

Abstract
Ferroptosis is an iron‐dependent form of non‐apoptotic cell death implicated in liver, brain, kidney, and heart pathology. How ferroptosis is regulated remains poorly understood. Here, we show that PPARα suppresses ferroptosis by promoting the expression of glutathione peroxidase 4 (Gpx4) and by inhibiting the expression of the plasma iron carrier TRF. PPARα directly induces Gpx4 expression by binding to a PPRE element within intron 3. PPARα knockout mice develop more severe iron accumulation and ferroptosis in the liver when fed a high‐iron diet than wild‐type mice. Ferrous iron (Fe2+) triggers ferroptosis via Fenton reactions and ROS accumulation. We further find that a rhodamine‐based "turn‐on" fluorescent probe(probe1) is suitable for the in vivo detection of Fe2+. Probe1 displays high selectivity towards Fe2+, and exhibits a stable response for Fe2+ with a concentration of 20 μM in tissue. Our data thus show that PPARα activation alleviates iron overload‐induced ferroptosis in mouse livers through Gpx4 and TRF, suggesting that PPARα may be a promising therapeutic target for drug discovery in ferroptosis‐related tissue injuries. Moreover, we identified a fluorescent probe that specifically labels ferrous ions and can be used to monitor Fe2+ in vivo.
Keywords: Ferroptosis, Gpx4, Liver, PPARα, TRF
Subject Categories: Autophagy & Cell Death, Molecular Biology of Disease
Gpx4 and TRF are PPARα target genes involved in ferroptosis regulation. Loss of PPARα aggravates iron overload‐induced ferroptosis in vivo, while activation of PPARα is sufficient to largely alleviate iron overload‐induced liver damage.
Introduction
Ferroptosis is an iron‐dependent cell death that involves iron accumulation and lipid peroxidation. Ferroptosis can be triggered by physiological conditions (e.g., high extracellular glutamate) or small molecules (e.g., sorafenib and sulfasalazine) that block system‐mediated cystine import (Xie et al, 2016). Recent studies indicate that ferroptosis contributes to pathological process in a variety of diseases and conditions, including acute organ failure secondary to ischemia/reperfusion, Huntington disease and other neurodegenerative diseases (Yang & Stockwell, 2016). Thus, inhibiting ferroptosis may represent a promising new approach for treating cell death‐related diseases. Loss of gene products, such as ACSL4, depletes the substrates for lipid peroxidation and increase resistance to ferroptosis (Dixon et al, 2015; Yuan et al, 2016; Doll et al, 2017; Kagan et al, 2017). The alteration in the transcription of iron regulation genes affects the sensibility of erastin‐induced ferroptosis and this sensibility is positively correlated with the abundance of intracellular iron. TRF, a serum glycoprotein secreted in liver, plays an essential role in the transport of iron from sites of absorption and storage to iron‐requiring cells (Muckenthaler et al, 2017). Ferroptosis can also be induced by genetic deletion of the Gpx4 (Lei et al, 2019). As an essential regulator of lipid peroxidation, Gpx4 is identified as the central regulator of ferroptosis, acting through the suppression of lipid peroxidation generation. Inactivation of Gpx4 by GSH depletion triggers ferroptosis by accumulation of ROS production from lipid peroxidation (Friedmann Angeli et al, 2014), indicating the protective role of Gpx4 as molecular target against ferroptosis related disease.
In order to investigate all the mechanisms that underlie iron‐induced ferroptosis, researchers have constructed iron overloaded models in mouse. It was found that enhanced iron levels in liver are associated with oxidative stress development and damage with increased fat accumulation (Wang et al, 2017). Iron‐rich diet induced liver steatosis and oxidative stress, mitochondrial dysfunction, loss of PUFAS, downregulation the expression of PPARα and caused liver damage (Barrera et al, 2018), suggesting the PUFAs and PPARα may play roles in the hepatic iron overload disorders.
PPARs are a group of nuclear receptor proteins that function as transcription factors regulating the expression of genes. Three isoforms of PPARs, which vary in their tissue distribution, have been confirmed and include PPARα, β, and γ (Tontonoz et al, 1994). PPARα stimulates the expression of target genes directly through binding to PPREs in the promoter regions of target genes. Upon ligand‐induced activation, PPARα regulates the expression of genes involved in lipid metabolism and peroxisome proliferation (Paumelle et al, 2019). PPARα alters lipid metabolism through multiple mechanisms that facilitate the transfer of fatty acids into mitochondria (Brandt et al, 1998; Cheng et al, 2004). PPARα overexpression or activation lowers plasma triglycerides, reduces adiposity, and improves hepatic steatosis, consequently improving insulin sensitivity (Guerre‐Millo et al, 2000; Kim et al, 2003), identifying new potential therapeutic areas by using metabolic nuclear receptors or their agonists.
Here, we sought to define the relationship between PPARα and ferroptosis and find that the expression of Gpx4 is upregulated and expression of TRF is reduced by PPARα activation. In addition, the absence of PPARα is sufficient to facilitate overloaded‐iron induced ferroptosis in vivo, suggesting that PPARα confers protection against ferroptosis during iron accumulation. We also find that a fluorescent probe (named as Probe1), that was shown to specifically label ferrous ions in vitro, can be applied in vivo to monitor Fe2+. Our research on the Fe2+ sensor provides an effective tool for future ferroptosis research in vitro and in vivo. Thus, our results suggest that PPARα is a potential therapeutic target for treating iron overload associated diseases by regulation the Gpx4 and iron metabolism.
Results and Discussion
Activation of PPARα suppresses iron overload‐induced ferroptosis in vivo
PPARs are a group of nuclear receptors which function as transcription factors and regulate gene expression by binding their heterodimeric partner RXRs at specific PPAR response elements (Tugwood et al, 1992). It was reported that iron‐rich diet for 21 days involving higher iron intake and liver accumulation, resulted in significant oxidative stress enhancement in the liver and many gene expression change such as SREBP‐1c and PPARα (Yang et al, 2014).
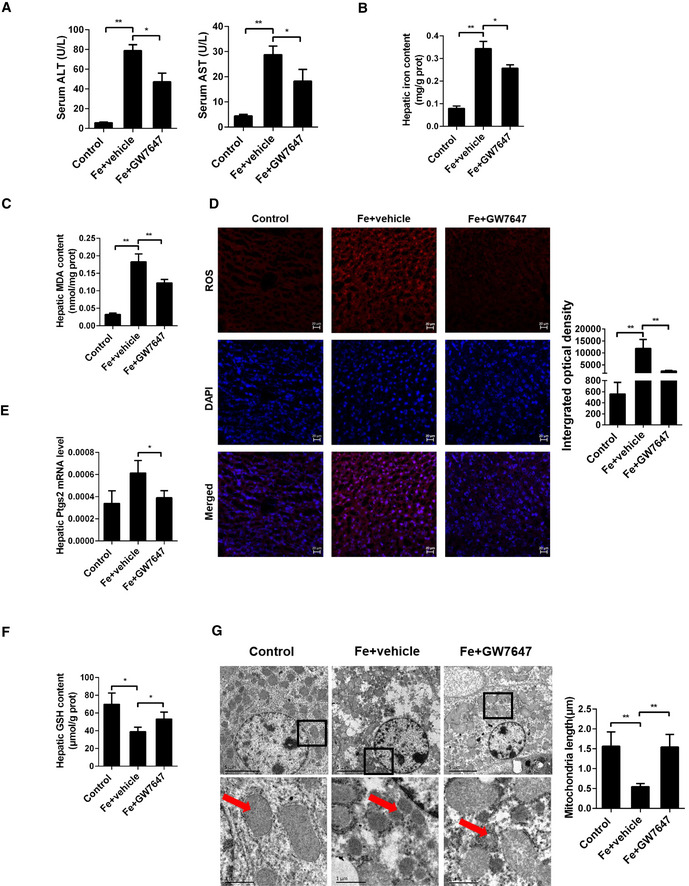
Ferroptosis is an iron‐dependent form of regulated cell death. Recent discoveries have revealed connections between ferroptosis and neurodegenerative and neoplastic diseases (Conrad et al, 2016; Hangauer et al, 2017). It also discovered that ferroptosis is a primary driver of ischemic injury in some models (Tonnus & Linkermann, 2016, 2017), and ferroptosis in liver hemochromatosis has been observed (Lei et al, 2019). Thus, inhibiting ferroptosis is a potential treatment for ferroptosis‐related diseases. Previous studies found that iron overload causes several forms of cell death, including apoptosis, necrosis, and ferroptosis. To investigate the exact role of PPARα in liver ferroptosis, we characterized ferroptosis in mouse models of liver iron‐overload. Mice were fed a high‐iron diet or injected dextriferron intraperitoneally, and were oral administration with either PPARα ligand (GW7647 or WY14643) or vehicle. Levels of ALT and AST were elevated significantly after iron administration in both high‐iron models, but the serum levels of ALT and AST were decreased significantly in the GW7647‐treated group (Figs 1A and EV1A), indicating the effect of the GW7647 treatment on protecting against iron‐overload induced liver injury. H&E staining showed that liver damages, including vacuoles caused by lipid accumulation and focal necrosis, occurred in WT mice. But less tissue damage morphology was observed by H&E staining in WY14643 combined with Fe‐treated mouse group comparing to the Fe alone group (Fig EV1B). Our results show that the liver injury of mice was elevated significantly after iron administration and it is consistent with previous reports (Lei et al, 2019). And pharmacological activation of PPARα significantly improved the situation. These findings indicate that the effect of the PPARα ligands on protecting against iron overload‐induced liver injury.
