Monocle 3 to generate cds object, along with the metadata information from Seurat. Cells were re-clustered by Monocle 3 using the default parameters. To learn trajectory graph and order cells in pseudotime, clustering information from Seurat object was projected into the cds object. Cells in the cluster with enriched homeostatic genes expression scores were defined as root cells.
shRNA knockdown
IFNAR1 shRNA (sc-35637-V), IFNAR2 shRNA (sc-40091-V), and non-targeting control shRNA (sc-108080) lentiviral particles were purchased from Santa Cruz Biotechnology. The IFNAR1 and IFNAR2 shRNA lentivirus particles carried three different shRNAs specifically targeted IFNAR1/IFNAR2 gene expression. DS2 and Tri-DS3 hiPSCs infected with lentivirus carrying the control shRNA (DS2 or Tri-DS3+ ContshRNA) or IFNAR1/2 shRNA (DS2 or Tri-DS3 + IFNAR1/2shRNA) were used in this study. Lentivirus was mixed with polybrene (5 mg/ml) in mTeSR plus and applied to undifferentiated hiPSCs overnight. The following day, the transduction medium was removed and replaced with fresh media. After three days of culture, a puromycin (0.75 mg/ml) selection was used for two weeks to select for transducing hiPSCs. Stable hiPSCs were then used for PMP differentiation. Knockdown efficiency was confirmed by examining IFNAR1 and IFNAR2 expression at both mRNA and protein levels.
Electrophysiology
Whole cell patch-clamp recording
Brain slice miniature excitatory postsynaptic current (mEPSC) recordings was carried out following an established protocol (Liu et al., 2017). Mice were anesthetized with Euthasol, and brains were quickly removed into ice cold (0 °C) oxygenated ACSF cutting solution (in mM): 50 sucrose, 2.5 KCl, 0.625 CaCl2, 1.2 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, and 2.5 glucose, pH to 7.3 with NaOH. 300 μm sagittal section slices were cut using a vibratome (VT 1200S; Leica). After 1 hour of recovery (30 °C) in artificial cerebrospinal fluid (ACSF) (in mM)125 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.25 NaH2PO4, 56 NaHCO3, and 10 glucose. For mEPSC recording, a Cs2+-based solution was used, which consisted of (in mM): 40 CsCl, 3.5 KCl, 10 HEPES, 0.05 EGTA, 90 K-gluconate, 1.8 NaCl, 1.7 MgCl2, 2 ATP-magnesium, 0.4 GTP-sodium, and 10 phosphocreatine. mEPSCs were recorded at a holding potential of −70 mV in the presence of 50 μM PTX and 1 μM tetrodotoxin (TTX; VWR, Catalog#89160–628). Electrophysiological data were analyzed using Clampfit 10.5 (Molecular Devices, USA). Brain slices after recordings were used for post hoc immunostaining with hTMEM119 to verify colocalization of xeno-grafted human microglia with Neurobiotin fluorescence-positive neurons labeled via recording electrodes.
Extracellular hippocampal slice recording
The extracellular recording of hippocampal slices was performed as described previously (Castillo et al., 2002). Mice brains were quickly immersed in cold (4°C) oxygenated cutting solution containing (in mM): 206 Sucrose, 11 D-Glucose, 2.5 KCl, 1 NaH2PO4, 10 MgCl2, 2 CaCl2, and 26 NaHCO3. Transverse 400 μm bilateral hippocampal sections were cut and transferred to an oxygenated ACSF contained (in mM): 125 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 2.5 glucose. After at least half hour 34°C of recovery, slices were transferred at room temperature and incubated for 1 hour. Field EPSPs were recorded with glass pipette (2–3MΩ). The stimulation electrode was placed in dorsal CA1b stratum radiatum equidistant from the CA1 stratum pyramidale. Single-pulse baseline stimulation was applied at 0.05 Hz with baseline intensity set to 40–50% of the maximum population spike-free fEPSP amplitude. LTP was induced by 4×100Hz stimulation with 200 ms interval between bursts.
Western Blotting
Western blotting was performed as described previously (Jin et al., 2018). Lysates from cells, S1 fractions, and human brain tissues were prepared using a RIPA buffer and the protein contents were measured using a Pierce™ BCA Protein Assay Kit (Thermo Scientific). Proteins were separated on 12% SDS-PAGE gradient gel and transferred onto a nitrocellulose membrane. The membrane was blocked with 5% non-fat milk for one hour at room temperature. After blocking, the membrane was incubated with primary antibody overnight at 4 °C overnight. Secondary antibodies conjugated to HRP were used in addition to ECL reagents (Pierce ECL Plus Western Blotting Substrate, Thermo scientific) for immunodetection. All the S1 fractions were derived from DSAD and Cont human brain tissues (Table S4). For quantification of band intensity, blots from three independent experiments for the molecule of interest were used. Signals were measured by ImageJ software and represented as relative intensity versus control. GAPDH was used as an internal control to normalize band intensity.
