

Abstract
In 1977, the world witnessed both the eradication of smallpox and the beginning of the modern age of genomics. Over the following half‐century, 7 epidemic viruses of international concern galvanized virologists across the globe and led to increasingly extensive virus genome sequencing. These sequencing efforts exerted over periods of rapid adaptation of viruses to new hosts, in particular, humans provide insight into the molecular mechanisms underpinning virus evolution. Investment in virus genome sequencing was dramatically increased by the unprecedented support for phylogenomic analyses during the COVID‐19 pandemic. In this review, we attempt to piece together comprehensive molecular histories of the adaptation of variola virus, HIV‐1 M, SARS, H1N1‐SIV, MERS, Ebola, Zika, and SARS‐CoV‐2 to the human host. Disruption of genes involved in virus–host interaction in animal hosts, recombination including genome segment reassortment, and adaptive mutations leading to amino acid replacements in virus proteins involved in host receptor binding and membrane fusion are identified as the key factors in the evolution of epidemic viruses.
Keywords: epidemic, membrane fusion, molecular adaptations, receptor‐binding domain, recombination
Subject Categories: Evolution & Ecology; Microbiology, Virology & Host Pathogen Interaction
This review comprehensively describes the molecular adaptation of variola virus, HIV‐1 M, SARS, H1N1‐SIV, MERS, Ebola, Zika, and SARS‐CoV‐2 to the human host and identifies the key factors in the evolution of epidemic viruses.
Introduction
The Fall of 1977 was a pivotal season for virology. In October, members of the World Health Organization eradication team identified the final naturally acquired Smallpox infection (Centers for Disease Control and Prevention, 2022a) marking the end to endemicity of the most devastating viral pathogen in recorded human history (Nicolas LePan, 2020). Two months later, on December 1st, an article was published in The Proceedings of the National Academy of Sciences, describing “a new method for determining nucleotide sequences in DNA” (Sanger et al, 1977), Sanger sequencing, which both empowered and galvanized molecular biologists across the globe to begin sequencing whole genomes, beyond the mere snippets of genes as was previously possible. This quest began with viruses. The first complete genome (Escherichia coli RNA bacteriophage MS2) was released in 1976 after years of painstaking assembly using existing methods (Fiers et al, 1976), whereas the seminal Sanger sequencing paper described the genome of Escherichia phage phiX174. These genomes were selected for practical reasons, principally, for being quite short (both below 4 kilobases, kb) and coming from viruses that easily grow to high titers in the laboratory. Over the next decade, dozens of genomes of viruses infecting diverse hosts were sequenced, including some much larger, laying the foundation for the study of comparative and evolutionary virus genomics. This burgeoning field provides a panoramic view of the genome evolution of viruses that have caused epidemics in humans. Although even at the time of this writing, Spring of 2022, global viral evolutionary genomics remains an enterprise of the future, several adaptive trends in the evolution of pathogenic viruses are readily decipherable.
Unlike cellular genomes that all share the same mechanisms for the expression of genetic information including about 100 genes that are conserved across the three domains of life (Tatusov et al, 2000; Rochman et al, 2020; Galperin et al, 2021), viruses employ effectively all possible routes for information transmission and lack universally shared genes (Iranzo et al, 2016). Moreover, cellular genomes can be considered essentially static compared with viral genomes. Large populations, short generation times, and high mutation rates enable viruses to undergo extremely rapid, adaptive molecular evolution (Drake & Holland, 1999; Sanjuán, 2012). Even at the species level, variations in selective pressures over time (Mutz et al, 2022) and, in particular, the overwhelming effect of purifying selection (Wertheim & Kosakovsky Pond, 2011) can obfuscate evolutionary histories.
Given these obstacles in the study of virus evolution, there is perhaps some poetic justice in the current state of the field. The only context, in which precise viral phylogenomics is feasible, is the same context where it is of the greatest practical value, namely, the molecular history of human viral epidemics. Epidemics begin with a sudden change in selective pressures, mainly caused by zoonoses or environmental changes, and result in brief, on the evolutionary scale, periods during which the size of the viral population dramatically increases. Consequently, analysis of virus phylodynamics is most likely to provide new biological insights into the molecular mechanisms of adaptive evolution when applied to data obtained during outbreaks, which is the time when intensified pathogen surveillance and sequencing efforts are critical to public health.
In this review, we attempt to piece together comprehensive molecular histories of the seven globally consequential epidemics (Nicolas LePan, 2020; Centers for Disease Control and Prevention EIS, 2022), which have occurred since 1977, and Smallpox. These epidemics all meet the classical definition of a pandemic, “an epidemic occurring worldwide, or over a very wide area, crossing international boundaries and usually affecting a large number of people” (Kelly, 2011); however, we will use the term epidemic throughout to avoid contradicting World Health Organization designations. Fig 1 summarizes key events across 8 separate timelines for each virus evolving through four periods relative to the point of zoonoses: viral “prehistory” inferred from comparative genomics; the period directly preceding zoonoses when “critical” adaptations mediating efficient human transmission are acquired, usually inferred through a combination of comparative genomics and model systems; “rapid” adaptations emerging at the very beginning of an epidemic; and signatures of “ongoing” molecular evolution throughout the initial outbreak and potentially into endemicity.
Figure 1. Molecular timelines of human virus epidemics.

Key molecular events are displayed for each virus across four time periods: viral prehistory, critical period preceding zoonosis, rapid adaptations following zoonosis, and ongoing human adaptation. Each timeline is represented as an arrow with large branches representing key molecular events, which may either promote human adaptation (forward) or viral diversity (reverse, with one prominent exception of Zika passenger mutations following a population bottleneck also illustrated by a reverse arrow), and small branches representing the conserved trend of decreasing diversity leading up to and increasing diversity following zoonosis. Events are categorized as related to receptor binding (Bind, yellow), membrane fusion (Fuse, pink), recombination (ReC, blue), and all others (Other, green). Groups of related events are numbered, and key sites, substitutions, or deletions are lettered. The absence of coloration indicates no significant molecular events for that virus in that period are identified or expected. The absence of an arrow (mainly around zoonosis) indicates a high probability that key molecular events are missing from the sequencing record.
These molecular features can be coarsely grouped into two categories: major genome remodeling in the form of recombination or reassortment (Fig 2) and point mutations. Many of the point mutations that have been established to dramatically affect human transmissibility result in amino acid replacement in surface proteins (Fig 3). Such amino acid changes reshape the major antigenic protein to stabilize the receptor interface, destabilize the antibody interface, or alter membrane fusion dynamics (Fig 4). While it seems reasonable to expect that adaptations within surface proteins play a primary role in virus adaptation to a novel host, it also should be noted that the functional impact of an amino acid change in a structural protein is easier to assess compared with such a change in a nonstructural protein, and consequently, surface proteins are likely to be overrepresented among the identified virus adaptations. Furthermore, even among the 8 well‐characterized viruses discussed here, yet unidentified molecular events occurring prior to zoonosis might have been key to supporting human transmissibility (see Fig 1). In what follows, the known molecular history of each epidemic virus is charted separately, with the exception of the three Betacoronaviruses, which are described together, and conserved trends are considered in the Concluding Remarks.
Figure 2. Modes of virus recombination.
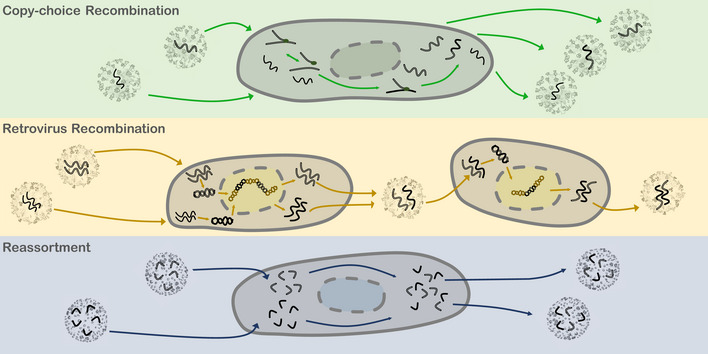
Top, Copy‐choice Recombination: Co‐infection of a single cell by more than one virus may result in chimeric viral progeny. Betacoronavirus are illustrated, and the caption describes RNA viruses in general. RNA‐dependent RNA polymerase (RdRP, green ovals) bound to nascent genomic RNA dissociates from a template of one virus and associates to the homologous position of another. Middle, Retrovirus Recombination: HIV is illustrated. Viral RNA is reverse transcribed into DNA and incorporated into the host cell genome. Mixed virions may be packaged with one copy of each genome. Upon cell entry and reverse transcription, this mixed virus produces chimeric DNA, which, when incorporated into the host genome and transcribed, produces chimeric virus progeny. Bottom, Reassortment: H1N1 is illustrated. Segmented viral RNA may be shuffled and packaged into chimeric virus progeny.
Figure 3. Virus receptor binding.

Virus receptors and primary antigens are detailed. For variola virus, the structure of the small molecule receptor chondroitin sulfate is shown. For SARS‐CoV‐2, the reversible conformational change between RBD‐accessible and inaccessible orientations (impacted by the residue in site S614) of the trimeric surface structure is highlighted. For H1N1, the structure of the small molecule receptor sialic acid is shown, with the host‐specific galactose linkages (human, α2,6Gal; waterfowl α2,3) emphasized. A public domain image for chondroitin sulfate was used: https://en.wikipedia.org/wiki/Chondroitin_sulfate.
Figure 4. Generalized virus lifecycle diagram.

Viral cell entry, replication, and dissemination are outlined, with the status of the host cell (light red) and virus membranes (dark red) highlighted throughout. The virus adheres to the cell surface, binding to host receptors, and is endocytosed, while the virus membrane remains intact. The virus membrane then fuses with the endosome membrane, often mediated by the same protein that is initially bound to the cell surface receptor. Virus and endosome membrane fusion admits viral entry to the cell cytoplasm, enabling replication and expression of the viral genome. Finally, virus surface proteins form a nascent structure within the cell membrane into which the viral genome is packaged, and the mature virion is released into the extracellular environment. Zika virus is used as a specific example.
Orthopoxvirus Variola virus
Speciation
Smallpox has played an important role in determining the course of recorded human history, with outbreaks having affected most if not all agricultural societies worldwide (Fenner et al, 1988; Geddes, 2006). As a result of the intolerable societal cost, vaccine development began in response to smallpox, with the practice of inoculation through exposure to the sores of convalescing patients potentially originating as early as the 2nd century BCE (The College of Physicians of Philadelphia, 2022). This led to safer inoculations by the 18th century mediated by a livestock Orthopoxvirus (historically referred to as “cowpox” but distinct from the viruses currently classified as Cowpox) and subsequently vaccinia virus, introducing the term vaccine itself (Geddes, 2006), and making Smallpox the first and so far only endemic human disease to be eradicated.
Until 2016, only two major diversification events in smallpox evolution were known. The first is the divergence of smallpox from its last common ancestor with the most closely related species of Orthopoxviruses, camelpox virus, and taterapox virus. Variola virus is strictly specific to the human host (Babkin & Babkina, 2015), and the speciation event that led to its emergence roughly coincided with the appearance of the earliest known agricultural societies with urban centers (Reba et al, 2016), about 2–4,000 years BCE. The second diversification event was the divergence of the Variola major strain (PI clade) causing the classic, severe disease (case fatality rates, CFR, of up to 30%) from the much milder Variola minor strain (PII clade) (CFR < 1%), about 300–500 years ago (Li et al, 2007; Babkin & Babkina, 2015; Babkin et al, 2022).
This history was substantially expatiated in 2016 with the isolation and sequencing of variola virus from a 17th‐century Lithuanian mummy (Duggan et al, 2016). Phylogenetic analysis suggested that the last common ancestor of the PI/II clades diverged from the common ancestor of this isolate approximately a century prior to the divergence of clades PI and PII. Subsequently, improved molecular clock analysis motivated the hypothesis that widespread vaccination in the 19th century played a role in the diversification of clades PI/II (Duggan et al, 2016). The known diversity of smallpox was again, even more dramatically, expanded in 2020 with the analysis of samples isolated from northern European individuals dated to the 6th or 7th century CE and belonging to a clade that likely diverged nearer the last common ancestor of all variola viruses than any other known branching event (Mühlemann et al, 2020). These results provide physical evidence of smallpox infection as early as the middle of the first millennium CE and deepen the understanding of the role played by gene loss in variola virus evolution.
Gene loss as the principal trend in Orthopoxvirus evolution
All Orthopoxvirus genomes are almost exclusively composed of subsets of genes contained within the Cowpox virus pangenome, with only 9 genes acquired by different individual Orthopoxvirus lineages (Senkevich et al, 2021). In contrast, extensive gene loss, via complete deletion or inactivation of genes, has been ubiquitously inferred across speciation events (Fig 1). The genomes of orthopoxviruses comprise about 200 kb, and the orthopoxvirus pangenome consists of 214 genes. Of these genes, 109 are conserved in all orthopoxviruses and encode proteins essential for virus replication and morphogenesis, whereas the remaining genes encode accessory proteins involved in virus–host interactions. Of these accessory genes, variola viruses retain only 53–55, having lost the other half, with 19 of these genes apparently lost after the divergence with the common ancestor of camelpox and taterapox. The essential genes occupy the middle part of Orthopoxvirus genomes whereas the accessory genes are located at both ends. A declining gradient of gene loss is observed from the ends toward the central portion of the genome.
Accessory gene content correlates with orthopoxvirus range. Cowpox viruses have the broadest host range whereas variola viruses (and similarly camelpox and taterapox) have both the narrowest host range and the smallest number of accessory genes, consistent with the hypothesis that gene reduction decreases the probability of cross‐species transmission (Hendrickson et al, 2010). The common ancestor of these three virus species is inferred to have had a broader host range and probably infected rodents. Several genes specifically implicated as host range determinants have been lost in the variola lineage. A similar pattern of gene loss or inactivation among variola viruses has been observed in modern and ancient isolates (Aguado et al, 1992; Duggan et al, 2016), with the number of disrupted or absent genes increasing roughly linearly over time (Mühlemann et al, 2020). The potential contributions of neutral evolution or positive selection associated with adaptation to narrow host ranges remain an open problem. The default explanation seems to be a ratchet of gene loss: if a complete set of accessory genes underlies the broad host range, the loss of even one of these genes eliminates the purifying selection that maintained the rest. Subsequently, these genes are randomly inactivated (pseudogenized) and are then deleted, arguably, under (weak) selection for an increased rate and reduced cost of virus genome replication and expression.
This process can, generally, lead to an increased reproduction rate in the single remaining host and, accordingly, increase virulence; however, more specific mechanisms benefitting individual gene loss cannot be ruled out as demonstrated by epistatic relationships between the loss of one gene with missense mutations in other genes in Vaccina virus model systems (Senkevich et al, 2020; Tak et al, 2021). The potentially complex role of gene loss, if any, in the emergence of the less virulent PII (Alastrim) clade is unclear and may involve gene‐specific pathways, which would shed light on the determinants of Orthopoxvirus pathogenicity when elucidated.
Lentivirus HIV‐1 M
Gateway zoonoses to apes
At least 13 subtypes of HIV resulting from independent Simian Immunodeficiency Virus (SIV) zoonoses have been identified, with 4 stemming from Apes (HIV‐1) and 9 stemming from monkeys (HIV‐2) (Sauter & Kirchhoff, 2019). Most HIV infections are HIV‐1, which accounts for more than 99.9% of HIV infections in the United States (Peruski et al, 2020), and the epidemic form of HIV, HIV‐1 M, accounts for nearly 99% of HIV‐1 infections worldwide (Sharp & Hahn, 2011). The evolutionary histories of progenitor SIV strains leading to HIV‐1 are known in detail. Chimpanzee SIVcpz, the progenitor of HIV‐1 M and N, is likely itself the result of cross‐species transfer from (prey) monkeys to (predator) chimpanzees. SIVcpz is a product of recombination between simian viruses SIVrcm and SIVgsn with the gene encoding the reverse transcriptase (Pol) of SIVcpz resembling that of SIVrcm and the gene encoding the viral envelope (Env) resembling that of SIVgsn (Bailes et al, 2003). Recombination provides a mechanism for generating viral diversity central to multiple epidemic evolutionary histories including HIV (Fig 2). Monkeys are more distantly related to chimpanzees than chimpanzees are to humans (Perelman et al, 2011) and symptom severity due to HIV infection among monkeys is believed to be substantially less than among chimpanzees (Chahroudi et al, 2012), which can be severe, resembling human infection (Keele et al, 2009) consistent with the substantial host adaptation required to cross the species barrier from monkeys to chimpanzees.
