

Abstract
Developmental neuron death plays a pivotal role in refining organization and wiring during neocortex formation. Aberrant regulation of this process results in neurodevelopmental disorders including impaired learning and memory. Underlying molecular pathways are incompletely determined. Loss of Bcl11a in cortical projection neurons induces pronounced cell death in upper‐layer cortical projection neurons during postnatal corticogenesis. We use this genetic model to explore genetic mechanisms by which developmental neuron death is controlled. Unexpectedly, we find Bcl6, previously shown to be involved in the transition of cortical neurons from progenitor to postmitotic differentiation state to provide a major checkpoint regulating neuron survival during late cortical development. We show that Bcl11a is a direct transcriptional regulator of Bcl6. Deletion of Bcl6 exerts death of cortical projection neurons. In turn, reintroduction of Bcl6 into Bcl11a mutants prevents induction of cell death in these neurons. Together, our data identify a novel Bcl11a/Bcl6‐dependent molecular pathway in regulation of developmental cell death during corticogenesis.
Keywords: Bcl11a, Bcl6, developmental cell death, neocortex, transcription factor
Subject Categories: Autophagy & Cell Death, Development, Neuroscience
Conditional mutagenesis in mice identifies a novel Bcl11a/Bcl6‐dependent pathway that regulates developmental cell death in upper‐layer cortical projection neurons.
Introduction
Developmental cell death (DCD) occurs in all animals and organs. It is part of a homeostatic balance between generation and elimination of cells. Developmental cell death provides a major checkpoint for quality control allowing selective removal of either defective, mis‐integrated or no longer required cells (Causeret et al, 2018; Wong & Marin, 2019). During the development of the mammalian neocortex, excess numbers of neurons are generated. Supernumerary neurons are eliminated during two distinct waves of apoptosis. In mice, the first wave of DCD occurs around E14 and affects predominantly proliferating neuron precursors (Blaschke et al, 1996; de la Rosa & de Pablo, 2000; Roth et al, 2000). During a second wave, corresponding to the first two postnatal weeks in rodents, approximately 30% of postmitotic cortical neurons are eliminated by DCD (Verney et al, 2000; Southwell et al, 2012). Within this period, entire neuron populations, as for example Cajal–Retzius cells, which transiently serve as signaling centers, are removed by DCD (Chowdhury et al, 2010; Ledonne et al, 2016), while in other neuron types, like cortical projection neurons (CPN), DCD adjusts definitive neuron numbers and refines immature synaptic circuits (Blanquie et al, 2017; Wong et al, 2018). In the neocortex, dysregulated DCD has been shown to be associated with a wide spectrum of neurodevelopmental disorders, including major structural changes as well as structurally more subtle defects, like autism‐spectrum disorders and intellectual disability (Kuida et al, 1996; Eriksson et al, 2001; Wei et al, 2014; Nakamura et al, 2016). Developmental cell death acts cell‐type specific and is spatio‐temporarily highly restricted suggesting complex molecular regulation. In contrast to the peripheral nervous system, where target‐derived neurotrophic signals have been extensively demonstrated to play a key role in regulation of neuron survival (Huang & Reichardt, 2001), the molecular controls of DCD within the central nervous system (CNS) are incompletely determined. Electrical and synaptic activity has been shown to confer survival signals onto postmitotic cortical neurons (Blanquie et al, 2017; Denaxa et al, 2018; Priya et al, 2018; Wong et al, 2018). Transcription factor cascades as well as secreted signaling molecules are of key importance for the development of the neocortex. It is, however, unclear, how these regulatory networks are connected to DCD.
Bcl11a (Ctip1) encodes a zinc‐finger protein that regulates transcription through interaction with COUP‐TF proteins as well as direct, sequence‐dependent DNA binding (Avram et al, 2002). We previously demonstrated that postmitotic upper‐layer CPN require expression of Bcl11a for early postnatal survival. Cre/loxP‐dependent ablation of Bcl11a in CPN results in massive increase in apoptosis between P4 and P6 selectively in upper‐layer CPN (Wiegreffe et al, 2015).
In this study, we employed Bcl11a mutation in CPN as a highly selective genetic tool to systematically identify downstream candidate genes involved in the regulation of DCD in postmitotic CPN. Using comparative transcriptome analysis, we found that Bcl6, previously reported to be involved in the transition of cortical neurons from progenitor to postmitotic differentiation state (Tiberi et al, 2012; Bonnefont et al, 2019), is downregulated in Bcl11a mutant upper‐layer CPN. Furthermore, we show Bcl11a to directly bind to a conserved promotor element and to activate transcription of the Bcl6 gene. Knockout of Bcl6 in postmitotic CPN induces their apoptosis. In turn, reintroduction of Bcl6 into Bcl11a mutant CPN prevents these neurons from apoptosis. Finally, we show Foxo1 to be downregulated in both, Bcl6 and Bcl11a mutant CPN. Normalization of Foxo1 expression is sufficient to suppress increased apoptosis in Bcl11a mutant CPN suggesting Foxo1 to participate in the regulation of DCD in CPN during postnatal neocorticogenesis. Taken together, in this study we demonstrate that DCD of postmitotic upper‐layer CPN is controlled by a novel Bcl11a/Bcl6‐dependent transcriptional pathway.
Results and Discussion
Identification of downstream candidate targets of Bcl11a
We used Bcl11a F/F ; Emx1 IRESCre brains as a model to identify genes that play a role in postnatal survival of projection neurons in the somatosensory neocortex. Bcl11a mutant brains display robust increase in apoptosis during the second wave of DCD in upper cortical layers at postnatal stages (Wiegreffe et al, 2015). Using laser capture microdissection, we specifically isolated cortical layers 2–4 of Bcl11a mutant and control brains at P2 (Fig 1A and B), a stage when the second wave of apoptosis has not yet been initiated (Blanquie et al, 2017) and cell death is not yet increased in Bcl11a mutants (Wiegreffe et al, 2015). We then performed a differential expression analysis using microarrays and identified a set of 137 differentially expressed (DE) genes that were subjected to a GO overrepresentation test as previously described (Mi et al, 2013; De Bruyckere et al, 2018), which revealed genes involved in axon guidance, cell–cell adhesion, and regulation of cell communication (Figs 1C, and EV1, EV2 and EV3A; Dataset EV1). To verify the validity of the experimental approach, selected candidate genes were tested by quantitative real‐time PCR and RNA in situ hybridization. Cdh6, Cdh12, Efna5, and Pcdh9 that were identified as downregulated were verified by this approach (Fig EV3B and C). In addition, Cdh13, Flrt2, Flrt3, and Slit2 were verified as upregulated (Fig EV3B and D). Together, these results show that our genetic approach consistently identified DE genes in upper cortical layers of Bcl11a mutant brains that could directly or indirectly be involved in the regulation of developmental apoptosis.
Figure 1. Identification of downstream candidate target genes of Bcl11a in superficial cortical layers at early postnatal development.
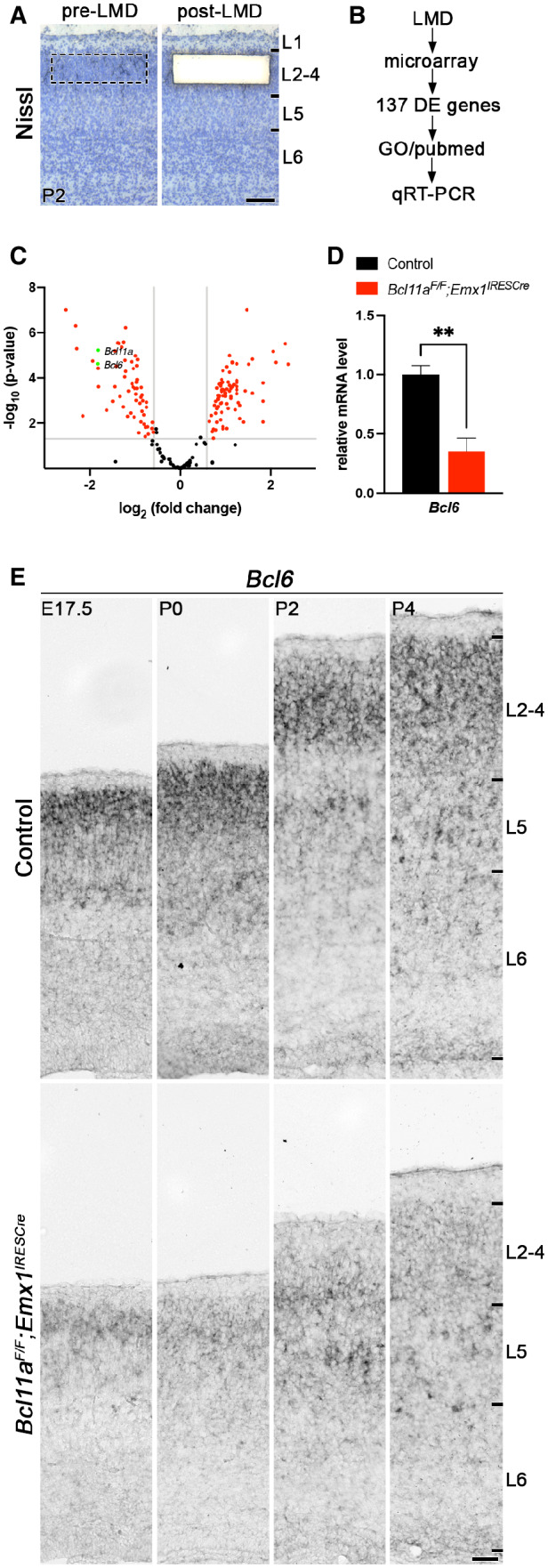
-
ACortical layers 2–4 were isolated by laser microdissection from Bcl11a F/F ;Emx1 IRESCre and control neocortex (n = 4).
-
BGene expression was compared using microarrays. From a set of 137 differentially expressed (DE) genes, candidate targets were selected based on gene ontology (GO) and PubMed analyses and verified by quantitative real‐time PCR and RNA in situ hybridization.
-
CVolcano plot showing DE genes (red). Those not significantly changed (fold change < 1.5; P > 0.05) are shaded black. Bcl11a and Bcl6 are highlighted in green.
-
DRelative Bcl6 mRNA expression level determined by quantitative real‐time PCR is decreased in laser‐microdissected cortical tissue of P2 Bcl11a F/F ;Emx1 IRESCre compared with control brains (n = 4). Results are expressed as mean ± s.e.m.; Student's t‐test; **P < 0.01.
-
ERNA in situ hybridization showing downregulation of Bcl6 expression in Bcl11a F/F ;Emx1 IRESCre compared with control neocortex at E17.5, P0, P2, and P4.
Data information: Scale bars, 100 μm (A) and 50 μm (E).
Figure EV1. Heat map of differentially expressed genes in upper layers of P2 Bcl11a F/F ;Emx1 IRESCre compared with control neocortex.
