

Abstract
Autophagy, an important cellular recycling process whereby macromolecules or organelles are encapsulated by autophagosome and degraded upon merging with lysosome, is recently shown to play an essential role in bone biology. However, the involvement of autophagy in bone and bone-related cells remains unclear. Here, we show that Beclin1, an autophagy-related (ATG) protein involved in autophagy initiation, plays a pivotal role in osteoclasts. Autophagy was activated during osteoclast differentiation in vitro. Beclin1 was enhanced and required for osteoclast differentiation. Mechanistically, we found that TRAF6-mediated ubiquitination of Beclin1 at K117, but not ULK1-mediated phosphorylation, is required for RANKL-stimulated osteoclast differentiation. In vivo, mice lacking Becn1 in CstK-expressing cells exhibited in increased cortical bone thickness due to impaired osteoclasts’ function. Interestingly, these mice also exhibited diminished trabecular bone mass, which was associated with defect in cartilage formation and chondrocyte differentiation. Collectively, our study highlights the functional importance of autophagy in osteoclasts and chondrocytes, and identifies autophagy as a potential therapeutic target for managing bone-related diseases.
Keywords: RANKL, autophagy, osteoclasts, chondrocytes, Beclin1, ubiquitination
INTRODUCTION
Bone is highly mineralized tissue that functions to support and protect various organs in the body. Although rigid, it continuously undergoes an active remodeling process primarily by two distinct cell types – osteoblasts that are responsible for bone formation and osteoclasts that resorb the bone (1). The imbalance between osteoblastic and osteoclastic activities, therefore, is of paramount importance in causing bone-related diseases such as osteoporosis (2).
Osteoclasts are the only known bone resorbing cells derived from monocyte-macrophage lineage precursors. Upon stimulation by receptor activator of NF-κB ligand (RANKL), osteoclast precursor cells such as bone marrow derived macrophages (BMMs) become large, polarized, multi-nucleated cells with a formation of F-actin ring around ruffled borders through which the cells release hydrogen ions, hydrolytic enzymes, and matrix metalloproteases (MMPs) that are required for bone resorption (3). These enzymes are induced by RANKL stimulation and transported to the ruffled border via lysozymes (4). As such, enlarged cytoplasm with massive formation of vacuoles such as lysosomes is the hallmark of osteoclast formation. However, detail mechanisms as to how RANKL stimulation leads to drastic structural alteration and functional changes remain largely elusive.
Autophagy is an important cellular degradation pathway whereby macromolecules or organelles are broken down to be recycled in cells (5). Autophagy pathways are characterized by the presence of double-membrane autophagosomes that ultimately fuse with lysosomes to form single-membrane autolysosomes in which macromolecules or organelles become degraded (6). Although autophagy become activated to conserve energy during cellular stress such as starvation, increasing numbers of evidence supports a notion that autophagy pathway can be activated by receptor-mediated signaling pathways and regulates normal cellular functions of cells (7,8).
During osteoclastogenesis, RANKL binds to its receptor, RANK, and elicit the downstream signaling pathways, including Src, JNK, and NF-κB, that are essential for osteoclast differentiation (9). Although studies demonstrated the essential role of autophagy-related (ATG) proteins in differentiation and functions of osteoclasts (10–15), the mechanisms by which RANKL regulates autophagic pathway is incompletely understood. Here, we report the pivotal role of ubiquitination of Beclin1, a mammalian homologue of yeast Atg6/Vps30 and a core component of autophagosome initiator, in RANKL-stimulated autophagy activation and osteoclast differentiation in vitro. To determine the role of Becn1 in osteoclasts in vivo, we generated CtsK-specific Becn1 knockout mice and analyzed their bone phenotype.
MATERIALS AND METHODS
Reagents and antibodies
The antibodies used for the western blot analysis were: anti-phosphorylated Beclin1 antibody from Abbiotec (San Diego, CA); anti-Ub-K63 (14–6077) antibody from eBioscience (San Diego, CA); anti-TRAF6 and anti-HA antibodies from Abcam (Cambridge, MA); anti-β-actin (C4) and anti-GAPDH (0411) antibodies from Santa Cruz Biotechnology (Santa Cruz, CA); anti-LC3B (2775), anti-Beclin1 (D40C5), anti-ULK1 (D9D7), anti-phosphorylated ULK1 (D1H4), anti-P62 (5114) and anti-NFATc1 (D15F1) antibodies from Cell Signaling Technology (Danvers, MA). Bafilomycin A1 was purchased from Sigma-Aldrich (St. Louis, MO).
Cells and cell culture
RAW 264.7 cells were obtained from ATCC (Rockville, MD). To obtain bone marrow macrophages, bone marrow cells were obtained from tibiae and femur of adult C57/BL6 mice (4–6 weeks old) or Beclin1f/f mice (provided by Dr. Binfeng Lu, University of Pittsburgh School of Medicine) and cultured in α-MEM (Invitrogen, Carlsbad, CA) containing10% fetal bovine serum (FBS, Invitrogen) in the presence of 10 4 U/ml M-CSF (R&D Systems, Inc. Minneapolis, MN). After culture for 16 hour, nonadherent cells were harvested, and 3 × 10 5 cells /ml were incubated with 10 4 U/ml M-CSF for 3 days in 60 mm plates. The adherent cells were used as bone marrow macrophages. This time point was designated as day 0 in all the experiments. Bone marrow macrophages were further cultured with or without 100 ng/ml RANKL (R&D Systems, Inc.) in the presence of 10 4 U/ml M-CSF for 3 days.
Primary culture of immature murine articular chondrocytes (iMACs) and quantification iMAC were isolated as described (16). Briefly, the isolated murine articular cartilages were plated in high-glucose DMEM (Invitrogen, Carlsbad, CA) containing10% fetal bovine serum (FBS, Invitrogen) and antibiotics in the presence of digestion solution (collagenase D solution at 3mg/ml; Sigma-Aldrich). Digested chondrocytes were seeded on a culture dish and incubated at 37°C in a humidified gas mixture containing 5% CO2. To test for expression of the markers of chondrocyte differentiation, Alcian blue staining which represents sulfated proteoglycans in the ECM was performed. For quantification, Alcian blue is extracted by guanidine solution containing 4M Guanidine-HCl, 33% isopropanol, and 0.25% Triton-X 100. Absorbance values of the supernatant were measured at 600nm.
Vectors and vector construction
The pBABEpuro GFP-LC3 (plasmid #22405), pQCXIH-Beclin1-HA WT (plasmid #46993), pQCXIH-Beclin1-HA S-A (plasmid #46994), and pQCXIH-Beclin1-HA S-D (plasmid #46995) were purchased from Addgene (Cambridge, MA) (17,18). The retroviral vectors pLPCX-Beclin1-HA WT was generated by inserting DNA fragment encoding BECN1 WT into a pLPCX retroviral expression vector. In situ mutagenesis was performed to generate pLPCX-Beclin1-HA K117R by targeting Beclin1 WT candidate sites using QuickChange Site-Directed Mutagenesis kit (Stratagene) according to the manufacturer’s protocol.
Virus production and transduction of cells
Briefly, retroviral vectors were transfected into GP2–293 universal packaging cells (Clonetech, Mountain View, CA, USA) along with pVSV-G envelope plasmid using Lipofectamine 2000 (Life Technologies, Grand island, NY). Two days after transfection, the virus supernatant was collected and concentrated by ultracentrifugation. The virus pellet was suspended in α-MEM and was used for infection immediately or stored in −80˚C for later use. Cells were infected with the retroviruses in the presence of 6 μg/ml polybrene for three hours. All of these viruses consistently gave more than 90% of infection efficiency (19). The infected cells were treated with puromycin (1 μg/ml puromycin) 48 hours after infection and maintained for additional 7 days.
