

Abstract
Gymnocypris eckloni is widely distributed in isolated lakes and the upper reaches of the Yellow River and play significant roles in the trophic web of freshwater communities. In this study, we generated a chromosome-level genome of G. eckloni using PacBio, Illumina and Hi-C sequencing data. The genome consists of 23 pseudo-chromosomes that contain 918.68 Mb of sequence, with a scaffold N50 length of 43.54 Mb. In total, 23,157 genes were annotated, representing 94.80% of the total predicted protein-coding genes. The phylogenetic analysis showed that G. eckloni was most closely related to C. carpio with an estimated divergence time of ~34.8 million years ago. For G. eckloni, we identified a high-quality genome at the chromosome level. This genome will serve as a valuable genomic resource for future research on the evolution and ecology of the schizothoracine fish in the Qinghai-Tibetan Plateau.
Subject terms: Genome, Ichthyology
| Measurement(s) | Genome |
| Technology Type(s) | Whole Genome Sequencing |
| Sample Characteristic - Organism | Gymnocypris eckloni |
| Sample Characteristic - Environment | fresh water |
| Sample Characteristic - Location | Little Yellow River |
Background & Summary
The Qinghai-Tibetan Plateau (QTP) is the highest and one of the biggest plateaus on earth, covering 2.5 × 106 square kilometers with an elevation of 3000–5000 m for most parts of the area. The intensive uplifts of QTP resulted from collision of the India plate and the Eurasia plate had a profound impact on the climate and environment1,2. Characterized by high altitude, low oxygen partial pressure (hypoxia), low temperatures, dramatic temperature fluctuations, and high UV radiation, the QTP environment posed harsh challenges to the endemic animals3,4. Recently, comparative genomic studies of animals endemic to the QTP provide valuable clues for scientists to understand the molecular mechanism of environmental adaptation4–8. However, the genome information of fish species in QTP is still lacking.
Schizothoracine fish (Teleostei: Cyprinidae) are the largest and most diverse taxon within the QTP ichthyofauna and their radiation has been correlated with the plateau’s rapid upheaval9,10. The schizothoracine fish, confined to regions at either high altitudes or high latitudes, have evolved a number of unique traits (i.e., degeneration of body scales, slow growth, and late sexual maturity) that adapt to the extreme environment of the QTP and play significant roles in the trophic web of QTP freshwater communities10–13. Therefore, the schizothoracine fish have been accepted as ideal models for studying the molecular mechanisms underlying the adaptation to harsh environments11–13.
The schizothoracine fish comprises 11 or 12 genera and approximately 100 species and are mainly distributed in cold tributaries and lakes of the QTP and adjacent areas at 2000 m above sea level10,11. The phylogenetic analysis based on morphological traits revealed that the schizothoracine fishes can be divided into three sub-groups including primitive, specialized and highly specialized group10, which was proposed to be associated with the tectonic upshifts of the QTP14–16. Previous studies have shown that the karyotypes of the schizothoracine fish range from 90 to 446 and that almost all species were polyploid17–20. A recent genomic study confirmed that Schizothorax o’connori of Schizothoracinae was a young tetraploid that underwent a fourth whole-genome duplication (4 R WGD) after the teleost-specific third WGD (3 R WGD)21. Other studies indicated that the globin gene superfamily, toll-like receptor family, and interferon regulatory factors in a representative species from this subfamily underwent adaptive evolution in response to the plateau environment, specifically gene loss, and gain events as a result of genome and/or gene duplications13,22–24. Gymnocypris eckloni is a representative species of the highly specialized schizothoracine fish that is widely distributed in isolated lakes and the upper reaches of the Yellow River, and is very well adapted to the plateau’s aqueous environment9,10. Investigating the genomic evolution of G. eckloni may shed light on the underlying molecular mechanisms involved in high-altitude adaptations in schizothoracine fish of the QTP.
In the present study, we integrated PacBio long-read sequencing, Illumina short-read sequencing, and high-throughput chromosome conformation capture (Hi-C) technology to generate a high-quality chromosome-level reference genome for G. eckloni. The reference genome obtained in this study will provide a foundation for future investigations on the evolution and adaptation of schizothoracine fish.