Figure 1. Activation of PPARα suppresses iron overload‐induced Ferroptosis in vivo .
-
ASerum ALT and AST levels were measured in 8‐week‐old WT mice that were fed a HID with or without GW7647 treatment; n = 5 mice/group.
-
B, CHepatic iron content (B) and hepatic MDA (C) content was measured in the indicated mice; n = 5 mice/group.
-
DMeasurement of intracellular ROS levels by fluorescent probe DCFH‐DA, and the fluorescence intensity of ROS was calculated; All scale bars are 20 μm. n = 3 biological replicates.
-
E, FHepatic Ptgs2 mRNA levels (E) and hepatic GSH content (F) were measured in the indicated mice; n = 5 mice/group. mRNA levels were normalized to 36b4 and are expressed relative to the mean value of the WT group.
-
GLiver tissues were obtained from the indicated mice and then examined using transmission electron microscopy (Arrowheads indicate mitochondria); Scale bars are 1 μm and 5 μm. n = 3 biological replicates.
Data information: Data are presented as means ± SD. *P < 0.05, **P < 0.01, determined by ANOVA.
Figure EV1. Activation of PPARα suppresses iron overload‐induced Ferroptosis in vivo .
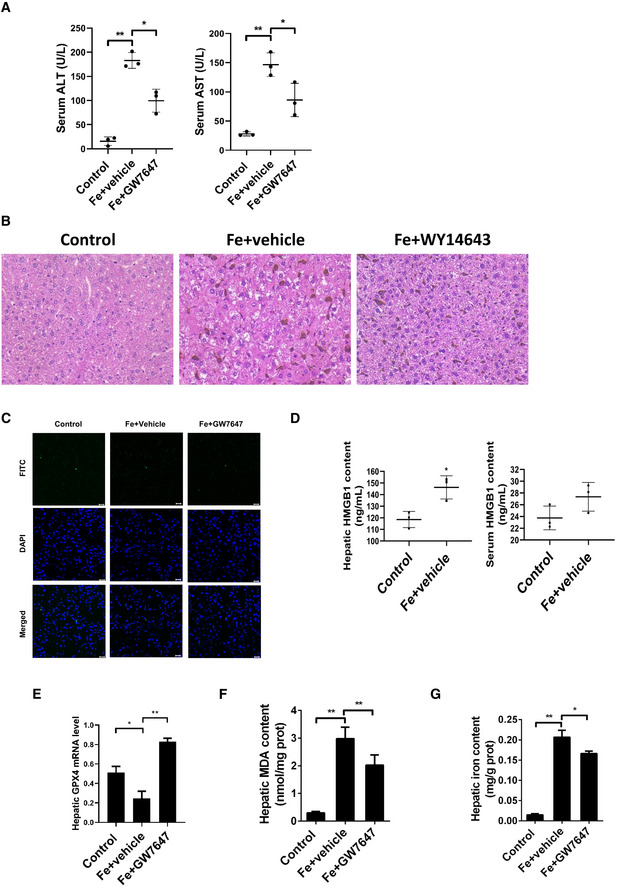
- Serum ALT and AST levels was measured in 8‐week‐old WT mice that were received intraperitoneal injections of dextriferron with or without GW7647 treatment.
- Liver sections were obtained from the indicated mice and stained with H&E. All scale bars are 50 μm.
- Liver sections were obtained from the indicated mice and stained with TUNEL. All scale bars are 20 μm.
- Hepatic and serum HMGB1 content were measured in the indicated mice.
- Hepatic Gpx4 mRNA levels were measured in the indicated mice.
- Hepatic MDA content was measured in 8‐week‐old WT mice that were received intraperitoneal injections of dextriferron with or without GW7647 treatment.
- Hepatic iron content was measured in 8‐week‐old WT mice that were received intraperitoneal injections of dextriferron with or without GW7647 treatment.
Data information: In (A–G), n = 3–5 mice/group. mRNA levels were normalized to 36B4 and are expressed relative to the mean value of the WT group; Data are presented as means ± SD. *P < 0.05, **P < 0.01, determined by ANOVA.
In order to verify whether apoptosis is involved liver injury caused by HID, we performed TUNEL staining, and results show that HID barely triggered apoptosis (Fig EV1C). Then, we investigated whether activation of PPARα suppresses iron overload‐induced ferroptosis in vivo or not. As shown in Fig 1B, iron administration strongly elevated the content of iron in liver, and as expected, GW7647 administration significantly decreased the iron content (Fig 1B). Lipid peroxidation (Fig 1C), ROS level (Fig 1D), Ptgs2 mRNA levels (Fig 1E), and HMGB1 content (Fig EV1D) were significantly increased. GSH content (Fig 1F) and Gpx4 mRNA levels (Fig EV1E) were decreased in mice fed with a high‐iron diet. In addition, mice injected with dextriferron showed significantly increased MDA content and iron content (Fig EV1F–G). Specific PPARα agonist GW7647 significantly reversed iron‐induced the rising MDA content (Fig 1C), ROS level (Fig 1D), Gpx4 mRNA levels (Fig EV1E) and Ptgs2 mRNA levels (Fig 1E) and the markedly decreased GSH content (Fig 1F). Compared to the GW7647‐treated group, livers of mice fed with iron‐rich diet had smaller, ruptured mitochondria (Fig 1G) which is the cellular morphological feature of ferroptosis.
Recent reports have shown that AIFM2/FSP1 can be induced in a PPARα dependent manner (Venkatesh et al, 2020), then we examined the expression of AIFM2/FSP1 in mouse liver during the development of HID‐induced ferroptosis. However, no significant changes were observed on AIFM2/FSP1 expression (Fig EV2A). Gpx4 and FSP1 (previously known as apoptosis‐inducing factor mitochondrial 2—AIFM2) were both identified as ferroptosis suppression factors, and AIFM2 has been hereafter renamed ferroptosis suppressor protein‐1 (FSP1) due to its critical role in a second FSP1‐Q10‐NADPH system, independent of the canonical GSH‐based Gpx4 pathway, which may regulate ferroptosis execution (Bersuker et al, 2019; Doll et al, 2019). For this reason, we speculate that AIFM2/FSP1 is not the main signaling pathway in HID‐induced ferroptosis.
Figure EV2. Activated PPARα inhibited Ferroptosis caused by iron overload.
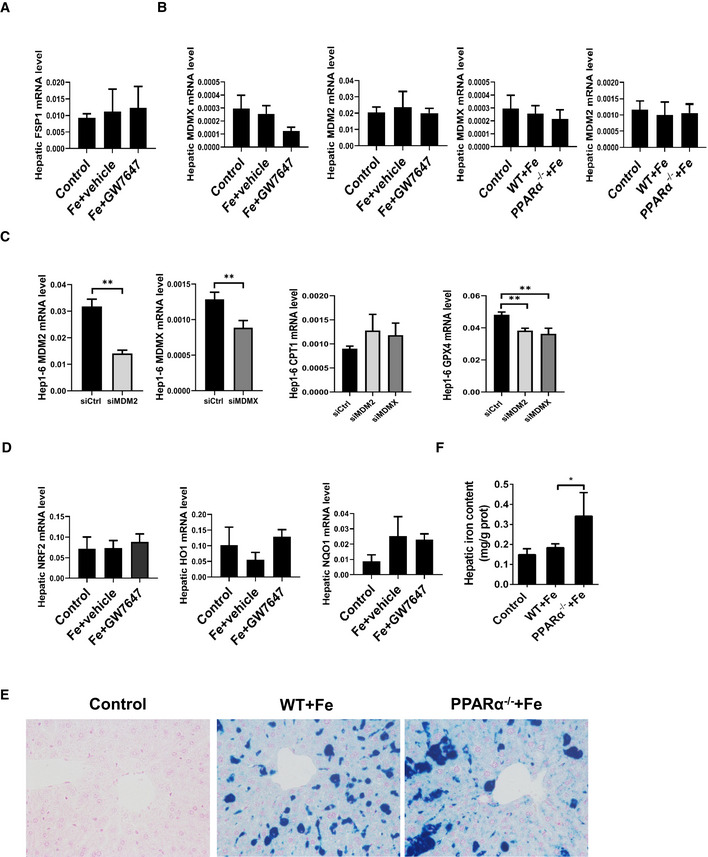
- Hepatic FSP1 mRNA levels were measured in the indicated mice.