A major component of the species barriers in the case of SIV and HIV (and in other viruses) is an ensemble of antiviral restriction factors, that is, proteins that inhibit viral replication. Restriction factors can play a pivotal role in preventing cross‐species transmission despite often being less effective against endemic viruses due to robust and often narrow host adaptation (Kluge et al, 2015). SIVcpz needed to overcome at least two restriction factors, APOBEC3G and Tetherin, to successfully reproduce in chimpanzees (Sauter & Kirchhoff, 2019). As demonstrated experimentally, most SIVs are unable to antagonize chimpanzee APOBEC3G (Etienne et al, 2015) and SIVcpz uses a distinct accessory protein (relative to its inferred ancestral mechanism) to antagonize Tetherin. The APOBEC3 family deaminases induce numerous (and hence lethal) mutations in the viral genome through deamination and also inhibit reverse transcription of HIV by binding to the virus genomic RNA and suppressing the primer tRNA3(Lys) (Kluge et al, 2015). As with smallpox, gene loss apparently played a major role in host adaptation. The recombination event(s) leading to the emergence of SIVcpz resulted in the loss of the entire gene coding for SIVrcm protein Vpx and the formation of a novel Vif protein (Etienne et al, 2013). Both Vpx and Vif limit the action of restriction factors. Vif inhibits chimpanzee APOBEC3D and APOBEC3G, at the cost of the loss of Vpx, which targets restriction factors HUSH and SAMHD1 affecting host cell tropism (Sauter & Kirchhoff, 2019). Tetherin tethers budding progeny virions to the plasma membrane of infected cells, preventing dissemination (Kluge et al, 2015). The adaptation of SIVcpz in chimpanzees for the Nef‐mediated Tetherin antagonism was apparently enabled by the separation of the Env and Nef genes through the elimination of an overlapping region (Sauter & Kirchhoff, 2019). In addition to the adaptations acquired to evade APOBEC3G and Tetherin, the cyclophilin‐binding loop of the capsid protein (encoded by Gag) differs from the ancestral SIVrcm motif and enables interaction with chimpanzee RanBP2, which mediates nuclear import and export (Meyerson et al, 2018).
Adaptation of SIV/HIV for reproduction in humans
Some adaptations acquired by SIVcpz enabling replication within chimpanzees lowered the species barrier for subsequent zoonosis to human hosts (Fig 1), perhaps most importantly, those mediating APOBEC3G evasion (Sauter & Kirchhoff, 2019). However, human APOBEC3H and human Tetherin are not counteracted by SIVcpz, requiring the acquisition of human‐specific adaptations for the efficient transmission of HIV‐1 M. Variability in APOBEC3H expression among the human population might have provided a susceptible subpopulation aiding zoonosis (Zhang et al, 2017; Warren & Sawyer, 2019) as the virus adapted its Vif protein to counteract this restriction factor (Ooms et al, 2013). In particular, sites 39 and 48 have been demonstrated to be key determinants of APOBEC3H degradability (Ooms et al, 2013). In HIV‐1M, the role of Tetherin antagonism was transferred from Nef to Vpu (similarly utilized by the progenitor virus SIVgsn) and appears to be determined by two small regions in the transmembrane domain of this protein. Additionally, a single amino acid in the matrix protein, Gag30, is highly conserved as Met or Leu in SIVcpz and as Arg or Lys in HIV‐1 M, N, O, and one of the two P strains, suggesting that this residue is important for reproduction in human hosts although its specific role remains to be determined(Sauter & Kirchhoff, 2019).
Although the clinical outcome of Human HIV infection has been rapidly changing through the start of the 21st century (Hughes, 2010); historically, active infections have been chronic and often produce distinct signatures of intra‐host adaptation (Holmes, 2001). Host polymorphisms, especially variability in APOBEC3H as mentioned above (Ooms et al, 2013); stochasticity associated with population bottlenecks and genetic drift (Theys et al, 2018); and the heterogeneity of the host response associated with disease progression itself result in pronounced heterogeneity among the mutational landscapes within individual hosts. Despite these complicating factors, conserved features have emerged including specific amino acid replacements in the HIV‐1 reverse transcriptase (Larder & Kemp, 1989; Shafer et al, 2007) and Env proteins (Borrow et al, 1997; Van Duyne et al, 2019; McCaul et al, 2021), which confer drug resistance. The recent discovery of a highly divergent and highly virulent HIV‐1 variant, which likely emerged in the 1990s as a result of de novo mutation, in contrast to recombination (Wymant et al, 2022), emphasizes the vast mutational repertoire accessible to this virus. Intra‐ and interhost adaptive signatures, in large part due to the immune‐compromising effects of infection, may widely differ, and prolonged evolution within individual hosts prior to transmission can result in significantly diverged variants that would not have emerged otherwise. Furthermore, co‐infection with HIV‐1 and a secondary virus can potentiate otherwise unlikely evolutionary trajectories for the latter virus, admitting passage over steep fitness barriers to inter‐host circulation. For example, prolonged SARS‐CoV‐2 co‐infection enabled the evolution of strains demonstrating substantial immune escape within individual patients (Cele et al, 2022) and the Omicron variant might have originated as the result of a co‐infection (see below).
Betacoronaviruses
Origins of Betacornaviruses in bats and accessory gene loss
Half of the viruses discussed here likely originated among bats and passed through intermediate hosts including all three Betacoronaviruses SARS, MERS, and SARS‐CoV‐2 and Ebola (Peiris et al, 2004; Wang & Eaton, 2007; Mohd et al, 2016; Jacob et al, 2020; Zhang & Holmes, 2020). While a deep understanding of viral immunity is limited to a few species across the tree of life and therefore many unusual features relevant to zoonotic potential most likely remain undiscovered, antivirus defense systems among bats display apparently unique features admitting persistent or latent infection for extended durations. Bats are able to mitigate what would otherwise be pathological inflammation as a result of active infection, reducing cytokine levels and elevating interferon expression (Subudhi et al, 2019). Consequently, bats often display high viral loads and shedding, while apparently eliciting few adverse host reactions. Conceivably, these distinct selective pressures triggered the evolution of viral accessory proteins, which interact with bat‐specific defense systems (Chu et al, 2018). The presence of these, often poorly conserved (Pereira, 2020), accessory proteins within the genomes of descendent viruses infecting different hosts could be neutral or even (weakly) deleterious to the virus.
This hypothesis is further motivated by the observation that large deletions or truncations among accessory proteins have repeatedly emerged over the course of all three Betacoronavirus epidemics (Guan et al, 2003; Chinese SARS Molecular Epidemiology Consortium, 2004; Chu et al, 2018; El‐Kafrawy et al, 2019; Zhang et al, 2021b; Zinzula, 2021). For SARS and SARS‐CoV‐2, this trend is most apparent within ORF8, for MERS ORFs 3, 4a, and 4b (Fig 1). At least three independent deletion events resulting in the loss of 29, 82, and 415 (encompassing the entire ORF8) nucleotide fragments were observed during the SARS outbreak. However, complicating the interpretation that these deletions are adaptive across a broad host range, the most commonly observed 29nt deletion (within ORF8) was shown to substantially decrease replication across multiple model systems (Muth et al, 2018); furthermore, a hairpin in the RNA secondary structure (Chinese SARS Molecular Epidemiology Consortium, 2004) could confer a high probability of deletion in this region. Large ORF8 deletions have also been observed for SARS‐CoV‐2 although none were detected in dominant strains as of the time of this writing. The products of ORF8 appear to be involved in a variety of pathways leading to immune evasion through the downregulation of MHC‐I and interferon suppression (Zhang et al, 2021b; Zinzula, 2021). In addition to decreased replication, deletions in ORF8 have been associated with milder symptoms. Therefore, it appears likely that selection pressure for decreased pathogen virulence played a role in the emergence of these deletions.
Spike protein malleability and membrane fusion dynamics
Comparative genomics has revealed conserved inserts that appear to be associated with zoonotic potential and pathogenicity among Betacoronaviruses (Gussow et al, 2020). SARS, SARS‐CoV‐2, and MERS each contain unique insertions within the receptor‐binding domain (RBD), which were likely acquired during the epidemic “prehistory” creating a more malleable Spike/receptor interface and enabling more rapid adaptation to novel hosts. In SARS, a direct hydrophobic interaction between the residue L472 within the insert and M82&L79 of the receptor is affected. In SARS‐CoV‐2, F486 within the insert provides a large, hydrophobic surface interacting with ACE2 residues M82, L79, and Y83. The insert also mediates a charge interaction between spike E484 and ACE2 K31, and multiple hydrogen bonds. These insert‐mediated interactions are stronger in SARS‐CoV‐2 than in SARS. MERS displays two inserts in similar positions in the RBD, within longer, more rigid β‐sheets. These differences could contribute to the apparent variations in zoonotic potential and transmissibility following zoonosis among MERS, SARS, and SARS‐CoV‐2.
Following cell attachment and endocytosis, the spike protein is then responsible for membrane fusion (viral and host cell endosome) admitting viral entry into the cytoplasm, which is accomplished through the insertion of the fusion peptide into the host membrane mediated by bundling of two heptad repeats (Huang et al, 2020). Two distinct, four amino acid insertions, one shared between SARS and SARS‐CoV‐2, were identified within an alpha‐helical structure connecting the first heptad repeat (HR1) and the fusion peptide. Although not demonstrated directly, the increased flexibility afforded by this insertion is likely to affect fusion dynamics, and the recurrence of this structural feature among high case fatality rate (CFR) coronaviruses implies epidemiological relevance (Gussow et al, 2020).
Subcellular localization and charge accumulation
Betacoronaviruses display complex subcellular localization patterns that differ among the virus proteins (Timani et al, 2005). The nucleocapsid (N) protein is a major structural protein participating in many functions displaying substantial antigenicity, is trafficked in and out of the host cell nucleus, and may act as a shuttle for other cargo (Lin et al, 2003). Nuclear localization of N may enable SARS (and by extension, other Betacoronaviruses) to regulate the cell cycle, delay cell growth, and promote viral protein synthesis (Timani et al, 2005) and the regulation of nuclear trafficking by the N protein could play an important role in admitting and maintaining efficient transmission within novel hosts following cross‐species transfer for Betacoronaviruses. Viral hijacking of host cell‐cycle regulation is often deleterious for the host and, indeed, phylogenetic analysis has revealed a strong correspondence between coronavirus pathogenicity and the presence of positive charges in the vicinity of nuclear localization and export motifs (Gussow et al, 2020). Removing positively charged residues in the vicinity of nuclear localization motifs is known to disrupt nuclear import and an accumulation of positive charge around these motifs has also been observed following zoonosis, during the early period of ongoing human adaptation of SARS‐CoV‐2 (Rochman et al, 2021c). In contrast, some of the characteristic ORF4b deletions observed during the MERS epidemic resulted in the loss of an NLS motif (Chu et al, 2018), emphasizing the complex relationship between nuclear localization strength, pathogenicity, and fitness of Betacoronaviruses.
HCoV‐OC43 and HKU1
Cross‐species transmission events are frequent among Betacoronaviruses, and three of the eight human epidemic viruses reviewed here are Betacoronaviruses. Although these three events, all of which occurred in the 21st century, are the only epidemics known to have been definitively caused by Betacoronaviruses, they likely do not represent the earliest Betacoronavirus epidemics. Two additional Betacoronaviruses, Betacoronavirus 1 (subspecies Human Coronavirus OC43, HCoV‐OC43) and Human coronavirus HKU1 are endemic among human populations (Vijgen et al, 2005; Woo et al, 2005). Molecular clock analysis has placed the last common ancestor of HCoV‐OC43 and Betacoronavirus 1, Bovine Coronavirus (BCoV), at the end of the 19th century (Vijgen et al, 2005) suggesting a zoonotic event coinciding with the 1889–1890 epidemic (also referred to as the Russian or Asiatic Flu). Both evolutionary and clinical records (Brüssow & Brüssow, 2021) suggest HCoV‐OC43 was likely the causative agent of this outbreak contradicting the historical attribution of Influenza. While not reported until 2005 (Woo et al, 2005), to our knowledge, there is no indication that HKU1 was not already endemic at that time; its origins are unclear; and it appears plausible that the emergence of HKU1 could also be mapped to an established epidemic.
Severe acute respiratory syndrome coronavirus
Severe acute respiratory syndrome coronavirus (SARS) likely first emerged in Guangdong (China) in 2002 (Peiris et al, 2004) as a result of zoonosis from a reservoir of SARS‐CoV‐like viruses among horseshoe bats (genus Rhinolophus) linked by an intermediate host(s) including masked palm civets (Paguma larvata) (Wang & Eaton, 2007). The host receptor for SARS, angiotensin 1‐converting enzyme 2 (ACE2), is highly conserved among vertebrates (Damas et al, 2020). Accordingly, only 6 amino acid residues in the RBD differ between the civet and human isolates, of which 3 are located at the ACE2 interface (Graham & Baric, 2010) including sites 479 (mainly N among human isolates and variably K, N, or R in civet) and 487 (T in human classic, epidemic isolates, 2002–3; S in human later, mild isolates 2003–4, likely the result of a limited independent zoonotic event (Peiris et al, 2004); S in civet)(Li, 2008). Structural modeling demonstrated that sites 479 and 487 are critical only for human ACE2 binding and that civet ACE2 binding is less sensitive to substitutions at these sites (Li, 2008).
Laboratory studies demonstrating the nonselective binding of both human and civet isolates to civet ACE2, in contrast to the selective binding of human isolates to human ACE2, further support a straightforward scenario in which adaptations in the RBD required for efficient human transmission were already acquired in the intermediate host (Graham & Baric, 2010). Remarkable efforts resulted in the containment of the SARS epidemic with limited opportunities for genomic sequencing; nonetheless, some signatures of continued molecular adaptation to human hosts occurring after zoonosis are apparent including evidence of significant positive selection within the S protein at the start of the epidemic (Chinese SARS Molecular Epidemiology Consortium, 2004).
Middle eastern respiratory syndrome coronavirus
As with SARS and SARS‐CoV‐2, Middle eastern respiratory syndrome coronavirus (MERS) likely originated among bats; however, unlike SARS and SARS‐CoV‐2 for which the identification of intermediate hosts is challenging, dromedary camels are well‐established as the primary animal reservoir supporting MERS zoonoses (Mohd et al, 2016). MERS is ubiquitous among camel populations in Africa and the Arabian Peninsula but limited among other livestock (Mohd et al, 2016). While the first human infections were not recognized until 2012 (Memish et al, 2020), diverse MERS lineages have infected humans (Sabir et al, 2016); MERS infections are common within the camel industry; and antibodies have been observed in up to 3% of associated individuals (El‐Kafrawy et al, 2019), suggesting the possibility of earlier human infections. Despite its ubiquity, MERS infections are geographically limited, suggesting MERS was not communicated to Asia via caravans; did not circulate among camels until modern trade practices supplanted the practice; and thus was introduced more recently than Smallpox (Mohd et al, 2016).
