

Abstract
Unexplained weight changes that occur in Parkinson’s disease (PD), are often neglected and remain a poorly understood non-motor feature in patients with PD. A specific ‘Park-weight’ phenotype with low body weight has been described, and our aim was to evaluate the clinical and prognostic trajectories and biomarkers of weight variability in PD. We evaluated body weight-related biomarkers in 405 de novo PD patients and 187 healthy controls (HC) over a 5-year follow-up period from the PPMI database. Body-weight variability was defined as intra-individual variability in body weight between visits. PD patients were categorized as weight losers, gainers, or patients with stable weight. The differential progression of motor and non-motor clinical variables between groups was explored using linear mixed-effects models. Finally, we estimated longitudinal changes in weight as a function of baseline and longitudinal striatal presynaptic dopaminergic transporter imaging. PD patients presented a greater weight variability compared to HC (p = 0.003). Patients who developed weight loss had lower CSF amyloid-beta 1–42 (p = 0.009) at baseline. In addition, patients with weight loss showed a faster cognitive decline (p = 0.001), whereas patients with weight gain showed a slower motor progression (p = 0.001), compared to patients with stable weight. Baseline right striatal denervation was a predictor of weight variability in both PD patients and HC (p < 0.001). Similarly, weight variability in PD patients was associated with the progression of right striatal denervation (p < 0.001). Weight variability and specifically weight loss are more frequent in PD compared to HC, and are associated with specific motor, non-motor and cognitive progression patterns. A greater CSF amyloid burden was present at baseline in patients with subsequent weight loss. Presynaptic dopaminergic imaging in the right striatum may serve as a predictor of future weight changes in PD and HC.
Subject terms: Parkinson's disease, Prognostic markers
Introduction
Weight variability, which can be pathological, is a relatively common clinical finding among patients with Parkinson’s disease (PD) and yet remains poorly researched1. Weight loss has been reported across all stages of PD1 and it has been proposed that it might be a prodromal feature of PD2–4. Weight loss has been associated with female sex5, levodopa daily dose6, dysautonomia7, and olfactory dysfunction8 as well as greater severity of motor features and dyskinesias9, more frequent occurrence of cognitive impairment, and increased disability and mortality in PD patients10–12. On the other hand, a considerable number of PD patients show weight gain associated with comorbidities13, chronic use of dopamine replacement therapy with associated binge-eating behaviour14, and ablative15 or functional neurosurgery16. Of note, contrasting data are available on the role of body weight as a risk factor for the development of PD2,3,17,18.
Although body weight is regulated by many variables including genetic, epigenetic, metabolic, and environmental factors, under physiological conditions, homoeostatic behavioural adaptations tend to preserve a stable body weight19. Weight fluctuations may involve changes in energy expenditure, perturbation of homoeostatic control, and eating behaviour modulated by the dopaminergic system1, which is known to be altered in PD. Ghrelin and leptin are peptides regarded as modulators of human energy balance, and lower plasma levels of the latter have been identified in patients with PD and weight loss20,21. In addition, a variety of additional factors might lead to reduced caloric intake and subsequent weight loss in PD including decreased appetite (due to depression as well as hyposmia), dysphagia, and gastrointestinal dysmotility with altered intestinal absorption. Interestingly, data from preclinical models of PD seems to suggest a possible role of central noradrenergic neurotransmission in weight variations in PD22.
Even though the occurrence of unintended weight changes in PD patients has long been recognized23, its underlying mechanisms need to be further elucidated. The lack of a clear pathophysiological understanding is accompanied by the scarcity of biomarkers for this complex clinical condition. In healthy elderly individuals, weight loss has been associated with increased PET amyloid uptake24, and lower body mass index (BMI) coinciding with decreased levels of amyloid-beta 1–42 (Aβ1–42) in cerebrospinal fluid (CSF)25, suggesting a higher amyloid burden in these individuals. However, the relationship between weight variability and CSF biomarkers in PD is still unknown.
Furthermore, since the dorsal striatum, which comprises of the putamen and caudate, has been implicated in maintaining caloric requirements26, there have been attempts to assess its relationship with weight variability. A recent study found that the putamen-to-caudate DAT ratio at the time of diagnosis predicted subsequent weight change in PD, but only in male patients27. In another recent study conducted on healthy individuals, it has been shown that body weight is linked to a higher dopamine receptor availability in the right putamen compared to the left28. This relationship in PD, however, remains unexplored. Finally, weight changes have also been associated with urate levels in men with a high cardiovascular risk profile, however, their associations have not been demonstrated in PD29. Interestingly, urate levels have been negatively associated with non-motor symptom burden in PD patients30.
In this study, we sought to characterize weight variability, their related clinical (motor and non-motor) trajectories, and associated clinical and imaging biomarkers in a large cohort of de novo patients with PD, with a 5-year follow-up with the above-cited possible biomarkers. Specifically, we aimed to explore whether weight variability might be associated with different clinical progression patterns, CSF Aβ1–42, urate levels and dopaminergic denervation in the striatum.
Results
Weight variability in Parkinson's Disease patients and healthy controls
Demographic and clinical baseline data are shown in Table 1. No demographic differences were found between the two groups. While BMI was not different between PD patients and HC, weight variability (as measured by average successive variability) was higher in patients with PD (1.38 ± 1.5 kg vs 1.08 ± 1.5 kg, p = 0.003).
Table 1.
Baseline Characteristics of Parkinson's Disease patients and healthy controls.
| Variables | Healthy controls (n = 187) |
Parkinson’s disease patients (n = 405) |
p |
|---|---|---|---|
| Age, years | 61.1 ± 11.1 | 61.5 ± 9.7 | 0.865 |
| Sex, male (%) | 65.4% | 64.7% | 0.863 |
| Education, years | 16.1 ± 2.9 | 15.5 ± 2.9 | 0.350 |
| MDS-UPDRS I | 2.9 ± 2.9 | 5.5 ± 4.0 | – |
| MDS-UPDRS II | 0.4 ± 1.0 | 5.8 ± 4.1 | – |
| MDS-UPDRS III | 1.2 ± 2.2 | 20.8 ± 8.8 | – |
| MDS-UPDRS total | 4.6 ± 4.4 | 32.1 ± 13.1 | – |
| BMI (Kg/m2) | 26.8 ± 4.4 | 27.1 ± 4.6 | 0.564 |
| Weight variabilitya | 1.08 ± 1.5 | 1.38 ± 1.5 | 0.003 |
| Mean Putamen, SBR | 2.14 ± 0.54 | 0.81 ± 0.28 | – |
| Mean Caudate, SBR | 2.97 ± 0.62 | 1.98 ± 0.55 | – |
Data are presented as number (%) or mean ± standard deviation.
aWeight variability calculated as average successive variability (ASV).
MDS-UPDRSMovement Disorders Society Unified Parkinson Disease Rating Scale. SBR striatal binding ratio.