- Hepatic MDM2 and MDMX mRNA levels were measured in the indicated mice.
- MDM2, MDMX, CPT1 and GPX4 mRNA levels were measured in the Hep1‐6 cells after MDM2 or MDMX knockdown.
- Hepatic NRF2 mRNA levels, hepatic HO1 mRNA levels, and hepatic NQO1 mRNA levels were measured in the indicated mice.
- Liver sections were obtained from the indicated mice and stained with Prussian blue All scale bars are 50 μm.
- Hepatic iron content was measured in 8‐week‐old WT, PPARα−/− mice that were fed a HID or received intraperitoneal injections of dextriferron; n = 3–5 mice/group.
Data information: In (A, B, D‐E), n = 3–5 mice/group. mRNA levels were normalized to 36B4 and are expressed relative to the mean value of the WT group; Data are presented as means ± SD. *P < 0.05, **P < 0.01, determined by ANOVA.
It was reported that PPARα is involved in MDM2 and MDMX‐related ferroptotic death (Venkatesh et al, 2020). To test whether MDM2 and MDMX have cross‐talking with PPARα signaling pathway in HID liver injury model, we checked MDM2 and MDMX expression in HID mouse model w/o PPARα agonists, but our results display no differential expression of MDM2 and MDMX among the three groups (Fig EV2B). Previous studies have reported that the MDM2–X complex may post‐translationally modify PPARα and alter its activity(Gopinathan et al, 2009; Venkatesh et al, 2020). To further determine whether knockdown of MDM2 and MDMX affects PPARα activity and GPX4 expression, siRNAs were used to knock down MDM2 or MDMX in Hep1‐6 cells, and then the expression levels of PPARα target gene CPT1 and Gpx4 were detected. Although not statistically significant, there was a trend of increasing of CPT1 expression after knockdown of MDM2 or MDMX. Knockdown of both MDM2 and MDMX resulted in a significant down‐regulation of GPX4 expression (Fig EV2C). The result suggests that knockdown of MDM2 and MDMX enhanced the activity of PPARα, but suppressed the expression of Gpx4 in an unknown regulatory manner. It requires further research to explore the mechanism.
Another important model of ferroptosis in the liver can be induced by sorafenib. In particular, the activation of NRF2 pathway diminishes sorafenib‐induced ferroptosis in vitro and in vivo (Sun et al, 2016). More importantly, Gpx4 and TRF are also target genes of NRF2 (Dai et al, 2020). In order to verify whether there is an interaction between the PPARα and NRF2 signaling pathways in liver, we detected expression of NRF2 and NRF2‐related genes in HID mouse liver. Our data show that there was no significant association between PPARα and NRF2 signaling in the model (Fig EV2D), suggesting PPARα and NRF2 pathways may be independent of each other in HID ferroptotic model.
Together, all the above data indicate that iron overload induced ferroptosis in vivo, and specific PPARα ligands prevented this process.
PPARα regulates Gpx4 transcriptional activity by binding to a PPRE in intron of Gpx4, induces Gpx4 gene expression in vivo
Although the regulatory mechanisms that underlie ferroptosis are poorly understood, several molecules that play a role in iron and redox metabolism have been implicated in ferroptosis. ACSL4 expression correlates with cellular sensitivity to erastin‐induced ferroptosis (Yuan et al, 2016). Knockdown of TFR1 by shRNA inhibits erastin‐induced ferroptosis in BJeLR cells, confirming that inhibition of iron uptake prevents iron‐dependent cell death (Yang & Stockwell, 2008).
Gpx4 is a negative regulator of ferroptosis (Friedmann Angeli et al, 2014). Ferroptosis occurs when the oxidation of membrane PUFAs can run out of control due to inactivation of the lipid hydroperoxidase Gpx4. Lipid peroxidation was observed in all knockout models, highlighting the importance of Gpx4 for protecting cells from detrimental effects of lipid peroxides (Seiler et al, 2008). Gpx4 uses GSH as a reducing agent in its peroxidase reaction cycle, and Gpx4 converts potentially toxic lipid hydroperoxides to non‐toxic lipid alcohols (Stockwell et al, 2017). The Gpx4‐catalyzed reaction between GSH and lipid peroxides can prevent ferroptosis, and overexpressing Gpx4 reduces ferroptosis (Friedmann Angeli et al, 2014).
To examine whether and how PPARα contributes to ferroptosis, WT and PPARα−/− mice were administered by oral administration with either vehicle or GW7647 twice a day for 2 days. Then, a range of ferroptosis‐related gene expression including Gpx4 was examined. As expected, GW7647 induced the known PPARα target, Acox1, expression in WT, but not in PPARα−/− mice. Gpx4 mRNA levels were dramatically increased in PPARα agonist treatment of WT mice, and the response was lost in PPARα−/− mice (Fig 2A). Meanwhile, PPARα agonist GW7647 caused the induction of Gpx4 protein expression in WT, but not in PPARα−/− mouse livers (Fig 2B).
Figure 2. PPARα regulates Gpx4 transcriptional activity by binding to an PPRE in Intron 3 of Gpx4, induces Gpx4 gene expression in vivo .

- Acox1 and Gpx4 mRNA was measured in liver of WT and PPARα−/− mice fed blank solvent (white bars, n = 5 mice/group) or GW7647 (black bars, n = 5 mice/group).
- Gpx4 protein was measured in liver of WT and PPARα−/− mice fed blank solvent or GW7647; n = 4 biological replicates.
- Schematic of the WT and mutant PPRE elements. The six nucleotides that were altered to form the mutant construct are underlined.
- A 123‐base pair fragment of intron 3 of mouse Gpx4 (position from +7679 to +7803 with respect to the transcription start site) was inserted into the pGL3 promoter vector to generate the pGL3 promoter intron 3 reporter constructs. The PPRE in intron 3 was mutated to create the mutant construct (pGL3 promoter intron 3 mut). These reporter constructs were transfected into Hep1‐6 cells. The indicated PPARα ligands were added to cell cultures 24 h before the reporter gene assay; n = 3 biological replicates. Data were calculated as the fold induction with respect to the empty vector (pGL3 promoter luciferase vector).
- Chromatin immunoprecipitation assays were performed on soluble formaldehyde‐crosslinked chromatin isolated from untreated and GW7647‐treated WT or PPARα−/− livers with polyclonal anti‐PPARα antibodies (anti‐PPARα) or control IgG. The final DNA extraction was polymerase chain reaction‐amplified with a primer pair that covered the sequence in intron 3 of Gpx4.
Data information: mRNA levels were normalized to 36B4 and are expressed relative to the mean value of the WT group. Data are presented as means ± SD. **P < 0.01, determined by ANOVA.
Next, we analyzed whether mouse Gpx4 is a direct target gene of PPARα. To determine the region within the mouse Gpx4 gene that confers transcriptional responsiveness to PPARα, we amplified a series of progressively deleted Gpx4 promoter and intron fragments. These fragments were inserted into a pGL3 promoter luciferase vector, and constructs were transiently transfected into Hep1‐6 cells. We detected a significant increase in luciferase expression after the activation of PPARα by GW7647 in cells transfected with pGL3‐intron 3 (inserted DNA fragment position from +7690 to +7712 with respect to the transcription starting site. Fig 2C) in comparison with cells that were not exposed to PPARα ligands. The Gpx4 intron 3 contains a putative PPRE. We then performed site‐directed mutagenesis of the PPRE (changing CAATGC to TTTTTT), which blocked the induction of reporter expression by PPARα activation (Fig 2D). These results suggest that Gpx4 may be directly regulated by PPARα activation that converges on the PPRE acting as an enhancer within intron 3 of Gpx4.
To further demonstrate that PPARα binds to the intronic enhancer in vivo, we performed ChIP assays on soluble formaldehyde‐crosslinked chromatin isolated from untreated and GW7647‐treated livers with a polyclonal anti‐PPARα antibody. PPARα was bound to the intronic enhancer in vivo (Fig 2E), and PPARα knockout displayed no association of PPARα with the intronic enhancer after the treatment.
Our data indicate GW7647 treatment strongly increased mRNA levels of Gpx4 in WT mice. And the GW7647‐treated group had decreased levels of lipid peroxidation and Ptgs2 mRNA in the liver, suggesting PPARα can restrain ferroptosis through the PPARα‐Gpx4 axis.