Among affected camel populations, the regionality of MERS subtypes is apparent with two distinct clades identified in Northern Africa and the Arabian Peninsula. Two Northern African subtypes, East and West, are further delineated and East Africa is the putative origin of the common ancestor of all extant lineages. Arabian MERS appears to be best adapted to transmission among camels (El‐Kafrawy et al, 2019). The molecular features distinguishing these regional subtypes are primarily deletions in accessory proteins, principally, ORF3, ORF4a, and ORF4b (Chu et al, 2018; El‐Kafrawy et al, 2019). However, deletions have independently emerged among both African and Arabian variants (Chu et al, 2018; El‐Kafrawy et al, 2019), and although differences in the replicative capacities of these subtypes have been demonstrated in model systems, no characteristic deletion has been identified as causative (Chu et al, 2018). As described above, repeated deletions in genes encoding accessory proteins are common among Betacoronaviruses.
Thus, the molecular features leading to increased transmissibility and zoonotic capacity among Arabian strains likely remain unrecognized (Chu et al, 2018). Phylogenetic analysis suggests that there have been 100s of independent zoonotic events from Arabian lineages to humans since 2012 without widespread human transmission (Dudas et al, 2018) despite sizable hospital outbreaks (Memish et al, 2020), suggesting MERS spread is strongly influenced by low host/social tolerance (Rochman et al, 2021a) in addition to constraints on infectivity. Furthermore, recombination among camel isolates of MERS is extensive (Dudas & Rambaut, 2016) (Fig 2) and each zoonotic event can result in distinct disease, ranging from the unremarkable endemicity within the camel industry to highly virulent outbreaks with a CFR of about 35% (Memish et al, 2020).
These high rates of recombination and numerous predicted independent zoonoses distinguish MERS from SARS and SARS‐CoV‐2. MERS also recognizes a different receptor, Dipeptidyl‐peptidase 4 (DPP4) (Fig 3) (Memish et al, 2020); nevertheless, similarly to the cases of SARS and SARS‐CoV‐2, signatures associated with human adaptation in the MERS spike protein are apparent. MERS S protein affinity for human DPP4 is substantially higher than that for the related bat coronavirus HKU4 (likely representative of the ancestral MERS bat coronavirus ancestor), and as expected, HKU4 S protein preferentially binds to bat DPP4 (Yang et al, 2014). Furthermore, as described above for SARS, MERS cell entry is accomplished through spike‐cleavage‐mediated membrane fusion. Human cellular proteases can mediate MERS but not HKU4 cell entry, whereas bat cellular proteases are compatible with both MERS and HKU4 spikes (Yang et al, 2014). As described above, MERS S protein contains a four amino acid insertion in the vicinity of the fusion peptide (Gussow et al, 2020), which could contribute to this compatibility. More generally, the MERS spike is capable of rapidly adapting to species variation in DPP4, in part facilitated by facile surface charge adjustment (Letko et al, 2018). These rapid adjustments could be mediated in part by insertions in the receptor‐binding motif, which likely increase the flexibility of the interface (also described above) (Gussow et al, 2020).
Severe acute respiratory syndrome coronavirus 2
The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐CoV‐2) epidemic is still ongoing at the time of this writing. Given that the molecular evolution of this virus is still rapidly unfolding, and that the literature devoted to its study is expanding at an unprecedented rate, the features described here will likely be incomplete by the time of publication. SARS‐CoV‐2 began circulating among human hosts in Hunan (China) by December 2019, likely following at least two independent zoonotic events (Data ref: Pekar et al, 2022; Data ref: Worobey et al, 2022). Like the other epidemic Betacoronaviruses, SARS‐CoV‐2 likely originated among bats with at least one, still unidentified, intermediate host mediating zoonosis (Zhang & Holmes, 2020). Unprecedented genomic sequencing efforts enabled the rapid identification and real‐time surveillance of the molecular evolution of SARS‐CoV‐2, with public data sharing facilitated by GenBank (Benson et al, 2012), the China National Center for Bioinformation (CNCB) (Zhao et al, 2020), and Gisaid (Elbe & Buckland‐Merrett, 2017). These efforts resulted in more sequences obtained for SARS‐CoV‐2 than any other virus by several orders of magnitude at the time of writing (Mutz et al, 2022). The SARS‐CoV‐2 genome is closely related to a bat coronavirus RaTG13, with a sequence identity of more than 96%. The S genes have a lower nucleotide identity (93%), and both SARS‐CoV‐2 and RaTG13 RBD are divergent (< 75% identity) from related Betacoronaviruses. SARS and SARS‐CoV‐2 both recognize ACE2 as the receptor (Fig 3) utilizing a largely conserved receptor‐binding motif, which, nonetheless, differs by at least 4 key residues (Lan et al, 2020).
As described above, both SARS‐CoV‐2 and RaTG13 contain insertions within the spike protein relative to related Betacoronaviruses (Zhou et al, 2020) including two associated with high pathogenicity among coronaviruses (Gussow et al, 2020). In addition to these shared inserts, SARS‐CoV‐2 differs from RaTG13 by an insert of a furin‐like cleavage site (Coutard et al, 2020), also in S protein, which could promote efficient virion egress and dissemination (Johnson et al, 2021; Peacock et al, 2021). The differential expression of furin might affect tissue tropism and the presence of a furin cleavage site in the S proteins of disparate Betacoronaviruses including MERS (but not SARS) suggests that this feature is an example of convergent evolution (Coutard et al, 2020; Zhang & Holmes, 2020).
Although the evidence of more than one independent zoonosis, mediated in part by the distinctive features of the S and possibly N proteins, indicates that SARS‐CoV‐2 progenitor viruses were likely already capable of sustained human transmission at the time of the first human infection, over the course of the epidemic, mutations resulting in amino acid replacements continue to accumulate within the spike protein admitting greater infectivity (for the sake of brevity, amino acids rather than nucleotides are indicated hereafter). The first to emerge was D614G, which became dominant worldwide by April 2020 (Rochman et al, 2021c). Although this residue is outside the RBD, it substantially impacts ACE2 binding by stabilizing the larger spike complex (Jackson et al, 2021). The S protein is organized into trimers on the virion surface (see Fig 3). In these trimers, the RBDs are present in two distinct conformations: the receptor‐accessible “up” conformation and the receptor‐inaccessible “down” conformation. A glycine in position 614 stabilizes the trimer and introduces a kinetic barrier decreasing the rate of conversion between up and down conformations (Mansbach et al, 2021). As described above for SARS and MERS, after receptor binding, S protein is responsible for membrane fusion (Fig 4). This stabilization results in an abundance of conformations in which only one of the three RBDs is in the up conformation. While reducing the number of accessible RBDs, 614G also reduces the rate at which the virus is shed and increases the probability of successful cell entry (Zhang et al, 2021a).
The remainder of 2020 and the beginning of 2021 saw the extensive circulation of multiple key nonsynonymous mutations within the SARS‐CoV‐2 RBD, most if not all of which emerged independently more than once, including single substitution variants 439K, 459F, 477N, 478K, 484K, and 501Y; double substitution variant 452R¦478K; and triple substitution variants 346K¦484K¦501Y, 417N¦484K¦501Y, and 417T¦484K¦501Y (Rochman et al, 2022). These RBD substitutions also primarily characterized the first four principal variants of concern as identified by the World Health Organization: Alpha (501Y) (Galloway et al, 2021), Beta (417N¦484K¦501Y) (Hoffmann et al, 2021), Gamma (417T¦484K¦501Y) (Hoffmann et al, 2021), and Delta (452R¦478K) (Liu & Rocklöv, 2021). 501Y dramatically increases ACE2 binding affinity, possibly through an aromatic interaction at the interface (Liu et al, 2021a), by decreasing the rate of dissociation whereas 484K slightly increases binding affinity through an increased rate of association (Laffeber et al, 2021). Likely more importantly, 484K disrupts neutralizing antibody binding (Collier et al, 2021). The 417N/T substitution both substantially decreases the receptor affinity through disruption of a salt bridge (Barton et al, 2021) and promotes antibody evasion (Greaney et al, 2021).
The 452R substitution is not at the RBD‐ACE2 interface and modestly increases binding affinity relative to 501Y (Deng et al, 2021), consistent with the observation that Delta variant receptor and antibody complexes are more similar to the wild type than Gamma variant structures (Rochman et al, 2022). This substitution might increase viral infectivity through enhanced membrane fusion and mediates immune evasion through altering cellular (HLA) responses (Motozono et al, 2021), as opposed to directly inhibiting neutralizing antibody binding, although it too decreases antibody affinity (Deng et al, 2021). Like 452R, 478K modestly increases ACE2 binding affinity by increasing positive charge and favorable electrostatic interactions (Xiong et al, 2022).
Over the course of a viral outbreak, as the number of hosts, which have previously been infected rises, selective pressures change, increasing the fitness benefit associated with the potential for immune evasion (Rochman et al, 2021b). This change in selective pressure might be reflected in the emergence of the Beta and Gamma variants, which both contain substitutions in site 417, disrupting both antibody and receptor interfaces and enabling immune evasion at the cost of receptor affinity relative to the Alpha variant. Unlike Beta and Gamma, both Delta RBD substitutions (452R¦478K) increase receptor‐binding affinity, which could contribute to the high infectivity reported for this variant while still apparently mediating some degree of immune evasion (Wilhelm et al, 2021). This connection between receptor binding and antibody evasion could be expected because neutralizing antibodies, by definition, physically block viral antigens from binding to host receptors (Ju et al, 2020).
Although other mechanisms are possible, this is often accomplished by antibody binding to the RBD itself (Niu et al, 2021). Sharing the same binding footprint, as is the case for the principal Sars‐CoV‐2 neutralizing antibodies, correlates the effect of RBD substitutions on receptor and antibody affinities, and limits the mutational repertoire, which both achieves antibody (and consequently, vaccine) escape and maintains infectivity. This correlation, observed in the wild type, may be broken in emergent variants through epistatic interactions among RBD amino acid substitutions; however, while epistasis has been observed among select RBD mutations (Laffeber et al, 2021; preprint: Nelson et al, 2021), computational analyses have demonstrated that the effects of most combinations are purely additive (Rochman et al, 2022). In the absence of epistasis, the effect of any substitution will be the same across all variants (Rochman et al, 2022) further limiting RBD evolvability under the intense selective pressures associated with inter‐host transmission.
As described above (in particular, for HIV), viral persistence within immunocompromised hosts, and under relaxed selective pressures, provides evolutionary paths not accessible during inter‐host transmission. These privileged environments enable the acquisition of multiple neutral or deleterious mutations, the combinatorial space of which is vast and the effects of which are not predictable at the time of writing through experimental or computational means. The dominant variants, as of June 2022, are all descendants of the Omicron variant, which first emerged in Fall 2021 and can be robustly classified into at least three sub‐lineages, BA.1, BA.2, and BA.3 (World Health Organization, 2021). Omicron sequences differ substantially from earlier variants and the human immune response to Omicron variant infection is measurably different (Sigal, 2022; Suryawanshi et al, 2022), further emphasizing the possibility that SARS‐CoV‐2 might evolve into regional subtypes (Rochman et al, 2021c) and eventually diverge into distinct serotypes with minimal cross‐protection (Shaffer et al, 2022).
The earliest omicron variants likely originated as a result of viral persistence among select, possibly immunocompromised, human hosts (as proposed for earlier variants as well) or within an alternative host species via a succession of reverse zoonosis and secondary zoonosis. At the time of writing, however, the origins of Omicron remain uncertain, and it cannot be ruled out that it emerged through a stepwise process with uninterrupted regional circulation, which went undetected (preprint: Martin et al, 2022). The Omicron variant contains 15 RBD substitutions: 339D, 371L, 373P, 375F, 417N, 440K, 446S, 477N, 478K, 484A, 493R, 496S, 498R, 501Y, 505H. This list includes 417N, 478K, and 501Y discussed above and 484A, which, like 484K discussed above, confers resistance to neutralization (Liu et al, 2021c) and 477N, the first RBD substitution to broadly circulate (Rochman et al, 2022), which modestly increases binding affinity by slowing the dissociation rate (Barton et al, 2021). Omicron additionally contains a nine nucleotide insertion in the N‐terminal domain (preprint: Martin et al, 2022). Insertions in this region have been demonstrated to promote immune escape (Garushyants et al, 2021).
Both antibody and receptor complexes more significantly differ from the wild type than all prior variants (Rochman et al, 2022); Omicron has been demonstrated to be both highly infectious and immune evasive (Liu et al, 2022); and unlike prior variants, structural analysis suggests a systematic bias toward antibody complex destabilization as a result of epistatic effects (Rochman et al, 2022). Although on the whole, as with prior variants, RBD epistasis appears limited, the epidemiological properties and likely origin of this variant highlight the evolutionary risk associated with viral reservoirs subject to relaxed or otherwise distinct selection pressures. Sars‐CoV‐2 has broadly circulated and likely continues to circulate among white‐tailed deer (Hale et al, 2022) and other mammals, which pose a high risk of repeated zoonoses (Fischhoff et al, 2021).
In addition to the S protein, adaptive signatures within the N protein are apparent. As discussed above, Betacoronaviruses display complex subcellular localization patterns. Increasing positive charge in the vicinity of nuclear localization signaling (NLS) motifs, a marker of NLS strength (Timani et al, 2005), is associated with the evolution of highly pathogenic coronaviruses, including Sars‐CoV‐2 (Gussow et al, 2020). Phylodynamic analysis demonstrated an accumulation of likely positively selected mutations in the vicinity of NLS within the N protein throughout 2021, most notably R(agg)203K(aaa) and G(gga)204R(cga), the result of three adjacent nucleotide substitutions, which almost always co‐occur and, as a result of the G>R substitution, increase positive charge (Rochman et al, 2021c). Finally, as described above, large deletions or truncations are repeatedly observed among accessory genes, in particular ORF8 (Zhang et al, 2021b; Zinzula, 2021).
Alphainfluenzavirus H1N1
Reassortment and viral diversity
Alphainfluenzaviruses are named according to surface protein subtypes defined by hemagglutinin (HA) and neuraminidase (N). The ancestry of the currently circulating, swine‐origin influenza A (H1N1) strain (S‐OIV), commonly referred to as “swine flu” can be mapped back to the epidemic of 1918 (Neumann et al, 2009; Zimmer & Burke, 2009). H1N1, like all viruses of the order Articulavirales, has a segmented genome, allowing for reassortment, that is, the exchange of genomic segments between viruses co‐infecting a single cell (Fig 2). Close physical proximity during replication is not required for reassortment, in contrast to typical recombination, and the rate of reassortment among Alphainfluenzaviruses is high (Pérez‐Losada et al, 2015) enabling these viruses to maintain high genetic diversity through modularity. The epidemic of 1918 was likely the result of zoonosis from an avian host reservoir ultimately resulting in antigenically similar viruses circulating among both North American swine and global human populations. Subsequently, these viruses rapidly diverged and acquired presumably host‐specific adaptations. Although descendants of the 1918 epidemic strain continued to circulate among pigs, it was rapidly displaced to the point of extinction by a novel reassortment between this virus and an avian virus, leading to the 1957 H2N2 epidemic (Zimmer & Burke, 2009). H2N2 was then displaced by H3N2, itself a result of a reassortment between H2N2 and an avian virus, resulting in the 1968 epidemic (Capua & Alexander, 2002).
In 1977, the flu landscape again rapidly changed with the reintroduction of H1N1 into the human population, in co‐circulation with H3N2, mostly likely originating with the release of a strain preserved for study in vitro (Rozo & Gronvall, 2015). Beginning around the same time, H1N1 spread from North American swine populations to Europe and Asia and began co‐circulating with “avian‐like” swine viruses originating from an independent cross‐species transmission event(s) from birds into pigs (Pensaert et al, 1981; Brown, 2000). Further cross‐species transmissions between humans, pigs, and birds also occurred so that by 1998, triple reassortant viruses with HA, NA, and PB1 genes of human H3N2 influenza origin; M, NP, and NS genes of swine H1N1 influenza origin; and PA and PB2 of avian influenza origin began circulating among pigs (Olsen, 2002; Zimmer & Burke, 2009). Finally in April 2009, H1N1 S‐OIV emerged in the human population, the result of reassortment between the triple reassortant swine virus and an “avian‐like” Eurasian H1N1 swine lineage (NA and matrix protein) (Zimmer & Burke, 2009).