Baseline differences between patients with weight loss, gain and stable weight
Patients with weight gain over the follow-up period of five years were younger at baseline compared to patients with stable weight (p = 0.033, Table 2). No other demographic differences were found. Patients with weight loss had more difficulties with their activities of daily living (ADL) (p = 0.003) and had lower CSF levels of Aβ1–42 (p = 0.009) at baseline compared to patients with a stable weight. Other CSF and imaging biomarkers were not different between the three groups (Table 2).
Table 2.
Baseline differences of de novo Parkinson’s disease patients with subsequent stable weight, weight loss or weight gain after five years of follow-up.
| Stable weight (n = 203) |
Weight loss (n = 134) |
Weight gain (n = 68) |
P stable vs loss |
P stable vs gain |
|
|---|---|---|---|---|---|
| Age, years | 62.0 ± 9.9 | 62.0 ± 9.7 | 59.4 ± 9.5 | 0.990 | 0.033 |
| Sex, male (%) | 65.5% | 67.9% | 60.3% | 0.649 | 0.437 |
| Education, years | 15.6 ± 2.8 | 15.6 ± 3.1 | 15.3 ± 2.9 | 0.560 | 0.481 |
| Binge eatinga | 7.4% | 9.7% | 11.8% | 0.460 | 0.268 |
| ADL | 93.9 ± 5.3 | 91.9 ± 6.1 | 93.3 ± 6.1 | 0.003* | 0.588 |
| HY | 1.5 ± 0.5 | 1.6 ± 0.5 | 1.5 ± 0.5 | 0.217 | 0.885 |
| MDS-UPDRS I | 5.4 ± 4.0 | 5.7 ± 3.9 | 5.3 ± 4.4 | 0.440 | 0.589 |
| MDS-UPDRS II | 5.4 ± 4.1 | 6.2 ± 4.3 | 6.3 ± 4.1 | 0.057 | 0.076 |
| MDS-UPDRS III | 19.4 ± 8.1 | 22.0 ± 9.2 | 22.7 ± 9.5 | 0.020 | 0.012 |
| MDS-UPDRS total | 30.2 ± 12.5 | 33.9 ± 13.1 | 34.3 ± 14.5 | 0.015 | 0.083 |
| MoCA | 26.9 ± 2.4 | 27.3 ± 2.2 | 27.2 ± 2.2 | 0.028 | 0.504 |
| UPSIT | 22.0 ± 8.0 | 22.8 ± 8.4 | 22.2 ± 8.1 | 0.501 | 0.759 |
| GDS | 2.1 ± 2.2 | 2.3 ± 2.3 | 2.8 ± 3.1 | 0.546 | 0.130 |
| STAI | 63.9 ± 18.0 | 64.9 ± 17.4 | 69.8 ± 20.3 | 0.559 | 0.033 |
| SCOPA-AUT | 8.9 ± 5.7 | 10.3 ± 6.6 | 8.9 ± 5.4 | 0.096 | 0.981 |
| ESS | 5.5 ± 3.1 | 6.4 ± 3.8 | 4.8 ± 2.8 | 0.066 | 0.130 |
|
Weight variabilityb |
0.00 ± 0.5 | -2.4 ± 1.6 | 2.2 ± 1.9 | – | – |
| Mean putamen, SBR | 0.84 ± 0.29 | 0.77 ± 0.25 | 0.84 ± 0.34 | 0.038 | 0.596 |
| Mean caudate, SBR | 2.03 ± 0.55 | 1.91 ± 0.54 | 2.03 ± 0.57 | 0.015 | 0.338 |
| CSF amyloid beta, pg/mL | 944.9 ± 389.7 | 845.1 ± 380.5 | 936.5 ± 531.0 | 0.009* | 0.356 |
| CSF alpha synuclein, pg/mL | 1556.1 ± 671.3 | 1407.0 ± 543.7 | 1550.9 ± 853.2 | 0.942 | 0.338 |
| CSF tau, pg/mL | 169.8 ± 53.4 | 167.2 ± 52.3 | 167.3 ± 68.8 | 0.647 | 0.928 |
| Serum urate, µmol/L | 313.5 ± 77.8 | 329.1 ± 83.8 | 307.3 ± 67.7 | 0.127 | 0.546 |
Data are n (%) and the mean ± standard deviation.
aItem of the Questionnaire for Impulsive-Compulsive Disorders.
bWeight variability calculated as average successive variability taking directionality into account.
HY Hoehn and Yahr scale, ADL Activities of Daily Living, MDS-UPDRS Movement Disorders Society Unified Parkinson Disease Rating Scale, MoCA Montreal Cognitive Assessment, SCOPA-AUT SCOPA-Autonomic, UPSIT University of Pennsylvania Smell Identification Test, GDS Geriatric Depression Scale, STAI State-Trait Anxiety Inventory, ESS Epworth Sleepiness Scale, SBR striatal binding ratio, CSF cerebrospinal fluid.
*Significant p-values after Bonferroni correction for multiple testing.
Clinical trajectories in patients with stable weight, weight loss and weight gain
The longitudinal clinical trajectories of the subgroups are shown in Table 3. Compared to patients with a stable weight, patients with weight loss showed faster progression in MDS-UPDRS II (p < 0.001), total MDS-UPDRS (p < 0.001), ADL (p = 0.001) and MoCA (p < 0.001) scores, whereas patients with weight gain showed slower progression in MDS-UPDRS III (p < 0.001), and anxiety (STAI, p = 0.004) scores.
Table 3.
Generalized linear mixed analysis for the comparison of the progression over time of clinical variables between de novo Parkinson’s patients with stable weight, weight loss, or weight gain.
