

Abstract
The hemizygous R47H variant of Triggering receptor expressed on myeloid cells 2 (TREM2), a microglia-specific gene in the brain, increases risk for late-onset Alzheimer’s disease (AD). Using transcriptomic analysis of single-nuclei from brain tissues of patients with AD carrying the R47H mutation or the common variant (CV)-TREM2, we found that R47H-associated microglial subpopulations had enhanced inflammatory signatures reminiscent of previously identified disease-associated microglia (DAM) and hyperactivation of AKT, one of the signaling pathways downstream of TREM2. We established a tauopathy mouse model with heterozygous knock-in of the human TREM2 with the R47H mutation or CV, and found that R47H induced and exacerbated TAU-mediated spatial memory deficits in female mice. Single-cell transcriptomic analysis of microglia from these mice also revealed transcriptomic changes induced by R47H that had substantial overlaps with R47H microglia in human AD brains, including robust increases in proinflammatory cytokines, activation of AKT signaling, and elevation of a subset of disease-associated microglial signatures. Pharmacological AKT inhibition with MK-2206 largely reversed the enhanced inflammatory signatures in primary R47H microglia treated with TAU fibrils. In R47H heterozygous tauopathy mice, MK-2206 treatment abolished a tauopathy-dependent microglial subcluster, and rescued tauopathy-induced synapse loss. By uncovering disease-enhancing mechanisms of the R47H mutation conserved in human and mouse, our study supports inhibitors of AKT signaling as a microglial modulating strategy to treat AD.
One-sentence Summary:
R47H-TREM2 mutation enhances AKT signaling in human AD microglia and mediates proinflammatory and synaptic toxicity in a tauopathy mouse model.
Introduction
Alzheimer’s disease (AD) is the most common form of late-onset dementia. Genome-wide association studies have identified many risk alleles for late-onset sporadic AD that are highly expressed in microglia (1, 2), providing compelling genetic evidence for important roles of microglia in AD pathogenesis. Among these risk genes, Triggering receptor expressed on myeloid cells 2 (TREM2) is the strongest immune-specific risk factor identified to date, with the heterozygous R47H point mutation substantially increasing the odds ratio of developing late-onset AD (1, 2).
TREM2 is a single transmembrane receptor expressed exclusively in cells of the myeloid lineage, especially microglia (3, 4). Upon ligand engagement, TREM2, together with its adaptor DNAX activating protein of 12 kDa (DAP12), recruits Spleen associated tyrosine kinase (SYK) and triggers several signaling cascades such as Phosphoinositide 3-kinase (PI3K)-AKT and Mitogen-activated protein kinase (MAPK) pathways (5, 6). These TREM2-dependent pathways in turn regulate many microglial functions, including inflammatory cytokine secretion, proliferation, phagocytosis, and cell survival (7–12).
In the context of neurodegenerative mouse models, TREM2 is required for the conversion of microglia into disease-associated microglia (DAM) or a microglial neurodegenerative phenotype (MGnD) (13, 14). This MGnD microglia-state can be activated by apoptotic cells and is partially mediated through TREM2’s interaction with Apolipoprotein E (APOE) (13). These microglia are characterized by downregulation of homeostatic genes, such as Purinergic receptor P2Y12 (P2ry12), Transmembrane protein 119 (Tmem119) and Spalt like transcription factor 1 (Sall1), and upregulation of pro-inflammatory signatures such as Apoe, Axl receptor tyrosine kinase (Axl), Toll-like receptor 2 (Tlr2), Cluster of differentiation 74 (Cd74), and Integrin subunit alpha X (Itgax). Currently, it is unclear whether this DAM state is neuroprotective or neurotoxic for disease progression. Deletion of mouse Trem2 (mTrem2) prevents microglial conversion to this disease-state and protects against tauopathy-induced atrophy (15, 16). mTrem2 deficiency in amyloid models, however, leads to increased amyloid toxicity, likely due to the role of TREM2 in plaque compaction (17–20). Furthermore, human AD-microglia seem to be enriched in some of these DAM genes, such as APOE and CD74 and show overlap in molecular pathways related to lipid and lysosomal biology. However, there is likely to be human-specific AD-microglia subpopulations since many gene signatures do not overlap between the mouse and human AD-associated microglia (21, 22). These observations suggest the role of TREM2 and DAMs in neurodegenerative diseases is context- and disease state-specific.
Little is known about how the R47H mutation of TREM2 contributes to AD. Previous studies reported that patients with AD carrying the heterozygous R47H variant show higher neuritic plaque densities, reduced microglial coverage of amyloid plaques and more severe plaque-associated neuritic dystrophy, as well as increased accumulation of autophagosomes in microglia (7, 20, 23). One bulk-tissue transcriptomic study showed that, several immune-related genes are decreased in R47H carriers such as Interferon regulatory factor 8 (IRF8) and Allograft inflammatory factor 1 (AIF1), suggesting either a decrease in the number of microglia or decreased expression of these genes on a per-cell basis (24). Transcriptomic studies at either the single-cell level or with a large sample size of patient brain tissues have not been done. In mouse models, homozygous knock-in of R47H human TREM2 (R47H-hTREM2) leads to deficits in microglial amyloid plaque compaction, similar to mTrem2-deficient mice, and increases TAU staining and dystrophic neurites bypassing plaques (20, 25). In a recent study, male P301S tauopathy mice expressing homozygous R47H-hTREM2 exhibited reduced TAU phosphorylation, brain atrophy, and synapse loss compared to mice expressing CV-hTREM2 (26), similar to the phenotype of mTrem2-deficient tauopathy mice. However, it remains a puzzling conundrum how the R47H mutation appears to protect against tauopathy in mice yet elevates AD risk in humans.
In the current study, we uncovered an R47H-enriched microglia subpopulation by performing single-nuclei RNA sequencing (snRNA-seq) analysis of brain tissue from 46 patients with AD carrying the common variant (CV) or the R47H mutation of TREM2. To investigate the functional changes induced by R47H in AD, we used a CRISPR-based genetic tool to replace one allele of mTrem2 with the common variant (CV)- or R47H-hTREM2, generating a heterozygous R47H-hTREM2 mouse model that was then crossed to the P301S tauopathy model. Our female heterozygous R47H-hTREM2 tauopathy mice had enhanced spatial memory deficits. In addition, R47H-associated microglia upregulated a subset of DAM signatures, increased expression of pro-inflammatory cytokines, and enhanced AKT signaling pathways in response to tau pathology. Pharmacological inhibition of AKT reversed the transcriptomic and pro-inflammatory cytokine profiles in TAU fibril-treated primary microglia, as well as decreased the R47H-associated microglial subpopulation and protected against synaptic toxicity in tauopathy mice. Together, our study uncovered disease-enhancing mechanisms of the R47H mutation and a potential therapeutic strategy for modulating brain immune responses to treat AD.
Results
R47H Induces Cell Type and Sex-Specific Transcriptional Changes in Human AD
To dissect the pathogenic mechanisms associated with TREM2R47H in patients with AD, we performed snRNA-seq of mid-frontal cortical tissues from 46 patients with AD harboring the TREM2 common-variant (CV) or a single allele of the R47H mutation (n=22 CV, 24 R47H samples, Fig. 1A, fig. S1, A and B, table S1). The samples were matched in age and TAU burdens (fig. S1, C and D), as well as clinical dementia rating, if known (table S1). Following an established human snRNA-seq protocol (27, 28), we sequenced 323,140 nuclei and used 263,672 nuclei for downstream analysis after removal of potential multiplets using DoubletFinder (29) and filtering for low-quality nuclei determined by thresholding gene counts, UMI counts, and percent mitochondrial genes per nuclei (fig. S1, E–I, table S2). Using reference gene sets for cluster annotations (30, 31), we identified the major cell types of the brain and observed that cell types were similarly represented in all samples sequenced, with the exception of some samples having very few excitatory neurons (Fig. 1, B and C, fig. S1, J and K).
Fig. 1. R47H Mutation Induces Cell Type- and Sex-Specific Transcriptional Changes in Brains of Patients with AD.
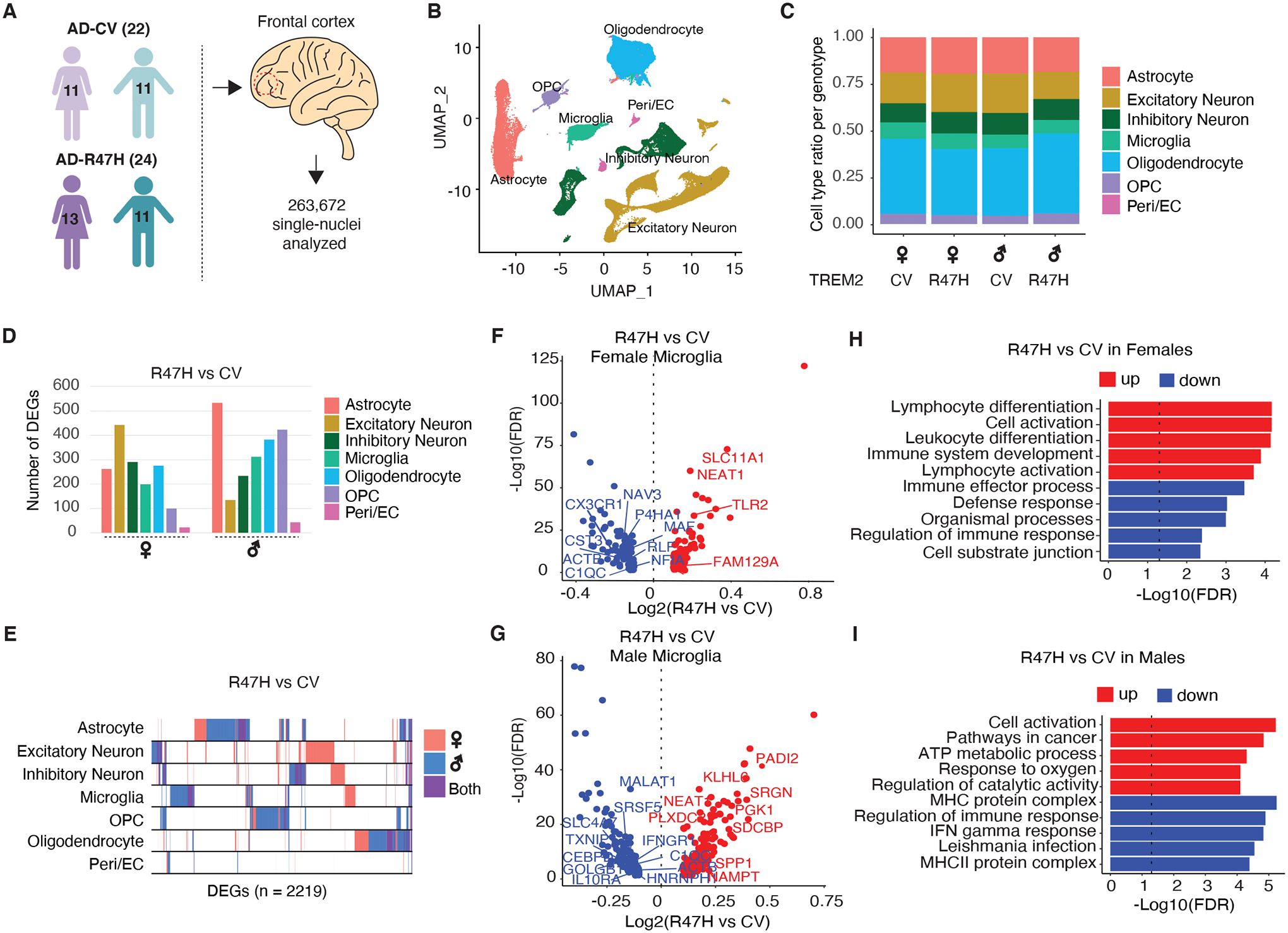
(A) Schematic showing the sex and genotypes of age-matched human donors used for snRNA-seq. n = 263,672 nuclei were isolated from the mid-frontal cortex of patients with AD carrying the CV-TREM2 variant (n=22) and patients with AD carrying the R47H-TREM2 variant (n=24). Purple and turquoise cartoons denote females and males, respectively. See also table S1.
