

 This work is licensed under a
This work is licensed under a Abstract
Summary
Complicated Rathke’s cleft cyst (RCC) is a rare occurrence of symptomatic bleeding or growth of a previously asymptomatic (and often undiagnosed) intrasellar cyst derived from remnants of Rathke’s pouch, situated on the midline between the adeno- and neurohypophysis. Symptoms may be identical to those of pituitary apoplexy: acute onset of headache, hypopituitarism, and neurological disturbances. Both syndromes may also exhibit a similar appearance of a large haemorrhagic sellar mass at initial radiological evaluation. We report on two patients who presented with headache and complete hypopituitarism. Based on the initial MRI, they were first diagnosed with pituitary apoplexy but managed conservatively with hormone therapy alone because of the absence of severe visual or neurological threat. Upon follow-up at 4 months, clinical evolution was good in both patients but their pituitary mass had not reduced in size and, after careful radiologic reviewing, was more indicative of a large midline complicated RCC. In conclusion, the diagnosis of complicated RCC is challenging because it can mimic pituitary apoplexy clinically, biologically, and radiologically. Clinicians should distinguish between the two entities using specific radiological signs or evolution of the mass at MRI if the patient does not undergo surgery. To our knowledge, we report conservative management of this rare condition for the first time, though it seems appropriate in the absence of neurological compromise or visual compression. Long-term follow-up is however mandatory.
Learning points
Complicated Rathke’s cleft cyst can mimic pituitary apoplexy, presenting with sudden onset of headache, hypopituitarism, and visual and neurological compromise in the most severe cases.
At diagnosis, pituitary MRI may not be able to differentiate between the two entities, showing a large haemorrhagic mass inside the sella, with little or no normal pituitary tissue visible. Patients are often diagnosed with apoplexy at this stage and may undergo pituitary surgery.
When surgery has not been performed initially in these patients, repeat imaging at 3–6 months is unchanged and does not show the expected involution usually seen after adenoma apoplexy.
Conservative management with hormonal replacement seems a valid option in the absence of visual or neurological deficits that would require trans-sphenoidal surgery.
Patient Demographics: Adult, Male, White, Belgium
Clinical Overview: Pituitary, Pituitary
Related Disciplines: Surgery
Publication Details: Error in diagnosis/pitfalls and caveats, August, 2022
Background
Pituitary apoplexy is defined by acute haemorrhage or infarction of an intrasellar adenoma. Among pituitary tumours, non-functioning macroadenomas most frequently lead to this complication (1). Symptoms occur suddenly and can include headache, visual field defects, oculomotor palsy, altered consciousness, and symptoms of hypopituitarism. Treatment consists of hormonal replacement and surgical decompression in cases of visual or neurological compromise.
Rathke’s cleft cysts (RCCs) are benign cystic lesions derived from remnants of Rathke’s pouch, situated between the anterior and posterior pituitary. Sudden haemorrhage or growth of a RCC can lead to the same clinical syndrome as classical adenoma-related apoplexy and thus be confused with apoplexy (2).
Furthermore, the limited published literature on complicated RCC’s reports only patients that were treated with trans-sphenoidal surgery (2, 3). However, conservative management with hormonal replacement and regular follow-up is accepted for patients who do not have neurological or visual compromise, in accordance with recent guidelines on the treatment of apoplexy (4).
We describe two cases of complicated RCCs mimicking pituitary apoplexy and treated conservatively, that might help clinicians to distinguish between these two entities and improve their management.
Case presentation
Case 1: A 59-year-old man, with a history of type 2 diabetes and hypertension, presented with intense and sudden headache along with the concomitant and rapid development of profound asthenia and malaise. His family doctor performed general biology that showed low thyroid-stimulating hormone (TSH) and asked for an endocrinological consultation. Glasgow coma score was normal at 15 and neurological examination was normal. He did not have visual complaints. Before this episode, the patient was well and had no signs of hormone deficiency. A normal TSH concentration was observed 4 months before the acute episode.
Case 2: A 27-year-old man, with no medical history, presented with sudden and violent headache not responding to analgesics. He also complained of the concomitant and rapid development of fatigue, loss of libido, hypotension, and vertigo as well as polydipsia and polyuria. He did not have visual complaints. His general practitioner identified low T4 and testosterone levels and referred him to an endocrinologist, who immediately suspected hypopituitarism and sent him to the emergency department for hospitalization. Clinical examination was normal except for hypotension at 105/65 mmHg.
