

ABSTRACT
Lung adenocarcinoma (LUAD) is associated with a poor prognosis due to early metastasis to distant organs. TGF-β potently induces epithelial-to-mesenchymal transition (EMT) and promotes invasion and metastasis of cancers. However, the mechanisms underlying this alteration are largely unknown. PTBP3 plays a critical role in RNA splicing and transcriptional regulation. Although accumulating evidence has revealed that PTBP3 exhibits a pro-oncogenic role in several cancers, whether and how PTBP3 mediates TGF-β-induced EMT and metastasis in LUAD remains unknown. The expression levels and prognostic value of PTBP3 were analyzed in human LUAD tissues and matched normal tissues. siRNAs and lentivirus-mediated vectors were used to transfect LUAD cell lines. Various in vitro experiments including western blot, qRT-PCR, a luciferase reporter assay, chromatin immunoprecipitation (ChIP), transwell migration and invasion assay and in vivo metastasis experiment were performed to determine the roles of PTBP3 in TGF-β-induced EMT and metastasis. PTBP3 expression was significantly upregulated in patients with LUAD, and high expression of PTBP3 indicated a poor prognosis. Intriguingly, we found that PTBP3 expression level in LUAD cell lines was significantly increased by exogenous TGF-β1 in a Smad-dependent manner. Mechanistically, p-Smad3 was recruited to the PTBP3 promoter and activated its transcription. In turn, PTBP3 knockdown abolished TGF-β1-mediated EMT through the inhibition of Smad2/3 expression. Furthermore, PTBP3 overexpression increased lung and liver metastasis of LUAD cells in vivo. PTBP3 is indispensable to TGF-β-induced EMT and metastasis of LUAD cells and is a novel potential therapeutic target for the treatment of LUAD.
KEYWORDS: LUAD, PTBP3, TGF-β/Smad signaling, EMT, tumor metastasis
Introduction
Non-small-cell lung cancer (NSCLC) is the most common cancer with the highest morbidity and mortality worldwide, accounting for approximately 25% of all cancer deaths [1]. Lung adenocarcinoma (LUAD) is the major pathological subtype of NSCLC and is characterized by high heterogeneity and invasiveness [2]. With the improvement of surgical techniques and emerging immunotherapy, the survival of patients with LUAD has improved, yet the therapy fails mostly due to local and distant metastasis [3]. Consequently, a better understanding of the mechanisms underlying LUAD metastasis is crucial for LUAD treatment.
Epithelial-mesenchymal transition (EMT) is a key cause of tumor metastasis and chemotherapy resistance [4,5]. Transforming growth factor-β (TGF-β) plays important roles in multiple cellular functions and has been demonstrated to be a potent promoter of EMT and tumor metastasis in various cancer types, including NSCLC [6–9]. TGF-β-induced EMT mainly depends on the activation of canonical Smad signaling, and in the canonical TGF-β/Smad signaling pathway, TGF-β ligand directly binds to TGF-β receptor II (TβRII), followed by phosphorylation of TGF-β receptor I (TβRI). The activated TβRI recruits and phosphorylates cytoplasmic effectors Smad2/3, which then forms a heterotrimeric complex with Smad4. Subsequently, the complex is transported into the nucleus, where it binds with smad binding elements (consensus sequence CAGAC or GTCTG, SBEs) in the promoter region of TGF-β target genes (Snail, Slug, PAI-1, and others) and regulates the transcription of EMT-related genes [10–12]. Considering the critical role of TGF-β in tumor invasion and metastasis, several inhibitors targeting the TGF-β pathway have been used for cancer interventions both preclinically and clinically [13,14]. However, none of these treatments have been successful, especially since TGF-β regulates gene expression depending on a highly specific cellular context [15]. Thus, a deeper understanding of the molecular mechanisms that mediate the pro-metastatic effects of TGF-β will facilitate the development of effective anti-metastasis approaches targeting TGF-β signaling.
Polypyrimidine tract-binding protein 3 (PTBP3), also known as ROD1, is a critical RNA-binding protein that interacts with RNA through one or multiple RNA-recognition motifs (RRMs) and plays key roles in RNA splicing, maturation, localization and translation [16]. The cellular functions and physiological roles of PTBP3 have been well studied, and previous studies have demonstrated that PTBP3 correlates with nonsense-mediated mRNA decay and functions as a splicing repressor [17,18]. In addition, recent studies have shown that dysregulation of PTBP3 is involved in tumorigenesis and tumor progression. For example, high expression of PTBP3 was confirmed to promote EMT and breast cancer progression by preventing ZEB1 mRNA degradation [19], and PTBP3 contributes to the metastasis of gastric cancer and hepatocellular carcinoma by mediating CAV1 alternative splicing [20] or modulating the splicing balance of NEAT1 and pre-miR-612 [21]. It was also found that PTBP3 promotes pancreatic cancer growth and chemoresistance by upregulating ATG12 [22]. By comparison, the role of PTBP3 in LUAD tumor invasion and metastasis remains largely unknown. Although previous reports determined that knockdown of PTBP3 impairs LUAD cell motility, possibly by regulating EMT [23,24], the specific molecular mechanisms involved in invasion and metastasis of LUAD cells are still not addressed.
In this study, we demonstrated that PTBP3 was markedly upregulated in LUAD tissues compared with adjacent normal lung tissues and was positively associated with the poor prognosis of patients with LUAD. In the several LUAD cell lines, including A549, H1792, H1299, and H838, the expression of PTBP3 was robustly induced by TGF-β1 in a Smad-dependent manner. By analyzing the PTBP3 gene promoter, we uncovered that p-Smad3 directly binds to SBEs within PTBP3ʹs promoter and activates its transcription. In addition, TGF-β-induced EMT and invasion of LUAD cells was nearly completely blocked by PTBP3 siRNA. The in vivo metastasis assays showed that overexpression of PTBP3 facilitated lung and liver metastasis of LUAD cells in nude mice. Moreover, we found that knockdown of PTBP3 abolished the effect of TGF-β-induced cell motility by suppressing Smad2/3 expression transcriptionally. Thus, our findings determined that PTBP3 acts as a novel, critical component of the TGF-β/Smad pathway that promotes LUAD cell invasion and metastasis.
Materials and methods
Cell lines, cell culture, and reagents
Human LUAD cell lines A549, H1299, H1975, H1792, PC9, H838, HCC827, immortalized human bronchial epithelial cell line BEAS-2B, and human embryonic lung epithelial cell line L132 used in the study were obtained from the Shanghai Cell Bank of the Chinese Academy of Sciences. All the cell lines had cell line certification and were not contaminated by mycoplasma. The cell lines were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS, Gibco, Australia) at 37°C in a 5% CO2 humidified incubator. Recombinant TGF-β1 was purchased from PeproTech (Rocky Hill, NJ, USA). The TGF-β type I inhibitor, SB431542, was purchased from Selleckchem (Houston, TX, USA). Following serum starvation for 24 h, human LUAD cell lines were treated with TGF-β1 for different durations to induce EMT.
Human LUAD tissue samples
Human LUAD samples and matched normal tissues enrolled in this study were obtained from the Shanghai Tongji Hospital (Shanghai, China). All patients were pathologically diagnosed, and none of the patients had received either chemotherapy or radiotherapy before surgical resection. All LUAD tissue specimens were evaluated by histopathological examination to determine the tumor size, histological grade, TNM stage, and lymphatic metastasis. Immunohistochemical (IHC) staining was conducted to analyze these samples by a clinical pathologist. This study was authorized by the Ethics Review Committees of Tongji Hospital, School of Medicine, Tongji University.
