

 This work is licensed under a
This work is licensed under a Abstract
Context
Idiopathic intracranial hypertension (IIH) is a disease of raised intracranial pressure (ICP) of unknown etiology. Reductions in glucocorticoid metabolism are associated with improvements in IIH disease activity. The basal IIH glucocorticoid metabolism is yet to be assessed.
Objective
The objective of this study was to determine the basal glucocorticoid phenotype in IIH and assess the effects of weight loss on the IIH glucocorticoid phenotype.
Design
A retrospective case–control study and a separate exploratory analysis of a prospective randomized intervention study were carried out.
Methods
The case–control study compared female IIH patients to BMI, age, and sex-matched controls. In the randomized intervention study, different IIH patients were randomized to either a community weight management intervention or bariatric surgery, with patients assessed at baseline and 12 months. Glucocorticoid levels were determined utilizing 24-h urinary steroid profiles alongside the measurement of adipose tissue 11β-HSD1 activity.
Results
Compared to control subjects, patients with active IIH had increased systemic 11β-hydroxysteroid dehydrogenase (11β-HSD1) and 5α-reductase activity. The intervention study demonstrated that weight loss following bariatric surgery reduced systemic 11β-HSD1 and 5α-reductase activity. Reductions in these were associated with reduced ICP. Subcutaneous adipose tissue explants demonstrated elevated 11β-HSD1 activity compared to samples from matched controls.
Conclusion
The study demonstrates that in IIH, there is a phenotype of elevated systemic and adipose 11β-HSD1 activity in excess to that mediated by obesity. Bariatric surgery to induce weight loss was associated with reductions in 11β-HSD1 activity and decreased ICP. These data reflect new insights into the IIH phenotype and further point toward metabolic dysregulation as a feature of IIH.
Introduction
Idiopathic intracranial hypertension (IIH) is a disease characterized by raised intracranial pressure (ICP), papilledema, and disabling headaches (1, 2). IIH predominantly affects obese women of the reproductive age, and its incidence is increasing in parallel with the obesity epidemic (3, 4). Disease modification of IIH is through weight management, and the IIH weight trial (IIH:WT) provided evidence that weight loss through bariatric surgery inferred long-term remission of ICP as compared to a community weight management (CWM) intervention (5, 6, 7).
Defining the pathophysiology of IIH and identifying biomarkers to guide diagnosis and target management were deemed top priorities for research by both healthcare professionals and people with IIH in a priority-setting partnership (8). IIH has long been thought to be a disease isolated to the CNS; however, recent evidence has demonstrated systemic metabolic features linked to but in excess of those seen in simple obesity (9, 10, 11). IIH patients have truncal obesity, an increased risk of cardiovascular disease and type 2 diabetes mellitus in addition to being more insulin resistant and having altered adipose tissue function with a greater magnitude of derangement than that mediated by obesity (10, 12, 13). Additionally, IIH patients have a unique phenotype of serum and cerebrospinal fluid (CSF) androgen excess, highlighting altered steroid metabolism (9).
Glucocorticoids (GC) and 11β-hydroxysteroid dehydrogenase (11β-HSD) are associated with disease activity in IIH. Pre-receptor corticosteroid availability is mediated by the bidirectional enzyme, 11β-HSD (14). This enzyme has two isoforms: 11β-HSD1 which acts predominantly as an oxoreductase and activates cortisol from cortisone and 11β-HSD2, which inactivates cortisol to cortisone. 11β-HSD1 is expressed in a wide variety of tissues but its principal role in humans is mediating local cortisol availability where, for example, in the adipose tissue, it can promote adipocyte differentiation and drive hepatic glucose output (15). In IIH, 11β-HSD1 activity is decreased in association with a reduction in ICP following a therapeutic diet (16). Change in systemic 11β-HSD1 activity correlates significantly with change in ICP. A randomized controlled trial in IIH demonstrated that specific therapeutic inhibition of 11β-HSD1 (AZD4017) reduces ICP (17). Cognitive impairment has been associated with GC excess (18, 19). Cognitive dysfunction is documented in patients with IIH and has been shown to significantly correlate with serum cortisol levels, a known driver of cognitive impairment (20, 21). Normalizing serum cortisol through weight loss improves the cognitive impairment in IIH (6, 20).
While these data suggest that cortisol secretion and its metabolism by 11β-HSD1 activity are relevant in IIH etiology, it is unknown if IIH patients have altered systemic 11β-HSD1 activity compared to weight-matched controls. Indeed, in there is no consensus on what simple obesity does to systemic 11β-HSD1 activity (15, 22, 23, 24). Furthermore, it is unclear what may be driving altered 11β-HSD1 activity in IIH.
Here, the urinary GC phenotype in IIH is defined, the role of bariatric surgery on the urinary GC phenotype is assessed, and the contribution of adipose tissue to the IIH GC phenotype is understood.
Methods
Study conduct
IIH subjects were identified from multiple UK centres, and samples were collected following informed, written consent. The trials received ethical approval for the IIH and control cohorts from the York and Humber-Leeds West Research Ethic committee (REC) (13/YH/0366), Dudley local REC (06/Q2702/64), and the Black Country REC (14/WM/0011) (collected as part of three separate ethical applications).
Control patients were recruited via advertisement, where sample collection occurred following informed written consent. Sample collection was approved by the South Birmingham Local REC and the Black Country REC (14/WM/0011). Control patients for adipose tissue experiments were recruited from elective National Health Service bariatric surgery procedures following written informed consent and was approved by the Black Country REC (14/WM/0011).
Study population
Adult (18–55 years) female IIH patients with active IIH (papilloedema ≥Frisén grade 1 and lumbar puncture opening pressure (LP OP) ≥ 25 centimeters of cerebrospinal fluid (cmCSF) on the date of research assessment visit) diagnosed in line with the International IIH Guideline criteria were recruited (5). IIH patients at any stage of their active disease were included. Control patients met the same inclusion criteria as the IIH patients, with the absence of an IIH diagnosis.
Exclusion criteria
Exclusion criteria for all patients included receiving hormone manipulating medication (including contraceptives and those exposed to GC therapy in the last 3 months), significant comorbidities including known endocrinopathies, and the inability to give informed consent. Additionally, IIH patients were excluded if they were pregnant during the visit.
Assessments
All participants underwent detailed medical history and examination. Anthropometric data were recorded. Lumbar punctures were carried out in all IIH patients and conducted in the left lateral decubitus position with knees bent at a 90° angle or more and (LP OP) recorded. Twenty-four-hour urine samples for urinary steroid metabolite profiling were also provided by patients in attendance at the research facility. Urine samples were stored at −80°C and analyzed after a maximum of one freeze––thaw cycle. Patients were fasted overnight for all visits.
Case–control study
A case–control study comparing IIH with matched controls was conducted to determine the urinary GC metabolome. The control subject cohort was group matched (retrospectively) to the IIH population for age, gender, and BMI.