(B) UMAP plots of all single nuclei and their annotated cell types. Peri/EC = pericyte/endothelial cells, OPC = Oligodendrocyte precursor cells.
(C) Proportion of cell types for each of the 4 genotypes.
(D) Number of DEGs between R47H vs. CV samples for each cell type and each sex. FDR<0.05. See also table S3.
(E) Binary plot indicating whether a gene (column) is a DEG or not in a given cell type (rows) or in each sex (pink: female; blue: male; purple: overlapping in both sexes) (n=2,219 DEGs).
(F and G) Volcano plots of significant DEGs (FDR < 0.05) between R47H-TREM2 and CV-TREM2 samples in females (F) and males (G). Genes overlapping with DAM signatures highlighted (14). See also table S3.
(H and I) Bar plots of Gene Ontology pathways enriched in DEGs identified in F and G for female (H) and male (I) samples. Dashed line indicates the threshold of significant enrichment for the pathway analysis (-Log10(FDR) ≥ 1.3).
We first performed differential expression analysis to compare the effects of the R47H mutation in each cell type and sex. The mutation was associated with many transcriptional changes in all cell types in both sexes (Fig. 1D, table S3). TREM2R47H carriers exhibited sex-specific transcriptomic changes, with a higher number of differentially expressed genes (DEGs) in male versus female glia, including microglia, astrocytes, and oligodendrocytes, but far fewer sex-specific alterations in excitatory neurons. We found little overlap of the DEGs among different cell types (rows, Fig. 1E). Specifically, in microglia, the R47H mutation induced sex-specific DEGs, with some of these genes reminiscent of those altered in DAM compared to control microglia, including upregulation of TLR2 and downregulation of C-X3-C motif chemokine receptor 1 (CX3CR1) in females and upregulation of Secreted phosphoprotein 1 (SPP1) and downregulation of Metastasis associated lung adenocarcinoma transcript 1 (MALAT1) in males (Fig. 1, F and G). Indeed, the molecular pathways enriched in these DEGs were also sex-specific, with R47H microglia from female samples upregulating immune activation pathways whereas male samples showing upregulation of metabolic and ATP pathways (Fig. 1, H and I).
Human R47H AD-Microglia Exhibit Hyperactivation of Inflammatory and AKT Signaling
To further dissect the transcriptomic changes in microglia induced by TREM2R47H, we subclustered the 20,461 microglia cells from all samples and identified 12 different transcriptional states (Fig. 2A, table S4) that had contributions from all samples (fig. S2). Based on subcluster marker genes, we identified 7 clusters that had high expression of microglial genes such as P2RY12, CD14, and TREM2, and low expression of other CNS cell type markers, such as Mannose receptor C-type 1 (MRC1) and Protein tyrosine phosphatase receptor type C (PTPRC) indicative of macrophages (MAC1 and MAC2), Synaptotagmin 1 (SYT1) and Neurexin 1 (NRXN1) for neurons (N1 and N2) and Myelin oligodendrocyte glycoprotein (MOG) and Proteolipid protein 1 (PLP1) for oligodendrocytes (OG1) (Fig. 2B, table S4). We focused our analyses on these 7 pure-microglia subclusters (MG1-MG7). When split by TREM2 genotype, we found subtle differential distributions of microglial subclusters between TREM2R47H and TREM2CV samples (Fig. 2C), with some variation between the sexes (fig. S2). We focused on MG4, which was the only cluster significantly more enriched in TREM2R47H samples (p=0.048; Fig. 2C), though no differences were noted when the sexes were analyzed separately (fig. S2).
Figure 2. R47H Mutation Increases TREM2-Signaling in a Unique Microglia Cluster in Brains of Patients with AD.
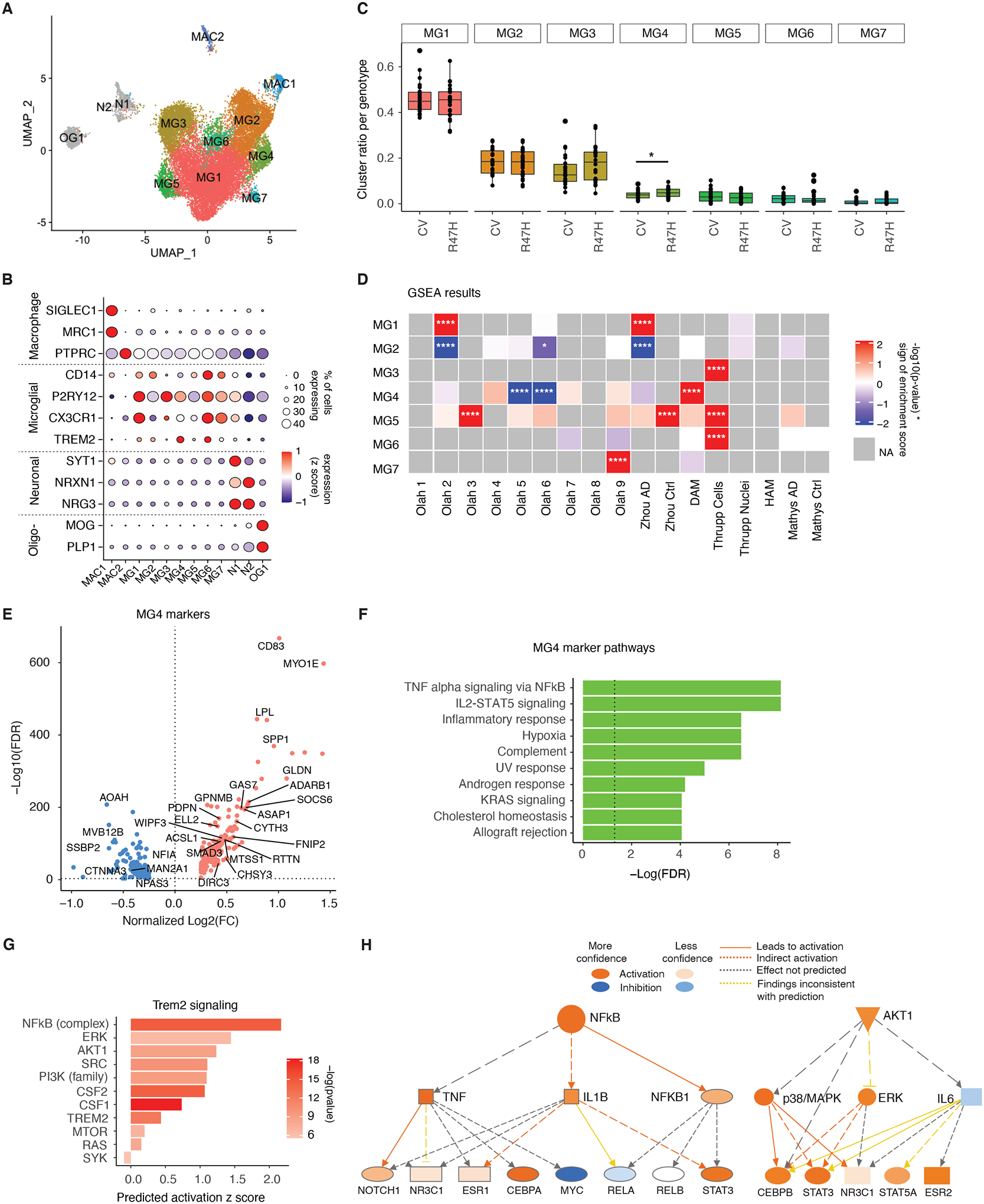
(A) UMAP of microglia subclusters for all nuclei. MG = microglia, MAC = macrophage, N = neuron, OG = oligodendrocyte.
(B) Dot plot of selected marker genes expressed by each subcluster identified in (A).
(C) Boxplot of proportion of microglia subcluster per genotype. *p=0.048, negative binomial generalized linear model adjusted for brain bank location, sex, age, and APOE genotype.
(D) Heatmap of gene set enrichment analysis results (GSEA) for subcluster gene signatures. Colors denote positive enrichment (+1, red) or negative enrichment (−1, blue) multiplied by the −log10(p-value). Comparison datasets used are the following: Olah (32), Zhou (24), DAM (14), Thrupp (34), HAM (33), Mathys (21). *p<0.05, ****p<0.0001.
(E) Volcano plot of significant DEGs (FDR < 0.05) between MG4 and all other clusters. Genes overlapping with DAM signatures highlighted (14). See also table S4.
(F) Bar plot of GSEA Hallmark pathways based on the unique, non-overlapping markers for MG4 identified in (E). Dashed line indicates the threshold of significant enrichment for the pathway analysis (-Log10(FDR) ≥ 1.3).
(G) IPA of genes involved in TREM2 signaling based on MG4 gene signatures identified in (E).
(H) Diagram of the NF-κB and AKT activation network predicted by IPA upstream regulator analysis in (G).
Gene set enrichment analysis showed some of our microglial subclusters overlapped with previously-published microglial datasets (Fig. 2D) (14, 21, 24, 32–34). MG4, enriched in TREM2R47H samples, was most reminiscent of the previously identified mouse DAM microglia (14), with genes such as Lipoprotein lipase (LPL), Cluster of differentiation 83 (CD83), and SPP1 being upregulated in these cells (Fig. 2, D and E, fig. S3). The R47H-enriched MG4 signatures were further analyzed using pathway enrichment analysis (Fig. 2F). The top pathway involved was Tumor necrosis factor (TNF)-α signaling via Nuclear factor kappa B (NF-κB), as well as other immune pathways such as Interleukin 2 (IL2)- Signal transducer and activator of transcription 5 (STAT5) signaling and inflammatory response, suggesting an elevated proinflammatory state (Fig. 2F). Upstream and downstream mediators of TREM2 signaling, including NF-κB, Colony-stimulating factors 1 and 2 (CSF1/2), and AKT, were predicted to be activated in human R47H-enriched microglia (Fig. 2, G and H). Together, in patients with AD, the R47H mutation expanded a unique microglial subpopulation reminiscent of DAMs and characterized by hyperactivation of TREM2-associated signaling molecules, including increases in pro-inflammatory and AKT pathways.