Western blotting
Cells were lysed in RIPA lysis buffer with 1% phosphatase and protease inhibitors (Beyotime, China). Same amount (~20ug) of protein was electrophoresed using 10% SDS-PAGE gel and was transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The membranes were placed in 5% skimmed milk for 1 h on a rotary shaker to block nonspecific binding sites, followed by incubation with primary specific antibodies overnight at 4°C. On the second day, after washing with 1× TBST buffer three times (10 min/time), anti-mouse or anti-rabbit HRP-labeled secondary antibodies were added and incubated at room temperature for 1 h. After washing again, the membranes were subjected to immunoblotting analysis using ECL detection reagent (Beyotime, China). The primary antibodies used in this study were as follows: PTBP3 (#sc-100845, Santa Cruz Biotechnology), EMT antibody sampler kit (#9782, CST), Smad2/3 antibody sampler kit (#12747, CST), PAI-1 (#11907, CST), TβRI (#sc-101574, Santa Cruz Biotechnology), TβRII (#sc-17791, Santa Cruz Biotechnology), Akt (#4691, CST), p-Akt (#4060, CST), MEK (#4694, CST), p-MEK (#9154, CST), Erk (#12950, CST), and p-Erk (#4370, CST).
Immunofluorescent staining
Cells that were cultured on glass slides were washed three times (5 min/time) with phosphate-buffered saline, fixed in 4% paraformaldehyde (PFA) for 15 min, and permeabilized with 0.3% Triton X-100 for 20 min, and then blocked for 1 h with 5% bovine serum albumin. Subsequently, the cells were incubated with primary antibodies overnight at 4°C and then probed with Alexa Fluor 594-conjugated anti-rabbit secondary antibody (#8889S, CST). The nuclei were stained with DAPI, and the fluorescence was visualized under a fluorescent microscope.
H&E staining and immunohistochemistry staining
Lung and liver tissues taken out of mice were fixed in Bouin’s solution and embedded in paraffin. Next, the paraffin was cut into tissue sections with a thickness of 3–5um and applied to H&E staining. Lung adenocarcinoma tissue microarrays were obtained from Outdo Biotech (Shanghai, China). For immunohistochemistry staining, tissue microarrays were heated to 100°C in ammonium citrate solution (10 mmol/L, pH 6.0) for antigen retrieval. After cooling to room temperature, the slides were immersed in 3% H2O2 for 15 min for blocking endogenous peroxidase activity. Subsequently, the slides were incubated with corresponding primary antibodies for 12 h at 4°C, followed by secondary antibodies for 1 h at room temperature. We then counterstained the slides with hematoxylin after incubating with DAB for immunoreactivity detection. Two experienced pathologists evaluated and calculated the Score of IHC staining based on the following formula: Score = stained intensity (negative staining: 0; light staining: 1; moderate staining: 2; strong staining: 3) × stained cell number (positive cells as ≤25% of the cells, 1; 26–50% of the cells, 2; 51–75% of the cells, 3; >75% of the cells, 4). When the staining score was <7, it was defined as low expression, otherwise high expression.
RNA interference and generation of stably transfected cell lines
A549 and H1299 cell lines were transiently transfected with 100 nM siRNA, which were designed and synthesized by GenePharma Biotech (Shanghai, China), using RFect Transfection Reagent according to the manufacturer’s procedure. After 24 h, cells were transfected again. At 48 h post-transfection, the cells were treated for further experiments or harvested. The sequences of siRNA were as follows: PTBP3-1 (sense: 5’-CCAAUCACAGAGAACUUAATT-3’, antisense: 5’-UUAAGUUCUCUGUGAUUGGTT-3’), PTBP3-2 (sense: 5’-CCCUGUUACCCUGGAAGUUTT-3’, antisense: 5’-AACUUCCAGGGUAACAGGGTT-3’), Smad4-1 (sense: 5’-UACUUACCAUCAUAACAUUTT-3’, antisense: 5’-AAUGUUAUGAUGGUAAGUAGC-3’) and Smad4-2 (sense: 5’-UCCAUUGCUUACUUUGAUUTT-3’, antisense: 5’-AAUCAAAGUAAGCAAUGGAAC-3’). To establish the PTBP3-overexpressing stable A549 cell line, the human full-length cDNA of PTBP3 was subcloned into the lentiviral vector pSLenti-EF1-F2A-Puro-CMV-3× FLAG-WPRE. The PTBP3 expression vector or empty vector was transfected into HEK 293 T cells to produce recombinant lentiviruses. Subsequently, we collected the packaged lentiviruses and transfected A549 cells for 72 h. Finally, Flag-tagged PTBP3-overexpressing A549 stable cells were selected using 2ug/ml puromycin (Beyotime, China).
Real-time quantitative reverse transcription PCR (qRT-PCR)
Total RNA was extracted with RNAfast2000 reagent (Fastagen Biotch, Shanghai, China), cDNA synthesis was performed with Evo M-MLV RT Kit (Accurate Biology, Shanghai, China), and qRT-PCR analysis was performed with SYBR Green Premix Pro Taq HS qPCR Kit (Accurate Biology, Shanghai, China) according to the supplier’s instructions. The primers used in the study are listed in Supplemental Table S1. All reactions were performed in triplicate and repeated three times.
Luciferase reporter gene assay
To make human PTBP3 promoter constructs, the wild-type (WT) and mutated (MUT) fragments, approximately 2kb sized of PTBP3 promoter were synthesized by Shanghai sangon biotech. and then subcloned into pGL3-basic vector. The Renilla luciferase reporter plasmid was used as an internal control. To perform the luciferase reporter assay, various constructs (pGL3-basic-empty, pGL3-basic-PTBP3 promoter-WT, pGL3-basic-PTBP3 promoter-MUT), and pRL-TK plasmids were transiently co-transfected into A549 cells. After 24 h post-transfection, cells were treated with 5 ng/ml of TGF-β1 for another 24 h. Subsequently, cells were harvested to measure luciferase activity using a dual-luciferase reporter assay system (Promega). Firefly luciferase values were normalized to Renilla luciferase activity and are presented as the mean ± SD of three independent experiments.
Cell counting kit 8 assays
CCK-8 Kit (Beyotime, Shanghai, China) was used to evaluate cell proliferation. Tumor cells were seeded into the 96-well cell culture plates at a density of 1000 cells per well and cultured under normal conditions for 24 h. Then, CCK-8 reagents were added to each well at various time points. After 2 h, the absorbance value was measured at 450 nm according to manufacturer’s instructions. Each experiment was performed in triplicate.
Transwell migration and invasion assays
Migration and invasion were measured using transwell plates (BIOFIL) coated with or without Matrigel (BD Biosciences). The cells resuspended in 200 µL serum-free RPMI 1640 medium were added to the upper chambers, and 500 µl RPMI 1640 medium containing 10% FBS with 5 ng/ml or 10 ng/ml TGF-β1 was added to the lower chambers. After 36 h, the transwell chambers were removed and the cells on the upper chambers were gently wiped with a cotton swab. Subsequently, the cells were fixed with 4% PFA for 15 min and stained with 2% crystal violet for 5 min, and at least four randomly selected fields were photographed and counted. Transwell migration and invasion assays were performed in triplicate.
Chromatin immunoprecipitation(ChIP) assay
ChIP assay was performed on A549 cells using the EpQuikTM Chromatin Immunoprecipitation Kit according to the manufacturer’s instructions, and the chromatin fraction from cell nuclei was precipitated with negative control IgG and p-Smad3 antibody (#9520, CST) in combination with protein A/G agarose beads overnight at 4°C. The protein/DNA complexes were washed, eluted, and extracted, and the extracted DNA was quantified by performing qPCR and DNA agarose gel. The primers used for amplifying the two SBEs in the promoter region of PTBP3 are as follows: SBE1 (sense: 5’- GGTGATCTGTGAGTTGAAAAAG-3’, antisense: 5’-GTACAAACATGGAATGGCTATA-3’), SBE2 (sense: 5’- TCCTTGTGGGACATTCCTTC-3’, antisense: 5’-CTGTAAGACCCCGCCTCTC −3’).