Methods
Experimental fish and sequencing
G. eckloni genomic DNA were extracted from the muscle samples of a healthy female individuals obtained from the Native Fish Artificial Proliferation and Release Station, Xunhua, Qinghai Province, China (Fig. s1). For genome assembly, two libraries with insert sizes of 300 bp and 20 kb were separately constructed using an Illumina TruSeq Nano DNA Library Prep Kit and SMRT bell Template Prep Kit. The two libraries were subsequently sequenced using an Illumina HiSeq X Ten instrument and a PacBio Sequel platform25. For the PacBio platform, a total of 312.2 Gb PacBio long sequencing reads were generated, and 239.0 Gb subreads (334.6 × coverage) with an average length of 23,706 bp were obtained after removing adaptors in polymerase reads (Table 1). For the Illumina HiSeq X Ten sequencing platform, a total of 251.7 Gb short sequencing reads were generated. After filtering, 215.2 Gb (231.2 × coverage) of clean Illumina data were retained to perform a genome survey.
Table 1.
Sequencing data used for the genome G. eckloni assembly.
| Library types | Insert size (bp) | Raw data (Gb) | Clean data (Gb) | Read length (bp) | Sequence coverage (X) |
|---|---|---|---|---|---|
| Illumina reads | 300 | 215.7 | 215.2 | 150 | 231.2 |
| PacBio reads | 20000 | 312.2 | 239.0 | 23706 | 334.6 |
| Hi-C reads | — | 257.3 | 257.3 | 300 | 275.8 |
| RNA reads | 300 | 67.76 | 66.43 | 150 | — |
| Total | — | 852.96 | 777.93 | — | — |
To conduct chromosome-level assembly of the G. eckloni genome, a Hi-C library was generated using the Mbo I restriction enzyme following previously described standard protocol with minor modifications26. In brief, the purified DNA from the fresh muscle sample was digested with Mbo I restriction enzyme and labelled by incubating with Biotin-14-dATP (Thermo Fisher Scientific, USA), and then ligated by T4 DNA Ligase. After incubating overnight to reverse crosslinks, the ligated DNA was sheared into 200–600 bp fragments, and then blunt-end repaired and A-tailed, followed by purification through biotin-streptavidin-mediated pull down. Finally, the Hi-C libraries were quantified and sequenced on the Illumina NovaSeq6000 platform (Illumina, USA) using a PE-150 module, generating a total of 257.3 Gb (275.8 × coverage) clean data after using the same filter criteria with short reads (Table 1).
To provide evidence of transcripts for genome structure annotation, we conducted RNA-seq for muscle, skin, gill, liver, gut, spleen, kidney, heart, eye and blood samples. RNA was extracted using Ambion MagMAX-96 total RNA isolation kit (Life Sciences, United States) for all samples, and DNase I treatment was performed to eliminate DNA contamination. After the quality assessment of the extracted RNAs using NanoPhotometer® spectrophotometer (Implen, United States), RNA-seq libraries were constructed according to the protoco and were sequenced by Illumina HiSeq4000 in paired-end 150 bp mode, resulting in a total of 66.43 Gb clean transcriptome data (Table 1).
De novo assembly of G. eckloni genome
We used the k-mer method to survey the genomic features of the G. eckloni. The k-mer count histogram was obtained from Illumina paired-end sequencing data using Jellyfish v2.9927. Based on the total number of 169,021,371,761 17-mers and a peak 17-mer depth of 181, the genome size of G. eckloni was estimated to be 927.13 Mb, and the estimated heterozygosity rate was approximately 1.82% (Table s1).
The 239.0 Gb subreads from the PacBio Sequel platform were used for genome assembly using wtdbg228 followed by Quiver29 and Pilon30 polishing using the 215.2 Gb of Illumina HiSeq clean reads, which produced a 918.45 Mb genome assembly, consisting of 3,170 contigs with a contig N50 size of 4.19 Mb (Table 2).
Table 2.