R47H-hTREM2 Exacerbates Inflammation in Female Tauopathy Mice
To further dissect the molecular pathways induced by the R47H mutation, we generated knock-in mouse lines expressing one copy of CV- (hTREM2CV/+) or R47H-hTREM2 (hTREM2R47H/+) cDNA at the mTrem2 locus using CRISPR (Fig. 3A). PCR and Sanger sequencing confirmed the correct recombination and insertion of human TREM2-CV and TREM2-R47H cDNA at the mTrem2 locus (fig. S4, A and C–F). We did not detect any non-specific integration in the hTREM2R47H/+ mouse line. However, a non-specific integration event occurred in hTREM2CV/+ mice at an unknown mouse genomic region (fig. S4, B, G, and H). Nevertheless, hTREM2CV/+ and hTREM2R47H/+ mice had equivalent amounts of hTREM2 protein (Fig. 3, B and C). TAU pathology strongly correlates with cognitive deficits in AD (35, 36). P301S mice, which express a human MAPT gene with the P301S mutation, develop hallmarks of tauopathy, including gliosis, TAU inclusions, and cognitive deficits, including hippocampal-dependent memory and spatial learning deficits seen in patients with AD (37). hTREM2R47H/+ mice were crossed with P301S mice to generate P301S hTREM2R47H/+ and their littermate P301S mTrem2+/+ controls; hTREM2CV/+ mice were crossed with P301S mice to generate their respective littermate controls (fig. S4I). The R47H mutation did not affect the quantity of hTREM2 and mTrem2 mRNA (Fig. 3, D and E), allowing us to assess the effects of the heterozygous R47H variant in vivo.
Figure 3. R47H-hTREM2 Increases Inflammatory Signatures and Exacerbates Spatial Learning and Memory Deficits in Female Tauopathy Mice.
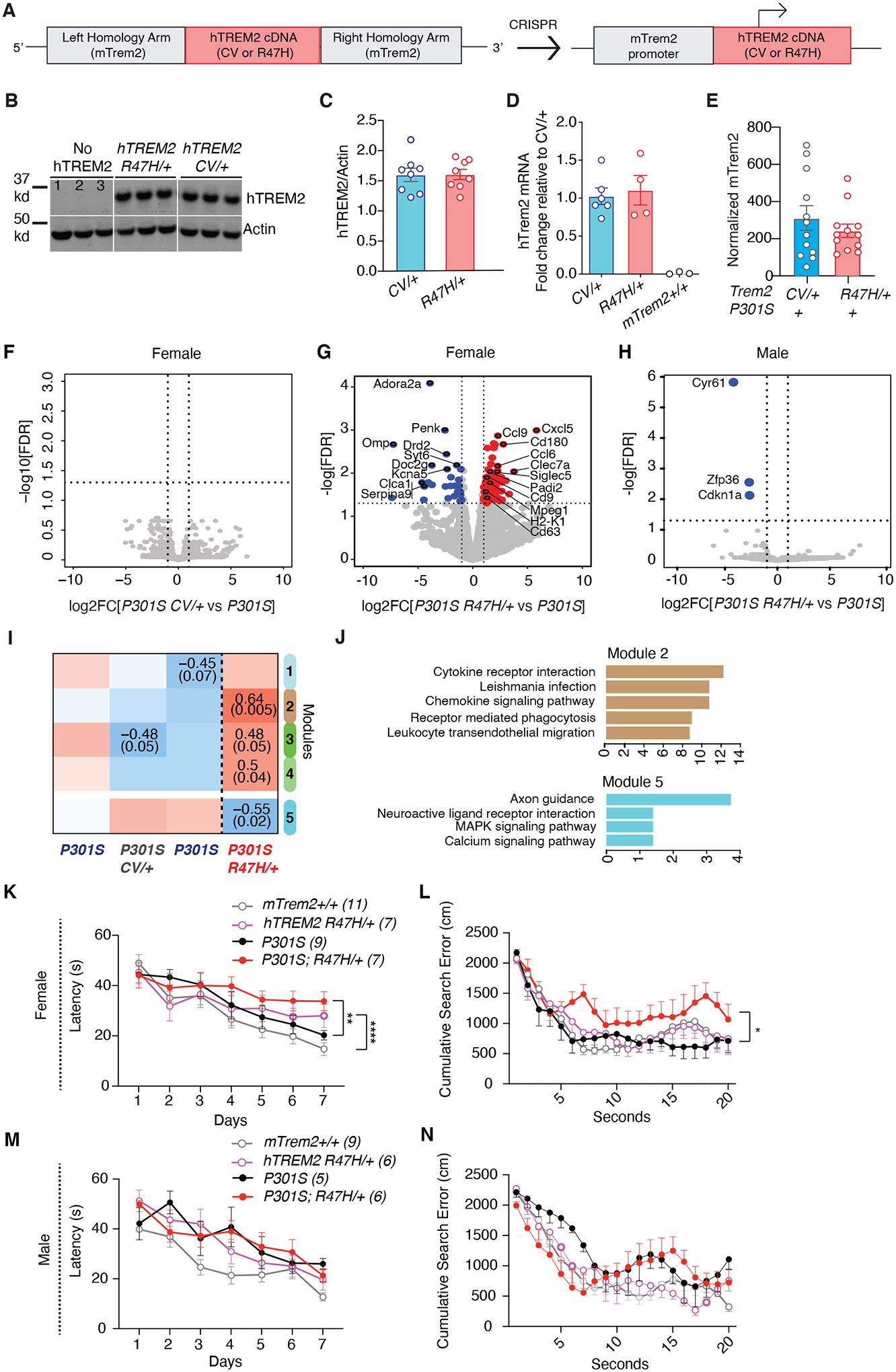
(A) The human TREM2 donor vector was designed with two 1-kilobase long arms homologous to mTrem2 flanking CV or R47H hTREM2 cDNA sequence. When inserted into the genome, hTREM2 cDNA is driven by the endogenous mTrem2 promoter.
(B) Representative western blot of RIPA-soluble cortical lysates from 8–9-month-old mice immunoblotted for hTREM2 and β-actin. Lane 1=mTrem2−/−, Lanes 2–3=mTrem2+/+.
(C) Quantification of hTREM2 normalized by β-actin of the entire cohort by western blot. Student’s two-tailed t-test.
(D) Quantitative real-time PCR analysis of cortical tissue from 3–4-month-old mice for hTREM2 mRNA. Samples were run in triplicate, and averages of the three wells were used for quantification. 2−ddCT calculation method used, normalized to Gapdh and relative to hTREM2CV/+. Each dot represents the average of three wells from one mouse. Two-tailed Mann-Whitney U-test comparing CV/+ and R47H/+.
(E) Bar plot of normalized mTrem2 RNA expression of bulk hippocampal tissue from 8–9-month-old P301S hTREM2R47H/+ and P301S hTREM2CV/+ mice. Student’s two-tailed t-test.
(F) Volcano plot of bulk RNA-seq data of hippocampal tissue from female P301S hTREM2CV/+ and line-specific female P301S control. Vertical dashed lines indicate log2FC ± 1. Horizontal dashed line indicates −log10(0.05). Wald test used. (n = 3 mice for P301S; n = 6 mice for P301S hTREM2CV/+).
(G) Volcano plot of bulk RNA-seq data of hippocampal tissue from 8–9-month-old female P301S hTREM2R47H/+ mice and line-specific female P301S littermate controls. Blue dots are genes with significantly higher normalized counts in P301S controls than in P301S hTREM2R47H/+ samples (28 mRNAs). Red dots are genes with significantly higher normalized counts in P301S hTREM2R47H/+ samples than P301S controls (94 mRNAs). Highlighted upregulated genes are disease-associated microglial (DAM) genes and genes involved in inflammation whereas highlighted downregulated genes are the most significantly downregulated genes. Vertical dashed lines indicate log2FC ± 1. Horizontal dashed line indicates −log10(0.05). Wald test used. (n = 3 mice for P301S; n = 5 mice for P301S hTREM2R47H/+). See also table S5.
(H) Volcano plot of bulk RNA-seq data of hippocampal tissue from male P301S hTREM2R47H/+ and line-specific male P301S littermate controls. Blue dots are genes with significantly higher normalized counts in P301S controls than P301S hTREM2R47H/+ samples (3 mRNAs). Vertical dashed lines indicate log2FC ± 1. Horizontal dashed line indicates −log10(0.05). Wald test used. (n = 2 mice for P301S; n = 5 mice for P301S hTREM2R47H/+).
(I) Heatmap of results from WGCNA of bulk RNA-seq data from (F and G), with only the significant module associations shown (top number: Pearson correlation, bottom number: adjusted p-value). Brown and cyan modules were the most significant. **p = 0.005, *p = 0.02.
(J) Top 5 enriched KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways of genes in brown and cyan modules from the WGCNA in (I). Colors of the bars represent the WGCNA module. See also table S6.
(K and M) Latency to reach the platform during hidden trials (d1-d7) for female (K) and male (M) hTREM2R47H/+ and P301S hTREM2R47H/+ and their mTrem2+/+ and P301S mTrem2+/+ littermate control mice. **p=0.003, ****p=0.0001, STATA mixed-effects modeling.
(L and N) Cumulative search error for female (L) and male (N) hTREM2R47H/+ and P301S hTREM2R47H/+ and their mTrem2+/+ and P301S littermate control mice. *U=9, p=0.0164, two-tailed Mann-Whitney U-test of area under the curve.
Behavioral data represent the combination of two behavioral cohorts that were run independently. Values are mean ± SEM. See also fig. S4–S6.
We first compared the hippocampal transcriptomes of 7- to 9-month-old male and female P301S hTREM2R47H/+ or P301S hTREM2CV/+ mice with their respective littermate P301S mTrem2+/+ controls. No transcriptomic changes were induced in female P301S hTREM2CV/+ mice compared with P301S mTrem2+/+ controls (Fig. 3F), indicating that CV-hTREM2 phenocopies mTrem2. In contrast, R47H induced upregulation of 94 genes, including several DAM genes (Ccl6, Clec7a, Siglec5, Cd9, Cd63) (14) and other inflammatory genes (Cxcl5, Ccl9), and 28 downregulated genes, including neuron-associated genes (Adora2a, Syt6, Serpina9, Penk) (Fig. 3G, table S5). These R47H-specific alterations in female tauopathy mice were not observed in male P301S R47H-hTREM2 mice, which exhibited only three downregulated genes compared with their male littermate P301S controls (Fig. 3H).
We further assessed the pathways induced by the R47H mutation in female tauopathy mice using weighted gene-correlation network analysis (WGCNA), and identified modules with statistically significant correlation to P301S hTREM2R47H/+ mice, including modules 5 and 2 (p=0.005 for module 2 and p=0.02 for module 5, Fig. 3I). Pathway analysis showed that module 2, which exhibited the most positive correlation with the P301S hTREM2R47H/+ genotype, was enriched with transcripts encoding cytokines/chemokines and cytokine receptors (Ccr5, Ccl5, Ccl3, Cxcl5) (Fig. 3J, table S6). The module 2, which negatively correlated with the P301S hTREM2R47H/+ genotype, was enriched in transcripts encoding axon guidance molecules (Sema6b, Sema3f, Epha8, Ephb6) (Fig. 3J, table S6). Together, these data suggest an upregulation of pro-inflammatory transcripts and a concomitant decrease in neuronal signaling genes in female P301S hTREM2R47H/+ mice compared to control animals.