PPARα deficiency influence iron metabolism in the liver
Fe is essential for life because it constitutes the required cofactor for a multitude of proteins of diverse biological functions. Small pools of labile Fe2+ reside in the cytosol and the mitochondrial matrix. These redox‐active iron pools are capable of directly catalyzing damaging free radical formation via Fenton chemistry (Dixon & Stockwell, 2014). It was speculated that such Fenton chemistry might account for the lethal effect of ferroptosis inducers. However, the current view favors a more biological route of iron‐mediated lipid peroxide generation. Thus, iron import, export, storage, and turnover all have an impact on ferroptosis. Then, an iron‐overloaded murine model was used to study the effects of iron in ferroptosis. When fed a high iron diet, mice develop severe tissue iron overload, and serum transferrin binding approaches saturation, suggesting the presence of non‐transferrin‐bound iron in the circulation (Yang et al, 2014). However, the precise role of iron in ferroptosis remains unclear. Therefore, a convenient and rapid method for the analysis of Fe2+ and Fe3+ in biological samples has important consequences in biological concerns. During past decades, many approaches have been designed for noninvasive detection of labile Fe3+ within living cells and other intact biological specimens (Weerasinghe et al, 2010). Fe2+ is the main cause of ferroptosis, but unfortunately, there are relatively few fluorescent probes that allow the detection of Fe2+ in vivo. As a consequence, the research on iron ions mainly focuses on ferric ions, for which various Fe3+ fluorescent probes are available (Luo et al, 2016).
Hou et al (2013) recently reported a rhodamine‐based “turn‐on” fluorescent probe(probe1) that displays high selectivity for Fe2+ (Hou et al, 2013). To further test the biologically relevant utility of the Fe2+ specific fluorescent probe, culturing Hep1‐6 were incubated with 20 μM probe1 in culture medium for 30 min at 37°C, and very weak intracellular fluorescence inside the living cells was observed (Fig 3A). Addition of external Fe2+ (20 μM) enhanced intracellular fluorescence intensity, while only a limited increase in intracellular fluorescence was observed with Cu2+ and Fe3+ at the same concentration (Fig 3A). These results suggest that the highly cell permeable probe1 specifically responds to labile Fe2+ iron pools.
Figure 3. PPARα deficiency influences iron metabolism in the liver.
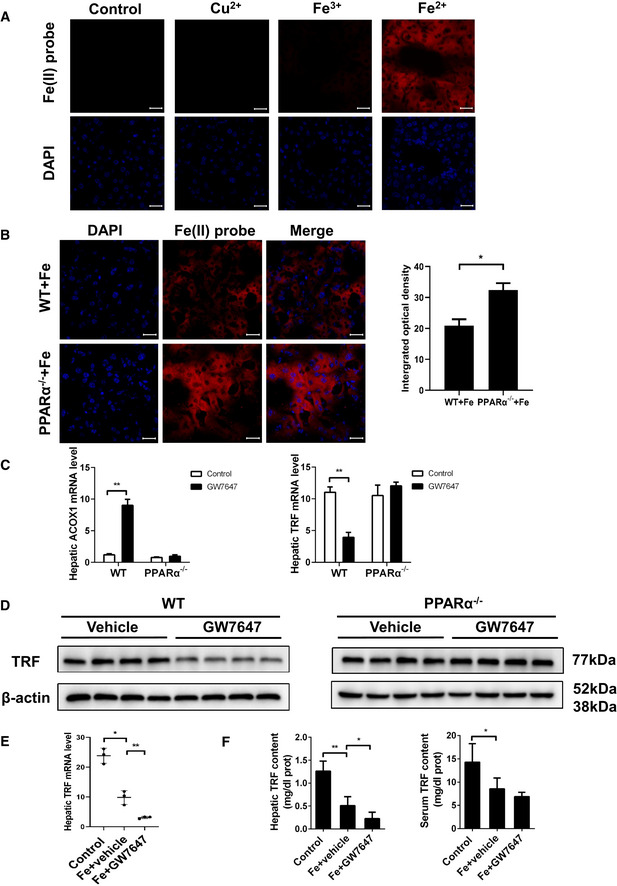
- Fluorescent images of labile Fe2+ in hep1‐6 cells. Hep1‐6 cells were incubated with control, 20 μM probe1 for 30 min; 20 μM Cu2+ for 30 min then 20 μM probe1 for another 30 min; 20 μM Fe3+ for 30 min then 20 μM probe1 for another 30 min; 20 μM Fe2+ for 30 min then 20 μM probe1 for another 30 min; All scale bars are 20 μm. n = 3 biological replicates.
- Liver sections were obtained from the indicated mice and stained with probe1. All scale bars are 20 μm. n = 3 biological replicates.
- Hepatic Acox1 and TRF mRNA levels were measured in the WT and PPARα−/− mice with or without GW7647 treatment. n = 3–5 mice/group.
- TRF protein was measured in liver of WT and PPARα−/− mice fed blank solvent or GW7647. n = 4 biological replicates.
- Hepatic TRF mRNA levels were measured in the indicated mice. n = 3–5 mice/group.
- Hepatic and serum TRF content were measured in the indicated mice. n = 3–5 mice/group.
Data information: mRNA levels were normalized to 36B4 and are expressed relative to the mean value of the WT group. Data are presented as means ± SD. *P < 0.05, **P < 0.01, determined by ANOVA.
To investigate the iron changes in metabolism, we used a HID diet eliciting substantial iron accumulation in the mouse liver. To study the physiological and pathological consequences of biological ferrous iron regulation, we utilize the Fe2+ fluorescent probes for monitoring labile Fe2+ pools in the liver. As shown in Fig 3B, a significant increase in intracellular fluorescence in the liver of PPARα−/− mice compare to WT mice was observed (Fig 3B). The hepatic iron content in PPARα−/− mice increased significantly comparing with the WT group (Fig EV2E and F), indicating severe iron accumulation in the liver of PPARα−/− mice. All above suggested that PPARα deficiency induced significant increasing of Fe2+ accumulation.
To examine whether and how PPARα deficiency contributes to iron accumulation. A range of iron metabolism‐related gene expression was examined in mice oral administration with either vehicle or GW7647, and TRF expression level was significantly reduced in GW7647‐treated mice (Fig EV3A). Further testing found TRF mRNA and protein levels were dramatically decreased in WT mice with PPARα agonist treatment but that change disappeared in the PPARα knockout mice (Fig 3C and D). TRF, the major plasma iron carrier, is synthesized and secreted mainly by the liver. TRF delivers iron to tissues through binding to its receptor and receptor‐mediated endocytosis. It is widely believed that iron delivery is the primary function of TRF. The hepatic TRF mRNA levels of mice were oral administration with PPARα ligand was significantly lower comparing to the mice with solvent comparison (Fig 3E). Hepatic and serum levels of TRF were detected by enzyme linked immunosorbent assay. Hepatic and serum levels of TRF were detected by enzyme linked immunosorbent assay. The TRF of hepatic and serum content of mice in Fe group was significantly lower than that in control group. And GW7647 administration significantly decreased the TRF content compared to Fe group (Fig 3F). Moreover, we analyzed whether the mouse TRF is a PPARα direct binding target gene or not. Putative PPREs in the TRF promoter region were predicted using an online algorithm (NUBIScan: http://www.nubiscan.unibas.ch/). The fragment was inserted into a pGL3 promoter luciferase vector, and constructs were transiently transfected into Hep1‐6 cells. We detected a significant decrease in luciferase expression after the activation of PPARα by GW7647 in cells transfected with pGL3 promoter (Fig EV3B and C) in comparison with cells that were not exposed to PPARα ligands. Next, we performed ChIP assays on soluble formaldehyde–crosslinked chromatin isolated from untreated and GW7647‐treated livers with a polyclonal anti‐PPARα antibody. PPARα was bound to the promoter in vivo (Fig EV3D), and PPARα knockout displayed no association of PPARα with the promoter after the treatment. This result suggests that TRF is down‐regulated by PPARα activation through binding to the PPRE in promoter in vivo and PPARα deficiency induced significant increase in iron turnover. Therefore, we think that inhibition of TRF is another important reason for reducing liver ferroptosis after activation of PPARα.
Figure EV3. PPARα regulates TRF transcriptional activity by binding to a PPRE in promoter of TRF.
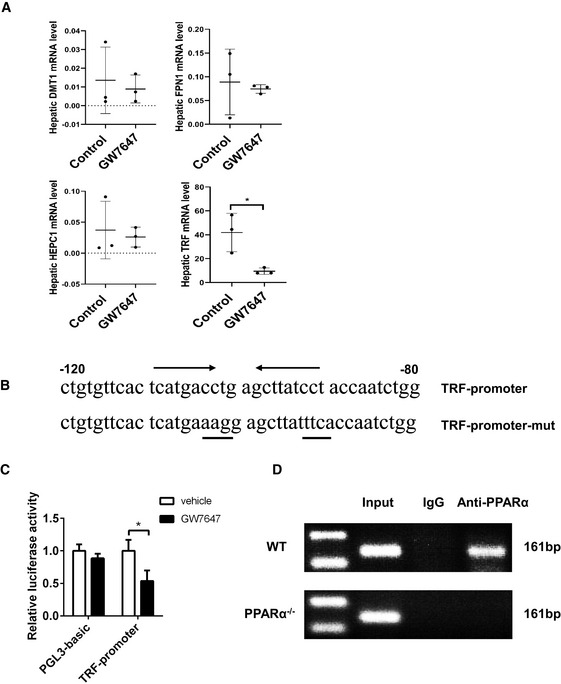
- Hepatic DMT1, FPN1, HEPC1 and TRF mRNA levels were measured in the indicated mice; n = 3 mice/group.