The statistics of length and number for the de novo assembled G. eckloni genome.
| Term | Length | No. | ||
|---|---|---|---|---|
| Contig (bp) | Scaffold (bp) | Contig | Scaffold | |
| Total | 918,450,624 | 918,681,488 | 3,170 | 711 |
| Max | 22,682,260 | 89,391,071 | — | — |
| Number > = 2000 | — | — | 3,058 | 711 |
| N50 | 4,192,824 | 43,543,958 | 56 | 8 |
| N60 | 2,476,204 | 34,715,927 | 85 | 11 |
| N70 | 1,500,513 | 32,896,108 | 133 | 13 |
| N80 | 641,416 | 29,129,546 | 229 | 16 |
| N90 | 146,685 | 25,669,045 | 553 | 20 |
Hi-C technology was applied to conduct the chromosome-level genome assembly of G. eckloni. Clean reads sequenced from the Hi-C library were aligned to the contig-level genome with an end-to-end algorithm implemented in Bowtie v2.3.5 according to the Hi-C-Pro strategy31,32. Juicer v1.6.2 and 3D de novo assembly (3D-DNA) pipelines were used to assemble the contigs into the chromosome-level genome33,34. Ultimately, the assembled sequences were further anchored and orientated onto 23 pseudo-chromosomes using Hi-C data. The 23 pseudo-chromosomes ranged in size from 15.91 to 89.39 Mb (Fig. 1 and Table s2), covering ~98.52% of the whole genome. Finally, the G. eckloni genome was obtained with 711 scaffolds and a total length of 918,681,488 bp, a contig N50 of 4.19 Mb, and scaffold N50 of 43.54 Mb (Table 2).
Fig. 1.

Characteristics of the G. eckloni genome. (a) Hi-C intra-chromosomal contact map of the G. eckloni genome assembly. (b) Circos plot of the G. eckloni genome assembly. 1) Pseudo-chromosomes; 2) gene distribution; 3) GC content; 4) repeat distribution; 5) rRNA distribution; 6) tRNA distribution; 7) miRNA distribution; 8) snRNA distribution. All data were obtained using a sliding window of 10 Kb.
The completeness of the genome assembly was assessed by the single copy orthologs (BUSCO, version 5.3.2)35 and CEGMA36 software. The BUSCO analysis based on the actinopterygii_odb10 database showed that 87.5% (single-copy genes: 83.0%, duplicated genes: 4.5%) of the 3,640 single-copy genes were identified as complete, 1.3% were fragmented, and 11.2% were missing from the assembled genome. The CEGMA analysis revealed that 221 conserved genes (89.11% of the core eukaryotic genes) supported the completeness of the assembled genome. Illumina short reads were mapped to the assembled genome using BWA37 software to evaluate completeness of the genome assembly. The results showed that 93.40% of the reads could be mapped, covering 96.34% of the assembled genome.
Repetitive element and non-coding gene annotation in the G. eckloni genome
A combined strategy using homology alignments and de novo searches to identify whole-genome repeats was applied in our repeat annotation pipeline. Tandem repeats were extracted using TRF (http://tandem.bu.edu/trf/trf.html) by ab initio prediction. For homolog prediction, Repbase (http://www.girinst.org /repbase) employing RepeatMasker (http://www.repeatmasker.org/) software and its in-house scripts (RepeatProteinMask) with default parameters was used to extract repeat regions. Additionally, ab initio prediction based on the de novo repetitive elements database was conducted by LTR_FINDER (http://tlife.fudan.edu.cn/ltr_finder/), RepeatScout (http://www.repeatmasker.org/), and RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html) with default parameters. Then, all repeat sequences with lengths > 100 bp and gap ‘N’ < 5% were used to construct the raw transposable element (TE) library. A custom library (a combination of Repbase and our de novo TE library, which was processed by uclust to yield a non-redundant library) was supplied to RepeatMasker for DNA-level repeat identification. The results showed revealed that 47.63% of the G. eckloni genome was annotated as repetitive elements (Table s3), of which LTRs were the most abundant with a total length of 356.79 Mb, accounting for 38.84% of the whole genome. SINEs were the rarest with a total length of 2.37 Mb and represented 0.26% of the whole genome (Table s4).
The tRNAs were predicted using tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE/), and the rRNA sequences were predicted using BLAST. The results showed that a total of 12,157 tRNAs were predicted using tRNAscan-SE, and 1,780 rRNA genes were annotated using BLASTN tool with an E-value of 1E-1032 against human rRNA sequence. Other ncRNAs, including miRNAs and snRNAs, were identified by searching against the Rfam database with default parameters using infernal software (http://infernal.janelia.org/) (Table s5).