R47H-hTREM2 Exacerbates Spatial Memory Deficits in Female Tauopathy Mice
We next used the Morris Water Maze test to assess how a single allele of R47H-hTREM2 and CV-hTREM2 may affect TAU-induced deficits in spatial learning and memory. Consistent with the downregulation of neuronal gene expression, female P301S R47H-hTREM2 exhibited significantly impaired spatial learning compared to their littermate P301S mTrem2+/+ controls (p=0.003; Fig. 3K). P301S hTREM2R47H/+ female mice also made significantly more search errors during the 72-hour probe trial than other groups (p=0.0164; Fig. 3L), suggesting that the R47H mutation enhances tauopathy-induced spatial learning and memory deficits. In contrast, male P301S hTREM2R47H/+ mice did not exhibit exacerbation in spatial learning and memory deficits compared to their littermate P301S mTrem2+/+ controls (Fig. 3, M and N), consistent with their similar transcriptomes (Fig. 3H). hTREM2CV/+ and mTrem2+/+ littermate mice behaved similarly to each other in both the absence and presence of tauopathy, regardless of sex, confirming that hTREM2CV/+ phenocopies mTrem2+/+ (fig. S5, A–D). No differences were observed between genotypes in locomotion in the open field (fig. S5, E–H) nor in the percentage of time spent in the open arms of the elevated plus maze (fig. S5, I–L), ruling out genotype differences in hyperactivity and anxiety, which could confound the spatial memory test results.
The R47H mutation did not impact the accumulation of insoluble TAU aggregates detected using a conformation-specific antibody, MC1 (38), suggesting that the disease-enhancing effects of R47H-hTREM2 in female P301S mice were not mediated by elevation in toxic TAU load (fig. S6). Indeed, even in the absence of TAU pathology, the R47H mutation led to modest spatial learning deficits in females (Fig. 3K). Taken together, our transcriptome and functional findings show that the R47H mutation worsens the inflammatory responses and the toxic effects induced by TAU irrespective of TAU pathology load, in a sex-dependent manner.
R47H-hTREM2 Enhances Disease-associated Microglial Signatures and AKT Signaling in Female Tauopathy Mice
Our snRNA-seq of human AD microglia revealed a modest expansion of the DAM-related microglial subpopulation in R47H carriers. We next specifically probed the effects of the R47H mutation on the microglial transcriptome in response to TAU pathology by performing single-cell RNA-seq (scRNA-seq) using the Smart-Seq2 platform (39). Microglia were isolated from the hippocampal tissue of 8-month-old female mTrem2+/+, hTREM2R47H/+, P301S mTrem2+/+, and P301S hTREM2R47H/+ mice, gating on CD45intCD11b+ cells (fig. S7, A and B). Out of the 1,480 cells that were sorted, 1,424 passed quality control thresholds (fig. S7, C–G). mTrem2 expression was decreased in hTREM2R47H/+ microglia compared to mTrem2+/+ microglia, confirming the replacement of one allele of mTrem2 (fig. S7H). Two distinct clusters were identified by unsupervised clustering of these 1,424 cells (Fig. 4A). Whereas cluster 1 microglia were found in all 4 genotypes, cluster 2 microglia were mainly associated with the expression of P301S TAU (Fig. 4B). hTREM2R47H/+ expression significantly increased the proportion of cluster 2 microglia in P301S mice (p<0.0001; Fig. 4, B and C). Compared to cells of cluster 1, cluster 2 cells upregulated several DAM transcripts, such as C-Type lectin domain containing 7A (Clec7a), Cathepsin B (Ctsb), Axl, Cystatin F (Cst7), Apoe, and Cd63 (Fig. 4, D and E, table S7), consistent with the increased transcripts observed in the bulk-tissue RNA-seq data (Fig. 3G). Cluster 2 cells also had expression of transcripts not seen in DAMs, including those involved in the interferon response pathway, such as Interferon regulatory factor 7 (Irf7), Interferon induced with helicase C domain 1 (Ifih1), Interferon induced transmembrane protein 3 (Ifitm3), MX Dynamin like GTPase 1 (Mx1), Interferon induced protein 44 (Ifi44), and Interferon induced protein with tetratricopeptide repeats 3 (Ifit3) (table S7). Whereas classical microglial genes, such as Hexosaminidase subunit Beta (Hexb), were present in both clusters, the homeostatic microglial gene P2ry12 was downregulated in cluster 2 cells (Fig. 4E). A direct comparison of cluster 2 marker genes versus DAM signature genes showed a significant positive correlation (R=0.7908; Fig. 4F). Thus, in the presence of TAU pathology, hTREM2R47H/+ enhances the DAM-like subpopulation and increases expression of Trem2-dependent microglial transcripts associated with neurodegeneration (MGnD), such as Apoe, Itgax, Lpl, Axl, and Cst7 (13, 14) (red, Fig. 4F). Given that activation of MGnD microglia-state is partially mediated through TREM2’s interaction with APOE (13), we further examined the microglial Apoe expression in brain sections of P301S hTREM2R47H/+ mice compared to P301S hTREM2+/+ mice by RNAscope. Indeed, the proportion of microglia expressing Apoe was significantly increased in P301S hTREM2R47H/+ mice (~90%) compared to P301S mTrem2+/+ (~60%) in the dentate gyrus of the hippocampus (p= 0.0254; Fig. 4, G–I).
Figure 4. R47H-hTREM2 Enhances the Disease-Associated Microglia Population and Elevates AKT Signaling.
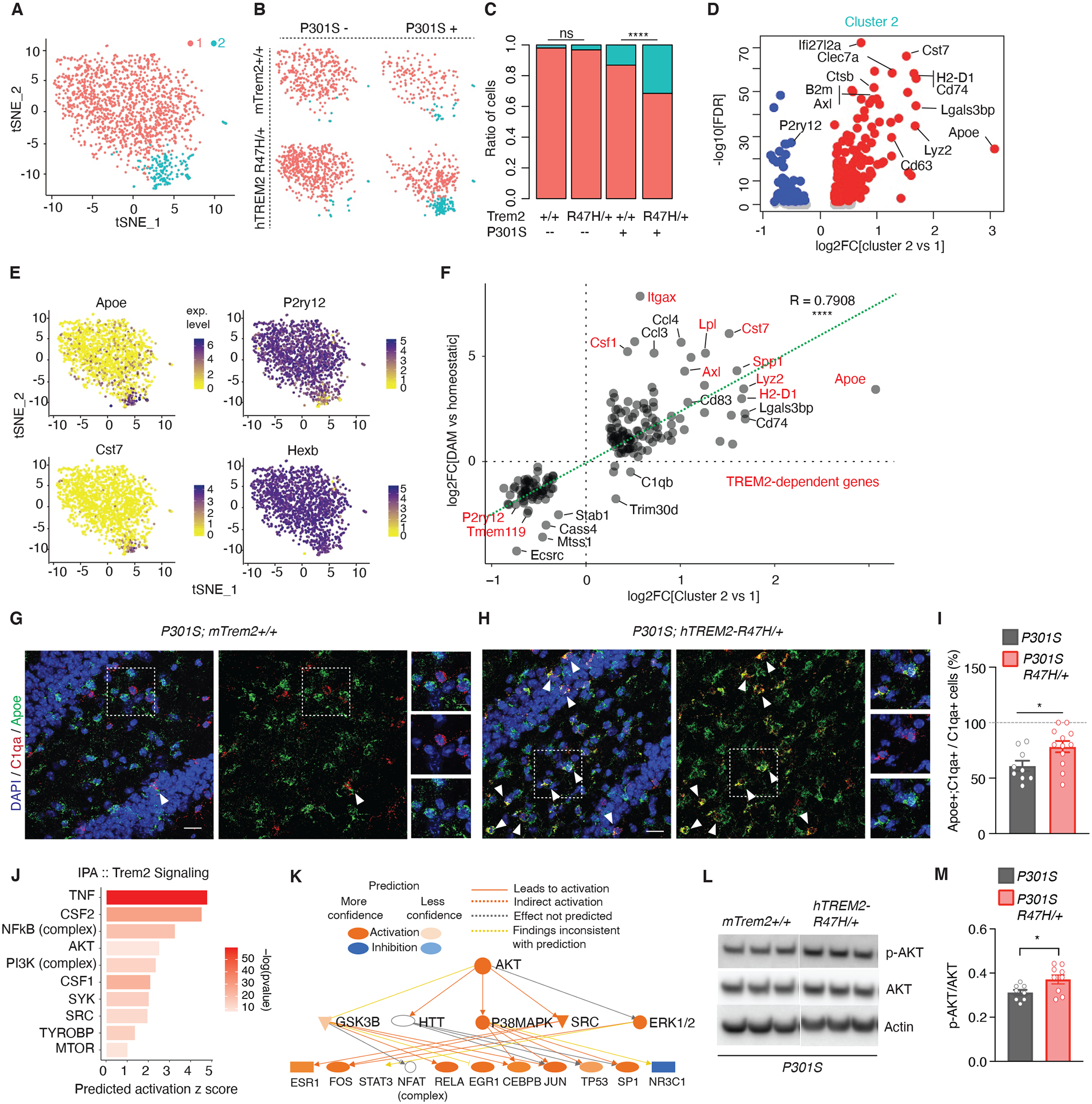
(A) t-SNE plot of all 1,424 microglial cells analyzed and clustered. (n = 3 mTrem2+/+, 2 hTREM2R47H/+, 1 P301S, 2 P301S hTREM2R47H/+, 8-month-old female mice).
(B) t-SNE plots based on clustering from (A) split by genotype.
(C) Ratio of cells in each cluster by genotype. ****p < 0.0001, two-sided Fisher’s exact test.
(D) Volcano plot of DEGs defining cluster 2 compared to cluster 1. See also table S7.
(E) Feature plots of transcript expression overlaid onto t-SNE of all microglial cells. Colored scale bar denotes normalized expression level.
(F) Correlation scatterplot of DEGs in the microglial cluster 2 vs cluster 1 comparison (x-axis) compared to disease-associated microglia (DAM/MGnD) versus homeostatic microglia (y-axis) previously published (14). Red genes are TREM2-dependent. r = 0.7908, ****p < 2.2e-16, Pearson’s correlation.
(G and H) Representative images of RNAscope using probes against C1qa (red) and Apoe (green) of P301S (G) and P301S hTREM2R47H/+ (H) dentate gyrus sections. White triangles highlight C1qa+;Apoe+ microglial cells. Dashed regions are zoomed in on the right side of the image. Scale bar = 20 μm, 10 μm for zoomed images.
(I) Quantification of RNAscope images for percent of cells that are C1qa+;Apoe+ over total C1qa+ cells. n = 9 sections, 3 mice for P301S; 12 sections, 4 mice for P301S hTREM2R47H/+. Student’s t-test, *p = 0.0254, t = 2.426, df = 19.
(J) IPA upstream regulator prediction for TREM2-signaling molecules based on cluster 2 markers from (D). Bar color denotes −log10(pvalue).
(K) IPA AKT activated network determined in (J) and its downstream predicted targets.
(L) Representative western blot of RIPA-soluble cortical lysates from 7- to 8-month-old mice immunoblotted for phospho-AKT, AKT, and β-actin. Lane 1–3= P301S mTrem2+/+, Lanes 4–6= P301S hTREM2R47H/+.
(M) Quantification of phospho-AKT levels normalized by total AKT levels of the entire cohort by western blot (n = 8 P301S mice, n = 9 P301S/R47H/+ mice). Student’s two-tailed t-test, * P < 0.05.
Values are mean ± SEM. Each sequencing dataset represents one independent sequencing experiment. See also fig. S7, fig. S8, and table S7.