Tissue immunostaining, image acquisition, and analysis
Mouse brains and organoids were fixed with 4% paraformaldehyde. The organoids were placed in 25% sucrose and mouse brains were placed in 20% and later in 30% sucrose for dehydration. Following dehydration, organoids and brain tissues were immersed in OCT and frozen for sectioning. The frozen tissues were cryo-sectioned with 30-μm thickness for immunofluorescence staining. The organoids and tissues were blocked with a blocking solution (5% goat or donkey serum in PBS with 0.2% or 0.8% Triton X-100) at room temperature (RT) for 1 hour. The primary antibodies were diluted in the same blocking solution and incubated with the organoids or tissues at 4 °C overnight (all the primary antibodies are listed in Supplementary Table 12). The sections were washed with PBS and incubated with secondary antibodies for 1 hour at RT. After washing with PBS, the slides were mounted with antifade Fluoromount-G medium containing 1, 40,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Southern Biotechnology).
All images were captured with a Zeiss 800 confocal microscope. Large scale images in Fig. 3A, 3I, S4B, and S5B were obtained by confocal tile scan by the Zen software (Zeiss), these images are composites of individual images that have been stitched together. To obtain a 3D reconstruction, images were processed by the Zen software (Zeiss). To visualize synaptic pruning and phagocytic function, super-resolution images in Fig. 2–7, Supplementary Fig. 1–3, and 5–6 were acquired by Zeiss Airyscan super-resolution microscope at 63X with 0.2mm z-steps. To generate 3D-surface rendered images, super-resolution images were processed by Imaris software (Bitplane 9.5). To visualize the engulfment of PSD95+, Synapsin I+, AT8+ and PHF-1+ p-tau within microglia, CD68+ phagolysosome inside microglia, fluorescence found outside of the microglia were subtracted from the image via the mask function in Imaris software. For hTMEM119+ human microglia and Iba-1+/ hN-negative mouse microglia morphology analysis, the Filament function was used to generate filaments for each cell in the images, and dendrites were automatically rendered based on the hTMEM119 and Iba1 signal. The number of positive cells from each section was counted after a Z projection. The dystrophic (Iba-1+hN+) microglial morphological changes after tau injection were assessed by calculating the soma size and process length using the software Fiji with MorpholibJ plugin and integrated library as described before (Flores-Aguilar et al., 2020; Legland et al., 2016). Soma size was measured with a closing morphological filter connecting dark pixels and process length was analyzed with Skeletonize function.
Behavioral tests
We examined three groups of mice: (1) Cont chimeric mice that received transplantation of Cont 2 and Cont 3 cells; (2) DS + ContshRNA chimeric mice that received transplantation of DS2+ContshRNA or Tri-DS3+ContshRNA cells; (3) DS + IFNAR1/2shRNA chimeric mice that received transplantation of DS2+ IFNAR1/2shRNA or Tri-DS3+ IFNAR1/2shRNA cells; All behavior tests were performed in a randomized order by two investigator double-blinded to treatment.
Open field test
Mice were placed into a clear chamber (40 long × 40 wide × 40 cm high) under dim ambient light conditions. The activity was monitored by overhead video camera and analyzed by Anymaze software (Stoelting Co., IL) for 5 min in a single trial. Four squares were determined as the center and the other twelve squares along the center as the periphery. The collected results included total distance traveled and the total number of entries into the center of the field.
Elevated plus maze
The elevated plus maze was performed in a grey, non-reflective base plate cross (arms 35 cm long × 5 cm wide) elevated 50 cm above the floor. Two opposite arms were enclosed by the grey walls (35 cm long × 15 cm high), and the two other arms were open. A mouse was placed in the center of the apparatus facing an enclosed arm and allowed to explore the apparatus for 5 min. The total traveling distance and the time spent on the open arms were recorded.
Novel object recognition
As previously described (Xu et al., 2019), two sample objects in one clear box were used to examine learning and memory with 24 hours delays. Before testing, mice were habituated in a clear chamber (40 long × 40 wide × 40 cm high) for 10 minutes on 2 consecutive days under ambient light conditions. The activities were monitored by an overhead video camera and analyzed by an Anymaze software (Stoelting Co., IL). First, two identical objects, termed as ‘familiar’, were placed into the chamber, and the mouse was placed at the mid-point of the wall opposite the objects. After 10 minutes to explore the objects, the mouse was returned to the home cage. After 24 hours, one of the ‘familiar’ objects used for the memory acquisition was replaced with a ‘new’ object similar to the ‘familiar’ one. The mouse was again placed in the chamber for 3 minutes to explore the objects; The total traveling distance and the time spent investigating the new objects was assessed. The preference to the novel or old object was calculated as Time exploring new or old object / (Time exploring new or old object + Time exploring familiar object). There were no differences between the preference to novel and old objects, indicating that the mice may have vision problems.
Golgi staining
The mice were anesthetized, and whole brains were collected for Golgi staining using the FD Rapid Golgi Stain Kit (FD Neuro Technologies, Columbia, MD, USA; PK401), according to the manufacturer’s introduction. Z-stack images were acquired with a widefield microscope (Olympus BX63), and spine density was quantified using Image J software (NIH).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are represented as mean ± SEM. When only two independent groups were compared, significance was determined by using two-tailed unpaired t test with Welch’s correction. When three or more groups were compared, one-way ANOVA with Bonferroni post-hoc test was used. A p-value of < 0.05 was considered significant. All the analyses were done in GraphPad Prism v.9. All experiments were independently performed at least three times with similar results.
Supplementary Material
Table S6. DAM gene list related to Fig. 6A, 6E, senescence gene list related to Fig. 6C, and ISG genes related to Fig. 6K.