Annotation of protein-coding genes
Gene predictions were conducted through a combination of homology, de novo, and transcriptome-based prediction methods. For homology-based predictions, the protein sequences of seven fish species, including Oryzias latipes, Ctenopharyngodon idellus, Ictalurus punctatus, Cyprinus carpio, Takifugu rubripes, Danio rerio, and Astyanax mexicanus, were downloaded from Ensembl database (http://asia.ensembl.org/index. html). Protein sequences were aligned to the genome using TblastN v2.2.26 with an e-value of 1e−5 38. Then, matching proteins were aligned to homologous genome sequences for accurate spliced alignments using GeneWise v2.4.139 (referred to “Homolog” in Table 3), which was subsequently used to predict gene structure of each protein region. RNA-sequencing data derived from nine tissues and blood samples were assembled using Trinity v2.1.140, and were aligned against the G. eckloni genome using Program to Assemble Spliced Alignment (PASA)41 (referred to “PASA” in Table 3). To optimize genome annotation, RNA-seq reads from different tissues were aligned to G. eckloni genome fasta using TopHat package v2.0.11 with default parameters to identify exons region and splice positions42. The alignment results were then used as inputs for Cufflinks package v2.2.1 with default parameters for genome-based transcript assembly43 (referred to “Cufflinks”in Table 3). Finally, EvidenceModeler v1.1.1 was used to combine the gene models into weighted consensus gene structures with masked repetitive elements41. Additionally, PASA was used to update the final gene models, thereby adding information of alternatively spliced sites and untranslated regions (UTR) (referred to “Pasa-update” in Table 3). Ultimately, a total of 24,430 protein-coding genes were predicted in the G. eckloni genome. The average transcript length was 16,219.34 bp with an average coding sequence (CDS) length of 1,536.71 bp. The average exon number per gene was 8.88 with an average exon length of 173.00 bp and average intron length of 1,862.69 bp (Table 3). The statistics of gene models, including lengths of a gene, CDS, intron, and exon in G. eckloni were comparable to those for close-related species (Table s6 and Fig. 2).
Table 3.
Gene annotation of G. eckloni genome via three methods.
| Method | Gene set | Number | Average length (bp) | Exons No. per gene | |||
|---|---|---|---|---|---|---|---|
| Transcript | CDS | Exon | Intron | ||||
| De novo | Augustus | 38,431 | 9,427.68 | 1,102.72 | 181.26 | 1,637.57 | 6.08 |
| GlimmerHMM | 88,372 | 9,368.19 | 580.21 | 146.67 | 2,973.05 | 3.96 | |
| SNAP | 47,478 | 20,534.02 | 796.89 | 143.55 | 4,336.44 | 5.55 | |
| Geneid | 32,716 | 17,045.39 | 1,223.45 | 216.31 | 3,398.15 | 5.66 | |
| Genscan | 32,712 | 19,569.14 | 1,429.87 | 189.78 | 2,775.94 | 7.53 | |
| Homolog | Oryzias latipes | 18,845 | 11,159.32 | 1,293.66 | 179.22 | 1,586.58 | 7.22 |
| Ctenopharyngodon idellus | 24,602 | 9,475.91 | 1,264.07 | 184.13 | 1,400.12 | 6.87 | |
| Ictalurus punctatus | 19,535 | 13,585.62 | 1,522.10 | 182.89 | 1,647.47 | 8.32 | |
| Cyprinus carpio | 23,776 | 10,240.81 | 1,276.54 | 182.36 | 1,494.04 | 7.00 | |
| Takifugu rubripes | 18,028 | 13,503.98 | 1,497.61 | 181.74 | 1,658.25 | 8.24 | |
| Danio rerio | 20,929 | 13,270.63 | 1,510.71 | 180.45 | 1,595.28 | 8.37 | |
| Astyanax mexicanus | 20,090 | 11,862.97 | 1,393.39 | 185.72 | 1,610.03 | 7.50 | |
| RNAseq | PASA | 91,220 | 14,128.66 | 1,240.08 | 165.12 | 1,979.72 | 7.51 |
| Transcripts | 66,837 | 31,133.65 | 2,702.91 | 300.16 | 3,551.63 | 9.00 | |
| EVM | 35,931 | 11,908.94 | 1,192.33 | 176.57 | 1,862.87 | 6.75 | |
| Pasa-update | 35,599 | 12,447.20 | 1,220.20 | 177.47 | 1,910.77 | 6.88 | |
| Final set | 24,430 | 16,219.34 | 1,536.71 | 173.00 | 1,862.69 | 8.88 | |
Note that CDS refers to coding sequence; GlimmerHMM was a new genefinder based on a Generalized Hidden Markov Model (GHMM); SNAP refers to Semi-HMM-based Nucleic Acid Parser; EVM refers to Evidence modeler.