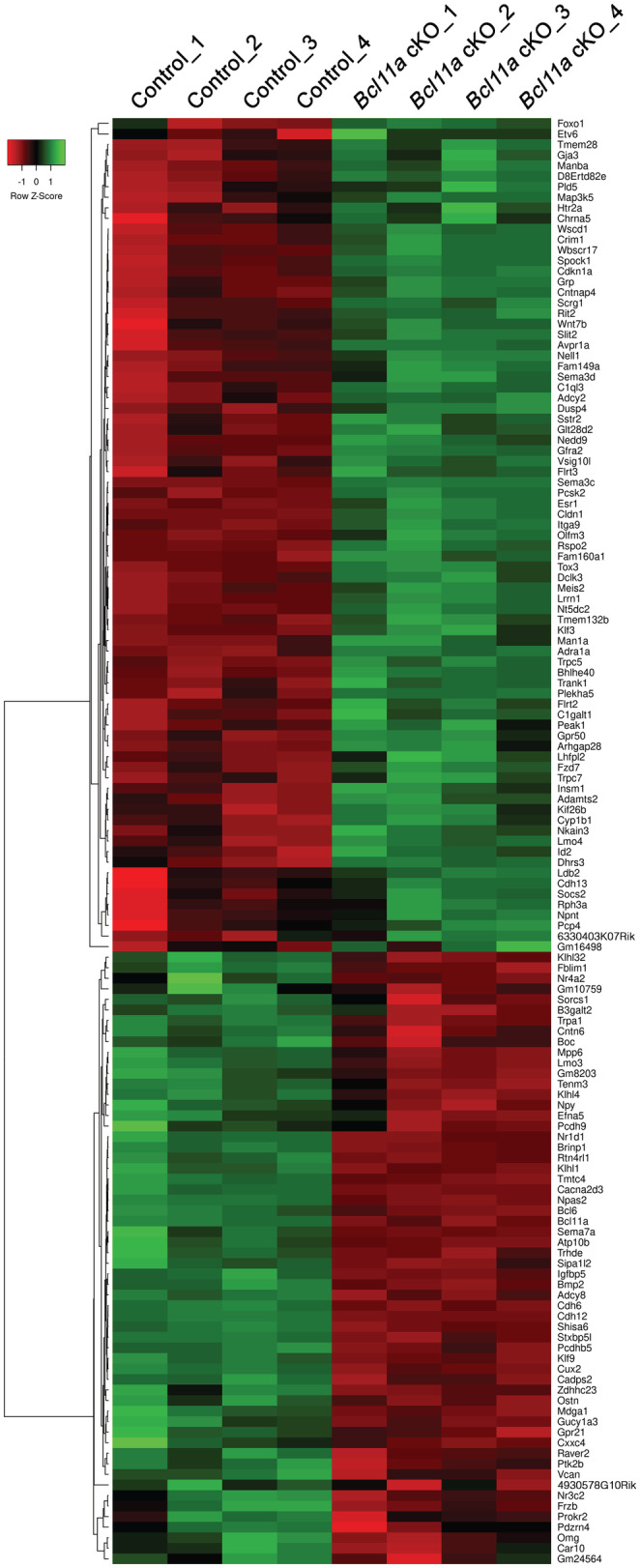
Heat map showing 79 upregulated genes (green) and 58 downregulated genes (red) in upper neocortical layers of conditional Emx1 IRESCre ;Bcl11a F/F mutants (Bcl11a cKO) compared with controls at postnatal day 2.
Figure EV2. Gene ontology enrichment analysis of differentially expressed genes in upper layers of P2 Bcl11a F/F ;Emx1 IRESCre compared with control neocortex.
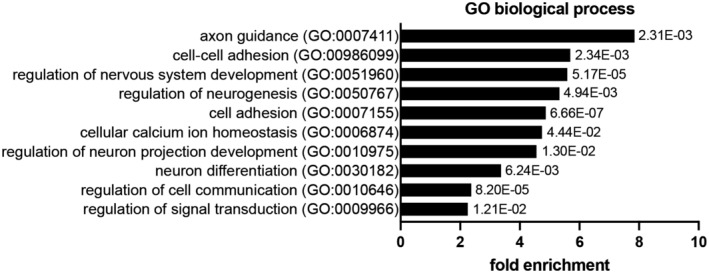
Graph shows selected GO terms for biological process, GO term accession numbers, fold enrichment, and P‐value. Data were obtained by GO overrepresentation test using PANTHER.
Figure EV3. Selected candidate downstream target genes of Bcl11a in superficial cortical layers at early postnatal development.
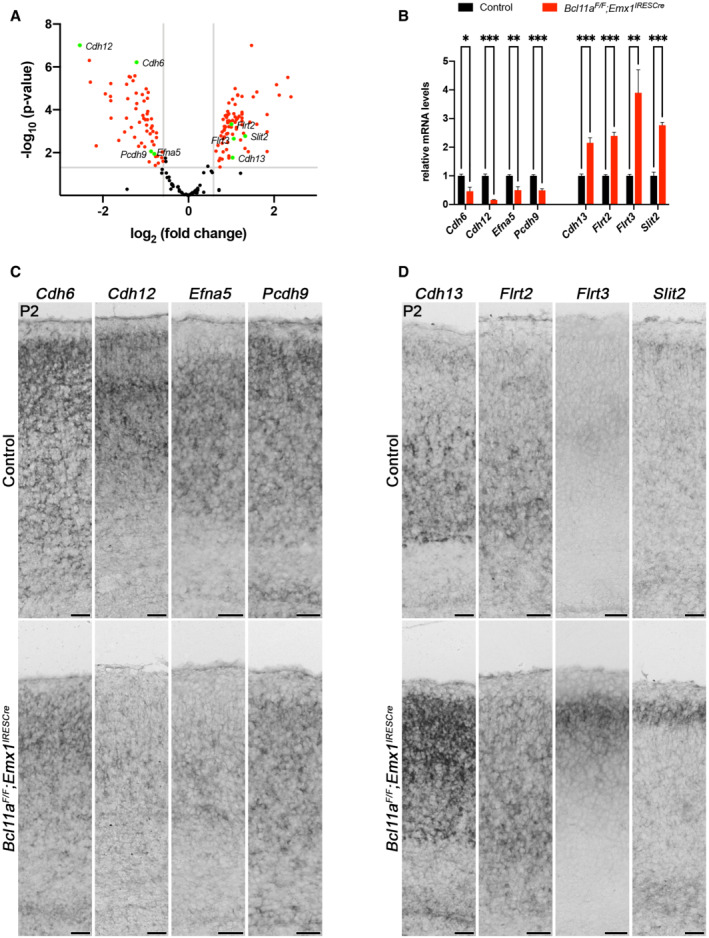
-
AVolcano plot showing differentially expressed (DE) genes in laser‐microdissected cortical layers 2–4 of Bcl11a F/F ;Emx1 IRESCre neocortex compared with controls. DE genes not significantly changed (fold change < 1.5; P > 0.05) are shaded black. Cdh6, Cdh12, Efna5, Pcdh9, Cdh13, Flrt2, Flrt3, and Slit2 are highlighted in green.
-
BRelative mRNA expression levels of selected DE genes determined by quantitative real‐time PCR in laser‐microdissected cortical tissue of P2 Bcl11a F/F ;Emx1 IRESCre and control brains (n = 4). Graph represents mean ± s.e.m.; Student's t‐test; *P < 0.05; **P < 0.01; ***P < 0.01.
-
CRNA in situ hybridization of selected DE genes with decreased expression in P2 Bcl11a F/F ;Emx1 IRESCre compared with control neocortex.
-
DRNA in situ hybridization of selected DE genes with increased expression in P2 Bcl11a F/F ;Emx1 IRESCre compared with control neocortex.
Data information: Scale bar, 50 μm.
Among the DE genes, we found Bcl6, a transcriptional repressor that was previously reported to regulate cortical neurogenesis (Tiberi et al, 2012; Bonnefont et al, 2019), to be downregulated by 64.8 ± 0.1% in Bcl11a mutant neocortex (Fig 1D). Using RNA in situ hybridization, we found robust expression of Bcl6 predominantly in upper and at low levels in deep cortical layers of controls between E17.5 and P4 (Fig 1E). In Bcl11a mutant neocortex, Bcl6 was downregulated in upper cortical layers at these stages (Fig 1E), suggesting this gene to be transcriptionally downstream of Bcl11a in upper cortical layers. Outside the CNS, Bcl6 exerts anti‐apoptotic functions by suppressing genes involved in DNA damage response (Phan & Dalla‐Favera, 2004; Phan et al, 2005; Ranuncolo et al, 2007), which could possibly be conserved in the developing neocortex as well. Therefore, we focused further analyses on Bcl6.
Bcl6 is a direct target of Bcl11a in upper‐layer cortical projection neurons
To better characterize the expression of Bcl6 protein in early postnatal somatosensory cortex we generated a polyclonal antibody in guinea pig raised against the N‐terminal 484 amino acids of mouse Bcl6. Specificity of the Bcl6 antibody was tested by immunohistochemistry using Bcl6 mutant brains, which lack exons 4–10 (Ye et al, 1997) and do not express Bcl6 protein (Tiberi et al, 2012). In comparison to wild‐type littermates, we did not detect Bcl6 protein in Bcl6 mutant brains at P0 (Appendix Fig S1), demonstrating our antibody to specifically detect Bcl6 protein. Coexpression analysis of Bcl6 together with Bcl11a and Satb2, a marker for callosal projection neurons (Alcamo et al, 2008; Britanova et al, 2008), showed 25.6 ± 1.9% Bcl6+ Bcl11a+ Satb2+, 1.3 ± 0.5% Bcl6+ Bcl11a+, and 0.8 ± 0.2% Bcl6+ Satb2+ cells in wild‐type brains. Only 0.3 ± 1.0% of cells exclusively expressed Bcl6 (Fig 2A and B). Coexpression analysis of Bcl6 together with Bcl11a and Cux1, a marker for cortical layers 2–4 (Nieto et al, 2004), showed 19.0 ± 0.7% Bcl6+ Bcl11a+ Cux1+, 11.6 ± 0.9% Bcl6+ Bcl11a+, and 1.0 ± 0.2% Bcl6+ Cux1+ cells. Again, only 0.9 ± 0.2% of cells exclusively expressed Bcl6 (Fig 2C and D). Thus, more than 90% of Bcl6+ cells coexpress Satb2 as well as Bcl11a and more than 61% of these cells are located in Cux1+ upper layers with distinct localization to cortical layers 2/3 (Fig 2C). Notably, a substantial proportion of Bcl6+ cells is located in deep cortical layers. Thus, Bcl6 is a marker for a subset of callosal projection neurons identified by coexpression of Bcl11a and that are located in cortical layers 2/3 as well as in deep cortical layers.
Figure 2. Bcl6 is expressed in superficial callosal projection neurons and a target gene of Bcl11a.
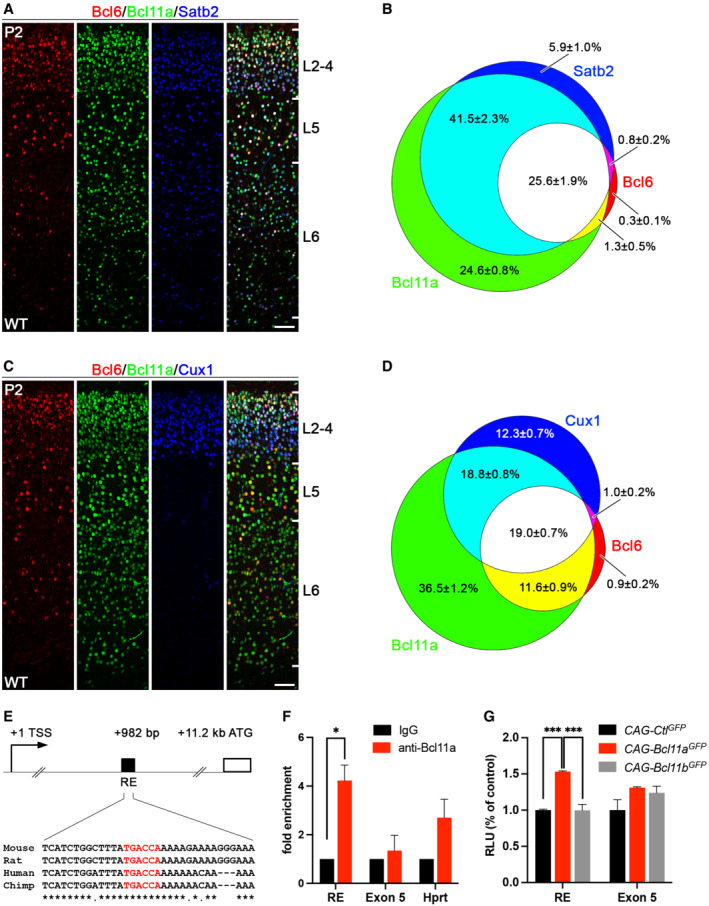
-
AImmunohistochemistry of Bcl6 (red), Bcl11a (green), and Satb2 (blue) in P2 wild‐type neocortex.