Upstream regulator analysis predicted activation of TREM2 pathway regulators such as TNF, Csf1 and Csf2, as well as downstream signaling molecules, such as NF-κB, and AKT signaling (5) (Fig. 4, J and K). Western blot against phospho-AKT normalized to AKT expression also demonstrated increased phosphorylation of AKT in P301S hTREM2R47H/+ compared to P301S mTrem2+/+ brains (Fig. 4, L and M). In sum, hTREM2R47H/+ expression in female tauopathy mice induced similar features observed in AD TREM2R47H human microglia (Fig. 2), including an expanded DAM-like subpopulation previously found to be Trem2-dependent, and enhanced inflammatory and AKT signaling.
Aside from modulating the microglial inflammatory response, TREM2 is also involved in other key microglial functions. Therefore, we assessed the microglial response to injury and phagocytosis (9, 16, 40, 41). The R47H mutation, however, did not alter the microglial response to laser-induced injury compared to hTREM2CV/+ or mTrem2+/+ controls (fig. S8, A–C, movie S1). The effects of R47H on phagocytosis were assessed by acquiring time-course images of primary microglia incubated with pHrodo-conjugated E. coli substrates. Consistent with a previous study in HEK293 cells (42), we did not detect differences in the dynamics of fluorescence intensity over time between hTREM2R47H/+ and mTrem2+/+ control cells (fig. S8, D and E), suggesting that heterozygotic R47H does not alter phagocytic activity of E. coli.
AKT Activation Underlies TAU-mediated Proinflammatory Signatures in R47H-hTREM2 Microglia
Our results so far showed that the R47H mutation enhances proinflammatory microglial responses in human AD and in female mouse tauopathy brains. We next investigated how the R47H mutation affects the microglial response to TAU by treating hTREM2R47H/+ and mTrem2+/+ primary microglia with TAU fibrils. Compared to mTrem2+/+ microglia, TAU fibril stimulation upregulated genes enriched in several signaling pathways in hTREM2R47H/+ microglia (Fig. 5, A and B). The cytokine–cytokine receptor interaction pathway was one of the top pathways altered by hTREM2R47H/+ (Fig. 5B), in agreement with our observation in female P301S hTREM2R47H/+ mice (Fig. 3J). Homozygotic hTREM2R47H/R47H microglia also exhibited similar exacerbation of cytokine response to TAU fibrils compared with mTrem2+/+ microglia (fig. S9, A and B). Moreover, TREM2-associated pathways, including TNF, NF-κB and AKT signaling, were again predicted to be activated in both hTREM2R47H/R47H (fig. S9C) and hTREM2R47H/+microglia (fig. S9, D and E), similar to our observations in our tauopathy mouse model and human AD tissues (Fig. 5C).
Figure 5. Pharmacological AKT-inhibition reverses TAU Fibril-induced Pro-inflammatory Signature in R47H-hTREM2 Primary Microglia.
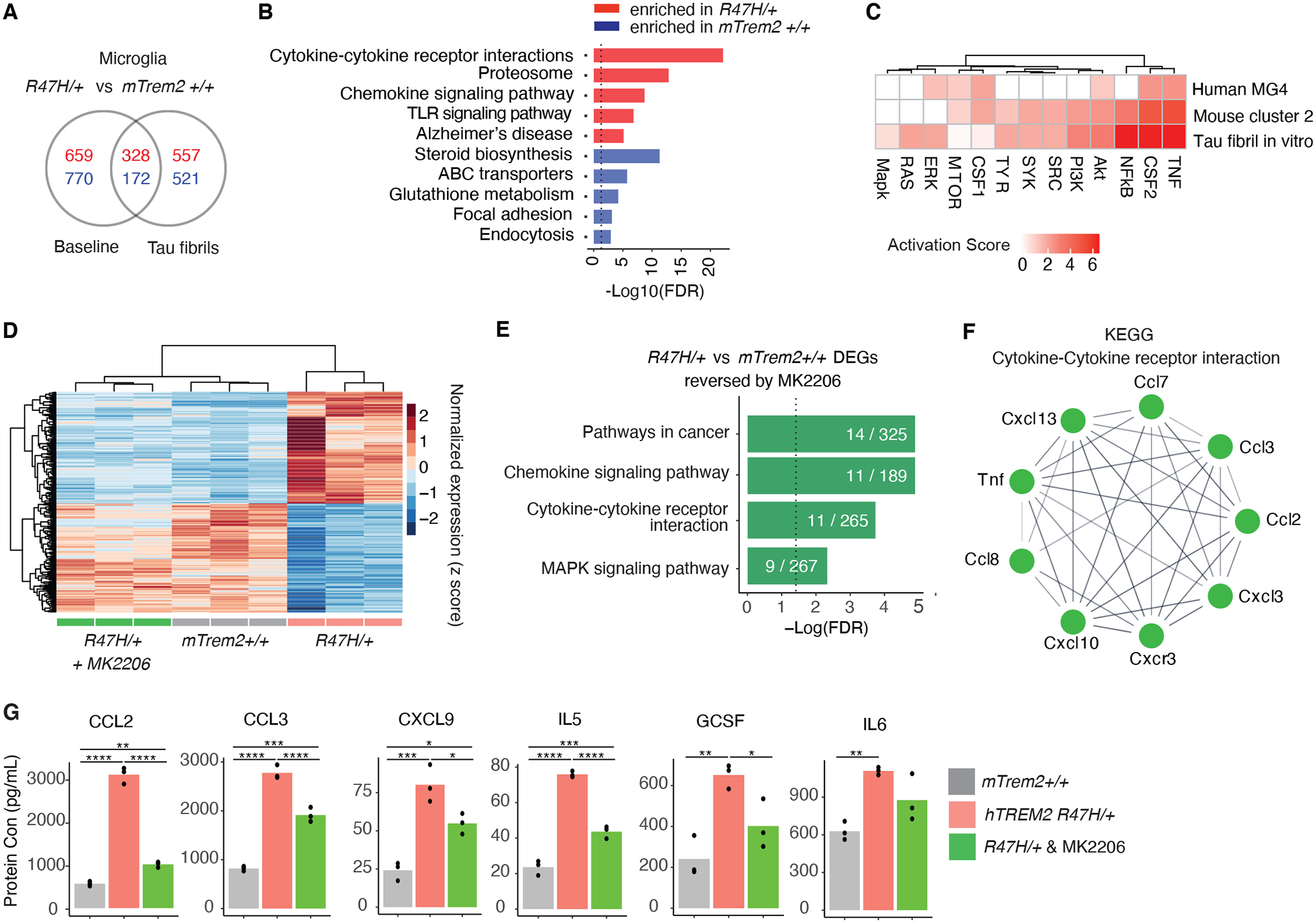
(A) Venn diagram of differentially expressed genes between mTrem2+/+ and hTREM2R47H/+ primary microglia with or without tau fibril stimulation. Red and blue numbers denote upregulated and downregulated genes, respectively. (n = 3 biological replicates for all conditions).
(B) KEGG pathway enrichment analysis of the genes from (A) that were uniquely changed in hTREM2R47H/+ microglia under TAU fibril stimulation conditions. Dashed line indicates the threshold of significant enrichment for the pathway analysis (-Log10(FDR) ≥ 1.3).
(C) Heatmap comparing the IPA predicted activation z score of TREM2 signaling molecules for all three models (Fig. 2, Fig. 4).
(D) Heatmap showing z scores of normalized expressions of 318 genes (adjusted p value < 0.05, log2FC > 0.5 or < −0.5) that are changed by hTREM2R47H/+ compared to mTrem2+/+ and are reversed towards control expression levels with MK-2206 treatment.
(E) KEGG pathway enrichment analysis of genes in heatmap from (D). Dashed line indicates the threshold of significant enrichment for the pathway analysis (-Log10(FDR) ≥ 1.3).
(F) STRING network representation of the genes in the “Cytokine-Cytokine receptor interaction” pathway from (E).
(G) Barplots of example cytokines measured by MAGPIX changed by hTREM2R47H/+ but reversed back to normal protein expression levels by MK-2206. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, One-way ANOVA with Tukey’s multiple comparisons correction.
Next, we directly tested the extent to which AKT signaling contributes to exaggerated inflammatory responses in TAU-treated hTREM2R47H/+ microglia. We acutely inhibited AKT in hTREM2R47H/+ microglia cultures with MK-2206, an allosteric AKT-specific inhibitor (43) before incubation with TAU fibrils. Transcriptomic analysis showed that MK-2206 specifically inhibited the AKT pathway (fig. S9F). Transcriptomic analysis demonstrated that, out of 1,578 DEGs between hTREM2R47H/+ and mTrem2+/+ microglia treated with TAU fibrils, 318 of them were reversed towards mTrem2+/+ control amounts upon AKT-inhibition (green columns, Fig. 5D, tables S8 and S9). These genes were enriched in pathways related to cytokine–cytokine receptor interaction (Fig. 5, E and F). Indeed, MK-2206 resulted in a predicted decrease in TNF signaling (fig. S9G). These transcriptional changes were further confirmed by measuring secreted cytokines in response to TAU fibrils with a multiplex immunoassay. Out of the 19 cytokines altered by the R47H mutation, 7 of them were rescued by MK-2206 (Fig. 5G). These results suggest that at both the RNA and protein levels, hyperactivation of AKT signaling mediates a portion of the R47H-induced pro-inflammatory signatures in response to TAU pathology.
Inhibition of AKT Signaling Rescues Synaptic Toxicity and Abolished the Proinflammatory Microglial Subpopulation in R47H Tauopathy Mice
To test the effects of AKT-inhibition on tauopathy-induced toxicity in vivo, we treated mice with MK-2206. Pharmacokinetic studies of MK-2206 in mice showed that the drug can readily enter the brain and maintain a stable concentration 18 hours after injection (Fig. 6A). For sustained treatment, MK-2206 was administered three times per week via oral gavage for 4-weeks. Brain target engagement was confirmed by western blot showing reduction of phospho-AKT normalized to AKT expression with MK-2206 treatment compared to vehicle control (Fig. 6B). We then treated 6–7-month-old female hTREM2R47H/+ tauopathy mice with MK-2206 and quantified protein expression of hippocampal synaptophysin, a presynaptic marker previously found to be reduced in P301S tauopathy mice compared to non-tauopathy controls (37). We found that chronic MK-2206 treatment rescued the loss of synaptophysin in the hippocampus of P301S hTREM2R47H/+ mice compared to vehicle-treated controls, confirmed by both western blot and IHC of the hippocampal CA3 region (Fig. 6, C–F). These findings provide direct evidence that hyperactivation of AKT signaling downstream of TREM2 signaling could underlie synaptic toxicity in tauopathy mice.
Figure 6. Pharmacological AKT-inhibition reverses Tauopathy-induced Pro-inflammatory Signature and Synapse Loss in R47H-hTREM2 Mice.