Highlights.
DS microglia over-prune synapses in hiPSC-based organoid and chimeric mouse models
DS microglia show dystrophic and senescent phenotypes in response to pathological tau
Type I IFN signaling is upregulated in DS microglia under pathophysiological conditions
Reducing IFNARs expression improves DS microglial functions and prevents senescence
Acknowledgements
This work was in part supported by grants from the NIH (R01NS102382, R01NS122108, and R01AG073779 to P.J.). We thank the UCI-ADRC, which is funded by NIH/NIA Grant P30AG066519 and the Brightfocus Foundation (BFF17-0008), for providing us with DSAD and Cont human brain tissues. PHF-1 antibody was a gift from Dr. Peter Davies at Albert Einstein College of Medicine, NY, USA. We thank Dr. Xiaobing Zhang from Florida State University and Dr. Zhengquan Tang from Anhui University in China for their suggestions on PPF and LTP recordings. We are also thankful to Mr. Haipeng Xue from Florida International University for the assistance with FISH experiment and Ms. Maharaib Syed from Rutgers University for the assistance with immunohistochemistry.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Financial Interests
The authors declare no competing financial interests.
References:
- Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, et al. (2017). iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 94, 278–293 e279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Lindo N, Baleriola J, de Los Rios V, Suarez T, and de la Rosa EJ (2019). RAG-2 deficiency results in fewer phosphorylated histone H2AX foci, but increased retinal ganglion cell death and altered axonal growth. Sci Rep 9, 18486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya P, Waugh KA, Sullivan KD, Nunez NG, Roselli E, Smith KP, Granrath RE, Rachubinski AL, Enriquez Estrada B, Butcher ET, et al. (2019). Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc Natl Acad Sci U S A 116, 24231–24241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avelar RA, Ortega JG, Tacutu R, Tyler EJ, Bennett D, Binetti P, Budovsky A, Chatsirisupachai K, Johnson E, Murray A, et al. (2020). A multidimensional systems biology analysis of cellular senescence in aging and disease. Genome Biol 21, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar E, and Barak B (2019). Microglia roles in synaptic plasticity and myelination in homeostatic conditions and neurodevelopmental disorders. Glia 67, 2125–2141. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Kleschevnikov AM, Becker A, Wagner GE, Lysenko LV, Yu YE, and Mobley WC (2015). Down Syndrome Cognitive Phenotypes Modeled in Mice Trisomic for All HSA 21 Homologues. PloS one 10, e0134861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito-Kwiecinski S, and Lancaster MA (2020). Brain Organoids: Human Neurodevelopment in a Dish. Cold Spring Harb Perspect Biol 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank T, and Prinz M (2017). Type I interferon pathway in CNS homeostasis and neurological disorders. Glia 65, 1397–1406. [DOI] [PubMed] [Google Scholar]
- Boluda S, Iba M, Zhang B, Raible KM, Lee VM, and Trojanowski JQ (2015). Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol 129, 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowski AB, Boesteanu AC, Mueller YM, Carafides C, Topham DJ, Altman JD, Jennings SR, and Katsikis PD (2007). Memory CD8+ T cells require CD28 costimulation. J Immunol 179, 6494–6503. [DOI] [PubMed] [Google Scholar]
- Braak H, and Del Tredici K (2015). The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain 138, 2814–2833. [DOI] [PubMed] [Google Scholar]
- Brownjohn PW, Smith J, Solanki R, Lohmann E, Houlden H, Hardy J, Dietmann S, and Livesey FJ (2018). Functional Studies of Missense TREM2 Mutations in Human Stem Cell-Derived Microglia. Stem cell reports 10, 1294–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, and Baker DJ (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, and Weiner HL (2018). Microglial signatures and their role in health and disease. Nat Rev Neurosci 19, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Sudhof TC, and Malenka RC (2002). RIM1alpha is required for presynaptic long-term potentiation. Nature 415, 327–330. [DOI] [PubMed] [Google Scholar]
- Chen C, Jiang P, Xue H, Peterson SE, Tran HT, McCann AE, Parast MM, Li S, Pleasure DE, Laurent LC, et al. (2014). Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat Commun 5, 4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, and Colonna M (2021). Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J Exp Med 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choong XY, Tosh JL, Pulford LJ, and Fisher EM (2015). Dissecting Alzheimer disease in Down syndrome using mouse models. Front Behav Neurosci 9, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello C, Yuan P, Schain A, and Grutzendler J (2015). Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun 6, 6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado E, and Barrena MJ (1996). Immune dysfunction in Down’s syndrome: primary immune deficiency or early senescence of the immune system? Clin Immunol Immunopathol 78, 209–214. [DOI] [PubMed] [Google Scholar]