Fig. 2.

The composition of gene elements in the G. eckloni genome to other species. (a) CDS length distribution and comparison with other species. (b) Exon length distribution and comparison with other species. (c) Exon number distribution and comparison with other species. (d) Gene length distribution and comparison with other species. (e) Intron length distribution and comparison with other species.
Public biological function databases of NR, SwissProt44, InterPro45, and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases46 were used for the functional annotation of protein-coding genes using BLASTX and BLASTN utilities46 with an e-value threshold of 1e−5. InterPro database was used to predict protein function based on the conserved protein domains by InterproScan tool47. A total of 23,157 genes (94.8%) were successfully annotated by at least one public database (Table s7 and Fig. 3).
Fig. 3.
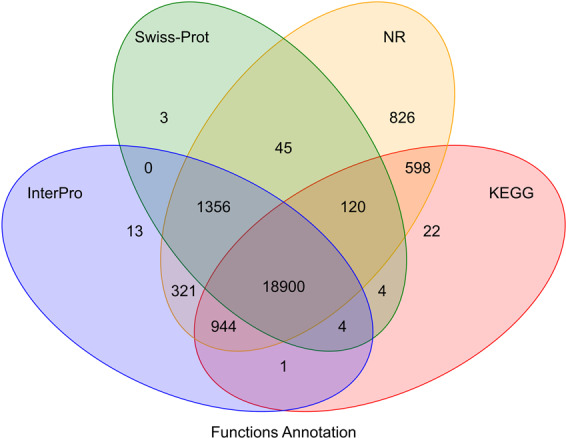
Venn diagram of number of genes with homology or functional classification by each method.
Evolutionary and comparative genomic analysis
To examine G. eckloni evolution, we used OthoMCL48 to cluster its genes with those from 13 other vertebrates: Astyanax mexicanus, Ictalurus punctatus, Danio rerio, C. carpio, Ctenopharyngodon idella, Oreochromis niloticus, Oryzias latipes, Takifugu rubripes, Gallus gallus, Homo sapiens, Mus musculus, Xenopus tropicalis, and Petromyzon marinus. From these 14 species, we identified 597 one-to-one single-copy genes that were used to construct a maximum likelihood (ML) tree using RaxML with the GTRGAMMA model49. Divergence times between species were calculated using the MCMC tree program implemented by PAML package50. According to the time-calibrated phylogeny, the age of the most recent common ancestor (MRCA) of the teleost fish was estimated to be 211.8–254.1 million years ago. The G. eckloni with the closest relationship to C. carpio shared an MRCA at ~ 34.8 million years ago (Fig. 4).
Fig. 4.
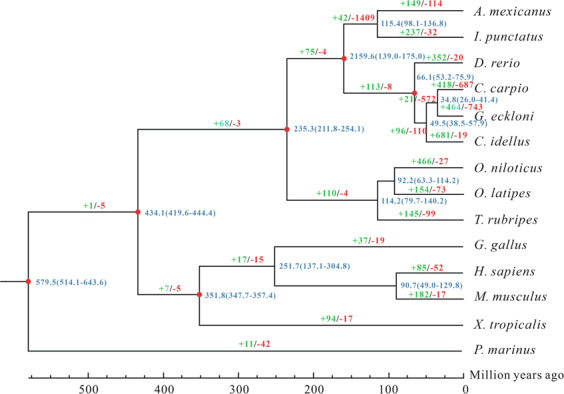
Phylogenetic tree based on single-copy genes from 14 species shows the estimated divergence time (blue numbers), topology and expansion (green numbers), and contraction (red numbers) of gene families.
