

Abstract
Diabetic foot ulcers (DFU) are a serious complication of diabetes mellitus and associated with reduced quality of life and high mortality rate. DFUs are characterized by a deregulated immune response with decreased neutrophils due to loss of the transcription factor, FOXM1. Diabetes primes neutrophils to form neutrophil extracellular traps (NETs), contributing to tissue damage and impaired healing. However, the role of FOXM1 in priming diabetic neutrophils to undergo NET formation remains unknown. Here, we found that FOXM1 regulates reactive oxygen species (ROS) levels in neutrophils and inhibition of FOXM1 results in increased ROS leading to NET formation. Next generation sequencing revealed that TREM1 promoted the recruitment of FOXM1+ neutrophils and reversed effects of diabetes and promoted wound healing in vivo. Moreover, we found that TREM1 expression correlated with clinical healing outcomes of DFUs, indicating TREM1 may serve as a useful biomarker or a potential therapeutic target. Our findings highlight the clinical relevance of TREM1, and indicates FOXM1 pathway as a novel regulator of NET formation during diabetic wound healing, revealing new therapeutic strategies to promote healing in DFUs.
Keywords: diabetic foot ulcers, FOXM1, neutrophil extracellular traps, neutrophils, TREM1
Subject Categories: Immunology, Molecular Biology of Disease
The FOXM1 transcription factor modulates NETs by regulating ROS during diabetic wound healing. TREM1 contributes to the clinical healing outcome and promotes FOXM1+ neutrophil recruitment to enhance diabetic wound healing.
Introduction
Diabetes Mellitus is associated with numerous debilitating comorbidities including cardiovascular disease, stroke, chronic kidney disease, and peripheral neuropathy (Brem & Tomic‐Canic, 2007; Alavi et al, 2014; Eming et al, 2014). A major complication of diabetes is the development of diabetic foot ulcers (DFUs), nonhealing ulcerative wounds present on the lower extremities that incur devastating clinical outcomes (Brem & Tomic‐Canic, 2007; Alavi et al, 2014; Eming et al, 2014). Approximately one in four diabetic patients will develop a DFU in their lifetime (Armstrong et al, 2017). Many DFUs necessitate lower limb amputation, which has a 5‐year survival rate of 40–50% (Armstrong et al, 2020; Soo et al, 2020). Despite the critical need for effective therapies to heal DFUs and reduce associated amputation rates, no new therapies have been FDA approved for efficacy since 1998. The pathogenesis of DFUs involves many intrinsic factors such as neuropathy, vasculopathy, ischemia, infection, fibrosis, and immune dysfunction (Alavi et al, 2014; Eming et al, 2014; Armstrong et al, 2017; Ramirez et al, 2018). In particular, the immune response in DFUs is permissive to a hyperproliferative and nonmigratory epidermis, biofilm formation, and infection (Eming et al, 2014; Ramirez et al, 2018).
Acute wound healing is a highly organized process that involves the sequential yet overlapping action of multiple process including hemostasis, inflammation, proliferation, and tissue remodeling (Pastar et al, 2014; Stone et al, 2017, 2020; Sawaya et al, 2019). Keratinocytes, macrophages, platelets, endothelial cells, fibroblasts, and inflammatory immunocytes are key cellular effectors of cutaneous healing and have stringently regulated roles in each healing stage (Eming et al, 2007; Koh & DiPietro, 2011). Studies comparing wounding in the oral mucosa, in which wound closure is rapid and occurs without scarring, with cutaneous wounding emphasize the importance of controlled inflammation to achieve optimal wound closure (Chen et al, 2010; Turabelidze et al, 2014; Iglesias‐Bartolome et al, 2018; Uchiyama et al, 2019). The proinflammatory cellular infiltrate of early stage acute wound healing is composed primarily of neutrophils that kill invading microbes, and macrophages that clear apoptotic neutrophils and restore tissue integrity for wound closure (Eming et al, 2007, 2014; Wilgus et al, 2013; Sawaya et al, 2020; Williams et al, 2021). Neutrophils use several antimicrobial mechanisms, including phagocytosis, reactive oxygen species (ROS) generation, and exocytosis of antimicrobial peptides from membrane‐bound granules (degranulation; Kaplan & Radic, 2012; Ley et al, 2018). An additional mechanism, the formation of neutrophil extracellular traps (NETs) is a distinct cell death program in which neutrophils extrude web‐like structures composed of decondensed chromatin decorated with antimicrobial peptides and enzymes (neutrophil elastase, cathepsin G, myeloperoxidase, and others; Brinkmann et al, 2004; Kaplan & Radic, 2012; Sollberger et al, 2018). Deregulated NET formation has been proposed to contribute to an array of inflammatory conditions including autoimmunity, thrombosis, malignancy, and sepsis (Fuchs et al, 2010; Amulic et al, 2012; Gupta & Kaplan, 2016; Jorch & Kubes, 2017; Byrd et al, 2019). Enhanced NET release and impaired NET clearance in the blood and tissues of systemic lupus erythematosus (SLE) patients are implicated in the progression of vascular damage and atherosclerosis as well as in lupus nephritis (Hakkim et al, 2010; Mistry et al, 2019; O'Neil et al, 2019). Similarly, unregulated NET formation contributes to an impaired healing response in diabetic wound healing (Wong et al, 2015; Fadini et al, 2016). Upon wounding, diabetic mice produce higher levels of NETs that is rescued by treatment with DNAse‐1 (Wong et al, 2015). An excess of other NET components, including neutrophil elastase and proteinase‐3, were found to be predictive indicators of poor healing outcome in patients with diabetes (Fadini et al, 2016). However, the priming of neutrophils to enhance NET formation during diabetic wound healing is poorly understood.
Forkhead Box M1 (FOXM1) is a transcriptional activator of proliferation in an array of cell types and is overexpressed in many cancers (Liao et al, 2018). FOXM1 is also involved in the acute wound resolution of hyperoxic lung injury (Xia et al, 2015), as well as hepatocyte proliferation (Gieling et al, 2010) and leukocyte function (Ren et al, 2010; Gage et al, 2018) during toxic injury of the liver. Transgenic mice with FOXM1 deletion in the myeloid cell lineage also show significantly delayed liver repair (Kalin et al, 2011). In addition, FOXM1 has been shown to regulate ROS levels by inducing expression of ROS scavenger genes to control oxidative stress (Park et al, 2009; Smirnov et al, 2016; Choi et al, 2020). We previously showed that inhibition of FOXM1 in diabetic mouse models of cutaneous wounding resulted in delayed wound closure and decreased recruitment of neutrophils and macrophages (Sawaya et al, 2020). Further, comparative transcriptomic analysis of human DFUs with acute wounds of skin and oral mucosa implicated FOXM1 as a regulator of the neutrophil response in diabetic wound healing (Sawaya et al, 2020).
Here, we investigated the role of FOXM1 in regulating NET formation in acute wounds and DFUs. Comparisons of neutrophil‐associated transcriptional signatures between DFUs and acute skin wounds showed partial or complete inhibition of transcripts involved in neutrophil function and response in DFUs. In addition, pharmacological inhibition of FOXM1 increased ROS levels and induced NET formation in human neutrophils, suggesting that loss of FOXM1 contributes to inhibition of healing in DFUs through increased NETs. Furthermore, we identified triggering receptor expressed on myeloid cell‐1 (TREM1) as a neutrophil‐specific regulator suppressed in DFUs. Activation of TREM1 promoted FOXM1+ neutrophil recruitment, decreased NETs, and enhanced diabetic wound healing in vivo, demonstrating a novel pathway for regulating NET formation in diabetic wounds. Moreover, TREM1 expression correlated with the clinical outcomes of healing in patients with DFUs. Our data identified a novel pathway for regulating NET formation during diabetic wound healing through TREM1/FOXM1. This regulatory pathway serves as a potential diagnostic biomarker to predict clinical patient outcomes and as a target for development of new therapies that can reprogram chronic, nonhealing DFUs into healing‐competent wounds.