- Das I, and Reeves RH (2011). The use of mouse models to understand and improve cognitive deficits in Down syndrome. Dis Model Mech 4, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss J, Richards C, Nelson L, and Oliver C (2013). Prevalence of autism spectrum disorder symptomatology and related behavioural characteristics in individuals with Down syndrome. Autism 17, 390–404. [DOI] [PubMed] [Google Scholar]
- Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, and Amit I (2018). Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 173, 1073–1081. [DOI] [PubMed] [Google Scholar]
- DiPatre PL, and Gelman BB (1997). Microglial cell activation in aging and Alzheimer disease: partial linkage with neurofibrillary tangle burden in the hippocampus. J Neuropathol Exp Neurol 56, 143–149. [DOI] [PubMed] [Google Scholar]
- Doran E, Keator D, Head E, Phelan MJ, Kim R, Totoiu M, Barrio JR, Small GW, Potkin SG, and Lott IT (2017). Down Syndrome, Partial Trisomy 21, and Absence of Alzheimer’s Disease: The Role of APP. J Alzheimers Dis 56, 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards FA (2019). A Unifying Hypothesis for Alzheimer’s Disease: From Plaques to Neurodegeneration. Trends Neurosci 42, 310–322. [DOI] [PubMed] [Google Scholar]
- Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjorn M, Kuss M, Liu Y, Porcu G, Kolkova K, Friis Rundsten C, et al. (2015). Lack of Neuronal IFN-beta-IFNAR Causes Lewy Body- and Parkinson’s Disease-like Dementia. Cell 163, 324–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Aguilar L, Iulita MF, Kovecses O, Torres MD, Levi SM, Zhang Y, Askenazi M, Wisniewski T, Busciglio J, and Cuello AC (2020). Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain 143, 3653–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PCG, Ngu H, et al. (2018). Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep 22, 832–847. [DOI] [PubMed] [Google Scholar]
- Frisch SM, and MacFawn IP (2020). Type I interferons and related pathways in cell senescence. Aging Cell 19, e13234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galatro TF, Holtman IR, Lerario AM, Vainchtein ID, Brouwer N, Sola PR, Veras MM, Pereira TF, Leite REP, Moller T, et al. (2017). Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci 20, 1162–1171. [DOI] [PubMed] [Google Scholar]
- Galloway A, Saveliev A, Lukasiak S, Hodson DJ, Bolland D, Balmanno K, Ahlfors H, Monzon-Casanova E, Mannurita SC, Bell LS, et al. (2016). RNA-binding proteins ZFP36L1 and ZFP36L2 promote cell quiescence. Science 352, 453–459. [DOI] [PubMed] [Google Scholar]
- Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, Balic A, Giladi A, Sheban F, Dutertre CA, et al. (2019). Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell 179, 1609–1622 e1616. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero A, De Strooper B, and Arancibia-Carcamo IL (2021). Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci 44, 714–727. [DOI] [PubMed] [Google Scholar]
- Haenseler W, Sansom SN, Buchrieser J, Newey SE, Moore CS, Nicholls FJ, Chintawar S, Schnell C, Antel JP, Allen ND, et al. (2017). A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem cell reports 8, 1727–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Chen M, Wang F, Windrem M, Wang S, Shanz S, Xu Q, Oberheim NA, Bekar L, Betstadt S, et al. (2013). Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 12, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Hanson JE, and Sheng M (2018). Microglia in Alzheimer’s disease. J Cell Biol 217, 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmann J, Coburn MA, England W, Figueroa Velez DX, Kiani Shabestari S, Tu CH, McQuade A, Kolahdouzan M, Echeverria K, Claes C, et al. (2019). Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron 103, 1016–1033 e1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head E, Lott IT, Hof PR, Bouras C, Su JH, Kim R, Haier R, and Cotman CW (2003). Parallel compensatory and pathological events associated with tau pathology in middle aged individuals with Down syndrome. J Neuropathol Exp Neurol 62, 917–926. [DOI] [PubMed] [Google Scholar]
- Heo JI, Kim W, Choi KJ, Bae S, Jeong JH, and Kim KS (2016). XIAP-associating factor 1, a transcriptional target of BRD7, contributes to endothelial cell senescence. Oncotarget 7, 5118–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman IR, Skola D, and Glass CK (2017). Transcriptional control of microglia phenotypes in health and disease. J Clin Invest 127, 3220–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li SM, Ramakrishnan S, Merry KM, Shi QQ, Rosenthal A, Barres BA, et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopp SC, Lin Y, Oakley D, Roe AD, DeVos SL, Hanlon D, and Hyman BT (2018). The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J Neuroinflammation 15, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, Di Blasio AM, Giuliani C, Tung S, Vinters HV, et al. (2015). Accelerated epigenetic aging in Down syndrome. Aging Cell 14, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini S, Michaelsen-Preusse K, Grigoryan G, Chhatbar C, Kalinke U, and Korte M (2020). Type I Interferon Receptor Signaling in Astrocytes Regulates Hippocampal Synaptic Plasticity and Cognitive Function of the Healthy CNS. Cell Rep 31, 107666. [DOI] [PubMed] [Google Scholar]