-
BVenn diagram displaying the proportions of Bcl6 neurons overlapping with Bcl11a and Satb2 expressing cells. The percentage of each labeled cell population is given in relation to all labeled cells (Bcl6+ and Bcl11a+ and Satb2+, in total 4,479 cells).
-
CImmunohistochemistry of Bcl6 (red), Bcl11a (green), and Cux1 (blue) in P2 wild‐type neocortex.
-
DVenn diagram displaying the proportions of Bcl6 neurons overlapping with Bcl11a and Cux1 expressing cells. The percentage of each labeled cell population is given in relation to all labeled cells (Bcl6+ and Bcl11a+ and Cux1+, in total 4,301 cells).
-
EScheme of the Bcl6 gene locus displaying the start codon (ATG) at +11.2 kb relative to the transcriptional start site (TSS). A regulatory element (RE) in the first intron at +982 bp contains a conserved binding motif (TGACCA, in red) of Bcl11a.
-
FChIP analysis using a Bcl11a antibody and P2 cortical tissue detects Bcl11a binding to the RE shown in (E). Negative controls include ChIP with unspecific IgG antibody and the precipitation of exon 5 of Bcl6 and the Hprt promoter. The experiment was independently repeated four times. Results are expressed as mean ± s.e.m.; Student's t‐test; *P < 0.05.
-
GLuciferase assays in HEK293 cells transfected with control (CAG‐Ctl GFP ), Bcl11a (CAG‐Bcl11a GFP ), or Bcl11b (CAG‐Bcl11b GFP ) expression vector show induction of luciferase activity of the RE reporter construct by Bcl11a. Negative controls include a reporter construct for a region of exon 5 of the Bcl6 gene and co‐transfection with the closely related transcription factor Bcl11b. The experiment was independently repeated four times. Results are expressed as mean ± s.e.m.; one‐way ANOVA followed by Tukey's post‐hoc test; ***P < 0.001. Data information: Scale bars, 50 μm.
By DNA sequence analysis, we found a TGACCA binding motif of Bcl11a (Liu et al, 2018) in the first intron that was located 982 bp downstream of the transcriptional start site and ~10.2 kb upstream of the first protein‐coding exon of the Bcl6 gene. This binding motif was embedded within a 55 bp long conserved region with a high degree of conservation between rat, human, and chimp (Fig 2E). Binding of Bcl11a to this motif was tested by chromatin immunoprecipitation (ChIP) followed by quantitative real‐time PCR using a primer pair flanking this region. An enrichment of more than fourfold was found using a Bcl11a‐specific antibody in comparison with an immunoglobulin G (IgG) control antibody (Fig 2F), demonstrating binding of Bcl11a to this region. As negative controls, binding of Bcl11a to exon 5 of Bcl6 and the Hprt promoter was tested, but no significant enrichment was found in comparison with the IgG control antibody (Fig 2F). The sequence containing the Bcl11a binding motif was further tested for its ability to activate gene expression. In luciferase assays, a 1.5‐fold induction was measured, indicating this element to convey functional activation upon Bcl11a binding (Fig 2G). As negative controls, we tested a region of exon 5 of the Bcl6 gene as well as activation in the presence of the closely related transcription factor Bcl11b, which both did not induce transcriptional activity in luciferase assays (Fig 2G).
Bcl6 is downregulated in upper layers of Bcl11a mutant neocortex
To confirm Bcl6 downregulation in the Bcl11a mutant neocortex on protein level, we performed immunohistochemistry with Bcl6 and neuron subtype‐specific antibodies. The overall expression of Bcl6 was reduced by 44.0 ± 7.0% compared with control neocortex at P2 (Fig 3A and B). We did not detect changes in the number of Satb2+ and Cux1+ cells that would normally coexpress with Bcl6 (c.f. Fig 2A–D), suggesting that these cells are born correctly, have for the most part migrated to their respective layers, and undergo neuron subtype‐specific differentiation (Fig 3A and B). Furthermore, the proportion of Cux1+ and Satb2+ cells coexpressing Bcl6 was reduced from 57.4 ± 2.3 to 24.2 ± 2.6% and 58.5 ± 2.8 to 31.8 ± 1.6%, respectively, in Bcl11a mutant compared with control neocortex (Fig 3A and C). As previously demonstrated, cortical thickness is reduced and layer 5 is increased at the expense of layer 6 in Bcl11a mutants at this stage (Wiegreffe et al, 2015; Woodworth et al, 2016). We did not detect significant changes in the number of cells coexpressing Bcl6 in deep cortical layers labeled by Bcl11b (layer 5) or Tbr1 (layer 6; Molyneaux et al, 2007; Fig EV4A–C), indicating a selective loss of Bcl6 in upper‐layer neurons. Together, these data are compatible with a function of Bcl6 in CPN survival, which is massively impaired in upper layers of the Bcl11a mutant neocortex after P2 (Wiegreffe et al, 2015).
Figure 3. Bcl6 expression is specifically downregulated in superficial cortical layers of Bcl11a F/F ;Emx1 IRESCre neocortex.
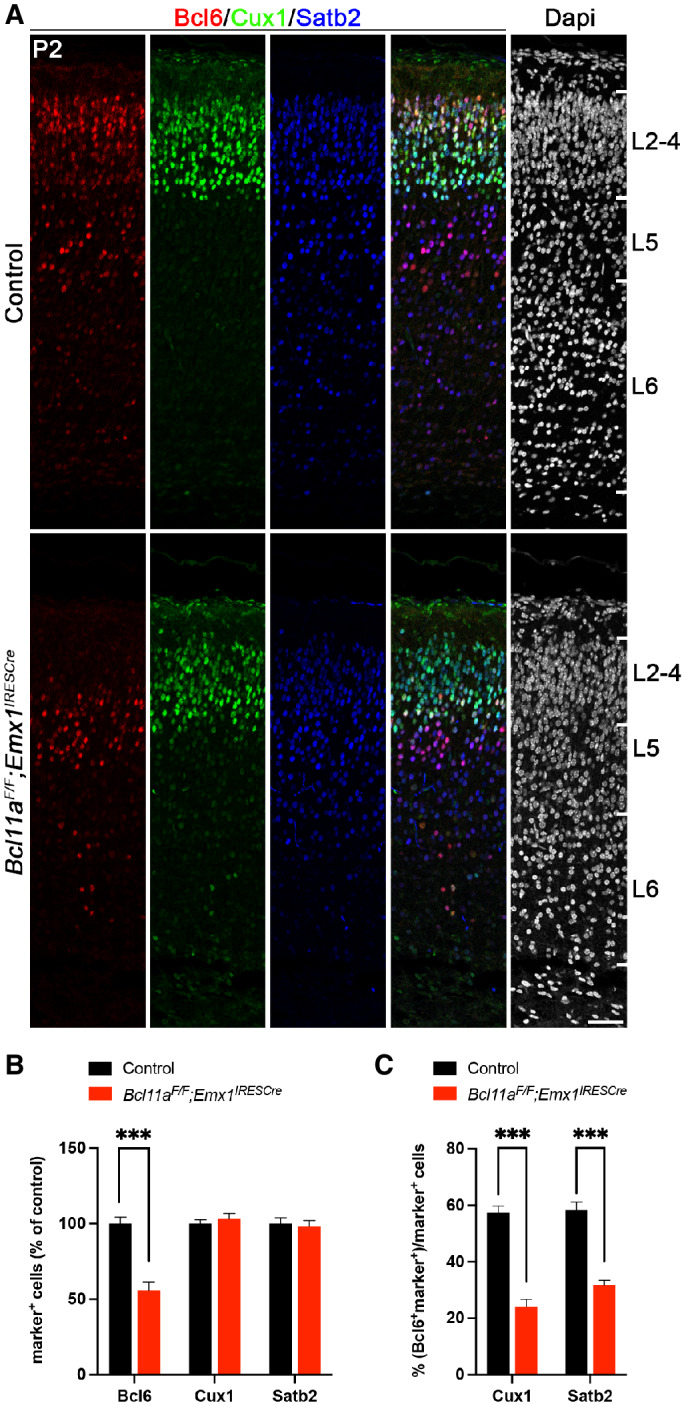
-
AImmunohistochemistry of Bcl6 (red), Cux1 (green), and Satb2 (blue) in P2 Bcl11a F/F ;Emx1 IRESCre and control neocortex. Nuclei are stained with Dapi (white).
-
BRelative quantification of Bcl6+, Satb2+, and Cux1+ cells in Bcl11a F/F ;Emx1 IRESCre and control neocortex (n = 4).
-
CNumbers of Cux1+ or Satb2+ cells that coexpress Bcl6 are reduced in Bcl11a F/F ;Emx1 IRESCre compared with control neocortex (n = 4).
Data information: All graphs represent the mean ± s.e.m.; Student's t‐test; ***P < 0.001. Scale bar, 50 μm.
Figure EV4. Relative Bcl6 expression is unchanged in deep cortical layers of Bcl11a F/F ;Emx1 IRESCre compared with control neocortex.
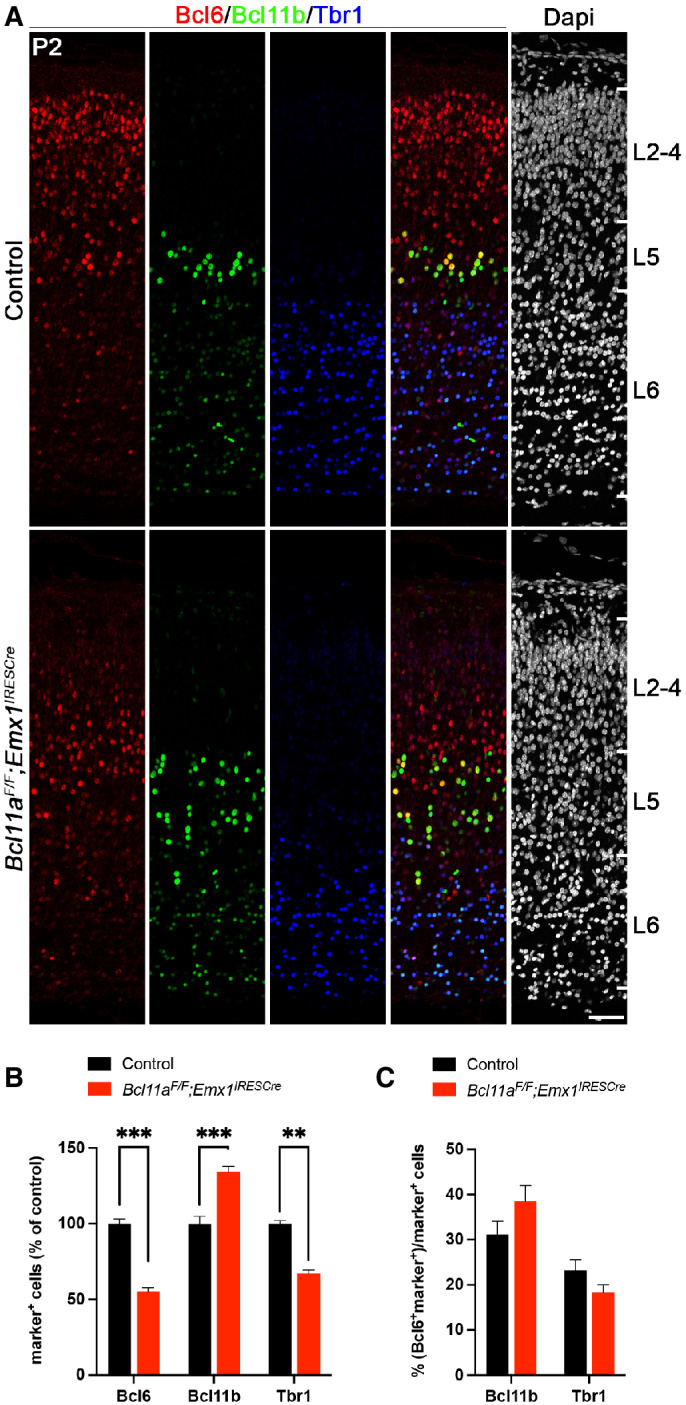
-
AImmunohistochemistry of Bcl6 (red), Bcl11b (green), and Tbr1 (blue) in P2 Bcl11a F/F ;Emx1 IRESCre and control neocortex. Bcl6 and Tbr1 expressions are downregulated, while Bcl11b expression is upregulated in Bcl11a F/F ;Emx1 IRESCre compared with control neocortex. Nuclei are stained with Dapi (white).