Animal experiments
Four-week-old female BALB/c nude mice were purchased from Shanghai JSJ Laboratory Animal Co., Ltd. (Shanghai, China) and fed under pathogen-free conditions. The mice were randomly divided into control vector groups and PTBP3 overexpression groups(6 mice per group). Then a suspension of 4.0 × 106 cells in 200ul of PBS were injected into the tail vein of mice in these two groups accordingly. To evaluate whether PTBP3 enhanced TGF-β1-induced tumor cell metastasis, every 5 days after intravenously tumor incubation, 4ug/kg TGF-β1 was intraperitoneally injected into these two groups of mice. The mice were euthanized after 8 weeks, the lung and the liver were taken out and photographed to assess macroscopically metastatic nodule. Lung and live tissues were histologically evaluated using Hematoxylin-eosin(H&E) staining for micrometastases lesions analysis. Animal studies were approved the by Animal Ethics Committee of Tongji Hospital, School of Medicine, Tongji University (Shanghai, China).
The cancer genome atlas, gene expression omnibus, GEPIA, and webtool
PTBP3 mRNA expression levels and survival data of patients with LUAD were downloaded from TCGA (https://cancergenome.nih.gov) and GEO database (https://www.ncbi.nlm.nih.gov/geo/). The gene expression profiling interactive analysis was obtained from http://gepia.cancer-pku.cn/.
Statistical analysis
All statistical analyses were performed using SPSS software (version 19.0; IBM, Chicago, IL, USA) and GraphPad Prism 8.0.2 (GraphPad Software Inc., San Diego, CA, USA). Statistical methods including ANOVA, Student’s t-test, Chi-square test, log-rank test, Pearson’s and Spearman’s correlation analysis were used to analyze the relationship between variables. Survival curves were calculated by the Kaplan–Meier method. Univariate and multivariate analyses were performed using the Cox regression model. Data were presented as the mean ± SD, and a value of P < 0.05 was considered statistically significant.
Results
PTBP3 expression was upregulated in patients with LUAD and was associated with poor outcome
To explore the PTBP3 expression levels in LUAD patients, we first searched the public databases, including GEO and TCGA, and found that PTBP3 mRNA had higher expression levels in LUAD tissues than in non-tumor lung tissues (Figure 1). We then performed IHC analysis to determine PTBP3 protein levels in LUAD specimens and normal lung specimens using LUAD tissue microarray and found that the expression of PTBP3 was significantly higher in LUAD specimens than in normal lung tissues (Figure 1). Additionally, we detected PTBP3 protein levels in normal alveolar and bronchial epithelial cells and LUAD cell lines, and found that PTBP3 was upregulated in LUAD cell lines (Figure 1).
Figure 1.
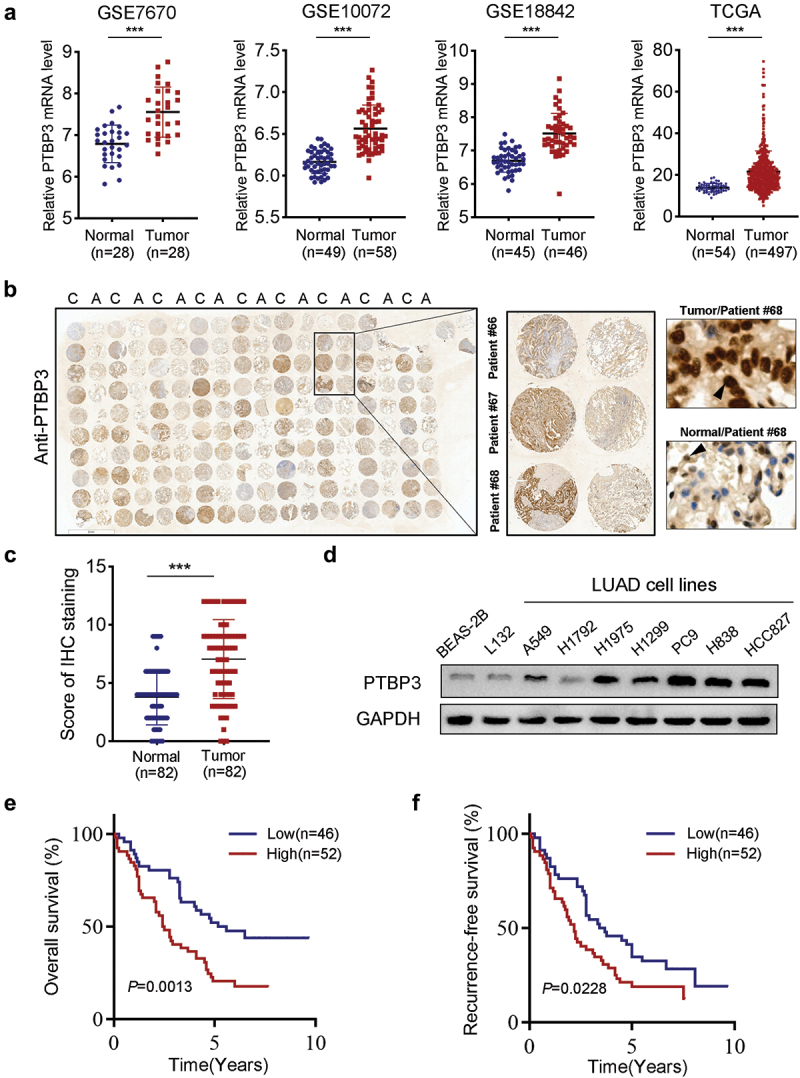
PTBP3 is upregulated in LUAD patients and indicates poor prognosis. (a) PTBP3 mRNA expression in normal lung tissues and primary LUAD tissues revealed by four different public databases, including GSE7670, GSE10072, GSE18842 and TCGA. ***P < 0.001 by Student’s t-test. (b) IHC staining showed that expression of PTBP3 in adjacent nontumor tissues and LUAD tissues (magnification ×400). (c) IHC scores of PTBP3 expression in adjacent nontumor tissues and primary LUAD tissues (n = 82). ***P < 0.001 by Student’s t-test. (d) Western-blot analysis of PTBP3 expression in human alveolar and bronchial epithelial cell lines (L132 and BEAS-2B), LUAD cell lines (A549, H1299, H1975, H1792, PC9, H838 and HCC827). GAPDH was used as an internal control. (e and f) Ninety-eight LUAD patients were divided into low (n = 46) and high (n = 52) PTBP3 expression groups as described in Materials and Methods. Kaplan-Meier analysis of overall survival (e) and recurrence-free survival (f) in LUAD patients stratified by PTBP3 protein level. Log-rank Test was used to analyze differences between groups.
Next, we further analyzed the correlation between PTBP3 expression and clinicopathological features in the LUAD tissue microarray cohort and found that the overexpression of PTBP3 was significantly correlated with lymph node metastasis and high tumor stage (Supplemental Table S2). Kaplan-Meier survival analysis showed that compared with patients with low PTBP3 expression, patients with high expression of PTBP3 had shorter overall survival (OS, P = 0.0013; Figure 1) and recurrence-free survival (RFS, P = 0.0228; Figure 1). Similar results were obtained in the GEO and TCGA LUAD mRNA dataset (Supplementary Figure S1). Next, univariate and multivariate Cox regression analyses were performed to evaluate whether the correlation of PTBP3 expression and patient survival was confounded by underlying clinicopathological features. The results showed that high expression levels of PTBP3 (P = 0.018) and TNM stage (P = 0.001) were independent and significant prognostic factors for patient survival (Supplementary Table S3). Collectively, results from both public databases and our clinical specimens indicated that PTBP3 was upregulated in patients with LUAD and was associated with poor clinical outcomes.