Weight loss study
This was a sub-study of the IIH:WT (a randomized controlled trial comparing CWM intervention to bariatric surgery in IIH), the full protocol and primary results have been published elsewhere (NCT02124486) (6, 25). Patients were randomized 1:1 to either a CWM intervention (utilizing weight watchers) or a bariatric surgery program (laparoscopic adjustable gastric banding, Roux-en-Y gastric bypass or laparoscopic sleeve gastrectomy). Randomization was done by a computer-generated randomization list via a phone line at the Birmingham Clinical Trials Unit. Patient visits occurred at baseline and 12 months after baseline as per the previously published protocol (25). This sub-study evaluated the steroid metabolome changes in 24-h urine samples collected at baseline and 12 months after baseline, following intervention. Patients randomized to surgery had a median time from randomization to bariatric surgery of 4.4 months (range, 2.2–10.3 months).
Urinary steroid profiling
Systemic steroid metabolism in 24-h urine samples was profiled using gas chromatography–mass spectrometry and liquid chromatography–mass spectrometry (LC-MS) as previously described (26). 11β-HSD1 activity was assessed using the ratio of the tetrahydrometabolites of cortisol (5α-THF+THF) to tetrahydrocortisone (THE). 11β-HSD2 activity was assessed via the free urinary cortisol to cortisone ratio (23). Total GC metabolite excretion was assessed as the sum of 5α-THF + THF + THE + cortolones + cortols + cortisol + cortisone and provided an accurate marker of 24-h cortisol secretion rate (23).
HOMA2-IR assessment
Fasting insulin was measured using commercially available assays (Mercodia), according to the manufacturer’s instructions. Homeostatic model assessment (HOMA2)-IR was calculated using the program HOMA calculator v2.2.3 (https://www.dtu.ox.ac.uk/homacalculator/).
Leptin ELISA
Leptin was quantified in serum using the human leptin DuoSet ELISA (DY-398, Bio-Techne, Minneapolis, MN, USA). The ELISA was carried out according to the manufacturer’s instructions using the recommended ancillary kit (Bio-Techne, DY008). Serum was diluted 1:100 in reagent diluent. Samples were run in duplicate.
Interleukin 6 ELISA
Interleukin 6 (IL-6) was quantified in serum by ELISA as per manufacturer's instructions using the Human IL-6 DuoSet ELISA (R&D Systems, Cat no. DY206, UK). The ELISA was carried out according to the manufacturer’s instructions using the recommended ancillary kit (Bio-Techne, DY008). Samples were run in duplicate.
Adipose tissue collection
At baseline, subcutaneous adipose tissue was collected from IIH and control subjects following an overnight fast (from midnight). Subcutaneous adipose tissue was biopsied and either placed immediately in RNA later or into phenol-free DMEM/F12 (Thermofisher), with no antibiotics.
Ex vivo adipose 11β-hydroxysteroid dehydrogenase activity
Subcutaneous abdominal adipose tissue explants (~100 mg, tested in triplicate) were incubated in phenol-free DMEM/F12 containing 100 nM cortisone for 24 h in glass tubes. Control experiments were also conducted with identical conditions but no adipose tissue. Steroid conversion was quantified using LC-MS as previously described and normalized to explant mass (27, 28).
Statistical analysis
Statistical analysis was performed using Graphpad Prism 9 (Graphpad Software Inc). Data are presented as mean ± s.d. unless otherwise stated. Data normality was assessed by a Shapiro–Wilk normality test. Where data were normally distributed, unpaired two-tailed t-tests (equal variance) or Welch’s test (unequal variance) were employed, whereas non-parametric data were assessed via Mann–Whitney U-test. For multigroup comparisons, two-way repeated-measures ANOVA followed by Sidak’s multiple comparisons test were utilized. Spearman’s rank correlation coefficient (ρ) and Pearson’s correlation coefficient (r) were used for assessing correlations in the IIH cohorts. Where data points are missing, data were not imputed. We did not correct for multiple comparisons as this would have increased the likelihood of type II errors. Results were judged significant at P < 0.05.
Results
Patient characteristics
Controls (n = 17) and IIH (n = 27) patients (all female) were matched for age (41.7 ± 4.2 vs 39.4 ± 5.9 years, P = 0.18) and BMI (34.9 ± 3.8 vs 38.4 ± 8.4 kg/m2, P = 0.1). IIH patients had a mean ICP of 35.1 ± 4.6 cmCSF (Table 1). There was no difference in the degree of insulin resistance between the groups (HOMA2-IR: 1.7 ± 1.1 vs 1.8 ± 2.0, P = 0.8) (Table 1).
Table 1.
Urinary steroid profiling characteristics in control vs IIH. Data are presented as mean ± s.d.
| Characteristics | Control (n = 17) | IIH (n = 27) |
|---|---|---|
| Age (years) | 41.7 ± 4.2 | 39.5 ± 6.0 |
| BMI (kg/m2) | 34.8 ± 3.8 | 38.4 ± 8.4 |
| ICP (cm CSF) | N/A | 40.6 ± 3.7 |
| Sex (female, %) | 100% | 100% |
| Fasting insulin (mIU/L) | 13.5 ± 9.6 | 13.39 ± 7.6 |
| HOMA2-IR | 1.7 ± 1.1 | 1.8 ± 2.0 |
| Urine (µg/24 h) | ||
| 5α-THF | 1177 ± 681 | 1421 ± 954 |
| THF | 1472 ± 817 | 1331 ± 597 |
| THE | 2960 ± 1581 | 2662 ± 1418 |
| Cortisol (F) | 46 ± 20 | 71 ± 36** |
| Cortisone (E) | 85 ± 32 | 107 ± 60 |
| Total GC metabolites | 8532 ± 4068 | 8039 ± 3807 |
| Derivative measurements | ||
| (5α-THF+THF/THE) | 0.9 1 ±0.1 | 1.13 ± 0.34* |
| F/E | 0.50 ± 0.16 | 0.53 ± 0.14 |
| 5α-THF/THF | 0.84 ± 0.33 | 1.20 ± 0.69* |
*P < 0.05, **P < 0.01.
cmCSF, centimetres of cerebrospinal fluid; GC, glucocorticoid; HOMA, homeostatic model assessment; ICP, intracranial pressure; THE, tetrahydrocortisone; THF, tetrahydrocortisol; 5α-THF, 5α-tetrahydrocortisol.