Reassortments among influenza viruses, like the many described above leading to the emergence of H1N1 S‐OIV (not including the additional intra‐subtype reassortments (Wolf et al, 2006)) underlie both zoonoses and ongoing adaptation within a fixed host population. Reassortments have the potential to generate chimeric viruses with a mix of gene segments adapted to two or more hosts (Lowen, 2017), likely often resulting in viruses with reduced fitness within any single host population but substantially reducing the cross‐species transmission barrier. Once efficient transmission is established within the population, variable selection pressures on different gene segments can lead to the dominance of a variant that harbors both adaptations conferring a strong fitness advantage and slightly deleterious hitchhiking mutations (Lowen, 2017) the latter of which can be removed through reassortment.
Point substitutions and transmissibility
Although most influenza life cycle characteristics are multigenic traits, HA is the principal determinant of host range (Neumann et al, 2009). HA is primarily responsible for cell attachment to the sialic acid receptor with preferential binding determined by galactose linkages, which are differentially expressed within the human trachea epithelium (SAα2,6Gal) and waterfowl intestinal tract (SAα2,3Gal, see Fig 3). In addition, N protein also binds to sialic acid and is responsible for receptor cleavage and progeny virus release. The functional balance of receptor binding and release requires compatibility between HA and N, which affects the mutational repertoires of both genes (Wagner et al, 2002). Adaptation to SAα2,6Gal can be mediated by just two amino acid residues within the HA receptor‐binding pocket: 226L/228S, H2&H3, and 190D/225D, H1 (numbered according to H3) (Stevens et al, 2004; Tumpey et al, 2007; Neumann et al, 2009). In fact, 190D alone apparently can admit SAα2,6Gal binding with some swine and human H1 matching the avian consensus, G, in site 225. Such 190D/225G variants likely bind both SAα2,3Gal and SAα2,6Gal receptors (Reid et al, 1999) suggesting that zoonosis and efficient human transmission was mediated through the stepwise appearance of 190D and 225D (Fig 1). Following cell attachment and endocytosis, HA is then responsible for membrane fusion (viral and host cell endosome, Fig 4) admitting viral entry into the cytoplasm (Bertram et al, 2010).
In addition to HA 226L/228S a third residue, 627K in PB2 appears to be a critical determinant of pathogenicity for H1N1 S‐OIV ancestors, H3N2, and 1918 H1N1 strain; however, H1N1 S‐OIV itself bears the avian residue 627E and substitution of 627K was not found to increase fitness in vitro (Zhu et al, 2010). Instead, H1N1 S‐OIV bears residues 590S/591R (Cauldwell et al, 2014), which, although apparently amounting to an incomplete determinant of function (Foeglein et al, 2011), alters the electrostatic charge of the neighborhood surrounding residue 627 in a similar way.
Viral mRNA synthesis requires the recruitment of host‐derived, capped primers through “cap‐snatching” (De Vlugt et al, 2018). PB2, along with polymerase PB1 and endonuclease PA, forms a heterotrimeric polymerase complex where PB2 binds to the cap structures of cellular pre‐mRNAs in a process dependent on host nuclear factors (Foeglein et al, 2011). In particular, small acidic nuclear phosphoproteins ANP32A and ANP32B are redundant but essential for influenza virus (alpha and beta) replication in human cells (Staller et al, 2019). Furthermore, in a Gammainfluenzavirus (influenza C) system, the crystal structure of ANP32A in complex with the heterotrimer was resolved demonstrating an interaction between ANP32A and the neighborhood surrounding residue 627 suggesting this interaction is a key determinant of host range (Carrique et al, 2020). These findings, along with the observation that pig, duck, and chicken ANP32A support (human) H3N2 polymerase activity (Staller et al, 2019), suggest that efficient human transmission of H1N1 S‐OIV was predicated in part by residues 590S/591R and that these residues might have been required for efficient transmission of the ancestral triple reassortant virus among pigs as well. This would place the first emergence of these residues before the start of the 2009 epidemic and potentially within ancestral avian strains.
Truncations and virulence
There are substantial differences between the PB1 proteins of H1N1 S‐OIV and the 1918 strain. Protein PB1‐F2 is encoded in the +1 reading frame and is a powerful immune modulator that mediates interferon response, immune cell death, and inflammation (Le Goffic et al, 2011). Consequently, PB1‐F2 is a major virulence factor (Cheung et al, 2020) and likely contributed to the severity of the 1918, H2N2, and H3N2 epidemics (Alymova et al, 2018). Multiple pro‐inflammatory or cytotoxic virulence‐promoting residues have been identified (in particular, S66) near the C‐terminus of PB1‐F2, and the frequency of these residues within each strain decreased over time while circulating within the human population. PB1 is truncated in H1N1 S‐OIV and contains no established virulence residues. This truncation was likely inherited from the ancestral H3N2 PB1 prior to reassortment, representing an event substantially predating the 2009 epidemic. Generally, the systematic loss of virulence residues throughout the evolution of human influenza viruses suggests that reduced pathogenicity and increased host tolerance provide a fitness advantage.
The H1N1 S‐OIV NS1 protein, along with PB1, is an interferon antagonist and also substantially differs from the corresponding protein of the 1918 strain as a result of truncation. Two major changes occurred during its evolution in pigs: the C‐terminal portion that includes a PDZ‐binding domain, a pathogenicity determinant in other Alphainfluenzaviruses (Neumann et al, 2009), was truncated in addition to the substitution K217E (which abrogates binding to host Crk/CrkL signaling adapters). However, the introduction of these features into H1N1 S‐OIV does not appear to substantially affect fitness, suggesting that NS1 is responsible for fewer functions in H1N1 S‐OIV relative to the 1918 ancestral strain (Hale et al, 2010).
Ebolavirus
End of evolutionary stasis
Ebola infection is characterized by extreme symptom severity compared with most human pathogens and has led to nearly 15,000 recorded deaths as the result of under 40,000 cases, with a CFR of more than 40% overall (Jacob et al, 2020) and up to 80% for select outbreaks (Wauquier et al, 2010). The largest recorded outbreak likely began in Guinea around January 2014 and stemmed from a single zoonotic event (resulting in the emergence of the Makona variant) although earlier outbreaks had likely been sustained in part through multiple zoonoses (Gire et al, 2014; Dudas et al, 2017). As in the case of MERS, there have been multiple, and likely many, prior zoonotic events leading to Ebola outbreaks, and most outbreaks appear to be linked to unique cross‐species transmission events. Like SARS, MERS, and SARS‐CoV‐2, bats appear to be a major natural reservoir for Ebolaviruses and, like SARS and SARS‐CoV‐2, unknown intermediate hosts might play a key role in facilitating zoonoses (Jacob et al, 2020).
Prior to 2014, Ebola outbreaks had been largely contained. Perhaps unsurprisingly, without sustained human transmission, the molecular evolution of Ebola was dominated by neutral drift (Azarian et al, 2015), with little evidence of functional change (Olabode et al, 2015). No sequences were obtained over the first few months during which the Makona variant spread, obscuring whatever features might have been essential for zoonosis and, although subsequent sequencing efforts enabled robust phylodynamic analysis, few mutations were observed repeatedly, hampering functional inference from sequence analysis alone (Ladner et al, 2015). Nonetheless, signatures of human adaptation are apparent as discussed below. Ebola inter‐outbreak evolutionary rate is lower than the intra‐outbreak rates (Mbala‐Kingebeni et al, 2019) consistent with the view that being endemic within the host reservoir, the viruses that were involved in each zoonosis were not separated by periods of rapid environmental change. Rather, introduction into the new, human host involved a dramatic change in selective pressures for each virus. Similarly, the evolutionary rate is estimated to have reduced over the course of the Makona outbreak (Park et al, 2015), suggestive of a period of rapid host adaptation shortly the following zoonosis.
Analysis of the ratio of nonsynonymous to synonymous substitution rates (dN/dS, a gauge of protein‐level selection (Hurst, 2002; Hejase et al, 2020)) revealed inter‐ and intra‐outbreak differences as well. Within the mucin‐like domain of the glycoprotein, the only protein that is present at the Ebola virion surface and primarily determines tissue and cell tropism, dN/dS was observed to be greater than 1 at both timescales but significantly higher intra‐outbreak (1.44 vs. 4.74) (Park et al, 2015). In addition to this domain, an excess of nonsynonymous mutations was observed at the C termini of both the nucleoprotein and the RNA‐dependent RNA polymerase (RdRP). Although such mutational hotspots and high dN/dS values could simply be a consequence of reduced functional constraints and relaxed purifying selection, select individual codons demonstrate support for positive selection as well, mostly in the glycoprotein (Ladner et al, 2015). Some of the implicated sites have been functionally validated in model systems described below.
Enhanced membrane fusion
The Ebola glycoprotein is primarily responsible for host cell attachment, binding to the Niemann–Pick C1 protein receptor (NPC1, Fig 3) (Jacob et al, 2020). Variations in NPC1 impact Ebolavirus susceptibility among bats (Ng et al, 2015) and, as mentioned above, variations in virus glycoprotein can determine host and cell tropism (Martinez et al, 2013). A82V resides within a short alpha‐helix of the RBD of the glycoprotein; emerged near the beginning of the epidemic (Ladner et al, 2015), Fig 1; and could have contributed to increased mortality (Diehl et al, 2016). Although A82V does not face NPC1, structural modeling has demonstrated that, during receptor binding, the native alpha helix is subject to displacement, which is impacted by this substitution (Urbanowicz et al, 2016). In model systems, A82V substantially increases infectivity in human cells (Diehl et al, 2016; Urbanowicz et al, 2016) although this might not depend on an increased affinity for the receptor (Wang et al, 2017).
As described above for influenza hemagglutinin and Betacoronavirus spike proteins, the Ebola glycoprotein is responsible for both cell attachment and entry, mediating membrane fusion (Fig 4). A82V increases membrane fusion activity, reducing virus dependence on host factors and potentially admitting cell entry in tissue types where NPC1 expression is low (Dietzel et al, 2017; Wang et al, 2017). Residue 544I has been shown to confer the same membrane fusion advantage (Hoffmann et al, 2017; Ueda et al, 2017; Wang et al, 2017) and was observed prior to the emergence of the Makona variant in at least three other outbreaks (Wang et al, 2017). However, 544I is a recognized tissue culture adaptation (Ruedas et al, 2017) and its presence in clinical isolates might, in many cases, reflect an artifact of cell passage. Positive (with respect to cell entry), epistatic effects between A82V and other glycoprotein residues are apparent as well, including R29K in the signal peptide and T206M&T230A in the glycan cap. All three substitutions modify glycosylation and, although a potential liability for neutralization, reducing glycosylation is an established mechanism for facilitating cell entry (Vigerust & Shepherd, 2007; Bagdonaite & Wandall, 2018). Some of these relationships are complex. I371V increases infectivity on top of the A82V background but decreases infectivity paired with the ancestral 82A, whereas P375S has the reverse effect with respect to the residue present at site 82. A compensatory relationship between P202L and L239S was observed as well, in addition to complex epistatic relationships involving site 480 that seem to have evolved under positive selection (Ladner et al, 2015; Urbanowicz et al, 2016). Residues Q638R/L (GP2) also appear to be positively selected and could impact pathogenesis by affecting tumor necrosis factor‐alpha converting enzyme‐mediated cleavage (and subsequently viral shedding) (Ladner et al, 2015). The finding of multiple, positively selected residues operating within an epistatic network strongly suggests adaptations increasing membrane fusion activity play a key role in Ebola cross‐species transfer or efficient human transmission.
Many mutations leading to amino acid replacements in and outside the glycoprotein, which emerged during the Makona outbreak, enhanced human cell entry (Dietzel et al, 2017) or otherwise demonstrated a fitness advantage in model systems, including D759G in the polymerase (Albariño et al, 2016; Dietzel et al, 2017) although the functional effects are not as well‐studied as those of residues affecting membrane fusion. Regardless of the specific function, most adaptations were found to be host‐specific. A82V was found to confer increased infectivity only in primate cells (Diehl et al, 2016) and, generally, bat infectivity was found to inversely correlate with that of human cell lines (Urbanowicz et al, 2016). Notably, the ancestral residues A82/T544 in the glycoprotein confer greater thermal stability than human adaptations 82V/544I, perhaps representing a trade‐off between stability and membrane fusion activity dependent on host internal temperature, which is likely to be substantially higher in bats (Wang et al, 2017).
Persistent infections
Ebola infection elicits a potent, complex immune response that includes both sustained immune activation after viral clearance and immunosuppression (McElroy et al, 2015). As mentioned above, the glycoprotein is the only protein present at the Ebola virion surface and, consequently, the primary antibody target. Selective pressures promoting antibody escape likely play a principal role in shaping the mutational signatures observed within the glycoprotein including an excess of amino acid substitutions within B cell epitopes (Park et al, 2015). This presumed host‐virus arms race is further revealed through patterns of excess U‐to‐C substitutions, often appearing within short genomic regions, and including B cell epitopes, a signature of RNA editing mediated by host ADAR adenosine deaminases. On the whole, U‐to‐C events are significantly enriched, to the extent that the inclusion of these putative editing signatures substantially increases the inferred evolutionary rate (Park et al, 2015; Whitmer et al, 2018). These mechanisms, which promote within‐host viral diversification, can dramatically alter between‐host transmission dynamics, further complicated by the possibility of persistent infection.
Live virus, primarily residing within the male genitourinary system, has been isolated from Ebola survivors up to two years after recovery from acute infection. In such instances, Ebola replication and transcription continued, indicative of viral persistence rather than latency, although latency in immune‐privileged tissues resulting in minimal genomic change is also possible (Bosworth et al, 2021). Persistent infections are critically relevant for public health as prior outbreaks have been sustained over periods without any active infections through transmission following persistent infection from apparently recovered individuals (Whitmer et al, 2018). Persistent infections provide a selective environment that enables the virus to explore weakly deleterious mutations. Due to epistasis, multiple such mutations, although only rarely sustained through between‐host transfer, can, in concert, result in a substantial fitness advantage. Multiple, nonsynonymous changes in the glycoprotein emerge over the course of persistent infection, likely due to relaxed selection constraints (Whitmer et al, 2018). Furthermore, sustained infection within an immune‐privileged environment (a “low‐dose” antibody regime) promotes antibody and vaccine escape (Rochman et al, 2021b). Although persistent infections display a reduced evolutionary rate relative to acute infections, perhaps impacted by variable tissue tropism (Blackley et al, 2016), the existence of these persistent viral niches within human hosts represents an evolutionary risk for the emergence of Ebola with epidemic potential as Ebola continues, at the time of writing, to circulate in the human population (Shaffer et al, 2022).
Flavivirus Zika virus
Early Asian spread and population bottlenecks
Zika virus likely originated in Africa and spread East, circulating within Africa prior to 1950; Asia by 1966; Pacific islands by 2007; and the Americas by 2015 (Musso et al, 2019). Over this period, the epidemiological properties of the virus appeared to change dramatically from modest, likely endemic circulation with mild symptoms (subject to reporting bias), towards epidemic cycles infecting up to 50% of affected communities with major neurological complications (Kindhauser et al, 2016; Liu et al, 2021b). Unique among the human epidemic‐causing viruses detailed in this work, Zika is primarily transmitted through a vector, the mosquito Aedes aegypti (“yellow fever mosquito”); however, sexual and maternal‐fetal transmission is also epidemiologically relevant (Musso et al, 2019).