Results and Discussion
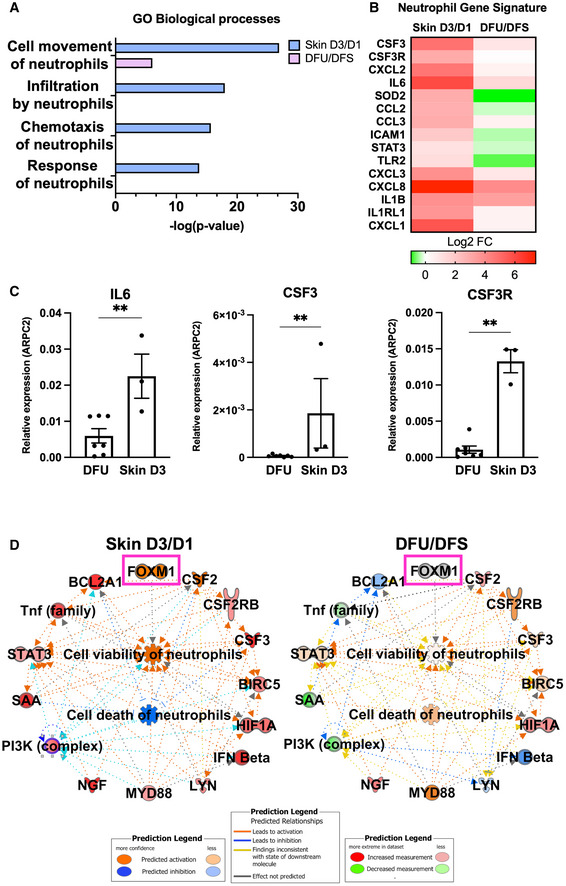
Deregulated neutrophil response in DFUs
We have previously demonstrated that decreased neutrophils in DFUs results in an overall poorly controlled inflammatory response contributing to inhibition of healing (Sawaya et al, 2020). To further investigate this, we assessed the transcriptomic differences by RNA‐seq of human DFU samples compared to human skin acute wounds at the day 3 and focused on processes involved in neutrophil function. Enriched GO biological processes in skin acute day 3 wounds included cell movement of neutrophils, infiltration by neutrophils, chemotaxis of neutrophils, and response of neutrophils that were either found absent or partially regulated in DFUs (Fig 1A). Several transcripts associated with a neutrophil gene signature were found to be either suppressed or partially regulated in DFUs compared to skin acute day 3 wounds (Fig 1B). Among them are included the cytokines CSF3, CSF3R, and IL6 and the chemokines CXCL2, CXCL3, CXCL8, and CCL2. In addition, the STAT3 transcription factor and TLR2 were found deregulated in DFUs compared to skin acute day 3 wounds. Next, we validated our findings by qPCR of several genes related to the neutrophil gene signature and determined these were induced in skin acute day 3 wounds, but inhibited in DFUs (Fig 1C). Moreover, Ingenuity Pathway Analysis (IPA) revealed cell viability of neutrophils' pathway to be upregulated and cell death of neutrophils' pathway to be inhibited in skin acute day 3 wounds, whereas the opposite was observed in DFUs (Fig 1D). Neutrophils are the first immune cells to arrive after injury and are involved in killing microbes and activating other cell types involved in the repair process (Brinkmann et al, 2004; Kaplan & Radic, 2012; Wilgus et al, 2013; Ley et al, 2018). We previously demonstrated that the immune landscape in DFUs is deficient in neutrophils due to lack of the FOXM1 transcriptional regulator. IPA analysis of cell death and viability pathways indicate that the absence of neutrophils in DFUs is due to increased neutrophil death linked to loss of FOXM1. Taken together, these results indicate that the neutrophil response present in skin acute wounds that facilitates healthy‐wound healing is deregulated in chronic nonhealing DFUs.
Figure 1. Deregulated neutrophil response in human tissue samples obtained from diabetic foot ulcers.
-
AEnriched GO processes from human skin acute day 3 wounds compared to human DFU demonstrates processes involved in neutrophil function to be deregulated in DFUs compared to acute wounds.
-
BNeutrophil gene signature comparing human skin acute day 3 wounds to human DFUs demonstrating decreased presence of neutrophils in human DFUs.
-
CqPCR validations of neutrophil genes. n = 7 DFUs and n = 3 skin acute day 3 wounds. **P < 0.01 (two‐tailed unpaired Student's t‐test). Data presented as mean ± SD.
-
DIngenuity Pathway Analysis of predicted network shows activation of cell viability of neutrophils and inhibition of cell death of neutrophils in human skin wounds compared to activation of cell death of neutrophils in human DFUs.
FOXM1 inhibition promotes NET formation
We have previously shown that blocking the function of FOXM1 inhibits neutrophil responses in DFUs (Sawaya et al, 2020). Neutrophils from patients with DFUs are known to undergo NET formation resulting in tissue damage and impaired healing (Fadini et al, 2016). Although apoptosis is the primary form of neutrophil cell death, cell death by NET formation does occur during acute wound healing as a mechanism for eliminating pathogens (Wong et al, 2015), which must be tightly regulated to ensure a proper immune response. Diabetes primes neutrophils to undergo NETosis (Wong et al, 2015); as a result, the balance between neutrophil apoptosis and NETosis in diabetic wounds shifts in favor of NETosis, producing an improper neutrophil response and inhibition of wound healing in DFUs. Therefore, we investigated if inhibition of FOXM1 regulates NET formation and contributes to the decreased neutrophil response. We isolated human peripheral blood neutrophils from healthy donors and utilized a pharmacological approach using a specific FOXM1 inhibitor, FDI‐6. Specificity of FDI‐6 inhibition of FOXM1 was validated by qPCR and showed decreased expression of FOXM1 and its target gene SOD2 (Fig EV1). Treatment with FDI‐6 resulted in increased NET release compared to vehicle control, implicating FOXM1 in the regulation of NET formation (Fig 2A). NETs were visualized by immunofluorescence staining of citrullinated histone‐3 (citH3), a marker of neutrophils undergoing NET formation (Wang et al, 2009), with the neutrophil marker elastase (Fig EV2A). We further validated NET formation after FOXM1 inhibition using the streptozoticin (STZ)‐induced diabetic mouse model and assessed citH3 by immunofluorescence staining. As expected, we found increased citH3 in diabetic wounds. Topical treatment of wounds with FDI‐6 resulted in increased citH3 compared to vehicle‐treated nondiabetic wounds (Fig EV2B). These findings demonstrate that inhibition of FOXM1 promotes NET formation.
Figure EV1. Inhibition of FOXM1 with FDI‐6 suppresses FOXM1 expression and downstream target genes in human neutrophils.
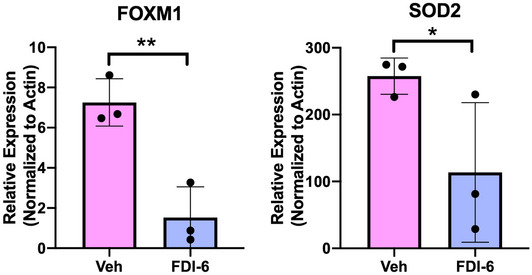
Human neutrophils were treated with FDI‐6 for 3 h. Vehicle (DMSO) served as a control. Treatment with FDI‐6 inhibits FOXM1 expression and its downstream target SOD2. n = 3 biological replicates. Data presented as mean ± SD. *P < 0.05. **P < 0.01 (two‐tailed unpaired Student's t‐test).
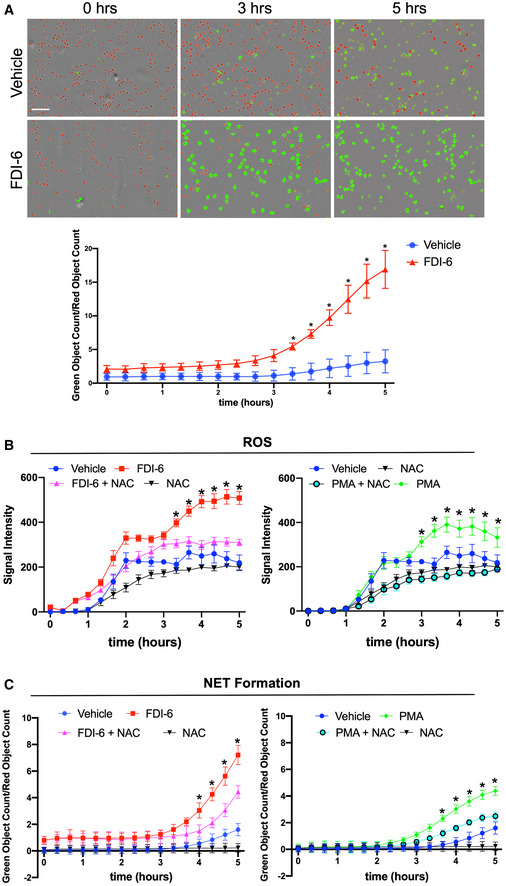
Figure 2. Inhibition of FOXM1 increases ROS and induces NET formation.