Basal glucocorticoid metabolome in IIH
We sought to investigate the systemic steroid metabolome in IIH patients using 24-h urine collections. Mass spectrometry analysis revealed a significant increase in systemic 11β-HSD1 activity in IIH (IIH 1.131 ± 0.34 vs control 0.91 ± 0.21 THF+THFα/THE; P = 0.019, Fig. 1A). Linked to this, IIH patients had increased urinary cortisol levels (71.2 ± 36.8 vs 46.5 ± 20.1 µg/24 h, P = 0.0069, Fig. 1D). Markers of 11β-HSD2 activity (Fig. 1B) and total GC excretion were not altered (Fig. 1C). As inferred from the urinary 5α-THF/THF ratio, we also demonstrated increased activity of the cortisol and testosterone metabolizing enzyme 5α-reductase in IIH (0.84 ± 0.33 vs 1.20 ± 0.69, P = 0.03) (Fig. 1E). All other measured steroids were found to be unchanged, with the exception of pregnanediol which was increased in IIH patients (391.1 ± 290.2 vs 155.5 ± 108.9 µg/24 h, P = 0.0018) (Supplementary Table 1, see section on supplementary materials given at the end of this article).
Figure 1.
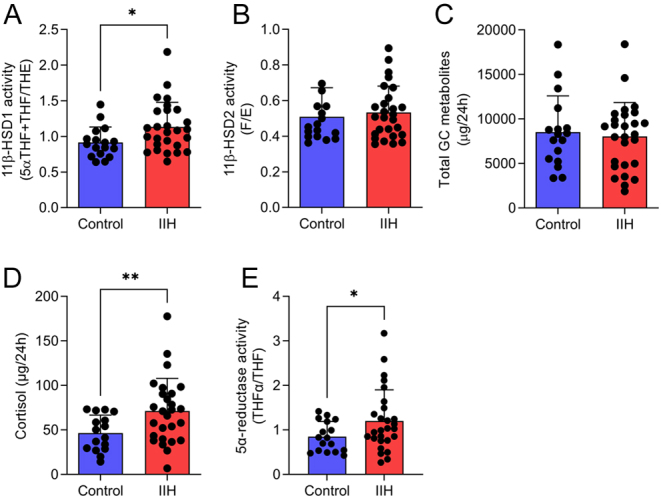
Altered glucocorticoid metabolism in patients with IIH. Twenty-four-hour urine steroid metabolomic assessment in control (17) and IIH patients (27). (A) 11β-HSD1 activity was denoted by 5α-THF+THF/THE. (B) 11β-HSD2 activity was denoted by urinary free cortisol/cortisone (F/E). (C) Total glucocorticoid metabolite excretion. (D) Cortisol secretion. (E) 5α-reductase activity. Data are presented as mean ± s.d., Mann–Whitney U-test for A and B, t-test for (C) and t-test with Welch’s correction for (D) and (E). *P < 0.05, **P < 0.01. IIH, idiopathic intracranial hypertension; 11β-HSD1, 11β-hydroxysteroid dehydrogenase; 5α-THF+THF/THE, 5α-tetrahydocortisol+tetrahydrocortisol/tetrahydrocortisone. A full color version of this figure is available at https://doi.org/10.1530/EJE-22-0108.
Markers of 11β-HSD1 activity (r = −0.13, P = 0.4) and 5α-reductase activity (r = −0.11, P = 0.59) did not correlate with ICP at baseline. Additionally, we demonstrated no correlation between 11β-HSD1 activity and BMI (ρ = −0.1, P = 0.6), fasting insulin (ρ = 0.29, P = 0.16) or HOMA2-IR (ρ = 0.22, P = 0.29). We observed no correlation between 5α-reductase activity and BMI (ρ = −0.25, P = 0.2), fasting insulin (ρ = 0.06, P = 0.77), and HOMA2-IR (ρ = 0.02, P = 0.87).
Weight loss study
The IIH:WT established that weight loss following bariatric surgery led to significant sustained reduction in ICP in IIH (6). In this sub-study, 24-h urine samples were collected at baseline and at 12 months (16 from the CWM intervention and 13 from the bariatric surgery arm). Among these participants at baseline, both IIH cohorts were well matched for gender (100% female), BMI (44.7 ± 8.2 vs 44.0 ± 7.3 kg/m2, P = 0.79), age (31.9 ± 7.9 vs 32.0 ± 7.5 years, P = 0.79), and ICP (32.6 ± 4.8 vs 35.0 ± 5.1 cmCSF, P = 0.18).
Twelve months following enrollment, the participants randomized to surgery had a lower BMI (44.1 ± 8.8 vs 34.8 ± 8.0 kg/m2, P = 0.007) and had a lower ICP (30.4 ± 5.6 vs 23.5 ± 6.6, P = 0.008) compared to participants randomized to the CWM intervention (Table 2).
Table 2.
Urinary steroid profiling characteristics table following intervention. Data are presented as mean ± s.d.. There was no statistical significance between CWM and surgery at baseline.
| Baseline | 12 Months | |||
|---|---|---|---|---|
| CWM (16) | Surgery (13) | CWM (16) | Surgery (13) | |
| Age (years) | 31.8 ± 7.9 | 32.6 ± 7.4 | ||
| BMI (kg/m2) | 44.7 ± 8.1 | 44.0 ± 7.6 | 44.1 ± 8.8 | 34.0 ± 8.0**,#### |
| ICP (cmCSF) | 32.6 ± 4.8 | 35.2 ± 5.1 | 30.3 ± 5.6 | 23.6 ± 6.6**,#### |
| Sex (female, %) | 100% | 100% | ||
| Fasting insulin (mIU/L) | 17.4 ± 7.6 | 10.7 ± 3.8 | 14.7 ± 6.1$$ | 4.3 ± 4.3*,### |
| HOMA2-IR | 1.9 ± 0.8 | 1.5 ± 0.6 | 1.1 ± 0.4 $$ | 0.4 ± 0.4*, ### |
| Leptin (ng/mL) | 89.3 ± 37.0 | 76.8 ± 32.2 | 79.3 ± 38.8 | 31.9 ± 24.0***, #### |
| IL-6 (pg/mL) | 5.8 ± 2.7 | 6.3 ± 2.1 | 5.4 ± 1.9 | 5.0 ± 2.1 |
| Urine (µg/24 h) | ||||
| 5α-THF | 518 ± 537 | 324 ± 304 | 897 ± 641$ | 403 ± 304* |
| THF | 2305 ± 1708 | 2058 ± 1406 | 2640 ± 1309 | 1967 ± 899 |
| THE | 3435 ± 2121 | 2666 ± 1741 | 5055 ± 2231$ | 3780 ± 1836 |
| Cortisol (F) | 69 ± 51 | 63 ± 47 | 65 ± 27 | 66 ± 25 |
| Cortisone (E) | 90 ± 70 | 73 ± 41 | 118 ± 52 | 106 ± 55 |
| Total GC metabolites | 10057 ± 5881 | 7502 ± 4806 | 14138 ± 6597 | 12195 ± 6026 |
| Derivative measurements | ||||
| 5α-THF+THF/THE | 0.799 ± 0.14 | 0.933 ± 0.22 | 0.699 ± 0.13 | 0.660 ± 0.16#### |
| F/E | 0.812 ± 0.56 | 0.816 ± 0.24 | 0.563 ± 0.11$$$$ | 0.683 ± 0.22# |
| 5α-THF/THF | 0.231 ± 0.10 | 0.188 ± 0.14 | 0.32 ± 0.13$$ | 0.22 ± 0.09 |
*Statistical comparisons between CWM and surgery at 12 months; #Statistical comparison between surgery baseline and surgery 12 months; $Statistical comparison between CWM baseline and surgery 12 months; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
cmCSF, centimeters of cerebrospinal fluid; GC, glucocorticoid; HOMA, homeostatic model assessment; ICP, intracranial pressure; IL-6, interleukin 6; THE, tetrahydrocortisone; THF, tetrahydrocortisol; 5α-THF, 5α-tetrahydrocortisol.