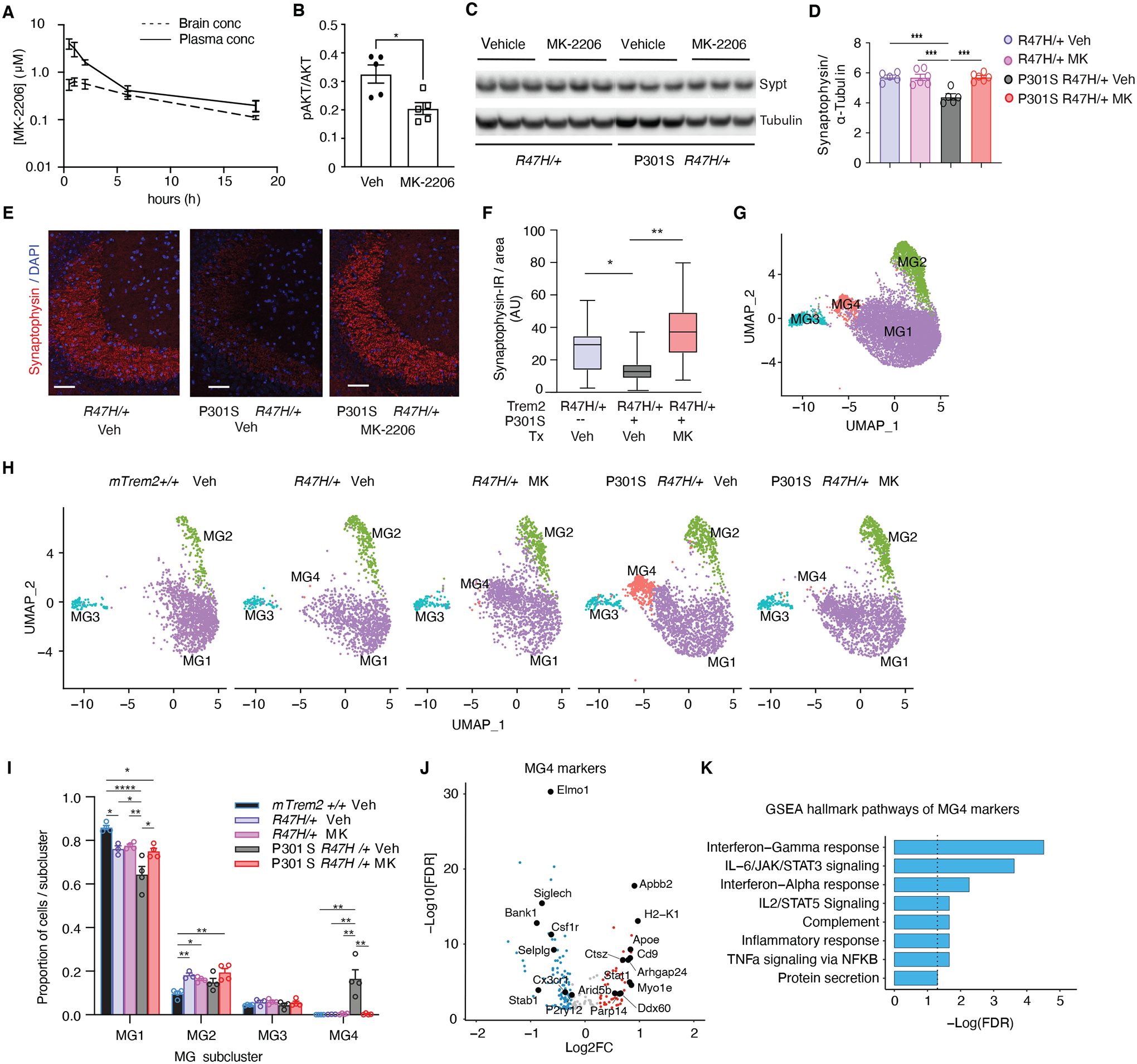
(A) MK-2206 concentrations in brain and plasma measured at different time points after oral gavage administration in mice. n = 3 per mouse per time point.
(B) Quantification of western blot showing protein levels of phospho-AKT normalized to total AKT in hippocampus of female hTREM2R47H/+ mice after 4 weeks of MK-2206 vs vehicle control (Veh) treatment. n = 5 mice/condition. *p < 0.05, unpaired student t-test.
(C) Representative western blot of RIPA-soluble cortical lysates from 7- to 8-month-old hTREM2R47H/+ and P301S hTREM2R47H/+ mice after 4-week MK-2206 vs vehicle treatment immunoblotted for synaptophysin (top bands) and α-tubulin (bottom bands). Lane 1–3 = hTREM2R47H/+ vehicle, Lanes 4–6 = hTREM2R47H/+ MK-2206, Lanes 7–9 = P301S hTREM2R47H/+ vehicle, Lanes 10–12 = P301S hTREM2R47H/+ MK-2206.
(D) Quantification of synaptophysin normalized by α-tubulin levels of the entire cohort by western blot. One-way ANOVA with Tukey’s multiple comparisons test. n = 5 mice/genotype/condition. ***p < 0.001.
(E) Representative images of synaptophysin immunostaining in CA3 hippocampal brain region of hTREM2R47H/+ mice treated with vehicle, P301S hTREM2R47H/+ mice treated with vehicle, and P301S hTREM2R47H/+ mice treated with MK-2206 for 9 weeks. Scale bar = 50 μm.
(F) Quantification of synaptophysin immunofluorescence in hTREM2R47H/+, P301S hTREM2R47H/+ treated with vehicle, and P301S hTREM2R47H/+ treated with MK-2206 (n = 5 per genotype). Pairwise linear mixed models. *p = 0.015, **p = 0.01.
(G) UMAP plots of 9,854 microglial single-nuclei analyzed and clustered.
(H) UMAP split by genotype and condition (Veh = vehicle, MK = MK-2206). (n = 4 mTrem2+/+, 3 hTREM2R47H/+ Veh, 4 hTREM2R47H/+ MK, 4 P301S hTREM2R47H/+ Veh, 4 P301S hTREM2R47H/+ MK, 8-month-old female mice).
(I) Ratio of cells in each cluster by genotype and condition. *p < 0.05, **p < 0.01, ****p < 0.0001, One-way ANOVA with Tukey’s multiple comparisons correction within each subcluster.
(J) Volcano plot of DEGs with FDR < 0.05 defining cluster MG4 compared to all other clusters.
(K) Bar plot of GSEA Hallmark pathways enriched in MG4 markers identified in (J). Dashed line indicates −Log10(FDR) ≥ 1.3.
To further dissect the effects of MK-2206 on microglia, we performed snRNA-seq of hippocampi from this cohort. 218,320 total nuclei were sequenced, with 198,741 nuclei analyzed after pre-processing (fig. S10). 9,854 nuclei expressed microglial markers (fig. S10, A and B), from which we identified 4 subclusters (Fig. 6G, table S10). Microglial subcluster 1 (MG1), the homeostatic cluster, was most enriched in non-transgenic mTrem2+/+ mice and reduced in vehicle-treated P301S hTREM2R47H/+ mice (Fig. 6, H and I). Meanwhile, the MG4 subcluster was observed almost exclusively in vehicle-treated P301S hTREM2R47H/+ mice (Fig. 6, H and I). Nine weeks of MK-2206 treatment eliminated the MG4 subcluster in P301S hTREM2R47H/+ mice, suggesting that AKT activation is required for inducing the tauopathy-dependent MG4 subcluster (Fig. 6, H and I). Microglia in this subcluster expressed markers reminiscent of DAMs (Fig. 6J), with enrichment of genes involved in inflammatory response pathways, including TNFα signaling and interferon pathways (Fig. 6K), consistent with our scRNA-seq and bulk-tissue RNA-seq analyses in P301S hTREM2R47H/+ mice (Fig. 3G, Fig. 4D). Taken together, our findings establish that AKT-dependent microglial responses underlie the disease-enhancing proinflammatory properties of the R47H mutation in tauopathy.
Discussion
Compelling human genetic studies strongly suggest that maladaptive innate immune responses are associated with elevated risk of developing late-onset AD. Recent single-cell transcriptomic findings suggest that a subpopulation of microglia is enriched in response to AD-related pathologies (DAM or MGnD) (13, 14). Nevertheless, among the DAM signature genes, the identity of those that are disease-enhancing microglial genes (DEMs), disease-mitigating microglial genes (DMMs), or mere bystanders remains elusive. As the strongest immune-specific risk gene, the R47H-TREM2 variant provides a unique model to help define drivers for DEMs in AD. Through single-nuclei transcriptomic analysis of mid-frontal cortical tissues from 46 patients with AD carrying the R47H or CV variant of TREM2, we uncovered a microglial subpopulation enriched in AD R47H-TREM2 carriers. This subpopulation had transcriptomic signatures reminiscent of DAMs and had predicted enhancement of TREM2 signaling, including AKT hyperactivation. To identify the mechanistic drivers for the disease-enhancing property of R47H microglia, we established the R47H-hTREM2 knock-in tauopathy mouse model. We showed that the R47H microglia in mouse tauopathy similarly exhibited a heightened inflammatory state and TREM2 signaling as those in human AD brains. Importantly, inhibition of AKT diminished TAU-induced inflammatory responses in R47H microglia and protected against synaptic loss in R47H tauopathy mice, establishing an essential role of microglial AKT hyperactivation in driving the toxic effects of DEMs in tauopathy.
In previous studies in amyloid mouse models, the homozygotic R47H mutation was found to dampen the microglial response to amyloid pathology, and correlated with increased neurotoxicity (20, 25). Paradoxically, in a tauopathy mouse model, the homozygotic R47H mutation was shown to be neuroprotective against neurodegeneration (26). Our current study provides evidence that the heterozygotic R47H mutation is disease-enhancing in the presence of TAU pathology. Whereas the R47H mutation did not impact general microglial functions such as phagocytosis and response to acute injury, the mutation exacerbated tauopathy-induced spatial learning and memory deficits in female tauopathy mice without affecting other cognitive domains, such as locomotion or anxiety. Importantly, this enhanced toxicity in our tauopathy model was not due to differences in TAU-pathology load. Instead, transcriptomic profiling revealed that the toxic effects of R47H-hTREM2 on TAU-mediated cognitive deficits in female mice were associated with substantial transcriptional changes, particularly involving increased expression of pro-inflammatory genes. Meanwhile, the lack of toxic cognitive effects of R47H in male tauopathy mice was associated with few transcriptional changes. These findings support the notion that R47H-induced cognitive-deficits are driven by disease-enhancing microglial responses to stimuli including pathogenic TAU, but not by directly influencing TAU load itself.
Previous bulk-tissue RNA-seq analyses of AD R47H-TREM2 brains yielded inconsistent findings. Although reduced microglial activation signatures (AIF1 and IRF8) were observed in one bulk-tissue RNA-seq study (24), a more recent study showed that pro-inflammatory immune networks and pathways are activated in TREM2 R47H AD brains compared with non-R47H AD (44). By using snRNA-seq, we were able to dissect the changes in the microglial-specific transcriptome at a higher-resolution. In addition, given the complexity of microglial states, the small number of alternations identified by bulk RNA-seq may not be able to capture the complexity of these diverse microglial activation states in human AD brains (45). A recent study showed that snRNA-seq is insufficient to capture microglial heterogeneity in human brain tissues, especially in detecting disease-associated activation genes (34). Our human snRNA-seq analysis was able to capture these heterogenous microglial states, as we included almost five times the number of microglia with a much greater number of genes sequenced per nuclei compared to the previously published results (56).