-
ARepresentative images of human neutrophils treated with the FOXM1 inhibitor, FDI‐6. Vehicle (DMSO) served as a control. Neutrophils undergoing NET formation are visualized in green and live neutrophils are visualized in red. Quantification was performed by normalizing the number of neutrophils undergoing NET formation to the number of live neutrophils. Neutrophils from n = 3 different blood donors were isolated, pooled, and performed in triplicate. *P < 0.05 (two‐way ANOVA followed by Tukey's post‐hoc test). Data presented as mean ± SD. (Scale bar: 100 μm).
-
BQuantification of ROS levels in human neutrophils treated with FDI‐6 or in combination with N‐acetylcysteine (NAC). Phorbol 12‐myristate 13‐acetate (PMA) treatment served as a positive control. Data presented as mean ± SD. Neutrophils from n = 3 different blood donors were isolated, pooled and performed in triplicate. *P < 0.05 (two‐way ANOVA followed by Tukey's post‐hoc test).
-
CQuantification of NET formation in human neutrophils treated with FDI‐6 or in combination with NAC. PMA treatment served as a positive control. Neutrophils from n = 3 different blood donors were isolated, pooled, and performed in triplicate. Data presented as mean ± SD. *P < 0.05 (two‐way ANOVA followed by Tukey's post‐hoc test).
Figure EV2. Inhibition of FOXM1 induces NET formation in vitro and in vivo .
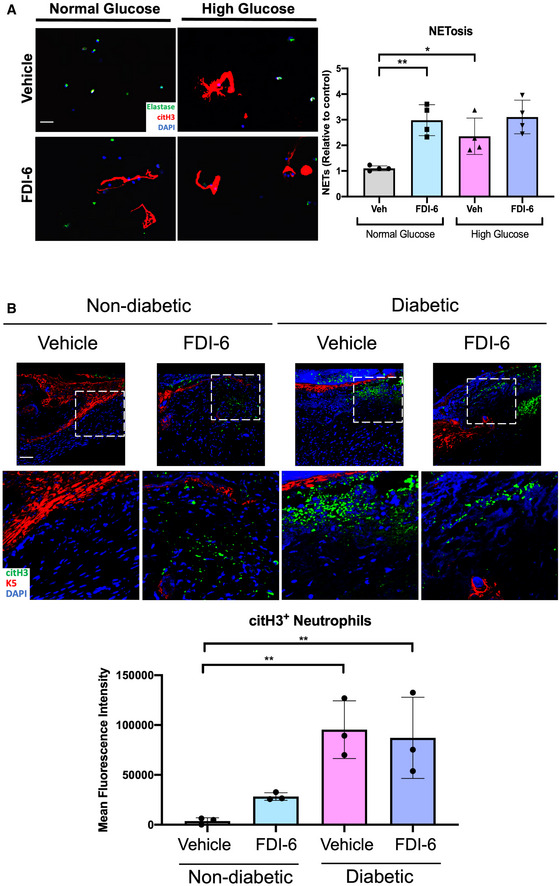
-
AHuman neutrophils cultured in either normal glucose media (5 mM glucose) or high glucose media (25 mM) to recapitulate diabetic conditions and treated with FDI‐6. Vehicle (DMSO) served as a control. NETs were visualized by immunostaining with citH3 and the neutrophil marker, elastase. (Scale bar: 50 μm). Quantification of n = 4 biological replicates per group were performed. Data presented as mean ± SD. *P < 0.05, and **P < 0.01 (one‐way ANOVA followed by Sidak post‐hoc test).
-
BImmunofluorescence staining of vehicle (DMSO) and FDI‐6 treated wounds in STZ‐induced diabetic mice at day 4 showing basal keratin marker K5, and NET marker citH3. Treatment of diabetic wounds with FDI‐6 resulted in increased citH3 compared to vehicle treated wounds. (Scale bar: 50 μm). Quantification of wound areas in n = 3 wounds per group were performed with Fiji software. Data presented as mean ± SD. **P < 0.01 (one‐way ANOVA followed by Sidak post‐hoc test).
To determine the mechanism by which FOXM1 regulates NET formation, we assessed reactive oxygen species (ROS) levels after FOXM1 inhibition. ROS generation is implicated in the pathways leading to NET formation (Brinkmann et al, 2004; Lood et al, 2016). Pathway analysis revealed suppression of FOXM1 in DFUs results in increased ROS compared to acute skin wounds (Fig EV3A). Therefore, we measured ROS levels and NET formation in neutrophils treated with FDI‐6 in the presence or absence of N‐acetylcysteine (NAC), a known inhibitor of ROS and NET formation (Fuchs et al, 2007; Lim et al, 2011). Neutrophils treated with phorbol 12‐myristate 13‐acetate (PMA), a known inducer of NET formation (Fuchs et al, 2007; Lim et al, 2011), in the presence or absence of NAC were used as controls. Inhibition of FOXM1 induced ROS and NET formation to levels comparable to PMA (Figs 2B and C, and EV3B). Combination treatment of FDI‐6 with NAC significantly inhibited ROS levels and NET formation (Fig 2B and C). Furthermore, IPA analysis identified FOXM1‐regulated genes known to be involved in ROS generation, including SOD2 and CAT that function to inhibit ROS accumulation in acute skin acute day 3 wounds (Fig EV3A). They were found to be inhibited in DFUs (Fig EV3A). These results support that inhibition of FOXM1 leads to increased ROS levels resulting in NET formation in neutrophils.
Figure EV3. FOXM1 regulates ROS levels during wound healing.

-
AFOXM1 regulates genes involved in reducing ROS levels and is activated in human skin acute day 3 wounds compared to suppression in human DFUs.
-
BRepresentative images of human neutrophils treated with the FOXM1 inhibitor, FDI‐6, in presence or absence of NAC. PMA served as positive control. Reactive oxygen species formation are visualized in green. Representative images of human neutrophils treated with the FOXM1 inhibitor, FDI‐6, in presence or absence of NAC. PMA served as positive control. NET formation are visualized in green. (Scale bar: 100 μm).
TREM1, inhibited in DFUs, stimulates neutrophil healing responses that are linked to FOXM1
Next, we performed IPA analysis to determine potential upstream regulators responsible for regulating FOXM1 pathway in neutrophils that could serve as a potential therapeutic target for patients with DFUs. To further investigate this, we assessed the transcriptomic differences in DFUs compared to skin acute day 3 wounds by RNA‐seq and performed IPA analysis to determine specific upstream regulators involved in regulating neutrophil responses during wound healing. Among them, the cytokine‐encoding transcripts included TNF, IL6, IL1B, IFNγ, and CSF2, which were found to be either suppressed or incompletely activated in DFUs (Fig 3A). We also found the transcription factors NFκB, STAT3, STAT1, and FOXM1 as well as P38 MAPK and ERK1/2 pathways to be suppressed or less activated in DFUs, in contrast to being found highly upregulated in human skin acute day 3 wounds. Moreover, we identified TREM1, a potent amplifier of the inflammatory response known to be highly expressed in neutrophils, to be significantly upregulated in skin acute day 3 wounds, but suppressed in DFUs (Fig 3A). TREM1 is a membrane‐bound receptor expressed on myeloid lineage of cells and is a potent stimulator of the inflammatory response (Bouchon et al, 2000, 2001; Colonna, 2003). TREM1 activation triggers release of pro‐inflammatory molecules such as IL‐8 and TNF (Colonna, 2003; Carrasco et al, 2019) that we found were inhibited in DFUs. We performed further IPA analysis to determine the role of TREM1 in neutrophil function during wound healing. We identified several enriched processes that are involved in regulation of neutrophil functions that include cell movement of neutrophils, recruitment of neutrophils, and immune response of cells (Fig 3B). These processes were strongly activated in skin day 3 acute wounds and suppressed in DFUs. To determine if TREM1 is linked to FOXM1, we performed analysis connecting TREM1 pathway to FOXM1 and found several genes previously described to be regulated by TREM1 that could promote FOXM1 activation, including IL1B, IL6, CDK1, AREG, SOD2, and CXCL8 (Fig 3C). Taken together, these results indicate that TREM1 may stimulate the neutrophil response present in skin acute wounds through FOXM1 activation to facilitate healthy wound healing. Furthermore, its downregulation in chronic DFUs may be important contributor of nonhealing wounds.