A total of 24,619 gene families were identified among the 14 species (Table s8), of which 2,739 core gene families were shared by all 14 species and 856 gene families is unique for G. eckloni including 1,488 genes. Analysis of the expansion and contraction of the gene families revealed that there were 464 (1650 genes) expanded and 743 (192 genes) contracted gene families in G. eckloni when compared to its MRCA (Fig. 4). The expanded gene families included ABC transporters, Peroxisome, Herpes simplex virus 1 infection, Staphylococcus aureus infection, Axon guidance, Dorso-ventral axis formation, Pertussis, Legionellosis, Rap1 signaling pathway and so on, and the contracted gene families included Tight junction, Systemic lupus erythematosus, Pathogenic Escherichia coli infection, Gap junction, Alcoholism, Pertussis, Ascorbate and aldarate metabolism, NOD-like receptor signaling pathway and so on.
Data Records
All raw data of the whole genome have been deposited into the National Center for Biotechnology Information (NCBI) SRA database (Experiments for SRP377513) under BioProject accession number PRJNA83561151. The assembled genome has been deposited at DDBJ/ENA/GenBank under the accession JAMHKY00000000052. Data of the expansion and contraction of the gene families, gene functional annotations, repeat annotation and results of evolutionary analysis had been deposited at Figshare53.
Technical Validation
RNA integrity
The transcriptomes for nine tissues and blood from three fish individuals were sequenced. Before constructing RNA-Seq libraries, RNA purity was analyzed with a NanoPhotometer Spectrophotometer (Implen, United States). The RNA concentration was quantified with a Qubit RNA Assay Kit in a Qubit 2.0 Fluorometer (Life Technologies, United States). RNA integrity was analyzed using a RNA Nano 6000 Assay Kit and an Agilent Bioanalyzer 2100 (Agilent Technologies, United States). The total amount of RNA, RNA integrity and rRNA ratio were used to estimate the quality, content and degradation level of RNA samples. In the present study, RNAs samples with a total RNA amount ≥ 10 μg, RNA integrity number ≥ 8, and rRNA ratio ≥ 1.5 were finally subjected to construct the sequencing library.
Comparative genomic analyses
The protein sequences of 13 vertebrates, including A. mexicanus, I. punctatus, D. rerio, C. carpio, C. idella, O. niloticus, O. latipes, T. rubripes, G. gallus, H. sapiens, M. musculus, X. tropicalis, and P. marinus, were downloaded from the Ensembl database (Release 98). Orthologous relationships between the genes from G. eckloni and the 13 other vertebrates were inferred through all-against-all protein sequence similarity searches using OthoMCL48. Only the longest predicted transcript per locus was retained. In the all-against-all BLASTP comparisons, a cutoff e-value of 1e−5 was used. The MCL inflation index was set to 1.5.
For each gene family, an alignment was produced using Muscle (http://www.drive5.com/muscle/), and ambiguously aligned positions were trimmed using Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks.html). The tree was inferred using RAxML49. The best-scoring ML tree was inferred by a rapid bootstrap algorithm and ML searches after performing 1000 rapid bootstrap replications. Divergence times between species were calculated using the MCMC tree program implemented by PAML package50. The divergence times for D. rerio vs C. idella (48–75 Ma), A. mexicanus vs C. carpio (137–174 Ma), C. carpio vs T. rubripes (206–252 Ma), G. gallus vs X. tropicalis (347.6–358.3 Ma), T. rubripes vs G. gallus (413–443 Ma), and G. gallus vs P. marinus (515–646 Ma) were obtained from the TimeTree database then used to calibrate divergence dates of other nodes on the phylogenetic tree54.
According to the divergence times and phylogenetic relationships, CAFÉ was used to analyze the expansion and constriction of gene families in the G. eckloni genome based on the gene families identified by OrthoMCL55. The phylogenetic tree topology and branch lengths were taken into account when inferring the significance of change in the gene family size of each branch. Enrichment analyses based on the Gene Ontology (GO) and KEGG annotations were performed to identify the functional implications of expanded and contracted genes (Fisher’s exact test, adjusted p-value < 0.05).
Supplementary information
Supplementary information of Chromosome-level assembly of Gymnocypris eckloni genome
Acknowledgements
This work was supported by grants from the Natural Science Foundation of Qinghai Science & Technology Department in China (No. 2020-ZJ-907) and the National Natural Science Foundation of China (No. 31960127).