In the bariatric surgery cohort, 11β-HSD1 activity was significantly reduced at 12 months compared to baseline (0.93 ± 0.2 vs 0.66 ± 0.1, P < 0.0001, Fig. 2A) while there was no significant reduction in the CWM arm. At 12 months, the change (Δ) in 11β-HSD1 activity from baseline was significantly greater in the bariatric surgery cohort compared to those in the CWM arm (−0.1 ± 0.15 vs −0.27 ± 0.19 Δ11β-HSD1 activity, P = 0.013, Supplementary Table 2). The change in 11β-HSD1 activity was significantly associated with change in ICP in all groups (r = 0.43, P = 0.02, Fig. 2B). 11β-HSD2 activity was reduced following bariatric surgery (0.82 ± 0.2 vs 0.68 ± 0.2, P = 0.013) and within the CWM arm (0.81 ± 0.2 vs 0.56 ± 0.1, P < 0.0001, Fig. 2C) with no difference between arms at 12 months (−0.25 ± 0.12 vs −0.13 ± 0.21 Δ11β-HSD2, P = 0.09, Supplementary Table 2). 5α-reductase activity remained unchanged at 12 months in the bariatric surgery group (0.18 ± 0.1 vs 0.22 ± 0.1, P = 0.39); however, the activity was noted to be increased in the CWM arm (0.23 ± 0.1 vs 0.32 ± 0.13, P = 0.0012, Fig. 2D) but there was no difference in the change at 12 months (0.09 ± 0.1 vs 0.03 ± 0.08 Δ5α-reductase activity, P = 0.1, Supplementary Table 2). Change in 5α-reductase activity was correlated positively with change in ICP (r = 0.39, P = 0.03, Fig. 2E). Changes in other analyzed steroids can be found in Supplementary Table 2.
Figure 2.

IIH urinary steroid metabolome following weight loss. Twenty-four-hour urine steroid metabolome profiling at baseline and 12 months in diet (16) and surgical (13) IIH patients. (A) Change in 11β-HSD1, (B), scatter graph of change in 11β-HSD1 activity vs change in intracranial pressure (ICP), (C) change in 11β-HSD2 activity, (D) change in 5α-reductase activity, and (E) scatter graph of change in 11β-HSD1 activity vs change in ICP. Two-way repeated measures ANOVA followed by Sidak’s multiple comparisons test for (A), (B), and (D). Pearson’s correlation for (B) and (E). Data are presented as mean ± s.d. *P < 0.05. cmCSF, centimetres of cerebrospinal fluid; IIH, idiopathic intracranial hypertension; 11β-HSD1, 11β-hydroxysteroid dehydrogenase. A full color version of this figure is available at https://doi.org/10.1530/EJE-22-0108.
The relationship of 11β-HSD1 and 5α-reductase activity to other markers of metabolic dysregulation and inflammation was then evaluated. The change in systemic 11β-HSD1 activity had a trend to correlation with change in fasting leptin (ρ = 0.37, P = 0.05), fasting insulin (ρ = 0.38, P = 0.085) and HOMA2-IR (ρ = 0.404, P = 0.07), with no correlation with change in the inflammation marker IL-6 (ρ = −0.004, P = 0.98). The change in 5α-reductase activity was not associated with changes in leptin, fasting insulin, HOMA2-IR, or IL-6.
Subcutaneous adipose 11β-HSD1 activity
We have shown increased systemic 11β-HSD1 activity in IIH reduced in parallel with decreased ICP in the context of weight loss. Next, we assessed the ability of 11β-HSD1 to generate cortisol in subcutaneous adipose tissue from IIH patients. We demonstrated a 2.2-fold increase in 11β-HSD1 activity in IIH subcutaneous adipose explants when compared to control participants (IIH 593.8 ± 79.3 vs control 261.9 ± 37.6 pg cortisol/h/100 mg; P = 0.015, Fig. 3).
Figure 3.
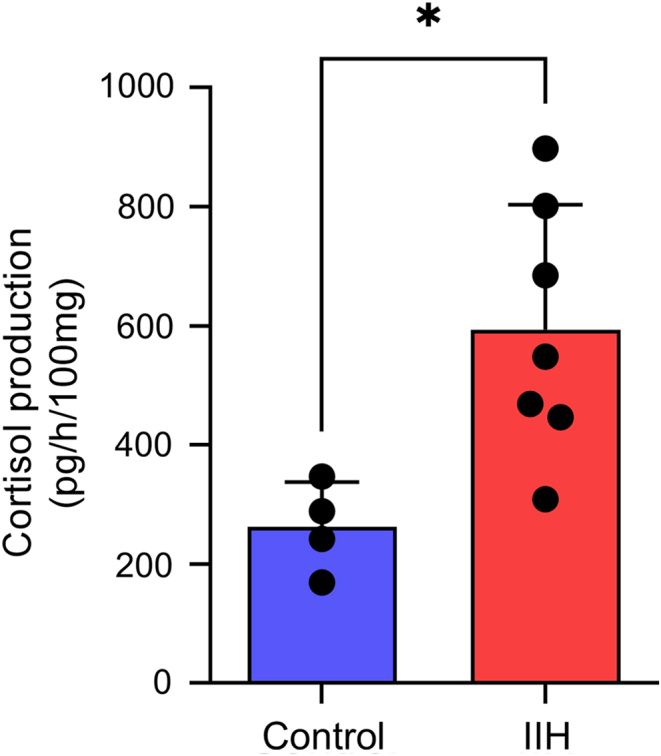
Increased 11β-HSD1 activity in subcutaneous adipose tissue explants from patients with IIH and controls. 11β-HSD1 activity as measured by cortisol production in control (4) and IIH (7) subcutaneous adipose explants (unpaired t-test). Data is presented as mean ± s.d.. *P < 0.05. IIH, idiopathic intracranial hypertension; 11β-HSD1, 11β-hydroxysteroid dehydrogenase. A full color version of this figure is available at https://doi.org/10.1530/EJE-22-0108.