-
BRelative quantification of Bcl6+, Bcl11b+, and Tbr1+ cells in Bcl11a F/F ;Emx1 IRESCre and control neocortex (n = 4).
-
CNumbers of Bcl11b+ or Tbr1+ cells that coexpress Bcl6 are normal in Bcl11a F/F ;Emx1 IRESCre compared with control neocortex (n = 4).
Data information: All graphs represent mean ± s.e.m.; Student's t‐test; **P < 0.01; ***P < 0.001. Scale bar, 50 μm.
Cell‐autonomous control of Bcl6 expression by Bcl11a
To further examine whether Bcl6 expression is directly regulated by Bcl11a in neurons, we created a mosaic mutant in vivo situation by using in utero electroporation. We generated Bcl11a‐deficient neurons in cortical layer 2/3 by electroporating Cre together with GFP (CAG‐Cre GFP ) or GFP alone (CAG‐Ctl GFP ) into conditional Bcl11a mutant (Bcl11a F/F ) brains at E15.5 and analyzed the transfected brains at P2 (Fig 4A and B). The proportion of GFP+ cells that coexpresses Bcl6 was reduced from 70.3 ± 4.0% in controls to 9.5 ± 1.5% in Bcl11a‐deficient cortical neurons (Fig 4C and F). In contrast, the proportions of GFP+ cells that coexpress Cux1 or Satb2 remained unchanged (Fig 4D–F). Thus, cell‐autonomous loss of Bcl11a in superficial cortical layers leads to a dramatic and specific reduction of Bcl6. Together with the direct binding of Bcl11a to the Bcl6 gene and its transcriptional activation through a conserved binding motif (Fig 2E–G), this suggests that Bcl11a directly controls Bcl6 expression in these cells.
Figure 4. Cell‐autonomous loss of Bcl11a in superficial cortical layers leads to reduced Bcl6 expression and reintroduction of Bcl6 into Bcl11a mutant superficial projection neurons rescues the Bcl11a mutant phenotype.
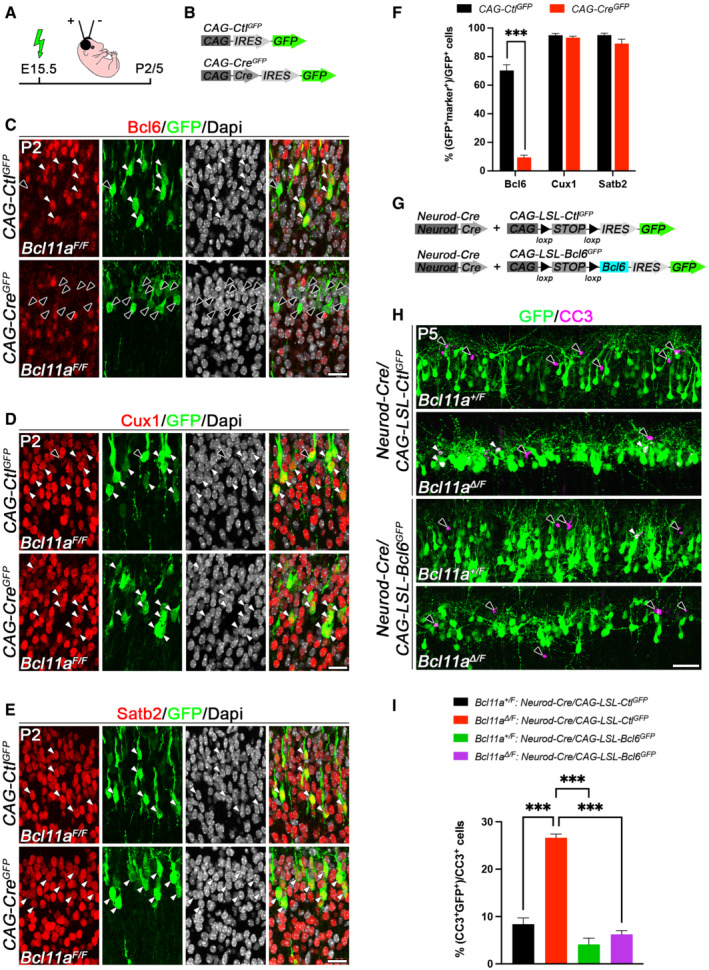
-
ASchematic representation of the experimental approach. Embryos are electroporated at E15.5 with the indicated DNA plasmids and sacrificed at either P2 or P5.
-
BDNA plasmids used in the experiment shown in (C–F).
-
C–EImmunohistochemistry of electroporated P2 Bcl11a F/F neurons in superficial cortical layers with GFP (green) and Bcl6 (red, C), Cux1 (red, D), or Satb2 (red, E) antibodies. Bcl6 expression is specifically downregulated Bcl11a F/F neocortex upon electroporation of CAG‐Cre GFP in comparison with CAG‐Ctl GFP control plasmid. Nuclei are stained with Dapi (white).
-
FQuantification of the percentage of electroporated cells expressing Bcl6 (n = 3), Satb2 (n = 3), and Cux1 (n = 5). Results are expressed as mean ± s.e.m.; Student's t‐test; ***P < 0.001.
-
GDNA plasmids used in the experiment shown in (H and I).
-
HImmunohistochemistry of electroporated P5 Bcl11a ∆/F and Bcl11a +/F neurons in superficial cortical layers with GFP (green) and cleaved caspase 3 (CC3, magenta) antibodies. Electroporation of Neurod‐Cre GFP plasmid together with CAG‐LSL‐Bcl6 GFP into Bcl11a F/F neocortex reduces the number of CC3+ cells to control levels.
-
IQuantification of the experiment shown in (H) (n = 4). Results are expressed as mean ± s.e.m.; one‐way ANOVA followed by Tukey's post‐hoc test; ***P < 0.001.
Data information: White arrowheads point at GFP+ cells that also express Bcl6, Cux1, Satb2, or CC3. Black arrowheads indicate cells expressing only GFP+. Scale bars, 20 μm (C–E), 50 μm (H).
Reintroduction of Bcl6 into Bcl11a mutants rescues neuron death
We next asked whether reintroduction of Bcl6 into Bcl11a mutant neurons located in upper cortical layers could rescue mutant neurons from undergoing apoptosis and thereby normalize the Bcl11a mutant phenotype. We generated Cre‐dependent control (CAG‐LSL‐Ctl GFP ) and Bcl6 (CAG‐LSL‐Bcl6 GFP ) expression constructs that were tested in HEK293 cells and by western blot (Appendix Fig S2A). Both constructs induced robust GFP expression in the presence or absence of Cre, indicating that the floxed stop (LSL) cassette did not prevent the GFP from being expressed. However, Bcl6 expression was only observed in the presence of Cre, indicating a tight regulation of Bcl6 expression from this construct (Appendix Fig S2B). We then overexpressed Bcl6 in Bcl11a mutant cortical neurons by in utero electroporation at E15.5 and analyzed the brains at P5 (Fig 4A). To circumvent functions of Bcl6 that could interfere with neurogenesis (Tiberi et al, 2012; Bonnefont et al, 2019), we directed Bcl6 expression to postmitotic neurons by electroporating CAG‐LSL‐Ctl GFP or CAG‐LSL‐Bcl6 GFP expression constructs together with Cre placed under the control of the postmitotically activated Neurod promoter (Neurod‐Cre) into Bcl11a Δ/F (i.e., conditional mutant) or Bcl11a +/F (i.e., control) brains (Fig 4G). Co‐electroporation of Neurod‐Cre together with CAG‐LSL‐Ctl GFP robustly induced cell death in Bcl11a Δ/F in comparison with control brains by more than threefold. In contrast, postmitotic reintroduction of Bcl6 into Bcl11a‐deficient neurons reduced apoptosis to control levels. Of note, overexpression of Bcl6 in control brains did not significantly reduce the number of cleaved caspase 3+ cells below control levels (Fig 4H and I). Together, these data strongly support a role for Bcl6 as a direct functional downstream target of Bcl11a that controls neuron survival during the second wave of DCD at the early postnatal stage.
Increased cell death in postnatal Bcl6 mutant neocortex
To further corroborate that Bcl6 confers survival of cortical projection neurons, we generated forebrain‐specific Bcl6 mutants by crossing conditional Bcl6 mutant mice (Bcl6 F/F ), in which exons 7–9 are flanked by loxP sites (Hollister et al, 2013), with Nex Cre mice (Goebbels et al, 2006) that induce recombination in postmitotic cortical projection neurons. Quantitative real‐time PCR showed that Nex Cre reduced Bcl6 expression by 80.0 ± 0.1% compared with controls at P0 (Fig 5B). Due to restricted activity of Nex Cre , incomplete reduction of Bcl6 is likely caused by residual expression in non‐neuronal cell types. We chose P5 to analyze DCD in Bcl6 mutant brains because Bcl11a mutants display massively increased cell death (Wiegreffe et al, 2015) and naturally occurring cell death in wild‐type brains peaks around this stage (Blanquie et al, 2017). We found a significant increase in cleaved caspase 3+ cells located predominantly in the upper cortical layers from 6.04 ± 0.02% in controls to 8.44 ± 0.77% cells/mm2 in Bcl6 mutant brains concomitant with a reduction of cortical area by 9.7 ± 2.0% (Fig 5A, C and D). We compared this increase in apoptosis in Bcl6 mutants to the phenotypes observed in Bcl11a F/F ;Nex Cre and the previously described Bcl11a F/F ;Emx1 IRESCre mutants (Fig EV5A and B; Wiegreffe et al, 2015). Both Bcl11a mutants display a similar extent of apoptosis. In both cases, increased neuron death was more pronounced as compared to the apoptosis rate observed in Bcl6 F/F ;Nex Cre mutants. This suggests that upstream of Bcl6, Bcl11a controls additional functions in neocortex development, which may indirectly and independently of Bcl6 contribute to cell survival control of CPN. For example, we and others have shown previously that morphogenesis and connectivity of CPN depend on Bcl11a (Simon et al, 2020).
Figure 5. Postnatal developmental cell death is increased in Bcl6 F/F ;Nex Cre neocortex.