- Schematic of the TRF‐promoter‐PPRE.
- A 2,000‐base pair fragment of the promoter of mouse TRF was inserted into the pGL3 promoter vector to generate the pGL3 promoter constructs. These reporter constructs were transfected into Hep1‐6 cells. The indicated PPARα ligands were added to cell cultures 24 h before the reporter gene assay. Data were calculated as the fold induction with respect to the empty vector (pGL3 promoter luciferase vector). n = 3 biological replicates.
- Chromatin immunoprecipitation assays were performed on soluble formaldehyde–crosslinked chromatin isolated from untreated and GW7647‐treated WT or PPARα−/− livers with polyclonal anti‐PPARα antibodies (anti‐PPARα) or control IgG. The final DNA extraction was polymerase chain reaction‐amplified with a primer pair that covered the sequence in the promoter of Gpx4.
Data information: mRNA levels were normalized to 36B4 and are expressed relative to the mean value of the WT group; Data are presented as means ± SD. *P < 0.05, determined by ANOVA.
PPARα deletion increases susceptibility to iron overload‐induced ferroptosis
Further, we examined whether the loss of PPARα expression increases iron overload‐induced ferroptosis in vivo. Feeding WT and PPARα−/− mice the HID for 3 weeks or injecting dextriferron intraperitoneally for 2 weeks led to significantly higher tissue iron content in PPARα−/− mice compared with WT mice (Figs EV2D and E, and EV4A). The levels of ALT and AST were elevated significantly after iron administration in both WT and PPARα−/− mice, but serum levels of ALT and AST were much higher in PPARα−/− mouse group than that of the WT mouse group (Figs 4A and EV4B), indicating the effect of the deleted PPAR on aggravated overload‐induced liver injury. H&E staining showed that liver damages, including vacuoles caused by lipid accumulation and focal necrosis, were more sever in PPARα−/− mice than that in WT mice (Fig EV4C). And as we expected that hepatic MDA content and ROS levels in PPARα−/− mice was higher than that of the WT mice after the HID and intraperitoneal injection (Figs 4B and EV4D and E). The hepatic GSH content of PPARα−/− mice was significantly lower comparing to the WT mice that fed with a HID (Fig 4C), and the abundance of Ptgs2 mRNA in the liver of PPARα−/− mice was much higher than that in WT mouse livers (Fig 4D). Compared to the WT mice, livers of PPARα−/− mice fed with HID had smaller, ruptured mitochondria (Fig 4E). These results indicated that deficiency PPARα markedly increases iron overload‐induced liver injury and the PPAR−/− mice are more sensitive to ferroptosis.
Figure EV4. PPARα deletion increases ferroptosis caused by iron overload.
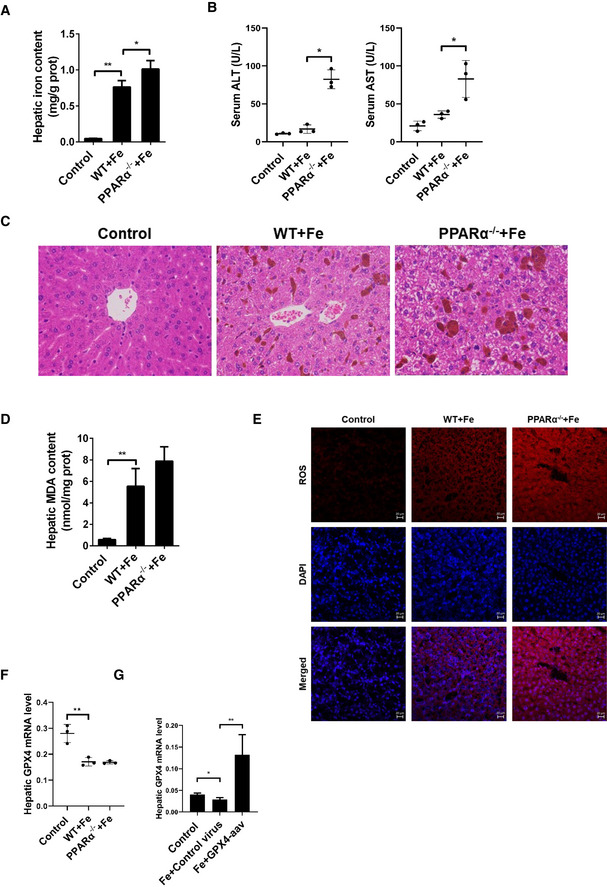
- Hepatic iron content was measured in 8‐week‐old WT, PPARα−/− mice that received intraperitoneal injections of dextriferron or saline.
- Serum ALT and AST levels was measured in 8‐week‐old WT, PPARα−/− mice that received intraperitoneal injections of dextriferron or saline.
- Liver sections were obtained from the indicated mice and stained with H&E. All scale bars are 50 μm. n = 3 biological replicates.
- Hepatic MDA content was measured in 8‐week‐old WT, PPARα−/− mice that received intraperitoneal injections of dextriferron or saline.
- Measurement of intracellular ROS levels by fluorescent probe DCFH‐DA, and the fluorescence intensity of ROS was calculated. All scale bars are 20 μm. n = 3 biological replicates.
- Hepatic Gpx4 mRNA levels were measured in the indicated mice.
- Hepatic Gpx4 mRNA levels were measured in the mice that were fed a HID with or without Gpx4‐AAV treatment.
Data information: In (A–G), n = 3–5 mice/group. mRNA levels were normalized to 36B4 and are expressed relative to the mean value of the WT group; Data are presented as means ± SD. *P < 0.05, **P < 0.01, determined by ANOVA.
Figure 4. PPARα deletion increases susceptibility to iron overload‐induced ferroptosis.
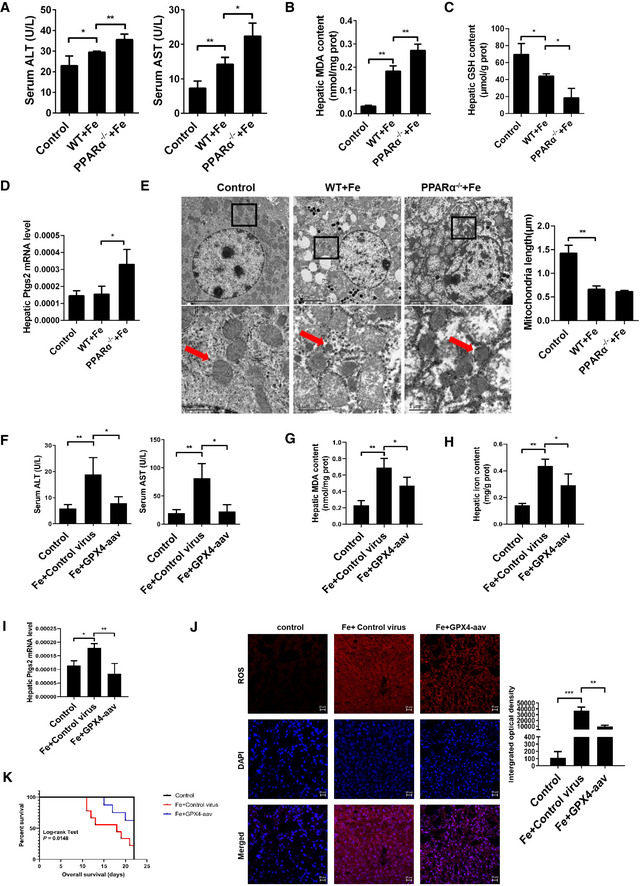
-
ASerum ALT and AST levels were measured in 8‐week‐old WT, PPARα−/− mice that fed a normal diet or a HID; n = 3–5 mice/group.
-
B–DHepatic MDA content (B), hepatic GSH content (C), hepatic Ptgs2 mRNA levels (D) were measured in the indicated mice. n = 3–5 mice/group.
-
ELiver tissue was obtained from the indicated mice and then examined using transmission electron microscopy (Arrowheads indicate mitochondria). Scale bars are 1 μm and 5 μm. n = 4 biological replicates.
-
FSerum ALT and AST levels were measured in 8‐week‐old PPARα−/− mice that were fed a HID with or without Gpx4‐AAV treatment; n = 8 mice/group.
-
G, IHepatic MDA content (G), hepatic iron content (H), hepatic Ptgs2 mRNA levels (I) were measured in the indicated mice; n = 8 mice/group.
-
JMeasurement of intracellular ROS levels by fluorescent probe DCFH‐DA, and the fluorescence intensity of ROS was calculated. All scale bars are 20 μm. n = 3 biological replicates.
-
KKaplan–Meier curves with univariate analysis of overall survival based on the Gpx4‐AAV treatment. n = 5–9 mice/group.
Data information: Data are presented as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, determined by ANOVA.