Discussion
IIH has traditionally been regarded as a disease of the neuro-ophthalmic axis (29). The link to obesity is long established but data are emerging identifying systemic metabolic perturbations in excess to that driven by simple obesity alone (7, 10, 11). Inhibition of 11β-HSD1 in IIH has been demonstrated to have therapeutic benefits including reduction of ICP, improved lipid profiles (decreased cholesterol, increased HDL, and cholesterol/HDL ratio), decreased markers of hepatic dysfunction, and increased lean muscle mass (17, 30). However, it is unknown if IIH patients have deranged GC metabolism. In this study, we conducted GC phenotyping in patients with IIH and then evaluated the impact of therapeutic weight loss on GC metabolism in IIH.
We have demonstrated increased urinary cortisol and urinary systemic 11β-HSD1 activity in IIH compared to matched controls with obesity. 11β-HSD1 activity has been previously shown to be modified in the context of obesity. There is no clear consensus on what happens to systemic 11β-HSD1 activity, where multiple studies report differing direction of change or no change at all (15, 22, 23, 24). However, it is clear that hepatic 11β-HSD1 activity is reduced in the context of obesity, where it is the main contributor to systemic 11β-HSD1 activity (31, 32, 33, 34, 35). In contrast, subcutaneous adipose 11β-HSD1 activity is increased in the context of obesity (22, 24). Our data suggest that systemic 11β-HSD1 activity in IIH is greater than in obesity alone. Thus, irrespective of the change of systemic 11β-HSD1 in simple obesity, IIH likely represents a novel 11β-HSD1 phenotype distinct to obesity. We did not compare IIH patients to lean controls, as such this could be an avenue of future investigation.
A number of historical case reports have linked IIH to GC therapy but GCs have in the past been prescribed to ameliorate IIH (36). Hence, the interrelationship between IIH and iatrogenic GCs is unclear but 11β-HSD1 maybe relevant (15). The regulation of 11β-HSD1 activity is known to be tissue-, gender-, and species-specific (35). Importantly, GCs themselves can regulate 11β-HSD1 activity (35). Cortisol, dexamethasone, and prednisolone have been noted to both activate and suppress 11β-HSD1 activity depending on the tissue studied (33, 37). The role of GCs in regulating 11β-HSD1 activity in the choroid plexus, the tissue that regulates ICP through CSF secretion, has not been evaluated. GCs are no longer routinely used in the management of IIH due to lack of evidence of efficacy and the significant risk of weight gain exacerbating the underlying disease (5, 38).
These data have also demonstrated increased systemic 5α-reductase activity in IIH. 5α-reductase is a pivotal enzyme involved in the breakdown of cortisol and conversion of testosterone to dihydrotestosterone with established sexual dimorphism (higher levels in females) demonstrated in rodents (39). 5α-reductase activity is enhanced in obesity and specifically in the adipose tissue (31). Here, we have illustrated increased systemic 5α-reductase activity in IIH compared to gender and BMI-matched controls. We have previously identified increased systemic and CSF testosterone in IIH (9). In this setting, the elevated 5α-reductase activity may reflect a compensatory mechanism to breakdown the excess cortisol in IIH. This may consequently increase the activation of testosterone to dihydrotestosterone, where androgens are potentially linked to the raised ICP in IIH (9).
To investigate whether weight loss improves the GC phenotype in IIH, the effects of bariatric surgery vs CWM were compared. The IIH:WT demonstrated that surgically mediated weight loss reduced systemic 11β-HSD1 activity; these results being similar to a previous very low-calorie diet study (16). This is, however, in contrast to a study on non-IIH obese individuals where bariatric surgery did not alter systemic 11β-HSD1 activity, despite weight loss (40). This could be potentially explained by the differential baseline hormonal and metabolic phenotype seen in IIH, as compared to those with obesity (but without IIH). Further evidence was provided when the 11β-HSD1 inhibitor AZD4017 was evaluated in a randomized control trial which reported a reduction in ICP in those with active IIH (17). In vivo studies have demonstrated that hydrocortisone increases CSF secretion in obese rats, suggesting a direct effect of active GCs on ICP dynamics (41). Indeed, we report that change in 11β-HSD1 activity correlates with change in ICP, further linking GCs to ICP levels. Together, our data and that of others suggest that 11β-HSD1 activity is likely linked to the raised ICP in IIH, although the potential mechanism underlying this requires further investigation.
Our findings of increased systemic 11β-HSD1 activity and the subsequent reduction in 11β-HSD1 activity with weight loss could be linked to the findings that patients with IIH have cognitive deficits that improve with weight loss (20). Within the CNS, 11β-HSD1 is involved in mediating mood and memory. An increase in cortisol exposure has been linked to cognitive decline and Alzheimer’s disease (42). Thus, it is interesting that in patients with IIH, cognitive improvement has been previously associated with decreased systemic 11β-HSD1 activity (20).
The association between weight loss and reduction in systemic 11β-HSD1 activity suggests that adipose tissue could be contributing to the increased 11β-HSD1 activity. Here, we demonstrated a doubling of subcutaneous adipose tissue 11β-HSD1 activity in IIH. Given that obesity confers an increase in adipose 11β-HSD1 activity, this increase in IIH is in excess to that driven by obesity alone (24). IIH adipose tissue has been previously demonstrated to have phenotypic features of GC excess, including depletion of ribosomal subunits, increased leptin secretion, and increased lipid turnover (10). Consequently, the current findings increased adipose 11β-HSD1 activity provides an explanation for the IIH adipose phenotype. It is already known that people with IIH have increased abdominal obesity, and with our demonstration of increased subcutaneous adipose 11β-HSD1 activity, it is therefore possible that subcutaneous adipose in IIH is directly contributing to the increased systemic measurement of 11β-HSD1 activity due to the compounding effects of increased 11β-HSD1 activity in a tissue that is more abundant in IIH (10, 13). This may also explain why other markers of GC metabolism remain unaltered in IIH. This is similar to rheumatoid arthritis, where tissue level 11β-HSD1 activity increases contribute to increased systemic 11β-HSD1 activity (43, 44). However, it is likely that there are other tissues contributing to increased systemic 11β-HSD1 that were not assessed in the present study. Indeed, although the contribution of subcutaneous adipose tissue to systemic 11β-HSD1 is unknown, visceral adipose is a major contributor and thus could be a focus of further research (45).
It has previously been demonstrated that inflammation increases 11β-HSD1 activity and thus could be a factor in IIH 11β-HSD1 increases. Previous studies demonstrated that IIH patients have unaltered circulating level of the pro-inflammatory cytokine IL-6 compared to obese controls (20, 46). Moreover, we demonstrate the IL-6 is unaltered following weight loss in this study. However, other pro-inflammatory cytokines which also can increase 11β-HSD1 activity such as IL-1B, IL-8, and TNF-α are increased in IIH suggesting that inflammation could play a role (46). However, this has not been assessed in the adipose of IIH patients. Consequently, the mechanisms underlying the increased subcutaneous adipose 11β-HSD1 activity remain unelucidated and should be a focus of further research.