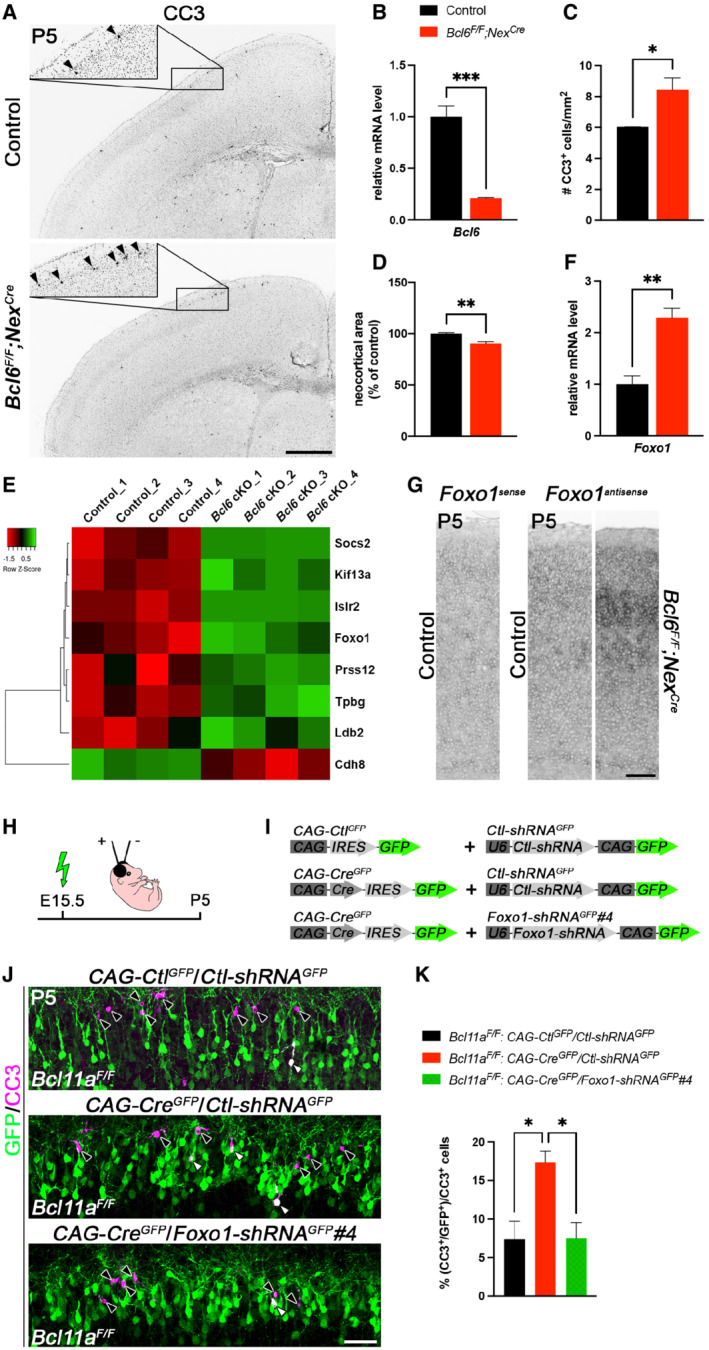
-
AImmunohistochemistry of cleaved caspase 3 (CC3) shows that the number of CC3+ cells (marked by black arrowheads) is increased in P5 Bcl6 F/F ;Nex Cre compared with control neocortex. Insets are enlargements of the boxed areas in corresponding panels.
-
BRelative Bcl6 mRNA expression level determined by quantitative real‐time PCR using primers targeting a region of exon 8 is decreased in P0 Bcl6 F/F ;Nex Cre compared with control brains (n = 4). Results are expressed as mean ± s.e.m.; Student's t‐test; ***P < 0.001.
-
CQuantification of the experiment shown in (A) (n = 3). Results are expressed as mean ± s.e.m.; Student's t‐test; *P < 0.05.
-
DQuantification of neocortical area in P5 Bcl6 F/F ;Nex Cre and control brains (n = 3). Results are expressed as mean ± s.e.m.; Student's t‐test; **P < 0.01.
-
EHeat map showing differentially expressed genes in laser‐microdissected superficial cortical layers of P5 Bcl6 F/F ;Nex Cre compared with control brains (n = 4).
-
FRelative Foxo1 mRNA expression level determined by quantitative real‐time PCR is increased in laser‐microdissected superficial cortical layers of P5 Bcl6 F/F ;Nex Cre compared with control brains (n = 4). Results are expressed as mean ± s.e.m.; Student's t‐test; **P < 0.01.
-
GRNA in situ hybridization showing upregulation of Foxo1 expression in P5 Bcl6 F/F ;Nex Cre compared with control neocortex.
-
HSchematic representation of the experimental approach. Embryos are electroporated at E15.5 and sacrificed at P5.
-
IDNA plasmids used in the experiment shown in (J and K).
-
JImmunohistochemistry of electroporated P5 Bcl11a F/F neurons in superficial cortical layers with GFP (green) and cleaved caspase 3 (CC3, magenta) antibodies. Electroporation of CAG‐Cre GFP plasmid together with Foxo1‐shRNA GFP #4 into Bcl11a F/F neocortex reduces the number of CC3+ cells to control levels. White and black arrowheads indicate GFP+ CC3+ and GFP+ cells, respectively.
-
KQuantification of the experiment shown in (J) (n = 4, CAG‐Ctl GFP /Ctl‐shRNA GFP ; n = 3, CAG‐Cre GFP /Ctl‐shRNA GFP ; n = 5, CAG‐Cre GFP /Foxo1‐shRNA GFP #4). Results are expressed as mean ± s.e.m.; one‐way ANOVA followed by Tukey's post‐hoc test; *P < 0.05.
Data information: Scale bars, 500 μm (A), 50 μm (G, J).
Figure EV5. Increased cell death in Bcl11a F/F ;Nex Cre and overlapping differentially expressed genes in upper cortical layers of Bcl11a F/F ;Emx1 IRESCre and Bcl6 F/F ;Nex Cre compared with controls.
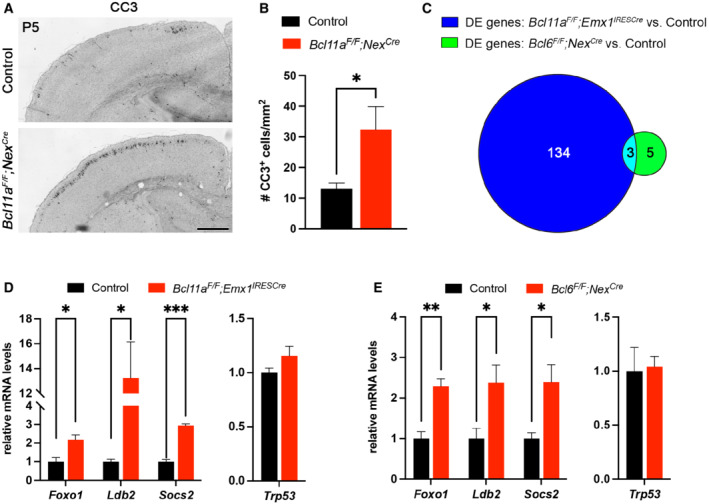
-
AImmunohistochemistry of cleaved caspase 3 (CC3) shows that the number of CC3+ cells is increased in P5 Bcl11a F/F ;Nex Cre compared with control neocortex.
-
BQuantification of the experiment shown in (A).
-
CVenn diagram showing the number of overlapping differentially expressed (DE) genes in P2 Bcl11a F/F ;Emx1 IRESCre and P5 Bcl6 F/F ;Nex Cre upper cortical layers.
-
D, ERelative mRNA expression levels of the overlapping DE genes Foxo1, Ldb2, and Socs2 as well as Trp53 determined by quantitative real‐time PCR in laser‐microdissected cortical tissue of P2 Bcl11a F/F ;Emx1 IRESCre compared with control brains (D) or P5 Bcl6 F/F ;Nex Cre compared with control brains (E).
Data information: All graphs represent mean ± s.e.m.; n = 4; Student's t‐test; *P < 0.05; **P < 0.01; ***P < 0.001. Scale bar, 500 μm.
Collectively, our data show that Bcl6 exerts functions in upper‐layer neuron survival during early postnatal neocortical development. To characterize downstream genetic pathways of Bcl6 responsible for the observed phenotype, we isolated upper cortical layers from Bcl6 mutant and control brains by laser capture microdissection at P5 and performed a differential expression analysis on this tissue using microarrays. This analysis revealed a small number of DE genes that were mostly upregulated in Bcl6 mutant upper cortical layers (Fig 5E, Dataset EV2). By systematic comparison of the differential transcriptomes of Bcl11a and Bcl6 mutants (Datasets EV1 and EV2, and Fig EV5C), we identified only three genes overlapping in both datasets that were verified by quantitative real‐time PCR (Fig EV5C–E). Of these genes, the cell death‐associated factor Foxo1 was found upregulated in both, Bcl6 and Bcl11a mutants (Fig 5F; Fig EV5D and E). Upregulation of Foxo1 was most apparent in upper layers of the Bcl6 mutant neocortex (Fig 5G). Bcl11a and Bcl6 were shown to physically interact and colocalize in nuclear paraspeckles suggesting common regulation of gene expression (Nakamura et al, 2000; Liu et al, 2006). Members of the Foxo family have been demonstrated to be involved in the control of neuron survival (Carter & Brunet, 2007; Santo & Paik, 2018). It might thus well be that Bcl6 together with Bcl11a exerts anti‐apoptotic functions in CPN through this pathway.
To further explore functions of Foxo1 in Bcl11a/Bcl6‐dependent DCD of CPN, we knocked down Foxo1 gene expression by the help of shRNA according to previously published experimental strategies (Wiegreffe et al, 2015). The shRNA sequences were selected according to published studies (Zhang et al, 2011; Park et al, 2019). Using western blot analysis, construct Foxo1‐shRNA GFP #4 was determined to be most efficient for its ability to reduce Foxo1 expression and employed for further experiments (Appendix Fig S3A–C). Using in utero electroporation, we introduced Foxo1‐shRNA GFP #4 together with CAG‐Cre GFP constructs into Bcl11a F/F upper‐layer CPN. As controls, Ctl‐shRNA GFP together with CAG‐Ctl GFP or CAG‐Cre GFP were used. The shRNA‐mediated knockdown of Foxo1 expression was sufficient to suppress, that is, rescue the Bcl11a‐dependent apoptosis phenotype in CPN in vivo. In contrast, co‐electroporation of Ctl‐shRNA GFP together with CAG‐Cre GFP did not affect increased apoptosis in Bcl11a mutant CPN (Fig 5H–K). This provides direct experimental evidence for a functional role of Foxo1 in Bcl11a/Bcl6‐dependent regulation of DCD of CPN. In lymphoid cells, Bcl6 regulates cell death through p53 function (Phan & Dalla‐Favera, 2004; Cerchietti et al, 2008). Using quantitative real‐time PCR, we did not detect changes in p53 expression in our expression analysis (Fig EV5D and E). Taken together, in this study we demonstrate that DCD of postmitotic upper‐layer CPN is controlled by a novel Bcl11a/Bcl6‐dependent transcriptional pathway that involves Foxo1 function.