Next, we assessed the in vivo potential of Gpx4 to prevent the consequences of inducible PPARα disruption in animals. In the previous experiment, we found the expression level of Gpx4 was significantly reduced, when the mice were fed a high‐iron diet for 3 weeks (Fig EV4F). PPARα−/− mice were injected with Gpx4‐AAV or control vector before the mice fed HID diet, at 3 weeks they were euthanized. As shown in Fig 4F, levels of ALT and AST were elevated significantly after iron administration, but the serum levels of ALT and AST dropped significantly in the Gpx4‐AAV treated group (Fig 4F), indicating the effect of the overexpression Gpx4 on protecting against iron‐overload induced PPARα−/− mice liver injury. Gpx4‐AAV treatment significantly reduced the iron‐induced increase in MDA content, iron content, Ptgs2 mRNA levels, and ROS levels, Gpx4 mRNA levels was increased in PPARα−/− mice (Figs 4G–J and EV4G). The overall survival curves of PPARα−/− mice were generated by Kaplan–Meier plotter based on the HID with or without Gpx4‐AAV treatment, and the results are illustrated in Fig 4K. Mice with Gpx4‐AAV treatment had a significantly reduced risk of death compared to those with control vector. Ferroptosis was inhibited in iron‐overload PPARα knockout mice with Gpx4‐AAV treatment. These results suggest that the in vivo potential of Gpx4 to prevent the consequences of PPARα disruption in high‐iron diet animals.
Activated PPARα and ferrostatin‐1 similarly protect against ferroptosis‐induced liver injury
Ferrostatin‐1 is a recognized ferroptosis inhibitor. Past studies have shown that Ferrostatin‐1 can significantly inhibit the ferroptosis. Now we have discovered another way to inhibit ferroptosis. The transcriptional regulation of PPARα can inhibit ferroptosis in the liver of mice. To confirm that activation of PPARα plays a considerable role in iron overload‐induced ferroptosis, WT mice were fed a high‐iron diet for 3 weeks. During this period, mice received with GW7647 administration or Fer‐1 administration for 3 weeks. First, serum ALT and AST were examined. Levels of ALT and AST were elevated significantly after a high‐iron diet in WT mice, but the serum levels of ALT and AST dropped significantly in both Fer‐1‐ and GW7647‐treated group (Fig 5A). This indicates that Fer‐1 and GW7647 treatments have similar effects in preventing liver damage caused by iron overload. Then, we investigated whether activation of PPARα or Fer‐1‐treated suppresses iron overload‐induced ferroptosis in vivo. As shown in Fig 5B, iron administration strongly elevated the hepatic iron content in WT mice, and as expected, Fer‐1‐ and GW7647‐treated significantly decreased the iron content (Fig 5B) in WT mice. Hepatic MDA (Fig 5C), Ptgs2 mRNA (Fig 5D) and ROS level (Fig 5E) were significantly increased in WT mice fed with a high‐iron diet. Fer‐1‐ and GW7647‐treated significantly reversed iron overload‐induced hepatic MDA, Ptgs2 mRNA levels, and ROS level in WT mice (Fig 5C–E).
Figure 5. Activated PPARα and ferrostatin‐1 have similar and important effects on inhibiting Ferroptosis damage caused by iron overload.
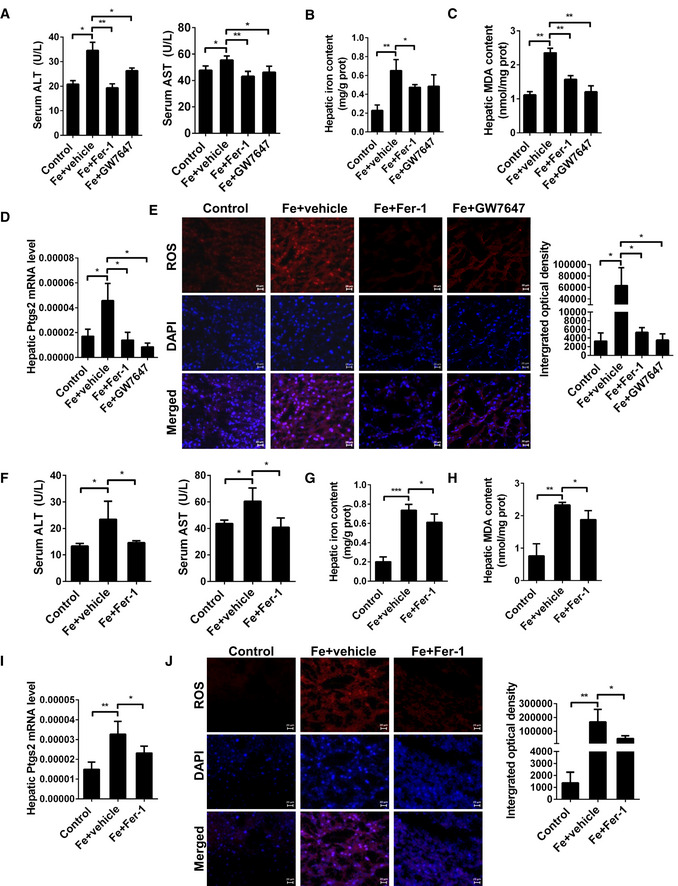
-
ASerum ALT and AST levels were measured in 8‐week‐old WT mice treated with or without Fer‐1 and GW7647 treatment; n = 6 mice/group.
-
B–DHepatic iron content (B), hepatic MDA content (C), hepatic Ptgs2 mRNA levels (D) were measured in the indicated mice.
-
EMeasurement of intracellular ROS levels by fluorescent probe DCFH‐DA, and the fluorescence intensity of ROS was calculated. n = 3 biological replicates. All scale bars are 20 μm.
-
FSerum ALT and AST levels were measured in 20‐week‐old PPARα−/− mice treated with or without Fer‐1; n = 6 mice/group.
-
G, IHepatic iron content (G), hepatic MDA content (H), hepatic Ptgs2 mRNA levels (I) were measured in the indicated mice. n = 6 mice/group.
-
JMeasurement of intracellular ROS levels by fluorescent probe DCFH‐DA, and the fluorescence intensity of ROS was calculated. n = 3 biological replicates. All scale bars are 20 μm.
Data information: mRNA levels were normalized to 36B4 and are expressed relative to the mean value of the WT group. Data are presented as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, determined by ANOVA.
In order to verify whether Fer‐1 plays a role in the liver ferroptosis of PPARα−/− mice, we fed PPARα knockout mice a high‐iron diet for 3 weeks, and then intraperitoneal injection Fer‐1 daily. Compared to untreated PPARα−/− mice, Fer‐1‐treated PPARα−/− mice had significantly decreasing of ALT and AST levels (Fig 5F).
This suggests that the liver damage of PPARα knockout mice was alleviated by Fer‐1. Moreover, Fer‐1 treatment significantly reduced the hepatic iron content (Fig 5G), MDA (Fig 5H), Ptgs2 mRNA levels (Fig 5I), and ROS level (Fig 5J) of PPARα knockout mice on HID. As we expected, Fer‐1 treatment resists ferroptosis in the liver of PPARα−/− mice caused by iron overload. Together, all the above data indicate that iron overload induced ferroptosis in vivo, and pharmacological activation PPARα or Fer‐1 has considerable effects on protecting the iron overload induced ferroptosis.
PPARα has the function of inhibiting peroxidation, while we found that PPARα inhibits ferroptosis through Gpx4 in this study. But we cannot rule out other unknown regulatory mechanisms, and it needs further study. In Hwang’s paper, PAPRδ rescues xCT‐deficient cells from ferroptosis by targeting peroxisomes (Hwang et al, 2021). Interestingly, Tao indicated CYP2J2‐produced epoxyeicosatrienoic acids contribute to the ferroptosis resistance of pancreatic ductal adenocarcinoma in a PPARγ‐dependent manner (Tao et al, 2021), whereas Han showed that PPARγ drives ferroptosis in dendritic cells (Han et al, 2021) indicating that the α and γ isoforms of PPAR have opposing cellular functions (Kersten, 2008). Considering that PPAR isotypes are expressed in different tissues and have functional differences and similarities, targeting PPARs for future clinical therapy need more investigation on ferroptosis‐related diseases.
In summary, our study identified the PPARα as a novel regulator in the suppression of iron overload‐induced liver ferroptosis, and results from this study thus provide important insights into the biological roles of PPARα in hepatic pathophysiology and hence provide new therapeutic targets for the drug discovery.