IIH is a rare disease, and consequently modest numbers of IIH patients were utilized in the present study (3, 4, 12). However, our IIH patients were meticulously phenotyped, thus this cohort is representative of female IIH patients with active disease. We also acknowledge that the control cohort was small due to finding an obesity-matched population with no comorbidities was challenging; however, the controls recruited to this study were valuable as they enabled us to infer biological insights into IIH. Adipose samples were not sought following weight loss due to ethical considerations. Therefore, we were not able determine if weight loss reduced the 11β-HSD1 activity of the adipose; however, this study represents the first demonstration of 11β-HSD1 activity in IIH adipose tissue (17).
Patients randomized to surgery had different surgical interventions based on clinical need (25). Each surgical type has been suggested to have subtle difference in their effect on the hypothalamic–pituitary–adrenal axis and thus on potential GC phenotype (47). However, this study was not powered to determine the effect of a particular surgical intervention on the GC phenotype. We did not assess the hepatic 11β-HSD1 activity in this study, although this has been evaluated previously in IIH, where hepatic 11β-HSD1 activity did not correlate with ICP (17). Future studies assessing hepatic 11β-HSD1 in IIH patients vs controls are warranted.
Conclusions
In summary, this study provides evidence that GC metabolism is dysregulated in IIH with 11β-HSD1 activity increased both systemically and within the subcutaneous adipose tissue. Therapeutic weight loss in IIH led to systemic reduction in the cortisol-metabolizing enzymes, 11β-HSD1 and 5α-reductase. The reduction in both 11β-HSD1 and 5α-reductase activity was associated with falling ICP levels suggesting a potential link to disease pathogenesis. It remains unclear if the altered GC phenotype in IIH is directly driving ICP dysregulation or a marker of systemic metabolic dysregulation.
Supplementary Material
Declaration of interest
SPM reports consulting and advisory board fees from Invex Therapeutics, teaching honoria from Heidelberg engineering during the conduct of the study; teaching honoria from Chugai-Roche Ltd, Allergan, Santen, Roche; advisory board fees from Janssen; consultancy fees from Neurodiem, outside the submitted work. A Y reports receiving speaker fees from Teva, UK outside the submitted work. A J S reports personal fees from Invex therapeutics, during the conduct of the study. J W T has received consultancy fees from Lumos and research funding from AstraZeneca. All other authors declare no competing interests.
Funding
A J S was funded by a National Institute for Health Research (NIHR) clinician scientist fellowship (NIHR-CS-011-028) and the Medical Research Council, UK (MR/K015184/1) for the duration of the study. A J S is funded by a Sir Jules Thorn Award for Biomedical Science. The trial was funded through the Medical Research Council Asset Sharing Scheme, UK (MR/K015184/1). A J S is funded by a National Institute for Health Research (NIHR) Clinician Scientist Fellowship (NIHR-CS-011-028). G G L is supported by a Wellcome Trust Senior Research Fellowship (104612/Z/14/Z). J W T is supported by MRC program (MR/P011462/1) and by the Oxford NIHR Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the UK National Health Service, the NIHR, or the UK department of Health and Social Care. Data from IIH:WT (trial beginning: July 25, 2014; date of registration: April 28, 2014; ClinicalTrials.gov: NCT02124486) and from IIH Drug trial (trial beginning: April 25, 2014; date of registration: December 20, 2013; ClinicalTrials.gov: NCT02017444).
Data availability statement
Anonymized individual participant data will be made available along with the trial protocol and statistical analysis plan. Proposals should be made to the corresponding author and will be reviewed by the Birmingham Clinical Trials Unit Data Sharing Committee in discussion with the Chief Investigator. A formal Data Sharing Agreement may be required between respective organisations once release of the data is approved and before data can be released. Restrictions apply to the availability of some or all data generated or analyzed during this study to preserve patient confidentiality or because they were used under license. The corresponding author will on request detail the restrictions and any conditions under which access to some data may be provided.
Author contribution statement
The NIHR and the MRC had no role in the design or conduct of the study; no role in collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; and no role in the decision to submit the manuscript for publication in the design, execution or write up of this trial.
References
- 1.Mollan SP, Grech O, Alimajstorovic Z, Wakerley BR, Sinclair AJ. New horizons for idiopathic intracranial hypertension: advances and challenges. British Medical Bulletin 2020136118–126. ( 10.1093/bmb/ldaa034) [DOI] [PubMed] [Google Scholar]
- 2.Mollan SP, Grech O, Sinclair AJ. Headache attributed to idiopathic intracranial hypertension and persistent post-idiopathic intracranial hypertension headache: a narrative review. Headache 202161808–816. ( 10.1111/head.14125) [DOI] [PubMed] [Google Scholar]
- 3.Mollan SP, Aguiar M, Evison F, Frew E, Sinclair AJ. The expanding burden of Idiopathic Intracranial hypertension. Eye 201833478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mollan SP, Mytton J, Tsermoulas G, Sinclair AJ. Idiopathic intracranial hypertension: evaluation of admissions and emergency readmissions through the hospital episode statistic dataset between 2002 and 2020. Life 202111 417. ( 10.3390/life11050417) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mollan SP, Davies B, Silver NC, Shaw S, Mallucci CL, Wakerley BR, Krishnan A, Chavda SV, Ramalingam S, Edwards Jet al. Idiopathic intracranial hypertension: consensus guidelines on management. Journal of Neurology, Neurosurgery, and Psychiatry 2018891088–1100. ( 10.1136/jnnp-2017-317440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mollan SP, Mitchell JL, Ottridge RS, Aguiar M, Yiangou A, Alimajstorovic Z, Cartwright DM, Grech O, Lavery GG, Westgate CSJet al. Effectiveness of bariatric surgery vs community weight management intervention for the treatment of idiopathic intracranial hypertension: a Randomized Clinical Trial. JAMA Neurology 202178 678–686. ( 