| Outcome | Baseline (n = 406) |
Year 1 (n = 363) |
Year 2 (n = 363) |
Year 3 (n = 359) |
Year 4 (n = 341) |
Year 5 (n = 314) |
Group × Time effect | |
|---|---|---|---|---|---|---|---|---|
| Est (SE) | p | |||||||
| MDS- UPDRS I | ||||||||
| Stable weight | 5.4 ± 4.0 | 6.7 ± 4.7 | 7.3 ± 4.8 | 7.8 ± 5.3 | 8.7 ± 5.7 | 8.9 ± 5.8 | ||
| Weight loss | 5.7 ± 3.9 | 7.3 ± 4.7 | 7.8 ± 5.5 | 9.1 ± 5.5 | 9.9 ± 6.7 | 10.0 ± 7.1 | 0.22 (0.09) | 0.013 |
| Weight gain | 5.9 ± 4.4 | 6.3 ± 4.3 | 8.0 ± 5.2 | 8.2 ± 5.7 | 8.9 ± 4.6 | 9.7 ± 5.7 | 0.25 (0.11) | 0.026 |
| MDS- UPDRS II | ||||||||
| Stable weight | 5.5 ± 4.1 | 7.0 ± 4.9 | 7.5 ± 5.2 | 8.1 ± 5.4 | 9.4 ± 6.6 | 9.4 ± 5.8 | ||
| Weight loss | 6.2 ± 4.3 | 8.4 ± 5.2 | 8.9 ± 5.5 | 10.3 ± 6.2 | 10.8 ± 7.2 | 11.5 ± 7.7 | 0.32 (0.09) | <0.001* |
| Weight gain | 6.3 ± 4.1 | 7.2 ± 4.4 | 7.4 ± 4.5 | 8.0 ± 4.6 | 9.4 ± 5.8 | 10.0 ± 5.7 | −0.01 (0.1) | 0.945 |
| MDS- UPDRS III | ||||||||
| Stable weight | 19.4 ± 8.2 | 22.7 ± 9.4 | 25.2 ± 11.5 | 27.6 ± 12.3 | 26.9 ± 11.4 | 30.6 ± 12.4 | ||
| Weight loss | 22.0 ± 9.2 | 27.4 ± 10.7 | 30.1 ± 10.5 | 33.1 ± 11.4 | 34.3 ± 12.4 | 33.7 ± 12.9 | 0.45 (0.2) | 0.854 |
| Weight gain | 22.7 ± 9.5 | 23.7 ± 12.0 | 27.3 ± 12.8 | 25.7 ± 11.2 | 29.5 ± 14.0 | 29.1 ± 11.8 | −0.81 (0.3) | <0.001* |
| MDS- UPDRS IV | ||||||||
| Stable weight | NA | 0.4 ± 1.3 | 0.7 ± 1.7 | 1.0 ± 1.8 | 1.6 ± 2.5 | 2.1 ± 2.8 | ||
| Weight loss | NA | 0.4 ± 1.3 | 0.9 ± 2.1 | 1.0 ± 2.2 | 1.6 ± 2.9 | 2.0 ± 3.2 | −0.003 (0.02) | 0.962 |
| Weight gain | NA | 0.6 ± 1.6 | 0.3 ± 0.9 | 0.5 ± 1.3 | 1.3 ± 2.4 | 2.4 ± 2.8 | 0.02 (0.09) | 0.696 |
| MDS-UPDRS | ||||||||
| Stable weight | 30.3 ± 12.5 | 36.1 ± 14.5 | 39.4 ± 17.4 | 42.6 ± 18.1 | 46.3 ± 17.9 | 48.0 ± 18.5 | ||
| Weight loss | 33.9 ± 13.1 | 43.6 ± 16.8 | 46.9 ± 16.6 | 52.7 ± 18.8 | 53.8 ± 22.1 | 53.8 ± 21.2 | 1.07 (0.32) | 0.001* |
| Weight gain | 34.3 ± 14.5 | 37.4 ± 16.4 | 42.8 ± 16.6 | 41.5 ± 16.6 | 46.8 ± 20.1 | 50.0 ± 18.9 | −0.40 (0.41) | 0.340 |
| ADL | ||||||||
| Stable weight | 94.0 ± 5.4 | 91.0 ± 7.0 | 89.2 ± 8.3 | 88.4 ± 7.8 | 86.6 ± 10.3 | 86.0 ± 10.4 | ||
| Weight loss | 91.9 ± 6.2 | 89.7 ± 6.3 | 87.4 ± 8.2 | 86.0 ± 8.5 | 83.0 ± 10.3 | 80.5 ± 16.4 | 0.07 (0.18) | <0.001* |
| Weight gain | 93.4 ± 6.2 | 91.2 ± 6.3 | 90.6 ± 6.4 | 89.5 ± 7.5 | 88.2 ± 3.7 | 86.9 ± 8.0 | 0.27 (0.23) | 0.237 |
| MoCA | ||||||||
| Stable weight | 26.9 ± 2.4 | 26.4 ± 2.7 | 26.3 ± 2.9 | 26.6 ± 2.8 | 26.8 ± 3.1 | 27.0 ± 2.9 | ||
| Weight loss | 27.3 ± 2.2 | 26.3 ± 3.0 | 26.2 ± 3.6 | 26.1 ± 3.3 | 25.7 ± 4.1 | 25.4 ± 4.6 | 0.35 (0.05) | <0.001* |
| Weight gain | 27.2 ± 2.2 | 26.1 ± 2.7 | 26.2 ± 3.2 | 26.5 ± 3.1 | 26.5 ± 3.7 | 27.2 ± 2.7 | 0.05 (0.06) | 0.448 |
| GDS | ||||||||
| Stable weight | 2.1 ± 2.2 | 2.3 ± 2.8 | 2.3 ± 2.6 | 2.3 ± 2.6 | 2.5 ± 2.6 | 2.5 ± 2.6 | ||
| Weight loss | 2.3 ± 2.3 | 2.7 ± 3.1 | 2.7 ± 2.9 | 3.1 ± 3.1 | 2.8 ± 3.1 | 3.3 ± 3.2 | 0.10 (0.05) | 0.061 |
| Weight gain | 2.8 ± 3.1 | 2.7 ± 2.7 | 3.4 ± 3.4 | 2.7 ± 2.7 | 2.6 ± 2.8 | 2.7 ± 2.6 | −0.10 (0.06) | 0.116 |
| STAI | ||||||||
| Stable weight | 63.9 ± 18.0 | 64.1 ± 18.5 | 62.9 ± 17.4 | 63.6 ± 17.9 | 64.2 ± 18.1 | 63.7 ± 18.1 | ||
| Weight loss | 64.7 ± 17.3 | 65.7 ± 18.3 | 67.3 ± 20.1 | 66.7 ± 20.7 | 65.7 ± 19.6 | 67.8 ± 22.7 | 0.42 (0.30) | 0.167 |
| Weight gain | 69.7 ± 20.3 | 68.0 ± 19.5 | 68.1 ± 19.0 | 65.3 ± 18.5 | 65.6 ± 19.1 | 63.4 ± 16.5 | −1.10 (0.39) | 0.004* |
| SCOPA | ||||||||
| Stable weight | 8.9 ± 5.7 | 10.5 ± 6.2 | 10.6 ± 6.3 | 11.3 ± 6.6 | 11.8 ± 7.1 | 12.7 ± 8.1 | ||
| Weight loss | 10.2 ± 6.6 | 12.3 ± 6.8 | 12.7 ± 7.0 | 14.2 ± 7.8 | 14.2 ± 8.2 | 15.2 ± 9.0 | 0.20 (0.10) | 0.061 |
| Weight gain | 8.8 ± 5.4 | 9.83 ± 6.0 | 11.2 ± 5.3 | 11.8 ± 6.2 | 12.9 ± 6.0 | 13.6 ± 5.5 | 0.18 (0.13) | 0.168 |
| ESS | ||||||||
| Stable weight | 5.5 ± 3.1 | 6.1 ± 4.1 | 6.7 ± 4.4 | 7.2 ± 4.4 | 7.4 ± 4.5 | 7.6 ± 4.5 | ||
| Weight loss | 6.4 ± 3.8 | 6.5 ± 4.0 | 6.6 ± 4.0 | 7.6 ± 4.7 | 7.6 ± 4.8 | 8.0 ± 4.7 | −0.06 (0.08) | 0.433 |
| Weight gain | 4.8 ± 2.8 | 5.6 ± 3.5 | 6.8 ± 4.0 | 7.0 ± 4.0 | 7.3 ± 4.8 | 7.7 ± 5.2 | 0.18 (0.09) | 0.060 |
NA not applicable, MDS-UPDRS Movement Disorders Society Unified Parkinson Disease Rating Scale, MoCA Montreal Cognitive Assessment. SCOPA-AUT SCOPA-autonomic, UPSIT University of Pennsylvania Smell Identification Test, GDS Geriatric Depression Scale, STAI State-Trait Anxiety Inventory, ESS Epworth Sleepiness Scale.