Real-time quantitative RT-PCR (qRT-PCR)
qRT-PCR was performed as described previously(19). Briefly, the cells were harvested with 1 mL Trizol Reagent (Invitrogen) per plate. mRNA was extracted with High Pure RNA Isolation Kit (Roche) and cDNA was synthesized with PCR (SuperScript III Reverse Transcriptase). Real time PCR was done with SYBR Green I Master (LightCycler 480, Roche). A total of 45 cycles were executed, and the second derivative of Cq value determination method was used to compare fold-differences. Primer sequences are listed in the Table 1.
Table 1.
Bone histomorphometric analysis in Becn1 WT and cKO mice.
| 6 months (femurs) | 6 months (vertebrae, L5) | |||||||
|---|---|---|---|---|---|---|---|---|
| male | female | male | female | |||||
| WT (n = 4) | cKO (n = 7) | WT (n = 5) | cKO (n = 5) | WT (n = 4) | cKO (n = 7) | WT (n = 5) | cKO (n = 5) | |
| BV/TV (%) | 16.37 ± 2.02 | 11.93 ± 1.66 ** | 4.31 ± 1.74 | 3.56 ± 2.34 | 25.85 ± 4.28 | 20.68 ± 2.77 * | N.A | N.A |
| Tb.N (mm-1) | 4.45± 0.56 | 2.99 ± 0.44 ** | 1.71 ± 0.47 | 1.08 ± 0.64 | 6.63 ± 0.38 | 6.14 ± 0.73 | N.A | N.A |
| Tb.Th (μm) | 36.86 ± 2.61 | 39.95 ± 2.67 | 24.86 ± 2.68 | 32.39 ± 2.5 ** | 38.83 ± 4.78 | 33.66 ± 1.74 * | N.A | N.A |
| Tb.Sp (μm) | 190.74 ± 31.25 | 299.1 ± 42.72 ** | 602.6 ± 191.75 | 1264.35 ±914.37 | 112.35 ± 13.18 | 131.05 ± 18.76 | N.A | N.A |
| Ob.S/BS (%) | 2.77 ± 2.05 | 3.46 ± 2.91 | 7.93 ± 5.6 | 3.69± 4.26 | 6.48 ± 4.38 | 3.93 ± 2.26 | N.A | N.A |
| N.Ob/BS (mm-1) | 2.34 ± 1.64 | 3.17 ± 2.42 | 5.76 ± 4.23 | 3.3 ± 2.86 | 4.84 ± 2.74 | 3.27 ± 1.6 | N.A | N.A |
| OS/BS (%) | 7.25 ± 5.05 | 4.18 ± 2.77 | 6.03 ± 3.68 | 6.22 ± 8.92 | 8.2 ± 3.77 | 2.94 ± 0.87 ** | N.A | N.A |
| Oc.S/BS (%) | 1.73 ± 0.61 | 1.51 ± 0.92 | 1.56 ± 0.97 | 0 ** | 1.15 ± 0.26 | 2.34 ± 1.54 | N.A | N.A |
| N.Oc/BS (mm-1) | 0.68 ± 0.25 | 0.62 ± 0.36 | 0.42 ± 0.07 | 0 *** | 0.4 ± 0.09 | 0.87 ± 0.56 | N.A | N.A |
| ES/BS (%) | 3.04 ± 0.88 | 2.69 ± 1.78 | 2.74 ± 1.93 | 0 * | 1.95 ± 0.73 | 4.15 ± 2.99 | N.A | N.A |
| MS/BS (%) | 5.66 ± 4.31 | 18.7 ± 6.34 * | 16.38 ± 6.52 | 13.67 ± 7.32 | 4.4 ± 3.9 | 7.72 ± 1.77 | N.A | N.A |
| MAR (μm day-1) | 0.48 ± 0.19 | 0.74 ± 0.34 | 0.99 ± 0.31 | 0.83 ± 0.24 | 0.54 ± 0.11 | 0.65 ± 0.04 | N.A | N.A |
| BFR/BV (% Year-1) | 61.7 ± 63.2 | 255.09 ± 96.3 * | 522.76 ± 360.57 | 237.75 ± 118.62 | 56.58 ± 61.94 | 109.93 ± 27.62 | N.A | N.A |
Male Becn1 WT versus male Becn1 cKO, and female Becn1 WT versus female Becn1 cKO; Student’s t test.
p <0.05
p < 0.01
p < 0.001
Western blotting
Western blotting was performed as described previously (20). Briefly, cells were lysed using 0.1% NP-40 lysis buffer [20 mM Tris (pH 7.5), 50 mM β-glycerophosphate, 150 mM NaCl, 1 m MEDTA, 25 mM NaF, 1 mMNa3VO4, 16 protease inhibitor cocktail (Sigma-Aldrich), 16 phosphatase inhibitor cocktail I (Sigma-Aldrich) and phosphatase inhibitor cocktail II (Sigma-Aldrich)]. Lysates (40–50 mg) from cells were used for immunoprecipitation and Western blot analysis and fractionated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto the Immobilon protein membrane (Millipore, Billerica, MA). The immobilized membrane was incubated with anti-ALP antibody (H-300, Santa Cruz Biotechnology, Inc. Santa Cruz, CA) and probed with the respective secondary antibody. The membrane was exposed to the HyGLO Chemiluminescent HRP antibody detection reagent (Denville Scientific, South Plainfield, NJ) and scanned using ChemiDoc System (Bio-Rad, Hercules, CA). To quantify the Western results, the ImageJ software was used (21).
Ubiquitination assay
RAW264.7 cells were infected with retroviruses expressing pLPCX-Beclin1-HA WT or pLPCX-Beclin1-HA K117R and selected with puromycin (1 μg/ml puromycin) 48 hours after infection. Selected cells were treated with RANKL for 0 to 6 hours. Whole lysate was prepared with a lysis buffer [20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA and 1% Triton X-100] containing with 1 mM N-ethylmaleimide (Sigma-Aldrich) and 1% SDS. Lysates were immediately heated for 5 min and then were diluted 1:10 in lysis buffer. Ubiquitinated protein was immunoprecipitated with HA-conjugated agarose at 4 °C overnight. Immunoprecipitated samples were washed five times with lysis buffer, then subjected to SDS-PAGE and analyzed by Western blotting using mouse anti-K63-linked polyubiquitin (HWA4C4; eBioscience) and mouse anti-HA (Sigma-Aldrich) antibodies.
Resorption pits assay
Raw 264.7 cells (2 × 104/ ml) plated on dentin slices in 96-well culture plates (0.2 ml/well) and culture with RANKL for 3 days. The dentin slices were then transferred into 48-wells culture plates and culture for 7 days in α-MEM containing 10% FBS (0.4 ml/well). Cells were thoroughly removed from dentin slices using cotton swabs. If the remainders of cells on the slices were detected under a microscopy, the slices were rubbed again with cotton swabs. Dentin slices were stained with Mayer’s hematoxylin to detect resorption pits (22). The numbers of resorption pit were visually counted. To measure the depth of resorption pits, BMMs (1 × 105/ ml) isolated from Becn1 WT or cKO mice plated on dentin slices in 48-well culture pates (0.5 ml/well) and were treated with RANKL/M-CSF for 4 days. Then dentin slices were moved to new 48-well culture plates, cells were cultured for 8 days in the presence of RANKL/M-CSF. After cells were removed from dentin slices, the pits were stained with FITC-wheat germ agglutinin (WGA) for 40 min and visualized by confocal microscopy (Olympus, Japan). The resorbed pit depth was quantitated using a Thermo Scientific Amira Software (Thermo Fisher Scientific, Hillsboro, OR) and Fluoview software (Olympus, Japan).