Figure 3. TREM1 stimulates the neutrophil response and is linked to FOXM1, that is inhibited in DFUs.
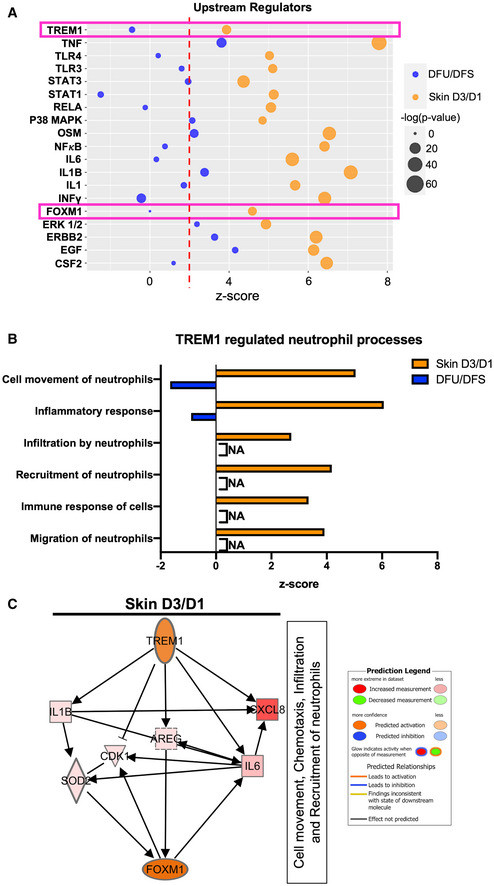
-
AUpstream regulators found to be activated in human skin wounds that are suppressed or partially regulated in human DFUs involved in neutrophil response.
-
BTREM1 functions related to neutrophil response shows activation in human skin wounds compared to suppression in human DFUs.
-
CTREM1 predicted network connecting downstream target genes to their downstream biological processes leading to FOXM1 activation.
TREM1 activation increases FOXM1+ neutrophil recruitment, decreases NETs, and enhances wound healing in diabetic mice
To corroborate the human data with an in vivo model, we investigated the effect of TREM1 activation on wound healing using the db/db diabetic mouse model of wound healing. We utilized a pharmacological approach for the activation with the ⍺‐TREM1 activator (Bouchon et al, 2000). ⍺‐TREM1 was topically applied to full‐thickness wounds created on dorsal skin of mice. We studied the kinetics of wound healing in wounds treated with ⍺‐TREM1 when compared to isotype‐matched IgG treatment (vehicle). Treatment with ⍺‐TREM1 significantly enhanced wound healing in diabetic wounds compared to control IgG at days 2 and 4 postwounding (Fig 4A), with day 4 being the time point of a peak inflammatory response.
Figure 4. Activation of TREM1 enhances wound healing and increases FOXM1+ neutrophils in the wounds of diabetic mice.
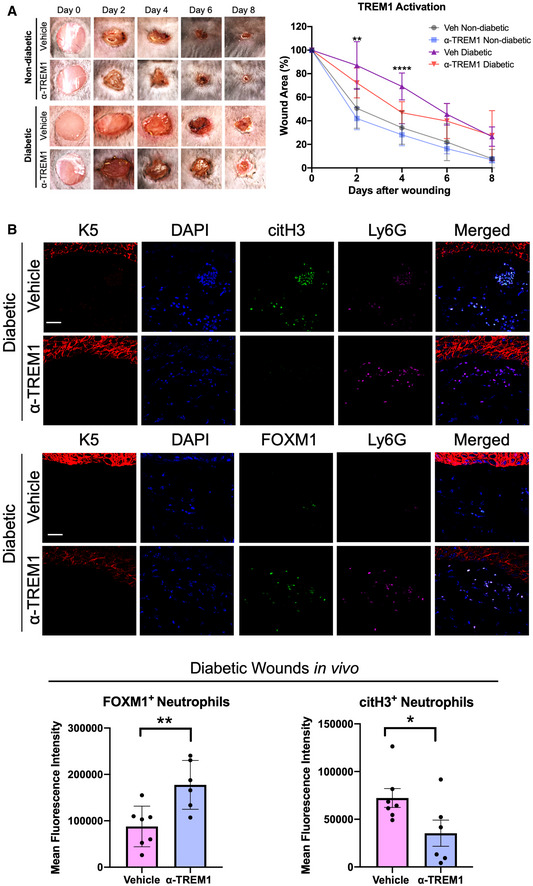
-
ARepresentative images of wounded skin after topical treatment with either vehicle (IgG isotype control) or ⍺‐TREM1 activator at 0, 2, 4, 6, and 8 days after wounding. Percent of wound area at each time following vehicle or ⍺‐TREM1 activator treatment relative to the original wound area. Quantification of wound areas in n = 10 for vehicle diabetic and 12 wounds for ⍺‐TREM1‐treated wounds were performed with Fiji software. Data presented as mean ± SD. **P < 0.01, and ****P < 0.0001 (two‐way ANOVA followed by Tukey's post‐hoc test).
-
BRepresentative pictures of vehicle (IgG isotype control) and ⍺‐TREM1‐treated diabetic wounds at day 4 show basal keratin marker K5, and neutrophil marker Ly6G, FOXM1, and citH3. Treatment of wounds with ⍺‐TREM1 resulted in increased FOXM1+ and decreased citH3+ neutrophils compared to vehicle‐treated diabetic wounds. (Scale bar: 50 μm). Quantification of mean fluorescence intensity was performed with Fiji software. n = 7 diabetic vehicle wounds and 6 diabetic ⍺‐TREM1 wounds. Data presented as mean ± SD. *P < 0.05, *P < 0.05, and **P < 0.01 (two‐tailed unpaired Student's t‐test).
Next, we tested the effect of TREM1 activation on neutrophil recruitment in vivo, by assessing the presence of FOXM1+ neutrophils in day 4 wounds in mice treated with either vehicle or ⍺‐TREM1. No significant differences were found in nondiabetic wounds treated with ⍺‐TREM1 compared to vehicle (Fig EV4). We found increased presence of FOXM1+ neutrophils in ⍺‐TREM1‐treated diabetic wounds compared to vehicle‐treated controls (Fig 4B). Moreover, to assess the NET formation, we quantified FOXM1+ and citH3+ neutrophils. We found FOXM1+ neutrophils inversely correlated with citH3+ neutrophils in ⍺‐TREM1‐treated diabetic wounds, supporting that FOXM1 inhibits NET formation (Fig 4B). These results indicate that TREM1 activation increases FOXM1+ neutrophil response in diabetic wounds by inhibiting NET formation and stimulating healing (Fig 4B).
Figure EV4. Treatment with ⍺‐TREM1 has no significant effect on FOXM1 and citH3 in nondiabetic wounds.