*Significant p-values after Bonferroni correction for multiple testing.
Binge eating disorder and dopaminergic medications
Binge eating disorder was more prevalent in weight gainers (Est: 0.35, SE: 0.15, p = 0.02) compared to those with a stable weight, although LEDD or the use of DAs was not different between the groups. However, in the whole sample, the clinical trajectory of binge eating disorder was significantly associated with the use of DAs (Est: 0.59, SE: 0.16, p < 0.001), with a trend towards statistical significance for LEDD (p = 0.051).
Urate and weight
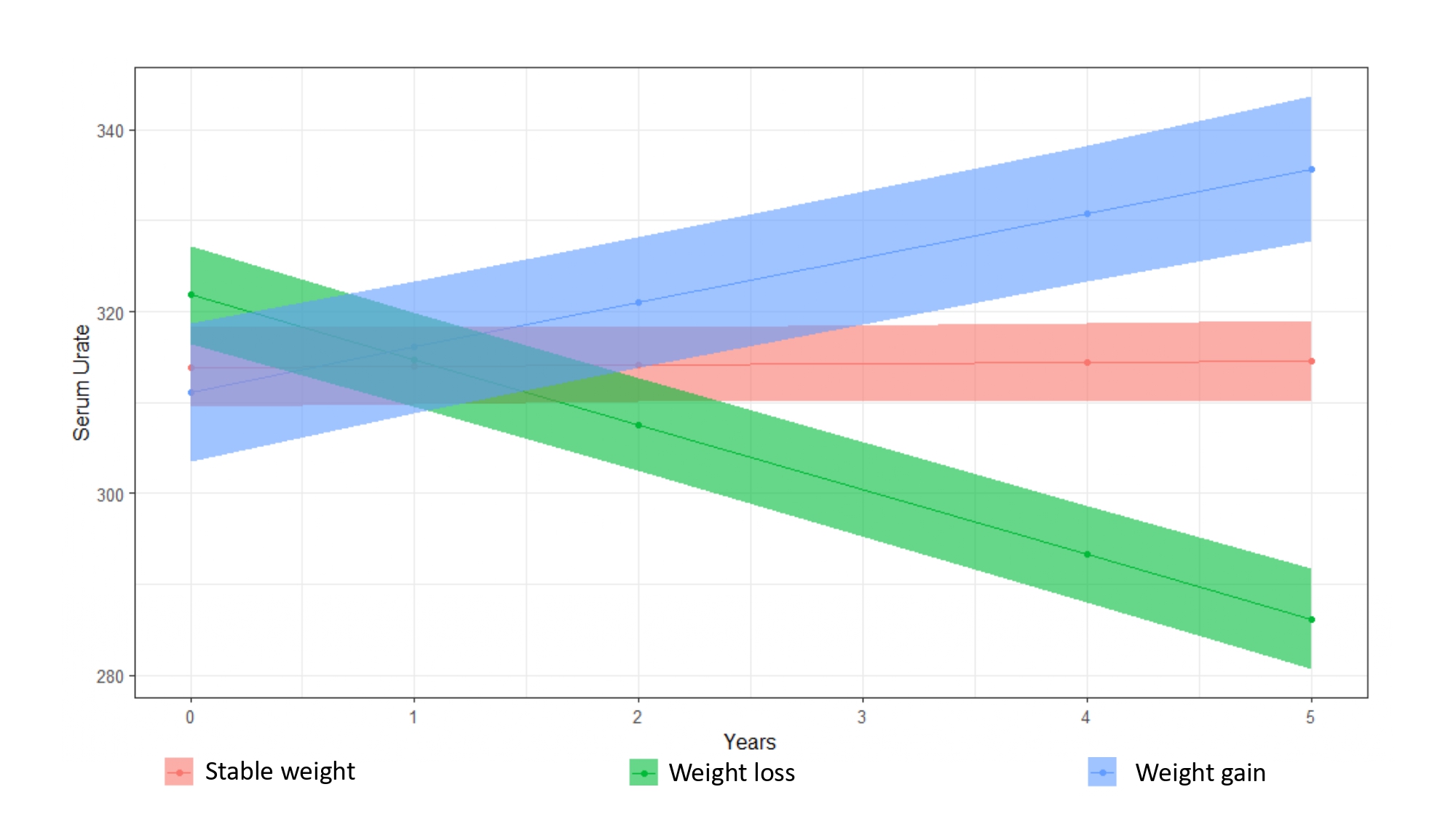
Serum urate leves were associated with BMI at baseline (r = 0.398, p < 0.001). Furthermore, compared to patients with a stable weight, patients with weight gain showed increasing levels of serum urate (p < 0.001, Supplementary Table 1, Supplementary Fig. 1), while patients with weight loss demonstrated decreasing serum urate levels (p < 0.001).
Weight variability and striatal dopaminergic integrity in Parkinson's Disease patients and healthy controls
We found a significant association between weight variability and right striatal DaT binding ratios (p < 0.001, Table 4), but not left striatum binding ratios. We also explored whether dopaminergic denervation at baseline could predict weight variability. Mean, as well as right striatum, putamen, and caudate DaT binding ratios were all predictive of weight variability (p < 0.001, Table 5), while the left-sided regions were not. Finally, we explored the directionality of this relationship in the different subgroups of PD patients (Supplementary Table 2). We found that in the group of patients with weight loss, weight variability was predicted by mean striatal (p < 0.005), right putamen (p < 0.001), left caudate (p = 0.003), and right striatal (p = 0.002) DaT binding ratios. In the group of patients with weight gain, weight was predicted by mean striatum (p = 0.001), putamen (p < 0.001), right putamen (p < 0.001) caudate (p < 0.001), and striatum (p < 0.001) DaT binding ratios. Finally, we found that in HC also, right putamen binding ratios (p < 0.001) were predictive of weight variability (Table 5).