TGF-β1 induces expression of PTBP3 via the Smad pathway in LUAD cell lines
To investigate the role of TGF-β1 in PTBP3 expression, we treated four LUAD cell lines with exogenous TGF-β1. We found that TGF-β1 induced PTBP3 protein exprssion with expected changes in EMT markers in A549, H1792, H1299, and H838 LUAD cell lines (Figure 2). The key pathway that regulated TGF-β-induced gene expression was the Smad-dependent pathway. To explore whether TGF-β1-induced PTBP3 expression was mediated by Smad signaling pathway, we used two siRNA to knock down Smad4 protein expression which was a key component of Smad2/3/4 complex that regulated TGF-β-induced gene expression in the A549 cell line. As expected, knockdown of Smad4 protein in the A549 cell line abolished TGF-β-induced increase in PTBP3 expression (Figure 2). Furthermore, when we used SB431542, a selective inhibitor of TGF-β type I receptor, to inhibit the Smad signaling pathway, we similarly found a marked reduction in TGF-β-induced PTBP3 protein levels in a dose-dependent manner (Figure 2). These data revealed that increased PTBP3 expression was induced by TGF-β1 via the canonical Smad pathway.
Figure 2.
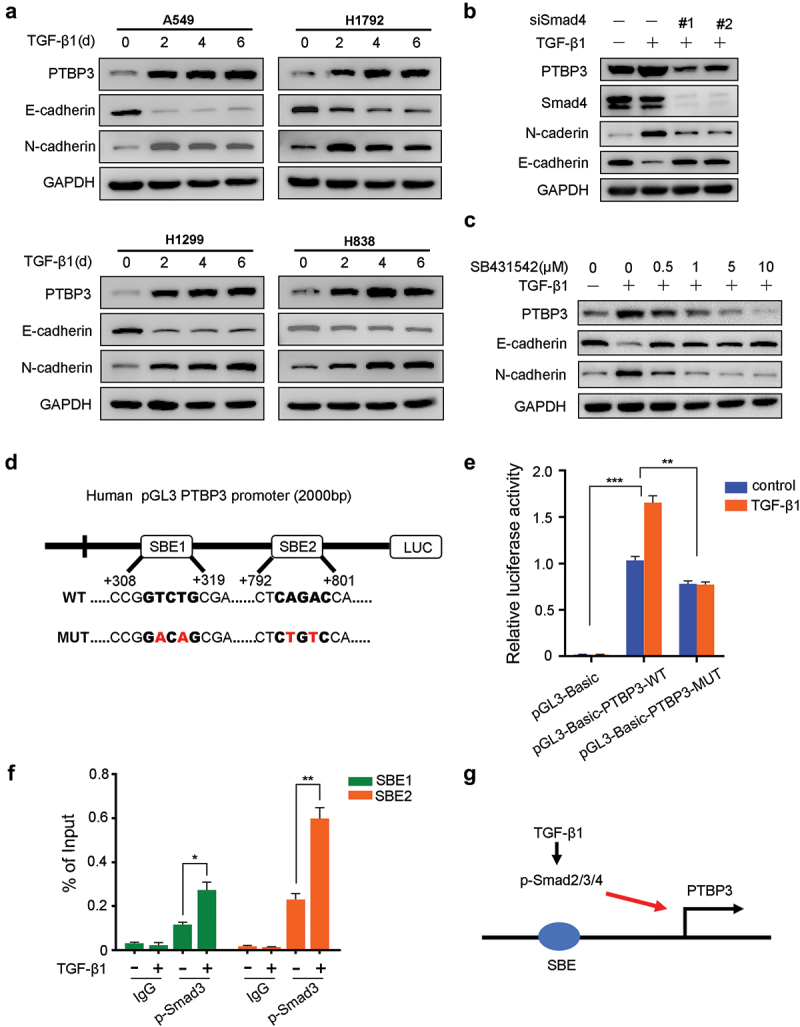
TGF-β1 increases PTBP3 protein levels of LUAD cell lines in a Smad-dependent manner. (a) Western-blot analysis of PTBP3 and EMT-related genes in four LUAD cell lines treated by TGF-β1 (5 ng/ml or 10 ng/ml) as indicated days. “d” represents the days for LUAD cell lines treated by TGF-β1. (b) Western-blot analysis of PTBP3, Smad4, N-cadherin and E-cadherin expression in A549 cells treated with control or Smad4 siRNA (100 nM) for 24 h and then treated by TGF-β1 (5 ng/ml) for another 24 h. (c) Western-blot analysis of PTBP3, N-cadherin and E-cadherin expression in A549 cells treated with 5 ng/ml TGF-β1 and 0–10 μM TGF-β type I receptor inhibitor SB431542 for 24 h. (d) A schematic diagram of the PTBP3 promoter cloned into pGL3 vector. Two predicted SBEs within the PTBP3 promoter are shown and PTBP3 promoter constructs containing mutations in these two SBEs are generated. The mutant bases of two predicted SBEs are shown in red bold letters. (e) A549 cells were co-transfected with pRL-TK plasmid and indicated constructs for 24 h, and then treated with or without TGF-β1 (5 ng/ml) for another 24 h. Then the cells were lysed to analyze luciferase activity. pRL-TK plasmid was used for internal control. (f) ChIP assay for p-Smad3 binding to two SBEs in the promoter of PTBP3. A549 cells left untreated or treated with 5 ng/ml TGF-β1 for 6 h were harvested and subjected to ChIP with isogenic IgG or anti-p-Smad3 antibody. The enrichment of the precipitated DNA by p-Smad3 antibody versus the IgG was analyzed by qPCR with the specific primers for SBE1 and SBE2. Data were presented as percent input value. (g) A schematic diagram shows that exogenous TGF-β1 induces transcription of PTBP3 in a Smad-dependent manner. GAPDH was used as an internal control for western-blot analysis. Error bars are mean ± SD from three independent experiments and Student’s t-test was used to analyze differences between groups. **P < 0.05, **P < 0.01, ***P < 0.001.
To further evaluate whether TGF-β directly regulated PTBP3 expression via the Smad pathway, we searched potential SBEs, CAGAC or GTCTG, in the PTBP3 promoter using the JASPAR. The schematic diagram identified two highly reliable SBEs within 2 kb of the PTBP3 transcriptional start site (Figure 2). Next, luciferase reporter gene assays were carried out by cloning the PTBP3 promoter constructs containing the two predicted SBEs (WT) and containing mutations in these two SBEs (MUT) into the pGL3 vector. Results of luciferase reporter assay showed that TGF-β1-induced pGL3-WT activity was almost abrogated when the two SBEs were mutated (Figure 2). These results indicate that the two SBEs were responsible for transcriptional regulation of the PTBP3 gene. We then performed a ChIP assay to verify whether Smad was directly bound to the two SBEs on the PTBP3 promoter and regulated PTBP3 transcription. We designed primers for the two Smad-binding sites and carried out a ChIP assay with p-Smad3 antibody followed by qPCR (ChIP-qPCR) in A549 cells. The results of qPCR revealed that p-Smad3 was significantly recruited and bound to the two SBEs in a TGF-β-dependent manner (Figure 2). Taken together, these data demonstrate that TGF-β activates PTBP3 gene transcription via the Smad pathway (Figure 2).
PTBP3 knockdown inhibits TGF-β1-induced EMT, migration, and invasion but has no effect on proliferation in LUAD cell lines
Since TGF-β-induced EMT, migration, and invasion have been implicated in LUAD cell lines, we decided to investigate whether PTBP3 is involved in TGF-β1-induced EMT. We used two distinct siRNAs to knock down PTBP3 expression in A549 and H1299 cells and found that PTBP3 knockdown abolished TGF-β-induced changes in the levels of EMT-related genes in A549 and H1299 cells (Figure 3). In addition, PTBP3 knockdown in A549 cells significantly decreased the mRNA and protein levels of Snail, Slug, and PAI-1, which are direct targets in response to TGF-β1 (Figure 3). Similar results were obtained for H1299 cells (Figure 3). These results indicate that PTBP3 deficiency significantly attenuates the regulation of EMT-related genes by TGF-β1. To further evaluate whether PTBP3 affects TGF-β1-induced migration and invasion, we performed transwell migration and invasion assays, which revealed that PTBP3 deficiency abrogated the migration and invasion enhanced by TGF-β1 in A549 and H1299 cells (Figure 3). After treatment with TGF-β1 for 24 h, A549 cells converted from a cobblestone-like morphology to a typical elongated mesenchymal morphology. Knockdown of PTBP3 completely blocked the morphological changes induced by TGF-β1 in A549 cells (Supplementary Figure S2).