Although Zika was first isolated from non‐human primates, the host reservoirs predating human circulation have not been conclusively identified (Kindhauser et al, 2016) and no specific molecular events are known to have been critical for the first human infection. Mutations that emerged as Zika spread East have, however, likely influenced infectivity and virulence. More than 50 nonsynonymous mutations distinguish the earliest African strains from the earliest isolates from Asia, but the functional relevance of these mutations remains unclear. At least 19 amino acid substitutions are associated with the first introduction of Zika into the Pacific Islands including substitutions in nonstructural proteins, which may have affected host interferon signaling. Two substitutions are particularly notable, D683E in the envelope protein receptor motif and V153M in prM, which affects cytotoxicity (Pettersson et al, 2016). Cleavage of prM (by host protease furin in contrast to capsid cleavage described below) into the mature membrane protein and the Pr protein leads to virion maturation. The complete prM and processed Pr can induce apoptosis; and ancestral African strains demonstrate greater cytotoxicity than (later, and considered part of the Asian lineage) Brazilian epidemic strains (Li et al, 2019). In addition to the characteristic V153M substitution, I110V, K143E, A148P, H157Y, and V158I observed in Asian but not African strains are predicted to amount to substantial structural change (Wang et al, 2016), and among these, A148P, V153M, H157Y, and V158I, when introduced into African strains, alleviate Pr‐induced apoptosis (Li et al, 2019).
It is inconclusive, however, which of these mutations, acquired early within the Asian lineage, represent an ongoing adaptation to human hosts, and which are founder effects. In addition to reduced cytotoxicity, Asian strains display reduced replicative capacity in neuronal cells (Bos et al, 2018), perhaps indicative of variable tissue tropism and potentially reduced fitness. Indeed, early, pre‐epidemic Asian strains demonstrate reduced replicative fitness in both mosquitos and mice due in part to four, likely founder, substitutions which were later reverted (Fig 1 and see below) to the ancestral states as the virus spread East, namely, A106T in the capsid protein, A1V in prM, V188A in nonstructural protein 1, NS1, and V872M in nonstructural protein 5, NS5 (Liu et al, 2021b). Thus, despite demonstrations of functional relevance, these and other substitutions (including T777M and V763M in the viral envelope protein (Pettersson et al, 2016)) acquired during the earliest outbreaks in Asia and the Pacific Islands (some of which affected large communities nonetheless (Liu et al, 2017)) are a mixed result of population bottlenecks and human host adaptation.
Neurotropism and antigenemia
The next wave of amino acid substitutions, which became apparent during the (primarily) French Polynesian outbreak in 2013 and later the Brazilian epidemic in 2015, although possibly emerging earlier (Delatorre et al, 2017), includes the four ancestral reversions (capsid T106A, prM V1A, NS1 A188V, and NS5 M872V), and prM S139N and V473M in the envelope protein. As described above, substitutions within prM can influence tissue tropism and cytotoxicity. S139N strongly affects Zika virulence by enhancing tropism for human neural progenitor cells (Yuan et al, 2017; Liu et al, 2019). This substitution has been associated with Guillain Barre syndrome (an ascending paralysis resulting from autoimmunity) and congenital Zika virus syndrome (Pettersson et al, 2016; Yuan et al, 2017; Liu et al, 2019). Although these complications had not been widely recognized prior to the emergence of S139N, this substitution is not essential for fetal harm or neurovirulence which have additionally been reported as consequences of ancestral African strain infection (Rosenfeld et al, 2017; Jaeger et al, 2019).
V473M is another substitution affecting virulence. Residue 473 of the envelope protein resides within a transmembrane domain, and V473M enhances virion assembly resulting in higher fetal neural viral loads in model systems. In addition to enhanced maternal‐fetal transmission, like NS1 A188V described below, V473M increases viremia, which is required for vector transmission (Liu et al, 2017; Shan et al, 2020). NS1 A188V is a critical, possibly dominant (Liu et al, 2021b), determinant of Zika viremia; NS1 antigenemia, which is critical for infectivity and does not directly correlate with viremia (Liu et al, 2017); and subsequent mosquito viral uptake (Liu et al, 2017, 2019; Liu et al, 2021b). A188V results in enhanced interferon inhibition (Xia et al, 2018; Liu et al, 2019) and increased viral titers in the brain, demonstrating that this adaptation generally increases replicative fitness. Nevertheless, the molecular mechanisms by which A188V selectively promotes viral confluence within and/or secretion of NS1 into host blood remain to be elucidated.
The mechanism by which T106A in the capsid protein promotes infectivity is better understood. Like prM, cleavage of the capsid protein is required for virion assembly. The capsid is cleaved by a viral‐encoded protease NS2B‐NS3. T106A is adjacent to the cleavage site and increases the efficiency of NS2B‐NS3 cleavage, thus promoting virion maturation; and increasing Zika infectivity (Yu et al, 2021). Finally, only a few characteristic substitutions distinguish Pacific Island and American outbreaks, suggesting that most of the molecular determinants of American strain infectivity were already present in prior outbreaks. The most common of these substitutions is M/T2634V in nonstructural protein 5, NS5, the RNA‐dependent RNA polymerase (RdRp). Although American outbreaks, along with this substitution, are associated with higher reported rates of neurological complications (Pettersson et al, 2016), it is likely that this substitution is not adaptive but rather results from a population bottleneck (Liu et al, 2019).
Elusive determinants of epidemic potential
In contrast to the clear, geographical directionality of Zika virus migration East across the globe, the molecular evolution of Zika virus does not clearly reveal a trend of increasing human infectivity and instead likely reflects a broader balance between infectivity in the vertebrate and mosquito hosts. Despite the functional relevance of the two epidemic‐associated substitutions that are not ancestral reversions, prM S139N and envelope V473M, and modified codon preference (see Concluding Remarks); in some model mouse and mosquito systems, ancestral African strains display higher fitness than more recent variants from the South Pacific and Americas (Xia et al, 2018).
These observations raise the question, was the recent global spread of Zika primarily driven by environmental factors? Epidemiological differences among regional communities including population density, vector range, and the degree of prior viral exposure likely determined the increasing epidemic potential of Zika and Chikungunya virus, which shares similar epidemiological and molecular evolutionary features (Liu et al, 2021b). These epidemiological factors are rapidly changing in many parts of the world as a result of modernization, globalization, and climate change. Phylodynamic analysis suggests that most human viruses form stable, noncompeting epidemiological niches marked by largely neutral evolution (Mutz et al, 2022) similar to the apparent landscape of Zika within Africa. The export of strains that have neutrally evolved within a small, stable epidemiological niche into a much larger community of susceptible hosts not only increases the epidemic potential in the short term but also introduces new selective pressures as a result of interstrain competition which can lead to more robust human adaptation. Furthermore, the landscape of intrahost adaptations observed within model systems studying maternal‐fetal transfer(Lemos et al, 2020) or other direct transmission chains (Riemersma et al, 2021) are different from those improving vector‐mediated transmission, suggesting the possibility that Zika could diversify into primarily vector‐mediated and sexually‐transmitted subtypes, dramatically increasing the complexity of its evolutionary landscape.
Concluding remarks
From smallpox to SARS‐CoV‐2, epidemics inescapably command the attention of virologists worldwide and provide large amounts of genomic information for the exploration of adaptive evolutionary processes. Spanning 6 widely different virus families and half a century of observation, the biological diversity that appears to underlie human transmission is vast. Which genomic features reflect adaptive evolution is not always clear, however, and key questions remain that are in need of answers (see Box 1), even for these 8, well‐studied, epidemic viruses.
Box 1. In need of answers.
-
1
To what extent is the trend of accessory gene loss across diverse viruses a result of positive selection (in contrast to neutral drift and subsequent pseudogenization)?
-
2
What processes promote the utilization of distinct host receptors among closely related viruses with conserved receptor‐binding domains?
-
3
To what degree is lasting immunity conferred by cellular immune responses distinct from humoral components (in particular, neutralizing antibodies) and what evolutionary routes are available for the direct viral antagonism of these cellular agents?
-
4
What molecular features could be predictive of host specialization or viral speciation?
-
5
How can we effectively explore viromes of non‐human species to reveal adaptations acquired in intermediate hosts, which are essential for human transmission?
Major genome remodeling
The changes that viruses undergo when adapting to a new host can be coarsely partitioned into two classes: major genome remodeling and point mutations in individual genes. A striking and, at least at first glance, an unexpected common trend in genome remodeling is gene loss. The complete loss or recurrent large deletions within multiple genes accompanies zoonoses not only in viruses with large genomes, such as smallpox, but also in those with small genomes including HIV, coronaviruses, and influenzaviruses. Predictably, in each case, the lost or inactivated genes are involved in various forms of virus–host interactions, in particular, host immunity suppression, rather than virus reproduction.
Whether or not the loss of virus genes is directly adaptive, that is, driven by positive selection, remains an open question (Box 1, Question 1). It appears likely that behind this trend is a combination of neutral and selective evolutionary processes whereby accessory virus genes are pseudogenized because in the new host they become neutral, that is, not maintained by purifying selection but then are deleted, under selection for the minimization of expression and replication costs. However, it cannot be ruled out that some of the interactions of the accessory proteins in the new host adversely affect virus fitness; and thus, the disruption of some virus genes may be driven by positive selection. Another important source of virus genome remodeling, which sometimes results in gene deletion, is recombination and, in particular, genome segment reassortment in viruses with segmented genomes, like influenza. Even if constrained by the requirement for segment compatibility, for example, HA and N, reassortment is a major source of diversity.
Amino acid substitutions in surface proteins
It is easier to assess the functional impact of amino acid substitutions in structural proteins, particularly virus surface proteins, than in nonstructural proteins; consequently, changes to surface proteins are likely overrepresented among those identified as adaptations. It is nonetheless clear, and hardly surprising, that changes in virion surface proteins are critical for zoonosis (Fig 3). Substitutions that improve binding to specific host receptors at the cost of receptors identified in intermediate hosts are common. Two canonical examples are H1N1 hemagglutinin 190D/225D, which results in preferential binding of sialic acid SAα2,6Gal (preferentially expressed within human respiratory tissues) (Stevens et al, 2004; Tumpey et al, 2007; Neumann et al, 2009), and SARS spike substitutions in sites 479 and 487 which are critical for human ACE2 binding (Li, 2008). While adaptations to each particular host receptor are often highly specific, closely related viruses often utilize distinct host proteins for cell entry. For example, despite both structural and sequence conservation of the RBD across diverse Betacoronaviruses (Tai et al, 2020), host receptors, although shared among diverse coronaviruses, are not deeply conserved and vary over short evolutionary distances (Li, 2016). The molecular determinants of receptor “choice” remain an open question (Box 1, Question 2).
Often, the same protein mediates receptor binding and membrane fusion, facilitating cell entry (Fig 4), and substitutions in surface proteins frequently provide selective advantages through enhanced membrane fusion activity, with only modestly increased, or even reduced receptor affinity. For example, the crucial SARS‐CoV‐2 spike 614G stabilizes the S trimer but reduces the number of accessible RBDs (essentially reducing receptor affinity); however, this substitution also reduces the rate at which the virus is shed and increases the probability of cell entry (Zhang et al, 2021a). Another influential substitution 452R (one of two characterizing the Delta variant) within the RBD modestly increases receptor affinity and alters membrane fusion properties (Motozono et al, 2021). Membrane fusion can be enhanced by improving virus compatibility with host proteases responsible for surface protein cleavage. This is apparent comparing MERS to HKU4, a bat Betacoronavirus likely related to ancestral strains. Human cellular proteases mediate MERS but not HKU4 cell entry, whereas bat proteases are compatible with both MERS and HKU4 spikes (Yang et al, 2014). The introduction of the furin cleavage site into the S protein of SARS‐CoV‐2 is another recent but widely cited example.
Adaptations beyond cell entry
Whether affecting receptor binding or membrane fusion, the surface protein adaptations highlighted in this review primarily mediate cell entry, marking only the beginning of the viral lifecycle. Cross‐species transfer might additionally require orchestrating new protein‐protein interactions for later events including viral mRNA synthesis as demonstrated by the key H1N1 host determinant, residue 627 in PB2 (Carrique et al, 2020). Similarly, while the impact of neutralizing antibody destabilizing changes in surface proteins can be systematically explored with relative ease, as discussed for SARS‐CoV‐2 (Rochman et al, 2022), it is far more challenging to predict which adaptations could enable nonstructural proteins to antagonize restriction factors, as exemplified by the gene deletion that mediated HIV‐1M zoonosis (Sauter & Kirchhoff, 2019).
In addition to the intrinsic immunity represented by restriction factors and adaptive responses epitomized by the production of neutralizing antibodies, viruses must contend with the various branches of innate immunity that present physical, chemical, and other nonspecific barriers to the introduction of foreign material. Adaptive immune responses are at least in part directed by pathogen‐sensing via pattern recognition receptors conducted by the innate immune system (Iwasaki & Medzhitov, 2015), which may be evaded by viruses (Beachboard & Horner, 2016; Kikkert, 2020), undercutting many “downstream” adaptive processes. For both innate and adaptive immune systems, the relative impact of cellular and humoral responses (macromolecular components including antibodies and complement proteins) remains an open question (Box 1, Question 3).
All the adaptations described above (enabling cell entry, protein synthesis, or immune evasion) were the result of nonsynonymous mutations, exerting their effects on protein structure and function. Synonymous mutations in genes coding for both structural and nonstructural proteins can also impact virus fitness. The degree to which virus synonymous codon usage is adapted to its host can impact gene expression and infectivity (Plotkin & Kudla, 2011) and may be reported with codon adaptation indices (CAI) (Sharp & Li, 1987). Virus adaptation to host synonymous codon preference is pervasive and likely factored into the molecular evolution of every human epidemic virus (Wong et al, 2010; Pandit & Sinha, 2011; RoyChoudhury et al, 2011; Cristina et al, 2015; Freire et al, 2018; Dilucca et al, 2020). In particular, Zika codon usage appears to be better adapted to human usage and Asian epidemic lineages display greater human adaptation than African lineages (Freire et al, 2018). Amidst the sea of mutations fixed as a result of population bottlenecks, which obscure the functional relevance of most point substitutions, this signature represents some of the best evidence for the continuous human adaptation of this virus.
Adaptive, neutral, or deleterious, host‐switches are associated with the rapid accumulation of mutations, in part, because the mutation rate itself is likely to be elevated when a virus invades a new host due to maladaptation of viral nonstructural proteins. For example, if RNA polymerase fidelity is host‐dependent, fidelity can be expected to be reduced in new hosts, elevating the mutation rate (Graham & Baric, 2010). Somewhat paradoxically, such maladaptation could contribute to virus evolvability in a manner analogous to the phenotypic switching observed in cellular organisms (Balaban et al, 2004; Rochman et al, 2016). As the virus becomes better adapted to the new host, and the strength of purifying selection increases, improved nonstructural protein compatibility can reduce the mutation rate.
Virus genomics outside epidemics
Some epidemic viruses, including Ebola and MERS, have caused many outbreaks, each from a distinct zoonotic event. This pattern of transmission requires viral compatibility with at least two hosts (as does vector‐mediated transmission) and precludes human adaptations that would lead to host specialization, for example, the extensive gene loss by variola virus which is predicted to prevent cross‐species transmission (Hendrickson et al, 2010). Notably, some impactful events in epidemic prehistory appear to promote generalizability to new hosts without clear signatures of species‐specific adaptations, for example, the Spike insertions identified among Betacoronaviruses (Gussow et al, 2020). The effect of a single mutation on a particular host‐virus relationship is often difficult to assess and determining how viral fitness is affected when a mutation may differentially impact multiple hosts is an even greater challenge. How to predict virus–host specialization and virus speciation remains an open question with clear repercussions for the prediction of future epidemics (Box 1, Question 4).