Table 4.
Longitudinal changes of weight in Parkinson’s patients are associated with longitudinal dopaminergic imaging.
| Variable × time effect | Est (SE) | P |
|---|---|---|
| Mean putamen | 0.74 (0.32) | 0.020 |
| Mean caudate | 0.28 (0.14) | 0.041 |
| Mean striatum | 0.46 (0.20) | 0.023 |
| Left putamen | 0.43 (0.29) | 0.142 |
| Right putamen | 0.71 (0.27) | 0.009 |
| Left caudate | 0.16 (0.13) | 0.223 |
| Right caudate | 0.34 (0.13) | 0.008 |
| Left striatum | 0.13(0.09) | 0.151 |
| Right striatum | 0.25 (0.09) | 0.001* |
Main and interaction effects of the linear mixed-effects models estimating the longitudinal changes of weight in PD patients as function of longitudinal presynaptic dopaminergic transporter imaging. The model was controlled for age, sex and disease duration.
*Significant p-values after Bonferroni correction for multiple testing.
Table 5.
Longitudinal changes of weight are predicted by baseline dopaminergic imaging in Parkinson’s patients and healthy controls (HC).
| Variable × time effect | PD | HC | ||
|---|---|---|---|---|
| Est (SE) | P | Est (SE) | P | |
| Mean putamen | 1.00 (0.18) | <0.001* | 0.30 (0.11) | 0.009 |
| Mean caudate | 0.34 (0.09) | <0.001* | 0.08 (0.10) | 0.421 |
| Mean striatum | 0.59 (0.13) | <0.001* | 0.189 (0.11) | 0.093 |
| Left putamen | 0.25 (0.15) | 0.109 | 0.25 (0.11) | 0.026 |
| Right putamen | 1.06 (0.15) | <0.001* | 0.31(0.11) | 0.004* |
| Left caudate | 0.16 (0.08) | 0.068 | 0.13(0.97) | 0.173 |
| Right caudate | 0.41 (0.08) | <0.001* | 0.01(0.09) | 0.858 |
| Left striatum | 0.11 (0.06) | 0.063 | 0.10 (0.05) | 0.060 |
| Right striatum | 0.35 (0.05) | <0.001* | 0.08 (0.05) | 0.132 |
Main and interaction effects of the linear mixed-effects models estimating the longitudinal changes of weight in PD patients and healthy controls as function of baseline presynaptic dopaminergic transporter imaging, while controlling for age, sex and disease duration (only in the PD group).
*Significant p-values after Bonferroni correction for multiple testing.
Discussion
In this longitudinal study we have demonstrated that (1) pathological weight loss may be associated with lower baseline levels of CSF Aβ1-42 in patients with early and de novo PD, (2) both weight loss and weight gain are associated with right striatal dopaminergic denervation in PD patients and HC, (3) there is a relationship between weight variability and serum urate levels, and (4) motor and non-motor longitudinal clinical trajectories of PD patients with either weight gain or weight loss are different.
Unintended weight changes and weight loss have long been recognized in PD patients. In the pre-levodopa era, PD was a disease associated with malnutrition and obesity was rarely observed, even up to a few decades ago31. More recently, in optimally treated patients, PD subjects can be overweight or even obese32, and this change of phenotype is probably related to modern pharmacotherapy (most likely the use of DAs), but may also be at least partly due to the overall increase of obesity in modern society1. However, in PD, weight changes, and especially weight loss, can become pathological and intrusive, and be prevalent from the prodromal to the advanced stage1. As such, this feature of PD needs a clearer research focus, as the pathophysiological basis remains an unmet need. Recent work has attempted a “deep dive” into the clinical associates of abnormal weight loss in PD, and weight loss appears to be associated with a more rapid decline in motor function, cognitive impairment, and disability and mortality in general. Cummings et al.33 recently showed that weight loss occurring within one year after PD diagnosis was independently associated with an increased risk of dependency, dementia and death. In another study from the NINDS Exploratory Trials, Wills and colleagues found that the weight loss group’s mean motor UPDRS score increased by 1.48 points per visit when compared to the weight-stable group’s mean motor UPDRS score11.
Currently, there is a scarcity of available biomarkers of weight variability in PD. In our cohort, de novo patients with subsequent weight loss had lower levels of CSF Aβ1–42 compared to patients with a stable weight. This association is in line with the finding that patients with weight loss have a faster progression in relation to cognitive dysfunction33, which has been linked with CSF Aβ1–4234. The presence of a greater amyloid burden has been previously demonstrated in healthy elderly individuals and individuals with mild cognitive impairment (MCI), where weight was associated with increased PET amyloid uptake24. Another study found a relationship between lower BMI and decreased levels of amyloid in CSF in cognitively normal and MCI individuals25. Furthermore, weight loss has been described in other neurodegenerative conditions, especially dementia35–37. Thus, amyloid burden, or protein misfolding in general, could explain both this commonality in neurodegeneration and a more rapid clinical progression, and suggests that weight loss is a condition that could be partially related to an underlying amyloid pathology load, already present during the first stages of clinically manifested PD38.
We found that both weight loss and weight gain are associated with right striatal dopaminergic denervation in PD patients and HC. While the dopaminergic ventral striatum has been involved in the reward system behind food intake in Binge Eating Disorder, dopamine in the dorsal striatum has been implicated in maintaining caloric requirements for survival and in “non-hedonic” food motivation1,26. Recent studies have demonstrated longitudinal associations of weight change with striatal dopaminergic degeneration. DaT binding at baseline seemed to predict weight changes, with putamen-to-caudate nucleus ratio as an independent predictor for weight change, although only in male subjects27,39. Whether right-left asymmetry in striatal binding ratios contributes to weight variability, as it has been shown in our study and one previous study28, remains to be further explored.
Urate, the soluble form of uric acid, is an important physiological antioxidant able to scavenge free oxygen radicals and interact with other antioxidant systems40,41. Increasing epidemiological and clinical evidence have supported the view that higher urate levels could be associated with a decreased risk of PD and a slower disease progression40. We found that weight variability was associated with urate level, since it increases in patients with weight gain and decreases in patients with weight loss. Interestingly, a recent observational study observed that urate levels were negatively associated with global NMS burden in PD patients, with a specific link to the miscellaneous domain of the NMS scale, which included weight variability30. A possible explanation for weight variability in PD could, therefore, be that these changes are mediated through urate. In this respect, it is interesting to note that in rats high urate diet is associated with the expression of pro-inflammatory cytokines and increased gliosis in the hypothalamus, especially in the mediobasal hypothalamus42, containing, for example, the infundibular and ventromedial nuclei involved in feeding and neuroendocrine control43.