Previously, we demonstrated Bcl6 to be required during early phases of neocortical development, where Bcl6 promotes the transition of neural progenitors into postmitotic neurons (Tiberi et al, 2012; Bonnefont et al, 2019). Our data suggest additional functions of Bcl6 in the postnatal development of postmitotic CPN. A conserved function of this factor in control of cell survival is supported by its well‐characterized functions in the lymphatic system. Bcl6 prevents apoptosis in germinal center B‐cells and exerts oncogenic activity in diffuse large B‐cell lymphoma both, through modulation of the p53 downstream pathway (Phan & Dalla‐Favera, 2004; Cerchietti et al, 2008). In the cerebellum, deletion of Bcl6 induces massively increased cell death of granule cell precursors but not postmitotic granule cells leading to reduction of organ size (Tiberi et al, 2014). Interestingly, activation of nuclear calcium pathway through synaptic NMDA‐receptor signaling induces Bcl6 expression in hippocampal neurons. In turn, upregulation of Bcl6 improves the survival of these neurons (Zhang et al, 2007). This suggests that activity‐dependent as well as activity‐independent, transcriptional regulatory pathways converge onto Bcl6 in the control of DCD.
Compared with Bcl11a mutants, we observed a milder increase in apoptosis in Bcl6 mutant CPN, raising the possibility of additional signals to contribute to apoptosis in Bcl11a mutants, for example, through regulation of alternative cell death pathways. Our systematic GO and transcriptome analyses, however, did not reveal further candidate target genes of Bcl11a commonly involved in alternative apoptosis pathways. Postnatally, Bcl11a mutant CPN display severe morphogenetic defects as characterized by shortened apical dendrites and disturbed dendritic branching pattern (Wiegreffe et al, 2015). This may result in impaired synaptic integration and electrical activity of Bc11a mutant neurons and in turn contribute to the severity of the phenotype.
Alternatively, additional, not yet characterized signals, might be involved. In our screen, we detected several axon guidance molecules, including Slit2, Efna5, Sema3c, −3d, −7a, Flrt2, −3 to be deregulated in Bcl11a mutant CPN. Semaphorins, for example, have extensively been demonstrated to influence neuronal connectivity (Koropouli & Kolodkin, 2014). Thus, differentially expressed guidance molecules, as observed in our study, might either directly or indirectly, through modulation of connectivity influence the severity of the apoptosis phenotype in Bcl11a mutants. In addition, we found cadherin 6, 12, 13 and protocadherin 9 to be deregulated in Bcl11a mutant CPN. Recent experimental evidence suggests cadherins, in addition to their well‐characterized functions in cell recognition and neural circuit assembly (Jontes, 2018; Sanes & Zipursky, 2020), to exert survival functions as well, for example, in neocortical interneurons (László et al, 2020). We previously demonstrated Bcl11a to be expressed in neocortical interneurons (Wiegreffe et al, 2015) raising the possibility that Bcl11a controls DCD also in these cells. During neocorticogenesis, DCD occurs in cell‐type‐specific and temporally distinct patterns. In mice, numbers of CPN are refined by DCD specifically between P4 and P6, whereas the time course of DCD in cortical interneurons is shifted to later developmental stages (Southwell et al, 2012; Blanquie et al, 2017; Wong et al, 2018). Emx1 IRESCre ‐/Nex Cre ‐dependent recombination as used in our study restricts Bcl11a mutation selectively to CPN. Thus, a role of Bcl11a in cortical interneuron survival remains to be determined by cell‐type‐specific mutation of Bcl11a in interneurons.
Bcl11a has been previously demonstrated to directly interact with Nr2f1 (COUP‐TFI). Moreover, in a very recent study Bcl1a was suggested to directly bind to the Nr2f1 gene locus and suppress its transcription (Du et al, 2021) raising the question whether Nr2f1 is involved in Bcl11a‐dependent control of late DCD in cortical projection neurons. Several lines of evidence argue against this assumption. (i) extensive phenotype analyses of Nr2f1 mutants from various laboratories have implicated this factor in control of cortical progenitor proliferation as well as cortical patterning, and laminar fate determination in postmitotic neurons (Tocco et al, 2021). Yet, a direct role for Nr2f1 in control of postmitotic neuron survival has not been reported. (ii) in our study, we did not detect deregulated Nr2f1 expression in Bcl11a nor in Bcl6 mutants as compared to controls. Interestingly, Bcl11a and Nr2f1 have been shown to be involved in establishing somatomotor versus somatosensory cortical area identity leading to a partial motorization of the mutant neocortex (Armentano et al, 2007; Greig et al, 2016). Interestingly, wild‐type Bcl6 expression is lower in the somatomotor cortex as in the somatosensory cortex. Correspondingly, numbers of neurons eliminated by DCD are substantially higher in the somatomotor as compared to the somatosensory cortex (Blanquie et al, 2017). Nr2f1 might thus indirectly participate in the control of Bcl6 expression through the control of cortical area identity. Whether this occurs through direct protein interaction with Bcl11a or indirectly through mechanisms independent of Bcl11a remains to be determined.
Materials and Methods
Animals
Mice carrying a conditional knockout allele of Bcl11a (Bcl11a F ) have previously been described (John et al, 2012). These mice were crossed to Emx1 IRESCre (Gorski et al, 2002), Nex Cre (Goebbels et al, 2006), or Deleter Cre (Schwenk et al, 1995) mice to generate conditional Bcl11a F/F ;Emx1 IRESCre , conditional Bcl11a F/F ;Nex Cre , and Bcl11a Δ/+ heterozygous mutants, respectively. Bcl11a F/+ ;Emx1 IRESCre littermates were phenotypically indistinguishable from Bcl11a +/+ ;Emx1 IRESCre animals (Appendix Fig S4A and B) and served as controls throughout the study. Mice carrying a conditional knockout allele of Bcl6 (Bcl6 F ; Hollister et al, 2013) were crossed to Nex Cre mice to generate conditional Bcl6 F/F ;Nex Cre mutants. Bcl6 F/F littermates without a Nex Cre allele served as controls. Bcl6 +/− mice have previously been described (Ye et al, 1997). Genotyping of the mice was performed by PCR. Animals were kept in a 12:12‐h light–dark cycle and at a constant temperature (22 ± 1°C) in IVC cages. All mouse experiments were carried out in compliance with German law and approved by the respective government offices in Tübingen, Germany.
Immunohistochemistry and RNA in situ hybridization
Brains were fixed in 4% PFA in 0.1 M phosphate buffer (pH 7.4), embedded in OCT compound (Polysciences), and frozen sections were prepared at 14 μm for immunohistochemistry or 18 μm for RNA in situ hybridization as previously described (John et al, 2012; Simon et al, 2012). Paraffin and vibratome sections were prepared at 7 and 50 μm, respectively. All clones for non‐radioactive RNA in situ hybridization, except for Flrt2 and Flrt3, which were a gift by Rüdiger Klein (Max Planck Institute of Neurobiology, Martinsried, Germany), were generated by reverse transcription PCR and oligonucleotides are listed in Table EV1.
The following antibodies were used: guinea pig anti‐Bcl11a at 1:1,000 dilution (John et al, 2012), mouse anti‐Bcl11a at 1:1,000 dilution (Abcam Cat# ab19487, RRID:AB_444947), rabbit anti‐Bcl11a at 1:1,000 dilution (John et al, 2012), rat anti‐Bcl11b at 1:1,000 dilution (Abcam Cat# ab18465, RRID:AB_2064130), rabbit anti‐cleaved Caspase 3 at 1:300 dilution (Cell Signaling Technology Cat# 9661, RRID:AB_2341188), rabbit anti‐Cux1 at 1:500 dilution (Santa Cruz Biotechnology Cat# sc‐13,024, RRID:AB_2261231), chicken anti‐GFP at 1:2,000 dilution (Abcam Cat# ab13970, RRID:AB_300798), mouse anti‐Satb2 at 1:1,000 dilution (Abcam Cat# ab51502, RRID:AB_882455), and rabbit anti‐Tbr1 at 1:500 dilution (Abcam Cat# ab31940, RRID:AB_2200219). To generate anti‐Bcl6 antiserum, guinea pigs were injected with a protein comprising amino acids 4–484 of mouse Bcl6 (NP_033874) and pooled sera were purified by affinity chromatography and used at 1:1,000 dilution. Biotin‐conjugated, HRP‐conjugated, and fluorescent secondary antibodies were purchased from Jackson ImmunoResearch and used at 1:500 dilution. Sections were counterstained with Dapi (Molecular Probes). Immunohistochemical detection of Bcl6 was performed on paraffin sections with antigen retrieval by boiling the section for 30 min in Tris‐EDTA buffer, pH 9.0 and enhanced using tyramide signal amplification (Invitrogen) according to the manufacturer's instructions or an avidin/biotin‐based peroxidase system and DAB substrate (Vector Laboratories). Cleaved caspase 3 was detected on frozen sections of conditional Bcl6 mutants using an avidin/biotin‐based peroxidase system and DAB substrate (Vector Laboratories). All fluorescent images were examined on a TCS SP5II confocal microscope (Leica) and processed with Adobe Photoshop (RRID:SCR_014199) software.
Laser microdissection
All procedures were performed in an RNase‐free environment. Cortical layers 2–4 were isolated from unfixed frozen sections via laser microdissection. Briefly, brains were quickly removed from the skull, washed in ice‐cold PBS, frozen in OCT compound (Polysciences), and stored at −80°C. Sections were prepared at 20 μm and mounted on membrane‐covered 1 mm PEN slides (Zeiss) that were UV‐treated and coated with poly‐L‐lysine. Sections were fixed in ice‐cold 70% EtOH for 1 min, incubated in 1% cresyl violet acetate solution (Waldeck) for 45 s, and washed in 70% EtOH and 100% EtOH for 1 min each. After a brief drying step on a 37°C warming plate, sections were immediately processed for laser microdissection using a PALM MicroBeam Rel.4.2 (Zeiss). Laser‐microdissected tissue was lysed in RLT lysis buffer (Qiagen) containing 2‐mercaptoethanol for 30 min on ice and stored at −80°C before total RNA extraction.
Plasmids
CAG‐Ctl GFP and CAG‐Cre GFP have previously been described (Hand et al, 2005; Wiegreffe et al, 2015). Bcl11a (NM_001242934) and Bcl11b (NM_001079883) were cloned by PCR using cDNA as template and inserted into CAG‐Ctl GFP to generate CAG‐Bcl11a GFP and CAG‐Bcl11b GFP , respectively. The recombinase Cre was from CAG‐Cre GFP and inserted into pNeuroD‐ires‐GFP (gift of Franck Polleux; RRID:Addgene_61403) to generate NeuroD‐Cre. The ires‐GFP cassette was cut from CAG‐Ctl GFP and inserted into pCALNL‐GFP (gift of Connie Cepko; RRID:Addgene_13770) to generate CAG‐LSL‐Ctl GFP . Bcl6 (NM_009744) was cloned by PCR using a cDNA clone (OriGene Tec. Cat.# MC203091) as template and inserted into CAG‐LSL‐Ctl GFP to generate CAG‐LSL‐Bcl6 GFP . CAG‐Cre was a gift of Connie Cepko (RRID:Addgene_13775). Foxo1 (NM_019739) was cloned by PCR using a cDNA clone (OriGene Tec. Cat.# MC203091) as template and inserted into pCAG‐DsRed2‐FLAG (gift of Franck Polleux) to generate CAG‐Foxo1 FLAG . Foxo1‐shRNA GFP #2 and #4 were generated by cloning published shRNAs directed against Foxo1 (Zhang et al, 2011; Park et al, 2019) into the short hairpin vector, pCA‐b‐EGFPm5‐silencer‐3 (gift of Matthieu Vermeren) using the oligonucleotide sequences listed in Table EV2. Ctl‐shRNA GFP was generated by cloning a scrambled sequence (5′‐TACGCGCATAAGATTAGGG‐3′) with no significant homology to any known gene sequence from mouse or human (Kawauchi et al, 2006) into pCA‐b‐EGFPm5‐silencer‐3.