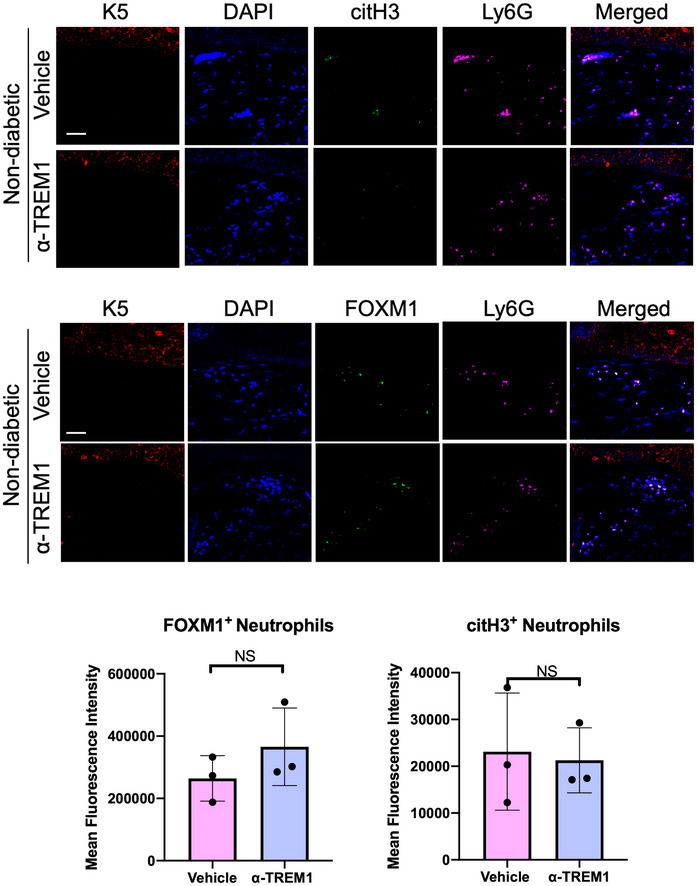
Representative pictures of vehicle (IgG isotype control) and ⍺‐TREM1‐treated nondiabetic wounds at day 4 show basal keratin marker K5, and neutrophil marker Ly6G, FOXM1, and citH3. Treatment of wounds with ⍺‐TREM1 resulted in no significant differences in FOXM1+ and citH3+ neutrophils compared to vehicle‐treated wounds. (Scale bar: 50 μm). Quantification of mean fluorescence intensity was performed with Fiji software. n = 3 wounds per group. Data presented as mean ± SD (two‐tailed unpaired Student's t‐test).
Although our data demonstrate TREM1 activation to be associated with increased wound healing in diabetic wounds by inhibiting NET formation, other factors are also known to contribute to inhibition of healing in diabetic wounds. In this complex and mutlifactorial disease, decreased angiogenesis and keratinocyte deregulation are among the hallmarks of DFUs that contribute to impaired wound healing (Eming et al, 2014). Reduced angiogenesis leads to increased cell death due to loss of balance between pro‐ and anti‐angiogenic factors (Eming et al, 2014). Furthermore, keratinocytes in DFUs lose their migratory capacity and fail to close the wound, contributing to increased risk of infection and amputation (Eming et al, 2014). TREM1 has been shown to be expressed on endothelial cells and keratinocytes (Hyder et al, 2013), suggesting that TREM1 activation may exert broad pro‐healing effects in diabetic wounds. Future studies are needed to address the role of TREM1 in regulating these processes during diabetic wound healing.
TREM1 expression and neutrophil recruitment contributes to the clinical healing outcome of DFUs
We next determined if inhibition of TREM1 and its downstream targets contribute to the nonhealing clinical outcome of patients with DFUs. We obtained tissue samples from patients in which the healing outcome was determined by a surrogate endpoint, the percent reduction in wound size after 4 weeks of standard wound care (Margolis et al, 2003; Stojadinovic et al, 2013). DFUs were grouped into two categories as either healing, in which wound reduction of wound size was greater than 50%; or nonhealing, in which wound reduction was less than 50%. We performed qPCR to quantify TREM1 and genes encoding for molecules involved in neutrophil chemotaxis and recruitment, CXCL8, CXCR1, and CXCR2, in healing and nonhealing. We found TREM1, CXCL8, CXCR1, and CXCR2 expression to be induced in healing, but suppressed in nonhealing (Fig 5A). To further validate our findings, we performed immunofluorescence staining for TREM1 the NET marker citH3. We found increased presence of TREM1 in tissue obtained from healing DFUs when compared to nonhealing DFUs (Fig 5B). Moreover, citH3 was found decreased in healing DFUs compared to nonhealing DFUs (Fig 5C). Taken together, our data support that TREM1 expression and NET formation are associated with the clinical healing outcome of patients with DFUs.
Figure 5. TREM1 expression is associated with the clinical outcome of healing in DFUs.
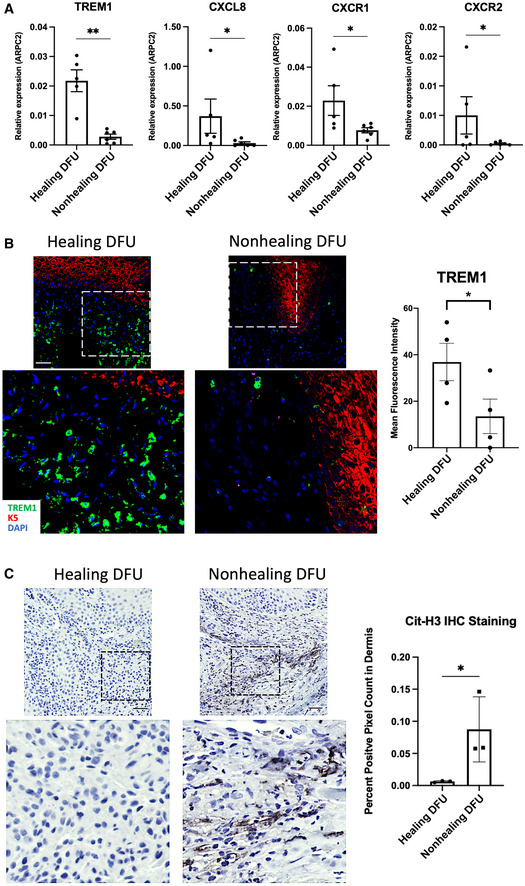
-
AqPCR of TREM1 and genes involved in neutrophil recruitment demonstrate increased expression in healing DFUs compared to nonhealing DFUs (n = 5 healing and n = 6 nonhealing). Data presented as mean ± SD. *P < 0.05. **P < 0.01 (two‐tailed unpaired Student's t‐test).
-
BRepresentative images of healing and nonhealing DFUs show basal keratin marker K5 and TREM1 and corresponding quantification from healing (n = 4) and nonhealing (n = 4) is shown in the graph. Data presented as mean ± SEM. *P < 0.05 (two‐tailed unpaired Student's t‐test). (Scale bar: 50 μm).
-
CRepresentative images of cit‐H3 immunohistochemistry show increase staining in nonhealing DFUs when compared to healing DFUs, which was confirmed by corresponding quantification from healing (n = 3) and nonhealing (n = 3), shown in the graph. Data presented as mean ± SD. *P < 0.05 (two‐tailed unpaired Student's t‐test). (Scale bar: 50 μm).
In this study, we investigated the role of TREM1 and FOXM1 signaling in regulating NET formation during diabetic wound healing. We performed a comprehensive comparative analysis between tissue biopsies derived from patients with DFUs and human skin acute wounds using next‐generation sequencing. We identified FOXM1 as a novel regulator of NET formation through modulation of ROS levels to promote a healthy‐neutrophil immune response during wound healing. In addition, we identified TREM1 to be linked to the FOXM1 pathway in this context. Activation of TREM1 increased recruitment of FOXM1+ neutrophils and enhanced diabetic wound healing in vivo. We previously found FOXM1 to be downregulated in DFUs, leading to decreased neutrophil response, suggesting modulation of the TREM1/FOXM1 network as a potential therapeutic target for restoring a healthy neutrophil response in diabetic wounds. Moreover, TREM1 expression correlated with the healing outcome of DFUs, further supporting TREM1 as an important regulator of promoting a proper neutrophil immune response during wound healing. Our data demonstrate that lack of TREM1/FOXM1 network in the DFU leads to increased NET formation of neutrophils contributing to inhibition of wound healing. In addition, we identify a novel regulation of NET formation during diabetic wound healing by the FOXM1 network, underscoring its role in pathophysiology of DFUs. As such the TREM1/FOXM1 regulatory network can serve as potential diagnostic biomarkers and as therapeutic targets. Targeting this network may achieve restoration of acute healing response in DFUs, reprogramming them into healing competent wounds.
Materials and Methods
Mice
All animal studies were carried out according to the protocol approved by the Animal and Care Committee at the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Db/db mice were purchased from Jackson Laboratory. Streptozotocin‐induced diabetic mice were generated as previously described (Sawaya et al, 2020). Both male and female mice were used in the wound healing studies. Treatments were performed at 7–9 weeks of age and all experiments were conducted using littermate controls.