We showed that R47H-TREM2 in human AD and in tauopathy mice exhibited heightened proinflammatory states, a diminished homeostatic signature, and an enrichment of DAM signature genes. These findings contrasted with data in Trem2-deficient microglia in amyloid and TAU mouse models, which exhibit microglia in homeostatic states with blocked induction of DAM/MGnD signatures (13, 14). Thus, heterozygotic R47H mutation does not phenocopy complete TREM2 deficiency, which results in Nasu-Hakola disease in humans. Although our findings are distinct from studies using homozygous R47H-hTREM2 in 5XFAD mice (25) and in P301S mice (26), the heterozygotic R47H-microglia from our tauopathy mice shared similar features to the R47H-associated human microglia, including similar enhancement of the TREM2-AKT-cytokine signaling and pro-inflammatory signatures. This distinction between the homozygous and heterozygous mutation is important, given that the vast majority of R47H carriers in AD are heterozygotes, and we previously demonstrated that Trem2 haploinsufficiency can have opposing effects on TAU pathology and microglial activation compared with mTrem2–/– (16).
AD has been shown to have sex-dependent differences, including in incidence, prevalence, pathological findings, and disease progression rates (46–49). The sex-specific effects of R47H-hTREM2 uncovered in our behavioral tests and transcriptomic studies in both mouse and human may be mediated by the differences between male and female microglial transcriptomes (50–52). Indeed, the R47H mutation led to disease-enhancing effects only in female mice. The lack of detrimental effects of R47H on male tauopathy mice is consistent with a recent study in which only male mice were used (26). Although we observed sex-specific alterations induced by R47H in both human AD brains and mouse tauopathy mice, there are important distinctions between the two conditions. Because our human samples came from patients with matched Cognitive Dementia Ratings, the distinct pathways induced by R47H microglia in male versus female samples reflect sex-specific responses to similar disease states. In contrast, age-matched male and female tauopathy mice exhibited different degrees of cognitive impairment, which could have contributed to the sex-specific effects of R47H in mouse tauopathy. Further longitudinal studies in both male and female tauopathy mice with matched disease and cognitive states are needed to correlate with the observations seen in our human samples.
In a previous study, induction of the MGnD-state by TREM2, including upregulation of Apoe, has been shown to be sex-specific (13). APOE4 increases risk for late-onset sporadic AD to a greater extent in females (53, 54), and female APOE4 knock-in mice have spatial memory deficits not seen in males (55). Microglia-derived APOE is a major source of plaque-associated APOE and is thought to be the driver of neurodegeneration in tauopathy mouse models (56, 57), suggesting that sex-specific differences in microglia may impact the sex-dependent effect of APOE4 in AD pathogenesis (58). However, how R47H-TREM2 and different APOE genotypes might interact to affect microglial function is unknown. Our analysis did not stratify by APOE genotype due to the limited number of human brain samples. Another limitation of the current study is that our mouse model expressed mouse Apoe, which differs substantially from human APOE. Further studies are needed to confirm and extend our observations related to the sex-differences induced by the R47H mutation and to investigate the effects of different APOE isoforms.
Using a small molecule inhibitor of AKT, MK-2206, we uncovered that AKT hyperactivation underlies the proinflammatory response and synaptic toxicity of R47H microglia in tauopathy. In cultured R47H microglia stimulated by TAU fibrils, we showed that MK-2206 corrected a substantial portion of the genes altered by R47H, including genes involved in the TNFα signaling pathway. Furthermore, chronic MK-2206 treatment abolished the tauopathy-induced DAM subpopulation while rescuing synaptic toxicity in vivo, suggesting the therapeutic potential of AKT inhibitors to reprogram disease-enhancing microglial states to reverse tauopathy-induced toxicity. MK-2206 potently inhibits AKT1 and AKT2, and to a lesser extent, AKT3. A limitation of the current study is that we did not establish isoform-specific roles of AKT nor did we address whether microglia-specific AKT is sufficient to reverse the tauopathy-induced toxicities. By exploring the disease mechanisms underlying the R47H-TREM2 variant in human and mouse, we discovered an essential driver of the disease-enhancing properties of microglia, which opens new avenues for developing microglia-targeted therapies in AD.
Materials and Methods
Study Design
The purpose of this study was to uncover the disease-enhancing pathways induced by the R47H-TREM2 mutation in AD. We used sequencing analysis of human patient brain samples as well as characterized a newly-developed tauopathy mouse model expressing the R47H-TREM2 mutation. Because the mutation is specifically expressed in microglia and increases the risk of late-onset AD, we hypothesized that the mutation would exacerbate spatial memory deficits in our tauopathy mouse model. We also hypothesized that this functional change would be correlated with unique transcriptomic changes in microglia, including alterations in downstream TREM2 signaling. Sample sizes for experiments were based on extensive prior experience with variability within the mouse lines and for each experimental assay (16). For human tissue, sample size was based on availability of tissue given the rarity of the mutation. For all experiments aside from behavioral assays and sequencing, we used GraphPad Prism’s outlier analysis to determine whether any samples were outliers and if so, they were excluded from that analysis. For behavioral assays, mice that were unable to perform the assay were excluded from that behavioral assay. For bulk-tissue RNA-Seq, three samples were excluded from further analysis based on hierarchical clustering algorithms. For snRNA-seq of human tissues, we focused our analysis on tissue from patients with AD and removed the 8 non-AD samples as well as one AD CV-TREM2 sample where we were only able to capture 24 microglial cells (0.056%). We also removed genes expressed in no more than 3 cells, cells with unique gene counts over 9,000 or less than 300, cells with unique molecular identifiers (UMI) count over than 50,000, and cells with high fraction of mitochondrial reads (> 5%). Potential doublet cells were predicted using DoubletFinder for each sample separately with high confidence doublets removed. For scRNA-seq of mouse microglia, we used the following criteria to filter out cells with low sequencing quality: The distribution of total reads (in logarithmic scale) was fitted by a truncated Cauchy distribution, and data points in two tails of the estimated distribution were considered as outliers and eliminated. Fitting and elimination were then applied to the remaining data. This process was run iteratively until the estimated distribution became stable. The threshold was set to the value where the cumulative distribution function of the estimated distribution reaches 0.05. Cells with small numbers of detected genes and poor correlation coefficients for ERCC (low sequencing accuracy) were dropped. 1,424 cells were retained for downstream analysis after filtering from 1,480 cells. Based on convention and due to high costs, RNA-seq experiments were performed once. Behavioral experiments were performed on two independent cohorts. All other experiments had at least three biological replicates. Mice were randomly assigned to groups for all behavioral assays and sequencing studies. Researchers were blinded during all experimental procedures and analyses.
Statistical Analysis
Data were analyzed with GraphPad Prism v.7 (GraphPad Software, San Diego, California USA, www.graphpad.com), STATA12 (StataCorp. 2011. Stata Statistical Software: Release 12. College Station, TX: StataCorp LP), or R (59). A multilevel mixed-effects linear regression model fitted with STATA12 was used to analyze latency in the Morris water maze. R was used to calculate the area under the curve for cumulative search errors in the Morris water maze. Outliers were removed with Prism’s outlier analysis algorithm. All statistical details can be found in the figure and figure legends. P < 0.05 and FDR < 0.05 was considered statistically significant, unless otherwise noted. All values are expressed as mean ± SEM, unless otherwise noted. A subset of mice from the behavior cohort was randomly selected for snRNA-seq and bulk RNA-seq studies. Data and visualizations were done using ggplot2 (60).
Supplementary Material
Acknowledgments
We thank the patients and their families for donating tissue samples used for this research; Dr. Junli Zhang from the Gladstone Institutes Transgenic Core for microinjections into embryos to generate knock-in human TREM2 mice; Allan Villanueva and Elizabeth Fan for assistance with genotyping the mice; Dr. Li-Huei Tsai and Dr. Yueming Li for their time discussing the data and feedback on the manuscript; Dr. Jason Gestwicki and his lab for synthesizing the 0N4R tau fibrils; Dr. Rosa Rademakers and Dr. Nilufer Taner for genotyping of the human brain tissues and Christopher Fernandez De Castro for assistance with the tissues; Silvie Lundgren for preparing the MK-2206 solution for PK analysis and drug treatment; and Stephen Ordway and Kathryn Claiborn for manuscript editing. All in vivo imaging was done by L.K., with helpful advice from Dr. Mario Merlini and Dr. Katerina Akassoglou, at the Gladstone Center for In Vivo Imaging Research. MC1 immunohistochemistry imaging was done by F.A.S. using the Gladstone Histology Core microscopes. Behavioral assays were done by F.A.S. and D.L. using equipment from the Gladstone Behavioral Core. RNA-sequencing was done by the UCSF Center for Advanced Technology (bulk-seq), and Weill Cornell Medicine Genomics Resources Core Facility (sc and snRNA-seq). The authors are also grateful to the Flow Cytometry Core and the Integrated Genomics Operation at MSKCC for their assistance.
Funding:
This work was supported by the National Institute of Health Grants R01AG051390 to L.G., U54NS100717 to L.G., R01AG054214 to L.G., Rainwater Foundation to L.G, the National Institute of Aging Grant F31 AG058505 to F.A.S., the National Institute of Aging Grant F30 AG062043-02 to L.K., the National Institute of Health Grant T32GM007618 to L.K., the National Institute of Health R01AG064239 to W.L., the Early Postdoctoral Mobility Fellowship from the Swiss National Science Foundation P2BSP3 151885 to H.M., the National Institute of Aging Grant K01 AG057862 to T.E.T., the National Institute of Mental Health Grant R01 MH110504 to G.Y., the National Institute of Aging Grant R01 AG057555 to L.X., the UK Dementia Research Institute, which receives its funding from DRI Ltd, funded by the UK Medical Research Council to J.H., Alzheimer’s Society and Alzheimer’s Research UK to J.H., Medical Research Council MR/N026004/1 to J.H., Wellcome Trust 202903/Z/16/Z to J.H., Dolby Family Fund to J.H., the National Institute for Health Research University College London Hospitals Biomedical Research Centre to J.H., Research Centre at University College London Hospitals NHS Foundation Trust and University College London to J.H., the National Institute of Aging Grants U01AG046170 to B.Z., RF1AG054014 to B.Z., RF1AG057440 to B.Z., R01AG057907 to B.Z., and the Alan and Sandra Gerry Foundation to L.M.
Footnotes
Supplementary Materials
Competing interests: L.G. is founder of Aeton Therapeutics, Inc. S.C.S. is a consultant of Aeton Therapeutics, Inc.
Data and materials availability:
All data associated with this study are in the paper or supplementary materials. Full western blots for Fig. 3 and Fig. 6 are available in data file S1 and S2, respectively, individual data values for the behavior experiments (Fig. 3 and Fig. S5) are in data files S3 – S10, individual data values for Fig. 6A and F are in data files S11 and S12, and RNA-seq gene lists with statistics are available in the respective supplementary tables accompanying this article. All RNA-seq data was deposited in the Gene Expression Omnibus (GEO) under the following series accession numbers: bulk-tissue RNA-seq of mouse hippocampus, GSE136389; mouse primary microglia, GSE181903; human single-nuclei, GSE183068; mouse single-cell, GSE140670; mouse MK-2206 single-nuclei, GSE181678. All codes used for data analysis are available on https://github.com/kozlama/Sayed-Kodama-Fan-et-al-2021. All new materials including the knockin mouse models will be available to academic and non-profit institutions by contacting Li Gan (lig2033@med.cornell.edu) to complete a standard Material Transfer Agreement.