In utero electroporation
In utero electroporation was performed as previously described (Saito & Nakatsuji, 2001; Wiegreffe et al, 2017) with minor modifications. Briefly, pregnant dams were anesthetized with Isoflurane (Abbott) and 1–2 μl of plasmid DNA were injected per embryo at a concentration of 0.5–1.0 μg/μl per construct. Five millimeter electrodes (Nepagene) and five pulses of 40 V (50 ms ON, 950 ms OFF) generated by a CUY21 EDIT electroporator (Nepagene) were used to transfect cells in the dorsolateral ventricular zone.
Microarray analysis, GO enrichment analysis, and quantitative real‐time PCR
Microarray analysis was performed as previously described (John et al, 2012; Simon et al, 2012) with minor modifications. Briefly, total RNA was isolated from laser‐microdissected control and mutant samples (n = 4) using the RNeasy Micro Plus Kit (Qiagen). The isolated RNA was checked for purity and integrity using Nanodrop spectrophotometer and TapeStation (Agilent), respectively. Transcriptome analysis was performed using GeneChip Mouse Gene 1.0 ST Arrays (Affymetrix) and BRB‐ArrayTools developed by Dr. Richard Simon and BRB‐ArrayTools Development Team (http://linus.nci.nih.gov/BRB‐ArrayTools.html).
DE genes identified by microarray analysis were subjected to a gene ontology (GO) enrichment analysis using PANTHER version 15.0 (released 2020‐02‐14) and overrepresentation test (released 2020‐02‐28) with default settings and mouse whole‐genome as reference list (Mi et al, 2013).
Total RNA was reverse transcribed using the SensiFast cDNA Synthesis Kit (Bioline), and quantitative real‐time PCR was performed using the LightCycler DNA Master SYBR Green I Kit in a LightCycler 480 System (Roche). Oligonucleotides used for quantitative real‐time PCR are listed in Table EV1. The relative copy number of Gapdh RNA was quantified and used for normalization. Data were analyzed using the comparative C T method (Schmittgen & Livak, 2008).
Chromatin immunoprecipitation and luciferase assay
Chromatin immunoprecipitation (ChIP) was carried out as previously described (Nelson et al, 2006) with minor modifications. Briefly, P0 cortical tissue was collected from wild‐type pups, flash‐frozen in liquid nitrogen, and stored at −80°C until ChIP. Tissue was disrupted in low sucrose buffer (320 mM sucrose, 10 mM HEPES, pH 8.0, 5 mM CaCl2, 3 mM Mg[CH3COO]2, 1 mM DTT, 0.1 mM EDTA, 0.1% Triton X‐100) and fixed for 15 min at RT in 1% formaldehyde. After quenching with glycine solution, nuclei were washed in Nelson buffer (140 mM NaCl, 20 mM EDTA, pH 8.0, 50 mM Tris, pH 8.0, 1% Triton X‐100, 0.5% NP‐40) and disrupted in RIPA buffer (140 mM NaCl, 10 mM Tris, pH 8.0, 1 mM EDTA, pH 8.0, 1% SDS, 1% Triton X‐100, 0.1% NaDOC). Chromatin was sonicated for 40 cycles (30 s ON/OFF) using a Bioruptor Plus (Diagenode) with high power settings. For each ChIP reaction, 15 μg of sheared chromatin was diluted 10 times with IP buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP‐40, 0.5% NaDOC, 20 mM EDTA, pH 8.0, 0.1% SDS) and incubated overnight at 4°C with 3 μl specific mouse monoclonal antibody recognizing Bcl11a (Abcam Cat# ab19487, RRID:AB_444947) or unspecific IgG1 antibody (Cell Signaling Technology Cat# 5415, RRID:AB_10829607), which served as a negative control. Twenty microliter of protein G magnetic beads (Invitrogen) were added to each ChIP reaction for 2 h at 4°C. After washing with IP buffer containing 0.1% SDS, LiCl buffer (500 mM LiCl, 100 mM Tris, pH 8.0, 1% NP‐40, 1% NaDOC, 20 mM EDTA, pH 8.0), and TE buffer (10 mM Tris, pH 8.0, 1 mM EDTA, pH 8.0), DNA was eluted from beads and purified by phenol‐chloroform extraction. The precipitated DNA was analyzed by quantitative real‐time PCR using oligonucleotides recognizing a region containing a conserved Bcl11a binding motif (TGACCA) in the first intron of Bcl6. As negative controls, oligonucleotides were used recognizing a region of exon 5 of Bcl6 and the Hprt promoter region, respectively. All oligonucleotide sequences are listed in Table EV1. ChIP quantitative real‐time PCR data were analyzed by the comparative C T method determining the fold enrichment of the immunoprecipitated DNA by the specific antibody versus IgG1 using the input as a reference.
The 93‐bp region containing the conserved Bcl11a binding motif in the first intron of Bcl6 was cloned into a Gaussia luciferase (GLuc) reporter vector containing a minimal CMV promoter (pEZX‐GN03; Genecopoeia). This construct was transfected into HEK293 cells (ATCC Cat# PTA‐4488, RRID:CVCL_0045) with CAG‐Ctl GFP or CAG‐Bcl11a GFP using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). A reporter vector containing a 112‐bp region of exon 5 of Bcl6 and CAG‐Bcl11b GFP was transfected as a control. pCMV‐SEAP (secreted alkaline phosphatase) was co‐transfected in each well as a transfection control. Supernatant from transfected cells was analyzed 48 h after transfection. Luciferase assays were performed using the Secrete‐Pair Dual Luminescence Assay Kit (Genecopoeia) in accordance with the manufacturer's instructions and a SpectraMax i3x instrument (Molecular Devices). Values are reported as the mean ratio of luminescence intensity of GLuc over SEAP and were collected from four independent experiments performed with at least two replicates per experiment.
Cell culture and western blot
HEK293 cells (ATCC Cat# PTA‐4488, RRID:CVCL_0045) were grown in DMEM with 10% fetal calf serum and 1% penicillin/streptomycin at 37°C under 5% CO2 atmosphere. Cells were transfected using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). After 48 h, total proteins were extracted with ice‐cold lysis buffer (1% NP‐40, 150 mM NaCl, 50 mM Tris, pH 8.0, 1 mM EDTA), separated by SDS–PAGE, and electrophoretically transferred onto PVDF membranes (Amersham). Membranes were blocked with 5% non‐fat milk (Bio‐Rad) and incubated with mouse anti‐β‐actin (Abcam Cat# ab8226, RRID:AB_306371), rabbit anti‐Bcl6 (Santa Cruz Biotechnology Cat# sc‐858, RRID:AB_2063450), rabbit anti‐FLAG (Sigma‐Aldrich Cat# F7425, RRID:AB_439687), rabbit anti‐Gapdh (Sigma‐Aldrich Cat# G9545, RRID:AB_796208), and chicken anti‐GFP (Abcam Cat# ab13970, RRID:AB_300798), followed by treatment with horseradish peroxidase‐conjugated secondary antibodies (Jackson ImmunoResearch) and ECL Plus western blotting detection reagents (ThermoScientific) or DAB substrate (Vector Laboratories) according to the manufacturers' instructions.
Cell counts and statistical analysis
For each experiment, at least three control and three mutant brains were analyzed, and three to five sections per brain were quantified. Anatomically matched sections were selected from an anterio‐posterior level between the anterior commissure and the dorsal hippocampus. Stained cells were counted in radial units of 100 μm (Figs 2, 3 and EV4), 350 μm (Fig 4C–F), or 750 μm (Figs 4H and I, and 5J and K) width in the presumptive somatosensory cortex or in the entire neocortex (Figs 5A and C, and EV5A and B, Appendix Fig S4). Cells were counted using ImageJ (RRID:SCR_003070) and Imaris (RRID:SCR:007370) software. Statistical analysis was done with Microsoft Excel (RRID:SCR_016137) or GraphPad Prism (RRID:SCR_002798) software. Venn diagrams were generated using MATLAB (RRID:SCR_001622) software. Significance between groups was assessed using a two‐tailed Student's t‐test or one‐way ANOVA, followed by Tuckey's post‐hoc test. P‐values < 0.05 were considered statistically significant.
Author contributions
Christoph Wiegreffe: Conceptualization; validation; investigation; visualization; methodology. Tobias Wahl: Investigation. Natalie Sophie Joos: Investigation. Jerome Bonnefont: Resources. Pentao Liu: Resources. Stefan Britsch: Conceptualization; supervision.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Dataset EV2
Source Data for Appendix
Acknowledgements
We are grateful to C. Cepko (Harvard Medical School, Boston), K.‐A. Nave (Max Planck Institute for Experimental Medicine, Göttingen), F. Polleux (Columbia University, New York) for the gift of mice and providing DNA plasmids. We thank K. Holzmann of the core facility “Genomics” and the staff of core facility “Laser Microdissection” of the Medical Faculty of Ulm University. We thank J. Andratschke, L. Schmid, and D. Krattenmacher for excellent technical assistance. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) to SB (BR 2215/1‐2), UULM | Medizinische Fakultät, Universität Ulm (Medical School, Ulm University) to CW (Bausteinprogramm 3.2) and the Studienstiftung des Deutschen Volkes (Studienstiftung) to TW. Open Access funding enabled and organized by Projekt DEAL.
EMBO reports (2022) 23: e54104
Data availability
The datasets produced in this study are available in the following databases:
-
i
microarray data: Gene Expression Omnibus GSE185287 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE185287).
-
ii
microarray data: Gene Expression Omnibus GSE185288 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE185288).