Wound healing assay in vivo
Full‐thickness wounds were created as previously described (Iglesias‐Bartolome et al, 2018; Uchiyama et al, 2019; Sawaya et al, 2020). Briefly, mice were anesthetized, and hair was shaved on dorsal skin and cleaned with 70% ethanol. Wounds were created using 6 mm full‐thickness excisional wounds with sterile punch biopsies (Integra Miltex) and topically treated with either 10 μg/ml anti‐TREM1 antibody (R&D Systems; MAB1187) or isotype control anti‐IgG (Invitrogen; 31903) dissolved in 1× sterile PBS. Treatments were applied every 2 days and digitally photographed at indicated time points and wound areas were measured using Fiji. Changes in wound area are expressed as percentages of initial wound area.
Patient demographics
Full‐thickness DFU (n = 13, mean age ± standard deviation = 56 ± 13, 13 males) and DFS (n = 8, mean age ± standard deviation = 66 ± 13, 7 males, 1 female) samples were obtained from patients receiving standard care at the University of Miami Hospital Wound Clinic, as previously described (Sawaya et al, 2020). The protocols including written informed consent were approved by the university Institutional Review Board (IRB #20140473; #20090709). Inclusion criteria for DFU included (i) diabetes mellitus; (ii) an ulcer on the plantar aspect of their foot that is larger than 0.5 cm2; (iii) neuropathy; (iv) age 21 years or older; (v) wound duration > 4 weeks; and (vi) hemoglobin A1c: ≤ 13.0%. Ulcers with clinical signs of infection were excluded. Exclusion criteria for DFU were (i) active cellulitis; (ii) osteomyelitis; (iii) gangrene; (iv) vascular insufficiency (defined as an ankle‐brachial index (ABI) < 0.7 and for those with an ABI > 1.3; (v) revascularization to the ipsilateral lower extremity in the last 6 weeks; and (vi) any experimental drugs taken or applied topically to the wound for 4 weeks preceding the study.
Real‐time reverse transcriptase PCR
Human skin, human DFU specimens, and human neutrophils were lysed with TRIAzol and RNA was isolated using the Qiagen RNeasy kit according to the manufacturer's instructions (Sawaya et al, 2020). RNA (1.0 μg) from human skin, human DFUs, or human neutrophils was reverse transcribed using a qScript cDNA kit (QuantaBio) and real‐time PCR was performed in triplicates using the Bio‐Rad CFX Connect thermal cycler and detection system and a PerfeCTa SYBR Green Supermix (QuantaBio). Relative expression was normalized for levels of ARPC2 or Actin where indicated. The primer sequences for ARPC2 are forward primer (5′‐TCCGGGACTACCTGCACTAC‐3′) and reverse primer (5′‐GGTTCAGCACCTTGAGGAAG‐3′); CSF3 forward primer (5′‐AAGCTGGTGAGTGAGTGTG‐3′) and reverse primer (5′‐GGGATGCCCAGAGAGTGTC‐3′); CSF3R forward primer (5′‐TTGCAGCCCCAACAGGAAG‐3′) and reverse primer (5′‐ATGATTGTGGGCACCCAGG‐3′); IL6 forward primer (5′‐CATCCTCGACGGCATCTCAG‐3′) and reverse primer (5′‐ACCAGGCAAGTCTCCTCATTG‐3′); TREM1 forward primer (5′‐TGCCCACTCTATACCAGCCC‐3′) and reverse primer (5′‐GTTGAACACCGGAACCCTGATG‐3′); CXCL8 forward primer (5′‐GAAGTTTTTGAAGAGGGCTGAGA‐3′) and reverse primer (5′‐TTGCTTGAAGTTTCACTGGCATC‐3′); CXCR1 forward primer (5′‐TGGCCGGTGCTTCAGTTAG‐3′) and reverse primer (5′‐AGGGGCTGTAATCTTCATCTGC‐3′); CXCR2 forward primer (5′‐CTAAGTGGCACCTGTCCTGG‐3′) and reverse primer (5′‐TTCTGACCTGGGTTGCAAGG‐3′); FOXM1 forward primer (5′‐CGTCGGCCACTGATTCTCAAA‐3′) and reverse primer (5′‐GGCAGGGGATCTCTTAGGTTC‐3′); SOD2 forward primer (5′‐GCACTAGCAGCATGTTGAGC‐3′) and reverse (5′‐TTGATGTGAGGTTCCAGGGC‐3′); Actin forward primer (5′‐CACCAACTGGGACGACAT‐3′) and reverse primer (5′‐ACAGCCTGGATAGCAACG‐3′).
Neutrophil isolation
Human peripheral blood from male and females was collected by venipuncture in heparinized tubes from healthy control subjects recruited at the Clinical Center, NIH, Bethesda, MD. Neutrophils were isolated by layering 20 ml of blood on top of 20 ml of PolymorphPrep™ (Progen) and centrifuged at 500 g for 35 min. The layer containing the neutrophil fraction was obtained and added to equal volume of 1× Hanks Balanced Salt Solution (HBSS) diluted with equal volumes of water to restore neutrophils to normal osmolality. Neutrophil suspension was centrifuged for 5 min at 400 g and resuspended with 0.2% NaCl for red blood cell lysis for 1 min. Equal volumes of 1× HBSS was added and neutrophil suspension was centrifuged for 5 min at 400 g and resuspended in RPMI media. Neutrophils from three different blood donors were isolated, pooled, and performed in triplicate per each condition.
Quantification of NET formation
Neutrophils were treated with either 5 μM of FDI‐6 (Sigma Aldrich), 0.5 μM of PMA (Sigma Aldrich), in the presence or absence of 5 mM of N‐acetylcysteine (Sigma Aldrich). Vehicle (DMSO) served as a control. Neutrophils were pretreated for 30 min with N‐acetylcysteine. For experiments involving TREM1, neutrophils were treated with 10 μg/ml anti‐TREM1 antibody (R&D Systems) in presence or absence of 5 μM of FDI‐6. Isotype control anti‐IgG (Bio‐Rad) served as a control. SYTOX green dye (Invitrogen) was added to a final concentration of 0.2 μM. NUCLEAR‐ID Red dye (Enzo Life Sciences) was added at a dilution of 1 μl into 1.5 ml media and was used for staining nuclei of live neutrophils. The IncuCyteS3 instrument software (Essen BioScience) was used to measure NET formation using a previously described protocol (Gupta et al, 2018). Three image sets from distinct regions per well using a 20× objective lens were taken every 20 min and each condition was run in quadruplets. The filters applied to the green channel excluded objects below the radius of 10 μm, fluorescence threshold of 1.00 green corrected units, and area of 100 μm2. This minimum area threshold for green was used to recognize the decondensed chromatin of cells. NET formation was quantified by normalizing the green object counts to the red object counts to obtain a ratio of the number of neutrophils undergoing NET formation to the total number of live neutrophils.
Immunofluorescence staining and visualization of NETs in vitro
Neutrophils were isolated from human blood and grown in either normal glucose (5 mM glucose) or high glucose (25 mM glucose) media and treated with vehicle (DMSO) or 5 μM of FDI‐6 for 3 h. Neutrophils were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.1% TritonX‐100 for 5 min. NETs were detected by immunofluorescence staining using anti cit‐Histone 3 antibody (Abcam, 1:300; ab5103) and anti Elastase (Novus Biologicals, 1:300; NBP2‐53193). NETs were visualized using a Leica TCS SP8 confocal microscopy. Quantification of NETs was carried out using the NETosis assay commercially available kit (Abcam) according to manufacturer's protocol.
Reactive oxygen species assay
Reactive oxygen species (ROS) was measured using ROS‐ID Total ROS/Superoxide Detection Kit (Enzo Life Sciences). Neutrophils were isolated as described above and the ROS‐ID Total ROS/Superoxide Detection Kit was used according the manufacturer's instructions. ROS levels were measured using IncuCyteS3 instrument software (Essen BioScience) and quantified by normalizing green object counts to the total number of live neutrophils.