- Hu Y, Fryatt GL, Ghorbani M, Obst J, Menassa DA, Martin-Estebane M, Muntslag TAO, Olmos-Alonso A, Guerrero-Carrasco M, Thomas D, et al. (2021). Replicative senescence dictates the emergence of disease-associated microglia and contributes to Abeta pathology. Cell Rep 35, 109228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel P, Urban C, Bergant V, Schneider WM, Knauer B, Stukalov A, Scaturro P, Mann A, Brunotte L, Hoffmann HH, et al. (2019). A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat Immunol 20, 493–502. [DOI] [PubMed] [Google Scholar]
- Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, and Lee VM (2013). Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33, 1024–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis I, McNally B, Willette M, Peeples ME, Chaussabel D, Durbin JE, Ramilo O, Mejias A, and Flano E (2012). Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J Virol 86, 5422–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Turkalj L, and Xu R (2020). High-Fidelity Modeling of Human Microglia with Pluripotent Stem Cells. Cell Stem Cell 26, 629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, and Vitorica J (2008). Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci 28, 11650–11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin MM, Wang F, Qi D, Liu WW, Gu C, Mao CJ, Yang YP, Zhao Z, Hu LF, and Liu CF (2018). A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Front Aging Neurosci 10, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581 e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legland D, Arganda-Carreras I, and Andrey P (2016). MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32, 3532–3534. [DOI] [PubMed] [Google Scholar]
- Li Q, and Barres BA (2018). Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18, 225–242. [DOI] [PubMed] [Google Scholar]
- Loh XY, Sun QY, Ding LW, Mayakonda A, Venkatachalam N, Yeo MS, Silva TC, Xiao JF, Doan NB, Said JW, et al. (2020). RNA-Binding Protein ZFP36L1 Suppresses Hypoxia and Cell-Cycle Signaling. Cancer Res 80, 219–233. [DOI] [PubMed] [Google Scholar]
- Long JM, and Holtzman DM (2019). Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 179, 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes KO, Sparks DL, and Streit WJ (2008). Microglial dystrophy in the aged and Alzheimer’s disease brain is associated with ferritin immunoreactivity. Glia 56, 1048–1060. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott IT, and Head E (2005). Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol Aging 26, 383–389. [DOI] [PubMed] [Google Scholar]
- Lott IT, and Head E (2019). Dementia in Down syndrome: unique insights for Alzheimer disease research. Nature reviews Neurology 15, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens JR, and Eyo UB (2022). Microglia and Neurodevelopmental Disorders. Annu Rev Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunnon K, Keohane A, Pidsley R, Newhouse S, Riddoch-Contreras J, Thubron EB, Devall M, Soininen H, Kloszewska I, Mecocci P, et al. (2017). Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol Aging 53, 36–47. [DOI] [PubMed] [Google Scholar]
- Martini AC, Helman AM, McCarty KL, Lott IT, Doran E, Schmitt FA, and Head E (2020). Distribution of microglial phenotypes as a function of age and Alzheimer’s disease neuropathology in the brains of people with Down syndrome. Alzheimers Dement (Amst) 12, e12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews S, Branch Woods A, Katano I, Makarov E, Thomas MB, Gendelman HE, Poluektova LY, Ito M, and Gorantla S (2019). Human Interleukin-34 facilitates microglia-like cell differentiation and persistent HIV-1 infection in humanized mice. Mol Neurodegener 14, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Luehmann M, and Prinz M (2015). Myeloid cells in Alzheimer’s disease: culprits, victims or innocent bystanders? Trends Neurosci 38, 659–668. [DOI] [PubMed] [Google Scholar]
- Navarro V, Sanchez-Mejias E, Jimenez S, Munoz-Castro C, Sanchez-Varo R, Davila JC, Vizuete M, Gutierrez A, and Vitorica J (2018). Microglia in Alzheimer’s Disease: Activated, Dysfunctional or Degenerative. Front Aging Neurosci 10, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmo J, Johnston DA, Dodart JC, MacGregor-Sharp MT, Weller RO, Nicoll JAR, Verma A, and Carare RO (2020). Peri-arterial pathways for clearance of alpha-Synuclein and tau from the brain: Implications for the pathogenesis of dementias and for immunotherapy. Alzheimers Dement (Amst) 12, e12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormel PR, Vieira de Sa R, van Bodegraven EJ, Karst H, Harschnitz O, Sneeboer MAM, Johansen LE, van Dijk RE, Scheefhals N, Berdenis van Berlekom A, et al. (2018). Microglia innately develop within cerebral organoids. Nat Commun 9, 4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmyer M, Helisalmi S, Soininen H, Laakso M, Riekkinen P Sr., and Alafuzoff I (1999). Reactive microglia in aging and dementia: an immunohistochemical study of postmortem human brain tissue. Acta Neuropathol 97, 383–392. [DOI] [PubMed] [Google Scholar]