Investigation
Case 1: Laboratory testing showed severe central hypothyroidism with low TSH and free T4. Further endocrine testing revealed very low morning serum cortisol, total testosterone, and LH, while the prolactin concentration was slightly elevated at 17.4 µg/L (Table 1).
Table 1.
Laboratory results at diagnosis.
| Laboratory serum results at initial work-up* | Patient 1 | Patient 2 |
|---|---|---|
| Sodium (135–145 mmol/L) | 136 | 147 |
| Serum osmolarity (275–295 mOsm/L) | Not available | 311 |
| Urine osmolarity (118–1041 mOsm/L) | Not available | 114 |
| Morning cortisol (150–450 nmol/L) | <13.8 | 26 |
| Morning ACTH (5–50 ng/L) | 6.9 | 6.2 |
| TSH (0.2–4 mU/L) | 0.02 | 3.59 |
| Free T4 (10–28 pmol/L) | 6.45 | 9.12 |
| Morning total testosterone (5–26 nmol/L) | 0.39 | 0.42 |
| LH (1.3–9.8 U/L) | <1 | <1 |
| Prolactin (5–15 µg/L) | 17.4 | 40.3 |
*Normal values and units are indicated in parentheses.
Hypopituitarism was diagnosed, and the patient underwent pituitary MRI, which revealed an intrasellar posterior cystic mass, measuring 10 × 10 × 13 mm, pushing the anterior pituitary gland forward and abutting the optic chiasm. There was a liquid-to-liquid level within the mass (Fig. 1A and B), with only the anterior section of the liquid appearing hyperintense on T1-weighted images, suggestive of the sedimentation of proteinous material. The diagnosis of subacute pituitary apoplexy was first evoked. Ophthalmological testing showed no visual deficit and no oculomotor alterations.
Figure 1.
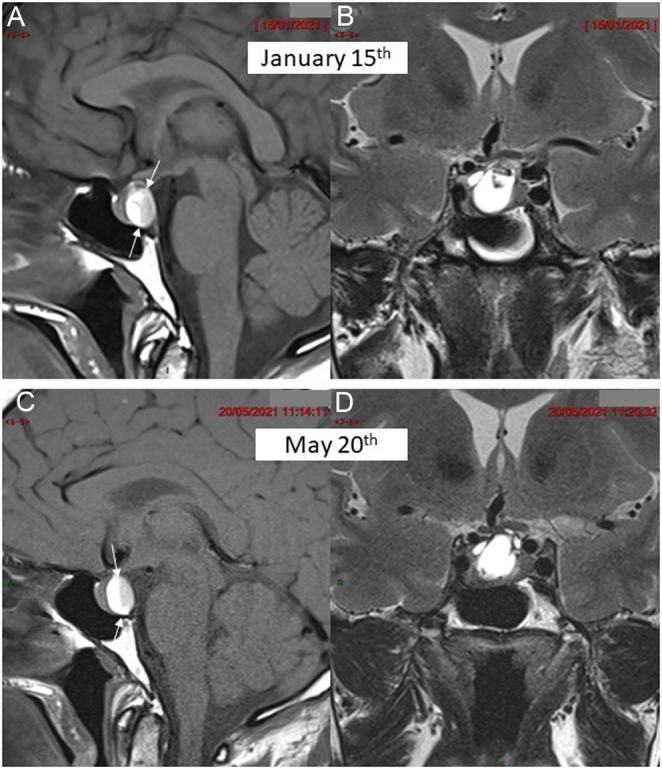
MRI in patient 1 at the time of diagnosis (upper arrow) and 4 months later (lower arrow). (A) Pre-contrast mid-sagittal T1-weighted view showing enlarged pituitary gland with a posterior cyst disclosing a fluid/fluid level (between arrows) with strongly hyperintense supernatant and a sediment in declivity (patient laying on the back) of moderately lower signal intensity. (B) Coronal T2-weighted view showing the hyperintense liquid content of the cyst with a small hypointense spot at its upper area. (C) Pre-contrast mid-sagittal T1-weighted view in similar slice location as (A) and (D) coronal T2-weighted view in similar slice location as (B) 4 months later failing to demonstrate significant change in the pituitary mass.
Case 2: Laboratory testing showed markedly reduced morning cortisol, adrenocorticotropic hormone (ACTH), total testosterone, and LH concentrations and a frankly increased prolactin level at 40.3 µg/L (Table 1). Serum sodium and osmolality were high while urine osmolality was low, all values indicative of diabetes insipidus (DI).