Figure 3.
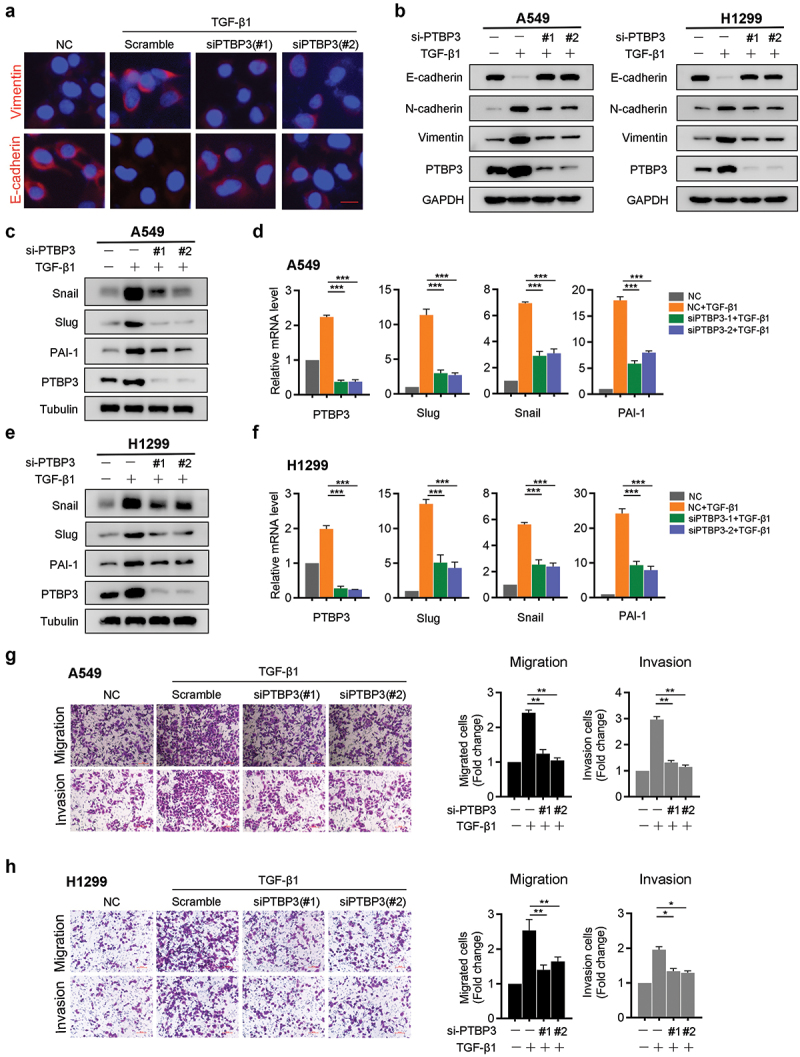
PTBP3 mediates TGF-β-induced EMT, migration and invasion of LUAD cell lines. (a) A549 cells were transfected with scramble and PTBP3 siRNA (100 nM) for 48 h and then treated with TGF-β1 (5 ng/ml) for 24 h, subsequently, the cells underwent immunofluorescence analysis by using E-cadherin and Vimentin antibodies (red). Cell nuclei were stained with DAPI (blue). scale bars, 20 μm. (b) A549 and H1299 cells transfected with scramble and PTBP3 siRNA were treated with 5 ng/ml or 10 ng/ml TGF-β1 for 24 h. Then EMT markers E-cadherin, N-cadherin and Vimentin expression were analyzed using Western blot. GAPDH was used for internal control. (c-f) A549 and H1299 cells were transfected as above. Whole cell lysates were analyzed by Western blot (c and e) and qRT-PCR (d and f) for the indicated genes. qRT-PCR data were normalized to GAPDH and presented as mean ± SD from three independent experiments. ***P < 0.001 by ANOVA. (g and h) PTBP3-knowdown A549 and H1299 cells and control cells were treated with or without TGF-β1, and were subjected to the transwell migration and invasion assay. Data were presented as the mean ±SD of triplicate experiments. *P < 0.05, **P < 0.01 by Student’s t-test.
Next, we further evaluated the effects of PTBP3 knockdown on proliferation of LUAD cells. CCK-8 assays showed no statistical difference between PTBP3 knockdown cells and control cells (Supplementary Figure S3). In summary, these data demonstrate that PTBP3 knockdown attenuates TGF-β-induced mesenchymal transition, migration, and invasion of LUAD cell lines but has no effect on proliferation in vitro.
PTBP3 enhances TGF-β1-induced cell motility by regulating Smad2/3 expression
To determine the molecular mechanism underlying the impact of PTBP3 on TGF-β-induced EMT and cell motility, we further investigated whether PTBP3 affected the key components of the TGF-β pathway. Next, we first performed qPCR assays to analyze changes in multiple Smads and found that Smad2 and Smad3 mRNA was decreased significantly in PTBP3-knockdown A549 cells compared with vector controls, but only small changes in the Smad1, Smad4, Smad5, and Smad7 transcripts (Figure 4). Similar changes were also observed in the protein expression levels (Figure 3). To further confirm our findings in H1299 cells, the same experiments were performed to corroborate the changes of Smad2 and Smad3 both at mRNA and protein expression levels in H1299 cells (Figure 4). Further investigation revealed that PTBP3 knockdown also attenuated TGF-β1-mediated Smad2 and Smad3 phosphorylation, while its overexpression using lentivirus transfection enhanced the phosphorylation in A549 cells (Figure 4). The Smad-independent signaling, mainly including MEK/ERK axis and PI3K/AKT pathway, had also been involved in TGF-β-induced EMT. We next determined whether PTBP3 affected the above-mentioned signaling pathway. Compared with vector control cells, the phosphorylation of MEK, ERK, and AKT was barely affected in PTBP3 overexpression cells in response to TGF-β1 (Figure 4). Collectively, these data indicate that PTBP3 mediates TGF-β-induced EMT by activating the Smad2/3 expression (Figure 4).
Figure 4.