Histochemical staining
For immunofluorescent staining, tissues were frozen in hexane using Liquid nitrogen and 2-methyl butane and embedded in 5% carboxymethyl cellulose (CMC) gel. Sections 5-um thick were prepared using Kawamoto’s film method (Cryofilm transfer kit; Finetec, Tokyo) (Kawamoto and Shimizu, 2000). The sections were fixed in ice-cold 5% acetic acid in ethanol. The sections were subjected to staining for RANK and LC3B using specific antibodies. Biotin-conjugated antibodies were used for RANK (R&D Systems. Horseradish peroxidase (HRP)-conjugated streptavidin (PerkinElmer, Boston, MA), HRP- conjugated anti-FITC (PerkinElmer), were used as the secondary antibodies. Nuclei were detected by 4,4’-diamidino-2-phenylindole (DAPI) staining (Vector Laboratories, Burlingame, CA).
Tartrate-resistant Acid Phosphatase (TRAP) staining was performed as described previously15. Briefly, EDTA-decalcified tissue sections were deparaffinized at 60ºC for 30 min. The slides were rehydrated and incubated for 1 hr in 37ºC with the TRAP staining solution according to manufacturer’s protocol (Acid Phosphatase Kit, Sigma-Aldrich). The slides were counterstained with hematoxylin solution and mounted with Permount (Fisher Scientific, Tustin, CA). Sections of mouse femur were stained with Safranin O-fast green according to manufacturer’s protocol (Safranin O staining kit, ScienCell) for visualization of cartilage and bone.
Confocal laser scanning microscopy
The cells were grown in the chamber slides. Confocal laser scanning microscopy was performed at the CNSI Advanced Light Microscopy/Spectroscopy Shared Resources Facilities at UCLA with technical assistance from Dr. Matthew Schibler. Briefly, images were captured using the Leica TCS-SP2-AOBS confocal microscope with x63 oil objective under different gain settings. The 488-line argon laser was used to capture GFP staining, and the diode 405 nm laser was used to capture DAPI nuclear stain. For mCherry staining, 594-line HeNe laser was used. Image acquisition was performed using Leica Confocal Software (LCS) version 2.61 Build 1537. Scans were performed with pinhole set at Airy unit 1.5. SmartGain were adjusted for each sample for optimal brightness. Fluorescent images of cells were taken as single channel images then converted to overlay images and all images were saved in TIFF format.
Generation of Becn1 knockout mice
Beclin1f/f mice were obtained from Dr. Binfeng Lu (University of Pittsburgh School of Medicine), RANKCre/+ mice from Dr. Yasuhiro Kobayashi (Matsumoto Dental University, Japan), and CtsKCre/+ mice from Dr. Nakamura Takashi (Keio University, Japan). These mice were crossbred to generate Becnf/f;RANKCre/+ and Becnf/f;CtsKCre/+ mice. Becnf/f;CtsK+/+ (Becn1 WT) mice and their littermates, Becnf/f;CtsKCre/+ (Becn1 cKO) mice were used in this study. The mice were maintained under specific pathogen free (SPF) condition. For animal study, all animal experiment protocols were approved (ARC protocol #2012–033 and #2011–062) and conducted according to the Animal Research Committee and the UCLA institutional animal care and use committee (IACUC).
Ligature-induced periodontitis model
Eight-week old female C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and kept in a pathogen-free vivarium in the UCLA Division of Laboratory Animal Medicine (DLAM). Ligature was placed on the second maxillary molar using 6–0 suture under general anesthesia using Ketamine/Xylazine (100 mg per kg and 5 mg per kg, respectively). Three weeks after placing the ligature, the mice were sacrificed, and maxillae were harvested. The harvested maxillae were fixed in 4% para-formaldehyde/1x PBS solution overnight. The fixed tissues were subjected to further analyses. All experiments were performed according to the approved institutional guidelines from the Chancellor’s Animal Research Committee (ARC # 2011–062).
Blood biochemistry
Serum C-telopeptide (CTX) and the tartrate-resistant acid phosphatase (TRAP) concentrations were determined with the use of a RatLaps EIA immunoassay kit (Immunodiagnostic systems) and a mouse TRAP ELISA immunoassay kit (Immunodiagnostic System), according to the manufacturer’s instructions.
Micro-computed tomography (µCT) scan and three-dimensional volumetric analysis
Areas of interest on the maxilla were subjected to μCT scanning (Scanco µCT 40, Scanco Medical, Brüttisellen, Switzerland) at 55 kVp and 145 μA using a voxel size of 20 μm3 and a 0.5 mm Aluminum filter with an integration time of 200 ms using a cylindrical tube (FOV/Diameter: 20.48 mm). Two-dimensional slices from each maxilla were combined using the Scanco software (μCT v6.1) to form a three-dimensional reconstruction. Reconstructions were oriented so that the viewing angles could provide an accurate representation of bone loss.
Statistical analysis
The outcome measurements were expressed as means ± standard deviation. To compare the outcome mean measurements among the different groups, we used one-way ANOVA with Tukey’s post-hoc test. For comparison between two groups, student t test was used. All statistical tests were performed using the SPSS 23 software (IBM Corp, Somers, NY). p values of less than 0.05 were considered statistically significant. * p < 0.05, ** p < 0.005, and *** p < 0.001.
RESULTS
RANKL induces autophagy during osteoclast differentiation
Increasing lines of evidence suggest that autophagy-related proteins are involved in osteoclast differentiation and functions (11–15). However, the extent to which autophagy pathway is involved in RANKL-induced osteoclastogenesis remains unclear. To examine whether autophagy is associated with the onset of osteoclastogenesis, we treated bone marrow derived macrophages (BMMs) with RANKL and macrophage colony-stimulating factor (M-CSF) to examine LC3B expression level and found that LC3B-II, activated form of LC3B, is induced in a time-dependent manner (Supplementary Fig. 1a). p62 protein, a autophagic cargo protein known to be induced by starvation (23–25), was also increased during osteoclastogenesis, (Supplementary Fig. 1a). To visually confirm autophagosome formation in cells, we treated RANKL onto GFP-LC3B-overexpressing RAW 264.7 cells and found progressive accumulation of green fluorescent puncta, an indicative of autophagosome formation in cells, in a time-dependent manner (Supplementary Fig. 1b, c). When autophagic flux was measured, there were no notable changes, suggesting that autophagy is activated without increasing the rate of autophagic process (Supplementary Fig. 1d,e). Furthermore, expression of genes associated with autophagy are significantly increased around day 2 (Supplementary Fig. 1f, g), indicating that RANKL induces autophagy during osteoclastogenesis. To examine whether activation of autophagy occurs in osteoclasts in vivo, we experimentally induced osteoclast differentiation in mice using the ligature-induced periodontitis model (Supplementary Fig. 1h,i). Histological examination revealed marked presence of TRAP+ osteoclasts around the resorbed alveolar bone around the ligated tooth (Supplementary Fig. 1j). Immunofluorescent staining showed co-expression of LC3B and RANK (Supplementary Fig. 1k), indicating that matured osteoclasts exhibit autophagy activation in vivo.
Beclin1 enhances RANKL-induced osteoclast differentiation
Beclin1 is a known to initiate autophagic process by forming initial autophagosome complex (26). During osteoclastogenesis, Becn1 mRNA expression was progressively induced (Fig. 1a). Similarly, Beclin1 protein expression was induced by RANKL treatment (Fig. 1b). To examine whether Beclin1 overexpression enhances RANKL-induced osteoclastogenesis, we retrovirally transduced Beclin1-HA into RAW 264.7 cells (Fig. 1c) and stimulated with RANKL. Upon RANKL treatment, RAW/Beclin1 cells underwent osteoclastic differentiation more readily (Fig. 1d, e) and exhibited increased bone resorption activity when compared to the RAW/EV cells (Fig. 1f, g). ATG genes such as Atg2a and Atg10 as well as Nfatc1, a master regulator of osteoclast differentiation, were all induced in RAW/Beclin1 cells when compared to RAW/EV cells (Fig. 1h, i, j). In line with this notion, the lipidation conversion of LC3B-I to LC3B-II forms occurred more in the presence of Beclin1 overexpression (Fig. 1k), indicating that Beclin1 enhanced osteoclastic differentiation and functions.