Much of the difficulty associated with predicting cross‐species transfers stems from the limited sequencing of virus genomes outside of an epidemic context. As has been perhaps best demonstrated throughout the COVID‐19 pandemic, robust sequencing efforts followed by comprehensive phylogenomic analysis can reveal recurrent molecular events, which lead to a succession of dominant virus variants with increasing fitness and enable an assessment of individual changes. While related, these events are distinct from those features that emerge as a result of rapid adaptation following a host switch, and elucidating the requirements (conserved or species‐specific) for human transmission will require commensurate sequencing efforts for non‐human species (Box 1, Question 5). Sequences from intermediate hosts or early human infections are missing for all but one (H1N1) of the epidemic viruses discussed here (See Fig 1), and varying selection pressures over time (Mutz et al, 2022) or even strong, constant purifying selection, characteristic of viral evolution (Wertheim & Kosakovsky Pond, 2011), make it impossible to uncover many adaptations to human hosts from the comparative genomics of human viruses alone.
Moreover, even diverse human viruses have not yet been robustly sequenced. At the time of this writing, there is an impactful international outbreak of Orthopoxvirus monkeypox (Centers for Disease Control and Prevention, 2022b), for which the case‐fatality rate could approach 10% (Bunge et al, 2022). However, fewer than 200 complete monkeypox genomes are currently available, precluding any genomic contribution to epidemiological forecasting. More generally and perhaps surprisingly, in this age of “big data,” there are fewer than 1000 complete genomes available for most of human viruses. Among Flaviviruses, only Dengue, with approximately 6000 genomes available, breaks 1000 despite the clinical relevance of Chikungunya and, as described above, Zika (< 500). There are multiple, diverse flaviviruses that share conserved viral life‐history traits and even molecular histories. For example, the spread of Chikungunya virus was marked by founder effects and subsequent compensatory mutations in a manner closely resembling the case of Zika as the virus, again quite similarly, spread from Africa, to Asia, to the Americas (Liu et al, 2021b). Identifying such conserved trends is paramount for predicting the evolutionary trajectories of pathogenic viruses.
The direct transmission of Zika (Lemos et al, 2020; Riemersma et al, 2021), persistent infections of HIV (Holmes, 2001) and Ebola (Blackley et al, 2016; Whitmer et al, 2018), and familiar mammalian host reservoirs of MERS (Mohd et al, 2016; El‐Kafrawy et al, 2019; Memish et al, 2020), SARS‐CoV‐2 (Fischhoff et al, 2021; Hale et al, 2022), and influenza viruses (Olsen, 2002; Zimmer & Burke, 2009) all provide avenues for viral diversification, speciation, and outbreaks. Extending sequencing efforts to divergent viruses and diverse hosts, combined with focused experimentation, can be expected to reveal the roots of past epidemics, and possibly predict and mitigate the next.
Author contributions
Nash D Rochman: Conceptualization; writing—original draft; writing—review and editing. Yuri I Wolf: Writing—review and editing. Eugene V Koonin: Conceptualization; writing—review and editing.
Disclosure and competing interests statement
The authors declare they have no conflict of interest.
Acknowledgements
The authors thank Eleanor August Rochman for her contribution to the design and preparation of the figures and Koonin group members for helpful discussions. NDR, YIW, and EVK are supported by the Intramural Research Program of the National Institutes of Health (National Library of Medicine).
EMBO reports (2022) 23: e55393
Contributor Information
Nash D Rochman, Email: nash.rochman@nih.gov.
Eugene V Koonin, Email: koonin@ncbi.nlm.nih.gov.
References
- Aguado B, Selmes IP, Smith GL (1992) Nucleotide sequence of 21.8 kbp of variola major virus strain Harvey and comparison with vaccinia virus. J Gen Virol 73: 2887–2902 [DOI] [PubMed] [Google Scholar]
- Albariño CG, Guerrero LW, Chakrabarti AK, Kainulainen MH, Whitmer SL, Welch SR, Nichol ST (2016) Virus fitness differences observed between two naturally occurring isolates of Ebola virus Makona variant using a reverse genetics approach. Virology 496: 237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alymova IV, McCullers JA, Kamal RP, Vogel P, Green AM, Gansebom S, York IA (2018) Virulent PB1‐F2 residues: effects on fitness of H1N1 influenza A virus in mice and changes during evolution of human influenza A viruses. Sci Rep 8: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azarian T, Lo Presti A, Giovanetti M, Cella E, Rife B, Lai A, Zehender G, Ciccozzi M, Salemi M (2015) Impact of spatial dispersion, evolution and selection on Ebola Zaire Virus epidemic waves. Sci Rep 5: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babkin IV, Babkina IN (2015) The origin of the variola virus. Viruses 7: 1100–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babkin IV, Babkina IN, Tikunova NV (2022) An update of orthopoxvirus molecular evolution. Viruses 14: 388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagdonaite I, Wandall HH (2018) Global aspects of viral glycosylation. Glycobiology 28: 443–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailes E, Gao F, Bibollet‐Ruche F, Courgnaud V, Peeters M, Marx PA, Hahn BH, Sharp PM (2003) Hybrid origin of SIV in chimpanzees. Science 300: 1713 [DOI] [PubMed] [Google Scholar]
- Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S (2004) Bacterial persistence as a phenotypic switch. Science 305: 1622–1625 [DOI] [PubMed] [Google Scholar]
- Barton MI, MacGowan SA, Kutuzov MA, Dushek O, Barton GJ, van der Merwe PA (2021) Effects of common mutations in the SARS‐CoV‐2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. eLife 10: e70658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beachboard DC, Horner SM (2016) Innate immune evasion strategies of DNA and RNA viruses. Curr Opin Microbiol 32: 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Cavanaugh M, Clark K, Karsch‐Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2012) GenBank. Nucleic Acids Res 41: D36–D42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram S, Glowacka I, Steffen I, Kühl A, Pöhlmann S (2010) Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev Med Virol 20: 298–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackley DJ, Wiley MR, Ladner JT, Fallah M, Lo T, Gilbert ML, Gregory C, D'ambrozio J, Coulter S, Mate S (2016) Reduced evolutionary rate in reemerged Ebola virus transmission chains. Sci Adv 2: e1600378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone M (1997) Antiviral pressure exerted by HIV‐l‐specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med 3: 205–211 [DOI] [PubMed] [Google Scholar]
- Bos S, Viranaicken W, Turpin J, El‐Kalamouni C, Roche M, Krejbich‐Trotot P, Desprès P, Gadéa G (2018) The structural proteins of epidemic and historical strains of Zika virus differ in their ability to initiate viral infection in human host cells. Virology 516: 265–273 [DOI] [PubMed] [Google Scholar]
- Bosworth A, Rickett NY, Dong X, Ng LF, García‐Dorival I, Matthews DA, Fletcher T, Jacobs M, Thomson EC, Carroll MW (2021) Analysis of an Ebola virus disease survivor whose host and viral markers were predictive of death indicates the effectiveness of medical countermeasures and supportive care. Genome Med 13: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown IH (2000) The epidemiology and evolution of influenza viruses in pigs. Vet Microbiol 74: 29–46 [DOI] [PubMed] [Google Scholar]
- Brüssow H, Brüssow L (2021) Clinical evidence that the pandemic from 1889 to 1891 commonly called the Russian flu might have been an earlier coronavirus pandemic. J Microbial Biotechnol 14: 1860–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunge EM, Hoet B, Chen L, Lienert F, Weidenthaler H, Baer LR, Steffen R (2022) The changing epidemiology of human monkeypox—A potential threat? a systematic review. PLoS Negl Trop Dis 16: e0010141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capua I, Alexander DJ (2002) Avian influenza and human health. Acta Trop 83: 1–6 [DOI] [PubMed] [Google Scholar]
- Carrique L, Fan H, Walker AP, Keown JR, Sharps J, Staller E, Barclay WS, Fodor E, Grimes JM (2020) Host ANP32A mediates the assembly of the influenza virus replicase. Nature 587: 638–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauldwell AV, Long JS, Moncorgé O, Barclay WS (2014) Viral determinants of influenza A virus host range. J Gen Virol 95: 1193–1210 [DOI] [PubMed] [Google Scholar]
- Cele S, Karim F, Lustig G, San JE, Hermanus T, Tegally H, Snyman J, Moyo‐Gwete T, Wilkinson E, Bernstein M (2022) SARS‐CoV‐2 prolonged infection during advanced HIV disease evolves extensive immune escape. Cell Host Microbe 30: 154–162.e155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2022a) History of smallpox. cdcgov .
- Centers for Disease Control and Prevention (2022b) U.S. Monkeypox outbreak 2022: situation summary.
- Centers for Disease Control and Prevention EIS (2022) A history of success: investigating and responding to public health threats since 1951.
- Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G (2012) Natural SIV hosts: showing AIDS the door. Science 335: 1188–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung PHH, Lee TWT, Chan CP, Jin DY (2020) Influenza A virus PB1‐F2 protein: An ambivalent innate immune modulator and virulence factor. J Leukoc Biol 107: 763–771 [DOI] [PubMed] [Google Scholar]
- Chinese SARS Molecular Epidemiology Consortium (2004) Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303: 1666–1669 [DOI] [PubMed] [Google Scholar]
- Chu DK, Hui KP, Perera RA, Miguel E, Niemeyer D, Zhao J, Channappanavar R, Dudas G, Oladipo JO, Traoré A (2018) MERS coronaviruses from camels in Africa exhibit region‐dependent genetic diversity. Proc Natl Acad Sci U S A 115: 3144–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier DA, De Marco A, Ferreira IA, Meng B, Datir RP, Walls AC, Kemp SA, Bassi J, Pinto D, Silacci‐Fregni C (2021) Sensitivity of SARS‐CoV‐2 B. 1.1. 7 to mRNA vaccine‐elicited antibodies. Nature 593: 136–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E (2020) The spike glycoprotein of the new coronavirus 2019‐nCoV contains a furin‐like cleavage site absent in CoV of the same clade. Antiviral Res 176: 104742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristina J, Moreno P, Moratorio G, Musto H (2015) Genome‐wide analysis of codon usage bias in Ebolavirus. Virus Res 196: 87–93 [DOI] [PubMed] [Google Scholar]
- Damas J, Hughes GM, Keough KC, Painter CA, Persky NS, Corbo M, Hiller M, Koepfli K‐P, Pfenning AR, Zhao H (2020) Broad host range of SARS‐CoV‐2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc Natl Acad Sci U S A 117: 22311–22322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vlugt C, Sikora D, Pelchat M (2018) Insight into influenza: a virus cap‐snatching. Viruses 10: 641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delatorre E, Mir D, Bello G (2017) Tracing the origin of the NS1 A188V substitution responsible for recent enhancement of Zika virus Asian genotype infectivity. Mem Inst Oswaldo Cruz 112: 793–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Garcia‐Knight MA, Khalid MM, Servellita V, Wang C, Morris MK, Sotomayor‐González A, Glasner DR, Reyes KR, Gliwa AS (2021) Transmission, infectivity, and neutralization of a spike L452R SARS‐CoV‐2 variant. Cell 184: 3426–3437.e38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl WE, Lin AE, Grubaugh ND, Carvalho LM, Kim K, Kyawe PP, McCauley SM, Donnard E, Kucukural A, McDonel P (2016) Ebola virus glycoprotein with increased infectivity dominated the 2013–2016 epidemic. Cell 167: 1088–1098.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzel E, Schudt G, Krähling V, Matrosovich M, Becker S (2017) Functional characterization of adaptive mutations during the West African Ebola virus outbreak. J Virol 91: e01913‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilucca M, Forcelloni S, Georgakilas AG, Giansanti A, Pavlopoulou A (2020) Codon usage and phenotypic divergences of SARS‐CoV‐2 genes. Viruses 12: 498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Holland JJ (1999) Mutation rates among RNA viruses. Proc Natl Acad Sci U S A 96: 13910–13913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas G, Carvalho LM, Bedford T, Tatem AJ, Baele G, Faria NR, Park DJ, Ladner JT, Arias A, Asogun D (2017) Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 544: 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas G, Carvalho LM, Rambaut A, Bedford T (2018) MERS‐CoV spillover at the camel‐human interface. eLife 7: e31257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas G, Rambaut A (2016) MERS‐CoV recombination: implications about the reservoir and potential for adaptation. Virus Evol 2: vev023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan AT, Perdomo MF, Piombino‐Mascali D, Marciniak S, Poinar D, Emery MV, Buchmann JP, Duchêne S, Jankauskas R, Humphreys M (2016) 17th century variola virus reveals the recent history of smallpox. Curr Biol 26: 3407–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe S, Buckland‐Merrett G (2017) Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall 1: 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Kafrawy SA, Corman VM, Tolah AM, Al Masaudi SB, Hassan AM, Müller MA, Bleicker T, Harakeh SM, Alzahrani AA, Alsaaidi GA (2019) Enzootic patterns of Middle East respiratory syndrome coronavirus in imported African and local Arabian dromedary camels: a prospective genomic study. Lancet Planet Health 3: e521–e528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne L, Bibollet‐Ruche F, Sudmant PH, Wu LI, Hahn BH, Emerman M (2015) The role of the antiviral APOBEC3 gene family in protecting chimpanzees against lentiviruses from monkeys. PLoS Pathog 11: e1005149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne L, Hahn BH, Sharp PM, Matsen FA, Emerman M (2013) Gene loss and adaptation to hominids underlie the ancient origin of HIV‐1. Cell Host Microbe 14: 85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner F, Henderson DA, Arita I, Jezek Z, Ladnyi ID (1988) Smallpox and its eradication. Geneva, Switzerland: World Health Organization; [Google Scholar]
- Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, Jou WM, Molemans F, Raeymaekers A, Van den Berghe A (1976) Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene. Nature 260: 500–507 [DOI] [PubMed] [Google Scholar]
- Fischhoff IR, Castellanos AA, Rodrigues JP, Varsani A, Han BA (2021) Predicting the zoonotic capacity of mammals to transmit SARS‐CoV‐2. Proc R Soc B 288: 20211651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foeglein A, Loucaides EM, Mura M, Wise HM, Barclay WS, Digard P (2011) Influence of PB2 host‐range determinants on the intranuclear mobility of the influenza A virus polymerase. J Gen Virol 92: 1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire CCM, Palmisano G, Braconi CT, Cugola FR, Russo FB, Beltrão‐Braga PC, Iamarino A, Lima Neto DF, Rosa‐Fernandes L, Larsen MR (2018) NS1 codon usage adaptation to humans in pandemic Zika virus. Mem Inst Oswaldo Cruz 113: e170385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway SE, Paul P, MacCannell DR, Johansson MA, Brooks JT, MacNeil A, Slayton RB, Tong S, Silk BJ, Armstrong GL (2021) Emergence of SARS‐CoV‐2 b. 1.1.7 lineage—United States, December 29, 2020–January 12, 2021. MMWR Morb Mortal Wkly Rep 70: 95–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY, Wolf YI, Makarova KS, Vera Alvarez R, Landsman D, Koonin EV (2021) COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res 49: D274–D281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garushyants SK, Rogozin IB, Koonin EV (2021) Template switching and duplications in SARS‐CoV‐2 genomes give rise to insertion variants that merit monitoring. Commun Biol 4: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes AM (2006) The history of smallpox. Clin Dermatol 24: 152–157 [DOI] [PubMed] [Google Scholar]