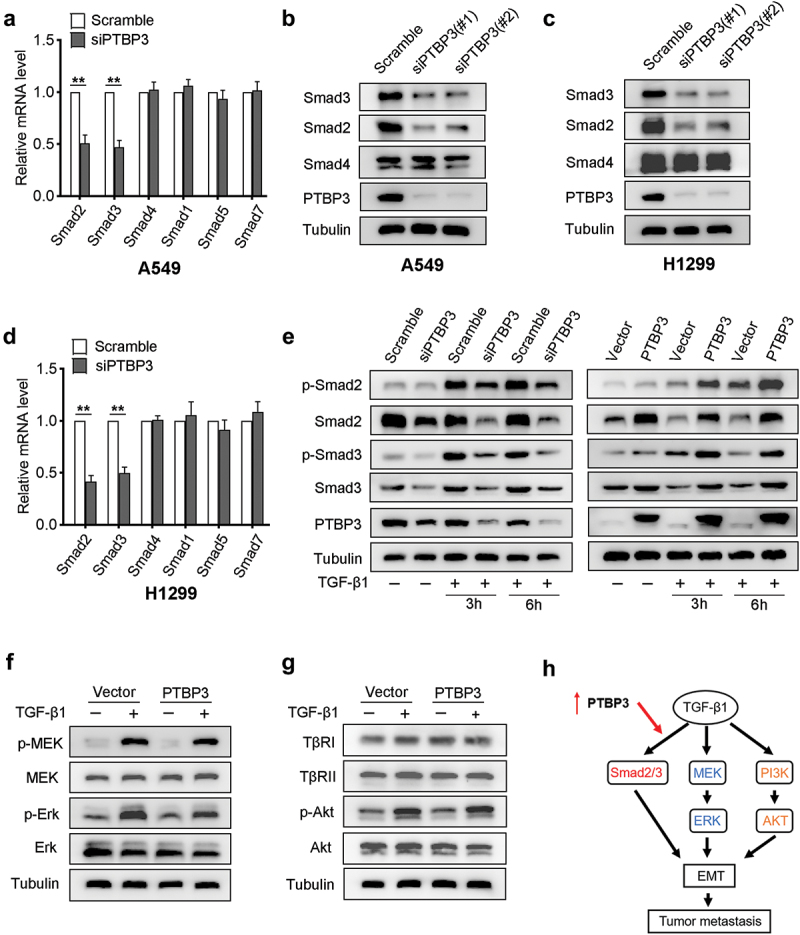
PTBP3 mediates TGF-β1-induced cell motility by activating Smad2/Smad3 expression. (a and d) qRT-PCR analysis of Smad family transcripts in PTBP3-knockdown A549 and H1299 cells and corresponding control cells. GAPDH was used for internal control. Error bars represent mean ± SD of three independent experiments. **P < 0.01 by Student’s t-test. (b and c) Western-blot analysis of Smad proteins in PTBP3-knockdown A549 and H1299 cells and corresponding control cells. Tubulin was used for internal control. (e) Western-blot analysis of the effects knowdown (left) and overexpression (right) of PTBP3 on TGF-β1-mediated Smad2 and Smad3 phosphorylation in A549 cells. Tubulin was used for internal control. (f and g) Western-blot analysis of TβRI/II, Akt, MEK, Erk and their phosphorylation derived from PTBP3 overexpression A549 cells and control cells treated with or without TGF-β1 (5 ng/ml). (h) A schematic diagram shows that PTBP3 enhanced TGF-β1-induced cell motility by activating Smad2/3 expression.
Smad2/3 expression in patient specimens is positively correlated with PTBP3
Using the GEPIA web tool, we found that the mRNA levels of Smad2 and Smad3 were positively correlated with PTBP3 in multiple cancer types (Figure 5). To further investigate the relationship between Smad2/3 and PTBP3 in patients with LUAD, tissue microarrays including 30 cases of LUAD tissue samples were subjected to IHC analysis to detect the expression of PTBP3 and Smad2/3. The staining scores of IHC were measured by the above-mentioned methods. Representative images of the staining of PTBP3 and Smad2/3 are shown in Figure 5. By analyzing the IHC scores of PTBP3 and Smad2/3 expression, we found a positive correlation between PTBP3 and Smad2/3 protein levels (Figure 5). These results indicate that there is a positive correlation between PTBP3 and Smad2/3 expression in the samples of patients with LUAD.
Figure 5.
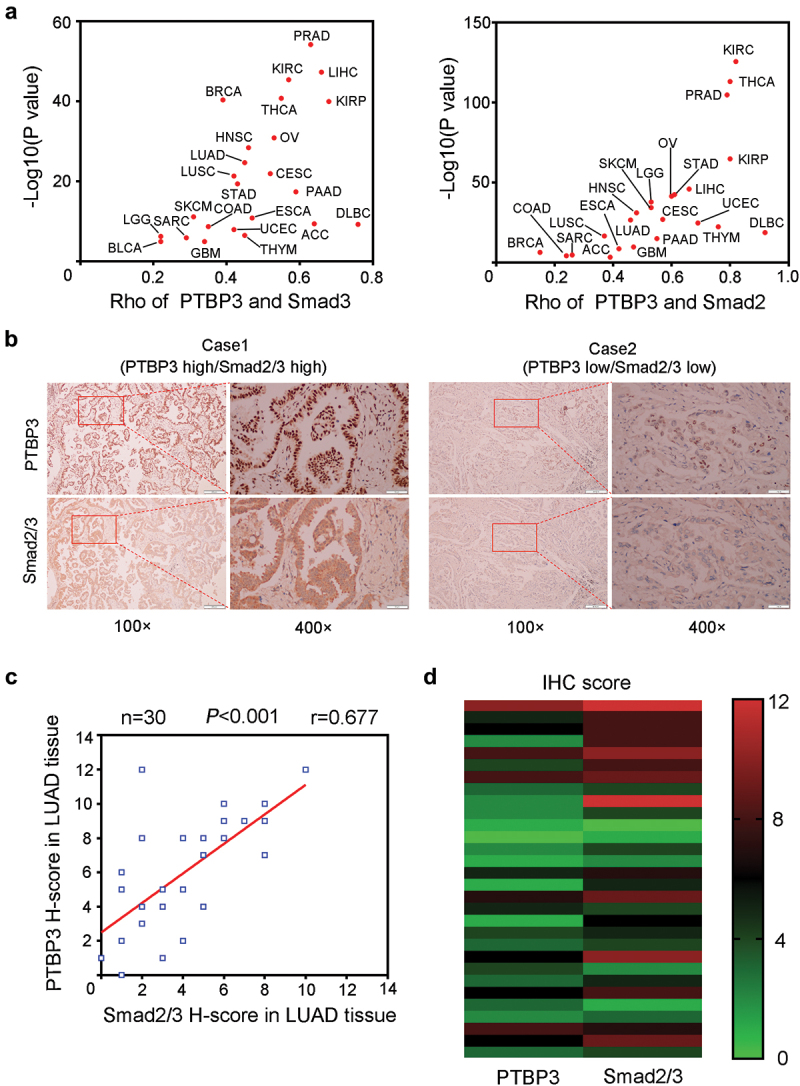
The correlation between the expression levels of PTBP3 and Smad2/3 in human LUAD specimens. (a) The correlation analysis of PTBP3 and Smad2/3 mRNA from GEPIA database. The correlation coefficient Rho and P values were calculated using Spearman’s correlation analysis. The red dots indicate P values < 0.05. (b) The representative images of immunohistochemistry staining of PTBP3 and Smad2/3 in LUAD tissue sections. Scale bars are shown as indicated. (c and d) the Pearson’s correlation analysis (c) and heatmap analysis (d) of PTBP3 and Smad2/3 protein levels from the IHC staining scores are shown (n = 30). The co-efficiency r and P values are shown as indicated.
PTBP3 promotes TGF-β-driven LUAD cell metastasis in vivo
Our above findings show that PTBP3 potently enhances TGF-β1-induced cell motility in vitro. To further explore the roles of PTBP3 in LUAD cell metastasis in vivo, we intravenously injected vector control and PTBP3 stably overexpressed A549 cells into BALB/c nude mice through the tail vein to establish an in vivo metastasis model (Figure 6), and TGF-β1 was injected intraperitoneally as described in Materials and Methods. Consistent with our findings in vitro, PTBP3 overexpression strikingly increased the number of lung metastatic nodules of A549 cells (Figure 6). No macroscopic liver metastatic nodules were detected in mice. However, the results of H&E staining show the mice injected with PTBP3 overexpressed A549 cells developed more micrometastatic foci in lung and liver than those injected with control A549 cells (Figure 6). Collectively, the in vivo metastasis assays show that PTBP3 overexpression promotes TGF-β-driven LUAD cell metastasis in vivo, further supporting our in vitro findings.
Figure 6.