Figure 1. Beclin1 enhances RANKL-induced osteoclast differentiation.
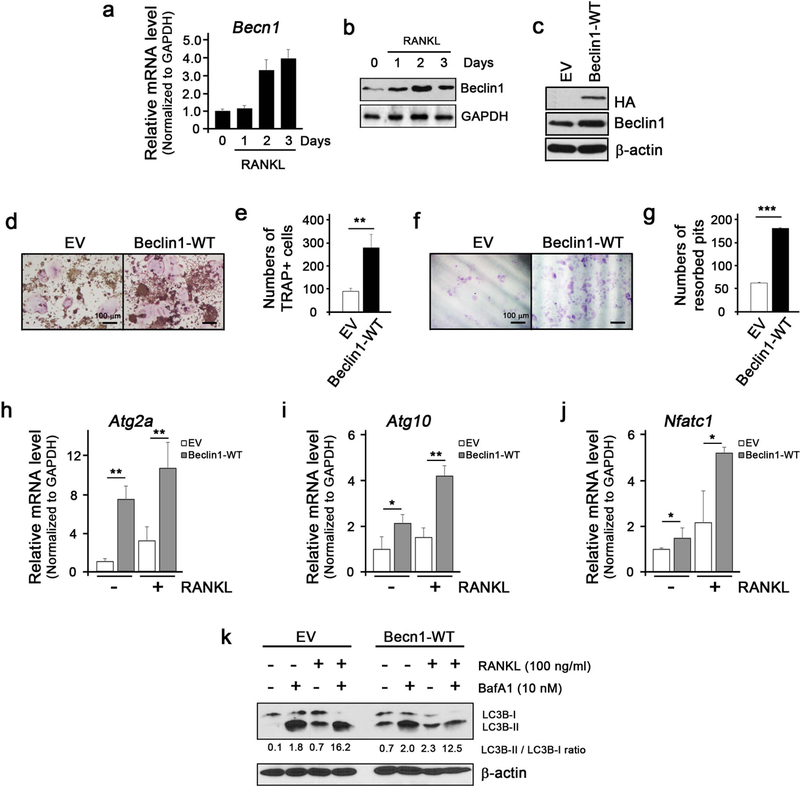
(a) Real-time qPCR for expression of Becn1 mRNA during RANKL-induced osteoclast differentiation in RAW 264.7 cells. (b) Western blotting against Beclin1 and GAPDH during RANKL-induced osteoclast differentiation in RAW 264.7 cells. (c) Western blotting against HA, Beclin1, and β-actin in RAW 264.7 cells infected with retroviruses harboring empty vector (EV) or wildtype Beclin1 (Beclin1-WT). (d) TRAP staining following RANKL treatment on RAW 264.7 cells for 3 days. Bar = 100 μm. (e) Quantification of TRAP+ cells. (f) Dentin slice assay following RANKL treatment on RAW 264.7 cells for 10 days. Cells were stained with hematoxylin, removed the cells with cotton swabs, and examined under the light microscope. Bar = 100 μm. (g) Quantification of resorbed pits. (h,i,j) Real-time qPCR for expression of Atg2a, Atg10, and Nfatc1 in EV- or Beclin1-WT-expressing RAW 264.7 cells treated with RANKL. (k) Western blotting against LC3B and β-actin during RANKL-induced osteoclast differentiation in RAW 264.7 cells with or without BafA1.
Beclin1 is required for autophagy induction and osteoclast differentiation.
To further show that Beclin1 is required for induction of autophagy and osteoclast differentiation, BMMs from Becnl f/f mice were isolated and transduced with lentivirus expressing Cre (LV-Cre). Transduction of LV-Cre suppressed Beclin1 expression during osteoclastogenesis (Fig. 2a). Upon RANKL treatment, LV-Cre-expressing BMMs exhibited suppression of osteoclast differentiation (Fig. 2b, c) and bone resorption activity (Fig. 2d, e) when compared to the control counterpart. Consistent with these findings, RANKL treatment on LV-Cre-expressing BMMs caused significant suppression in expression of autophagy-related genes (Fig. 2f–k) and osteoclast-related genes (Fig. 2l, m, n), indicating that Beclin1 is required for RANKL-mediating autophagy activation and osteoclast differentiation.
Figure 2. Beclin1 is required for autophagy induction and osteoclast differentiation.
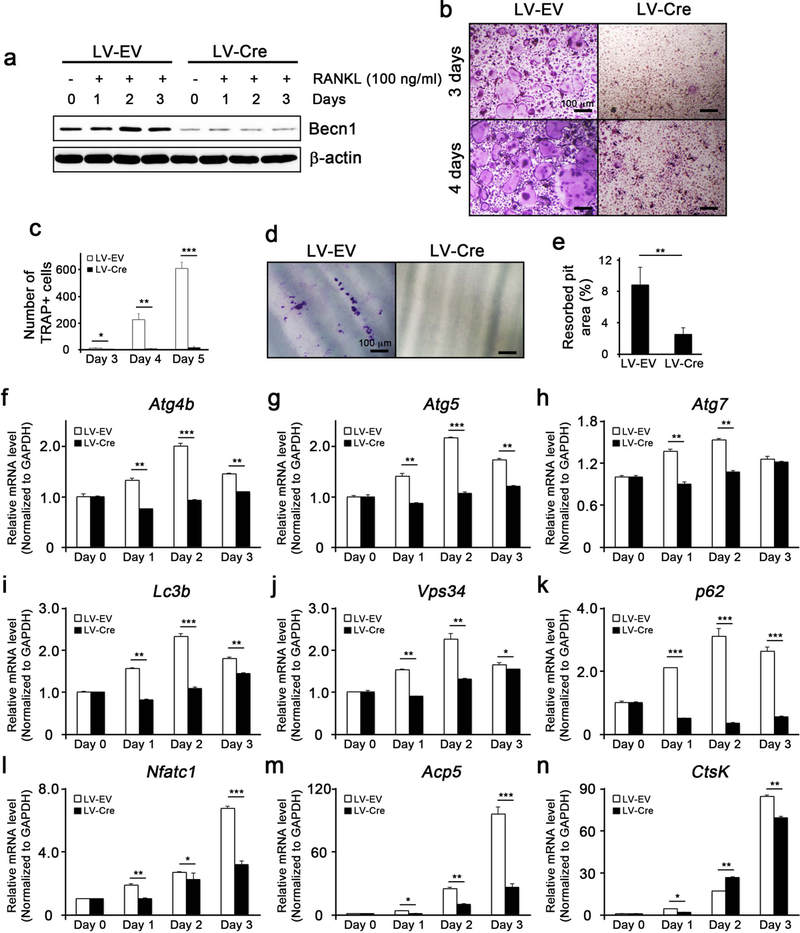
(a) Western blotting against Beclin1 and β-actin in RANKL/M-CSF-treated bone marrow macrophages (BMMs) isolated from Becn1 f/f mice and infected with lentiviruses harboring empty vector (LV-EV) or Cre (LV-Cre). (b) TRAP staining following RANKL/M-CSF treatment on EV- or Cre-expressing BMMs for 3 and 4 days. Bar = 100 μm. (c) Quantification of TRAP+ cells. (d) Dentin slice assay following RANKL/M-CSF treatment on EV- or Cre-expressing BMMs. Cells were stained with hematoxylin, removed with cotton swabs, and examined under the light microscope. Bar = 100 μm. (e) Quantification of resorbed pits. (f-k) Real-time qPCR for the expression of autophagy-related genes in EV- or Cre-expressing BMMs. (l,m,n) Real-time qPCR for the expression of osteoclast-related genes in EV- or Cre-expressing BMMs.