Pituitary MRI (Fig. 2A, B, C, and D) showed a 16 mm pituitary mass, without contrast uptake in T1-weighted sequences, suggestive of necrosis following recent bleeding. A diagnosis of acute pituitary apoplexy secondary to haemorrhagic non-functioning adenoma was made. Visual testing was normal.
Figure 2.

MRI in patient 2 at the time of diagnosis (upper arrow) and 5 months later (lower arrow). (A) Pre-contrast mid-sagittal T1-weighted view revealing a pituitary mass of intermediate and homogeneous signal intensity. (B) Pre-contrast T1-weighted view showing similar findings. (C) Post-contrast T1-weighted view in similar slice location as (B) revealing only a very thin marginal enhancement of the mass with complete devascularisation of its core. (D) Coronal T2-weighted view in similar slice location as (B, C) revealing strong hypointensity of the mass. (E) Pre-contrast mid-sagittal T1-weighted view in similar slice location as (A) revealing only a qualitative change with appearance of hypersignal intensity at anterior border of the mass (arrow) due to increase in protein concentration. (F) Pre-contrast T1-weighted view in similar slice location as (B) confirming subtle intensity changes (arrows). (G) Post-contrast T1-weighted view in similar slice location as (C) failing to reveal any change when compared to (C). (H) Coronal T2-weighted view in similar slice location as (D) revealing appearance of subtle areas of slightly increased signal intensity (arrows) mainly due to technical differences between (D) and (H).
Treatment
Case 1: The patient was started on hydrocortisone, levothyroxine, and testosterone therapy. His headaches and fatigue rapidly improved. In the absence of visual compromise and headache, surgery was not performed because complete and severe hypopituitarism was already present and would probably not have improved after surgery.
Case 2: The patient was started on hydrocortisone, levothyroxine, and desmopressin, while testosterone supplementation was delayed. He rapidly improved and was managed conservatively, for the same reasons as in case 1.
Outcome and follow-up
Case 1: Follow-up MRI at 4 months revealed stable images (Fig. 1C and D), without any changes in size or signal, ruling out the diagnosis of apoplexy as a haemorrhagic adenoma would have shown some involutive changes after 4 months. We therefore concluded that the patient harboured a large RCC that became symptomatic, despite the lack of current radiological signs of haemorrhage.
Case 2: At follow-up after 4 months, pituitary MRI (Fig. 2E, F, G, and H) showed no change in the size of the lesion and new and spontaneous T1 hyperintensity in the anterior segment of the lesion, possibly because of methaemoglobin deposition or changes in protein concentration. Retrospectively, the diagnosis of RCC with initial intracystic haemorrhage was made.
In both cases, hypopituitarism persisted (data not shown) and requested continuing hormonal substitution at the last follow-up visit (at 10 months for patient 1 and 8 months for patient 2). Of note, patient 2 had recurrence of DI symptoms when stopping desmopressin for 2 weeks, suggesting that the vasopressin deficit was likely permanent.
Discussion
Our two cases illustrate the difficulty of diagnosing complicated RCC, as in both patients the diagnosis of pituitary apoplexy was first made by the clinician and the radiologist. As well described in a review by Boellis et al. (5), the radiological features of pituitary haemorrhage change over time and can be separated into three periods: acute (0–7 days), subacute (7–21 days), or chronic (>21 days). Pituitary apoplexy typically appears as a large sellar mass, that will present the succession of these different MRI aspects corresponding to the timing of haemorrhage. After a few months, due to the necrosis of the cells constituting the adenoma, a size decrease of even complete disappearance of the adenoma is usually observed (6).
Initial distinction between haemorrhagic cystic adenoma and RCC is difficult. Signs that characterize RCCs are most visible on axial T1-weighted sections: a strict midline location, regular convex symmetric anterior surface, close contact with the posterior lobe, and little or no mass effect (7). It is also important to note that the radiological anomalies related to conservatively treated apoplexy usually show tumour shrinkage after several months (1) whereas haemorrhagic RCCs remain stable or even grow at imaging. Hints for the presence of RCC also include T1-hyperintensity related to protein content within the cyst (8) and the absence of nodular enhancement (9).
The initial presence of central DI (CDI) is also suggestive of RCC as it is rare in pituitary adenomas. Even very large adenomas do not lead to CDI because the compression or destruction of axon terminals in the posterior pituitary still allows the release of vasopressin in the region of the stalk, median eminence or infundibulum (10). Thus, CDI, if present initially like in our second case, should prompt clinicians to consider a diagnosis other than a pituitary adenoma. On the other hand, complicated RCC has been linked with CDI, through local lymphocytic infiltration. Oishi et al reported three cases of acute onset DI with headache in patients with RCC, putatively caused by a posterior rupture of the cyst wall towards the posterior pituitary, followed by a local inflammation of the neuropituitary (11).