Materials and Methods
Animals
PPARα−/− mice were purchased from the Jackson Laboratory (strain name: B6; 129S4‐Pparαtm1Gonz/J, stock number 008154). C57BL/6J SPF mice were purchased from Huazhong Agricultural University Experimental Animal Center. Unless stated otherwise, male mice between 6 and 8 weeks old were used in each group of experiments. Wild‐type and PPARα null mice were used to determine the role of PPARα. Unless stated otherwise, the mice were fed with a standard AIN‐76A diet containing 50 mg iron/kg body weight for 3 weeks and then sacrificed. The HID (8.3 g carbonyl iron/kg) were egg white‐based AIN‐76A diets. Mice were given tertian intraperitoneal injections of either PBS (control) or dextriferron (500 mg/kg body weight) for 2 weeks and then sacrificed. Mice were given a daily intraperitoneal injection of either vehicle or ferrostatin‐1 (Fer‐1, 1 mg/kg body weight) for 3 weeks before sacrificed. AAV carrying cDNA of mouse Gpx4 injected to the PPARα null mice induces the presence of Gpx4 inclusions. All experimental protocols were approved by the animal ethical and welfare committee of Huazhong Agricultural University.
In vivo treatment with GW7647
Mice were gavaged with either vehicle (4:1 of PEG‐400 and Tween 80) or GW7647 (10 mg/kg body weight) once every 2 days for 3 weeks.
Fe2+ probe incubation and imaging
The original Fe2+ probe named as “probe1” was designed and published by Hou et al (2013) and was synthesized in Beyotime, and the structure is provided in Appendix Fig S1.
Hep1‐6 cells were purchased from the National Infrastructure of Cell Line Resource (NICR, CHN). These cells were grown in Eagle's Minimum Essential Medium, with 10% fetal bovine serum, and 100 U/ml of penicillin/streptomycin in a humidified incubator with 5% CO2 and 95% air. The concentration of counted cells was adjusted to 1.0 × 106 cells ml−1 and cells were passed and plated on glass slide at 37 °C, 5% CO2 for 4 h. Adherent cells were incubated with 20 μM probe1 for 0.5 h at 37 °C under 5% CO2 and then washed with PBS three times before incubating with 20 μM FeSO4, CuSO4 or Fe2(SO4)3 for another 30 min. The fluorescence imaging of intracellular Fe2+ of the cells was observed under a Leica TCS confocal laser scanning microscope after three times PBS washing.
The liver tissue was frozen in optimal cutting temperature (OCT) solution for cryosections. A set of 20‐μm‐thick liver sections was generated from the selected liver region. After washing with PBS three times, the sections were incubated with 0.3% Triton X‐100. Then, the 20 μM probe1 was added onto the sections. Following by 20 min hours min incubation and thorough washing, each section was photographed under the Leica TCS confocal laser scanning microscope to visualize the probe1‐labeled Fe2+. To confirm that the fluorescence was from Fe2+, 50 μM DFO was used to treat the sections 1 h before probe1 addition.
Small interfering RNAs and transfection
Lipofectamine 2000 (Invitrogen) was used as the transfection reagent for the initial set of siRNA experiments (according to the manufacturer's instructions).
Sequences of siRNAs used in this study were: SiCtrl sense strand, 5′‐UUCUUCGAACGUGUCACGUTT‐3′; antisense strand, 5′‐ACGUGACACGUUCGGAGAATT‐3′; siMDMX sense strand, 5′‐GCCAGUAUAUAAUGGUGAATT‐3′; antisense strand, 5′‐UUCACCAUUAUAUACUGGCTT‐3′; siMDM2 sense strand, 5′‐CCUUCGUGAGAACUGGCUUTT‐3′; antisense strand, 5′‐AAGCCAGUUCUCACGAAGGTT‐3′.
The cells were plated prior to transfection such that they are only 80% confluent prior to RNA isolation. RNA was extracted 24 h after cell transfection.
Western blot analysis
Proteins were extracted using the Protein Extraction Kit (Beyotime; P0033). The protein concentration of the samples was determined by BCA method. 20 μg of cell lysates was loaded and separated using 10% SDS‐PAGE and transferred onto PVDF membrane. Blots were incubated with indicated primary antibodies in 5% non‐fat dry milk in PBS plus 0.1% Tween‐20 overnight at 4°C. Primary antibodies were: anti‐Gpx4 (dilution 1:1,000) (Abcam; no. 125066), anti‐TRF (dilution 1:2,000) (Hangzhou HuaAn Biotechnology; R1212‐1). Detection was achieved using horseradish peroxidase‐conjugate secondary antibody for 2 h at room temperature, and visualized with ECL plus (Juneng, K‐12045‐D10). Images were acquired by using imaging system (SYNGENE, G: Box).
Real‐time PCR analysis
Trizol reagent (Takara; 9109) was used to extract total RNA as indicated by the supplier, and the Prime Script RT kit (Takara; RR047A) was used to produce cDNA following the manufacturer's recommendations. Quantitative real‐time PCR was performed using SYBR Green PCR Master Mix (TOYOBO, QPK‐201) and ABI CFX Connect TM Real‐Time PCR Detection System (ABI). Appendix Table S1 shows the primer pair sequences for all amplicons, designed by using the Primer Premier 5. A relative gene‐expression quantification method was used to calculate the fold change of mRNA expression according to the comparative Ct method using 36b4 for normalization.
PPARα transcriptional activity assay
Mouse intron 3 fragments of Gpx4 were amplified by PCR using primers: forward: 5′‐CGAGCTCGTGTGTGGCTGTTCCCCAGG‐3′, reverse: 5′‐CCAAGCTTCAGGAAGCAACATTTACTTG‐3′. And mouse promoter of TRF were amplified by PCR using primers: forward:5′‐GGGGTACCAACTGGTGTAGTCTTCGCTGCTG‐3′, reverse: 5′‐CCAAGCTTTCAGACCCTTACATAAGGAGGTG‐3′. The product was cloned into pGL3‐basic luciferase reporter vector. For the determination of Gpx4 intron and TRF promoter activity, hep1‐6 cells were transfected with pGL3‐Gpx4 or pGL3‐TRF and Renilla luciferase plasmid, followed by treatment with 5 μM GW7647 for 24 h. The promoter activity was determined using the Dual‐Luciferase Reporter Assay Kit (Promega, E1910) according to the manufacturer’s instructions. The plasmid expressing Renilla luciferase was used for normalization of luciferase activity.
ChIP assay
ChIP assays for GW7647‐treated WT and PPARα−/− mouse livers were performed according to the protocol of the kit (Beyotime, P2078). GW7647‐treated WT and PPARα−/− mouse livers were cross‐linked with 1% formaldehyde at 37°C for 10 min. Livers were rinsed with ice‐cold PBS and harvested in SDS lysis buffer followed by sonication five times for 15 s each. Livers were centrifuged for 10 min, and supernatants were collected and diluted in ChIP dilution buffer. An aliquot of each sample was used as “Input” in the PCR analysis. The remainder of the soluble chromatin was immunoprecipitated with normal IgG, PPARα antibodies (Santa Cruz; sc‐398394) at 4°C for 18 h. Protein A‐Magnetic Beads were added and incubated for another 2 h at 4°C with a gentle rotation to collect the immune complexes. The complexes were sequentially washed in the following washing buffers: low salt immune complex washing buffer, high‐salt immune complex washing buffer and LiCl immune complex washing buffer. After wash with Tris‐EDTA buffer two times, the complexes were eluted twice for 100 μl aliquots of elution buffer each. The cross‐linked chromatin complex was reversed by incubation with 0.2 mol/l NaCl and heating at 65°C for 4 h. DNA was purified using GPTM DNA purification spin columns. PCR was performed using PCR Master Mix, according to the manufacturer's protocol. Ten percent of the total purified DNA was used for the PCR in a 50‐µL reaction mixture. The PCR products were analyzed by 1.5% agarose gel electrophoresis.
Liver damage
Serum ALT and AST were measured using an enzymatic assay kit (Jiancheng, C0092‐1; C010‐2‐1). The livers were fixed in 4% formaldehyde for 24 h, and subsequently were embedded in paraffin for the histologic assessment. Liver sections (4 μM) were deparaffinized, fixed, and stained with H & E and Prussian blue.
Measurement of malondialdehyde, and GSH content, iron content, ROS level, TRF content
The ROS was detected by Reactive Oxygen Species Fluorogenic Probe (BestBio; BB470513) and was observed with confocal microscopy. The frozen liver tissue (100 mg) was homogenized in 0.9 ml PBS. The lysate was centrifuged and the supernatant was collected. Hepatic MDA content, GSH content, iron content, TRF content was detected using the Malondialdehyde assay kit (Jiancheng, A003‐1‐2), Reduced glutathione assay kit (Jiancheng, A006‐2‐1), tissue iron assay kit (Jiancheng, A039‐2‐1), and Transferrin Assay Kit (Jiancheng, E028‐1‐1) following the manufacturer's instructions. The values for the levels of GSH in the liver tissues were measured using a microplate reader at the absorption wavelengths of 405nm. The values for the levels of MDA, iron, TRF in the liver tissues were measured using a spectrophotometer at the absorption wavelengths of 532, 520, and 340 nm.