References and Notes:
- 1.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis G, TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jonsson T, Stefansson K, TREM2 and neurodegenerative disease. N Engl J Med 369, 1568–1569 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL, Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17, 131–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J, The microglial sensome revealed by direct RNA sequencing. Nature neuroscience 16, 1896–1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colonna M, Wang Y, TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci 17, 201–207 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Deczkowska A, Weiner A, Amit I, The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 181, 1207–1217 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M, TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 170, 649–663 e613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M, TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi K, Rochford CD, Neumann H, Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 201, 647–657 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng Q, Danao J, Talreja S, Wen P, Yin J, Sun N, Li CM, Chui D, Tran D, Koirala S, Chen H, Foltz IN, Wang S, Sambashivan S, TREM2-activating antibodies abrogate the negative pleiotropic effects of the Alzheimer’s disease variant Trem2(R47H) on murine myeloid cell function. J Biol Chem 293, 12620–12633 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng H, Jia L, Liu CC, Rong Z, Zhong L, Yang L, Chen XF, Fryer JD, Wang X, Zhang YW, Xu H, Bu G, TREM2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J Neurosci 37, 1772–1784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE, A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem 109, 1144–1156 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O, The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581 e569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I, A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, Robinson GO, Anderson E, Colonna M, Holtzman DM, TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci USA 114, 11524–11529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sayed FA, Telpoukhovskaia M, Kodama L, Li Y, Zhou Y, Le D, Hauduc A, Ludwig C, Gao F, Clelland C, Zhan L, Cooper YA, Davalos D, Akassoglou K, Coppola G, Gan L, Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proceedings of the National Academy of Sciences of the United States of America 115, 10172–10177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, Holtzman DM, Altered microglial response to Abeta plaques in APPPS1–21 mice heterozygous for TREM2. Mol Neurodegener 9, 20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M, TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213, 667–675 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE, Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J Neurosci 37, 637–647 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J, TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 90, 724–739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai LH, Single-cell transcriptomic analysis of Alzheimer’s disease. Nature, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van der Brug MP, Foreman O, Paw JS, Modrusan Z, Beach TG, Serrano GE, Hansen DV, Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep 31, 107843 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roussos P, Katsel P, Fam P, Tan W, Purohit DP, Haroutunian V, The triggering receptor expressed on myeloid cells 2 (TREM2) is associated with enhanced inflammation, neuropathological lesions and increased risk for Alzheimer’s dementia. Alzheimers Dement 11, 1163–1170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, Beausoleil SA, Ulrich JD, Holtzman DM, Artyomov MN, Colonna M, Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nature medicine 26, 131–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, Colonna M, Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med 215, 745–760 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gratuze M, Leyns CE, Sauerbeck AD, St-Pierre MK, Xiong M, Kim N, Remolina Serrano J, Tremblay M, Kummer TT, Colonna M, Ulrich JD, Holtzman DM, Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J Clin Invest, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, Pflueger J, Lister R, Rackham OJL, Petretto E, Polo JM, A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nature neuroscience 22, 2087–2097 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, Weitz DA, Rozenblatt-Rosen O, Zhang F, Regev A, Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGinnis CS, Murrow LM, Gartner ZJ, DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337 e324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, Zhang K, Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, Clarke D, Gu M, Emani P, Yang YT, Xu M, Gandal MJ, Lou S, Zhang J, Park JJ, Yan C, Rhie SK, Manakongtreecheep K, Zhou H, Nathan A, Peters M, Mattei E, Fitzgerald D, Brunetti T, Moore J, Jiang Y, Girdhar K, Hoffman GE, Kalayci S, Gumus ZH, Crawford GE, Psych EC, Roussos P, Akbarian S, Jaffe AE, White KP, Weng Z, Sestan N, Geschwind DH, Knowles JA, Gerstein MB, Comprehensive functional genomic resource and integrative model for the human brain. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, Grün D, Kroshilina AA, Dionne D, Sarkis RA, Cosgrove GR, Helgager J, Golden JA, Pennell PB, Prinz M, Vonsattel JPG, Teich AF, Schneider JA, Bennett DA, Regev A, Elyaman W, Bradshaw EM, De Jager PL, Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun 11, 6129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PCG, Ngu H, Modrusan Z, Larson JL, Kaminker JS, van der Brug MP, Hansen DV, Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep 22, 832–847 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, Fourne Y, Matthews PM, Theys T, Mancuso R, de Strooper B, Fiers M, Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep 32, 108189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT, Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41, 17–24 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR, Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM, Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Weaver CL, Espinoza M, Kress Y, Davies P, Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol Aging 21, 719–727 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE, Marschallinger J, Yu G, Quake SR, Wyss-Coray T, Barres BA, Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 101, 207–223.e210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, Krasemann S, Capell A, Trümbach D, Wurst W, Brunner B, Bultmann S, Tahirovic S, Kerschensteiner M, Misgeld T, Butovsky O, Haass C, TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep 18, 1186–1198 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao H, Coppola K, Schweig JE, Crawford F, Mullan M, Paris D, Distinct Signaling Pathways Regulate TREM2 Phagocytic and NFκB Antagonistic Activities. Front Cell Neurosci 13, 457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleo A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sanchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C, TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6, 243ra286 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, Kotani H, MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9, 1956–1967 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Korvatska O, Kiianitsa K, Ratushny A, Matsushita M, Beeman N, Chien WM, Satoh JI, Dorschner MO, Keene CD, Bammler TK, Bird TD, Raskind WH, Triggering Receptor Expressed on Myeloid Cell 2 R47H Exacerbates Immune Response in Alzheimer’s Disease Brain. Front Immunol 11, 559342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, Grun D, Kroshilina AA, Dionne D, Sarkis RA, Cosgrove GR, Helgager J, Golden JA, Pennell PB, Prinz M, Vonsattel JPG, Teich AF, Schneider JA, Bennett DA, Regev A, Elyaman W, Bradshaw EM, De Jager PL, Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun 11, 6129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA, Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry 62, 685–691 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Benke T, Delazer M, Sanin G, Schmidt H, Seiler S, Ransmayr G, Dal-Bianco P, Uranüs M, Marksteiner J, Leblhuber F, Kapeller P, Bancher C, Schmidt R, Cognition, gender, and functional abilities in Alzheimer’s disease: how are they related? J Alzheimers Dis 35, 247–252 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Gamberger D, Lavrač N, Srivatsa S, Tanzi RE, Doraiswamy PM, Identification of clusters of rapid and slow decliners among subjects at risk for Alzheimer’s disease. Sci Rep 7, 6763 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koran MEI, Wagener M, Hohman TJ, Sex differences in the association between AD biomarkers and cognitive decline. Brain Imaging Behav 11, 205–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanamsagar R, Bilbo SD, Environment matters: microglia function and dysfunction in a changing world. Curr Opin Neurobiol 47, 146–155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, Lolli F, Marcello E, Sironi L, Vegeto E, Maggi A, Sex-Specific Features of Microglia from Adult Mice. Cell Rep 23, 3501–3511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guneykaya D, Ivanov A, Hernandez DP, Haage V, Wojtas B, Meyer N, Maricos M, Jordan P, Buonfiglioli A, Gielniewski B, Ochocka N, Comert C, Friedrich C, Artiles LS, Kaminska B, Mertins P, Beule D, Kettenmann H, Wolf SA, Transcriptional and Translational Differences of Microglia from Male and Female Brains. Cell Rep 24, 2773–2783.e2776 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM, Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama 278, 1349–1356 (1997). [PubMed] [Google Scholar]
- 54.Altmann A, Tian L, Henderson VW, Greicius MD, Alzheimer’s Disease Neuroimaging Initiative Investigators, Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol 75, 563–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, Mahley RW, Mucke L, Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci USA 95, 10914–10919 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, Hoyle R, Holtzman DM, Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med 216, 2546–2561 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Alzheimer’s Disease Neuroimaging I, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM, ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kodama L, Gan L, Do microglial sex differences contribute to sex differences in neurodegenerative diseases? Trends Mol Med, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.R. C. Team, R: A language and environment for statistical computing. R Foundation for Statistical Computing, (2017). [Google Scholar]
- 60.Wickham H, ggplot2 - Elegant Graphics for Data Analysis. (Springer, 2009). [Google Scholar]
- 61.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, Linsley PS, Gottardo R, MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP, Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benjamini Y, Hochberg Y, Controlling the false discovery rate - a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B-Statistical Methodology 57, 289–300 (1995). [Google Scholar]
- 66.Lun ATL, Richard AC, Marioni JC, Testing for differential abundance in mass cytometry data. Nature methods 14, 707–709 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014) 10.1186/s13059-014-0550-8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Langfelder P, Horvath S, WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ, An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.The Gene Ontology C, Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 45, D331–D338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kanehisa M, Goto S, KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C, The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45, D362–d368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R, Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9, 171–181 (2014). [DOI] [PubMed] [Google Scholar]
- 77.Schmieder R, Edwards R, Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anders S, Pyl PT, Huber W, HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsai H-F, Gajda J, Sloan TFW, Rares A & Shen AQ, Usiigaci: Instance-aware cell tracking in stain-free phase contrast microscopy enabled by machine learning. SoftwareX 9, 230–237 (2019). [Google Scholar]
- 82.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Fiji: an open-source platform for biological-image analysis. Nature methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abadi M, Barham P, Chen J, Chen Z, Davis A, Dean J, Devin M, Ghemawat S, Irving G, Isard M, Kudlur M, Levenberg J, Monga R, Moore S, Murray DG, Steiner B, Tucker P, Vasudevan V, Warden P, Wicke M, Yu Y, and Zheng X, Google Brain, TensorFlow: A system for large-scale machine learning. USENIX Association 12th USENIX Symposium on Operating Systems Design and Implementation, (2016). [Google Scholar]
- 84.Chollet F, others, Keras: The Python Deep Learning library. Astrophysics Source Code Library; (2018), pp. ascl:1806.1022. [Google Scholar]
- 85.He K, Gkioxari G, Dollár P, Girshick R, Mask R-CNN. arXiv:1703.06870, (2017). [DOI] [PubMed] [Google Scholar]
- 86.Abdulla W, Mask R-CNN for object detection and instance segmentation on Keras and TensorFlow. GitHub repository, (2017). [Google Scholar]
- 87.Lombardot B, Manual drift correction ImageJ plugin. Fiji, (2016). [Google Scholar]
- 88.Young MD, Behjati S, SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or supplementary materials. Full western blots for Fig. 3 and Fig. 6 are available in data file S1 and S2, respectively, individual data values for the behavior experiments (Fig. 3 and Fig. S5) are in data files S3 – S10, individual data values for Fig. 6A and F are in data files S11 and S12, and RNA-seq gene lists with statistics are available in the respective supplementary tables accompanying this article. All RNA-seq data was deposited in the Gene Expression Omnibus (GEO) under the following series accession numbers: bulk-tissue RNA-seq of mouse hippocampus, GSE136389; mouse primary microglia, GSE181903; human single-nuclei, GSE183068; mouse single-cell, GSE140670; mouse MK-2206 single-nuclei, GSE181678. All codes used for data analysis are available on https://github.com/kozlama/Sayed-Kodama-Fan-et-al-2021. All new materials including the knockin mouse models will be available to academic and non-profit institutions by contacting Li Gan (lig2033@med.cornell.edu) to complete a standard Material Transfer Agreement.