Immunohistochemistry
Paraffin embedded tissue sections of discarded DFUs were used for staining with anti‐Keratin 5 (1:750; LSBio; LS‐C22715), and anti TREM1 (1:1,000; Abcam; ab225861). Murine wounds were excised at day 4 postwounding and fixed in 4% paraformaldehyde overnight at 4°C and sections were used for anti Ly6G (1:500; BD Pharmingen; 551459), anti FOXM1 (1:500; Cell Signaling; 20459S), and anti citH3 (1:1,000; Abcam; ab5103) staining. Stainings were visualized with either Alexa Fluor 488‐conjugated goat anti‐rabbit antibody (1:500; Invitrogen; A21206), Alexa Fluor 555‐conjugated goat anti‐guinea pig antibody (1:500; Invitrogen; A21435), Alexa Fluor 647‐conjugated goat anti‐mouse antibody (1:500; Invitrogen; A21235) and mounted with VECTASHIELD antifade mounting media with DAPI (Vectorlabs) to visualize cell nuclei. Specimens were analyzed using a Leica TCS SP8 confocal microscope.
For peroxidase stainings, slides were deparaffinized and rehydrated in xylene and decreasing ethanol solutions. Peroxidase was quenched using 0.3% H2O2 in methanol for 20 min. Antigen retrieval was performed for 30 min in 95° water bath in 10 mM of TRIS, 1 mM of EDTA, 0.1% tween pH = 9 buffer. Slides were washed in PBS and blocked/permeabilized for 10 min with 10 mg/ml gelatin and 1.25% Triton in PBS. Slides were blocked 10 min in 5% FBS, 5% Goat Serum, and 7.5% Bockhen in 1× PBS. Slides were blocked for additional 10 min using 50% Background Sniper and 50% Background Punisher solution. Slides were incubated overnight in anti citH3 (1:10,000; Abcam; ab5103) in Background Enhancer at 4°C. Slides were washed 3× in PBST and then blocked with 5% goat serum for 10 min. Rabbit on pharma polymer (secondary) was added for 20 min and washed 3× with PBST. Immunoperoxidase reaction was stimulated with DAB substrate. Slides were counterstained with Mayers Hemoxatylin and washed/dehydrated with successive ethanol solutions and xylene washes. Dermis peroxidase staining was quantified using deprecated, positive pixel count function in QuPath software.
Statistical analysis
Pathway enrichment statistics were calculated within the Ingenuity software package using Fisher's exact test with Benjamini‐Hochberg correction for multiple testing. Upstream regulators and gene ontology enrichment P‐values were similarly calculated within IPA using Fisher's exact test. Statistics for NET and ROS assays studies were performed using one‐way ANOVA followed by Tukey's posthoc test. Statistics for qPCR validations comparing DFU to acute skin wounds were performed using Mann–Whitney U two‐tailed test. Statistics for qPCR comparing healing and nonhealing DFUs were performed using two‐tailed unpaired t test. Statistics for wound healing assay and NET formation assays were performed using either a one‐way ANOVA followed by Sidak post‐hoc test or a two‐way ANOVA followed by Tukey's post‐hoc test where indicated.
Author contributions
Andrew P Sawaya: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Rivka C Stone: Data curation; formal analysis; funding acquisition; investigation; visualization; methodology; writing – original draft; writing – review and editing. Spencer Mehdizadeh: Data curation; formal analysis; validation; investigation; writing – original draft. Irena Pastar: Data curation; formal analysis; validation; investigation. Stephen Worrell: Data curation; formal analysis; validation; investigation. Nathan C Balukoff: Data curation; formal analysis; validation; investigation. Mariana J Kaplan: Resources; formal analysis; validation. Marjana Tomic‐Canic: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Maria I Morasso: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH (ZIA‐AR041124 to MIM), (R01NR015649; U01DK119085; R01NR01388; RC1DK086364 to MT‐C), the Office of Science and Technology, NIH Bench‐to‐Bedside award made possible by the NIH Office of Clinical Research (to MIM and MT‐C), University of Miami SAC‐2016‐9R1 award to RCS and SAC 2013‐19 to MT‐C), and University of Miami Skin Disease Resource‐based Center. The authors thank members of the NIAMS Light Imaging Core Facility. They thank Niki Moutsopoulos and Teresa Wild of the NIDCR. The authors also thank University of Miami wound healing clinical research team and all members of their laboratories for their continuous support.
EMBO reports (2022) 23: e54558
Contributor Information
Marjana Tomic‐Canic, Email: mtcanic@med.miami.edu.
Maria I Morasso, Email: morassom@mail.nih.gov.
Data availability
Raw and analyzed RNA‐Seq data regarding skin acute human wounds and DFUs have been deposited in the Gene Expression Omnibus (GEO) site (GSE97615, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97615; GSE134431, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134431 as previously described (Iglesias‐Bartolome et al, 2018; Sawaya et al, 2020).
References
- Alavi A, Sibbald RG, Mayer D, Goodman L, Botros M, Armstrong DG, Woo K, Boeni T, Ayello EA, Kirsner RS (2014) Diabetic foot ulcers: Part I. pathophysiology and prevention. J Am Acad Dermatol 70: 1–18 [DOI] [PubMed] [Google Scholar]
- Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A (2012) Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30: 459–489 [DOI] [PubMed] [Google Scholar]
- Armstrong DG, Boulton AJM, Bus SA (2017) Diabetic foot ulcers and their recurrence. N Engl J Med 376: 2367–2375 [DOI] [PubMed] [Google Scholar]
- Armstrong DG, Swerdlow MA, Armstrong AA, Conte MS, Padula WV, Bus SA (2020) Five year mortality and direct costs of care for people with diabetic foot complications are comparable to cancer. J Foot Ankle Res 13: 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchon A, Dietrich J, Colonna M (2000) Cutting edge: inflammatory responses can be triggered by TREM‐1, a novel receptor expressed on neutrophils and monocytes. J Immunol 164: 4991–4995 [DOI] [PubMed] [Google Scholar]
- Bouchon A, Facchetti F, Weigand MA, Colonna M (2001) TREM‐1 amplifies inflammation and is a crucial mediator of septic shock. Nature 410: 1103–1107 [DOI] [PubMed] [Google Scholar]
- Brem H, Tomic‐Canic M (2007) Cellular and molecular basis of wound healing in diabetes. J Clin Invest 117: 1219–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535 [DOI] [PubMed] [Google Scholar]
- Byrd AS, Carmona‐Rivera C, O'Neil LJ, Carlucci PM, Cisar C, Rosenberg AZ, Kerns ML, Caffrey JA, Milner SM, Sacks JM et al (2019) Neutrophil extracellular traps, B cells, and type I interferons contribute to immune dysregulation in hidradenitis suppurativa. Sci Transl Med 11: eaav5908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco K, Boufenzer A, Jolly L, Le Cordier H, Wang G, Heck AJ, Cerwenka A, Vinolo E, Nazabal A, Kriznik A et al (2019) TREM‐1 multimerization is essential for its activation on monocytes and neutrophils. Cell Mol Immunol 16: 460–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Arbieva ZH, Guo S, Marucha PT, Mustoe TA, DiPietro LA (2010) Positional differences in the wound transcriptome of skin and oral mucosa. BMC Genomics 11: 471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Jhe YL, Kim J, Lim JY, Lee JE, Shin MK, Cheong JH (2020) FoxM1‐dependent and fatty acid oxidation‐mediated ROS modulation is a cell‐intrinsic drug resistance mechanism in cancer stem‐like cells. Redox Biol 36: 101589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M (2003) TREMs in the immune system and beyond. Nat Rev Immunol 3: 445–453 [DOI] [PubMed] [Google Scholar]
- Eming SA, Krieg T, Davidson JM (2007) Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol 127: 514–525 [DOI] [PubMed] [Google Scholar]
- Eming SA, Martin P, Tomic‐Canic M (2014) Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med 6: 265sr266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadini GP, Menegazzo L, Rigato M, Scattolini V, Poncina N, Bruttocao A, Ciciliot S, Mammano F, Ciubotaru CD, Brocco E et al (2016) NETosis delays diabetic wound healing in mice and humans. Diabetes 65: 1061–1071 [DOI] [PubMed] [Google Scholar]
- Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA 107: 15880–15885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage MC, Becares N, Louie R, Waddington KE, Zhang Y, Tittanegro TH, Rodriguez‐Lorenzo S, Jathanna A, Pourcet B, Pello OM et al (2018) Disrupting LXRalpha phosphorylation promotes FoxM1 expression and modulates atherosclerosis by inducing macrophage proliferation. Proc Natl Acad Sci USA 115: E6556–E6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieling RG, Elsharkawy AM, Caamano JH, Cowie DE, Wright MC, Ebrahimkhani MR, Burt AD, Mann J, Raychaudhuri P, Liou HC et al (2010) The c‐Rel subunit of nuclear factor‐kappaB regulates murine liver inflammation, wound‐healing, and hepatocyte proliferation. Hepatology 51: 922–931 [DOI] [PubMed] [Google Scholar]
- Gupta S, Chan DW, Zaal KJ, Kaplan MJ (2018) A high‐throughput real‐time imaging technique to quantify NETosis and distinguish mechanisms of cell death in human neutrophils. J Immunol 200: 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kaplan MJ (2016) The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol 12: 402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A (2010) Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA 107: 9813–9818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyder LA, Gonzalez J, Harden JL, Johnson‐Huang LM, Zaba LC, Pierson KC, Eungdamrong NJ, Lentini T, Gulati N, Fuentes‐Duculan J et al (2013) TREM‐1 as a potential therapeutic target in psoriasis. J Invest Dermatol 133: 1742–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias‐Bartolome R, Uchiyama A, Molinolo AA, Abusleme L, Brooks SR, Callejas‐Valera JL, Edwards D, Doci C, Asselin‐Labat ML, Onaitis MW et al (2018) Transcriptional signature primes human oral mucosa for rapid wound healing. Sci Transl Med 10: eaap8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorch SK, Kubes P (2017) An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med 23: 279–287 [DOI] [PubMed] [Google Scholar]
- Kalin TV, Ustiyan V, Kalinichenko VV (2011) Multiple faces of FoxM1 transcription factor: lessons from transgenic mouse models. Cell Cycle 10: 396–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MJ, Radic M (2012) Neutrophil extracellular traps: double‐edged swords of innate immunity. J Immunol 189: 2689–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh TJ, DiPietro LA (2011) Inflammation and wound healing: the role of the macrophage. Expert Rev Mol Med 13: e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, Catz SD (2018) Neutrophils: new insights and open questions. Sci Immunol 3: eaat4579 [DOI] [PubMed] [Google Scholar]
- Liao GB, Li XZ, Zeng S, Liu C, Yang SM, Yang L, Hu CJ, Bai JY (2018) Regulation of the master regulator FOXM1 in cancer. Cell Commun Signal 16: 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MB, Kuiper JW, Katchky A, Goldberg H, Glogauer M (2011) Rac2 is required for the formation of neutrophil extracellular traps. J Leukoc Biol 90: 771–776 [DOI] [PubMed] [Google Scholar]
- Lood C, Blanco LP, Purmalek MM, Carmona‐Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ (2016) Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus‐like disease. Nat Med 22: 146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DJ, Gelfand JM, Hoffstad O, Berlin JA (2003) Surrogate end points for the treatment of diabetic neuropathic foot ulcers. Diabetes Care 26: 1696–1700 [DOI] [PubMed] [Google Scholar]
- Mistry P, Nakabo S, O'Neil L, Goel RR, Jiang K, Carmona‐Rivera C, Gupta S, Chan DW, Carlucci PM, Wang X et al (2019) Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci USA 116: 25222–25228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil LJ, Kaplan MJ, Carmona‐Rivera C (2019) The role of neutrophils and neutrophil extracellular traps in vascular damage in systemic lupus erythematosus. J Clin Med 8: 1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, Lau LF, Costa RH, Raychaudhuri P (2009) FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J 28: 2908–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, Stojadinovic O, Yin NC, Ramirez H, Nusbaum AG, Sawaya A, Patel SB, Khalid L, Isseroff RR, Tomic‐Canic M (2014) Epithelialization in wound healing: a comprehensive review. Adv Wound Care 3: 445–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez HA, Pastar I, Jozic I, Stojadinovic O, Stone RC, Ojeh N, Gil J, Davis SC, Kirsner RS, Tomic‐Canic M (2018) Staphylococcus aureus triggers induction of miR‐15B‐5P to diminish DNA repair and deregulate inflammatory response in diabetic foot ulcers. J Invest Dermatol 138: 1187–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Zhang Y, Snyder J, Cross ER, Shah TA, Kalin TV, Kalinichenko VV (2010) Forkhead box M1 transcription factor is required for macrophage recruitment during liver repair. Mol Cell Biol 30: 5381–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya AP, Jozic I, Stone RC, Pastar I, Egger AN, Stojadinovic O, Glinos GD, Kirsner RS, Tomic‐Canic M (2019) Mevastatin promotes healing by targeting caveolin‐1 to restore EGFR signaling. JCI Insight 4: e129320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya AP, Stone RC, Brooks SR, Pastar I, Jozic I, Hasneen K, O'Neill K, Mehdizadeh S, Head CR, Strbo N et al (2020) Deregulated immune cell recruitment orchestrated by FOXM1 impairs human diabetic wound healing. Nat Commun 11: 4678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov A, Panatta E, Lena A, Castiglia D, Di Daniele N, Melino G, Candi E (2016) FOXM1 regulates proliferation, senescence and oxidative stress in keratinocytes and cancer cells. Aging (Albany NY) 8: 1384–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollberger G, Tilley DO, Zychlinsky A (2018) Neutrophil extracellular traps: the biology of chromatin externalization. Dev Cell 44: 542–553 [DOI] [PubMed] [Google Scholar]
- Soo BP, Rajbhandari S, Egun A, Ranasinghe U, Lahart IM, Pappachan JM (2020) Survival at 10 years following lower extremity amputations in patients with diabetic foot disease. Endocrine 69: 100–106 [DOI] [PubMed] [Google Scholar]
- Stojadinovic O, Yin N, Lehmann J, Pastar I, Kirsner RS, Tomic‐Canic M (2013) Increased number of Langerhans cells in the epidermis of diabetic foot ulcers correlates with healing outcome. Immunol Res 57: 222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone RC, Stojadinovic O, Rosa AM, Ramirez HA, Badiavas E, Blumenberg M, Tomic‐Canic M (2017) A bioengineered living cell construct activates an acute wound healing response in venous leg ulcers. Sci Transl Med 9: eaaf8611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone RC, Stojadinovic O, Sawaya AP, Glinos GD, Lindley LE, Pastar I, Badiavas E, Tomic‐Canic M (2020) A bioengineered living cell construct activates metallothionein/zinc/MMP8 and inhibits TGFbeta to stimulate remodeling of fibrotic venous leg ulcers. Wound Repair Regen 28: 164–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turabelidze A, Guo S, Chung AY, Chen L, Dai Y, Marucha PT, DiPietro LA (2014) Intrinsic differences between oral and skin keratinocytes. PLoS ONE 9: e101480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama A, Nayak S, Graf R, Cross M, Hasneen K, Gutkind JS, Brooks SR, Morasso MI (2019) SOX2 epidermal overexpression promotes cutaneous wound healing via activation of EGFR/MEK/ERK signaling mediated by EGFR ligands. J Invest Dermatol 139: 1809–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA et al (2009) Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 184: 205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilgus TA, Roy S, McDaniel JC (2013) Neutrophils and wound repair: positive actions and negative reactions. Adv Wound Care 2: 379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DW, Greenwell‐Wild T, Brenchley L, Dutzan N, Overmiller A, Sawaya AP, Webb S, Martin D, Genomics NN, Computational Biology C et al (2021) Human oral mucosa cell atlas reveals a stromal‐neutrophil axis regulating tissue immunity. Cell 184: 4090–4104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SL, Demers M, Martinod K, Gallant M, Wang YM, Goldfine AB, Kahn CR, Wagner DD (2015) Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med 21: 815–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Ren X, Bolte CS, Ustiyan V, Zhang Y, Shah TA, Kalin TV, Whitsett JA, Kalinichenko VV (2015) Foxm1 regulates resolution of hyperoxic lung injury in newborns. Am J Respir Cell Mol Biol 52: 611–621 [DOI] [PMC free article] [PubMed] [Google Scholar]