Author contributions
D.L.Q. and K.Z. planned the project. F.Y.W., L.H.W., M.M.N., R.G.Y., and W.L.N. performed the experiments. D.L.Q., Q.G., C.J.Y. and F.T. performed the data analyses. C.F.Z., S.H.Z. and Y.C. assisted with sampling and experimentation. D.L.Q. and F.Y.W. wrote and revised the manuscript. Also, all authors read, edited and approved the final manuscript.
Code availability
All software used in this work is in the public domain, with parameters being clearly described in Methods. If no detail parameters were mentioned for a software, default parameters were used as suggested by developer.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Fayan Wang, Lihan Wang.
Supplementary information
The online version contains supplementary material available at 10.1038/s41597-022-01595-w.
References
- 1.Li J, Fang X. Uplift of the Tibetan Plateau and environmental changes. Chinese Science Bulletin. 1999;44:2117–2124. doi: 10.1007/BF03182692. [DOI] [Google Scholar]
- 2.Favre A, et al. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol Rev Camb Philos Soc. 2015;90:236–253. doi: 10.1111/brv.12107. [DOI] [PubMed] [Google Scholar]
- 3.Scheinfeldt LB, Tishkoff SA. Living the high life: high-altitude adaptation. Genome Biol. 2010;11:133. doi: 10.1186/gb-2010-11-9-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiu Q, et al. The yak genome and adaptation to life at high altitude. Nat Genet. 2012;44:946–949. doi: 10.1038/ng.2343. [DOI] [PubMed] [Google Scholar]
- 5.Chen N, et al. Ancient genomes reveal tropical bovid species in the Tibetan Plateau contributed to the prevalence of hunting game until the late Neolithic. Proc Natl Acad Sci USA. 2020;117:28150–28159. doi: 10.1073/pnas.2011696117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qu Y, et al. Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau. Nat Commun. 2013;4:2071. doi: 10.1038/ncomms3071. [DOI] [PubMed] [Google Scholar]
- 7.Ge RL, et al. Draft genome sequence of the Tibetan antelope. Nat Commun. 2013;4:1858. doi: 10.1038/ncomms2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu, H. et al. Genomic evidence for the Chinese mountain cat as a wildcat conspecific (Felis silvestris bieti) and its introgression to domestic cats. Sci Adv7 (2021). [DOI] [PMC free article] [PubMed]
- 9.Chen, Y. F. & Cao, W. Y. in Fauna Sinica, Osteichthyes, Cypriniformes III. (ed P.Q. Yue) 273-390. (Science Press, 2000).
- 10.Wu, Y. F. & Wu, C. Z. The fishes of the Qinghai – Xizang plateau. (Science and Technology Press, 1992).
- 11.Qi D, et al. Convergent, parallel and correlated evolution of trophic morphologies in the subfamily schizothoracinae from the Qinghai-Tibetan plateau. PLoS One. 2012;7:e34070. doi: 10.1371/journal.pone.0034070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qi D, et al. Transcriptome Analysis Provides Insights Into the Adaptive Responses to Hypoxia of a Schizothoracine Fish (Gymnocypris eckloni) Front Physiol. 2018;9:1326. doi: 10.3389/fphys.2018.01326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xia M, et al. Changes of hemoglobin expression in response to hypoxia in a Tibetan schizothoracine fish, Schizopygopsis pylzovi. J Comp Physiol B. 2016;186:1033–1043. doi: 10.1007/s00360-016-1013-1. [DOI] [PubMed] [Google Scholar]
- 14.Cao, W. X., Chen, Y. Y., Wu, Y. F. & Zhu, S. Q. in Studies on the Period, Amplitude and Type of the Uplift of the Qinghai–Xizang Plateau (ed Chinese Academy of Sciences The Team of the Comprehensive Scientific Expedition to the Qinghai-Xizang Plateau) 118-130 (Science Press, 1981).