Transmission electron microscopy
The liver tissues were fixed with 2.5% glutaraldehyde in phosphoric acid buffer for 2 h, followed by post‐fixation in 1% osmium acid for 2 h. Then we sectioned and stained the tissue samples. The sections were dehydrated in ethanol, and embedded in acetone. The Sections were stained with 3% uranium acetate and lead citrate. Transmission electron microscopy was performed using a Tecnai 10 microscope (HT7700, HITACHI) at the Electron Microscopy Core Facility, XiangYa Hospital Central South University.
Apoptosis measurement
The number apoptotic cells were determined using the In Situ Cell Death Detection Kit (Roche, No. 11684795910) for cryopreserved tissue sections according to the manufacturer's instructions, which is for detection and quantification of apoptosis (programmed cell death) at single cell level, based on labeling of DNA strand breaks (TUNEL technology).
Statistical analysis
All experiments were performed at least in triplicate. Data in figure legends are presented as mean ± SD values. Statistical analyses were performed with GraphPad Prism 6.0 software using one‐way ANOVA for multiple comparison or Mantel‐Cox test for survival analyses. Statistical significance was assessed at *P < 0.05, **P < 0.01.
Author contributions
Guowei Xing: Conceptualization; Data curation; Formal analysis; Investigation; Methodology. Lihua Meng: Formal analysis; Investigation. Shiyao Cao: Supervision; Investigation. Shenghui Liu: Supervision. Jiayan Wu: Investigation. Qian Li: Supervision. Wendong Huang: Supervision. Lisheng Zhang: Conceptualization; Resources; Supervision; Funding acquisition; Validation; Visualization; Project administration.
In addition to the CRediT author contributions listed above, the contributions in detail are:
LZ, WH and GX conceived the project, designed the experiments, and wrote the manuscript. GX and LM performed the majority of the experiments. SC, SL, JW and QL performed experiments and analyzed the data generated. LZ supervised the project. All authors gave input on the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Acknowledgements
We would like to appreciate our workmates, Chenxia Lu, Dan Qin, and Yi Yan who gave us their help on discussion or writing of this thesis. We would also like to thank the core facilities for they help in EM image treatment. The project is supported by MOST|National Key Research and Development Program of China No. 2017YFA0103200 and 2017YFA0103202, and National Natural Science Foundation of China 32071143.
EMBO reports (2022) 23: e52280
Data availability
The data in this manuscript did not require deposition in a public repository. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
References
- Barrera C, Valenzuela R, Rincon MA, Espinosa A, Echeverria F, Romero N, Gonzalez‐Manan D, Videla LA (2018) Molecular mechanisms related to the hepatoprotective effects of antioxidant‐rich extra virgin olive oil supplementation in rats subjected to short‐term iron administration. Free Radic Biol Med 126: 313–321 [DOI] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R et al (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575: 688–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt JM, Djouadi F, Kelly DP (1998) Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator‐activated receptor alpha. J Biol Chem 273: 23786–23792 [DOI] [PubMed] [Google Scholar]
- Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y et al (2004) Cardiomyocyte‐restricted peroxisome proliferator‐activated receptor‐delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med 10: 1245–1250 [DOI] [PubMed] [Google Scholar]
- Conrad M, Angeli JP, Vandenabeele P, Stockwell BR (2016) Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 15: 348–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Chen X, Li J, Comish P, Kang R, Tang D (2020) Transcription factors in ferroptotic cell death. Cancer Gene Ther 27: 645–656 [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Stockwell BR (2014) The role of iron and reactive oxygen species in cell death. Nat Chem Biol 10: 9–17 [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti‐Furga G, Stockwell BR (2015) Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol 10: 1604–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH et al (2019) FSP1 is a glutathione‐independent ferroptosis suppressor. Nature 575: 693–698 [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A et al (2017) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13: 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E et al (2014) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16: 1180–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinathan L, Hannon DB, Peters JM, Vanden Heuvel JP (2009) Regulation of peroxisome proliferator‐activated receptor‐alpha by MDM2. Toxicol Sci 108: 48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerre‐Millo M, Gervois P, Raspe E, Madsen L, Poulain P, Derudas B, Herbert JM, Winegar DA, Willson TM, Fruchart JC et al (2000) Peroxisome proliferator‐activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J Biol Chem 275: 16638–16642 [DOI] [PubMed] [Google Scholar]
- Han L, Bai L, Qu C, Dai E, Liu J, Kang R, Zhou D, Tang D, Zhao Y (2021) PPARG‐mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem Biophys Res Commun 576: 33–39 [DOI] [PubMed] [Google Scholar]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL et al (2017) Drug‐tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551: 247–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou GG, Wang CH, Sun JF, Yang MZ, Lin D, Li HJ (2013) Rhodamine‐based "turn‐on" fluorescent probe with high selectivity for Fe(2+) imaging in living cells. Biochem Biophys Res Commun 439: 459–463 [DOI] [PubMed] [Google Scholar]
- Hwang JS, Kim E, Lee HG, Lee WJ, Won JP, Hur J, Fujii J, Seo HG (2021) Peroxisome proliferator‐activated receptor delta rescues xCT‐deficient cells from ferroptosis by targeting peroxisomes. Biomed Pharmacother 143. 112223 [DOI] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB et al (2017) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13: 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S (2008) Peroxisome proliferator activated receptors and lipoprotein metabolism. PPAR Res 2008: 132960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Haluzik M, Asghar Z, Yau D, Joseph JW, Fernandez AM, Reitman ML, Yakar S, Stannard B, Heron‐Milhavet L et al (2003) Peroxisome proliferator‐activated receptor‐alpha agonist treatment in a transgenic model of type 2 diabetes reverses the lipotoxic state and improves glucose homeostasis. Diabetes 52: 1770–1778 [DOI] [PubMed] [Google Scholar]
- Lei P, Bai T, Sun Y (2019) Mechanisms of ferroptosis and relations with regulated cell death: a review. Front Physiol 10: 139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo A, Wang H, Wang Y, Huang Q, Zhang Q (2016) A novel colorimetric and turn‐on fluorescent chemosensor for iron(III) ion detection and its application to cellular imaging. Spectrochim Acta A Mol Biomol Spectrosc 168: 37–44 [DOI] [PubMed] [Google Scholar]
- Muckenthaler MU, Rivella S, Hentze MW, Galy B (2017) A red carpet for iron metabolism. Cell 168: 344–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumelle R, Haas JT, Hennuyer N, Bauge E, Deleye Y, Mesotten D, Langouche L, Vanhoutte J, Cudejko C, Wouters K et al (2019) Hepatic PPARalpha is critical in the metabolic adaptation to sepsis. J Hepatol 70: 963–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W et al (2008) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15‐lipoxygenase dependent‐ and AIF‐mediated cell death. Cell Metab 8: 237–248 [DOI] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE et al (2017) Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171: 273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D (2016) Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63: 173–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao P, Jiang Y, Wang H, Gao G (2021) CYP2J2‐produced epoxyeicosatrienoic acids contribute to the ferroptosis resistance of pancreatic ductal adenocarcinoma in a PPARgamma‐dependent manner. Zhong Nan Da Xue Xue Bao Yi Xue Ban 46: 932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonnus W, Linkermann A (2016) "Death is my Heir"–ferroptosis connects cancer pharmacogenomics and ischemia‐reperfusion injury. Cell Chem Biol 23: 202–203 [DOI] [PubMed] [Google Scholar]
- Tonnus W, Linkermann A (2017) The in vivo evidence for regulated necrosis. Immunol Rev 277: 128–149 [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM (1994) mPPAR gamma 2: tissue‐specific regulator of an adipocyte enhancer. Genes Dev 8: 1224–1234 [DOI] [PubMed] [Google Scholar]
- Tugwood JD, Issemann I, Anderson RG, Bundell KR, McPheat WL, Green S (1992) The mouse peroxisome proliferator activated receptor recognizes a response element in the 5' flanking sequence of the rat acyl CoA oxidase gene. EMBO J 11: 433–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh D, O'Brien NA, Zandkarimi F, Tong DR, Stokes ME, Dunn DE, Kengmana ES, Aron AT, Klein AM, Csuka JM et al (2020) MDM2 and MDMX promote ferroptosis by PPARalpha‐mediated lipid remodeling. Genes Dev 34: 526–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, An P, Xie E, Wu Q, Fang X, Gao H, Zhang Z, Li Y, Wang X, Zhang J et al (2017) Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 66: 449–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerasinghe AJ, Schmiesing C, Varaganti S, Ramakrishna G, Sinn E (2010) Single‐ and multiphoton turn‐on fluorescent Fe(3+) sensors based on bis(rhodamine). J Phys Chem B 114: 9413–9419 [DOI] [PubMed] [Google Scholar]
- Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D (2016) Ferroptosis: process and function. Cell Death Differ 23: 369–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB et al (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156: 317–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR (2008) Synthetic lethal screening identifies compounds activating iron‐dependent, nonapoptotic cell death in oncogenic‐RAS‐harboring cancer cells. Chem Biol 15: 234–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR (2016) Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26: 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Li X, Zhang X, Kang R, Tang D (2016) Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun 478: 1338–1343 [DOI] [PubMed] [Google Scholar]