References
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Farinas I, Grosschedl R, McConnell SK (2008) Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 57: 364–377 [DOI] [PubMed] [Google Scholar]
- Armentano M, Chou SJ, Tomassy GS, Leingartner A, O'Leary DD, Studer M (2007) COUP‐TFI regulates the balance of cortical patterning between frontal/motor and sensory areas. Nat Neurosci 10: 1277–1286 [DOI] [PubMed] [Google Scholar]
- Avram D, Fields A, Senawong T, Topark‐Ngarm A, Leid M (2002) COUP‐TF (chicken ovalbumin upstream promoter transcription factor)‐interacting protein 1 (CTIP1) is a sequence‐specific DNA binding protein. Biochem J 368: 555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanquie O, Yang JW, Kilb W, Sharopov S, Sinning A, Luhmann HJ (2017) Electrical activity controls area‐specific expression of neuronal apoptosis in the mouse developing cerebral cortex. eLife 6: e27696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke AJ, Staley K, Chun J (1996) Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 122: 1165–1174 [DOI] [PubMed] [Google Scholar]
- Bonnefont J, Tiberi L, van den Ameele J, Potier D, Gaber ZB, Lin X, Bilheu A, Herpoel A, Velez Bravo FD, Guillemot F et al (2019) Cortical neurogenesis requires Bcl6‐mediated transcriptional repression of multiple self‐renewal‐promoting extrinsic pathways. Neuron 103: e1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan RC, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D et al (2008) Satb2 is a postmitotic determinant for upper‐layer neuron specification in the neocortex. Neuron 57: 378–392 [DOI] [PubMed] [Google Scholar]
- Carter ME, Brunet A (2007) FOXO transcription factors. Curr Biol 17: R113–R114 [DOI] [PubMed] [Google Scholar]
- Causeret F, Coppola E, Pierani A (2018) Cortical developmental death: selected to survive or fated to die. Curr Opin Neurobiol 53: 35–42 [DOI] [PubMed] [Google Scholar]
- Cerchietti LC, Polo JM, Da Silva GF, Farinha P, Shaknovich R, Gascoyne RD, Dowdy SF, Melnick A (2008) Sequential transcription factor targeting for diffuse large B‐cell lymphomas. Cancer Res 68: 3361–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury TG, Jimenez JC, Bomar JM, Cruz‐Martin A, Cantle JP, Portera‐Cailliau C (2010) Fate of cajal‐retzius neurons in the postnatal mouse neocortex. Front Neuroanat 4: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bruyckere E, Simon R, Nestel S, Heimrich B, Katzel D, Egorov AV, Liu P, Jenkins NA, Copeland NG, Schwegler H et al (2018) Stability and function of hippocampal mossy fiber synapses depend on Bcl11b/Ctip2. Front Mol Neurosci 11: 103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa EJ, de Pablo F (2000) Cell death in early neural development: beyond the neurotrophic theory. Trends Neurosci 23: 454–458 [DOI] [PubMed] [Google Scholar]
- Denaxa M, Neves G, Burrone J, Pachnis V (2018) Homeostatic regulation of interneuron apoptosis during cortical development. J Exp Neurosci 12: 1179069518784277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Wang Z, Guo R, Yang L, Liu G, Zhang Z, Xu Z, Tian Y, Yang Z, Li X et al (2021) Transcription factors Bcl11a and Bcl11b are required for the production and differentiation of cortical projection neurons. Cereb Cortex bhab437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson SH, Thom M, Heffernan J, Lin WR, Harding BN, Squier MV, Sisodiya SM (2001) Persistent reelin‐expressing Cajal‐Retzius cells in polymicrogyria. Brain 124: 1350–1361 [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA (2006) Genetic targeting of principal neurons in neocortex and hippocampus of NEX‐Cre mice. Genesis 44: 611–621 [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR (2002) Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1‐expressing lineage. J Neurosci 22: 6309–6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Greppi C, Macklis JD (2016) Ctip1 controls acquisition of sensory area identity and establishment of sensory input fields in the developing neocortex. Neuron 90: 261–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, Boutt E, Peters E, Barnes AP, Parras C et al (2005) Phosphorylation of Neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron 48: 45–62 [DOI] [PubMed] [Google Scholar]
- Hollister K, Kusam S, Wu H, Clegg N, Mondal A, Sawant DV, Dent AL (2013) Insights into the role of Bcl6 in follicular Th cells using a new conditional mutant mouse model. J Immunol 191: 3705–3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 677–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John A, Brylka H, Wiegreffe C, Simon R, Liu P, Juttner R, Crenshaw EB 3rd, Luyten FP, Jenkins NA, Copeland NG et al (2012) Bcl11a is required for neuronal morphogenesis and sensory circuit formation in dorsal spinal cord development. Development 139: 1831–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jontes JD (2018) The cadherin superfamily in neural circuit assembly. Cold Spring Harb Perspect Biol 10: a029306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M (2006) Cdk5 Phosphorylates and stabilizes p27kip1 contributing to Actin organization and cortical neuronal migration. Nat Cell Biol 8: 17–26 [DOI] [PubMed] [Google Scholar]
- Koropouli E, Kolodkin AL (2014) Semaphorins and the dynamic regulation of synapse assembly, refinement, and function. Curr Opin Neurobiol 27: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA (1996) Decreased apoptosis in the brain and premature lethality in CPP32‐deficient mice. Nature 384: 368–372 [DOI] [PubMed] [Google Scholar]
- László ZI, Bercsényi K, Mayer M, Lefkovics K, Szabó G, Katona I, Lele Z (2020) N‐cadherin (Cdh2) maintains migration and postmitotic survival of cortical interneuron precursors in a cell‐type‐specific manner. Cereb Cortex 30: 1318–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledonne F, Orduz D, Mercier J, Vigier L, Grove EA, Tissir F, Angulo MC, Pierani A, Coppola E (2016) Targeted inactivation of Bax reveals a subtype‐specific mechanism of Cajal‐Retzius neuron death in the postnatal cerebral cortex. Cell Rep 17: 3133–3141 [DOI] [PubMed] [Google Scholar]
- Liu H, Ippolito GC, Wall JK, Niu T, Probst L, Lee BS, Pulford K, Banham AH, Stockwin L, Shaffer AL et al (2006) Functional studies of BCL11A: characterization of the conserved BCL11A‐XL splice variant and its interaction with BCL6 in nuclear paraspeckles of germinal center B cells. Mol Cancer 5: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias‐Trevino C, Rogers JM, Kurita R et al (2018) Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell 173: e417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, Thomas PD (2013) Large‐scale gene function analysis with the PANTHER classification system. Nat Protoc 8: 1551–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD (2007) Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci 8: 427–437 [DOI] [PubMed] [Google Scholar]
- Nakamura A, Swahari V, Plestant C, Smith I, McCoy E, Smith S, Moy SS, Anton ES, Deshmukh M (2016) Bcl‐xL is essential for the survival and function of differentiated neurons in the cortex that control complex behaviors. J Neurosci 36: 5448–5461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Yamazaki Y, Saiki Y, Moriyama M, Largaespada DA, Jenkins NA, Copeland NG (2000) Evi9 encodes a novel zinc finger protein that physically interacts with BCL6, a known human B‐cell proto‐oncogene product. Mol Cell Biol 20: 3178–3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JD, Denisenko O, Bomsztyk K (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1: 179–185 [DOI] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA (2004) Expression of Cux‐1 and Cux‐2 in the subventricular zone and upper layers II‐IV of the cerebral cortex. J Comp Neurol 479: 168–180 [DOI] [PubMed] [Google Scholar]
- Park MK, Yao Y, Xia W, Setijono SR, Kim JH, Vila IK, Chiu HH, Wu Y, Billalabeitia EG, Lee MG et al (2019) PTEN self‐regulates through USP11 via the PI3K‐FOXO pathway to stabilize tumor suppression. Nat Commun 10: 636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan RT, Dalla‐Favera R (2004) The BCL6 proto‐oncogene suppresses p53 expression in germinal‐Centre B cells. Nature 432: 635–639 [DOI] [PubMed] [Google Scholar]
- Phan RT, Saito M, Basso K, Niu H, Dalla‐Favera R (2005) BCL6 interacts with the transcription factor Miz‐1 to suppress the cyclin‐dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol 6: 1054–1060 [DOI] [PubMed] [Google Scholar]
- Priya R, Paredes MF, Karayannis T, Yusuf N, Liu X, Jaglin X, Graef I, Alvarez‐Buylla A, Fishell G (2018) Activity regulates cell death within cortical interneurons through a calcineurin‐dependent mechanism. Cell Rep 22: 1695–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranuncolo SM, Polo JM, Dierov J, Singer M, Kuo T, Greally J, Green R, Carroll M, Melnick A (2007) Bcl‐6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA‐damage sensor ATR. Nat Immunol 8: 705–714 [DOI] [PubMed] [Google Scholar]
- Roth KA, Kuan C, Haydar TF, D'Sa‐Eipper C, Shindler KS, Zheng TS, Kuida K, Flavell RA, Rakic P (2000) Epistatic and independent functions of caspase‐3 and Bcl‐X(L) in developmental programmed cell death. Proc Natl Acad Sci USA 97: 466–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N (2001) Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol 240: 237–246 [DOI] [PubMed] [Google Scholar]
- Sanes JR, Zipursky SL (2020) Synaptic specificity, recognition molecules, and assembly of neural circuits. Cell 181: 536–556 [DOI] [PubMed] [Google Scholar]
- Santo EE, Paik J (2018) FOXO in neural cells and diseases of the nervous system. Curr Top Dev Biol 127: 105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108 [DOI] [PubMed] [Google Scholar]
- Schwenk F, Baron U, Rajewsky K (1995) A cre‐transgenic mouse strain for the ubiquitous deletion of loxP‐flanked gene segments including deletion in germ cells. Nucleic Acids Res 23: 5080–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Brylka H, Schwegler H, Venkataramanappa S, Andratschke J, Wiegreffe C, Liu P, Fuchs E, Jenkins NA, Copeland NG et al (2012) A dual function of Bcl11b/Ctip2 in hippocampal neurogenesis. EMBO J 31: 2922–2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Wiegreffe C, Britsch S (2020) Bcl11 transcription factors regulate cortical development and function. Front Mol Neurosci 13: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell DG, Paredes MF, Galvao RP, Jones DL, Froemke RC, Sebe JY, Alfaro‐Cervello C, Tang Y, Garcia‐Verdugo JM, Rubenstein JL et al (2012) Intrinsically determined cell death of developing cortical interneurons. Nature 491: 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiberi L, Bonnefont J, van den Ameele J, Le Bon SD, Herpoel A, Bilheu A, Baron BW, Vanderhaeghen P (2014) A BCL6/BCOR/SIRT1 complex triggers neurogenesis and suppresses medulloblastoma by repressing sonic hedgehog signaling. Cancer Cell 26: 797–812 [DOI] [PubMed] [Google Scholar]
- Tiberi L, van den Ameele J, Dimidschstein J, Piccirilli J, Gall D, Herpoel A, Bilheu A, Bonnefont J, Iacovino M, Kyba M et al (2012) BCL6 controls neurogenesis through Sirt1‐dependent epigenetic repression of selective notch targets. Nat Neurosci 15: 1627–1635 [DOI] [PubMed] [Google Scholar]
- Tocco C, Bertacchi M, Studer M (2021) Structural and functional aspects of the neurodevelopmental gene NR2F1: From animal models to human pathology. Front Mol Neurosci 14: 767965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verney C, Takahashi T, Bhide PG, Nowakowski RS, Caviness VS Jr (2000) Independent controls for neocortical neuron production and histogenetic cell death. Dev Neurosci 22: 125–138 [DOI] [PubMed] [Google Scholar]
- Wei H, Alberts I, Li X (2014) The apoptotic perspective of autism. Int J Dev Neurosci 36: 13–18 [DOI] [PubMed] [Google Scholar]
- Wiegreffe C, Feldmann S, Gaessler S, Britsch S (2017) Time‐lapse confocal imaging of migrating neurons in organotypic slice culture of embryonic mouse brain using in utero electroporation. J Vis Exp 55886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegreffe C, Simon R, Peschkes K, Kling C, Strehle M, Cheng J, Srivatsa S, Liu P, Jenkins NA, Copeland NG et al (2015) Bcl11a (Ctip1) controls migration of cortical projection neurons through regulation of Sema3c. Neuron 87: 311–325 [DOI] [PubMed] [Google Scholar]
- Wong FK, Bercsenyi K, Sreenivasan V, Portales A, Fernandez‐Otero M, Marin O (2018) Pyramidal cell regulation of interneuron survival sculpts cortical networks. Nature 557: 668–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong FK, Marin O (2019) Developmental cell death in the cerebral cortex. Annu Rev Cell Dev Biol 35: 523–542 [DOI] [PubMed] [Google Scholar]
- Woodworth MB, Greig LC, Liu KX, Ippolito GC, Tucker HO, Macklis JD (2016) Ctip1 regulates the balance between specification of distinct projection neuron subtypes in deep cortical layers. Cell Rep 15: 999–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye BH, Cattoretti G, Shen Q, Zhang J, Hawe N, de Waard R, Leung C, Nouri‐Shirazi M, Orazi A, Chaganti RS et al (1997) The BCL‐6 proto‐oncogene controls germinal‐Centre formation and Th2‐type inflammation. Nat Genet 16: 161–170 [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Steijaert MN, Lau D, Schutz G, Delucinge‐Vivier C, Descombes P, Bading H (2007) Decoding NMDA receptor signaling: Identification of genomic programs specifying neuronal survival and death. Neuron 53: 549–562 [DOI] [PubMed] [Google Scholar]
- Zhang X, Yalcin S, Lee DF, Yeh TY, Lee SM, Su J, Mungamuri SK, Rimmele P, Kennedy M, Sellers R et al (2011) FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat Cell Biol 13: 1092–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]