- Gire SK, Goba A, Andersen KG, Sealfon RS, Park DJ, Kanneh L, Jalloh S, Momoh M, Fullah M, Dudas G (2014) Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345: 1369–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham RL, Baric RS (2010) Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross‐species transmission. J Virol 84: 3134–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaney AJ, Starr TN, Gilchuk P, Zost SJ, Binshtein E, Loes AN, Hilton SK, Huddleston J, Eguia R, Crawford KH (2021) Complete mapping of mutations to the SARS‐CoV‐2 spike receptor‐binding domain that escape antibody recognition. Cell Host Microbe 29: 44–57.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Zheng B, He Y, Liu X, Zhuang Z, Cheung C, Luo S, Li PH, Zhang L, Guan Y (2003) Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302: 276–278 [DOI] [PubMed] [Google Scholar]
- Gussow AB, Auslander N, Faure G, Wolf YI, Zhang F, Koonin EV (2020) Genomic determinants of pathogenicity in SARS‐CoV‐2 and other human coronaviruses. Proc Natl Acad Sci U S A 117: 15193–15199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale VL, Dennis PM, McBride DS, Nolting JM, Madden C, Huey D, Ehrlich M, Grieser J, Winston J, Lombardi D (2022) SARS‐CoV‐2 infection in free‐ranging white‐tailed deer. Nature 602: 481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale BG, Steel J, Manicassamy B, Medina RA, Ye J, Hickman D, Lowen AC, Perez DR, García‐Sastre A (2010) Mutations in the NS1 C‐terminal tail do not enhance replication or virulence of the 2009 pandemic H1N1 influenza A virus. J Gen Virol 91: 1737–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejase HA, Dukler N, Siepel A (2020) From summary statistics to gene trees: methods for inferring positive selection. Trends Genet 36: 243–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson RC, Wang C, Hatcher EL, Lefkowitz EJ (2010) Orthopoxvirus genome evolution: the role of gene loss. Viruses 2: 1933–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Arora P, Groß R, Seidel A, Hörnich BF, Hahn AS, Krüger N, Graichen L, Hofmann‐Winkler H, Kempf A (2021) SARS‐CoV‐2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 184: 2384–2393.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Crone L, Dietzel E, Paijo J, González‐Hernández M, Nehlmeier I, Kalinke U, Becker S, Pöhlmann S (2017) A polymorphism within the internal fusion loop of the Ebola virus glycoprotein modulates host cell entry. J Virol 91: e00177–e00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EC (2001) On the origin and evolution of the human immunodeficiency virus (HIV). Biol Rev 76: 239–254 [DOI] [PubMed] [Google Scholar]
- Huang Y, Yang C, Xu X‐f, Xu W, Liu S‐w (2020) Structural and functional properties of SARS‐CoV‐2 spike protein: potential antivirus drug development for COVID‐19. Acta Pharmacol Sin 41: 1141–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes V (2010) The outlook for a cure. Nature 466: S11–S13 [DOI] [PubMed] [Google Scholar]
- Hurst LD (2002) The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet 18: 486 [DOI] [PubMed] [Google Scholar]
- Iranzo J, Krupovic M, Koonin EV (2016) The double‐stranded DNA virosphere as a modular hierarchical network of gene sharing. mBio 7: e00978‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R (2015) Control of adaptive immunity by the innate immune system. Nat Immunol 16: 343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CB, Zhang L, Farzan M, Choe H (2021) Functional importance of the D614G mutation in the SARS‐CoV‐2 spike protein. Biochem Biophys Res Commun 538: 108–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob ST, Crozier I, Fischer WA, Hewlett A, Kraft CS, de La Vega M‐A, Soka MJ, Wahl V, Griffiths A, Bollinger L (2020) Ebola virus disease. Nat Rev Dis Primers 6: 1–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger AS, Murrieta RA, Goren LR, Crooks CM, Moriarty RV, Weiler AM, Rybarczyk S, Semler MR, Huffman C, Mejia A (2019) Zika viruses of African and Asian lineages cause fetal harm in a mouse model of vertical transmission. PLoS Negl Trop Dis 13: e0007343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Xie X, Bailey AL, Kalveram B, Lokugamage KG, Muruato A, Zou J, Zhang X, Juelich T, Smith JK (2021) Loss of furin cleavage site attenuates SARS‐CoV‐2 pathogenesis. Nature 591: 293–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju B, Zhang Q, Ge J, Wang R, Sun J, Ge X, Yu J, Shan S, Zhou B, Song S (2020) Human neutralizing antibodies elicited by SARS‐CoV‐2 infection. Nature 584: 115–119 [DOI] [PubMed] [Google Scholar]
- Keele BF, Jones JH, Terio KA, Estes JD, Rudicell RS, Wilson ML, Li Y, Learn GH, Beasley TM, Schumacher‐Stankey J (2009) Increased mortality and AIDS‐like immunopathology in wild chimpanzees infected with SIVcpz. Nature 460: 515–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly H (2011) The classical definition of a pandemic is not elusive. Bull World Health Organ 89: 540–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkert M (2020) Innate immune evasion by human respiratory RNA viruses. J Innate Immun 12: 4–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindhauser MK, Allen T, Frank V, Santhana RS, Dye C (2016) Zika: the origin and spread of a mosquito‐borne virus. Bull World Health Organ 94: 675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluge SF, Sauter D, Kirchhoff F (2015) SnapShot: antiviral restriction factors. Cell 163: 774.e1 [DOI] [PubMed] [Google Scholar]
- Ladner JT, Wiley MR, Mate S, Dudas G, Prieto K, Lovett S, Nagle ER, Beitzel B, Gilbert ML, Fakoli L (2015) Evolution and spread of Ebola virus in Liberia, 2014–2015. Cell Host Microbe 18: 659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffeber C, de Koning K, Kanaar R, Lebbink JH (2021) Experimental evidence for enhanced receptor binding by rapidly spreading SARS‐CoV‐2 variants. J Mol Biol 433: 167058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L (2020) Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature 581: 215–220 [DOI] [PubMed] [Google Scholar]
- Larder BA, Kemp SD (1989) Multiple mutations in HIV‐1 reverse transcriptase confer high‐level resistance to zidovudine (AZT). Science 246: 1155–1158 [DOI] [PubMed] [Google Scholar]
- Le Goffic R, Leymarie O, Chevalier C, Rebours E, Da Costa B, Vidic J, Descamps D, Sallenave J‐M, Rauch M, Samson M (2011) Transcriptomic analysis of host immune and cell death responses associated with the influenza A virus PB1‐F2 protein. PLoS Pathog 7: e1002202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos D, Stuart JB, Louie W, Singapuri A, Ramírez AL, Watanabe J, Usachenko J, Keesler RI, Sanchez‐San Martin C, Li T (2020) Two sides of a coin: a Zika virus mutation selected in pregnant rhesus macaques promotes fetal infection in mice but at a cost of reduced fitness in nonpregnant macaques and diminished transmissibility by vectors. J Virol 94: e01605‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letko M, Miazgowicz K, McMinn R, Seifert SN, Sola I, Enjuanes L, Carmody A, Van Doremalen N, Munster V (2018) Adaptive evolution of MERS‐CoV to species variation in DPP4. Cell Rep 24: 1730–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F (2008) Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J Virol 82: 6984–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F (2016) Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol 3: 237–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Bos S, Tsetsarkin KA, Pletnev AG, Desprès P, Gadea G, Zhao RY (2019) The roles of prM‐E proteins in historical and epidemic Zika virus‐mediated infection and neurocytotoxicity. Viruses 11: 157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Carroll DS, Gardner SN, Walsh MC, Vitalis EA, Damon IK (2007) On the origin of smallpox: correlating variola phylogenics with historical smallpox records. Proc Natl Acad Sci U S A 104: 15787–15792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Shen X, Yang RF, Li YX, Ji YY, He YY, Shi MD, Lu W, Shi TL, Wang J (2003) Identification of an epitope of SARS‐coronavirus nucleocapsid protein. Cell Res 13: 141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Iketani S, Guo Y, Chan JF‐W, Wang M, Liu L, Luo Y, Chu H, Huang Y, Nair MS (2022) Striking antibody evasion manifested by the Omicron variant of SARS‐CoV‐2. Nature 602: 676–681 [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu J, Du S, Shan C, Nie K, Zhang R, Li X‐F, Zhang R, Wang T, Qin C‐F (2017) Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 545: 482–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang Q, Wei P, Chen Z, Aviszus K, Yang J, Downing W, Jiang C, Liang B, Reynoso L (2021a) The basis of a more contagious 501Y. V1 variant of SARS‐CoV‐2. Cell Res 31: 720–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liu Y, Shan C, Nunes BT, Yun R, Haller SL, Rafael GH, Azar SR, Andersen CR, Plante K (2021b) Role of mutational reversions and fitness restoration in Zika virus spread to the Americas. Nat Commun 12: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, VanBlargan LA, Bloyet L‐M, Rothlauf PW, Chen RE, Stumpf S, Zhao H, Errico JM, Theel ES, Liebeskind MJ (2021c) Identification of SARS‐CoV‐2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 29: 477–488.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Rocklöv J (2021) The reproductive number of the Delta variant of SARS‐CoV‐2 is far higher compared to the ancestral SARS‐CoV‐2 virus. J Travel Med 28: taab124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z‐Y, Shi W‐F, Qin C‐F (2019) The evolution of Zika virus from Asia to the Americas. Nat Rev Microbiol 17: 131–139 [DOI] [PubMed] [Google Scholar]
- Lowen AC (2017) Constraints, drivers, and implications of influenza A virus reassortment. Annu Rev Virol 4: 105–121 [DOI] [PubMed] [Google Scholar]
- Mansbach RA, Chakraborty S, Nguyen K, Montefiori DC, Korber B, Gnanakaran S (2021) The SARS‐CoV‐2 Spike variant D614G favors an open conformational state. Sci Adv 7: eabf3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Lytras S, Lucaci AG, Maier W, Gruning B, Shank SD, Weaver S, MacLean OS, Orton RJ, Lemey P (2022) Selection analysis identifies unusual clustered mutational changes in Omicron lineage BA 1 that likely impact Spike function. bioRxiv 10.1101/2022.01.14.476382 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez O, Ndungo E, Tantral L, Miller EH, Leung LW, Chandran K, Basler CF (2013) A mutation in the Ebola virus envelope glycoprotein restricts viral entry in a host species‐and cell‐type‐specific manner. J Virol 87: 3324–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbala‐Kingebeni P, Aziza A, Di Paola N, Wiley MR, Makiala‐Mandanda S, Caviness K, Pratt CB, Ladner JT, Kugelman JR, Prieto K (2019) Medical countermeasures during the 2018 Ebola virus disease outbreak in the North Kivu and Ituri Provinces of the Democratic Republic of the Congo: a rapid genomic assessment. Lancet Infect Dis 19: 648–657 [DOI] [PubMed] [Google Scholar]
- McCaul N, Quandte M, Bontjer I, van Zadelhoff G, Land A, Crooks ET, Binley JM, Sanders RW, Braakman I (2021) Intramolecular quality control: HIV‐1 envelope gp160 signal‐peptide cleavage as a functional folding checkpoint. Cell Rep 36: 109646 [DOI] [PubMed] [Google Scholar]
- McElroy AK, Akondy RS, Davis CW, Ellebedy AH, Mehta AK, Kraft CS, Lyon GM, Ribner BS, Varkey J, Sidney J (2015) Human Ebola virus infection results in substantial immune activation. Proc Natl Acad Sci U S A 112: 4719–4724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memish ZA, Perlman S, Van Kerkhove MD, Zumla A (2020) Middle East respiratory syndrome. Lancet 395: 1063–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson NR, Warren CJ, Vieira DA, Diaz‐Griferro F, Sawyer SL (2018) Species‐specific vulnerability of RanBP2 shaped the evolution of SIV as it transmitted in African apes. PLoS Pathog 14: e1006906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohd HA, Al‐Tawfiq JA, Memish ZA (2016) Middle East respiratory syndrome coronavirus (MERS‐CoV) origin and animal reservoir. Virol J 13: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motozono C, Toyoda M, Zahradnik J, Saito A, Nasser H, Tan TS, Ngare I, Kimura I, Uriu K, Kosugi Y (2021) SARS‐CoV‐2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 29: 1124–1136.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlemann B, Vinner L, Margaryan A, Wilhelmson H, de la Fuente CC, Allentoft ME, de Barros DP, Hansen AJ, Nielsen SH, Strand LM (2020) Diverse variola virus (smallpox) strains were widespread in northern Europe in the Viking Age. Science 369: eaaw8977 [DOI] [PubMed] [Google Scholar]
- Musso D, Ko AI, Baud D (2019) Zika virus infection—after the pandemic. N Engl J Med 381: 1444–1457 [DOI] [PubMed] [Google Scholar]
- Muth D, Corman VM, Roth H, Binger T, Dijkman R, Gottula LT, Gloza‐Rausch F, Balboni A, Battilani M, Rihtarič D (2018) Attenuation of replication by a 29 nucleotide deletion in SARS‐coronavirus acquired during the early stages of human‐to‐human transmission. Sci Rep 8: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutz P, Rochman ND, Wolf YI, Faure G, Zhang F, Koonin EV (2022) Human pathogenic RNA viruses establish noncompeting lineages by occupying independent niches. Proceedings of the National Academy of Sciences 119. 10.1073/pnas.2121335119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Buzko O, Spilman P, Niazi K, Rabizadeh S, Soon‐Shiong P (2021) Molecular dynamic simulation reveals E484K mutation enhances spike RBD‐ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y. V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. bioRxiv 10.1101/2021.01.13.426558 [PREPRINT] [DOI] [Google Scholar]
- Neumann G, Noda T, Kawaoka Y (2009) Emergence and pandemic potential of swine‐origin H1N1 influenza virus. Nature 459: 931–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M, Ndungo E, Kaczmarek ME, Herbert AS, Binger T, Kuehne AI, Jangra RK, Hawkins JA, Gifford RJ, Biswas R (2015) Filovirus receptor NPC1 contributes to species‐specific patterns of ebolavirus susceptibility in bats. eLife 4: e11785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas LePan VC (2020) A visual history of pandemics. Geneva, Switzerland: World Economic Forum; [Google Scholar]
- Niu L, Wittrock KN, Clabaugh GC, Srivastava V, Cho MW (2021) A structural landscape of neutralizing antibodies against SARS‐CoV‐2 receptor binding domain. Front Immunol 12: 647934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olabode AS, Jiang X, Robertson DL, Lovell SC (2015) Ebolavirus is evolving but not changing: No evidence for functional change in EBOV from 1976 to the 2014 outbreak. Virology 482: 202–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen CW (2002) The emergence of novel swine influenza viruses in North America. Virus Res 85: 199–210 [DOI] [PubMed] [Google Scholar]
- Ooms M, Brayton B, Letko M, Maio SM, Pilcher CD, Hecht FM, Barbour JD, Simon V (2013) HIV‐1 Vif adaptation to human APOBEC3H haplotypes. Cell Host Microbe 14: 411–421 [DOI] [PubMed] [Google Scholar]
- Pandit A, Sinha S (2011) Differential trends in the codon usage patterns in HIV‐1 genes. PLoS One 6: e28889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DJ, Dudas G, Wohl S, Goba A, Whitmer SL, Andersen KG, Sealfon RS, Ladner JT, Kugelman JR, Matranga CB (2015) Ebola virus epidemiology, transmission, and evolution during seven months in Sierra Leone. Cell 161: 1516–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC, Kugathasan R, Penn R, Brown JC, Sanchez‐David RY (2021) The furin cleavage site in the SARS‐CoV‐2 spike protein is required for transmission in ferrets. Nat Microbiol 6: 899–909 [DOI] [PubMed] [Google Scholar]
- Peiris JS, Guan Y, Yuen K (2004) Severe acute respiratory syndrome. Nat Med 10: S88–S97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekar JE, Magee A, Parker E, Moshiri N, Izhikevich K, Havens JL, Gangavarapu K, Malpica Serrano LM, Crits‐Christoph A, Matteson NL (2022) SARS‐CoV‐2 emergence very likely resulted from at least two zoonotic events. Zenodo 10.5281/zenodo.6291628 [DATASET] [DOI] [Google Scholar]