Non-motor endophenotyping of PD is a recent, albeit controversial, concept of great clinical focus, and may aid subtype-specific medicine44,45. Weight variability is an essential constituent of the recently described “circle of personalised medicine”46. Weight loss is also the underpinning anchor in the proposed ‘Park-weight’ PD phenotype9. These patients have been shown to be affected by severe loss of olfaction, a symptom that could increase their risk of unexplained weight loss, developing dyskinesia, and worse disease prognosis9. However, the full range of symptoms associated with this phenotype has not yet been extensively explored or defined, and our report attempts to unravel some of the clinical associates of this endophenotype. Firstly, this analysis, with datamining from an independent cohort of de novo PD patients, suggests that a subgroup of patients with PD have more pronounced weight variability compared to HC, confirming the validity of the original description of the park-weight phenotype and nonmotor subtype of PD. We can also confirm the observations of Sharma et al.9, suggesting that PD patients with weight loss have a more rapid progression of motor symptoms, cognitive decline, and disability. Interestingly, we found that patients with weight gain had a slower progression of motor function. Therefore, we confirm that weight variability may have a critical clinical significance in PD, with weight loss as a driver of poor outcomes, and weight gain associated with slower motor progression in the long term. We also found that patients with weight loss had more motor disability measured by the Schwab and England Activities of Daily Living (ADL) scale at baseline compared with patients with stable weight, which is in line with a previous study showing an association between difficulty in eating and drinking and weight loss11. As previously reported14, binge eating disorder, a manifestation of Impulsive Control Disorder, was associated with the use of DAs.
Strengths of the current study include the use of a large sample from an international cohort of patients of early de novo PD and HC with a follow-up of up to 5 years. Contrary to other studies where weight changes have been defined as a change between the baseline and last visits, here we used body-weight variability by calculating the average successive variability, which takes into account intra-individual variability in body weight between each visit47. Moreover, we have comprehensively characterized weight variability, confirming that this is more prevalent in PD compared with the HC, describing the motor and non-motor trajectories of patients with weight loss and weight gain. In addition, we included in our analysis both right and left striatal regions to explore possible lateralization of function. Nevertheless, some limitations should be recognised in this study. Data on other potential confounders, including intentional weight change, dietetic interventions, non-dopaminergic medication use, gut motility, potential external stressors, exercise and nutritional status, were unavailable. Secondly, olfactory function was only assessed at baseline preventing us from exploring its longitudinal association with weight variability. Further longitudinal studies using more comprehensive nutritional assessments, including specific clinical scales and anthropometric measurements of body composition, are required to improve the characterization and the identification of biomarkers of weight variability in PD.
In conclusion, in this longitudinal study, we found that weight loss was associated with poor clinical outcomes and with a lower level of CSF Aβ1–42 at baseline, while a more favourable progression of motor function was observed in patients with weight gain. Weight variability was associated with urate levels in PD and with right striatal dopaminergic integrity in both PD patients and HC. As such, presynaptic dopaminergic imaging and urate levels may serve as a predictor of weight variability in PD.
Methods
Subjects
Data used in the preparation of this article were obtained from the Parkinson Progression Marker Initiative (PPMI)48. The PPMI is an ongoing prospective, observational, international, multicentre study aimed at identifying clinical biomarkers of PD in a large cohort of participants with early PD at enrolment alongside healthy controls. The aims and methodology of the study have been extensively published elsewhere and are available at www.ppmi-info.org/study-design. Inclusion criteria for PD patients were age 30 years or older, diagnosis of PD (based on one of the following: the presence of (1) asymmetrical resting tremor or (2) asymmetrical bradykinesia or (3) at least two of either of resting tremor, bradykinesia, and rigidity), and a disease duration of 1–24 months, Hoehn and Yahr (H&Y) stage of 1 to 2, and presence of striatal dopamine transporter deficit on 123I-FP-CIT SPECT. The data were collected from more than 33 clinical sites in 11 countries. The PPMI study was approved by the local Institutional Review Boards of all participating sites and written informed consent for imaging data and clinical questionnaires was obtained from each participant at the time of enrolment. All methods were performed in accordance with the relevant guidelines and regulations. We obtained data from the PPMI database on 4 May 2020 in compliance with the PPMI Data Use Agreement.
Clinical assessment
We included data from 405 de novo PD patients and 187 HC with complete information on weight at baseline and throughout the 5-year follow-up. Follow-up visits were performed annually. Data extracted included demographics, age at onset, disease duration, baseline and longitudinal body weight and height, Hoehn and Yahr (HY) staging, Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), SCOPA-Autonomic (SCOPA-AUT), Montreal Cognitive Assessment (MoCA), University of Pennsylvania Smell Identification Test (UPSIT), Geriatric Depression Scale (GDS), State-Trait Anxiety Inventory (STAI), Epworth Sleepiness Scale (ESS), The Schwab and England Activities of Daily Living (ADL) scale. Binge eating was evaluated using the Questionnaire for Impulsive-Compulsive Disorders in Parkinson’s Disease (QUIP). We also extracted data on medications including levodopa equivalent daily dosage (LEDD) and the use of dopamine agonist (DA). UPSIT scores were only available at baseline.
Biomarkers assessment
SPECT images of DAT radioligand binding were acquired at baseline and years 1, 2, and 4 in accordance with PPMI neuroimaging protocols49. After pre-processing, regions of interest were placed on the left and right caudate and putamen. Occipital cortex was used as the reference region. Striatal binding ratios (SBRs) were calculated as target ROI binding intensities normalized by the reference region. Biochemical analyses of uric acid have been carried out in Covance laboratories in a uniform fashion, as per the study protocol50. Measurements of Aβ1–42, total tau and p-tau were obtained for CSF samples at the University of Pennsylvania using the multiplex Luminex xMAP platform (Luminex Corp: Austin, Texas, USA) with research-use-only Fujirebio-Innogenetics INNO-BIA AlzBio3 immunoassay kit-based reagents (Innogenetics Inc: Harvard, MA, USA)49. CSF α-synuclein was analyzed at a central laboratory (Covance, MA, US) using a commercially available enzyme-linked immunosorbent assay kit51. This kit was developed and optimized for PPMI.
Body mass index and weight variability
Weight and height have been measured at baseline and annually. BMI was calculated as weight in kilograms divided by height in square metres. Weight variability was calculated by the average successive variability (ASV) method47. In detail, weight variability was determined by calculating the averaged absolute values of the differences in weight between visits. PD patients were then stratified according to the median value of ASV into patients with Stable Weight (below the median ASV) or Unstable Weight (above the median ASV)47. Unstable Weight patients were further divided according to the directionality of AVS into the weight loss group (negative ASV) and weight gain group (positive ASV).