A recent study classified headaches in patients with RCCs according to their onset, duration, and associated histological observations (12). In acute headache, inflammatory cell invasion was most often identified, and apoplectic headaches were most often associated with haemorrhage. In patient 1, no radiological sign of haemorrhage was indeed visible, but the onset of pain was rapid, classifying it as acute headache. We believe that a local inflammatory reaction caused by cyst expansion or rupture was responsible for the symptom in this patient. In patient 2, radiological signs of intracystic haemorrhage were observed, thus suggesting that intra-cystic bleeding could have led to an apoplexy-mimicking phenotype.
The differential diagnosis should also include craniopharyngioma, partially thrombosed carotid aneurysms, dermoid cysts and lipomas (5). However, several clues plead against these diagnoses in our cases: the location of the cyst and lack of involvement of carotid arteries, the absence of any fleshy portion or calcification in the cyst wall, the T1 and T2 characteristics of the tumour which were not suggestive of a dermoid cyst or a lipoma.
We acknowledge that the major limitation of our report is the lack of definitive histological diagnosis. Surgical management of such cases is certainly a valid treatment option, with certain advantages: definitive histological diagnosis, volume reduction to prevent future damage to the optic chiasm and surrounding structures, and in certain conditions partial endocrine recovery. However, we felt that conservative management was also a viable option, from a diagnostic and therapeutic standpoint. First of all, a radiological key-feature supporting the hypothesis of a RCC was the absence of evolution of the pituitary status within short (4 months) follow-up MR examination with the knowledge that apoplectic adenomas rapidly shrink and strong signal intensity changes are observed according to the degree of haemoglobin degradation products. To support this, a case series including 19 conservatively managed apoplectic adenomas revealed significant short term reductions in size in 18/19 (95%) patients (13). Moreover, the midline localization of the cyst between the adeno and neuro-pituitary in patient 1, the initial presence of DI in patient 2, and the absence of nodular enhancement in both patients were all suggestive of RCC.
As endocrine deficiencies were already well installed for several weeks, we considered that the chance of recovery of the endocrine functions was very low. Oishi et al. reported two cases of acute complicated RCC with DI that underwent surgery: both had persistent DI after surgery, although controlled with a smaller dose of desmopressin, indicating that surgery does not improve CDI when this has already become chronic (11). Likewise, a recent study comparing surgery and observation in the early management of RCC found that when pituitary deficits were installed, surgery failed to improve endocrine function, although it did relieve headache and hyperprolactinemia (14). Finally, an older study of 28 surgically managed symptomatic RCCs showed that among patients with panhypopituitarism, only 15% (2/13) showed any endocrine improvement after surgery (15). On the other hand, the risks of neurosurgery should not be forgotten, as recent surgical series show a 11–35% rate of complications, notably a 16% risk of new-onset DI or CSF leak after gross total removal of RCC (16). The availability of experienced neurosurgeons and patient preference should also be taken into account.
Nevertheless, we consider close follow-up to be paramount to detect further cyst enlargement or worsening pituitary function. Overall, management of complicated RCC should be in fact very similar as the one which is now well accepted for true pituitary apoplexy (1), based on studies showing similar outcomes between early surgery and surveillance in cases without neurological or visual compromise (4).
In conclusion, the diagnosis of complicated RCC is challenging because it can mimic pituitary apoplexy clinically, biologically, and radiologically. Clinicians should distinguish between the two entities using specific radiological signs or evolution of the mass at MRI if the patient did not undergo surgery. To our knowledge, we report conservative management of this rare condition for the first time, though it seems appropriate in the absence of neurological compromise or visual compression. Long-term follow-up is however mandatory.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work did not receive any specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Patient consent
Written informed consent was obtained from both patients for the publication of the submitted article and accompanying images.
Author contribution statement
S M Constantinescu and D Maiter wrote the manuscript with important remarks from all authors. T Duprez created and described all figures. All authors read and approved the final manuscript.