- Pandya H, Shen MJ, Ichikawa DM, Sedlock AB, Choi Y, Johnson KR, Kim G, Brown MA, Elkahloun AG, Maric D, et al. (2017). Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat Neurosci 20, 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, et al. (2010). Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004–2006. Birth Defects Res A Clin Mol Teratol 88, 1008–1016. [DOI] [PubMed] [Google Scholar]
- Pascoal TA, Benedet AL, Ashton NJ, Kang MS, Therriault J, Chamoun M, Savard M, Lussier FZ, Tissot C, Karikari TK, et al. (2021). Microglial activation and tau propagate jointly across Braak stages. Nat Med 27, 1592–1599. [DOI] [PubMed] [Google Scholar]
- Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, Duytschaever H, Ver Donck L, Torremans A, Sluydts E, et al. (2015). Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis 73, 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto B, Morelli G, Rastogi M, Savardi A, Fumagalli A, Petretto A, Bartolucci M, Varea E, Catelani T, Contestabile A, et al. (2020). Rescuing Over-activated Microglia Restores Cognitive Performance in Juvenile Animals of the Dp(16) Mouse Model of Down Syndrome. Neuron 108, 887–904 e812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy ER, Chiu G, Li S, Propson NE, Kanchi R, Wang B, Coarfa C, Zheng H, and Cao W (2022). Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid beta plaques. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, and Hertzog PJ (2013). Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res 41, D1040–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Mejias E, Navarro V, Jimenez S, Sanchez-Mico M, Sanchez-Varo R, Nunez-Diaz C, Trujillo-Estrada L, Davila JC, Vizuete M, Gutierrez A, et al. (2016). Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol 132, 897–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidehpour RK, Higdon RE, Crawford NG, Neltner JH, Ighodaro ET, Patel E, Price D, Nelson PT, and Bachstetter AD (2021). Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol Aging 99, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, and Griffin WS (1997). Glial-neuronal interactions in Alzheimer disease: progressive association of IL-1alpha+ microglia and S100beta+ astrocytes with neurofibrillary tangle stages. J Neuropathol Exp Neurol 56, 285–290. [PubMed] [Google Scholar]
- Simmons DA, Casale M, Alcon B, Pham N, Narayan N, and Lynch G (2007). Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 55, 1074–1084. [DOI] [PubMed] [Google Scholar]
- Smith AM, and Dragunow M (2014). The human side of microglia. Trends Neurosci 37, 125–135. [DOI] [PubMed] [Google Scholar]
- Smith LK, He Y, Park JS, Bieri G, Snethlage CE, Lin K, Gontier G, Wabl R, Plambeck KE, Udeochu J, et al. (2015). beta2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat Med 21, 932–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobue A, Komine O, Hara Y, Endo F, Mizoguchi H, Watanabe S, Murayama S, Saito T, Saido TC, Sahara N, et al. (2021). Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer’s disease. Acta Neuropathol Commun 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van der Brug MP, Foreman O, Paw JS, Modrusan Z, Beach TG, Serrano GE, et al. (2020). Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep 31, 107843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, and Bechmann I (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 118, 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Khoshbouei H, and Bechmann I (2020). Dystrophic microglia in late-onset Alzheimer’s disease. Glia 68, 845–854. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Sammons NW, Kuhns AJ, and Sparks DL (2004). Dystrophic microglia in the aging human brain. Glia 45, 208–212. [DOI] [PubMed] [Google Scholar]
- Suizu F, Hiramuki Y, Okumura F, Matsuda M, Okumura AJ, Hirata N, Narita M, Kohno T, Yokota J, Bohgaki M, et al. (2009). The E3 ligase TTC3 facilitates ubiquitination and degradation of phosphorylated Akt. Dev Cell 17, 800–810. [DOI] [PubMed] [Google Scholar]
- Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, Smith KP, Liggett LA, Gomez EB, Galbraith MD, et al. (2016). Trisomy 21 consistently activates the interferon response. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Suzuki S, Millar DG, Unno M, Hara H, Calzascia T, Yamasaki S, Yokosuka T, Chen NJ, Elford AR, et al. (2006). A critical role for the innate immune signaling molecule IRAK-4 in T cell activation. Science 311, 1927–1932. [DOI] [PubMed] [Google Scholar]