Statistical analysis
Between-group comparisons were performed by one-way ANOVA or Mann–Whitney U test for normally or non-normally distributed variables, respectively. Categorical variables were compared using Pearson Chi-square. Correlations were performed using the Pearson correlation coefficient test. The differential progression of clinical variables between groups was calculated using linear mixed effects (LME) or mixed effects logistic regression methods. Finally, LME models estimated the longitudinal changes in weight as a function of baseline or longitudinal presynaptic dopaminergic transporter imaging. All models were controlled for age, sex, disease duration and LEDD. Values of p < 0.05 were considered as statistically significant, and Bonferroni post-hoc correction was used for multiple comparisons.
Supplementary information
{kind=link}
Acknowledgements
Data used in the preparation of this article were obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (www.ppmi-info.org/data). For up-to-date information on the study, visit www.ppmi-info.org. PPMI (a public–private partnership) is funded by the Michael J Fox Foundation for Parkinson’s Research and funding partners, including AbbVie, Avid, Biogen, Bristol-Myers Squibb, Covance, GE Healthcare, Genentech, GlaxoSmithKline, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Roche, Servier, Teva, and UCB. The current data analysis was not supported by funding. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author contributions
D.U. and K.R.C. conceived the idea, planned, and designed the study; D.U., D.vW., L.B., V.L. and J.P. planned the data management and statistical analysis; D.U. wrote the first draft; J.S. and G.L. provided critical insights. D.U. D.vW., L.B., V.L., J.S., J.P. and K.R.C. reviewed the manuscript. All the authors have approved and contributed to the final written manuscript.
Data availability
All data used in this study are available from the PPMI database (www.ppmi-info.org/data).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41531-022-00362-3.
References
- 1.Kistner A, Lhommée E, Krack P. Mechanisms of body weight fluctuations in Parkinson’s disease. Front. Neurol. 2014;5:84–84. doi: 10.3389/fneur.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Logroscino G, Sesso HD, Paffenbarger RS, Jr., Lee IM. Body mass index and risk of Parkinson’s disease: a prospective cohort study. Am. J. Epidemiol. 2007;166:1186–1190. doi: 10.1093/aje/kwm211. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Zhang SM, Hernán MA, Willett WC, Ascherio A. Weight loss in Parkinson’s disease. Ann. Neurol. 2003;53:676–679. doi: 10.1002/ana.10577. [DOI] [PubMed] [Google Scholar]
- 4.Song, S. et al. Changes in body composition before and after Parkinson’s disease diagnosis. Mov. Disord. n/a(n/a), e28536 (2021). [DOI] [PMC free article] [PubMed]
- 5.Lorefält B, et al. Factors of importance for weight loss in elderly patients with Parkinson’s disease. Acta Neurol. Scand. 2004;110:180–187. doi: 10.1111/j.1600-0404.2004.00307.x. [DOI] [PubMed] [Google Scholar]
- 6.Bachmann CG, Zapf A, Brunner E, Trenkwalder C. Dopaminergic treatment is associated with decreased body weight in patients with Parkinson’s disease and dyskinesias. Eur. J. Neurol. 2009;16:895–901. doi: 10.1111/j.1468-1331.2009.02617.x. [DOI] [PubMed] [Google Scholar]
- 7.Umehara T, Nakahara A, Matsuno H, Toyoda C, Oka H. Body weight and dysautonomia in early Parkinson’s disease. Acta Neurol. Scand. 2017;135:560–567. doi: 10.1111/ane.12633. [DOI] [PubMed] [Google Scholar]
- 8.Sharma JC, Turton J. Olfaction, dyskinesia and profile of weight change in Parkinson’s disease: identifying neurodegenerative phenotypes. Parkinsonism Relat. Disord. 2012;18:964–970. doi: 10.1016/j.parkreldis.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Sharma JC, Vassallo M. Prognostic significance of weight changes in Parkinson’s disease: the Park-weight phenotype. Neurodegener. Dis. Manag. 2014;4:309–316. doi: 10.2217/nmt.14.25. [DOI] [PubMed] [Google Scholar]
- 10.Akbar U, et al. Weight loss and impact on quality of life in Parkinson’s disease. PLoS ONE. 2015;10:e0124541. doi: 10.1371/journal.pone.0124541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wills AM, et al. Association Between Change in Body Mass Index, Unified Parkinson’s Disease Rating Scale Scores, and Survival Among Persons With Parkinson Disease: Secondary Analysis of Longitudinal Data From NINDS Exploratory Trials in Parkinson Disease Long-term Study 1. JAMA Neurol. 2016;73:321–328. doi: 10.1001/jamaneurol.2015.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HJ, et al. Relationship between changes of body mass index (BMI) and cognitive decline in Parkinson’s disease (PD) Arch. Gerontol. Geriatr. 2012;55:70–72. doi: 10.1016/j.archger.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 13.Ikeda K, et al. Body mass index and the risk of Parkinson disease. Neurology. 2007;68:2156. doi: 10.1212/01.wnl.0000269477.49238.ec. [DOI] [PubMed] [Google Scholar]
- 14.Weintraub D, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch. Neurol. 2010;67:589–595. doi: 10.1001/archneurol.2010.65. [DOI] [PubMed] [Google Scholar]
- 15.Ondo WG, et al. Weight gain following unilateral pallidotomy in Parkinson’s disease. Acta Neurologica Scandinavica. 2000;101:79–84. doi: 10.1034/j.1600-0404.2000.101002079.x. [DOI] [PubMed] [Google Scholar]
- 16.Krack P, et al. Five-year follow-up of bilateral stimulation of the subthalamic nucleus in advanced Parkinson’s disease. N. Engl. J. Med. 2003;349:1925–1934. doi: 10.1056/NEJMoa035275. [DOI] [PubMed] [Google Scholar]
- 17.Hu G, et al. Body mass index and the risk of Parkinson disease. Neurology. 2006;67:1955–1959. doi: 10.1212/01.wnl.0000247052.18422.e5. [DOI] [PubMed] [Google Scholar]
- 18.Ma K, et al. Weight loss and malnutrition in patients with Parkinson’s disease: current knowledge and future prospects. Front. Aging Neurosci. 2018;10:1–1. doi: 10.3389/fnagi.2018.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 20.Abbott RA, Cox M, Markus H, Tomkins A. Diet, body size and micronutrient status in Parkinson’s disease. Eur. J. Clin. Nutr. 1992;46:879–884. [PubMed] [Google Scholar]
- 21.Beyer PL, Palarino MY, Michalek D, Busenbark K, Koller WC. Weight change and body composition in patients with Parkinson’s disease. J. Am. Diet. Assoc. 1995;95:979–983. doi: 10.1016/S0002-8223(95)00269-3. [DOI] [PubMed] [Google Scholar]