10.1001/jamaneurol.2021.0659) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mollan SP, Tahrani AA, Sinclair AJ. The potentially modifiable risk factor in idiopathic intracranial hypertension: body weight. Neurology. Clinical Practice 202111e504–e507. ( 10.1212/CPJ.0000000000001063) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mollan SP, Hemmings K, Herd CP, Denton A, Williamson S, Sinclair AJ. What are the research priorities for idiopathic intracranial hypertension? A priority setting partnership between patients and healthcare professionals. BMJ Open 20199 e026573. ( 10.1136/bmjopen-2018-026573) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Reilly MW, Westgate CS, Hornby C, Botfield H, Taylor AE, Markey K, Mitchell JL, Scotton WJ, Mollan SP, Yiangou Aet al. A unique androgen excess signature in idiopathic intracranial hypertension is linked to cerebrospinal fluid dynamics. JCI Insight 20194 e125348. ( 10.1172/jci.insight.125348) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Westgate CSJ, Botfield HF, Alimajstorovic Z, Yiangou A, Walsh M, Smith G, Singhal R, Mitchell JL, Grech O, Markey KAet al. Systemic and adipocyte transcriptional and metabolic dysregulation in idiopathic intracranial hypertension. JCI Insight 20216 e145346. ( 10.1172/jci.insight.145346) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hornby C, Mollan SP, Botfield HF, OʼReilly MW, Sinclair AJ. Metabolic concepts in idiopathic intracranial hypertension and their potential for therapeutic intervention. Journal of Neuro-Ophthalmology 201838522–530. ( 10.1097/WNO.0000000000000684) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adderley NJ, Subramanian A, Nirantharakumar K, Yiangou A, Gokhale KM, Mollan SP, Sinclair AJ. Association between idiopathic intracranial hypertension and risk of cardiovascular diseases in women in the United Kingdom. JAMA Neurology 2019761088–1098. ( 10.1001/jamaneurol.2019.1812) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hornby C, Botfield H, O’Reilly MW, Westgate C, Mitchell J, Mollan SP, Manolopoulos K, Tomlinson J, Sinclair A. Evaluating the fat distribution in idiopathic intracranial hypertension using dual-energy X-ray absorptiometry scanning. Neuro-Ophthalmology 20184299–104. ( 10.1080/01658107.2017.1334218) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hewitt KN, Walker EA, Stewart PM. Minireview: Hexose-6-Phosphate dehydrogenase and redox control of 11β-hydroxysteroid dehydrogenase type 1 activity. Endocrinology 20051462539–2543. ( 10.1210/en.2005-0117) [DOI] [PubMed] [Google Scholar]
- 15.Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM. 11β-Hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocrine Reviews 200425831–866. ( 10.1210/er.2003-0031) [DOI] [PubMed] [Google Scholar]
- 16.Sinclair AJ, Walker EA, Burdon MA, van Beek AP, Kema IP, Hughes BA, Murray PI, Nightingale PG, Stewart PM, Rauz Set al. Cerebrospinal fluid corticosteroid levels and cortisol metabolism in patients with idiopathic intracranial hypertension: a link between 11β-HSD1 and intracranial pressure regulation? Journal of Clinical Endocrinology and Metabolism 2010955348–5356. ( 10.1210/jc.2010-0729) [DOI] [PubMed] [Google Scholar]
- 17.Markey K, Mitchell J, Botfield H, Ottridge RS, Matthews T, Krishnan A, Woolley R, Westgate C, Yiangou A, Alimajstorovic Zet al. 11β-Hydroxysteroid dehydrogenase type 1 inhibition in idiopathic intracranial hypertension: a double-blind randomized controlled trial. Brain Communications 20202fcz050. ( 10.1093/braincomms/fcz050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naber D, Sand P, Heigl B. Psychopathological and neuropsychological effects of 8-days’ corticosteroid treatment. A prospective study. Psychoneuroendocrinology 19962125–31. ( 10.1016/0306-4530(9500031-3) [DOI] [PubMed] [Google Scholar]
- 19.Wolkowitz OM, Reus VI, Weingartner H, Thompson K, Breier A, Doran A, Rubinow D, Pickar D. Cognitive effects of corticosteroids. American Journal of Psychiatry 19901471297–1303. ( 10.1176/ajp.147.10.1297) [DOI] [PubMed] [Google Scholar]
- 20.Grech O, Clouter A, Mitchell JL, Alimajstorovic Z, Ottridge RS, Yiangou A, Roque M, Tahrani AA, Nicholls M, Taylor AEet al. Cognitive performance in idiopathic intracranial hypertension and relevance of intracranial pressure. Brain Communications 20213fcab202. ( 10.1093/braincomms/fcab202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP, Thakur M, McEwen BS, Hauger RL, Meaney MJ. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nature Neuroscience 1998169–73. ( 10.1038/271) [DOI] [PubMed] [Google Scholar]
- 22.Rask E, Olsson T, Söderberg S, Andrew R, Livingstone DE, Johnson O, Walker BR. Tissue-specific dysregulation of cortisol metabolism in human obesity. Journal of Clinical Endocrinology and Metabolism 2001861418–1421. ( 10.1210/jcem.86.3.7453) [DOI] [PubMed] [Google Scholar]
- 23.Stewart PM, Boulton A, Kumar S, Clark PMS, Shackleton CHL. Cortisol metabolism in human obesity: impaired cortisone→cortisol conversion in subjects with central adiposity 1. Journal of Clinical Endocrinology and Metabolism 1999841022–1027. ( 10.1210/jcem.84.3.5538) [DOI] [PubMed] [Google Scholar]
- 24.Rask E, Walker BR, Söderberg S, Livingstone DE, Eliasson M, Johnson O, Andrew R, Olsson T. Tissue-specific changes in peripheral cortisol metabolism in obese women: increased adipose 11β-hydroxysteroid dehydrogenase type 1 activity. Journal of Clinical Endocrinology and Metabolism 2002873330–3336. ( 10.1210/jcem.87.7.8661) [DOI] [PubMed] [Google Scholar]
- 25.Ottridge R, Mollan SP, Botfield H, Frew E, Ives NJ, Matthews T, Mitchell J, Rick C, Singhal R, Woolley Ret al. Randomised controlled trial of bariatric surgery versus a community weight loss programme for the sustained treatment of idiopathic intracranial hypertension: the Idiopathic Intracranial Hypertension Weight Trial (IIH:WT) protocol. BMJ Open 20177 e017426. ( 10.1136/bmjopen-2017-017426) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arlt W, Walker EA, Draper N, Ivison HE, Ride JP, Hammer F, Chalder SM, Borucka-Mankiewicz M, Hauffa BP, Malunowicz EMet al. Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet 20043632128–2135. ( 10.1016/S0140-6736(0416503-3) [DOI] [PubMed] [Google Scholar]
- 27.Jühlen R, Idkowiak J, Taylor AE, Kind B, Arlt W, Huebner A, Koehler K. Role of ALADIN in human adrenocortical cells for oxidative stress response and steroidogenesis. PLoS ONE 201510 e0124582. ( 10.1371/journal.pone.0124582) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mooij CF, Parajes S, Rose IT, Taylor AE, Bayraktaroglu T, Wass JA, Connell JM, Ray DW, Arlt W, Krone N. Characterization of the molecular genetic pathology in patients with 11β-hydroxylase deficiency. Clinical Endocrinology 201583629–635. ( 10.1111/cen.12834) [DOI] [PubMed] [Google Scholar]