- Pensaert M, Ottis K, Vandeputte J, Kaplan MM, Bachmann P (1981) Evidence for the natural transmission of influenza A virus from wild ducks to swine and its potential importance for man. Bull World Health Organ 59: 75–78 [PMC free article] [PubMed] [Google Scholar]
- Pereira F (2020) Evolutionary dynamics of the SARS‐CoV‐2 ORF8 accessory gene. Infect Genet Evol 85: 104525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuánez HN, Horvath JE, Moreira MA, Kessing B, Pontius J, Roelke M, Rumpler Y (2011) A molecular phylogeny of living primates. PLoS Genet 7: e1001342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Losada M, Arenas M, Galán JC, Palero F, González‐Candelas F (2015) Recombination in viruses: mechanisms, methods of study, and evolutionary consequences. Infect Genet Evol 30: 296–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruski AH, Wesolowski LG, Delaney KP, Chavez PR, Owen SM, Granade TC, Sullivan V, Switzer WM, Dong X, Brooks JT (2020) Trends in HIV‐2 diagnoses and use of the HIV‐1/HIV‐2 differentiation test—United States, 2010–2017. Morb Mortal Wkly Rep 69: 63–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson JH‐O, Eldholm V, Seligman SJ, Lundkvist Å, Falconar AK, Gaunt MW, Musso D, Nougairède A, Charrel R, Gould EA (2016) How did Zika virus emerge in the Pacific Islands and Latin America? mBio 7: e01239‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12: 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reba M, Reitsma F, Seto KC (2016) Spatializing 6,000 years of global urbanization from 3700 BC to AD 2000. Sci Data 3: 160034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AH, Fanning TG, Hultin JV, Taubenberger JK (1999) Origin and evolution of the 1918 “Spanish” influenza virus hemagglutinin gene. Proc Natl Acad Sci U S A 96: 1651–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemersma KK, Jaeger AS, Crooks CM, Braun KM, Weger‐Lucarelli J, Ebel GD, Friedrich TC, Aliota MT (2021) Rapid evolution of enhanced Zika virus virulence during direct vertebrate transmission chains. J Virol 95: e02218‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochman ND, Faure G, Wolf YI, Freddolino PL, Zhang F, Koonin EV (2022) Epistasis at the SARS‐CoV‐2 Receptor‐Binding Domain Interface and the Propitiously Boring Implications for Vaccine Escape. mBio: e00135‐22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochman N, Si F, Sun SX (2016) To grow is not enough: impact of noise on cell environmental response and fitness. Integr Biol 8: 1030–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochman N, Wolf Y, Koonin EV (2021a) Evolution of human respiratory virus epidemics. F1000Res 10: 447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochman N, Wolf Y, Koonin EV (2021b) Substantial impact of post‐vaccination contacts on cumulative infections during viral epidemics. F1000Res 10: 315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochman ND, Wolf YI, Faure G, Mutz P, Zhang F, Koonin EV (2021c) Ongoing global and regional adaptive evolution of SARS‐CoV‐2. Proc Natl Acad Sci U S A 118: e2104241118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochman ND, Wolf YI, Koonin EV (2020) Deep phylogeny of cancer drivers and compensatory mutations. Commun Biol 3: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld AB, Doobin DJ, Warren AL, Racaniello VR, Vallee RB (2017) Replication of early and recent Zika virus isolates throughout mouse brain development. Proc Natl Acad Sci U S A 114: 12273–12278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- RoyChoudhury S, Pan A, Mukherjee D (2011) Genus specific evolution of codon usage and nucleotide compositional traits of poxviruses. Virus Genes 42: 189–199 [DOI] [PubMed] [Google Scholar]
- Rozo M, Gronvall GK (2015) The reemergent 1977 H1N1 strain and the gain‐of‐function debate. mBio 6: e01013‐15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruedas JB, Ladner JT, Ettinger CR, Gummuluru S, Palacios G, Connor JH (2017) Spontaneous mutation at amino acid 544 of the Ebola virus glycoprotein potentiates virus entry and selection in tissue culture. J Virol 91: e00392‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabir JS, Lam TT‐Y, Ahmed MM, Li L, Shen Y, Abo‐Aba SE, Qureshi MI, Abu‐Zeid M, Zhang Y, Khiyami MA et al (2016) Co‐circulation of three camel coronavirus species and recombination of MERS‐CoVs in Saudi Arabia. Science 351: 81–84 [DOI] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain‐terminating inhibitors. Proc Natl Acad Sci U S A 74: 5463–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R (2012) From molecular genetics to phylodynamics: evolutionary relevance of mutation rates across viruses. PLoS Pathog 8: e1002685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter D, Kirchhoff F (2019) Key viral adaptations preceding the AIDS pandemic. Cell Host Microbe 25: 27–38 [DOI] [PubMed] [Google Scholar]
- Senkevich TG, Yutin N, Wolf YI, Koonin EV, Moss B (2021) Ancient gene capture and recent gene loss shape the evolution of orthopoxvirus‐host interaction genes. mBio 12: e01495‐21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich TG, Zhivkoplias EK, Weisberg AS, Moss B (2020) Inactivation of genes by frameshift mutations provides rapid adaptation of an attenuated vaccinia virus. J Virol 94: e01053‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer RW, Rhee S‐Y, Pillay D, Miller V, Sandstrom P, Schapiro JM, Kuritzkes DR, Bennett D (2007) HIV‐1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS 21: 215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer KC, Hui S, Bratcher A, King LB, Mutombe R, Kavira N, Kompany JP, Tambu M, Musene K, Mukadi P (2022) Pan‐ebolavirus serology study of healthcare workers in the Mbandaka Health Region, Democratic Republic of the Congo. PLoS Negl Trop Dis 16: e0010167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan C, Xia H, Haller SL, Azar SR, Liu Y, Liu J, Muruato AE, Chen R, Rossi SL, Wakamiya M (2020) A Zika virus envelope mutation preceding the 2015 epidemic enhances virulence and fitness for transmission. Proc Natl Acad Sci U S A 117: 20190–20197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Hahn BH (2011) Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med 1: a006841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li W‐H (1987) The codon adaptation index‐a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15: 1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal A (2022) Milder disease with Omicron: is it the virus or the pre‐existing immunity? Nat Rev Immunol 22: 69–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staller E, Sheppard CM, Neasham PJ, Mistry B, Peacock TP, Goldhill DH, Long JS, Barclay WS (2019) ANP32 proteins are essential for influenza virus replication in human cells. J Virol 93: e00217‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J, Corper AL, Basler CF, Taubenberger JK, Palese P, Wilson IA (2004) Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 303: 1866–1870 [DOI] [PubMed] [Google Scholar]
- Subudhi S, Rapin N, Misra V (2019) Immune system modulation and viral persistence in bats: understanding viral spillover. Viruses 11: 192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryawanshi RK, Chen IP, Ma T, Syed AM, Brazer N, Saldhi P, Simoneau CR, Ciling A, Khalid MM, Sreekumar B (2022) Limited cross‐variant immunity from SARS‐CoV‐2 Omicron without vaccination. Nature: 1–3 10.1038/s41586-022-04865-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, Zhou Y, Du L (2020) Characterization of the receptor‐binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol 17: 613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak AI, Americo JL, Diesterbeck US, Moss B (2021) Loss of the vaccinia virus 35‐amino‐acid hydrophobic O3 protein is partially compensated by mutations in the transmembrane domains of other entry proteins. J Virol 95: e02228‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome‐scale analysis of protein functions and evolution. Nucleic Acids Res 28: 33–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The College of Physicians of Philadelphia (2022) The history of vaccines. Philadelphia, PA: The College of Physicians of Philadelphia; [Google Scholar]
- Theys K, Libin P, Pineda‐Pena A‐C, Nowé A, Vandamme A‐M, Abecasis AB (2018) The impact of HIV‐1 within‐host evolution on transmission dynamics. Curr Opin Virol 28: 92–101 [DOI] [PubMed] [Google Scholar]
- Timani KA, Liao Q, Ye L, Zeng Y, Liu J, Zheng Y, Ye L, Yang X, Lingbao K, Gao J (2005) Nuclear/nucleolar localization properties of C‐terminal nucleocapsid protein of SARS coronavirus. Virus Res 114: 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumpey TM, Maines TR, Van Hoeven N, Glaser L, Solórzano A, Pappas C, Cox NJ, Swayne DE, Palese P, Katz JM (2007) A two‐amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science 315: 655–659 [DOI] [PubMed] [Google Scholar]
- Ueda MT, Kurosaki Y, Izumi T, Nakano Y, Oloniniyi OK, Yasuda J, Koyanagi Y, Sato K, Nakagawa S (2017) Functional mutations in spike glycoprotein of Zaire ebolavirus associated with an increase in infection efficiency. Genes Cells 22: 148–159 [DOI] [PubMed] [Google Scholar]
- Urbanowicz RA, McClure CP, Sakuntabhai A, Sall AA, Kobinger G, Müller MA, Holmes EC, Rey FA, Simon‐Loriere E, Ball JK (2016) Human adaptation of Ebola virus during the West African outbreak. Cell 167: 1079–1087.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyne R, Kuo LS, Pham P, Fujii K, Freed EO (2019) Mutations in the HIV‐1 envelope glycoprotein can broadly rescue blocks at multiple steps in the virus replication cycle. Proc Natl Acad Sci U S A 116: 9040–9049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigerust DJ, Shepherd VL (2007) Virus glycosylation: role in virulence and immune interactions. Trends Microbiol 15: 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijgen L, Keyaerts E, Moës E, Thoelen I, Wollants E, Lemey P, Vandamme A‐M, Van Ranst M (2005) Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J Virol 79: 1595–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R, Matrosovich M, Klenk HD (2002) Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev Med Virol 12: 159–166 [DOI] [PubMed] [Google Scholar]
- Wang L‐F, Eaton BT (2007) Bats, civets and the emergence of SARS. Curr Top Microbiol Immunol 315: 325–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MK, Lim S‐Y, Lee SM, Cunningham JM (2017) Biochemical basis for increased activity of Ebola glycoprotein in the 2013–16 epidemic. Cell Host Microbe 21: 367–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Valderramos SG, Wu A, Ouyang S, Li C, Brasil P, Bonaldo M, Coates T, Nielsen‐Saines K, Jiang T (2016) From mosquitos to humans: genetic evolution of Zika virus. Cell Host Microbe 19: 561–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CJ, Sawyer SL (2019) How host genetics dictates successful viral zoonosis. PLoS Biol 17: e3000217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauquier N, Padilla C, Becquart P, Leroy E, Vieillard V (2010) Association of KIR2DS1 and KIR2DS3 with fatal outcome in Ebola virus infection. Immunogenetics 62: 767–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim JO, Kosakovsky Pond SL (2011) Purifying selection can obscure the ancient age of viral lineages. Mol Biol Evol 28: 3355–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmer SL, Ladner JT, Wiley MR, Patel K, Dudas G, Rambaut A, Sahr F, Prieto K, Shepard SS, Carmody E (2018) Active Ebola virus replication and heterogeneous evolutionary rates in EVD survivors. Cell Rep 22: 1159–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm A, Toptan T, Pallas C, Wolf T, Goetsch U, Gottschalk R, Vehreschild MJ, Ciesek S, Widera M (2021) Antibody‐mediated neutralization of authentic SARS‐CoV‐2 B. 1.617 variants harboring L452R and T478K/E484Q. Viruses 13: 1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Viboud C, Holmes EC, Koonin EV, Lipman DJ (2006) Long intervals of stasis punctuated by bursts of positive selection in the seasonal evolution of influenza A virus. Biol Direct 1: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EH, Smith DK, Rabadan R, Peiris M, Poon LL (2010) Codon usage bias and the evolution of influenza A viruses. Codon usage biases of influenza virus. BMC Evol Biol 10: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo PC, Lau SK, Chu C‐m, Chan K‐h, Tsoi H‐w, Huang Y, Wong BH, Poon RW, Cai JJ, Luk W‐k (2005) Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79: 884–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2021) Enhancing readiness for Omicron (B.1.1.529): technical brief and priority actions for member states. Geneva, Switzeland: World Health Organization; [Google Scholar]
- Worobey M, Levy JI, Serrano LMM, Crits‐Christoph A, Pekar JE, Goldstein SA, Rasmussen AL, Kraemer MU, Newman C, Koopmans MP (2022) The Huanan market was the epicenter of SARS‐CoV‐2 emergence. Zenodo 10.5281/zenodo.6299600 [DATASET] [DOI] [Google Scholar]
- Wymant C, Bezemer D, Blanquart F, Ferretti L, Gall A, Hall M, Golubchik T, Bakker M, Ong SH, Zhao L (2022) A highly virulent variant of HIV‐1 circulating in the Netherlands. Science 375: 540–545 [DOI] [PubMed] [Google Scholar]
- Xia H, Luo H, Shan C, Muruato AE, Nunes BT, Medeiros D, Zou J, Xie X, Giraldo MI, Vasconcelos PF (2018) An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat Commun 9: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong D, Zhao X, Luo S, Duan L (2022) Insights from computational analysis: how does the SARS‐CoV‐2 Delta (B. 1.617. 2) variant hijack ACE2 more effectively? Phys Chem Chem Phys 24: 8683–8694 [DOI] [PubMed] [Google Scholar]
- Yang Y, Du L, Liu C, Wang L, Ma C, Tang J, Baric RS, Jiang S, Li F (2014) Receptor usage and cell entry of bat coronavirus HKU4 provide insight into bat‐to‐human transmission of MERS coronavirus. Proc Natl Acad Sci U S A 111: 12516–12521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Shan C, Zhu Y, Ma E, Wang J, Wang P, Shi P‐Y, Cheng G (2021) A mutation‐mediated evolutionary adaptation of Zika virus in mosquito and mammalian host. Proc Natl Acad Sci U S A 118: e2113015118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Huang X‐Y, Liu Z‐Y, Zhang F, Zhu X‐L, Yu J‐Y, Ji X, Xu Y‐P, Li G, Li C (2017) A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 358: 933–936 [DOI] [PubMed] [Google Scholar]
- Zhang J, Cai Y, Xiao T, Lu J, Peng H, Sterling SM, Walsh RM Jr, Rits‐Volloch S, Zhu H, Woosley AN (2021a) Structural impact on SARS‐CoV‐2 spike protein by D614G substitution. Science 372: 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen Y, Li Y, Huang F, Luo B, Yuan Y, Xia B, Ma X, Yang T, Yu F (2021b) The ORF8 protein of SARS‐CoV‐2 mediates immune evasion through down‐regulating MHC‐Ι. Proc Natl Acad Sci U S A 118: e2024202118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gu Q, de Manuel MM, Bravo IG, Marques‐Bonet T, Häussinger D, Münk C (2017) Stably expressed APOBEC3H forms a barrier for cross‐species transmission of simian immunodeficiency virus of chimpanzee to humans. PLoS Pathog 13: e1006746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y‐Z, Holmes EC (2020) A genomic perspective on the origin and emergence of SARS‐CoV‐2. Cell 181: 223–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W‐M, Song S‐H, Chen M‐L, Zou D, Ma L‐N, Ma Y‐K, Li R‐J, Hao L‐L, Li C‐P, Tian D‐M (2020) The 2019 novel coronavirus resource. Yi Chuan 42: 212–221 [DOI] [PubMed] [Google Scholar]
- Zhou P, Yang X‐L, Wang X‐G, Hu B, Zhang L, Zhang W, Si H‐R, Zhu Y, Li B, Huang C‐L (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579: 270–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wang J, Wang P, Song W, Zheng Z, Chen R, Guo K, Zhang T, Peiris JS, Chen H (2010) Substitution of lysine at 627 position in PB2 protein does not change virulence of the 2009 pandemic H1N1 virus in mice. Virology 401: 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer SM, Burke DS (2009) Historical perspective—emergence of influenza A (H1N1) viruses. N Engl J Med 361: 279–285 [DOI] [PubMed] [Google Scholar]
- Zinzula L (2021) Lost in deletion: the enigmatic ORF8 protein of SARS‐CoV‐2. Biochem Biophys Res Commun 538: 116–124 [DOI] [PMC free article] [PubMed] [Google Scholar]