- Svoboda DS, Barrasa MI, Shu J, Rietjens R, Zhang S, Mitalipova M, Berube P, Fu D, Shultz LD, Bell GW, et al. (2019). Human iPSC-derived microglia assume a primary microglia-like state after transplantation into the neonatal mouse brain. Proc Natl Acad Sci U S A 116, 25293–25303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, and Crack PJ (2014). Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol Aging 35, 1012–1023. [DOI] [PubMed] [Google Scholar]
- Taylor JM, Moore Z, Minter MR, and Crack PJ (2018). Type-I interferon pathway in neuroinflammation and neurodegeneration: focus on Alzheimer’s disease. J Neural Transm (Vienna) 125, 797–807. [DOI] [PubMed] [Google Scholar]
- Teipel SJ, and Hampel H (2006). Neuroanatomy of Down syndrome in vivo: a model of preclinical Alzheimer’s disease. Behav Genet 36, 405–415. [DOI] [PubMed] [Google Scholar]
- Vasileiou PVS, Evangelou K, Vlasis K, Fildisis G, Panayiotidis MI, Chronopoulos E, Passias PG, Kouloukoussa M, Gorgoulis VG, and Havaki S (2019). Mitochondrial Homeostasis and Cellular Senescence. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verina T, Kiihl SF, Schneider JS, and Guilarte TR (2011). Manganese exposure induces microglia activation and dystrophy in the substantia nigra of non-human primates. Neurotoxicology 32, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Yue H, Hu Z, Shen Y, Ma J, Li J, Wang XD, Wang L, Sun B, Shi P, et al. (2020). Microglia mediate forgetting via complement-dependent synaptic elimination. Science 367, 688–694. [DOI] [PubMed] [Google Scholar]
- Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, Chen L, Chen Y, Zhu G, Yin W, et al. (2019). Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 176, 334–347 e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh KA, Araya P, Pandey A, Jordan KR, Smith KP, Granrath RE, Khanal S, Butcher ET, Estrada BE, Rachubinski AL, et al. (2019). Mass Cytometry Reveals Global Immune Remodeling with Multi-lineage Hypersensitivity to Type I Interferon in Down Syndrome. Cell Rep 29, 1893–1908 e1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner G, Moss J, Smith P, and Howlin P (2014). Autism characteristics and behavioural disturbances in ~ 500 children with Down’s syndrome in England and Wales. Autism Res 7, 433–441. [DOI] [PubMed] [Google Scholar]
- Xu R, Boreland AJ, Li X, Erickson C, Jin M, Atkins C, Pang ZP, Daniels BP, and Jiang P (2021). Developing human pluripotent stem cell-based cerebral organoids with a controllable microglia ratio for modeling brain development and pathology. Stem cell reports 16, 1923–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Brawner AT, Li S, Liu JJ, Kim H, Xue H, Pang ZP, Kim WY, Hart RP, Liu Y, et al. (2019). OLIG2 Drives Abnormal Neurodevelopmental Phenotypes in Human iPSC-Based Organoid and Chimeric Mouse Models of Down Syndrome. Cell Stem Cell 24, 908–926 e908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Li X, Boreland AJ, Posyton A, Kwan K, Hart RP, and Jiang P (2020). Human iPSC-derived mature microglia retain their identity and functionally integrate in the chimeric mouse brain. Nat Commun 11, 1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue QS, and Streit WJ (2011). Microglial pathology in Down syndrome. Acta Neuropathol 122, 455–466. [DOI] [PubMed] [Google Scholar]
- Yamada T, and Yamanaka I (1995). Microglial localization of alpha-interferon receptor in human brain tissues. Neurosci Lett 189, 73–76. [DOI] [PubMed] [Google Scholar]
- Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, Guha M, Li N, Chen Q, Yang T, et al. (2015). DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep 11, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Chen L, and Qu H (2016). CSGene: a literature-based database for cell senescence genes and its application to identify critical cell aging pathways and associated diseases. Cell Death Dis 7, e2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S6. DAM gene list related to Fig. 6A, 6E, senescence gene list related to Fig. 6C, and ISG genes related to Fig. 6K.
Data Availability Statement
The RNA-seq and scRNA-seq datasets generated in this study have been deposited at NCBI GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Mouse anti-β-amyloid | Biolegend | RRID:AB_2715854 |
| Goat anti-PSD95 | Abcam | RRID:AB_298846 |
| AF700 anti-human IFNAR1 | R&D Systems | RRID:AB_2920857 |
| FITC anti-human CD45 | BioLegend | RRID:AB_2566368 |
| PE anti-human IFNAR2 | Miltenyi Biotec | RRID:AB_2652221 |
| Mouse anti-AT8 | Invitrogen | RRID:AB_223647 |
| Mouse anti-CD43 | Invitrogen | RRID:AB_763493 |
| Mouse anti-CD45 | Invitrogen | RRID:AB_11063696 |
| Mouse anti-CD68 | Invitrogen | RRID:AB_10987212 |
| Mouse anti-GAPDH | Santa Cruz Biotechnologies | RRID:AB_627679 |
| Mouse anti-human nuclei | Millipore | RRID:AB_827439 |
| Mouse anti-Ki67 | Thermo Fisher Scientific | RRID:AB_2341197 |
| Mouse anti-MAP2 | Santa Cruz Biotechnologies | RRID:AB_1126219 |
| Mouse anti-PHF1 | Dr. Peter Davies, Albert Einstein College of Medicine | N/A |
| Rabbit anti-Synapsin 1 | Millipore | RRID:AB_90757 |
| Rabbit anti-CD235 | Invitrogen | RRID: AB_2544630 |
| Rabbit anti-Ferritin | Sigma-Aldrich | RRID:AB_259684 |
| Rabbit anti-Iba1 | Wako | RRID:AB_839504 |
| Rabbit anti-IFNAR2 | Invitrogen | RRID:AB_2545779 |