- 29.Virdee J, Larcombe S, Vijay V, Sinclair AJ, Dayan M, Mollan SP. Reviewing the recent developments in idiopathic intracranial hypertension. Ophthalmology and Therapy 20209767–781. ( 10.1007/s40123-020-00296-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardy RS, Botfield H, Markey K, Mitchell JL, Alimajstorovic Z, Westgate CSJ, Sagmeister M, Fairclough RJ, Ottridge RS, Yiangou Aet al. 11βHSD1 inhibition with AZD4017 improves lipid profiles and lean muscle mass in idiopathic intracranial hypertension. Journal of Clinical Endocrinology and Metabolism 2021106174–187. ( 10.1210/clinem/dgaa766) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrew R, Phillips DIW, Walker BR. Obesity and gender influence cortisol secretion and metabolism in man. Journal of Clinical Endocrinology and Metabolism 1998831806–1809. ( 10.1210/jcem.83.5.4951) [DOI] [PubMed] [Google Scholar]
- 32.Reynolds RM, Walker BR, Syddall HE, Andrew R, Wood PJ, Whorwood CB, Phillips DI. Altered control of cortisol secretion in adult men with low birth weight and cardiovascular risk factors 1. Journal of Clinical Endocrinology and Metabolism 200186245–250. ( 10.1210/jcem.86.1.7145) [DOI] [PubMed] [Google Scholar]
- 33.Tomlinson JW, Sinha B, Bujalska I, Hewison M, Stewart PM. Expression of 11β-hydroxysteroid dehydrogenase type 1 in adipose tissue is not increased in human obesity. Journal of Clinical Endocrinology and Metabolism 2002875630–5635. ( 10.1210/jc.2002-020687) [DOI] [PubMed] [Google Scholar]
- 34.Paulmyer-Lacroix O, Boullu S, Oliver C, Alessi MC, Grino M. Expression of the mRNA coding for 11β-hydroxysteroid dehydrogenase type 1 in adipose tissue from obese patients: an in situ hybridization study. Journal of Clinical Endocrinology and Metabolism 2002872701–2705. ( 10.1210/jcem.87.6.8614) [DOI] [PubMed] [Google Scholar]
- 35.Li X, Lindquist S, Chen R, Myrnäs T, Angsten G, Olsson T, Hernell O. Depot-specific messenger RNA expression of 11β-hydroxysteroid dehydrogenase type 1 and leptin in adipose tissue of children and adults. International Journal of Obesity 200731820–828. ( 10.1038/sj.ijo.0803470) [DOI] [PubMed] [Google Scholar]
- 36.Sinclair AJ, Ball AK, Burdon MA, Clarke CE, Stewart PM, Curnow SJ, Rauz S. Exploring the pathogenesis of IIH: an inflammatory perspective. Journal of Neuroimmunology 2008201–202212–220. ( 10.1016/j.jneuroim.2008.06.029) [DOI] [PubMed] [Google Scholar]
- 37.Bujalska IJ, Kumar S, Stewart PM. Does central obesity reflect ‘Cushing’s disease of the omentum’? Lancet 19973491210–1213. ( 10.1016/S0140-6736(9611222-8) [DOI] [PubMed] [Google Scholar]
- 38.Hoffmann J, Mollan SP, Paemeleire K, Lampl C, Jensen RH, Sinclair AJ. European Headache Federation guideline on idiopathic intracranial hypertension. Journal of Headache and Pain 201819 93. ( 10.1186/s10194-018-0919-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mode A, Norstedt G. Effects of gonadal steroid hormones on the hypothalamo-pituitary-liver axis in the control of sex differences in hepatic steroid metabolism in the rat. Journal of Endocrinology 198295181–187. ( 10.1677/joe.0.0950181) [DOI] [PubMed] [Google Scholar]
- 40.Woods CP, Corrigan M, Gathercole L, Taylor A, Hughes B, Gaoatswe G, Manolopoulos K, Hogan AE, O’Connell J, Stewart PMet al. Tissue specific regulation of glucocorticoids in severe obesity and the response to significant weight loss following bariatric surgery (BARICORT). Journal of Clinical Endocrinology and Metabolism 20151001434–1444. ( 10.1210/jc.2014-4120) [DOI] [PubMed] [Google Scholar]
- 41.Alimajstorovic Z, Pascual-Baixauli E, Hawkes CA, Sharrack B, Loughlin AJ, Romero IA, Preston JE. Cerebrospinal fluid dynamics modulation by diet and cytokines in rats. Fluids and Barriers of the CNS 202017 10. ( 10.1186/s12987-020-0168-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meaney MJ, O’Donnell D, Rowe W, Tannenbaum B, Steverman A, Walker M, Nair NP, Lupien S. Individual differences in hypothalamic-pituitary-adrenal activity in later life and hippocampal aging. Experimental Gerontology 199530229–251. ( 10.1016/0531-5565(9400065-b) [DOI] [PubMed] [Google Scholar]
- 43.Gathercole LL, Lavery GG, Morgan SA, Cooper MS, Sinclair AJ, Tomlinson JW, Stewart PM. 11β-Hydroxysteroid dehydrogenase 1: translational and therapeutic aspects. Endocrine Reviews 201334525–555. ( 10.1210/er.2012-1050) [DOI] [PubMed] [Google Scholar]
- 44.Hardy R, Rabbitt EH, Filer A, Emery P, Hewison M, Stewart PM, Gittoes NJ, Buckley CD, Raza K, Cooper MS. Local and systemic glucocorticoid metabolism in inflammatory arthritis. Annals of the Rheumatic Diseases 2008671204–1210. ( 10.1136/ard.2008.090662) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andrew R, Westerbacka J, Wahren J, Yki-Järvinen H, Walker BR. The contribution of visceral adipose tissue to splanchnic cortisol production in healthy humans. Diabetes 2005541364–1370. ( 10.2337/diabetes.54.5.1364) [DOI] [PubMed] [Google Scholar]
- 46.Samancl B, Samancı Y, Tüzün E, Altıokka-Uzun G, Ekizoğlu E, İçöz S, Şahin E, Küçükali Cİ, Baykan B. Evidence for potential involvement of pro-inflammatory adipokines in the pathogenesis of idiopathic intracranial hypertension. Cephalalgia 201737525–531. ( 10.1177/0333102416650705) [DOI] [PubMed] [Google Scholar]
- 47.Cornejo-Pareja I, Clemente-Postigo M, Tinahones FJ. Metabolic and endocrine consequences of bariatric surgery. Frontiers in Endocrinology 201910626. ( 10.3389/fendo.2019.00626) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized individual participant data will be made available along with the trial protocol and statistical analysis plan. Proposals should be made to the corresponding author and will be reviewed by the Birmingham Clinical Trials Unit Data Sharing Committee in discussion with the Chief Investigator. A formal Data Sharing Agreement may be required between respective organisations once release of the data is approved and before data can be released. Restrictions apply to the availability of some or all data generated or analyzed during this study to preserve patient confidentiality or because they were used under license. The corresponding author will on request detail the restrictions and any conditions under which access to some data may be provided.