- 15.Li Y, et al. High altitude adaptation of the schizothoracine fishes (Cyprinidae) revealed by the mitochondrial genome analyses. Gene. 2013;517:169–178. doi: 10.1016/j.gene.2012.12.096. [DOI] [PubMed] [Google Scholar]
- 16.Yonezawa T, Hasegawa M, Zhong Y. Polyphyletic origins of schizothoracine fish (Cyprinidae, Osteichthyes) and adaptive evolution in their mitochondrial genomes. Genes Genet Syst. 2014;89:187–191. doi: 10.1266/ggs.89.187. [DOI] [PubMed] [Google Scholar]
- 17.Zan RG, Liu WG, Song Z. Tetraploid-hexaploid relationship in Schizothoracinae. Acta Genet. Sin. 1985;12:137–142. [Google Scholar]
- 18.Yu XY, Li YC, Zhou T. Karyotype studies of cyprinid fishes in China -Comparative study of the karyotypes of 8 species of schizothoracine fishes. Journal of Wuhan University. 1990;2:97–104. [Google Scholar]
- 19.Yang S, et al. Morphogenesis of blood cell lineages in Ya-fish (Schizothorax prenanti) Chinese Journal of Zoology. 2015;50:231–242. [Google Scholar]
- 20.Dai Y, Han H. Karyological analysis of two species in the subfamily schizothoracinae (Cypriniformes: Cyprinidae) from China, with notes on karyotype evolution in schizothoracinae. Turkish Journal of Fisheries and Aquatic Sciences. 2018;18:175–186. doi: 10.4194/1303-2712-v18_1_20. [DOI] [Google Scholar]
- 21.Xiao S, et al. Genome of Tetraploid Fish Schizothorax o’connori Provides Insights into Early Re-diploidization and High-Altitude Adaptation. iScience. 2020;23:101497. doi: 10.1016/j.isci.2020.101497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi D, et al. Adaptive evolution of interferon regulatory factors is not correlated with body scale reduction or loss in schizothoracine fish. Fish Shellfish Immunol. 2018;73:145–151. doi: 10.1016/j.fsi.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 23.Qi D, et al. Duplication of toll-like receptor 22 in teleost fishes. Fish Shellfish Immunol. 2019;94:752–760. doi: 10.1016/j.fsi.2019.09.067. [DOI] [PubMed] [Google Scholar]
- 24.Chen QC, et al. A new pattern of hemoglobin switching in teleost fish-study of the embryonic hemoglobin in the Schizopygopsis pylzovi. Acta Hydrobiologica Sinica. 2020;44:1199–1207. [Google Scholar]
- 25.Peng, Y. et al. Chromosome-level genome assembly of the Arctic fox (Vulpes lagopus) using PacBio sequencing and Hi-C technology. Mol Ecol Resou (2021). [DOI] [PubMed]
- 26.Belton JM, et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2020;17:155–158. doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chin CS, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 30.Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Servant N, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Durand NC, et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016;3:95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dudchenko O, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–95. doi: 10.1126/science.aal3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 36.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gertz EM, Yu YK, Agarwala R, Schaffer AA, Altschul SF. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 2006;4:41. doi: 10.1186/1741-7007-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stanke M, et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.UniProt Consortium, T. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2018;46:2699. doi: 10.1093/nar/gky092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn RD, et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017;45:D190–D199. doi: 10.1093/nar/gkw1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanehisa M, et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L, Stoeckert CJ, Jr., Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 50.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 51.2022. NCBI Sequence Read Archive. SRP377513
- 52.Qi D. 2022. Gymnocypris eckloni isolate SKLPE_202101. NCBI Assembly. GCA_024082105.1
- 53.Qi D. 2022. Chromosome-level assembly of Gymnocypris eckloni genome. figshare. [DOI] [PMC free article] [PubMed]
- 54.Kumar S, Stecher G, Suleski M, Hedges SB. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol Biol Evol. 2017;34:1812–1819. doi: 10.1093/molbev/msx116. [DOI] [PubMed] [Google Scholar]
- 55.De Bie T, Cristianini N, Demuth JP, Hahn MW. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22:1269–1271. doi: 10.1093/bioinformatics/btl097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- 2022. NCBI Sequence Read Archive. SRP377513
- Qi D. 2022. Gymnocypris eckloni isolate SKLPE_202101. NCBI Assembly. GCA_024082105.1
- Qi D. 2022. Chromosome-level assembly of Gymnocypris eckloni genome. figshare. [DOI] [PMC free article] [PubMed]
Supplementary Materials
Supplementary information of Chromosome-level assembly of Gymnocypris eckloni genome
Data Availability Statement
All software used in this work is in the public domain, with parameters being clearly described in Methods. If no detail parameters were mentioned for a software, default parameters were used as suggested by developer.