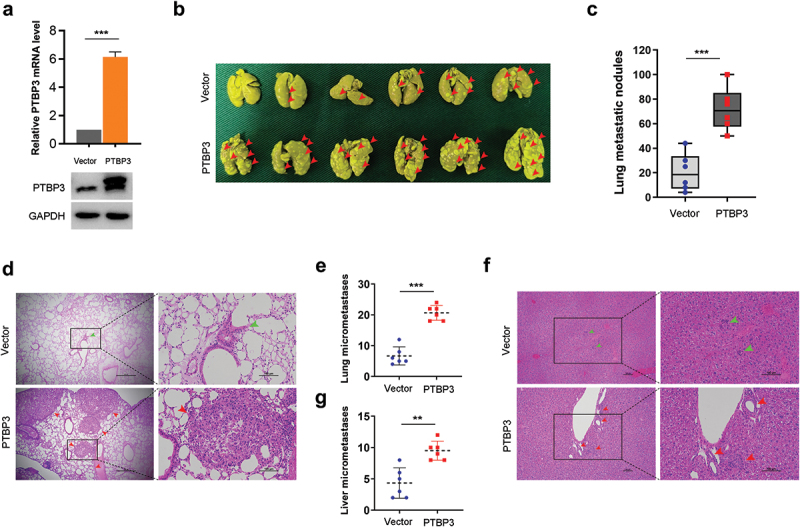
Overexpression of PTBP3 promotes TGF-β-driven LUAD cell metastasis in vivo. (a) qRT-PCR and Western-blot analysis of PTBP3 expression levels in A549 cells with stable PTBP3 overexpression. qRT-PCR data were presented as mean ± SD from three independent experiments. ***P < 0.001 by ANOVA. (b) The representative images of metastatic nodules formed in lungs removal from BALB/c nude mice at 8 weeks after intravenously injection of A549 control cells (Vector group) and PTBP3 stably overexpressed cells (PTBP3 group), with 6 mice per group in vector + TGF-β1 and PTBP3 + TGF-β1. Red arrowheads indicate lung metastatic nodules. (c) Lung metastatic nodules were counted and compared between vector group and PTBP3 overexpressed group. ***P < 0.001 by Student’s t-test. (d-g) The representative images of H&E staining for the evalution of lung (d) and liver (f) micrometastases. The arrowheads indicate metastatic nodules and micrometastases developed in lung and liver of vector group (green) and PTBP3 overexpressed group (red). Lung (e) and liver (g) micrometastases were counted and compared between vector group and PTBP3 overexpressed group. Scale bars are shown as indicated. **P < 0.01, ***P < 0.001 by Student’s t-test.
Discussion
LUAD, one of the most common subtypes of NSCLC, is characterized by rapid metastasis to distant organs [25]. EMT plays a critical role in tumor recurrence and metastasis [26]. TGF-β signaling is a key pathway reported to potently induce EMT [9]. However, the gene and molecular mechanisms that mediate TGF-β-induced EMT remain largely unknown. In the present study, we demonstrated that PTBP3 represents a novel modulator of TGF-β/Smad signaling that promotes LUAD cell invasion and metastasis. TGF-β robustly upregulates the expression of PTBP3, which, in turn, enhances TGF-β1-induced EMT by activating Smad2/3 expression.
Previous studies have suggested that PTBP3 contributes to lung cancer development and progression by regulating EMT [24]. It has also been reported that TGF-β is highly expressed in NSCLC tissues [27,28] and can promote EMT and invasion of NSCLC [7,29]. However, whether PTBP3 is involved in TGF-β-induced EMT and invasion in LUAD cells remains to be determined. In the current study, we found that PTBP3 expression is significantly increased in LUAD tissues and cell lines, and is positively correlated with TNM stage and indicates poor prognosis of patients with LUAD. In addition, we found that PTBP3 expression was potently induced by exogenous TGF-β1 in LUAD cell lines, and it has been reported that TGF-β could directly modulate EMT-related gene expression in a Smad-dependent manner [30]. Thus, we identified two potential Smad-binding motifs on the PTBP3 promoter using the JASPAR webtool. By performing the luciferase reporter assay and ChIP assay, we demonstrated that p-Smad3 binds to the promoter region of PTBP3 and positively controls PTBP3 accumulation; Moreover, when we knocked down Smad4 expression in A549 cells or used the TGF-β type I receptor inhibitor SB431542 to interfere with the TGF-β/Smad pathway, the results showed that the TGF-β1-induced increase in PTBP3 expression was entirely blocked, implying that TGF-β transcriptionally induced PTBP3 expression in LUAD cell lines via the canonical Smad pathway. However, we cannot exclude the possibility that other Smad binding sites outside of promoter-proximal regions at putative enhancer elements is involved in PTBP3 expression, especially since there is a large proportion of Smad-binding sites found in these distal loci and confirmed to be responsible for TGF-β-induced gene expression [31]. Further studies are required to elucidate this possibility.
Previous studies have shown that TGF-β-induced EMT plays a major role in LUAD cell metastasis [8] and multiple molecules involve in TGF-β signaling. For example, DOCK4, induced by TGF-β in a Smad-dependent manner, is indispensable for TGF-β-driven LUAD metastasis and blockade of DOCK4 abolishes the ability of LUAD cells to extravasate into distant organs in mice [32]. TGF-β increases PREP1 expression, which, in turn, activates the transcription of Smad3, a downstream effector of TGF-β/Smad signaling, and enhances TGF-β-induced EMT and cell motility in A549 cells [33]. In addition, it has also been reported that the expression of HtrA3-L was significantly downregulated by TGF-β, and overexpression of HtrA3-L abrogated TGF-β-mediated invasion-metastasis cascades via suppression of Smad2/3 in NSCLC [34]. In this study, we showed that PTBP3 was robustly induced by exogenous TGF-β1 in a Smad-dependent manner, and knockdown of PTBP3 attenuates TGF-β1-induced EMT and invasion of LUAD cells as well as expression of mesenchymal genes, such as Snail and Slug.
How does PTBP3 regulate TGF-β-induced EMT? Changes in TGF-β signaling components have been observed during tumor progression [35] and current studies primarily focus on the regulation of TGF-β receptors and Smads. It has been widely reported that various molecules are involved in the regulation of TGF-β receptors and Smads either at the transcriptional level [33,36,37] or post-transcriptional level [38–41]. In the present study, we found that overexpression of PTBP3 in A549 cells had no obvious effect on the expression of TβRI/II. However, PTBP3 knockdown and overexpression significantly decreased and increased, respectively, the expression of Smad2/3 and levels of TGF-β1-mediated Smad2/3 phosphorylation. Due to MEK/ERK and PI3K/AKT pathway, all of which have been confirmed to be involved in TGF-β-induced EMT [42], we next detected the expression changes of these pathways upon PTBP3 overexpression. Compared with control cells, the phosphorylation of MEK, ERK, and AKT was barely affected in PTBP3 overexpression A549 cells treated with TGF-β1. Unfortunately, we could not determine molecular mechanisms that PTBP3 regulates the expression of Smad2/3. Considering the critical role of PTBP3 in RNA alternative splicing, we speculate that PTBP3 may regulate the Smad2/3 expression transcriptionally in LUAD cells by RNA alternative splicing, which is an interesting issue to explore in the future. Despite its limitation, our study does demonstrate that PTBP3 is a novel, key component involved in TGF-β-induced EMT and identify a positive feedback loop between PTBP3 and Smad2/3 driving TGF-β-induced EMT.
In conclusion, our data unveiled that PTBP3 is significantly upregulated in LUAD tissues and high expression of PTBP3 was associated with poor OS and RFS of patients with LUAD. Furthermore, we demonstrated that PTBP3 is a novel mediator of TGF-β/Smad signaling and promotes invasion and metastasis of LUAD cells by activating Smad2/3 expression. Our findings provide a deepened insight into how PTBP3 regulates TGF-β/Smad signaling in LUAD and reveal that PTBP3 is a promising therapeutic target for the treatment of patients with LUAD.
Supplementary Material
Acknowledgments
We gratefully acknowledge contributions from TCGA and GEO databases and would like to thank Editage (www.editage.cn) for English language editing.