TRAF6-dependent ubiquitination of Beclin1 at K117 is required for RANKL-induced osteoclast differentiation
Autophagy is originally termed to describe a structural alterations and cellular degradation pathway whereby macromolecules are broken down to be recycled in autolysosomes during stressed conditions such as starvation (5). Recent finding showed that starvation-induced autophagy is mediated by ULK1/ULK2, which phosphorylates Beclin1 at the S14 (S15 in human) residue that is required for activation of ATF14-containing VPS34 complexes (17). To gain mechanistic insights, we further examined whether ULK plays a functional role in RANKL-mediated activation of autophagy and osteoclastogenesis by phosphorylating Beclin1 at S14. Upon RANKL treatment, p-ULK was slightly induced; however, expression of ULK was diminished rather than increased (Supplementary Fig. 2a). Consistent with this finding, there was no differences in numbers of osteoclasts when RANKL was treated on RAW 264.7 cells overexpressing either wild type (Beclin1-WT), constitutively phosphorylated (Beclin1-S14D) or phosphorylation incompetent Beclin1 (Beclin1-S14A) (Supplementary Fig. 2b, c, d), implying that phosphorylation of Becn1 at S14 is dispensable in RANKL-induced osteoclastogenesis
Previous study demonstrated a critical role of Beclin1 ubiquitination in activating ligand-induced autophagy. In particular, knockdown of Beclin1 in macrophages caused complete abolishment of TLR7-mediated removal of Bacille Calmette-Guerin (BCG) in macrophages (8). Similarly, LPS-mediated activation of TLR4 in macrophages was shown to cause upregulation of pro-inflammatory cytokine gene expression via Beclin1 ubiquitination (7). Because K63-linked ubiquitination of Beclin1 is known to mediate lipopolysaccharide (LPS)-induced autophagy (7), we then asked whether K63-linked ubiquitination of Beclin1 at K117 plays a role in RANKL-induced osteoclastogenesis. When RANKL was treated, K63-linked ubiquitination of Beclin1 increased progressively in a time-dependent manner (Fig. 3a). Consistent with this finding, K63-linked ubiquitination and Becn1 co-localized after RANKL treatment as determined by confocal microscopy (Fig. 3b). When Beclin1-K117R, an ubiquitination incompetent Beclin1 at K117, was overexpressed in RAW264.7 cells, osteoclastogenesis was significantly suppressed when compared to the control counterpart (Fig. 3c–e). Furthermore, ubiquitination of Beclin1 was abolished in cells expressing Beclin1-K117R (Fig. 3f), suggesting the critical involvement of Beclin1 and underscoring the importance of autophagy pathway during osteoclastogenesis.
Figure 3. TRAF6-dependent ubiquitination of Beclin1 at K117 is required for RANKL-induced osteoclast differentiation.
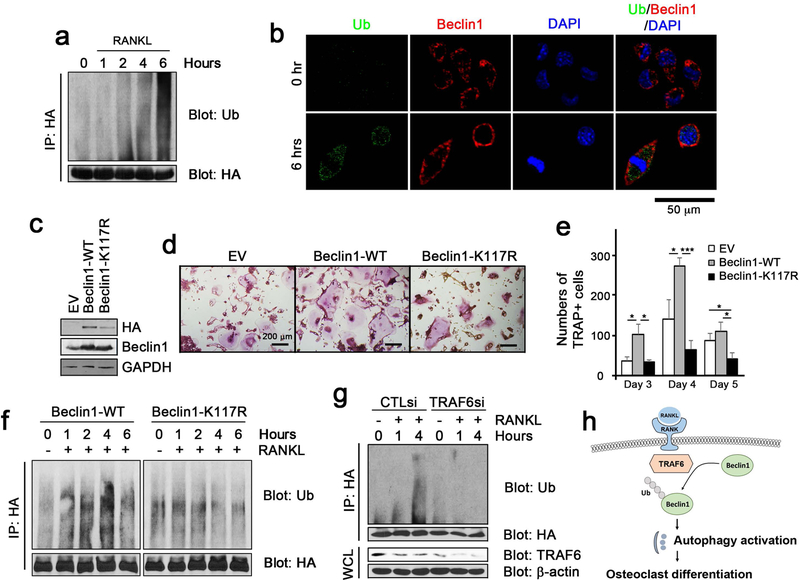
(a) The ubiquitination assay in RANKL-treated RAW 264.7 cells in the indicated time points. (b) Fluorescent staining for ubiquitin (green) and Beclin1 (red) visualized using confocal microscopy. (c) Western blotting against HA, Beclin1, and β-actin in RAW 264.7 cells infected with retroviruses harboring empty vector (EV) or wildtype Beclin1 (Beclin1-WT), and ubiquitination incompetent mutant Beclin1 at K117 (Beclin1-K117R). (d) TRAP staining following RANKL treatment on RAW 264.7 cells for 3, 4 and 5 days. Bar = 200 μm. (e) Quantification of TRAP+ cells. (f) The ubiquitination assay in RANKL-treated RAW 264.7 cells expressing wildtype Beclin1 (Beclin1-WT) or ubiquitination incompetent mutant Beclin1 at K117 (Beclin1-K117R) in the indicated time points. (g) The ubiquitination assay in RANKL-treated RAW 264.7 cells expressing wildtype Beclin1 (Beclin1-WT) following transfection with CTL siRNA (CTLsi) and TRAF6 siRNA (TRAF6si). (h) A model for RANKL-induced autophagy activation via Beclin1 ubiquitination and osteoclast differentiation.
TRAF6 is an essential adaptor protein for osteoclast differentiation as TRAF6 knockout mice exhibited osteopetrotic phenotypes (27–29). Upon RANKL stimulation, TRAF6 is recruited to the RANK and interacts with multiple binding proteins including Limd1, p62, FHL2, and Smad2/3 (9,30–33). Such interactions subsequently lead to activation of the downstream signaling pathways such as Src, JNK, and NF-κB that are essential for osteoclast differentiation (9). TRAF6 is an E3 ubiquitin ligase and, although TRAF6 is known to self-ubiquitinate upon RANKL stimulation (34), it is also known to ubiquitinate Beclin1 at K117 upon LPS stimulation (7). To examine whether RANKL-induced ubiquitination of Becn1 is mediated through TRAF6, we knocked down TRAF6 and examined the ubiquitination status of Beclin1. We found that ubiquitination of Beclin1 by RANKL stimulation in RAW 264.7 cells was inhibited when TRAF6 was knocked down (Fig. 3g). These data indicate that TRAF6 is required for Beclin1 ubiquitination and that TRAF6-mediated ubiquitination of Beclin1 is critically important in initiating autophagy at the earlier stages and subsequent osteoclast differentiation by RANKL stimulation (Fig. 3h).