- 22.Guimarães J, et al. Locus coeruleus is involved in weight loss in a rat model of Parkinson’s disease: an effect reversed by deep brain stimulation. Brain Stimul. 2013;6:845–855. doi: 10.1016/j.brs.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 23.Durrieu G, et al. Parkinson’s disease and weight loss: A study with anthropometric and nutritional assessment. Clin. Autonomic Res. 1992;2:153–157. doi: 10.1007/BF01818955. [DOI] [PubMed] [Google Scholar]
- 24.Jimenez A, et al. Weight loss in the healthy elderly might be a non-cognitive sign of preclinical Alzheimer’s disease. Oncotarget. 2017;8:104706–104716. doi: 10.18632/oncotarget.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vidoni ED, Townley RA, Honea RA, Burns JM. Alzheimer disease biomarkers are associated with body mass index. Neurology. 2011;77:1913–1920. doi: 10.1212/WNL.0b013e318238eec1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volkow ND, et al. “Nonhedonic” food motivation in humans involves dopamine in the dorsal striatum and methylphenidate amplifies this effect. Synapse. 2002;44:175–180. doi: 10.1002/syn.10075. [DOI] [PubMed] [Google Scholar]
- 27.Pak K, et al. Prediction of future weight change with dopamine transporter in patients with Parkinson’s disease. J. Neural Transm. 2019;126:723–729. doi: 10.1007/s00702-019-02016-w. [DOI] [PubMed] [Google Scholar]
- 28.Cho SS, Yoon EJ, Kim SE. Asymmetry of dopamine D2/3 receptor availability in dorsal putamen and body mass index in non-obese healthy males. Exp. Neurobiol. 2015;24:90–94. doi: 10.5607/en.2015.24.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y, Zhang Y, Choi HK. The serum urate-lowering impact of weight loss among men with a high cardiovascular risk profile: the Multiple Risk Factor Intervention Trial. Rheumatology. 2010;49:2391–2399. doi: 10.1093/rheumatology/keq256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Wamelen DJ, et al. Serum uric acid levels and non-motor symptoms in Parkinson’s disease. J. Parkinsons Dis. 2020;10:1003–1010. doi: 10.3233/JPD-201988. [DOI] [PubMed] [Google Scholar]
- 31.Jankovic J, Wooten M, Van der Linden C, Jansson B. Low body weight in Parkinson’s disease. South Med. J. 1992;85:351–354. doi: 10.1097/00007611-199204000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Barichella M, Marczewska A, Vairo A, Canesi M, Pezzoli G. Is underweightness still a major problem in Parkinson’s disease patients? Eur. J. Clin. Nutr. 2003;57:543–547. doi: 10.1038/sj.ejcn.1601581. [DOI] [PubMed] [Google Scholar]
- 33.Cumming K, Macleod AD, Myint PK, Counsell CE. Early weight loss in Parkinsonism predicts poor outcomes. Evid. Incid. Cohort Study. 2017;89:2254–2261. doi: 10.1212/WNL.0000000000004691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siderowf A, et al. CSF amyloid β 1-42 predicts cognitive decline in Parkinson disease. Neurology. 2010;75:1055–1061. doi: 10.1212/WNL.0b013e3181f39a78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stewart R, et al. A 32-year prospective study of change in body weight and incident dementia: the Honolulu-Asia Aging Study. Arch. Neurol. 2005;62:55–60. doi: 10.1001/archneur.62.1.55. [DOI] [PubMed] [Google Scholar]
- 36.Atti AR, et al. Late-life body mass index and dementia incidence: nine-year follow-up data from the Kungsholmen Project. J. Am. Geriatr. Soc. 2008;56:111–116. doi: 10.1111/j.1532-5415.2007.01458.x. [DOI] [PubMed] [Google Scholar]
- 37.Besser LM, et al. Body mass index, weight change, and clinical progression in mild cognitive impairment and Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2014;28:36–43. doi: 10.1097/WAD.0000000000000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fereshtehnejad S-M, Zeighami Y, Dagher A, Postuma RB. Clinical criteria for subtyping Parkinson’s disease: biomarkers and longitudinal progression. Brain. 2017;140:1959–1976. doi: 10.1093/brain/awx118. [DOI] [PubMed] [Google Scholar]
- 39.Pak K, et al. Weight loss is associated with rapid striatal dopaminergic degeneration in Parkinson’s disease. Parkinsonism Relat. Disord. 2018;51:67–72. doi: 10.1016/j.parkreldis.2018.02.044. [DOI] [PubMed] [Google Scholar]
- 40.Gao X, Chen H, Choi HK, Curhan G, Schwarzschild MA, Ascherio A. Diet, urate, and Parkinson’s disease risk in men. Am. J. Epidemiol. 2008;167:831–838. doi: 10.1093/aje/kwm385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu W, et al. Uric acid produces an inflammatory response through activation of NF-κB in the hypothalamus: implications for the pathogenesis of metabolic disorders. Sci. Rep. 2015;5:12144. doi: 10.1038/srep12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swaab, D. F. Handbook of Clinical Neurology, Vol 79 (ed. Swaab, D. F.) Ch. 11, 249–261 (Elsevier, 2003).
- 44.Sauerbier A, Jenner P, Todorova A, Chaudhuri KR. Non motor subtypes and Parkinson’s disease. Parkinsonism Relat. Disord. 2016;22:S41–46. doi: 10.1016/j.parkreldis.2015.09.027. [DOI] [PubMed] [Google Scholar]
- 45.Marras C, Chaudhuri KR. Nonmotor features of Parkinson’s disease subtypes. Mov. Disord. 2016;31:1095–1102. doi: 10.1002/mds.26510. [DOI] [PubMed] [Google Scholar]
- 46.Titova N, Chaudhuri KR. Non-motor Parkinson disease: new concepts and personalised management. Med J. Aust. 2018;208:404–409. doi: 10.5694/mja17.00993. [DOI] [PubMed] [Google Scholar]
- 47.Bangalore S, et al. Body-weight fluctuations and outcomes in coronary disease. N. Engl. J. Med. 2017;376:1332–1340. doi: 10.1056/NEJMoa1606148. [DOI] [PubMed] [Google Scholar]
- 48.Martinez-Martin P, et al. Chronic subcutaneous infusion therapy with apomorphine in advanced Parkinson’s disease compared to conventional therapy: a real life study of non motor effect. J. Parkinsons Dis. 2011;1:197–203. doi: 10.3233/JPD-2011-11037. [DOI] [PubMed] [Google Scholar]
- 49.Marek K, et al. The Parkinson’s progression markers initiative (PPMI) - establishing a PD biomarker cohort. Ann. Clin. Transl. Neurol. 2018;5:1460–1477. doi: 10.1002/acn3.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parkinson Progression Marker Initiative. The Parkinson Progression Marker Initiative (PPMI). Prog. Neurobiol. 95, 629–635 (2011). [DOI] [PMC free article] [PubMed]
- 51.Mollenhauer B, et al. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with Parkinsonism: a cohort study. Lancet Neurol. 2011;10:230–240. doi: 10.1016/S1474-4422(11)70014-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this study are available from the PPMI database (www.ppmi-info.org/data).