Funding Statement
This work was supported by the National Natural Science Foundation of China (No.81974053) and Shanghai Committee of Science and Technology (No 18441902300, 16441907800).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- [2].Kuhn E, Morbini P, Cancellieri A, et al. Adenocarcinoma classification: patterns and prognosis. Pathologica. 2018;110(1):5–11. [PubMed] [Google Scholar]
- [3].Inamura K, Ishikawa Y.. Lung cancer progression and metastasis from the prognostic point of view. Clin Exp Metastasis. 2010;27(6):389–397. [DOI] [PubMed] [Google Scholar]
- [4].Du B, Shim JS.. Targeting Epithelial-Mesenchymal Transition (EMT) to overcome drug resistance in cancer. Molecules. 2016;21(7):965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29(3):212–226. [DOI] [PubMed] [Google Scholar]
- [6].Zhang HJ, Wang HY, Zhang HT, et al. Transforming growth factor-β1 promotes lung adenocarcinoma invasion and metastasis by epithelial-to-mesenchymal transition. Mol Cell Biochem. 2011;355(1–2):309–314. [DOI] [PubMed] [Google Scholar]
- [7].Wang L, Tong X, Zhou Z, et al. Circular RNA hsa_circ_0008305 (circPTK2) inhibits TGF-β-induced epithelial-mesenchymal transition and metastasis by controlling TIF1γ in non-small cell lung cancer. Mol Cancer. 2018;17(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tong X, Wang S, Lei Z, et al. MYOCD and SMAD3/SMAD4 form a positive feedback loop and drive TGF-β-induced epithelial-mesenchymal transition in non-small cell lung cancer. Oncogene. 2020;39(14):2890–2904. [DOI] [PubMed] [Google Scholar]
- [9].Massagué J. TGFbeta in Cancer. Cell. 2008;134(2):215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dennler S, Itoh S, Vivien D, et al. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. Embo J. 1998;17(11):3091–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development. 2009;136(22):3699–3714. [DOI] [PubMed] [Google Scholar]
- [12].Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yingling JM, Blanchard KL, Sawyer JS. Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov. 2004;3(12):1011–1022. [DOI] [PubMed] [Google Scholar]
- [14].Connolly EC, Freimuth J, Akhurst RJ. Complexities of TGF-β targeted cancer therapy. Int J Biol Sci. 2012;8(7):964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mullen AC, Orlando DA, Newman JJ, et al. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 2011;147(3):565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tan LY, Whitfield P, Llorian M, et al. Generation of functionally distinct isoforms of PTBP3 by alternative splicing and translation initiation. Nucleic Acids Res. 2015;43(11):5586–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Xue Y, Zhou Y, Wu T, et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell. 2009;36(6):996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Brazão TF, Demmers J, van IW, et al. A new function of ROD1 in nonsense-mediated mRNA decay. FEBS Lett. 2012;586(8):1101–1110. [DOI] [PubMed] [Google Scholar]
- [19].Hou P, Li L, Chen F, et al. PTBP3-Mediated regulation of ZEB1 mRNA stability promotes epithelial-mesenchymal transition in breast cancer. Cancer Res. 2018;78(2):387–398. [DOI] [PubMed] [Google Scholar]
- [20].Liang X, Chen W, Shi H, et al. PTBP3 contributes to the metastasis of gastric cancer by mediating CAV1 alternative splicing. Cell Death Dis. 2018;9(5):569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang X, Qu S, Wang L, et al. PTBP3 splicing factor promotes hepatocellular carcinoma by destroying the splicing balance of NEAT1 and pre-miR-612. Oncogene. 2018;37(50):6399–6413. [DOI] [PubMed] [Google Scholar]
- [22].Ma J, Weng L, Jia Y, et al. PTBP3 promotes malignancy and hypoxia-induced chemoresistance in pancreatic cancer cells by ATG12 up-regulation. J Cell Mol Med. 2020;24(5):2917–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tano K, Mizuno R, Okada T, et al. MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett. 2010;584(22):4575–4580. [DOI] [PubMed] [Google Scholar]
- [24].Wu Q, Zhang B, Li B, et al. PTBP3 promotes migration of non-small cell lung cancer through regulating E-cadherin in EMT signaling pathway. Cancer Cell Int. 2020;20(1):172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Provencio M, Isla D, Sánchez A, et al. Inoperable stage III non-small cell lung cancer: current treatment and role of vinorelbine. J Thorac Dis. 2011;3(3):197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gopal SK, Greening DW, Rai A, et al. Extracellular vesicles: their role in cancer biology and epithelial-mesenchymal transition. Biochem J. 2017;474(1):21–45. [DOI] [PubMed] [Google Scholar]
- [27].Asselin-Paturel C, Echchakir H, Carayol G, et al. Quantitative analysis of Th1, Th2 and TGF-beta1 cytokine expression in tumor, TIL and PBL of non-small cell lung cancer patients. Int J Cancer. 1998;77(1):7–12. [DOI] [PubMed] [Google Scholar]
- [28].Saji H, Nakamura H, Awut I, et al. Significance of expression of TGF-beta in pulmonary metastasis in non-small cell lung cancer tissues. Ann Thorac Cardiovasc Surg. 2003;9(5):295–300. [PubMed] [Google Scholar]
- [29].Cao L, Qi L, Zhang L, et al. Human nonsense-mediated RNA decay regulates EMT by targeting the TGF-ß signaling pathway in lung adenocarcinoma. Cancer Lett. 2017;403:246–259. [DOI] [PubMed] [Google Scholar]
- [30].Jeong MH, Park SY, Lee SH, et al. EPB41L5 mediates TGFβ-induced metastasis of gastric cancer. Clin Cancer Res. 2019;25(12):3617–3629. [DOI] [PubMed] [Google Scholar]
- [31].Schlenner SM, Weigmann B, Ruan Q, et al. Smad3 binding to the foxp3 enhancer is dispensable for the development of regulatory T cells with the exception of the gut. J Exp Med. 2012;209(9):1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yu JR, Tai Y, Jin Y, et al. TGF-β/Smad signaling through DOCK4 facilitates lung adenocarcinoma metastasis. Genes Dev. 2015;29(3):250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Risolino M, Mandia N, Iavarone F, et al. Transcription factor PREP1 induces EMT and metastasis by controlling the TGF-β-SMAD3 pathway in non-small cell lung adenocarcinoma. Proc Natl Acad Sci U S A. 2014;111(36):E3775–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhao J, Feng M, Liu D, et al. Antagonism between HTRA3 and TGFβ1 contributes to metastasis in non-small cell lung cancer. Cancer Res. 2019;79(11):2853–2864. [DOI] [PubMed] [Google Scholar]
- [35].Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17(1–2):41–58. [DOI] [PubMed] [Google Scholar]
- [36].Tang YN, Ding WQ, Guo XJ, et al. Epigenetic regulation of Smad2 and Smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat Commun. 2015;6(1):8230. [DOI] [PubMed] [Google Scholar]
- [37].Lei B, Wang D, Zhang M, et al. miR-615-3p promotes the epithelial-mesenchymal transition and metastasis of breast cancer by targeting PICK1/TGFBRI axis. J Exp Clin Cancer Res. 2020;39(1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pefani DE, Pankova D, Abraham AG, et al. TGF-β targets the hippo pathway scaffold RASSF1A to Facilitate YAP/SMAD2 nuclear translocation. Mol Cell. 2016;63(1):156–166. [DOI] [PubMed] [Google Scholar]
- [39].Deng C, Lin YX, Qi XK, et al. TNFRSF19 Inhibits TGFβ Signaling through Interaction with TGFβ receptor type i to promote tumorigenesis. Cancer Res. 2018;78(13):3469–3483. [DOI] [PubMed] [Google Scholar]
- [40].Zhang Q, Xiao M, Gu S, et al. ALK phosphorylates SMAD4 on tyrosine to disable TGF-β tumour suppressor functions. Nat Cell Biol. 2019;21(2):179–189. [DOI] [PubMed] [Google Scholar]
- [41].Liao Z, Chen L, Zhang X, et al. PTPRε acts as a metastatic promoter in hepatocellular carcinoma by facilitating recruitment of SMAD3 to TGF-β receptor 1. Hepatology. 2020;72(3):997–1012. [DOI] [PubMed] [Google Scholar]
- [42].Voulgari A, Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta. 2009;1796(2):75–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.