Becn1 conditional knockout mice exhibited increased cortical bone thickness but diminished trabecular bone mass
To examine the role of Becn1 in osteoclasts in vivo, we generated Becn1 conditional knockout mice by crossbreeding Becn1f/f mice with RANK-Cre or CtsK-Cre mice. Of 62 pups generated from breeding Becn1f/f and RANK-Cre mice, only 2 Becn1f/f;RANKCre/+ pups were born, both of which died few days after the birth (data not shown), presumably due to important functions of Becn1 in RANK-expressing cells other than osteoclasts during development (35). When we crossbred Becn1f/f and CtsKCre/+ mice, Becn1f/f;CtsKCre/+ (herein Becn1 cKO) mice were generated but with small in size compared with their littermates, Becn1f/f;CtsK+/+ (herein Becn1 WT) mice (Fig. 4a, b). These mice exhibited splenomegaly (Fig. 4c, d) and short limbs (Fig. 4e, f), typical phenotypes of mice lacking osteoclast functions (36,37). Consistent with our observations, we found a thickening of the cortical bone in the femur (Fig. 4g, h). Interestingly, the trabecular bone mass was markedly diminished with less trabecular numbers and more trabecular spacing (Fig. 4i–m). The diminished trabecular bone mass was also observed in vertebrae, L5, and such trend was also observed in both male and female (Table 1). Histological analysis revealed disorganized osteocartilaginous structures, complete absence of growth plate remnants, and increased bone marrow cavities filled with hematopoietic cells in Becn1 cKO mice (Fig. 4n). TRAP staining showed comparable numbers of osteoclasts in the trabecular bone areas in both Becn1 WT and cKO mice (Fig. 4o, p); however, in the cortical bone area, less osteoclasts were found in Becn1 cKO mice (Fig. 4q, r). These data suggest that Becn1 cKO mice exhibit increased thickness of cortical bone and decreased amount of trabecular bone in mice.
Figure 4. Becn1 cKO mice exhibited increased cortical bone thickness but diminished trabecular bone mass.

(a) Photographs of Becn1 WT and cKO mice. (b) Quantification of body weights at 6th month (n = 5 WT, n = 12 cKO, male). (c) Photographs of spleens from Becn1 WT and cKO mice. (d) Quantification of spleen at 6th month (n = 5 WT, n = 12 cKO, male) normalized to the body weights. (e) µCT of the femurs from Becn1 WT and cKO mice. (f) Quantification of femur lengths measured at 6th month (n = 5 WT, n = 12 cKO, male). (g) µCT of the femur cross-sections in Becn1 WT and cKO mice. (h) Quantification of femur cortical bone thickness measured at 6th month (n = 5 WT, n = 12 cKO, male). (i) µCT of the trabecular bone from Becn1 WT and cKO mice. Quantification of BV/TV (j), trabecular thickness (k), trabecular number (l), and trabecular spacing (m) measured at 6th month (n = 5 WT, n = 12 cKO, male). (n) H&E staining of the femur head. Bar = 200 µm. (o) TRAP staining in the femur trabecular bone. Bar = 100 µm. (p) Quantification of TRAP+ osteoclasts per bone perimeter. (q) TRAP staining in the femur cortical bone. Bar = 100 µm. (r) Quantification of the TRAP+ osteoclasts per bone perimeter.
Bone-resorbing function of osteoclasts is impaired in Becn1 cKO mice
To further evaluate functions of osteoclasts in these mice, we isolated BMMs and induced them to undergo osteoclast differentiation. As expected, upon stimulation, these BMMs progressively increased CtsK-driven Cre expression (Fig. 5a) and subsequently decreased Beclin1 protein level (Fig. 5b), confirming the specificity of this Cre system. In vitro osteoclast differentiation revealed that there is no difference in osteoclast formation (Fig. 5c, d), which is most likely due to intact Beclin1 expression at the early stage until Cre expression are induced. However, functional study using dentin slice assay showed a decrease in the resorbed pit depth (Fig. 5e, f) and TRAP activity (Fig. 5g), suggesting that loss of Beclin1 expression at the late stage of differentiation inhibited resorbing function of osteoclasts. Indeed, Western blotting from osteoclasts obtained at the end of the dentin slice assay confirmed the absence of Beclin1 (Fig. 5h). When osteoclastogenesis was induced in BMMs isolated from Becn1+/− mice, lipidation conversion of LC3B-I as well as the amount of LC3B-II were diminished, indicating that RANKL-induced autophagy is suppressed (Fig. 5i). Collectively, these data suggest that bone-resorbing function of osteoclasts is impaired in Becn1 cKO mice both in vitro and in vivo.
Figure 5. Bone-resorbing function of osteoclasts is impaired in Becn1 cKO mice.
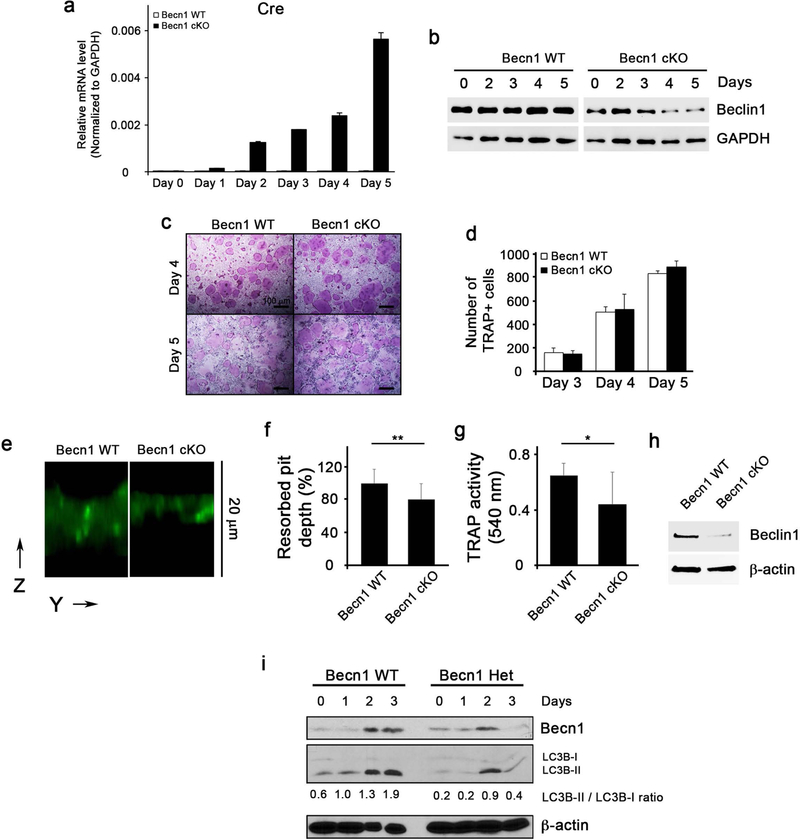
(a) qRT-PCR for Cre gene expression during osteoclast differentiation in BMMs isolated from Becn1 WT or cKO mice. (b) Western blotting for Beclin1 expression level during osteoclast differentiation. (c) TRAP staining following RANKL/M-CSF treatment on BMMs for 4 and 5 days. Bar = 100 μm. (d) Quantification of TRAP+ cells. (e) Dentin slice assay following RANKL/M-CSF treatment on BMMs isolated from Becn1 WT or cKO mice. Cells were removed with cotton swabs, and stained with FITC-wheat germ agglutinin (WGA). The resorbed pit depth on dentin slice was visualized in z stack by confocal microscopy. The image was reconstructed by Amira software. (f) Normalized resorbed pit depth excavated by Becn1 WT and cKO osteoclasts. Three different experiments (cell number = 100) were normalized to the average depth. (g) TRAP activity measurement from cell culture supernatant from dentin slices. (h) Western blotting for Beclin1 expression level from osteoclasts harvested from the dentin slices. (i) Western blotting against Beclin1, LC3B and β-actin during RANKL-induced osteoclast differentiation in BMMs from Becn1+/+ (WT) or Becn1+/− (Het) mice.
Chondrocyte differentiation is impaired in Becn1 cKO mice.
Unexpectedly, deletion of Becn1 in CtsK expressing cells showed reduced trabecular bone mass in femurs (Fig. 4i and 4k), which was associated with disorganized osteocartilaginous structures and complete absence of growth plate remnants by 6th month. CtsK is also heavily expressed in the hypertrophic zone in the growth plate (Supplementary Fig. 4). Further characterization of the growth plate revealed that hypertrophic zone was significantly reduced in Becn1 cKO mice (Fig. 6a, b, c, d). Consistent with this observation, expression of MMP13, an indicative of terminal hypertrophic zone, was absent in Becn1 cKO mice, whereas Sox9 and ColX expressions were not affected (Fig. 6e). Consistent with this finding, chondrocyte differentiation was also impaired (Fig. 6f, g), suggesting that reduced trabecular bone mass is associated with impaired growth plate formation and chondrocyte differentiation.