| Rabbit anti-Ki67 | Cell signaling | RRID:AB_2687446 |
| Rabbit anti-TMEM119 | Invitrogen | RRID:AB_2648507 |
| Donkey anti-goat 488 | Invitrogen | RRID:AB_2534102 |
| Donkey anti-goat 594 | Invitrogen | RRID:AB_142540 |
| Donkey anti-goat 647 | Invitrogen | RRID:AB_2535864 |
| Goat anti-mouse 488 | Invitrogen | RRID:AB_2534088 |
| Goat anti-mouse 594 | Invitrogen | RRID:AB_2534091 |
| Goat anti-mouse 647 | Invitrogen | RRID:AB_2535804 |
| Goat anti-rabbit 488 | Invitrogen | RRID:AB_2536097 |
| Goat anti-rabbit 594 | Invitrogen | RRID:AB_2534095 |
| Goat anti-rabbit 647 | Invitrogen | RRID:AB_2535813 |
| Bacterial and Virus Strains | ||
| IFNAR1 shRNA | Santa Cruz | sc-35637-V |
| IFNAR2 shRNA | Santa Cruz | sc-40091-V |
| Control shRNA | Santa Cruz | sc-108080 |
| Biological samples | ||
| Human brain tissue samples | NIH NeuroBioBank | See Table S3 for detailed information |
| Human brain tissue samples | UCI-ADRC | See Table S4 for detailed information |
| Chemicals, peptides, and recombinant proteins | ||
| 2-Mercaptoethanol | Gibco | Catalog# 21985-023 |
| 4% paraformaldehyde | Thermo Fisher Scientific | Catalog# J19943-K2 |
| Adult brain dissociation kit | Miltenyi Biotec | Catalog# 130-107-677 |
| Ascorbic acid | Sigma | Catalog# A4403 |
| B27-RA | Thermo Fisher Scientific | Catalog# 12587010 |
| BDNF | Peprotech | Catalog# 450-02 |
| BMP4 | Peprotech | Catalog# 120-05ET |
| CHIR99021 | Biogems | Catalog# 2520691 |
| Compensation beads | Miltenyi Biotec | Catalog# 01-2222-41 |
| Dibutyryl cAMP | Sigma | Catalog# D0260 |
| DMEM/F12 | HyClone | Catalog# SH3002201 |
| D-Sucrose | Fisher Bioreagents | Catalog# BP220-212 |
| FGF-2 | Peprotech | Catalog# 100-18B |
| GDNF | Peprotech | Catalog# 450-10 |
| GlutaMAX | Gibco | Catalog# 35050-061 |
| GM-CSF | Peprotech | Catalog# 300-03 |
| Growth factor reduced matrigel | Corning | Catalog# 354230 |
| hLIF | Millipore | Catalog# LIF1005 |
| IL-3 | Peprotech | Catalog# 200-03 |
| IL-34 | Peprotech | Catalog# 200-34 |
| Matrigel | Corning | Catalog# 354277 |
| M-CSF | Invitrogen | Catalog# PHC9501 |
| Mini-PROTEAN TGX Gels | Bio-rad | Catalog# 4561043 |
| Mouse cell depletion cocktail | Miltenyi Biotec | Catalog# 130-104-694 |
| M-PER Mammalian protein extraction reagent | Thermo Fisher Scientific | Catalog# 78501 |
| mTeSR 1 | Stemcell Technologies | Catalog# 85850 |
| mTeSR plus | Stemcell Technologies | Catalog# 100-0276 |
| N2 | Thermo Fisher Scientific | Catalog# 17502048 |
| Neurobasal | Thermo Fisher Scientific | Catalog# 21103-049 |
| Pierce BCA Protein assay kit | Thermo Fisher Scientific | Catalog# 23225 |
| Pierce ECL Western blotting substrate | Thermo Fisher Scientific | Catalog# 32106 |
| ReLeSR | Stemcell Technologies | Catalog# 100-0484 |
| RNeasy Kit | Qiagen | Catalog# 74004 |
| SB431542 | Stemgent | Catalog# 04-0010-05 |
| SCF | Peprotech | Catalog# 300-07 |
| Taqman Gene expression Master Mix | Applied Biosystem | Catalog# 4369016 |
| Tau (total) human ELISA kit | Invitrogen | Catalog# KHB0041 |
| Triton X-100 | Sigma | Catalog# T9284 |
| TRIzol | Thermo Fisher Scientific | Catalog# 15596026 |
| TrypLE Express | Thermo Fisher Scientific | Catalog# 12605028 |
| Tween 20 | Fisher Bioreagents | Catalog# BP337-100 |
| VEGF | Peprotech | Catalog# 100-20 |
| X-Vivo 15 | Lonza | Catalog# 04-418Q |
| Y-27632 | Tocris | Catalog# 1254 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3' GEM, Library & Gel Bead Kit v3.1 | 10x Genomics | Catalog# 1000128 |
| Chromium Next GEM Chip G Single Cell Kit | 10x Genomics | Catalog#1000127 |
| Deposited data | ||
| RNA-seq data | This paper | NCBI GEO: GSE189227 |
| scRNA-seq data | This paper | NCBI GEO: GSE189227 |
| Single Index Kit T Set A | 10x Genomics | Catalog#1000213 |
| Experimental Model | ||
| Cell lines | Coriell Institute for Medical Research | See Table S1 for detailed information |
| Mice: Rag2-/-hCSF1 | The Jackson Laboratory | Stock no:017708 |
| Oligonucleotides | ||
| qPCR primers | Thermo Fisher Scientific | See Table S11 for detailed information |
| Software and algorithms | ||
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| Adobe Photoshop | Adobe | https://www.adobe.com/products/photoshop.html |
| BioRender | BioRender.Inc | https://biorender.com/ |
| Cell Ranger v6.1.1 | 10x Genomics | https://github.com/10XGenomics/cellranger |
| Cluster Profiler 4.0.5 | https://guangchuangyu.github.io/software/clusterProfiler/ | |
| Clampfit 10.5 | Molecular Devices | https://support.moleculardevices.com/s/ |
| FlowJo_V10 | TreeStar | https://www.flowjo.com/solutions/flowjo/ |
| g:Profiler | e104_eg51_p15_3922dba | https://biit.cs.ut.ee/gprofiler/gost |
| GraphPad Prism 9.2.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.net/software/fiji/ |
| Imaris 9.5.1 | Bitplane | https://imaris.oxinst.com |
| Monocle v 1.0.0 | https://cole-trapnell-lab.github.io/monocle3/ | |
| R 4.1.0 | https://www.r-project.org | |
| RStudio | RStudio | https://www.rstudio.com/products/rstudio/download/ |
| Seurat v4.0.4 | https://satijalab.org/seurat/ | |
| Zen 2.3 | Carl Zeiss | N/A |