References
- 1.Briet C, Salenave S, Bonneville JF, Laws ER, Chanson P. Pituitary apoplexy. Endocrine Reviews 201536622–645. ( 10.1210/er.2015-1042) [DOI] [PubMed] [Google Scholar]
- 2.Schooner L, Wedemeyer MA, Bonney PA, Lin M, Hurth K, Mathew A, Liu CJ, Shiroishi M, Carmichael JD, Weiss MH, et al. Hemorrhagic presentation of Rathke cleft cysts: a surgical case series. Operative Neurosurgery 18470–479. ( 10.1093/ons/opz239) [DOI] [PubMed] [Google Scholar]
- 3.Chaiban JT, Abdelmannan D, Cohen M, Selman WR, Arafah BM. Rathke cleft cyst apoplexy: a newly characterized distinct clinical entity. Journal of Neurological Surgery 2011114318–324. ( 10.3171/2010.5.JNS091905) [DOI] [PubMed] [Google Scholar]
- 4.Capatina C, Inder W, Karavitaki N, Wass JAH. Management of endocrine disease: pituitary tumour apoplexy. European Journal of Endocrinology 2015172R179–R190. ( 10.1530/EJE-14-0794) [DOI] [PubMed] [Google Scholar]
- 5.Boellis A, di Napoli A, Romano A, Bozzao A. Pituitary apoplexy: an update on clinical and imaging features. Insights into Imaging 20145753–762. ( 10.1007/s13244-014-0362-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong MR, Douek M, Schellinger D, Patronas NJ. Regression of pituitary macroadenoma after pituitary apoplexy: Ct and mr studies. Journal of Computer Assisted Tomography 199115832–834. ( 10.1097/00004728-199109000-00021) [DOI] [PubMed] [Google Scholar]
- 7.Bonneville JF.Hemorrhagic pituitary adenoma versus Rathke cleft cyst: a frequent dilemma. AJNR. American Journal of Neuroradiology 201637E27–E28. ( 10.3174/ajnr.A4653) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goyal P, Utz M, Gupta N, Kumar Y, Mangla M, Gupta S, Mangla R. Clinical and imaging features of pituitary apoplexy and role of imaging in differentiation of clinical mimics. Quantitative Imaging in Medicine and Surgery 20188219–231. ( 10.21037/qims.2018.03.08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung HN, Kim ST, Kong DS, Suh il SI, Ryoo I. Rathke cleft cysts with apoplexy-like symptoms: clinicoradiologic comparisons with pituitary adenomas with apoplexy. World Neurosurgery 142e1–e9. ( 10.1016/j.wneu.2020.03.086) [DOI] [PubMed] [Google Scholar]
- 10.Verbalis JG.Acquired forms of central diabetes insipidus: mechanisms of disease. Best Practice and Research: Clinical Endocrinology and Metabolism 34101449. ( 10.1016/j.beem.2020.101449) [DOI] [PubMed] [Google Scholar]
- 11.Oishi M, Hayashi Y, Sasagawa Y, Kita D, Tachibana O, Nakada M. Outcome of diabetes insipidus in patients with Rathke’s cleft cysts. Clinical Neurology and Neurosurgery 2018167141–146. ( 10.1016/j.clineuro.2018.02.031) [DOI] [PubMed] [Google Scholar]
- 12.Hayashi Y, Kita D, Iwato M, Fukui I, Sano Het al. Classification of Headaches associated with Rathke’s cleft cyst according to their onset and duration. Austin Neurosurg Open Access 20141 1009. [Google Scholar]
- 13.Gruber A, Clayton J, Kumar S, Robertson I, Howlett TA, Mansell P. Pituitary apoplexy: retrospective review of 30 patients - is surgical intervention always necessary? British Journal of Neurosurgery 200620379–385. ( 10.1080/02688690601046678) [DOI] [PubMed] [Google Scholar]
- 14.Truong LUF, Bazin C, Gomis P, Decoudier B, Delemer B, Litré CF. Surgery versus conservative care for Rathke’s cleft cyst. Neuro-Chirurgie 67104–111. ( 10.1016/j.neuchi.2020.12.010) [DOI] [PubMed] [Google Scholar]
- 15.El-Mahdy W, Powell M. Transsphenoidal management of 28 symptomatic Rathke’s cleft cysts, with special reference to visual and hormonal recovery. Neurosurgery 1998427–16. ( 10.1097/00006123-199801000-00003) [DOI] [PubMed] [Google Scholar]
- 16.Higgins DM, Van Gompel JJ, Nippoldt TB, Meyer FB. Symptomatic Rathke cleft cysts: extent of resection and surgical complications. Neurosurgical Focus 201131 E2. ( 10.3171/2011.5.FOCUS1175) [DOI] [PubMed] [Google Scholar]