Figure 6. Chondrocyte differentiation is impaired in Becn1 cKO mice.

(a) Safranin O staining at the cartilage of Becn1 WT or cKO mice harvested at week 4, 8, and 24. (b) Quantification of the columnal zone at week 4 and 8. (c) Quantification of the hypertrophic zone at week 4 and 8. (d) Quantification of the total zone at week 4 and 8. (e) Immunohistochemical staining of Sox9, ColX, MMP13, and MMP9 at the cartilage. Bar = 100 µm. (f) Alcian blue staining of the chondrocytes isolated from the Becn1 WT and cKO mice at week 1, 2, and 3. (g) Quantification of Alcian blue staining.
DISCUSSION
In this study, we examined the role of Beclin1, one of the autophagy-related proteins and a known autophagy initiator that forms the initial autophagosome complex (26), and provide direct evidence that autophagy is a critical event that occurs during osteoclast differentiation and functions. We demonstrated that ubiquitination of Beclin1 plays a pivotal role in initiating autophagy by RANKL stimulation during osteoclastogenesis, and showed that targeted deletion of Becn1 in osteoclasts using CtsK-Cre exhibited thickening of the cortical bone in mice. Unexpectedly, these mice exhibited diminished trabecular bone mass due to defect in chondrocyte differentiation, suggesting the complexity of autophagy involvement in multiple bone-related cells.
Previous study reported that deficiency in autophagy proteins (e.g., Atg4b, Atg5, Atg7 and Lc3) impairs bone resorptive functions of osteoclasts but not osteoclast development and differentiation (14). These autophagy-related proteins, however, are primarily associated with phagophore expansion rather than the initiation of autophagy (5,38). Indeed, studies showed that the formation of autophagosomes has been shown to occur in the absence of Atg5 and Atg7 (39). Beclin1, on the other hand, plays a pivotal role in initiation of phagophore formation by recruiting autophagy-related protein at the phagophore-assembly site (5). In the absence of Beclin1, the presence of autophagosomes is very few or none (40). Our study suggested that Beclin1 is immediately ubiquitinated by TRAF6 as early as 2 hours after RANKL stimulation (Fig. 5) and that BMMs from Becn1 f/f mice infected with lentivirus-Cre exhibited suppression of multinucleated osteoclasts (Fig. 2). It is also noteworthy that when overexpression or deletion of Becn1 prior to RANKL treatment caused increased or decreased osteoclast numbers, respectively (Fig 1 or Fig 2). In sharp contrast, CtsK-mediated deletion of Becn1 occurred as early as day 3 post RANKL treatment, which only impaired osteoclast functions but not differentiation (Fig. 5). These data suggests that the RANKL-induced autophagy activation in osteoclasts is an early event that is required for not only osteoclast’s functions but also osteoclast’s differentiation.
It is interesting to note that notable increases in autophagy-related gene expression occurred around day 2 upon RANKL stimulation. Multi-nucleation and vacuolization also start occurring in the osteoclast pre-cursors around day 2 in vitro. Such observation suggests that significant increase in expression of autophagy-related genes may be associated with the structural alterations including enlargement and vacuolization of maturing osteoclasts in preparation for multi-nucleation to produce large numbers of autolysosomes to release H+ and proteolytic enzymes.
Our in vivo studies showed that deletion of Becn1 by crossbreeding Becn1f/f mice with Cre-expressing mice driven by RANK promoter, an early expressing gene in osteoclasts, resulted in only 2 viable Becn1f/f;RANKCre/+ mice that were prematurely died in 2 days after birth. Such results suggest that Beclin1 may play important roles in other RANK-expressing cells during development (35). On the other hand, deletion of Becn1 by crossbreeding Becn1f/f mice with Cre-expressing mice driven by CtsK promoter, a late expressing gene in osteoclasts, resulted in viable mice. Consistent with our in vitro findings, these mice exhibited thickened cortical bone, which is a direct suggestive of impaired osteoclast functions.
Intriguingly, these Becn1 cKO mice did not exhibit a classic osteopetrotic phenotype in trabecular bone; although cortical bone thickness was increased, trabecular bone mass was decreased (Fig. 4). Previous study suggested that CtsK are also expressed in the hypertrophic zone in the growth plate (41). Our further examinations confirmed that CtsK is expressed at the edge of the hypertrophic zone and that growth plate is severely disrupted (Supplementary Fig. 4). As such, the lack of trabecular bone may suggest that Becn1 may play a key role in CtsK-expressing cells other than osteoclast precursors at the growth plate regions, such as hypertrophic chondrocytes (42) (43). Indeed, autophagy is known to play a critical role in the growth plate(44). Therefore, this such seemingly paradoxical bone phenotype of our Becn1 cKO mice is due to strong expression of CtsK in both osteoclasts and chondrocytes at their late stages of differentiation such that increased cortical bone thickness is due to the inhibitory effects of beclin1 knockout in osteoclasts, while decreased trabecular bone amounts may be due to the inhibitory effects of beclin1 knockout in chondrocytes. It is noteworthy that the absence of changes in serum TRAP and CTX-I (Supplementary Figure 3) does not seem to support the reductions observed in osteoclast numbers in the cortical surface in vivo (Figure 4). This discrepancy may be due as yet unidentified role of Beclin1 in a site-specific manner. The precise role of Beclin1 in chondrocytes warrants closer examination.
In the context of cancers, Beclin1 is known as a tumor suppressor (45). Initially identified as a haplo-insufficient tumor suppressor (46,47), Beclin1 mediates a crosstalk between autophagy and apoptosis pathways by interacting with anti-apoptotic proteins such as Bcl-2, Bcl-XL and Mcl-1 through BH3 domain (48). These anti-apoptotic proteins becomes phosphorylated and disassociated from Beclin1 upon activation of JNK1 pathway (49). Not surprisingly, these anti-apoptotic proteins are known to suppress the functions of osteoclasts (50,51). Because JNK pathway is also activated upon RANKL stimulation, it is tempting to speculate that the activated JNK pathway may function in osteoclastogenesis by dissociating Beclin1 from these anti-apoptotic proteins, causing Beclin1 to be “primed” for the sustained autophagy activation during osteoclastogenesis.
In conclusion, we showed that RANKL, a potent activator of osteoclast differentiation, induces autophagy through Beclin1, an important autophagy-initiating protein that is directly becomes ubiquitinated and activated by TRAF6. Based on our study, we propose that, unlike traditionally known function of autophagy which becomes activated under stressed condition such as starvation to renew and recycle macromolecules for energy preservation, autophagy in osteoclasts are potently activated not to recycle but to mediate structural alterations that eventually lead to active purge of organelles such as lysozymes that contain H+ ions and degrading proteins to extracellular bone matrixes. Further examination is required to elucidate functional roles of different autophagy-related proteins in osteoclast biology and to examine how other known signaling pathways activated by RANKL are associated with the autophagy pathway.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by R01DE023348 (R.H.K.) and R01DE019412 (C.-Y.W.) from NIDCR/NIH. The authors thank Dr. Binfeng Lu (University of Pittsburgh School of Medicine) for providing Beclin1f/f mice, Dr. Yasuhiro Kobayashi (Matsumoto Dental University, Japan) for RANK-Cre mice, and Dr. Nakamura Takashi (Keio University, Japan) for CtsK-Cre mice. We also thank the UCLA Translational Procurement Core Laboratory and Dr. Renata Cristina Pereira for their expedited and cooperative service.
REFERENCES
- 1.Katagiri T, Takahashi N. Regulatory mechanisms of osteoblast and osteoclast differentiation. Oral Dis May 2002;8(3):147–59. [DOI] [PubMed] [Google Scholar]
- 2.Boyce BF, Rosenberg E, de Papp AE, Duong le T. The osteoclast, bone remodelling and treatment of metabolic bone disease. Eur J Clin Invest Dec 2012;42(12):1332–41. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med Jan 17 2000;191(2):275–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blair HC, Teitelbaum SL, Ghiselli R, Gluck S. Osteoclastic bone resorption by a polarized vacuolar proton pump. Science Aug 25 1989;245(4920):855–7. [DOI] [PubMed] [Google Scholar]
- 5.Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol Jul 2013;15(7):713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res Jan 2014;24(1):24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 2010;3(123):ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J Apr 9 2008;27(7):1110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyce BF. Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res Oct 2013;92(10):860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiu Y, Xu H, Zhao C, Li J, Morita Y, Yao Z, et al. Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation. J Clin Invest Jan 2014;124(1):297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnett TR, Gibbons DC, Utting JC, Orriss IR, Hoebertz A, Rosendaal M, et al. Hypoxia is a major stimulator of osteoclast formation and bone resorption. J Cell Physiol Jul 2003;196(1):2–8. [DOI] [PubMed] [Google Scholar]
- 12.Chung YH, Jang Y, Choi B, Song DH, Lee EJ, Kim SM, et al. Beclin-1 is required for RANKL-induced osteoclast differentiation. J Cell Physiol Dec 2014;229(12):1963–71. [DOI] [PubMed] [Google Scholar]
- 13.Chung YH, Yoon SY, Choi B, Sohn DH, Yoon KH, Kim WJ, et al. Microtubule-associated protein light chain 3 regulates Cdc42-dependent actin ring formation in osteoclast. Int J Biochem Cell Biol Jun 2012;44(6):989–97. [DOI] [PubMed] [Google Scholar]
- 14.DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell Nov 15 2011;21(5):966–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y, Chen G, Zhang W, Xu N, Zhu JY, Jia J, et al. Autophagy regulates hypoxia-induced osteoclastogenesis through the HIF-1alpha/BNIP3 signaling pathway. J Cell Physiol Feb 2012;227(2):639–48. [DOI] [PubMed] [Google Scholar]
- 16.Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc 2008;3(8):1253–60. [DOI] [PubMed] [Google Scholar]
- 17.Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol Jul 2013;15(7):741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell Mar 2008;19(3):797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sohn S, Park Y, Srikanth S, Arai A, Song M, Yu B, et al. The Role of ORAI1 in the Odontogenic Differentiation of Human Dental Pulp Stem Cells. J Dent Res Nov 2015;94(11):1560–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arai A, Mizoguchi T, Harada S, Kobayashi Y, Nakamichi Y, Yasuda H, et al. Fos plays an essential role in the upregulation of RANK expression in osteoclast precursors within the bone microenvironment. J Cell Sci Jun 15 2012;125(Pt 12):2910–7. [DOI] [PubMed] [Google Scholar]
- 21.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods Jul 2012;9(7):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takami M, Suda K, Sahara T, Itoh K, Nagai K, Sasaki T, et al. Involvement of vacuolar H+ -ATPase in incorporation of risedronate into osteoclasts. Bone Apr 2003;32(4):341–9. [DOI] [PubMed] [Google Scholar]
- 23.Duran A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP, et al. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell Feb 2004;6(2):303–9. [DOI] [PubMed] [Google Scholar]
- 24.Lee HM, Shin DM, Yuk JM, Shi G, Choi DK, Lee SH, et al. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J Immunol Jan 15 2011;186(2):1248–58. [DOI] [PubMed] [Google Scholar]
- 25.Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy Mar 2014;10(3):431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M, et al. Beclin1: a role in membrane dynamics and beyond. Autophagy Jan 2012;8(1):6–17. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J Mar 15 2001;20(6):1271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev Apr 15 1999;13(8):1015–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells Jun 1999;4(6):353–62. [DOI] [PubMed] [Google Scholar]
- 30.Yasui T, Kadono Y, Nakamura M, Oshima Y, Matsumoto T, Masuda H, et al. Regulation of RANKL-induced osteoclastogenesis by TGF-beta through molecular interaction between Smad3 and Traf6. J Bone Miner Res Jul 2011;26(7):1447–56. [DOI] [PubMed] [Google Scholar]
- 31.Bai S, Zha J, Zhao H, Ross FP, Teitelbaum SL. Tumor necrosis factor receptor-associated factor 6 is an intranuclear transcriptional coactivator in osteoclasts. J Biol Chem Nov 7 2008;283(45):30861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J Apr 3 2000;19(7):1576–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng Y, Zhao H, Luderer HF, Epple H, Faccio R, Ross FP, et al. The LIM protein, Limd1, regulates AP-1 activation through an interaction with Traf6 to influence osteoclast development. J Biol Chem Jan 5 2007;282(1):39–48. [DOI] [PubMed] [Google Scholar]
- 34.Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J Biol Chem Feb 9 2007;282(6):4102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell Sep 29 2000;103(1):41–50. [DOI] [PubMed] [Google Scholar]
- 36.Gowen M, Lazner F, Dodds R, Kapadia R, Feild J, Tavaria M, et al. Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J Bone Miner Res Oct 1999;14(10):1654–63. [DOI] [PubMed] [Google Scholar]
- 37.Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is essential for osteoclast and lymph node development. Genes Dev Sep 15 1999;13(18):2412–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy Apr 2012;8(4):445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature Oct 1 2009;461(7264):654–8. [DOI] [PubMed] [Google Scholar]
- 40.Xing S, Zhang Y, Li J, Zhang J, Li Y, Dang C, et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy Jan 2012;8(1):63–76. [DOI] [PubMed] [Google Scholar]
- 41.Rantakokko J, Aro HT, Savontaus M, Vuorio E. Mouse cathepsin K: cDNA cloning and predominant expression of the gene in osteoclasts, and in some hypertrophying chondrocytes during mouse development. FEBS Lett Sep 16 1996;393(2–3):307–13. [DOI] [PubMed] [Google Scholar]
- 42.Soderstrom M, Salminen H, Glumoff V, Kirschke H, Aro H, Vuorio E. Cathepsin expression during skeletal development. Biochim Biophys Acta Jul 07 1999;1446(1–2):35–46. [DOI] [PubMed] [Google Scholar]
- 43.Tchetina EV, Mwale F, Poole AR. Changes in gene expression associated with matrix turnover, chondrocyte proliferation and hypertrophy in the bovine growth plate. Acta Naturae Jul 2014;6(3):89–97. [PMC free article] [PubMed] [Google Scholar]
- 44.Kang X, Yang W, Feng D, Jin X, Ma Z, Qian Z, et al. Cartilage-Specific Autophagy Deficiency Promotes ER Stress and Impairs Chondrogenesis in PERK-ATF4-CHOP-Dependent Manner. J Bone Miner Res Oct 2017;32(10):2128–41. [DOI] [PubMed] [Google Scholar]
- 45.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ Apr 2011;18(4):571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A Dec 9 2003;100(25):15077–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu X Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. Journal of Clinical Investigation 2003;112(12):1809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J May 16 2007;26(10):2527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell Jun 20 2008;30(6):678–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lagasse E, Weissman IL. Enforced expression of Bcl-2 in monocytes rescues macrophages and partially reverses osteopetrosis in op/op mice. Cell Jun 27 1997;89(7):1021–31. [DOI] [PubMed] [Google Scholar]
- 51.Iwasawa M, Miyazaki T, Nagase Y, Akiyama T, Kadono Y, Nakamura M, et al. The antiapoptotic protein Bcl-xL negatively regulates the bone-resorbing activity of osteoclasts in mice. J Clin Invest Oct 2009;119(10):3149–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.